 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into structure and dynamics of (Mn,Fe)Ox-promoted Rh nanoparticles†
Maria
Dimitrakopoulou
a,
Xing
Huang
 a,
Jutta
Kröhnert
a,
Detre
Teschner
ab,
Sebastian
Praetz
c,
Christopher
Schlesiger
c,
Wolfgang
Malzer
c,
Christiane
Janke
d,
Ekkehard
Schwab
d,
Frank
Rosowski
de,
Harry
Kaiser
f,
Stephan
Schunk
f,
Robert
Schlögl
ab and
Annette
Trunschke
*a
a,
Jutta
Kröhnert
a,
Detre
Teschner
ab,
Sebastian
Praetz
c,
Christopher
Schlesiger
c,
Wolfgang
Malzer
c,
Christiane
Janke
d,
Ekkehard
Schwab
d,
Frank
Rosowski
de,
Harry
Kaiser
f,
Stephan
Schunk
f,
Robert
Schlögl
ab and
Annette
Trunschke
*a
aFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: trunschke@fhi-berlin.mpg.de
bMax Planck Institute for Chemical Energy Conversion, Stiftstr. 34-36, 45470 Mülheim, Germany
cTechnical University of Berlin, Institute of Optics and Atomic Physics, Hardenbergstraße 36, D-10587 Berlin, Germany
dBASF SE, Process Research and Chemical Engineering, Heterogeneous Catalysis, Carl-Bosch-Straße 38, 67056, Ludwigshafen, Germany
eBasCat, UniCat BASF JointLab, Technische Universität Berlin, Hardenbergstraße 36, 10623 Berlin, Germany
fhte GmbH, Kurpfalzring 104, 69123 Heidelberg, Germany
First published on 20th December 2017
Abstract
The mutual interaction between Rh nanoparticles and manganese/iron oxide promoters in silica-supported Rh catalysts for the hydrogenation of CO to higher alcohols was analyzed by applying a combination of integral techniques including temperature-programmed reduction (TPR), X-ray photoelectron spectroscopy (XPS), X-ray absorption spectroscopy (XAS) and Fourier transform infrared (FTIR) spectroscopy with local analysis by using high angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) in combination with energy dispersive X-ray spectroscopy (EDX). The promoted catalysts show reduced CO adsorption capacity as evidenced through FTIR spectroscopy, which is attributed to a perforated core–shell structure of the Rh nano-particles in accordance with the microstructural analysis from electron microscopy. Iron and manganese occur in low formal oxidation states between 2+ and zero in the reduced catalysts as shown by using TPR and XAS. Infrared spectroscopy measured in diffuse reflectance at reaction temperature and pressure indicates that partial coverage of the Rh particles is maintained at reaction temperature under operation and that the remaining accessible metal adsorption sites might be catalytically less relevant because the hydrogenation of adsorbed carbonyl species at 523 K and 30 bar hydrogen essentially failed. It is concluded that Rh0 is poisoned due to the adsorption of CO under the reaction conditions of CO hydrogenation. The active sites are associated either with a (Mn,Fe)Ox (x < 0.25) phase or species at the interface between Rh and its co-catalyst (Mn,Fe)Ox.
Introduction
The need for utilization of alternative energy resources has led to a revitalization of research in the field of synthesis gas conversion applying transition metal nanoparticles as catalysts.1,2 The hydrogenation of CO/CO2 mixtures results in a broad product spectrum including methane, saturated and unsaturated hydrocarbons with varying chain length, and oxygenates depending on the active metal.3–5 Supported rhodium is an attractive model system, since product selectivity is tunable in a wide range through the selection of specific supports6,7 or the addition of promoters.8 Silica-supported Rh mainly produces methane,9 but significant amounts of acetaldehyde and acetic acid are also formed.10 The addition of Fe shifted the selectivity towards ethanol without any effect on the activity, while Mn increased the rate of syngas conversion significantly.10,11The promising performance of Mn–Fe-promoted silica-supported Rh catalysts in the synthesis of higher alcohols initiated a number of studies on the promoter effects of Fe8–10,12–18 and Mn9,11,17,19–25 additives. However, it appears that these investigations do not converge in consistent conclusions. The lack of clarity might be ascribed to different procedures and materials that have been used for catalyst synthesis and activation, and the varying promoter content applied resulting in different catalytic properties.8
Beyond that, density functional calculations based on single crystals indicate substantial structure sensitivity in CO hydrogenation over pure Rh.8,26–29 Both molecular and dissociative adsorption of CO is needed for C2 oxygenate formation.22,29,30 The complex reaction mechanism for CO hydrogenation over Rh-based catalysts basically involves CO dissociation, hydrogenation of adsorbed carbon species into CHx (x = 0–3), CO insertion into adsorbed CHx to form acyl species (CHxCO) and their subsequent hydrogenation to ethanol. Defect sites are responsible for the dissociation of CO28,29 with a particular low barrier on stepped Rh(211) surfaces,8,28 leading to the selective formation of methane on this facet due to fast CO dissociation, while the rates for ethanol and acetaldehyde are comparable on both stepped Rh(211) and terrace Rh(111) sites.8 However, the barrier of CO dissociation increases with decreasing particle size,28 accounting for an optimum particle size in view of activity and C2 oxygenate formation. The Rh particles in a real catalyst are characterized by a certain particle size distribution and the particles contain both terrace (111)-like and stepped (211)-like sites. Altogether, support, promoter and catalyst synthesis31 affect particle size and preferred termination resulting in a broad distribution of catalytic properties even for nominal similar catalyst compositions. In addition, changes in the reaction network of CO hydrogenation may be initiated through the chemistry of the promoter element.
