 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoengineering ABO3 active sites from low-energy routes (TX100-stabilised water-in-oil microemulsions, surface segregation and surface complexation on colloidal AlOOH/sol–gel Al2O3 surfaces) for pollution control catalysis
M. P.
Worsley
a,
P. N.
Forrest
b,
S.
Roesch
b,
C.
Thatcher
b,
P. A.
Sermon
*a and
P.
Kaur
c
aNanomaterials Laboratory, Wolfson Centre, Brunel University, Uxbridge, Middx. UB8 3PH, UK. E-mail: paul.sermon@brunel.ac.uk
bDepartment of Chemistry, University of Surrey, Guildford, GU2 7XH, UK
cHoriba MIRA Ltd, Nuneaton, CV10 0TU, UK
First published on 8th May 2018
Abstract
It is shown that water-in-oil microemulsions (m/e or μE) can produce BaCeO3 (BCO) and LaCoO3 (LCO) precursors. The nanoparticles (NPs) adsorb on AlOOH sols, in much the same way as Turkevich previously immobilised platinum group metal sols. BCO is active in CO and propane oxidation and NO removal under stoichiometric exhaust conditions, but LCO is a better oxidation catalyst. Activity was also seen when Ba,Ce and La,Co are inserted into/segregate at the surface of AlOOH/Al2O3. However, there is only formation of low levels of BCO, CAIO3 (CAO), LCO and LaAIO3 (LAO) perovskites, along with aluminates and separate oxides. The complexing of cations by AlOOH surface-held oxalate ions, albeit with different efficiencies, has also been explored. All three routes yield active catalysts with micro-domains of crystallinity; microemulsions produce the best defined perovskite NPs, but even those from surface segregation have higher turnover numbers than traditional Pt catalysts. Perovskite NPs may open up green chemistry for air pollution control that is consistent with a circular economy.
1. Introduction
The authors were intrigued by Turkevich’s historic reports of adsorbing and supporting precious metal colloidal particles on AlOOH boehmite,1 taking advantage of electrostatic attraction, after which transmission electron microscopy (TEM) characterised the surface-held discrete NPs and in parallel catalytic activity could be measured directly. They wanted to see whether the approach might be applied to perovskites.2Routes to perovskites can be high-3 and low-temperature/energy4–8 (e.g. sol–gel, microwave-assisted sol–gel, electrospinning, hydrothermal synthesis, microemulsion) and mechanochemical.9 Some offer opportunities to prepare nanoparticles (NPs; d < 100 nm) with different structures: ultrasonics,10 microwaves (MW)11 and chelators (oxalic acid,12 citric acid13 and tartrate14), sometimes in one-step preparations.15 Oxide, halide and oxyhalide perovskite16 nanoparticles (NPs) can all be produced by such routes. They are used in a wide range of applications:17–33 (i) photovoltaic solar cells, (ii) LEDs, photoelectrodes, battery anodes, H2O and CO2 electrochemistry, (iii) photocatalysis, (iv) catalysis (e.g. combustion of pollutant soot particles with MW regeneration, water gas shift, toluene oxidation, CO methanation and CO oxidation (where activity can be MW-enhanced)), (v) gas sensing, (vi) dielectric heating, (vii) magnetotransport and (viii) biomedicine (e.g. hyperthermia and drug delivery). In all these areas perovskite NPs are especially advantageous34 and may be enhanced further by zero-valent NPs,35 supports36 or templates (both inorganic37 or polymeric38).
Green catalytic chemistry and resource efficiency are at the heart of a minimum waste circular economy.39 It has long been thought that ABO3 perovskites40 had an advantage in a circular economy over precious metals, provided that their surface areas could be raised above a very modest 1.4 m2 g−1 (ref. 41) as reported by Libby, celebrated for his work on 14C radiocarbon dating, and this might be helped by supporting36 or incorporating a polymer or Al2O3.
Previously some of the present authors have attempted to nanoengineer catalysts: (a) produced colloidal PtxAu1−xAu and Pt–SnOy alloy sols that were adsorbed on carbon or Al2O3 surfaces,42 (b) synthesised43 sol–gel Pt–Sn–K/SiO2–Al2O3 hydrocarbon conversion catalysts containing K2Pt(OH)6 perovskite precursors44 and (c) taken [AlO4Al12(OH)24(H2O)12]7+ Keggin ions (whose formation was followed by 27Al solution NMR in terms of 4-coordinate AlO4 at about 60 ppm and 6-coordinate AlO6 at about 2.5 ppm as a function of prevailing OH−/Al3+ ratios) and partially replaced Al3+ with Fe3+ or Au3+ (ref. 45). These were adsorbed to a 5% loading on SiO2 surfaces. They showed a similar light-off temperature (LOT) for CO oxidation (870 K) to commercial platinum-group metal three-way catalysts (TWCs) (812 K) in a stoichiometric gas stream. Maximum% conversion of CO at 633 K was seen at Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe = 25:75, but these were thermally unstable.
:Fe = 25:75, but these were thermally unstable.
Now we have investigated whether the BaCeO3 (BCO) and LaAl1−xCoxO3 (LACO) perovskite active sites and the sol–gel host could be designed simultaneously to form intriguing environmental catalysts by:
(i) forming these as NPs in aqueous droplets in water-in-oil microemulsions and harvesting by a phase inversion step at 273 K (appreciating that these might be unstable during calcination),
(ii) causing them to form at the surface of colloidal AlOOH or sol–gel Al2O3 as the component concentrations were progressively raised beyond the point of maximum solubility in the supporting sol–gel matrix. Alumina was chosen because it is a frequent catalyst support that comes in γ, δ and θ forms.46 In the latter, nanowires can be seen in high resolution TEM (HRTEM). Boehmite AlOOH converts to γ-alumina at 723 K, then δ, then θ and finally the α-phase at 1473 K. It can form a defective cubic O2− close-packing spinel structure with 16 possible octahedral sites and 8 tetrahedral sites for Al3+ and other stabilising cations. One knows that there is a maximum solubility of such stabilising cations (La3+/Fe2+, Ba2+/Ce4+, and La3+/Co2+, etc.) beyond which phases are formed at the alumina surface. Alumina can be stabilised by Ce3+/4+ or Ba2+;47 the authors appreciated that dispersed CeO2 NPs (including octapods48) could be formed and Mx+-alumina reactions could produce spinel BaAl2O4,49 barium hexaluminates, Ce3+/4+AlOy phases50 or perovskite CeAlO3 (CAO)51 phases, and
(iii) adsorbing oxalate ions on the surface of peptized AlOOH needles and reacting these with the perovskite cations in aqueous solution to give an insoluble surface oxalate that decomposes on calcination.
Goldschmidt46 in the 1920s explored ABX3 perovskites. Orthorhombic BCO forms at 1170–1223 K (ref. 52) and LaFeO3 (LFeO) exhibited its most intense {002} X-ray reflections53 at 2θ = 28.68° and 2θ = 32.20°. Rhombohedral LCO prepared at 1013 K (ref. 54 and 55) has its most intense {110} and {104} reflections at 2θ = 34°.56 But HRTEM is useful in detecting nanoscale perovskite domains (e.g. in LCO57). Surface titrations (COT![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) –OT
–OT![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) )58 help probe active sites and oxygen-storage capacity (OSC)59 that may be introduced by CeO2 (ref. 60 and 61) or Tb4O7–CeO2,62,63 sometimes promoted by platinum-group metals.64 Here it is perovskite nanoparticles that are characterised by X-ray diffraction (XRD), Raman, HRTEM, UV/vis, temperature-programmed reduction/oxidation (TPR/TPO) and XPS. It is important that these methods differentiate perovskite NP phases from other by-product phases (e.g. aluminates65). After characterising these they were evaluated for CO (and hydrocarbon) oxidation (where rates might be different on different facets66). 28–118 nm LaAl1−xMnxO3 (LAMnO) and LaAl1−xFexO3 (LAFeO) perovskites are good catalysts.67,68 Cu-doped LaAlO3 (LAO)69 and LaCoO3 (LCO)70 are photocatalysts and electrochemically active.71 Partial Pd-replacement of Al3+ in LAO produces three-way catalytic (TWC) activity in vehicle exhaust conditions.72 LCO and LAO are formed at higher La and Co loadings in sol–gel alumina73 (where some say that LAO is an inert perovskite, but others suggest citrate-derived LAO is active in the oxidative-coupling of methane (OCM)74), but 42.2 nm La2CoAlO6 double perovskite is more active and has a lower light-off temperature (LOT; 707.1 K) in CH4 catalysed combustion75 than either LCO (763.6 K) or LAO (845.3 K).
)58 help probe active sites and oxygen-storage capacity (OSC)59 that may be introduced by CeO2 (ref. 60 and 61) or Tb4O7–CeO2,62,63 sometimes promoted by platinum-group metals.64 Here it is perovskite nanoparticles that are characterised by X-ray diffraction (XRD), Raman, HRTEM, UV/vis, temperature-programmed reduction/oxidation (TPR/TPO) and XPS. It is important that these methods differentiate perovskite NP phases from other by-product phases (e.g. aluminates65). After characterising these they were evaluated for CO (and hydrocarbon) oxidation (where rates might be different on different facets66). 28–118 nm LaAl1−xMnxO3 (LAMnO) and LaAl1−xFexO3 (LAFeO) perovskites are good catalysts.67,68 Cu-doped LaAlO3 (LAO)69 and LaCoO3 (LCO)70 are photocatalysts and electrochemically active.71 Partial Pd-replacement of Al3+ in LAO produces three-way catalytic (TWC) activity in vehicle exhaust conditions.72 LCO and LAO are formed at higher La and Co loadings in sol–gel alumina73 (where some say that LAO is an inert perovskite, but others suggest citrate-derived LAO is active in the oxidative-coupling of methane (OCM)74), but 42.2 nm La2CoAlO6 double perovskite is more active and has a lower light-off temperature (LOT; 707.1 K) in CH4 catalysed combustion75 than either LCO (763.6 K) or LAO (845.3 K).