Intrinsically, pure silica-supported Rh seems to catalyze the formation of methane and acetaldehyde.8–10,30 Iron generally promotes ethanol formation.10 This effect of Fe has been attributed to the formation of a Rh–FeOx interface14,32 or the inhibition of Rh reduction by iron oxide14,15,33 leading to a higher concentration of Rh+ in the catalyst, stabilization of acetyl/acetaldehyde intermediates and an enhanced availability of hydrogen.9 Based on X-ray absorption spectroscopy performed in presence of synthesis gas at reaction temperature the direct interaction of metallic Rh with partially reduced Fe oxides (Fe2+) was proposed to be responsible for the high activity and selectivity of Fe–Rh/TiO2 for oxygenate production from syngas.32 In contrast, infrared studies suggested a geometric ensemble effect tentatively ascribed to a Rh–Fe alloy, because the formation of bridging CO, and consequently its dissociation into C and O, was suppressed.12,20 The disruption of large Rh ensembles by Fe may control CO dissociation and thus the CH4 formation rate. Experimental hints that can be interpreted in terms of Rh–Fe alloy formation have been found through infrared spectroscopy particularly on titania-supported Rh catalysts.18,34
Frequently, the effect of Mn is attributed to a stabilization of Rh+ ions.11,19,35–38 Rh+ sites33,39,40 or both zero-valent Rh0 sites in close contact with Rh+ ions20,41 have been proposed to serve as active sites in CO hydrogenation to oxygenates through the insertion of CO associated with Rh+ into a Rh0–alkyl bond. The stabilization of Rh+ ions by Mn2+ has been discussed alternatively in terms of a decrease in the heat of CO chemisorption resulting in an increase in H2 uptake and, consequently, reaction rate.19 Furthermore, Mn oxide has been suggested to facilitate CO dissociation through the formation of a tilted CO species adsorbed in di-hapto configuration at the Rh–MnOx interface and to stabilize acyl species, known as the precursors for oxygenates.12,20,24 On the other hand, the modification of metallic Rh with MnOx was interpreted in terms of a stabilization of non-dissociatively adsorbed CO on the Rh surface, which enhanced the CO insertion to form CHxCO intermediates.23,24 Recently, model catalysts have been prepared by using atomic layer deposition.25 The results suggest that MnOx over-layers on Rh appear to suffer from poor stability upon CO adsorption and are less effective than a thin MnOx sandwich layer in between the silica support and the Rh particle in enhancing the Rh–MnOx interface that facilitates CO dissociation and stabilizes the transition state of C2+ oxygenate formation.25 Alternative concepts have been proposed based on DFT calculations, including the direct modification of step edges at the Rh(211) surface by Mn atoms,27 and alloy formation that facilitates CO insertion into the CH surface intermediate balancing hydrogenation to CH4 and the formation of CHCO as an ethanol precursor.22
Synergistic effects in terms of ethanol productivity are usually observed when both Fe and Mn are added as promoters to silica-supported rhodium.17,42–44 Description of the state of Mn and Fe using X-ray photoelectron spectroscopy, electron microscopy and X-ray diffraction is challenging due to the low promoter content and the complexity of the chemistry under reaction conditions.17 Density functional theory calculations suggest that the state of the promoter metals depends on the reaction conditions. Manganese may be present as Mn(I) and Mn(II) oxide on the Rh (211) step edges and Fe may be present as Fe(I) oxide on the step edge and metallic subsurface iron on both Rh steps and terraces disabling CO dissociation and hydrogenation to CH4.17 Iron and manganese in oxidation states below 2+ occur in particular in carbonyl compounds of the elements.
In the current work the nanostructure of silica-supported unpromoted and Mn–Fe promoted rhodium catalysts in calcined and reduced states was analyzed by using a combination of spectroscopic methods with electron microscopy. The adsorption of carbon monoxide and co-adsorption of CO and H2 were investigated by using transmission and diffuse reflectance Fourier transform infrared spectroscopy (DRIFTS). The results suggest a perforated core–shell structure of silica-supported metal-promoter-sub-oxide nano-composites. The relevance of metallic Rh and/or Rh–Fe alloy sites versus metal-suboxide interphase sites in CO hydrogenation will be discussed reconciling the diverging views on structure and function of the complex catalyst.
Experimental
Catalysts
The catalysts were prepared through co-impregnation using aqueous metal nitrate solutions followed by calcination in air at 623 K for 2 h resulting in the calcined catalyst precursors Rh–Mn–Fe/SiO2 (ID 21102), Rh–Mn/SiO2 (ID 20818), Rh–Fe/SiO2 (ID 22436), Rh/SiO2 (ID 21104), Mn/SiO2 (ID 20508), and Fe/SiO2 (ID 21103). Davisil 636, 250–500 μm (ID 20510) was used as support. The metal content analyzed by using inductively coupled plasma optical emission spectrometry (ICP-OES) is presented in Table 1. The catalysts were tested in CO hydrogenation at 533 K and 54 bar in a reaction mixture containing 60% H2 and 20% CO (GHSV = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 667 h−1), nitrogen as balance. Under these reaction conditions Rh/SiO2, Rh–Mn/SiO2 and Rh–Mn–Fe/SiO2 showed an initial space-time yield of EtOH of 0.16, 1.8, and 2.5 mmol ml−1 s−1, respectively.
667 h−1), nitrogen as balance. Under these reaction conditions Rh/SiO2, Rh–Mn/SiO2 and Rh–Mn–Fe/SiO2 showed an initial space-time yield of EtOH of 0.16, 1.8, and 2.5 mmol ml−1 s−1, respectively.
| Rh/SiO2 | Rh–Mn/SiO2 | Rh–Mn–Fe/SiO2 | |
|---|---|---|---|
| a Based on ICP-OES. b Under the assumption Rh3+ → Rh0, Mn3+ → Mn2+, Fe3+ → Fe0. c Assumed oxidation state of Fe to calculate prospective oxidation state of Mn in Rh–Mn–Fe/SiO2. | |||
| Rh content/wt%a | 2.5 | 2.6 | 2.5 |
| Mn content/wt%a | 0 | 1.4 | 1.4 |
| Fe content/wt%a | 0 | 0 | 0.49 |
| Exp. H2 consumption/mmol g−1 | 0.3703 | 0.7323 | 0.7372 |
| Theor. H2 consumptionb/mmol g−1 | 0.3641 | 0.6335 | 0.6628 |
| Theor. H2 consumptionc/mmol g−1 | — | — | 0.7532 |
| H2 exp./H2 theor.b | 1.02 | 1.16 | 1.11 |
Temperature-programmed reduction (TPR)
TPR experiments were carried out in a quartz micro-reactor where the catalyst (0.1 g) was heated in 5% H2 diluted in Ar at a rate of 5 K min−1 up to 623 K. The sample was pretreated in flowing Ar at room temperature for 30 min before reduction started. The H2-uptake was monitored by means of a thermal conductivity detector (TCD). The TCD signal was calibrated by reducing pure copper oxide (CuO).X-ray photoelectron spectroscopy (XPS)
Samples were investigated as pressed pellets. Spectra were recorded at room temperature, using non-monochromatized Al Kα (1486.6 eV) excitation and a hemispherical analyzer (Phoibos 150, SPECS). Charging effects were corrected by internally referencing the Si2p peak of SiO2 to 103.3 eV. The catalysts were reduced ex situ in the reactor for TPR experiments and transferred without air contact via a glove box to the spectrometer.X-ray absorption spectroscopy (XAS)
Mn K-edge X-ray absorption near edge structure (XANES) spectra were collected using a von Hamos laboratory XAFS spectrometer,45 which operates in transmission mode. As the source an air-cooled micro focus X-ray tube with a power of 30 W, a spot size of 60 μm and molybdenum as the anode material was used. A cylindrically bent highly annealed pyrolytic graphite (HAPG) crystal with a size of 5 × 5 cm2 and a bending radius of 30 cm was applied as a dispersive element and detection of the X-rays was accomplished using an indirectly detecting X-ray CCD camera of the type Andor Newton A-DY940P-FO-CSI with a spatial resolution of 50 μm and a size of 1 × 0.25 inch2. With this setup a spectral resolving power of E/ΔE = 4000 can be achieved in an energy range from 4 keV up to 12 keV. The ground powder samples of the reference materials MnO, MnO2 and Mn2O3 have been applied on scotch tape and stacked in order to adapt the adequate thickness for the transmission mode measurements. Self-supported pellets (diameter 1.2 cm, thickness of about 1 mm) of the catalysts Rh–Mn/SiO2 and Rh–Mn–Fe/SiO2 were prepared by using a hydraulic pellet press applying a pressure force of 5–7 t. Energy calibration was performed by using a synchrotron measurement of the Mn K-edge of MnO as a reference.46 The absorption spectra were normalized and analyzed using the XAS evaluation software ATHENA.47Electron microscopy
Samples for electron microscopy were prepared by drop-casting a suspension of the catalyst powder in ethanol on Cu TEM grids. Images were taken on an abreaction-corrected JEOL JEM-ARM200F TEM equipped with a high angle Silicon Drift EDX detector with a detection area of 100 mm2 and a GIF Quantum energy filter. High angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were collected in order to gain information about the morphology and structure of the catalysts. Energy dispersive X-ray spectroscopy was used to study the homogeneity of the catalysts. Particle size distributions have been obtained by measuring around 100 particles.In situ Fourier transform infrared (FTIR) spectroscopy of adsorbed CO
FTIR spectra in transmission mode were recorded using a Perkin-Elmer Spectrum 100 FTIR spectrometer equipped with a nitrogen-cooled MCT detector. The spectra were collected at a resolution of 4 cm−1 with an accumulation of 128 scans, and normalized by dividing the recorded intensities by the corresponding pellet weight area (mg mm−2). An amount of approximately 15 mg of powder of the investigated sample was pressed to a disc and placed into a quartz IR cell, which allows adsorption at beam temperature and liquid-nitrogen temperature, separately, after thermal treatments under a controlled atmosphere. Prior to CO adsorption the catalysts were reduced in situ. During the reduction process, the samples were treated in H2 (0.005 MPa) at 623 K (heating rate 10 K min−1) for 2 hours with intermediate evacuation to remove the produced water followed by cooling down to room temperature under vacuum. In the last step, the pretreated samples were subjected to probe adsorption. For adsorption at 77 K helium was admitted up to an equilibrium pressure of 0.0002 MPa at 293 K before the cell was cooled to 77 K. A spectrum of the empty cell at 77 K served as a background for the measurements. The probes were adsorbed progressively (pCO = 0–0.002 MPa).In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) during CO hydrogenation
In situ DRIFTS measurements were conducted using an Agilent Cary 680 FTIR spectrometer equipped with a MCT detector at a spectral resolution of 2 cm−1 and an accumulation of 512 scans. An in situ cell (Harrick Praying MantisTM diffuse reflectance attachment DRP-P72 in combination with a low temperature CHC-CHA reaction chamber with ZnSe windows) was used. Spectra were taken at 77 K, 313 K or 523 K, the latter being the typical reaction temperature in CO hydrogenation. The amount of the catalyst used was approximately 50 mg. The catalyst was first reduced in 13% H2 in Ar (flow rate 20 ml min−1) at 623 K for 1 hour under atmospheric pressure with subsequent cooling to the desired adsorption temperature. For in situ experiments performed at 523 K the cell was flushed with pure Ar to remove the H2 followed by a pressure increase to 30 bar (reaction pressure). At this point CO diluted in Ar (22.5% CO–77.5% Ar, flow rate 20 ml min−1) was introduced into the reactor with subsequent purging with Ar for ca. 30 min to remove the gas phase and physically adsorbed CO molecules. Finally, the sample was treated with H2 in Ar (87% H2–13% Ar, flow rate 77.5 ml min−1).Results and discussion
Oxidation state of the supported elements before and after catalyst reduction
Temperature-programmed reduction was performed up to a temperature of 623 K for the Rh-containing catalysts, which is the temperature applied for reductive treatment before catalysis. The TPR profile of Rh/SiO2 exhibits a peak at 355 K that is assigned to the reduction of highly dispersed rhodium oxide species (Fig. 1, left bottom). The peak shape suggests a homogeneous distribution of the Rh2O3 precursor on the silica support, but a weak feature near 500 K indicates that small amounts of Rh oxide occur in a different nanostructure. Hydrogen consumption through the reduction of impurities present in the silica support can be excluded based on a reference experiment with pure silica and the satisfying agreement between experimentally measured and theoretically calculated hydrogen consumption for Rh/SiO2. The measured hydrogen consumption is in agreement with complete reduction of Rh from oxidation state 3+ to zero (Table 1). X-ray photoelectron spectroscopy (XPS) confirms the presence of basically oxidized Rh2O3 in all calcined catalysts due to the comparatively narrow peak with a binding energy at 308.9 eV, which shifts to 307.3 eV after reduction indicative for Rh0 in all reduced catalysts (Fig. S1†). No reliable information regarding the oxidation state of Mn and Fe was available from XPS due to the low loadings of the promoter elements.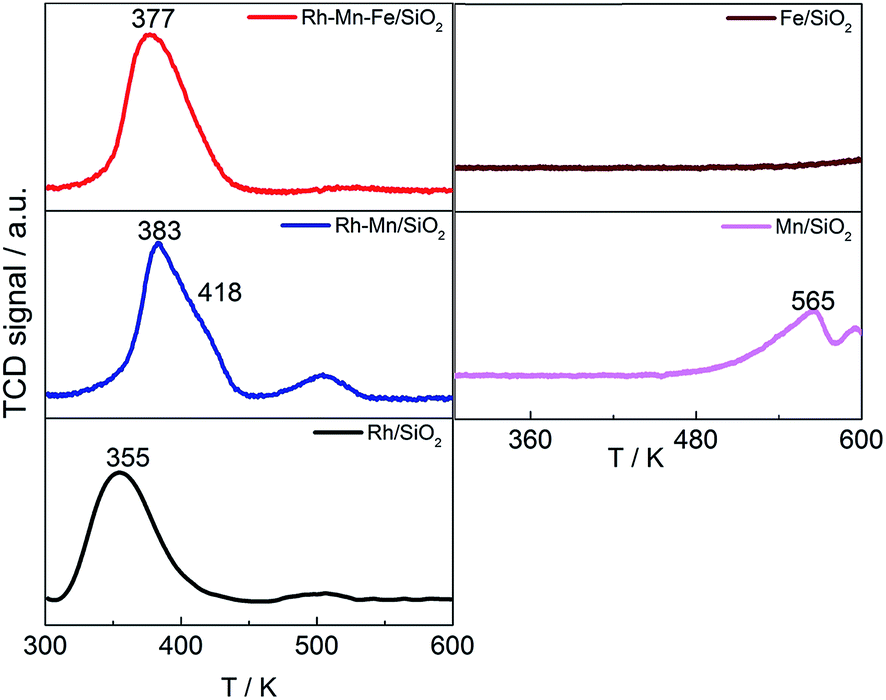 | ||
| Fig. 1 TPR profiles of Rh catalysts (left) and Rh-free references (right) as indicated in the legend. | ||
X-ray absorption spectroscopy was used to determine the oxidation state of Mn in the calcined Mn-promoted catalysts (Fig. 2). We used a weak beam laboratory instrument for the purpose of minimizing possible beam reduction by exposure to a synchrotron X-ray source. The position of the Mn K-edge of bulk oxides is sensitive to the formal oxidation state of Mn, while the pre-edge feature contains important electronic and structural information.48 In the present work, the position of the K-edge was determined using the 1st derivative of the spectrum. The edge positions for the references MnO, Mn2O3 and MnO2 were 6545.2 eV ± 0.2 eV, 6551.8 eV ± 0.1 eV and 6554.9 eV ± 0.2 eV, respectively, which is in good agreement with measurements at synchrotron radiation sources.46 The two catalysts investigated show edge positions at 6551.4 eV ± 0.2 eV for Rh–Mn/SiO2 and 6551.3 eV ± 0.3 eV for Rh–Mn–Fe/SiO2. Edge positions and shape of the spectra near the edge suggest the predominance of Mn3+ in the calcined Mn-containing catalysts. Linear combinations of the three reference spectra were used to fit the spectra of the catalysts in the region of 20 eV before and 50 eV after the absorption edge. The resulting linear combination basically agrees with the reference spectrum of Mn2O3. However, due to the low signal-to-noise ratio the presence of other oxidation states of Mn in the samples cannot be excluded.