2. Experimental
AlOOH synthesis
0.236 g AlOOH was prepared in 100 cm3 distilled H2O (initially at 353–358 K) by adding 3.9 mmol aluminium-tri-sec-butoxide (ASB, Aldrich, 97%), raising the reflux temperature to 403–413 K, adding 3.9 mmol HCl and refluxing for 48 h to reach a pH of 3.1. One fraction of this AlOOH dispersion was used directly; another was freeze-dried to an AlOOH powder that was calcined at 773 K and then 923 K. Dynamic light scattering (DLS) showed that the average particle size of the AlOOH sol was 60 nm (Fig. 1c); as this was calcined to higher temperatures it would inevitably transform (γ → δ → θ → α).46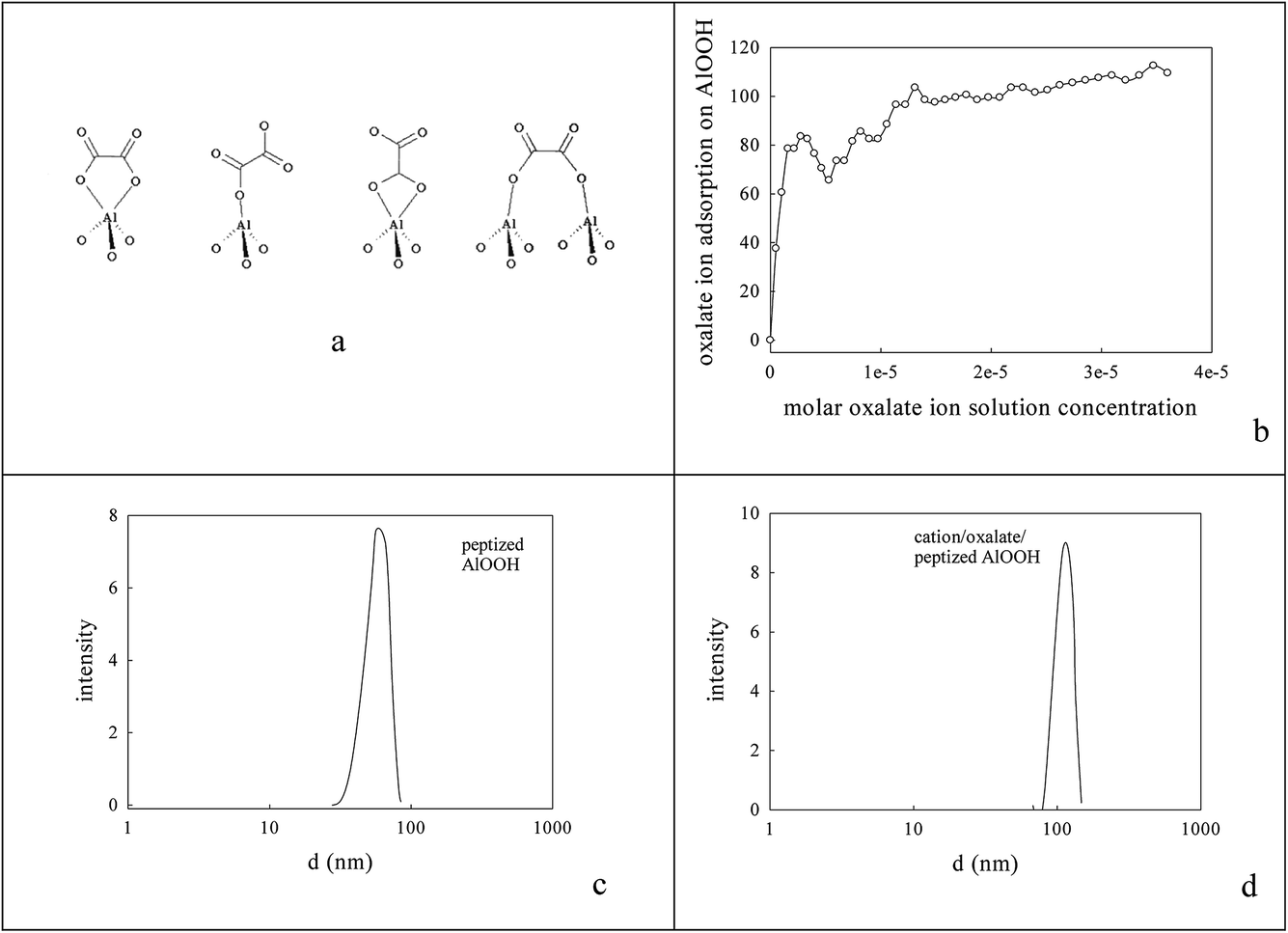 | ||
| Fig. 1 Nature of oxalate anion adsorption on AlOOH8,85 (a, b). DLS estimate of the average particle size in aqueous dispersions of (c) the peptized AlOOH (60 nm) and (d) after oxalate adsorption and formation of 3.65% cation/oxalate/AlOOH (114 nm). | ||
BCO and LCO synthesis by phase inversion at 273 K in a TX100-stabilised water-in-oil microemulsion
Here two homogenous 0.2 M TX100-stabilised water-in-cyclohexane (90%)/2-methyl-2-propanol (10%) microemulsions were used. In one μE a slight excess of ammonium oxalate was introduced to the dispersed water droplets. In another μE mixed 0.02 M La,Co nitrates (or mixed 0.02 M Ba,Ce nitrates) were introduced to the dispersed water droplets.76 The two w/o μEs at 298 K were then mixed to produce (as a result of droplet coalescence/re-dispersion mixed 5–10 nm (defined by the diameter of the aqueous droplet size)) Ba–Ce or La–Co oxalate nanoparticles (NPs). The combined microemulsion was then mixed with 50 cm3 of the pre-prepared AlOOH suspension to give Ba–Ce oxalate/AlOOH or La–Co oxalate/AlOOH that were harvested by lowering the combined μE to 273 K (defined by component melting points (280 K, 279.5 K, 273 K and 298–299 K for TX100, cyclohexane, water and 2-methyl-2-propanol)), when two phases appeared. The NP product was found in the upper organic phase. NPs adsorbed on AlOOH, were harvested, washed, dried and calcined (900 K).Surface segregation using AlOOH/Al2O3, Ce–AlOOH/Al2O3 and Ba,Ce–AlOOH/Al2O3
Surface segregation was attempted using dispersed colloidal AlOOH, where mixed metal cation pairs (Ba2+,Ce4+ or La3+,Co3+) were present in the AlOOH forming/peptizing solution/suspension sufficient to cause 25% Ba,Ce–AlOOH/Al2O3 and 25% La,Co–AlOOH/Al2O3 to form after calcination at 1173 K. Sol–gel–AlOOH/Al2O3 derived Ce- and Ba,Ce-containing catalysts77,78 were prepared from ASB, 2-methylpentane-2,4-diol (MPD, Aldrich, 99%), Ba(NO3)2–Ce(NO3)3 (Aldrich, 99.999%; dissolved in MPD) and deionised H2O78,79 as reported previously.45 For Ba,Ce–AlOOH/Al2O3 the Ba and Ce nitrates were dissolved in MPD at 360 K, followed by ASB and refluxing at 393 K for 6 h under N2. After cooling to 363 K, H2O was added dropwise over 25 min and refluxing was continued (2 h), followed by ageing at 373 K for 7 days, vacuum-drying (353 K) and then calcining (1173 K; 2 h; static air). Citrates, tartrates or acetates80,81 could have been used but nitrates were preferred. MPD was both a solvent82,83 and complexing agent80/pore templating agent. Fuentes83 derived La2O3–Al2O3 using MPD; here the metal:MPD:H2O ratio was 1:5:25. Sol–gel AlOOH/Al2O3 (BET surface area 179 m2 g−1) had a higher surface area than when Ce was added (e.g. 16.6% (ICP-MS derived) Ce–AlOOH/Al2O3 (BET surface area 168 m2 g−1)).
Surface complexation
The peptized AlOOH dispersion had a final pH of 3.1 (below its isoelectric point/point of zero charge (7.8–8.6)) and so it adsorbed oxalate anions84 on its surface e.g. see Fig. 1a and b; the % adsorption was deduced from the % differential change in suspension conductivity (100% × (σH2O − σAlOOH(aq))/σH2O) on sequential addition of 1 cm3 portions of 10 mM oxalic acid to 50 cm3 H2O or AlOOH(aq) respectively). After Ba2+,Ce4+ or La3+,Co3+ addition the average DLS particle size rose, almost doubling to 114 nm (Fig. 1c and d). This pre-adsorbed oxalate was titrated with Ba2+,Ce4+ or La3+,Co3+ forming a surface layer of low solubility mixed oxalates84 at an intended 5% loading. However, XPS (using Ba (780.7 eV), Ce (897.7 eV), La (834.7 eV) and Co (781.6 eV) peaks) and FAAS suggested that only a 3.65% metal loading on the oxalate/AlOOH was achieved with Ba:Ce and La:Co ratios on oxalate/AlOOH of 25:75 and 30:70. This was equivalent to the formation of 0.91% Ba, 2.74% Ce–AlOOH/Al2O3 and 1.095% La, 2.56% Co–AlOOH/Al2O3.