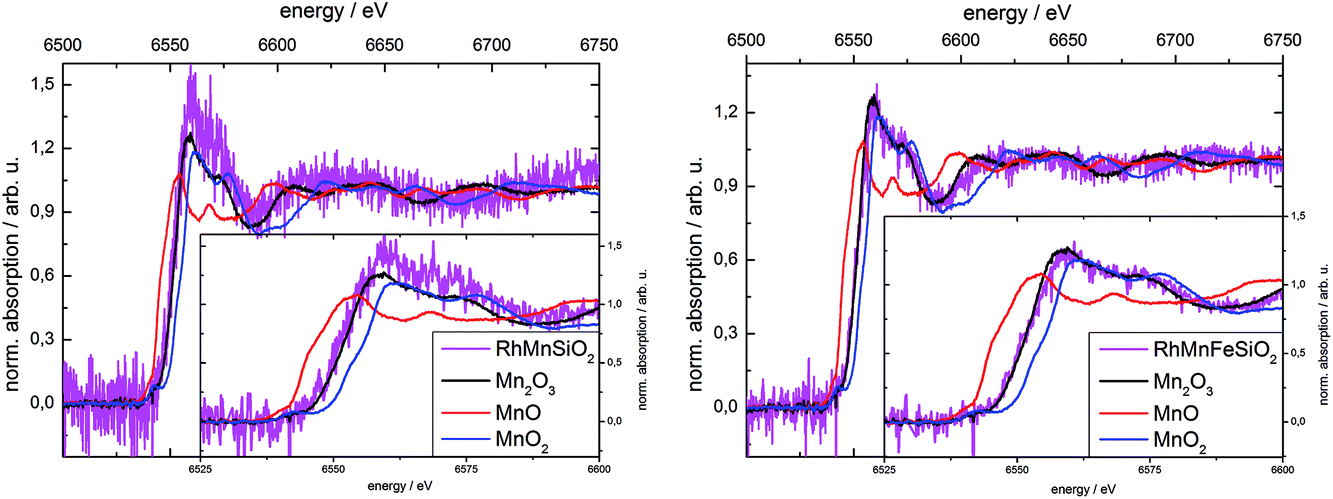 | ||
| Fig. 2 X-ray absorption spectra at the Mn K-edge of Rh–Mn/SiO2 (left) and Rh–Mn–Fe/SiO2 (right) recorded in air under ambient pressure; spectra of binary manganese oxides with Mn in different oxidation states are shown for reference. | ||
The low concentration of Fe of about 0.5 wt% made it impossible to acquire an absorption spectrum of the Fe K-edge. Referring to the literature, the chemical nature of iron after calcination may approximate to Fe2O3. In an Fe-promoted Rh/TiO2 catalyst with higher Fe content the oxidation state of iron in the calcined catalyst was determined by using X-ray absorption spectroscopy to be Fe3+ (Fe2O3).32 The oxidation state of 3+ is therefore assumed for Fe in the calcined catalyst in the subsequent discussion.
The TPR profiles revealed an onset of reduction below 623 K on both Rh-free reference samples Mn/SiO2 and Fe/SiO2 (Fig. 1, right), but the hydrogen consumption of Fe/SiO2 is hardly measurable below 623 K. The addition of Mn to Rh/SiO2 results in an increase in the reduction temperature of Rh from 355 K to 383 K with a pronounced shoulder at 418 K, which is in agreement with previous reports (Fig. 1, left middle).23,37,49 The peak shift suggests that Mn is in close contact with Rh, which apparently impedes the reduction of Rh oxide species. The second peak located at 500 K is increased in intensity compared to the unpromoted catalyst indicating that Mn has an impact on the nanostructure of the calcined precursor. The addition of Fe to the bimetallic Rh–Mn catalyst again shifts the main peak slightly to a lower temperature (377 K).15 The high-temperature reduction peak disappears almost completely in the TPR profile of Rh–Mn–Fe/SiO2 implying a homogeneous nanostructure of the catalyst that contains the two promoter elements.
The shift of the reduction temperature of Rh2O3 to higher temperature upon adding Mn- and Mn–Fe-oxide may be interpreted in terms of coverage of the Rh2O3 oxide particles with the promoter oxides, making H2 adsorption more difficult.
Reduced Rh–Mn–Fe/SiO2 contains crystalline metal nanoparticles with an fcc structure (Fig. 3). The lattice spacing in Rh–Mn–Fe/SiO2 is slightly reduced (d = 2.19 Å) compared to Rh/SiO2 and Rh–Mn/SiO2 (d = 2.23–2.25 Å) (Fig. S2†), which could be interpreted in terms of a Rh–Fe alloy formation.50 The deviation of approximately 2% is, however, rather small and may have other reasons, such as size-dependent reconstruction or interfacial strain.51–53 A clear statement regarding Rh–Fe alloy formation in Rh–Mn–Fe/SiO2 requires, therefore, further investigations.
 | ||
| Fig. 3 (a) HAADF-STEM image of the reduced Rh–Mn–Fe/SiO2 catalyst, showing Rh nano-particles; (b) colorized HAADF-STEM image corresponding to the dotted square region in (a) to highlight the locations of Rh (red) and the promoters (green). | ||
The HAADF-STEM image and EDX mapping presented in Fig. 4 are representatives of the entire reduced Rh–Mn–Fe/SiO2 catalyst. Elemental mapping indicates that all elements appear conjoined, but both promoter elements, in particular manganese, can be found in regions between the Rh particles (Fig. 4). Comparison of Rh and Mn elemental mapping in Fig. 4 indicates that the Rh particles in this particular catalyst are not generally anchored on a thin layer of manganese oxide. Such a configuration has been considered to be beneficial in terms of the promoting effect and maximization of the Rh–MnOx interface.25
 | ||
| Fig. 4 HAADF-STEM image and corresponding EDX elemental mapping of Fe, Mn and Rh, and a superimposed elemental map of the reduced Rh–Mn–Fe/SiO2 catalyst showing an intimate contact of the three elements. | ||
The TPR experiments were used to acquire information about the oxidation state of Mn and Fe in the reduced catalysts, but, in contrast to Rh/SiO2 and Rh–Mn/SiO2, the interpretation of hydrogen consumption for the Mn–Fe-promoted catalyst is less straightforward (Table 1).
Based on the information that (i) Rh occurs in oxidation state 3+ in the calcined catalysts as shown using XPS, (ii) the degree of reduction is 100% to metallic Rh as confirmed using XPS, and (iii) the formal oxidation state of manganese in the calcined promoted catalysts is 3+ as evidenced by XAS, the hydrogen consumption in TPR significantly exceeds the demand necessary to reduce Mn3+ to just Mn2+ in Rh–Mn/SiO2. In the literature the formal oxidation state of Mn in reduced Rh–Mn/SiO2 catalysts has been postulated to vary between 2+ and 0. Manganese in its oxidation state 2+ has been detected using EPR spectroscopy in reduced Rh–Mn/SiO2.11 In contrast, Mn–Rh alloy formation was proposed based on DFT calculations.22 The supplemental amount of hydrogen consumed in the TPR experiments of Rh–Mn/SiO2 and the information about the dominant oxidation state of Mn3+ in the calcined catalyst derived from XAS (Fig. 2) clearly indicate the formation of manganese sub-oxides with manganese in an average formal oxidation state of +0.22 (Table 1). However, the hydrogen consumption could be also in agreement with hydrogen storage in the catalyst during the temperature-programmed experiment. Absorption bands in the range between 2000 and 1500 cm−1 due to potential metal-hydride formation54,55 were observed occasionally on the freshly reduced catalysts in infrared experiments, but neither band position, nor appearance were proven reproducibly.
For calculation of the Mn oxidation state in the Rh–Mn–Fe/SiO2 catalyst formation of Fe0 was deliberately presumed.17,18,22,34,56 However, also in case of complete reduction of Fe to metallic iron the measured hydrogen consumption suggests the formation of manganese in an oxidation state below 2+. Complete reduction of Fe is, however, a limiting case that does not apply necessarily, because Fe was found by using HAADF-STEM (Fig. 4) not to be exclusively in contact with Rh, but also in contact with the support where it occurs most likely in sub-oxidic form. The hydrogen consumption in TPR of Rh–Mn–Fe/SiO2 suggests that Rh facilitates reduction of the promoter oxides and the formation of a Fe–Mn compound that contains residual oxygen and Mn and Fe in very low oxidation states, i.e., (Fe,Mn)Ox with x < 0.25.
In summary, a combination of temperature-programmed reduction experiments, XPS, XAS, and electron microscopy reveals that rhodium is completely reduced to metallic particles after treatment in hydrogen at 623 K. The Rh nanoparticles are in close contact with the promoters. Manganese forms sub-oxides in the Mn-promoted catalyst, whereas the oxidation state of Mn and Fe was estimated based on TPR, XPS and XAS to vary formally between zero and +2. Contraction of the lattice of the metal nano-particles determined using HAADF-STEM supports the formation of an alloy, but only in the case of Rh–Mn–Fe/SiO2. However, the identification of alloy formation is not unambiguous, because lattice contraction may have other reasons as well. Since different nanostructures are in agreement with these observations, infrared spectroscopy of adsorbed carbon monoxide has been used to probe the adsorption sites on the surface of the reduced catalysts.
Infrared spectroscopy of adsorbed CO in the absence and presence of hydrogen
Adsorption isotherms and infrared spectra of CO adsorbed at beam temperature on the reduced Rh-containing catalysts are shown in Fig. 5a–b. The two bands at 2058 cm−1 and 1881 cm−1 on Rh/SiO2 are due to linearly (Rh0–CO) and bridged bonded CO ((Rh0)x–CO), respectively. The peak at 2058 cm−1 is accompanied by two shoulders which appear on its high and low frequency side with absorption frequencies at 2095 cm−1 and 2015 cm−1 referring to the symmetric and asymmetric C–O stretching vibrations, respectively, of gem-dicarbonyl species Rh+(CO)2.57 Rh+ is formed due to either the oxidation of Rh via the dissociation of adsorbed CO or the oxidative addition of mobile Rh0–CO species to support hydroxyl groups.58 The observations are in full agreement with the typical spectra of CO adsorbed on supported Rh reported in the literature.59–61 Neither after calcination nor after reduction, were any absorption bands detected for both Mn/SiO2 and Fe/SiO2 due to the low stability of the carbonyl complexes of manganese and iron ions.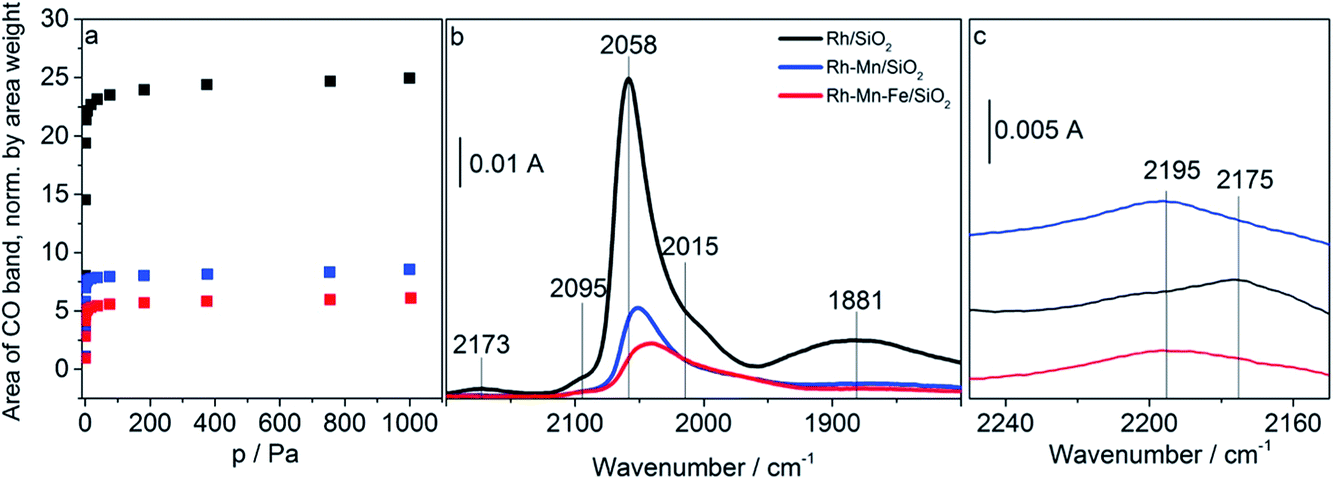 | ||
| Fig. 5 (a) Adsorption isotherms of adsorbed CO determined by using infrared spectroscopy at beam temperature (313 K); (b) FTIR spectra of adsorbed CO on Rh/SiO2, Rh–Mn/SiO2 and Rh–Mn–Fe/SiO2 as indicated in the figure at 10% coverage with CO, and (c) at 100% coverage with CO in the range between 2250 and 2150 cm−1. | ||
The addition of the promoter oxides induces three major changes in the infrared spectra: (i) reduction of the overall CO adsorption capacity (Fig. 5a and b), (ii) suppression of the band due to bridge-bonded CO (Fig. 5b), and (iii) shift of the band position of linearly adsorbed CO to lower wavenumbers. Furthermore, in the presence of gas-phase CO an additional band appears at low coverage (10%) already on Rh/SiO2 at 2175 cm−1 (Fig. 5b) and with increasing coverage (100%, Fig. 5c) on the promoted catalysts as well at approximately 2195 cm−1 due to the adsorption of CO on cationic sites formed either by reaction of the supported metals with CO or due to incomplete reduction of the catalyst.