Characterization
Malvern DLS, Jeol HRTEM, X’Pert Pro XRD, Supra SEM-EDX and XPS characterization was carried out. TPR,85 titration and catalysis was followed as now described. Setaram and Thermo Scientific DSC profiles were used for CO (40 μmol) and O2 (47 μmol) titrations of catalysts in a flowing N2 (50 cm3 min−1) inert gas stream held isothermally at a selected temperature. Catalytic measurements were carried out as described previously.59 Samples (200 mg) were placed in a silica reactor (6 mm diameter) and heated in flowing N2 to 773 K for 20 min. Then after cooling to 298 K the stoichiometric reactant stream (NO + O2 oxidant/CO + propane reductant (R) ratio = 1.13, where the reactant stream contained 1000 ppm NO, 5890 ppm O2, 6000 ppm CO, 520 ppm propane, 10% CO2 in a N2 balance) was introduced at 500 cm3 min−1 (equivalent to a 60000 h−1 space velocity in a gasoline-fuelled engine exhaust) at 101 kPa under Brooks smart 5850S mass flow controllers. NDIR, FID-GC and CLD analysers were used for CO/CO2, propane and NOx concentrations in real time during heating from 300 K to 900 K at 10 K min−1.
3. Results
BCO and LCO microemulsion precipitation
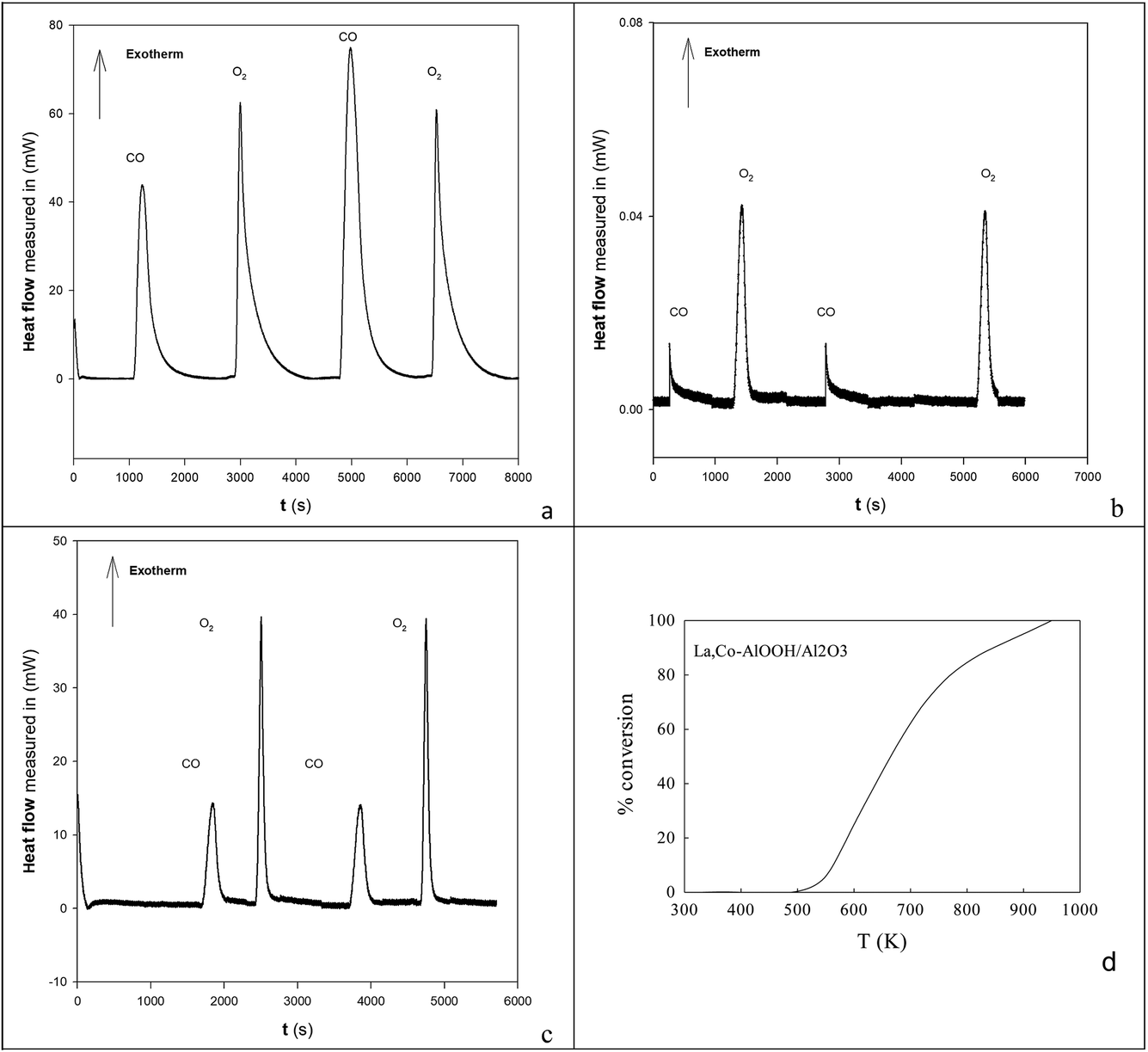
Fig. 2a shows the effect of the water content of TX100-stabilised water-in-cyclohexane/2-methyl-2-propanol μE on the partitioning of the surfactant between the water and hydrocarbon phases. There is continuous coalescence and separation of nm-sized water droplets, some containing a slight excess of ammonium oxalate and some metal cations. Fig. 2b and c show TEM evidence of the sizes of microemulsion-derived 10–20 nm BCO NPs (with 40–121 nm fractal nanothreads) and 10–21 nm LCO NPs (with and 43–108 nm fractal nano-chains) after phase inversion at 273 K. In Fig. 2d and e one sees the activity of such AlOOH-supported perovskite NPs under stoichiometric conditions as a function of temperature. Clearly BCO has activity in CO and propane oxidation and more surprisingly in NO conversion (under stoichiometric conditions), but LCO has higher CO and propane activity, but no NO reduction activity. This is promising.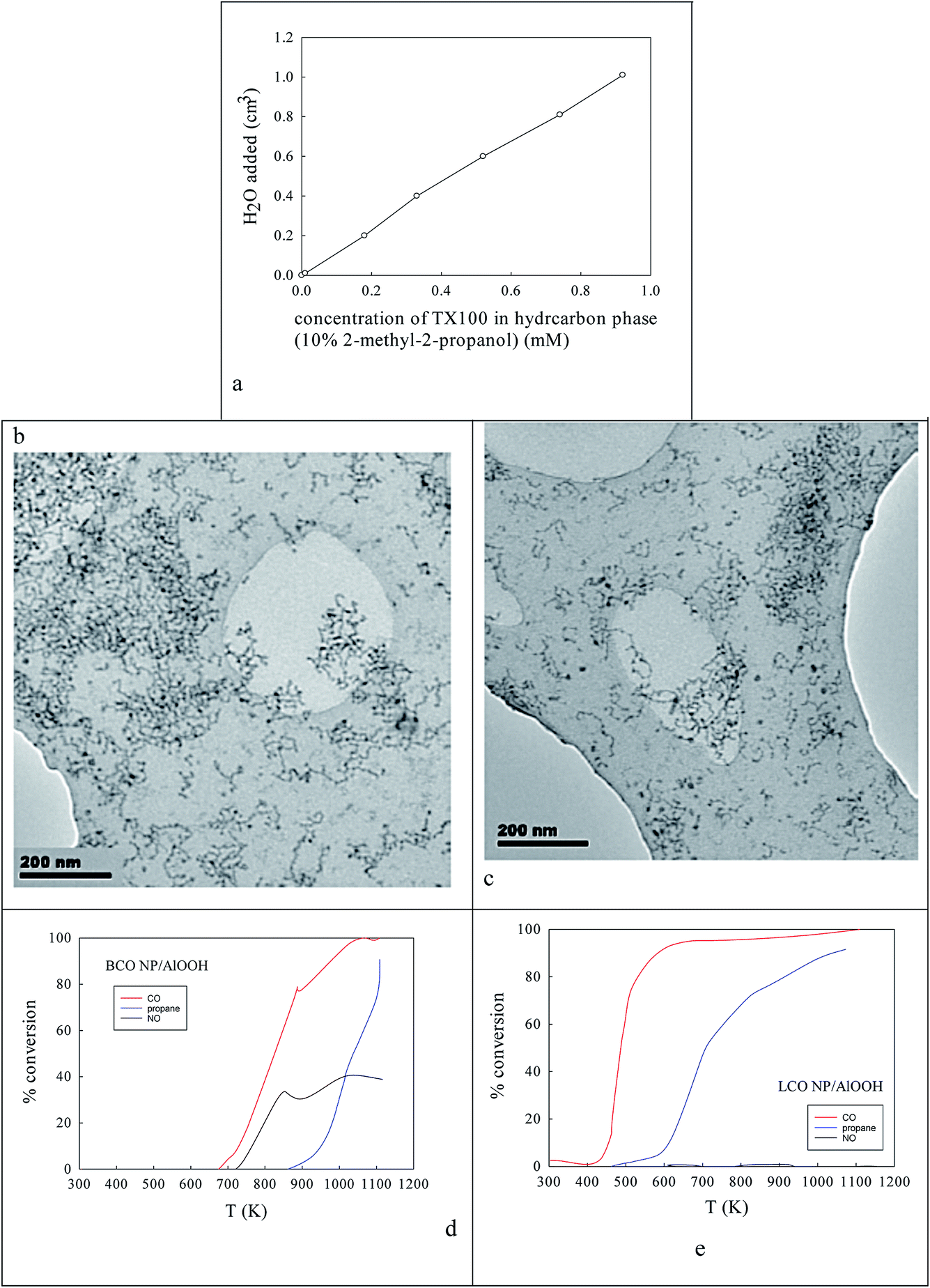 | ||
| Fig. 2 (a) Effect of H2O content on TX100 in the oil phase of a H2O-in-cyclohexane/2-methyl-2-propanol m/z. (b, c) TEM of m/z-derived BCO and LCO NPs and fractal aggregates. (d, e) The activity of such NPs adsorbed onto AlOOH. | ||
Ba,Ce and La,Co surface segregation59 on AlOOH/Al2O3
Clearly the ionic radius of Ce4+/3+ (87/101 pm) relative to O2− and Al3+ in part defines its solubility in AlOOH/Al2O3. Fig. 3 shows some characterization data before and after introduction of 20% Ce to the AlOOH/Al2O3. The sample surface area dropped by 6.1% on this Ce introduction, but HRTEM (Fig. 3a) saw 5 nm nano-crystallites with lattice spacings of 0.19 nm. XRD found after calcination the presence of θ-Al2O3, low levels of 10–12 nm γ-Al2O3 and CeO2, but there was no evidence of CAO. Interestingly, in H2-TPR a 720 K peak (α; H/CeO2 ratio = 0.156) was associated with alumina-dispersed CeO2,85,86 while a 900 K + (β) peak was thought to relate to CAO88 (that was too fine to be seen XRD) or Ce2O3. Fig. 3c shows that CeO2 addition raises the activity in CO conversion modestly above that of the alumina support only, despite the loss of surface area, but there is no HC or NO conversion.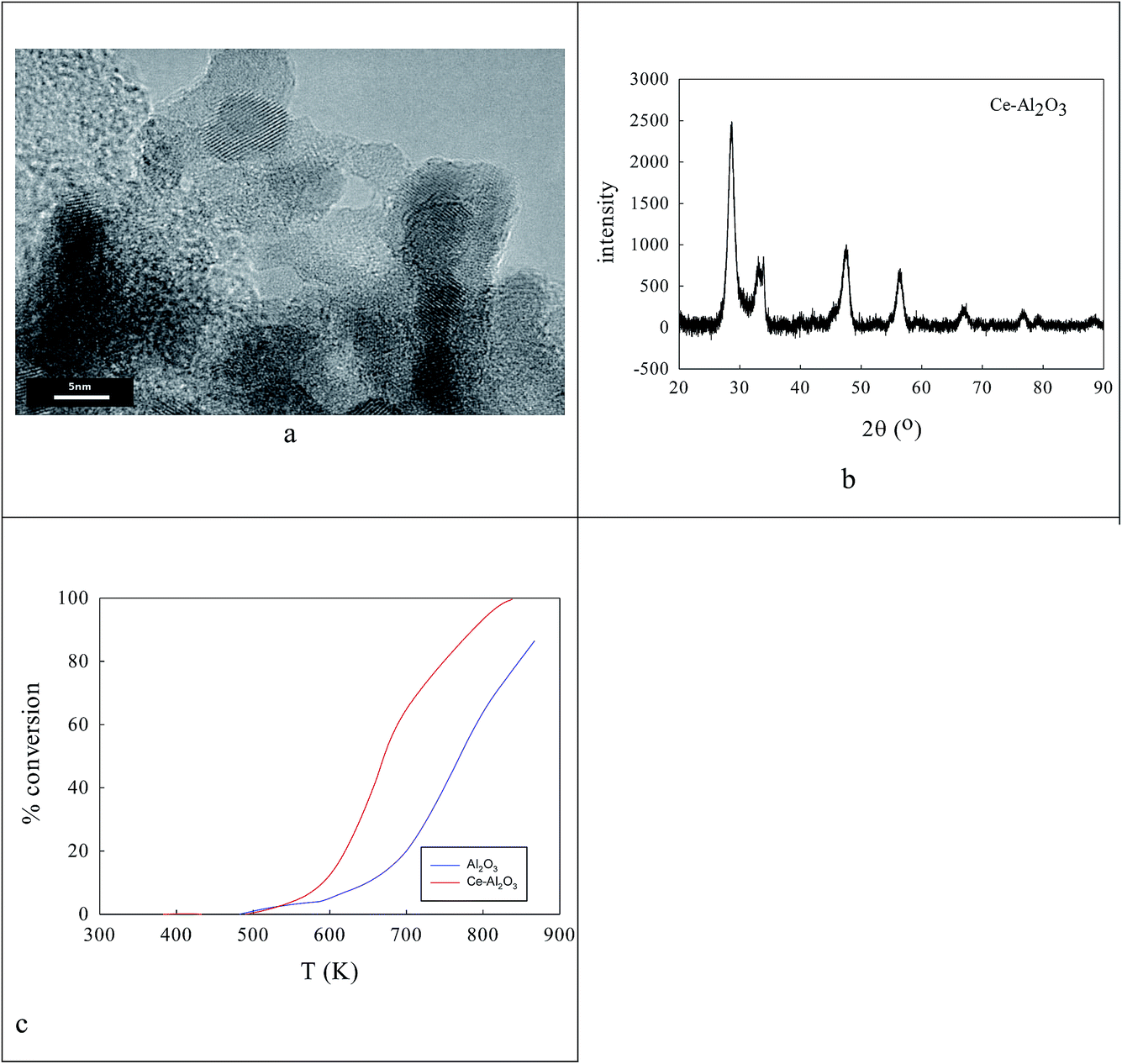 | ||
| Fig. 3 Well-dispersed sol–gel 20% CeO2–AlOOH (before calcination)/Al2O3 (after calcination) (168 m2 g−1) seen in (a) HRTEM and (b) XRD was slightly more active in CO oxidation under stoichiometric conditions (c) than the pure Al2O3 despite a 6.1% loss of surface area. HRTEM (a) shows 5 nm nano-crystallites (b). XRD (c) shows θ-Al2O3 and CeO2 (but not CAO). H2-TPR did, however, provide evidence in terms of a 900 K + β-peak for CAO. | ||
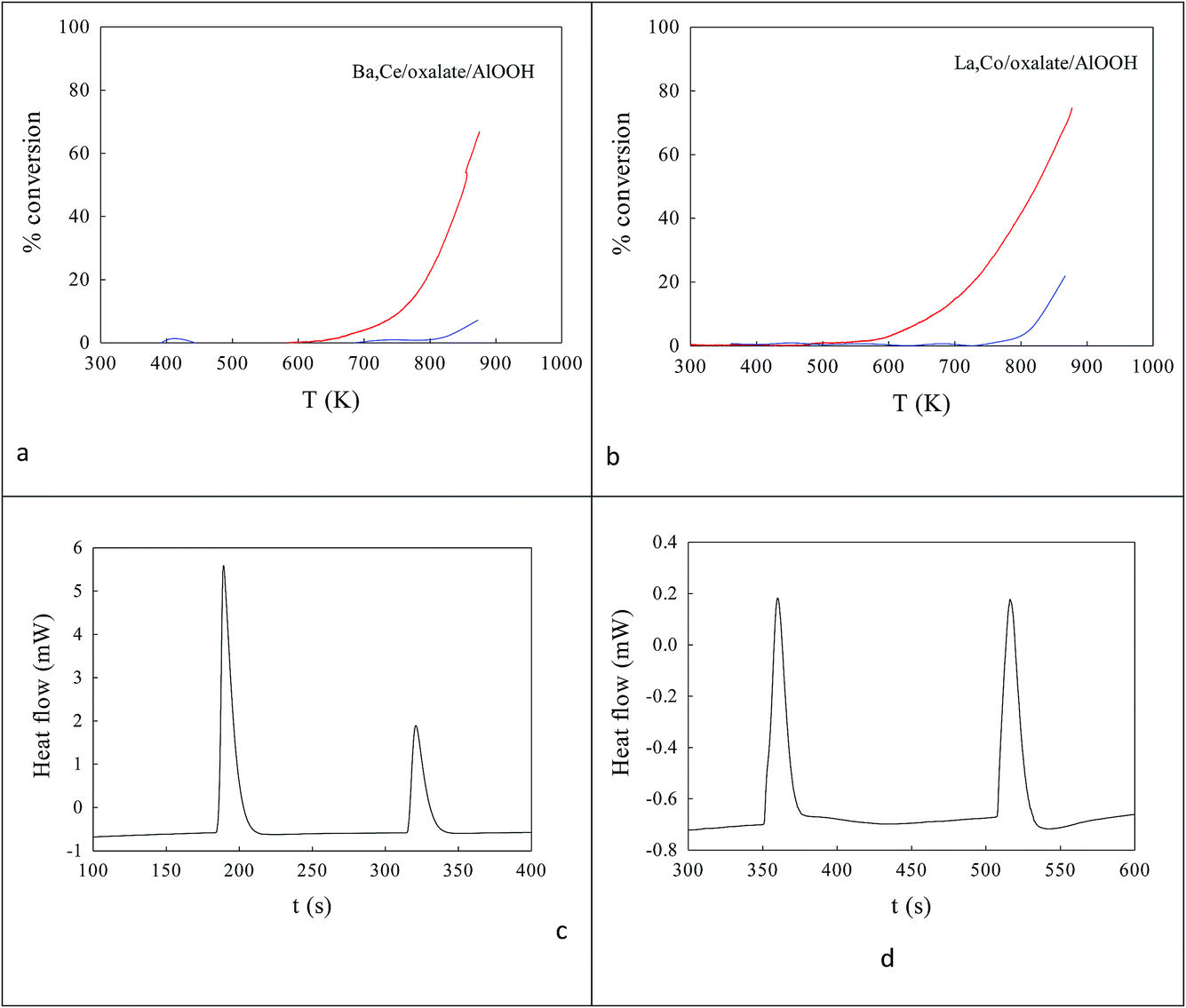
The ionic radius of Ba2+ (135 pm) is larger than that of Ce4+. As the Ba2+ and Ce4+ contents inserted into the AlOOH/Al2O3 each rise from 0 to 25%, it was expected that BaAl2O4 would appear, that CeO2 would segregate, but also that CAO or BCO might form in or at the surface of the host sol–gel matrix more strongly. It was found (see Fig. 4) that as the Ba,Ce content increased so HRTEM always showed 5–20 nm micro-domains of crystallinity (see Fig. 4a and b) and X-ray diffraction line broadening (XRDLB) (Fig. 4c) revealed unchanging and small average XRDLB crystallite sizes (dXRDLB) while MASNMR saw 6-coordinate Al3+ gradually replaced by 4-coordinate (as BaAl2O4 appeared), XRD peak intensities for BaAl2O4, CeO2 and CAO (Fig. 4c and d) increased, surface areas decreased by 8% (Fig. 4e), and total CO (and propane, but not NO) conversion activity at 650 K increased (Fig. 4f).