The loss in adsorption capacity is in agreement with a partial coverage of the Rh particles with the promoter oxides resulting in less freely available Rh0 sites. This in turn suggests a lack of space needed for successful CO bridging with a pair of Rh0 atoms, which explains why the bridge-bonded CO species are hardly detectable for the promoted catalysts. In the case of the Rh–Mn catalyst the absorption bands show higher intensity than those observed for Rh–Mn–Fe/SiO2 probably due to the formation of extended oxide coverage in the latter case. A simple particle size effect can be ruled out as the reason for the differences in the intensity of the Rh0 carbonyl peaks, since TEM data revealed a narrow particle-size distribution with a maximum at approximately 2 nm for all the Rh-based catalysts as depicted in Fig. 6. No significant changes in the electronic properties of the Rh particles due to the presence of the promoters were observed by using XPS (Fig. S1†) and also the STEM-EDX mapping (Fig. 4) may be interpreted in terms of the coverage of the Rh particles by the promoter elements as one reason for the reduction of the CO absorption capacity.
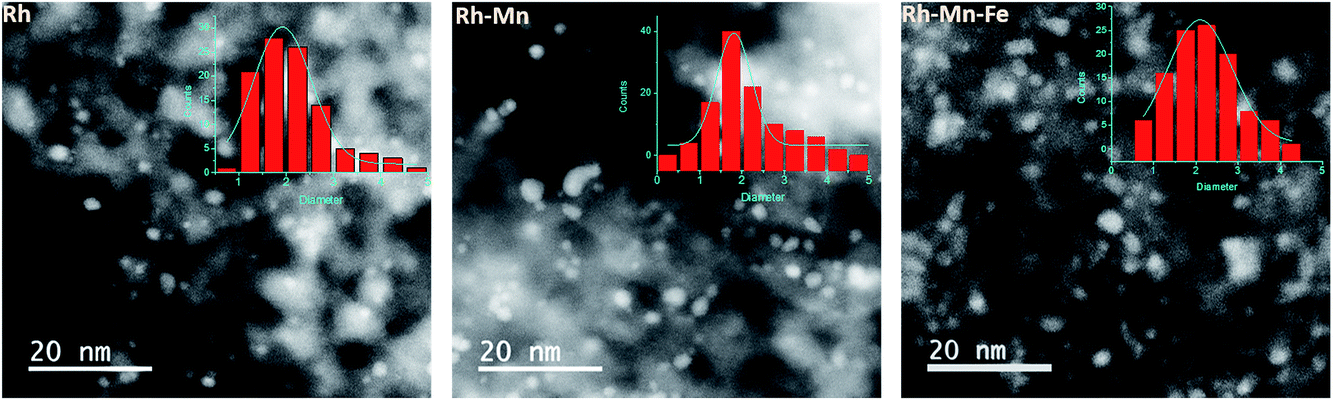 | ||
| Fig. 6 Scanning transmission electron microscope (STEM) images of the reduced Rh, Rh–Mn and Rh–Mn–Fe catalysts with particle size distribution histogram data. | ||
The formation of an alloy may suppress CO adsorption as well due to changes in the electronic structure.62,63 Since the CO adsorption is significantly reduced on both Rh–Mn/SiO2 and Rh–Mn–Fe/SiO2 the formation of a Rh–Fe alloy appears less likely to be the reason for the reduced adsorption capacity. The changes in the lattice constant observed by using HAADF-STEM only in the case of Rh–Mn–Fe/SiO2 (Fig. 3 and S2) may also have other reasons.
The explanation of the band shifts is less straightforward. The band due to Rh0–CO that appears at 2058 cm−1 for Rh/SiO2 (Fig. 5b) is shifted to 2051 cm−1 and 2036 cm−1 for the Mn- and Mn–Fe-containing catalysts, respectively (see Fig. S3† for the shift in the entire range of coverage). In particular the spectrum of Rh–Mn–Fe/SiO2 exhibits a distinct tail towards lower energies that cannot be ascribed exclusively to the low-energy component of the Rh+(CO)2 doublet. Similar features in the range between 2000 and 1950 cm−1 have been attributed to CO adsorption on a RhFe surface alloy.34 The very broad peak with a maximum near 1880 cm−1 for (Rh0)x–CO in the spectrum of Rh/SiO2 is hardly observable, but also shifted to lower vibrational frequencies when the promoters are present (Fig. S3†). The shift of the carbonyl bands to lower wavenumbers indicating a weakening of the C–O bond in adsorbed CO may arise from various factors. This shift primarily suggests an increase in the electron density at the Rh adsorption sites resulting in increased π-bonding due to a larger back donation of electrons from Rh to CO.64,65 The electronic effect is superimposed by shifts due to dipole–dipole coupling of adsorbed CO molecules. A decrease in dipole–dipole coupling also results in a red-shift (absorption band shifting toward the lower frequency side).66 Changes in the dipole–dipole coupling can be related to particle-size effects. Larger particles cause a shift of the bands toward higher wavenumbers since the local density of adsorbed CO molecules is higher than that which occurred in the smaller particles thus leading to a stronger dipolar coupling.61 However, as outlined above, all three catalysts exhibit a similar particle size distribution, which implies that the detected band shifts cannot be explained by changes in particle size. Reduced dipole–dipole coupling, in particular with regards to the band due to linearly adsorbed CO, would be in agreement with a partial coverage of the Rh0 surface by promoter oxides resulting in a higher fraction of single (isolated) Rh0 adsorption sites (holes in the promoter sub-oxide cover). By increasing the CO pressure (coverage) a shift of the band at 2059 cm−1 was observed to higher wavenumbers (by 14 cm−1) in the spectra of Rh/SiO2 (Fig. S3†). The shift is less distinct for the Mn- and Mn–Fe-promoted catalysts (10 cm−1 and 9 cm−1, respectively), which is also in agreement with a partial coverage. However, the shift of the band due to bridged bonded CO to lower wavenumbers seems to indicate indeed an impact of the promoters on the electronic properties of the metallic Rh particles, for example by alloy formation, which is in agreement with the reduced lattice spacing determined by electron microscopy (Fig. 3 and S2†). Alternatively, the low wavenumber of the bridged bonded CO could have its origin in the oxophilic behavior of the Mn and Fe promoter oxides, which can lead to an interaction of the oxygen atom in the CO ligands with the promoter oxides at the Rh0–MOx interface resulting in a weakening of the C–O bond. However, distinct bands below 1800 cm−1 have not been detected in the FTIR experiments performed at beam temperature.66
CO adsorption at liquid nitrogen temperature was used to study the interaction of CO with cationic sites potentially present on the surface of the promoted catalysts after reduction in hydrogen at 623 K. Adsorption isotherms and FTIR spectra recorded upon adsorption of CO (red data points and lines) and sequential adsorption of hydrogen followed by CO adsorption (blue data points and lines) on Rh–Mn–Fe/SiO2 at 77 K are shown in Fig. 7a–b. At 77 K CO molecules adsorbed on the promoted catalyst Rh–Mn–Fe/SiO2 give rise to a very broad absorption spectrum in the range between 2000 and 2120 cm−1 with two distinct maxima at 2090 and 2109 cm−1. The range above 2120 cm−1 cannot be probed due to the presence of a very intense band at 2158 cm−1 caused by van der Waals interactions between CO and silanol groups of the free silica support.67,68 The broadness of the signal in Fig. 7b indicates the presence of a high number of different adsorption sites that include also weak interactions between CO and the surface, which can only be detected by adsorption at low temperature. A clear assignment of surface Mn and Fe ions in low oxidation states is not possible based on this spectrum due to unclear assignments provided in the literature.69–72 However, the peak at 2109 cm−1 can certainly be attributed to CO adsorption on metal cations in a low oxidation state, either Rh+ or Mn/Fe sub-oxides (Mn and/or Fe in formal oxidation state < +2) as suggested through TPR and electron microscopy.