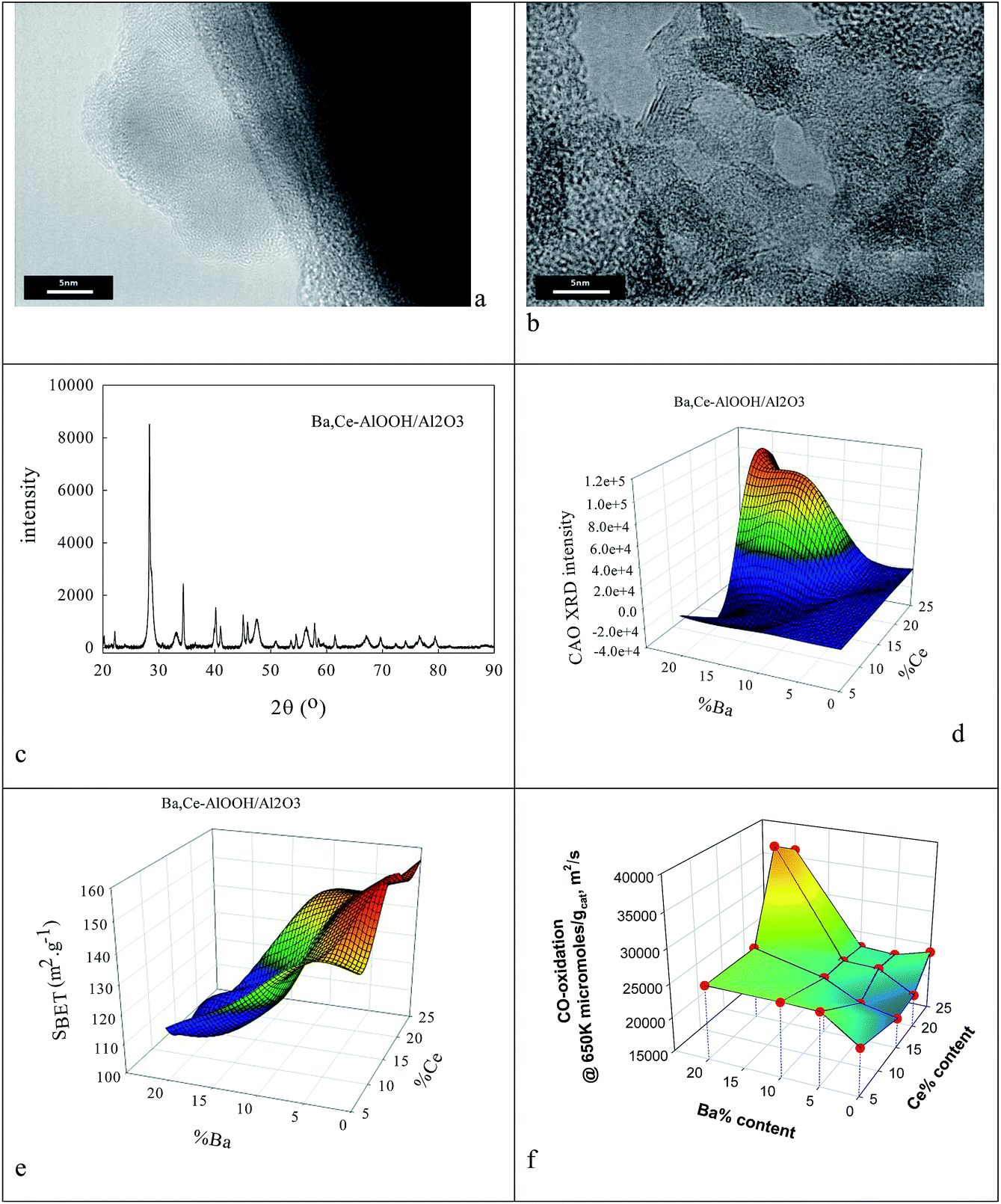 | ||
| Fig. 4 Effect of Ba,Ce insertions at increasing levels into AlOOH/Al2O3 after calcination at 1223 K. HRTEM (a, b), XRD structure (c) and CAO level (d), surface area (e) and CO oxidation rate at 650 K in a stoichiometric reactant stream (R = 1.13) (f). | ||
To understand the nature of the highest CO oxidation catalytic activity (at high Ba,Ce addition levels) COT–OT titrations of the surface were undertaken on 20% Ba,20% Ce–AlOOH/Al2O3 at 773 K and compared to EuroPt-1 at 573 K (see DSC profiles in Fig. 5 and integrated peak areas in Table 1) by DSC. Exothermic titration peaks increased in area or were stable with titration cycling, but the authors concentrated on the first CO titrations of surface (COT![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) ). On EuroPt-1 at 573 K (COT = 126.43 J g−1) COT was 4.5× higher than for 88 m2 g−1 20% Ba,20% Ce–AlOOH/Al2O3 at 773 K (COTO1 = 28.043 J g−1) and these were 57.2× higher than for 170 m2 g−1 undoped-AlOOH/Al2O3 (COT = 0.490 J g−1) at 773 K. Interestingly, COT can be converted to a number of active sites (*) adsorbing CO/g catalyst and hence to a turnover frequency (TOF); the TOF for 20% Ba,20% Ce–AlOOH/Al2O3 was 35% higher than for EuroPt-1 (see Table 1). Furthermore, the 20% Ba,20% Ce–AlOOH/Al2O3 showed a lower COT/OT titration ratio than EuroPt-1 (suggesting less potential for CO poisoning). At high CeO2 and BaO concentrations, peaks from low concentrations of CAO and BCO (presumably at the interface between BaAl2O4 and CeO2) appear in low concentration and may be significant contributors to catalytic activity.
). On EuroPt-1 at 573 K (COT = 126.43 J g−1) COT was 4.5× higher than for 88 m2 g−1 20% Ba,20% Ce–AlOOH/Al2O3 at 773 K (COTO1 = 28.043 J g−1) and these were 57.2× higher than for 170 m2 g−1 undoped-AlOOH/Al2O3 (COT = 0.490 J g−1) at 773 K. Interestingly, COT can be converted to a number of active sites (*) adsorbing CO/g catalyst and hence to a turnover frequency (TOF); the TOF for 20% Ba,20% Ce–AlOOH/Al2O3 was 35% higher than for EuroPt-1 (see Table 1). Furthermore, the 20% Ba,20% Ce–AlOOH/Al2O3 showed a lower COT/OT titration ratio than EuroPt-1 (suggesting less potential for CO poisoning). At high CeO2 and BaO concentrations, peaks from low concentrations of CAO and BCO (presumably at the interface between BaAl2O4 and CeO2) appear in low concentration and may be significant contributors to catalytic activity.
 | ||
| Fig. 5 DSC profiles for CO (40 μmol) and O2 (47 μmol) titrations on (a) EuroPt-1 6.3% Pt/SiO2 at 573 K (where COT = 126.43 J g−1; OT = 182.37 J g−1); (b) AlOOH/Al2O3 (170 m2 g−1) at 773 K (COT = 0.490 J g−1) and (c) 20% Ba,20% Ce–AlOOH/Al2O3 at 773 K (88 m2 g−1) at 773 K (COT = 28.043 J g−1; OT = 37.886 J g−1). (d) CO oxidation activity profile for 25% La,Co–AlOOH/Al2O3. | ||
La,Co doping was also achieved by replacing the 100 cm3 water for AlOOH by a mixed metal nitrate aqueous solution. This was used to produce 25%La,Co–AlOOH/Al2O3. After calcination (923 K) this gave the catalytic profile in Fig. 5d, which is only slightly poorer than Fig. 2e. Hence both Ba,Ce and La,Co cation pairs can be inserted into AlOOH/Al2O3 and will produce activity in atmospheric pollution control reactions.
Surface complexation with pre-adsorbed oxalate to give 5% Ba,Ce– and 5% La,Co–AlOOH/Al2O3
Fig. 1c gave the average size of AlOOH dispersions seen in DLS (60 nm), on which oxalate anions were adsorbed,85 followed by Ba,Ce or La,Co cations to form surface oxalates. Fig. 1d shows that this sequential coating caused the average particle size to rise to 114 nm after drying and calcination at 1223 K that was judged to be necessary to decompose oxalate species. Its oxidation activity was very modest (Fig. 6b).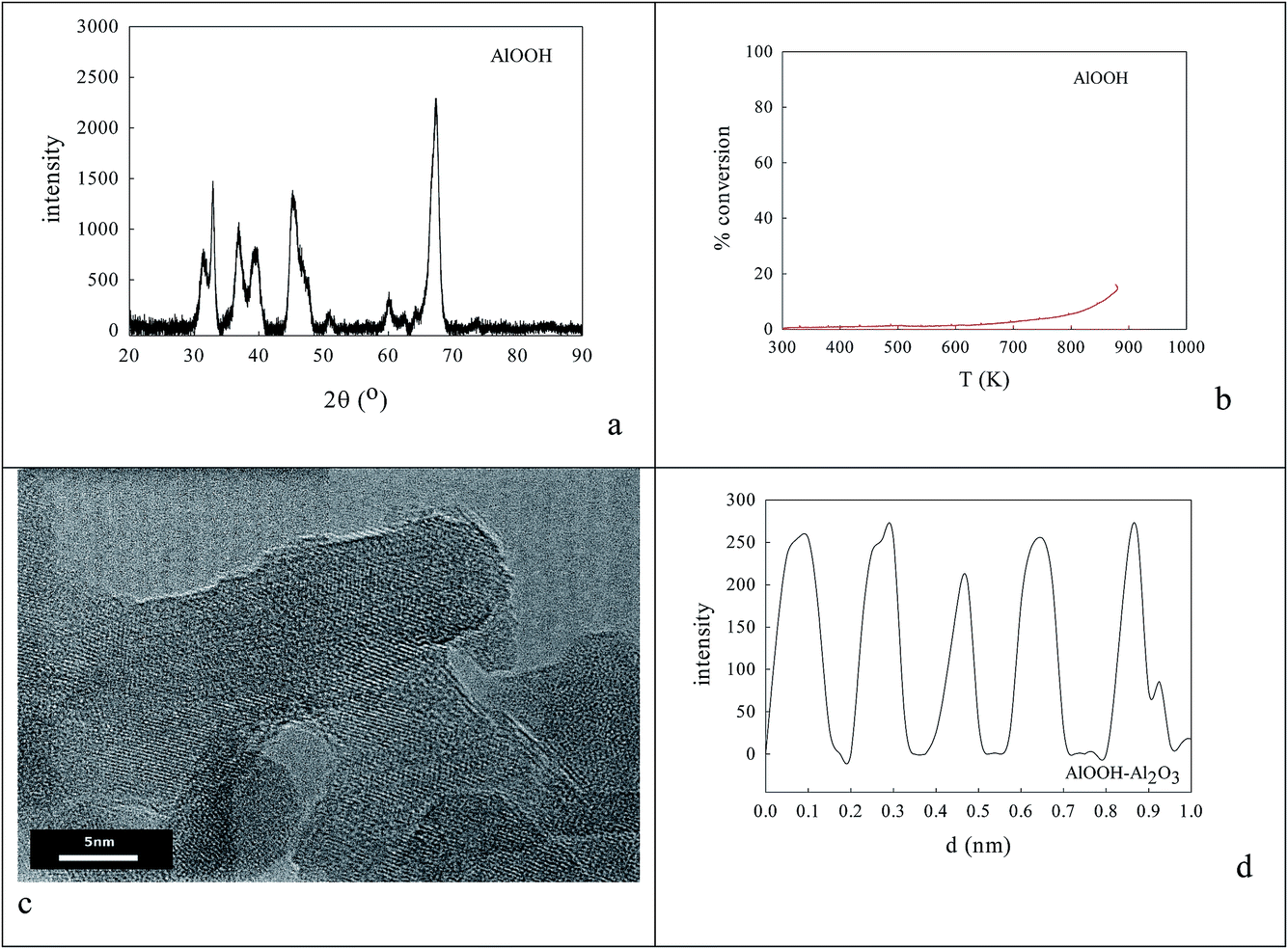 | ||
| Fig. 6 AlOOH (dDLS = 60 nm; see Fig. 2c)/Al2O3 samples onto which oxalate anions had been adsorbed,84 then dried and calcined at 1223 K to decompose oxalate species, gave XRD (a) and HRTEM (c, d) evidence of 8–20 nm micro-domains of crystallinity. The most frequent XRD peaks appeared to be 11.70 nm θ-Al2O3 (with perhaps low levels of γ-Al2O3). The line spacing in micro-crystals was AlOOH (0.19 nm spacing), but activity in CO or propane oxidation or NO reduction (b) was even lower than with sol–gel alumina (Fig. 4f). | ||
TGA indicated that AlOOH lost 22% of its mass on heating, but this increased to 36% in 5% Ba,Ce–AlOOH/Al2O3 and 37% in 5% La,Co–AlOOH/Al2O3. XPS showed that the surface Ba:Ce surface atomic ratio was 25:75, while the La:Co XPS ratio on the surface was found to be 85:15. XPS-FAAS suggests these samples may be 0.91% Ba, 2.74% Ce–AlOOH/Al2O3 or 1.095% La,2.56% Co–AlOOH/Al2O3. Fig. 7a and b show that these samples contain micro-domains of crystallinity 3–5 nm in diameter. Thus XRD (a) and HRTEM (c, d) found 8–20 nm micro-domains of crystallinity. Analysis of HRTEM lattice images for Ba,Ce–AlOOH/Al2O3 (0.295 nm and 0.32 nm spacings) and La,Co–AlOOH/Al2O3 (0.225 nm and 0.296 nm spacings) gave line spacings after calcination greater than for AlOOH alone (0.19 nm; Fig. 6d). XRD of Ba,Ce–AlOOH/Al2O3 (Fig. 7e) shows that the dominant phase is probably 48.3 nm BaAl2O4, but there is also evidence from a (311) peak for 14.0 nm CeO2 and traces of BCO and CeO2/CAO. XRD for La,Co–AlOOH/Al2O3 (Fig. 7f) suggests that the dominant phases are 36–41.5 nm LAO and 17.68 nm Co3O4 but with LCO also present in small amounts. This ion preference may relate to oxalate insolubility, but would not help perovskite formation. CO and propane oxidation activity (Fig. 8a and b) was even lower than with sol–gel alumina (Fig. 3c).
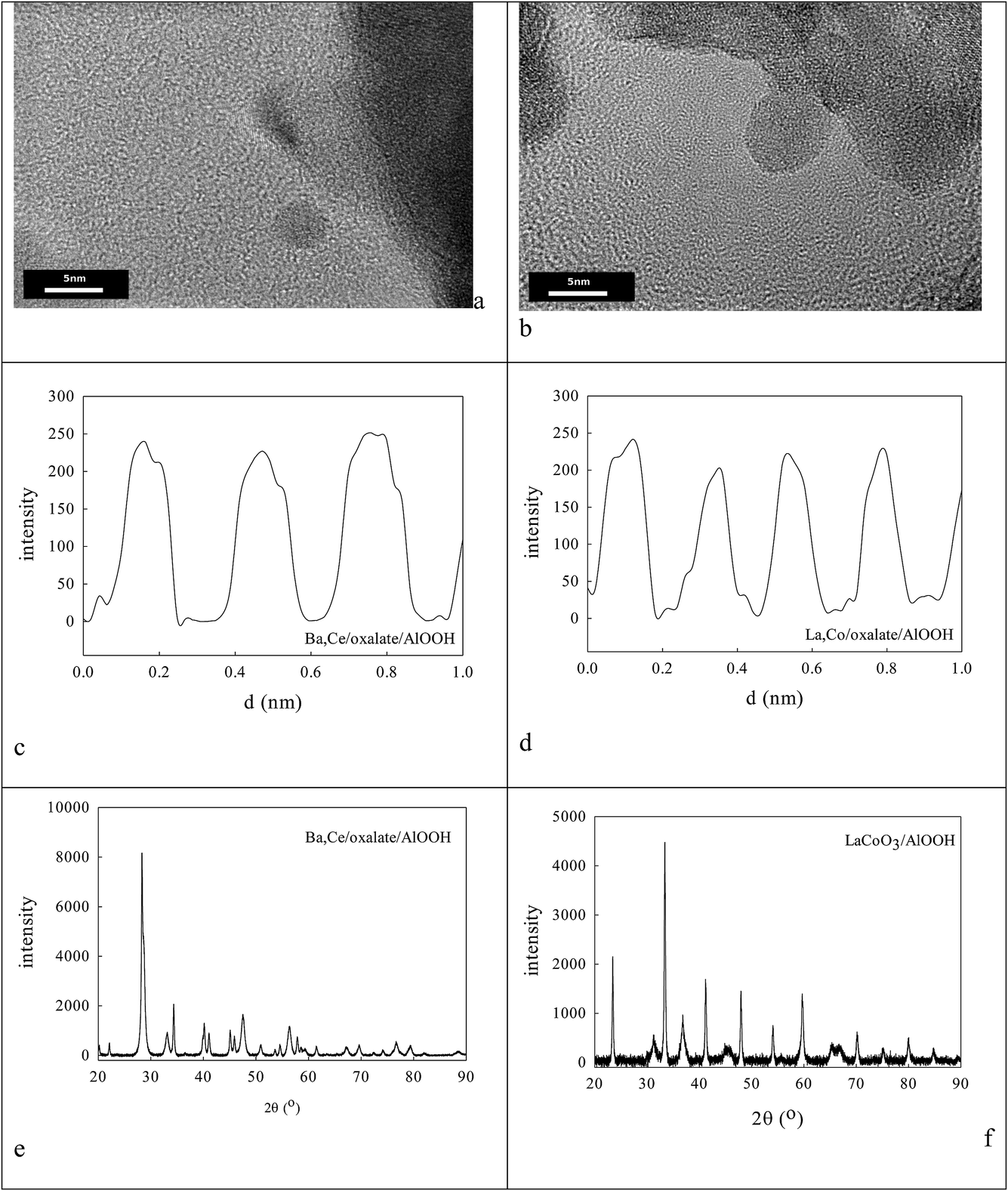 | ||
| Fig. 7 (a–d) HRTEM of Ba,Ce/oxalate/AlOOH (a, c) and La,Co/oxalate/AlOOH (b, d) produced from Ba2+ and Ce4+ cations adsorbed on oxalate/AlOOH, drying and then calcination at 1223 K. XRD of Ba,Ce/oxalate/AlOOH (e) and La,Co/oxalate/AlOOH (f) show the dominant and minor phases present. | ||
 | ||
| Fig. 8 Catalytic activities (a, b) and DSC analysis of COT and OT titrations in flowing N2 (c, d)58 of Ba,Ce/oxalate/AlOOH (a, c) and La,Co/oxalate/AlOOH (b, d). Catalytic activities were modest for both these samples. The summed heat flows in the first COT from Ba,Ce/oxalate/AlOOH (1.920 J g−1 catalyst) was a factor of 14× lower than that for the 20% Ba,20% Ce–AlOOH/Al2O3 sample (28.043 J g−1 catalyst) which is not surprising, given the lower loading in the surface complexation samples. COT was even lower for La,Co/AlOOH (0.661 J g−1 catalyst). | ||
DSC measurements of the first COT on EuroPt-1 at 423 K (ref. 58) was 143.01 kJ g−1 catalyst (2270 kJ g−1 Pt).58 The summed heat flows in the first COT from Ba,Ce/AlOOH (1.920 J g−1 catalyst) (and even more so La,Co/AlOOH (660.7 mJ g−1 catalyst)) (see Fig. 8c and d) were much smaller than those for EuroPt-1. That for COT from Ba,Ce–AlOOH/Al2O3 (1.920 J g−1 catalyst) was a factor of 14 × lower than that for 20% Ba,20% Ce–AlOOH/Al2O3 sample (28.04 J g−1 catalyst), but that is not surprising given the imbalance of cations and the lower loading in the surface complexation samples.