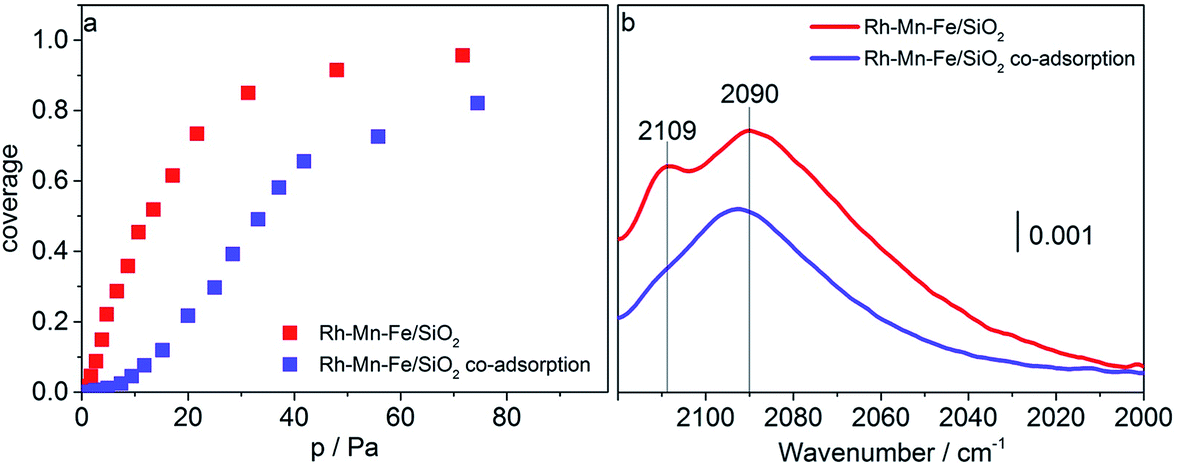 | ||
| Fig. 7 Adsorption isotherms of CO adsorbed on Rh–Mn–Fe/SiO2 determined using infrared spectroscopy at 77 K without (red) and with (blue) pre-adsorption of H2 (a), and FTIR spectra of adsorbed CO (coverage 10%) on Rh–Mn–Fe/SiO2 at 77 K without (red) and with (blue) pre-adsorption of H2 (b). The ratio of partial pressures of H2 and CO corresponds to H2:CO = 3:1. | ||
Most interestingly, the peak at 2109 cm−1 is suppressed when hydrogen is pre-adsorbed on the catalyst at 77 K (Fig. 7b, blue line). We may speculate at this point that hydrogen is adsorbed on manganese/iron sub-oxide species that partially cover the Rh particles. Generally, higher partial pressures of CO are required to reach comparable coverage of CO when hydrogen is pre-adsorbed (Fig. 7a, blue data points). The adsorption of hydrogen is certainly very weak (molecular) at this low temperature, because peaks due to hydride formation in the range 2000–1500 cm−1 were not observed. The present experiment supports the postulations that the promoter elements facilitate hydrogenation of the reaction intermediate in CO hydrogenation by promoting H2 adsorption.9,19
To probe the surface of the reduced Rh–Mn–Fe, Rh–Mn and Rh catalysts at 30 bar pressure and 523 K reaction temperature, in situ DRIFTS measurements were carried out. In the feed of synthesis gas the signals due to linearly adsorbed CO and the high-frequency peak of Rh dicarbonyl species are superimposed by the strong absorption of gas-phase CO (Fig. S4†). Only one broad band at 1872 cm−1 belonging to bridge-bonded CO species on Rh0 was detectable. In the experiments presented in Fig. 8, a flow of CO was first applied, which was then exchanged by pure argon. The obtained spectra for all the catalysts at 523 K and a pressure of 30 bar in flowing Ar (Fig. 8a) show the stable carbonyl species adsorbed at a reaction temperature of 523 K and p = 30 bar in the absence of gas phase CO. A major peak at 2036 cm−1 for the unpromoted catalyst, which is slightly shifted towards lower wavenumbers for the promoted ones, is visible and associated to linearly adsorbed CO on Rh0. For the promoted catalysts, the existence of these modes clearly indicates that metallic Rh is not entirely covered by the promoter oxides. In agreement with the low temperature adsorption experiments performed in transmission, DRIFTS provides compatible results pointing to the suppression of CO adsorption, which can be attributed to a partial coverage of the rhodium metal by promoter sub-oxides also at reaction temperature. A weak broad peak around 1900 cm−1 as evidence of bridge bonded CO modes on Rh0 was recorded with a signal intensity clearly stronger for Rh/SiO2 than that of the promoted catalysts. An additional band at 1564 cm−1 is only observed on Rh–Mn–Fe/SiO2. The signal may be attributed to carbonates or CO adsorbed in a tilted mode at the interface between Rh and the promoter sub-oxide.66 The promoter effect of manganese oxide has been frequently attributed to a stabilization of Rh+ sites.11,19,37,38,73 However, no Rh+ carbonyl species were detectable at reaction temperature excluding these species from being the active sites in CO hydrogenation. The present experiment clearly demonstrates that Rh+ does not exist on the surface of the promoted catalyst under reaction conditions (Fig. S4†).