4. Discussion and conclusions
It seems from the present work that microemulsion routes to BCO and LCO are effective and that the products adsorb on AlOOH sols, in much the same way as Turkevich immobilised platinum group metal sols.1 BCO is active in CO and propane oxidation and NO removal under stoichiometric exhaust conditions, but LCO is a better oxidation catalyst. In calcination, the surfactant TX100 is removed, overcoming a frequent impurity disadvantage of μE routes.Activity was also seen when Ba,Ce and La,Co are (i) inserted into AlOOH/Al2O3 and segregated at the surface, but in addition to formation of low levels of BCO, CAO, LCO and LAO perovskites, one also sees aluminates (e.g. BaAl2O4) and separate oxide (e.g. CeO2), although this might be minimised by fine tuning of cation loadings and (ii) when the cations were complexed with AlOOH surface-held oxalate ions, albeit with different efficiencies.
Therefore all three routes yield active catalysts with micro-domains of crystallinity.
The longer term aim of the present work was to design and produce nanoparticulate perovskites, with a variety of cations,46 stabilised by alumina (i.e. where there might be stabilisation by Al3+ substitution47). Of course one might still wish to use activation by Au nanoparticles/nanocrystals yielding green synergies66 in catalysis67 and gas sensing.68
Some have seen higher perovskite surface areas (e.g. LCO (22 m2 g−1) than LaMnO3 (LMnO) (10 m2 g−1)13). Some have compared the propane oxidation activity for 6–16 m2 g−1 (SBET) and 15–25 nm crystallite size (dXRDLB-Scherrer) LMnO perovskites with 1% Pt/Al2O3 (ref. 89) and found that the temperatures of 10%, 50% and 90% oxidation (T10%, T50% and T90%) were higher for the perovskites than the platinum group metal (PGM). The same authors found the perovskite-like trigonal X-ray diffraction patterns.89 Others90 have seen primary perovskite particle sizes, shapes, aggregation and structure by high resolution TEM. The present work should be seen against this background.
Here we have explored BaCeO3 (BCO; that has been used in CO2 conversion and characterised for structure by XRD48 and embedded in hosts49) and LCO (that is useful in catalysed phenol removal from water50). We believe that the present routes offer an opportunity to develop green nanoparticle perovskite catalysts for a circular economy and a better environment, and that these might ultimately find use for other perovskite applications.2
Acknowledgements
The financial support for SR through a Star studentship award by MIRA is gratefully acknowledged.Notes and references
- J. Turkevich and G. Kim, Science, 1970, 169, 873–879 CrossRef PubMed; K. Aika, L. L. Ban, I. Okura, S. Namba and J. Turkevich, J. Res. Inst. Catal., Hokkaido Univ., 1976, 25, 54–64 Search PubMed; J. Turkevich, Gold Bull., 1985, 18(4), 125–131 CrossRef.
- R. J. D. Tilley, Perovskites: structure-property relationships, 2016, Wiley Search PubMed.
- S. R. Noh, D. Lee, S. J. Park, D. S. Kim and M. Choi, Aerosol Sci. Technol., 2017, 51, 116–122 CrossRef.
- V. Solanki, S. Das, S. Kumar, M. M. Seikh, B. Raveaun and A. K. Kundu, J. Sol-Gel Sci. Technol., 2017, 82, 536–540 CrossRef.
- N. Bhardwaj, A. Gaur and K. Yadav, Appl. Phys. A: Mater. Sci. Process., 2017, 123, 429 CrossRef.
- S. D. Li, Z. X. He, X. L. Wang and K. Gao, Appl. Phys. A: Mater. Sci. Process., 2014, 117, 1381–1386 CrossRef.
- S. Ifrah, A. Kaddouri, P. Gelin and D. Leonard, C. R. Chim., 2007, 10, 1216–1226 CrossRef.
- S. A. E. Abdulla, P. A. Sermon, M. Worsley and I. R. Collins, Nanostructured Mater. Nanotechnol. IV, Ceram. Eng. Sci. Proc., 2008, 28, 131 Search PubMed.
- L. M. Chao, Y. D. Hou, M. P. Zheng, Y. G. Yue and M. K. Zhu, J. Alloys Compd., 2017, 695, 3331–3338 CrossRef.
- M. Yousefi and M. Ranjbar, J. Inorg. Organomet. Polym. Mater., 2017, 27, 633–640 CrossRef.
- T. Tabari, D. Singh, A. Calisan, M. Ebadi, H. Tavakkoli and B. Caglar, Ceram. Int., 2017, 43, 15970–15977 CrossRef.
- M. M. Hessien, G. A. M. Mersal, Q. Mohsen and D. Alosaimi, J. Mater. Sci.: Mater. Electron., 2017, 28, 4170–4178 CrossRef.
- E. Krupicka, A. Reller and A. Weidenkaff, Cryst. Eng., 2002, 5, 195–202 CrossRef.
- R. N. Das and P. Pramanik, Mater. Lett., 2000, 46, 7–14 CrossRef.
- S. A. Kulkarni, S. Muduli, G. C. Xing, N. Yantara, M. J. Li, S. Chen, T. C. Sum, N. Mathews, T. J. White and S. G. Mhaisalkar, ChemSusChem, 2017, 10, 3818–3824 CrossRef PubMed.
- M. E. F. Bouduban, A. Burgos-Caminal, R. Ossola, J. Teuscher and J. E. Moser, Chem. Sci., 2017, 8, 4371–4380 RSC.
- G. Kakavelakis, K. Petridis and E. Kymakis, J. Mater. Chem. A, 2017, 5, 21604–21624 RSC.
- Y. H. Kim, G. H. Lee, Y. T. Kim, C. Wolf, H. J. Yun, W. Kwon, C. G. Park and T. W. Lee, Nano Energy, 2017, 38, 51–58 CrossRef.
- R. Zhou, Y. Z. Huang, L. Wan, H. H. Niu, F. W. Ji and J. Z. Xu, J. Alloys Compd., 2017, 716, 162–170 CrossRef.
- L. Durai, B. Moorthy, C. I. Thomas, D. K. Kim and K. K. Bharathi, Mater. Sci. Semicond. Process., 2017, 68, 165–171 CrossRef.
- L. T. Ye, M. Y. Zhang, P. Huang, G. C. Guo, M. C. Hong, C. S. Li, J. T. S. Irvine and K. Xie, Nat. Commun., 2017, 8, 14785 CrossRef PubMed.
- N. Afifah and R. Saleh, Internat. Conf. Eng. Sci. Nanotechnol. (ICESNANO 2016), AIP Conf. Proc., 2017, 1788, 030080 CrossRef.
- H. Wang, Z. Zhao, C. M. Xu, J. Liu and Z. X. Lu, Chin. Sci. Bull., 2005, 50, 1440–1444 CrossRef.
- Y. Zhang-Steenwinkel, L. M. van der Zande, H. L. Castricum, A. Bliek, R. W. van den Brink and G. D. Elzinga, Chem. Eng. Sci., 2005, 60, 797–804 CrossRef.
- T. Maneerung, K. Hidajat and S. Kawi Internat, Int. J. Hydrogen Energy, 2017, 42, 9840–9857 CrossRef.
- M. J. Suh, Y. K. Park and S. K. Ihm, Catal. Today, 2016, 265, 210–217 CrossRef.
- S. S. Li, H. G. Tang, D. D. Gong, Z. Ma and Y. Liu, Catal. Today, 2017, 297, 298–307 CrossRef.
- H. Einaga, Y. Nasu, M. Oda and H. Saito, Chem. Eng. J., 2016, 283, 97–104 CrossRef.
- A. Lay-Ekuakille, S. Ikezawa, M. Mugnaini, R. Morello and C. De Capua, Measurement, 2017, 98, 49–59 CrossRef.
- Y. Zhang-Steenwinkel, H. L. Castricum, A. Bliek and E. Esveld, J. Mater. Sci., 2007, 42, 5851–5859 CrossRef.
- H. L. T. N’Goc, L. D. N. Mouafo, C. Etrillard, A. Torres-Pardo, J. F. Dayen, S. Rano, G. Rousse, C. Laberty-Robert, J. G. Calbet, M. Drillon, C. Sanchez, B. Doudin and D. Portehault, Adv. Mater., 2017, 29, 1604745 CrossRef PubMed.
- W. Zhang, X. D. Zuo, Y. Niu, C. W. Wu, S. P. Wang, S. Guan and S. R. P. Silva, Nanoscale, 2017, 9, 13929–13937 RSC.
- C. O. Ehi-Eromosele, B. I. Ita and E. E. J. Iweala, Colloids Surf., A, 2017, 530, 164–171 CrossRef.
- Q. W. Shen, Y. D. Zhang, H. R. Ding, L. J. Wu, Y. Q. Xu, B. C. Shi, Y. Zheng and J. L. Yuan, Energies, 2017, 10, 164 CrossRef.