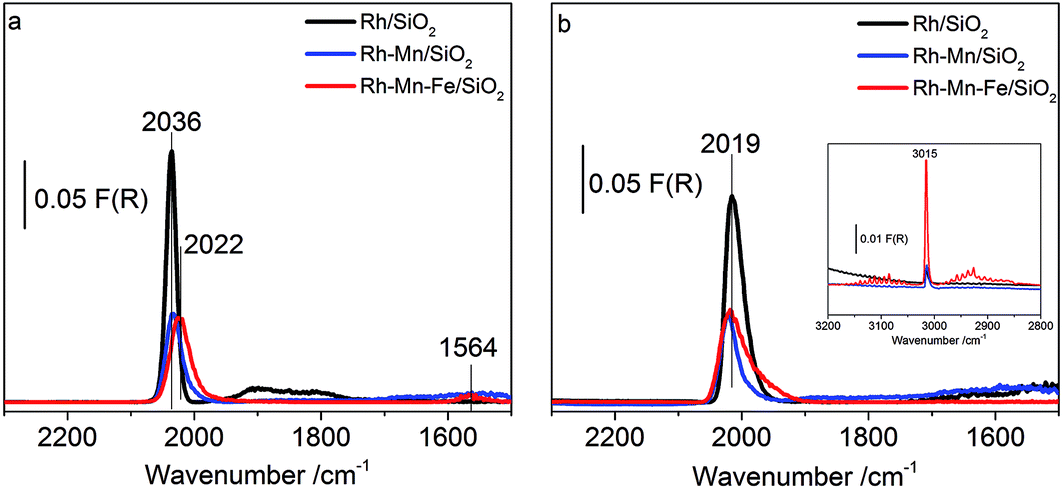 | ||
| Fig. 8 DRIFT spectra of the catalysts as indicated in the legend at p = 30 bar and T = 523 K after (a) contact with CO diluted in Ar (22.5% CO/77.5% Ar, flow rate 20 ml min−1) and subsequent purging with Ar (20 ml min−1) for ca. 30 min to remove gas-phase CO, and (b) treatment in H2 (87% H2/13% Ar, flow rate 77.5 ml min−1). | ||
Subsequent co-feeding of argon and hydrogen at 30 bar and 523 K on Rh/SiO2 (Fig. 8b) caused a red-shift of the signal due to linearly bonded CO to 2019 cm−1 and the disappearance of the band due to bridged bonded CO at 1900 cm−1. The peak shift indicates that hydrogen insertion into the Rh carbonyl bond occurs under formation of (Rh(H)CO and/or Rh(H2)CO).74,75 The corresponding Rh–H stretching vibrations are expected in the range between 2100 and 1900 cm−1 and may contribute to the broadening of the signal near 2000 cm−1.54 Hydrogen insertion into CO bonded to metallic Rh causes an increase in π-donation from the metal towards the antibonding π-orbital of the π-acceptor ligand (i.e., CO) and thus a red shift of the CO stretch occurs.76,77
The peak associated with the bridged bonded CO disappears immediately upon addition of hydrogen, but the intensity of the peak due to linearly adsorbed CO decreases only slightly on Rh/SiO2. In contrast, no changes were observed on the promoted catalysts even after prolonged treatment in flowing hydrogen at 30 bar and 523 K, except the disappearance of the band at 1564 cm−1. The formation of methane (inset in Fig. 8b, 3015 cm−1) was recorded as well over all catalysts. Knözinger et al., argued that it is rather difficult to distinguish which carbonyl species are responsible for the formation of the C2 oxygenates, since all species, such as the linear, bridged and tilted CO species, are removed upon reaction with hydrogen,66 unlike Ichikawa et al., who postulated that the bridged CO carbonyls (tilted-mode) are the active CO species for higher oxygenate synthesis.12 The results of this study provide a strong indication that the accessible metallic rhodium atoms on the surface of silica-supported Rh catalysts might be poisoned under the reaction conditions due to the strong adsorption of CO, which reacts only slowly with hydrogen at 523 K. Thus, taking into consideration all of the above results, it is concluded that the metallic Rh sites can be discarded as reactive sites and either mixed promoter sub-oxide(s) or species located at the perimeter between Rh and the promoter sub-oxide(s) should be considered.
Conclusions
It has been well known for decades that higher alcohols can be formed in CO hydrogenation through promoting supported Rh nanoparticles with Fe and Mn. The nanostructure of the promoted catalyst is, however, still debated. We resolved the mutual interaction between Rh nanoparticles and manganese/iron oxide promoters and dynamic structural changes at the atomic scale through applying a combination of integral techniques including temperature-programmed reduction, XPS, X-ray absorption and infrared spectroscopy with local observations of atomic arrangements by using HAADF-STEM imaging and EDX.The addition of promoters has no significant impact on the particle size distribution of rhodium, but the nano-structure of the metal particles is affected. Changes in the lattice constant of the metal particles as observed by using HAADF-STEM only in the case of Rh–Mn–Fe/SiO2 may be interpreted in terms of Rh–Fe alloy formation, but a clear interpretation requires further investigations. A close contact between Rh and the promoter elements is confirmed by using HAADF-STEM. Manganese and iron exist in very low formal oxidation states in the reduced promoted catalysts as evidenced through TPR and XAS. The comparable suppression of the CO adsorption capacity observed likewise on Rh–Mn/SiO2 as well as on Rh–Mn–Fe/SiO2 by using FTIR spectroscopy is attributed to the formation of perforated core–shell structures as illustrated in Scheme 1 for Rh–Mn–Fe/SiO2.
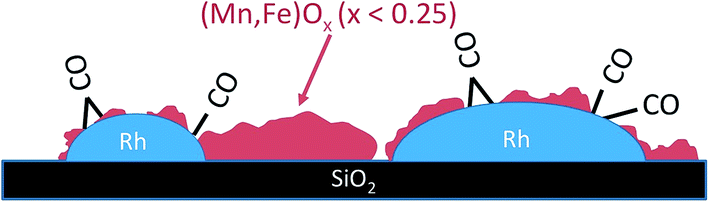 | ||
| Scheme 1 Schematic representation of promoter sub-oxide–metal interactions in reduced Rh–Mn–Fe/SiO2. Rh might be modified by alloy formation with Fe. | ||
The partial coverage of metallic Rh is stable at the reaction temperature of 523 K in the presence of hydrogen and under production of methane that comprises water formation as confirmed through DRIFTS. Rh+ cations have not been detected in the presence of hydrogen and are, therefore, excluded from playing an important role as active sites in the hydrogenation of CO to alcohols. Metallic Rh sites form strongly bound carbonyl complexes that react only very slowly at 523 K and 30 bar hydrogen. It is concluded that Rh0 is poisoned due to the adsorption of CO implying that the active sites in CO hydrogenation are associated either with (Mn,Fe)Ox (x < 0.25) or species at the interface between Rh and its co-catalyst (Mn,Fe)Ox.
Acknowledgements
This work was conducted in the framework of the BasCat collaboration between BASF SE, Technical University Berlin, Fritz-Haber-Institut der Max-Planck-Gesellschaft, and the cluster of excellence “Unified Concepts in Catalysis” (UniCat http://www.unicat.tu-berlin.de). The authors thank Maike Hashagen and Jasmin Allan for technical assistance, and Dr Andrey Tarasov and Dr Marc Willinger for discussions. Open Access funding provided by the Max Planck Society.Notes and references
- H. T. Luk, C. Mondelli, D. C. Ferre, J. A. Stewart and J. Perez-Ramirez, Chem. Soc. Rev., 2017, 46, 1358–1426 RSC.
- J. J. Spivey and A. Egbebi, Chem. Soc. Rev., 2007, 36, 1514–1528 RSC.
- I. Wender, Fuel Process. Technol., 1996, 48, 189–297 CrossRef.
- G. V. d. Lee and V. Ponec, Catal. Rev., 1987, 29, 183–218 Search PubMed.
- A. Medford, A. Lausche, F. Abild-Pedersen, B. Temel, N. Schjødt, J. Nørskov and F. Studt, Top. Catal., 2014, 57, 135–142 CrossRef.
- Z. Fan, W. Chen, X. Pan and X. Bao, Catal. Today, 2009, 147, 86–93 CrossRef.
- F. Solymosi, I. Tombácz and M. Kocsis, J. Catal., 1982, 75, 78–93 CrossRef.
- N. Yang, A. J. Medford, X. Liu, F. Studt, T. Bligaard