- Y. C. Ye, H. Yang, R. S. Li and X. X. Wang, J. Sol-Gel Sci. Technol., 2017, 82, 509–518 CrossRef.
- S. S. Li, H. G. Tang, D. D. Gong, Z. Ma and Y. Liu, Catal. Today, 2017, 297, 298–307 CrossRef.
- Y. Janbutrach, S. Hunpratub and E. Swatsitang, Nanoscale Res. Lett., 2014, 9, 498 CrossRef PubMed.
- D. G. Shchukin, A. A. Yaremchenko, M. G. S. Ferreira and V. V. Kharton, Chem. Mater., 2005, 17, 5124–5129 CrossRef.
- R. A. Sheldon, Green Chem., 2016, 18, 3180–3183 RSC.
- E. Grabowska, Appl. Catal., B, 2016, 186, 97–126 CrossRef.
- W. F. Libby, Science, 1971, 171(3970), 499–500 CrossRef PubMed.
- K. A. Grant, K. M. Keryou and P. A. Sermon, Faraday Discuss., 2008, 138, 257–271 RSC.
- M. R. Soames, PhD thesis, Brunel University, 2000.
- P. Klonowski, J. C. Goloboy, F. J. Uribe-Romo, F. R. Sun, L. Y. Zhu, F. Gandara, C. Wills, R. J. Errington, O. M. Yaghi and W. G. Klemperer, Inorg. Chem., 2014, 53, 13239–13246 CrossRef PubMed.
- C. Thatcher, PhD thesis, University of Surrey, 2005.
- M. V. Goldschmidt, Naturwissenschaften, 1926, 14(21), 477–485 CrossRef.
- Z. R. Ismagilov, R. A. Shkrabina, N. A. Koryabkina and F. Kapteijn, Catal. Today, 1995, 24, 269–271 CrossRef; R. A. Shkrabina, N. A. Koryabkina, O. A. Kirichenko, V. A. Ushakov, F. Kapteijn and Z. R. Ismagilov, Stud. Surf. Sci. Catal., 1995, 91, 1145–1152 CrossRef.
- S. Cimino, L. Lisi, S. De Rossi, M. Faticanti and P. Porta, Appl. Catal., B, 2003, 43, 397–406 CrossRef.
- I. Pettiti, S. Colonna, S. De Rossi, M. Faticanti, G. Minelli and P. Porta, Phys. Chem. Chem. Phys., 2004, 6, 1350–1358 RSC.
- M. A. Malecka and L. Kepinski, CrystEngComm, 2015, 17, 2273–2278 RSC; M. A. Malecka and L. Kepinski, CrystEngComm, 2015, 17, 8282–8288 RSC.
- A. Piras, S. Colussi, A. Trovarelli, V. Sergo, J. Llorca, R. Psaro and L. Sordelli, J. Phys. Chem. B, 2005, 109, 11110–11118 CrossRef PubMed.
- S. I. Lopatin, S. M. Shugurov, A. V. Fedorova, A. V. Utina and A. I. Panin, J. Alloys Compd., 2017, 693, 1028–1034 CrossRef.
- JCPDS 22-74; 15–148.
- L. Tepech-Carrillo, A. Escobedo-Morales, A. Perez-Centeno, E. Chigo-Anota, J. F. Sanchez-Ramirez, E. Lopez-Apreza and J. Gutierrez-Gutierrez, J. Nanomater., 2016, 6917950 Search PubMed.
- D. P. Belanger, T. Keiber, F. Bridges, A. M. Durand, A. Mehta, H. Zheng, J. F. Mitchell and V. Borzenets, J. Phys.: Condens. Matter, 2016, 28, 025602 CrossRef PubMed.
- JCPDS 9–358.
- G. J. Liu, X. T. Li, Y. Q. Wang, W. S. Liang, B. Liu, H. L. Feng, H. W. Yang, J. Zhang and J. R. Sun, Appl. Surf. Sci., 2017, 425, 121–129 CrossRef.
- M. A. M. Luengo, P. A. Sermon and A. T. Wurie, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 1651–1665 RSC.
- S. Roesch, P. A. Sermon, A. Wallum, P. N. Forrest and P. Kaur, Top. Catal., 2001, 16, 115–118 CrossRef; T. Salvesen, S. Roesch, P. A. Sermon and P. Kaur, Top. Catal., 2001, 16, 381–384 CrossRef.
- J. Z. Shyu, W. H. Weber and H. S. Gandhi, J. Phys. Chem., 1988, 92, 4964–4970 CrossRef.
- C. Morterra, V. Bolis and G. Magnacca, J. Chem. Soc., Faraday Trans., 1996, 92, 1991–1999 RSC.
- B. Harrison, A. F. Diwell and C. Hallett, Platinum Met. Rev., 1988, 32, 73 Search PubMed.
- A. F. Diwell, R. R. Rajaram, H. A. Shaw and T. J. Truex, Stud. Surf. Sci. Catal., 1991, 71, 139–152 CrossRef.
- L. Matějová, P. Topka, L. Kaluža, S. Pitkäaho, S. Ojala, J. Gaálová and R. L. Keiski, Appl. Catal., B, 2013, 142–143, 54–64 CrossRef.
- S. K. Saji, R. Vinodkumar, S. M. Eappen and P. R. S. Wariar, Mater. Chem. Phys., 2017, 193, 189–195 CrossRef.
- A. M. Harzandi, J. N. Tiwari, H. S. Lee, H. Jeon, W. J. Cho, G. Lee, J. Baik, J. H. Kwak and K. S. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 2495–2499 CrossRef PubMed.
- P. Ciambelli, S. Cimino, G. Lasorella, L. Lisi, S. De Rossi, M. Faticanti, G. Minelli and P. Porta, Appl. Catal., B, 2002, 37, 231–241 CrossRef.
- S. Cimino, L. Lisi, S. De Rossi, M. Faticanti and P. Porta, Appl. Catal., B, 2003, 43, 397–406 CrossRef.
- H. H. Wang, L. L. Zhang, C. Hu, X. K. Wang, L. Lyu and G. D. Sheng, Chem. Eng. J., 2018, 332, 572–581 CrossRef.
- L. Q. Wang, Q. Pang, Q. Q. Song, X. W. Pan and L. S. Jia, Fuel, 2015, 140, 267–274 CrossRef.
- M. M. Natile, G. Eger, P. Batocchi, F. Mauuy and A. Glisenti, Int. J. Hydrogen Energy, 2017, 42, 1724–1735 CrossRef.
- D. Y. Yoon, Y. J. Kim, J. H. Lim, B. K. Cho, S. B. Hong, I. S. Nam and J. W. Choung, J. Catal., 2015, 330, 71–83 CrossRef.
- L. F. Liotta, G. Di Carlo, A. Longo, G. Pantaleo, G. Deganello, G. Marci and A. Martorana, J. Non-Cryst. Solids, 2004, 345, 620–623 CrossRef.
- G. Lee, I. Kim, I. Yang, J. M. Ha, H. B. Na and J. C. Jung, Appl. Surf. Sci., 2018, 429, 55–61 CrossRef.
- H. M. Zhang, R. S. Hu, J. N. Hu and Y. L. Zhang, Acta Phys.-Chim. Sin., 2011, 27, 1169–1175 Search PubMed.
- J. Cai, K. Laubernds, F. S. Galasso, S. L. Suib, J. Liu, X. F. Shen, E. Begge, H. R. Kunz and J. M. Fenton, J. Am. Ceram. Soc., 2005, 88, 2729–2735 Search PubMed.
- P. Fornasiero, G. Balducci, J. Kaspar, S. Meriani, R. diMonte and M. Graziani, Catal. Today, 1996, 29, 47–52 CrossRef.
- K. Masuda, T. Sano, F. Mizukami and M. Watanabe, Catal. Lett., 1995, 33, 229–235 CrossRef.
- B. E. Yoldas, Am. Ceram. Soc. Bull., 1975, 54, 286–290 Search PubMed; B. E. Yoldas, J. Mater. Sci., 1975, 10, 1856–1857 CrossRef.
- T. Maunula, Y. Kintaichi, M. Haneda and H. Hamada, Catal. Lett., 1999, 61, 121–130 CrossRef.
- K. Maeda, F. Mizukami, M. Watanabe, N. Arai, S. Nniwa, M. Toba and K. Shimizu, J. Mater. Sci. Lett., 1990, 9, 522–523 CrossRef.
- K. Maeda, F. Mizukami, S. Niaw, M. Toba, M. Watanabe and K. Masuda, J. Chem. Soc., Faraday Trans., 1992, 88, 97–104 RSC.
- N. E. Bogdanchikova, S. Fuentes, M. Avalos-Borja, M. H. Farias, A. Boronin and G. Diaz, Appl. Catal., B, 1998, 17, 221–231 CrossRef.
- M. J. D’ Aniello, J. Catal., 1981, 69, 9–17 CrossRef.
- M. F. L. Johnson and J. Mooi, J. Catal., 1987, 103, 502–505 CrossRef.
- E. Rogemond, R. Frety, V. Perrichon, M. Primet, S. Salasc, M. Chevrier, C. Gauthier and F. Mathis, J. Catal., 1997, 169, 120–131 CrossRef.
- J. Z. Shyu, K. Otto, W. L. H. Watkins, G. W. Graham, R. K. Belitz and H. S. Gandhi, J. Catal., 1988, 114, 23–33 CrossRef.
- J. Wang, C. X. Huang, X. L. Chen, H. T. Zhang, Z. S. Li and Z. G. Zou, Appl. Surf. Sci., 2015, 358, 463–467 CrossRef.
- N. Miniajluk, J. Trawczynski and M. Zawadzki, Appl. Catal., A, 2017, 531, 119–128 CrossRef.
- M. Ogasawara, S. Kato, H. Tsukidate, T. Akaogi, Y. Moriya and S. Nakata, Chem. Lett., 2005, 34, 208–209 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |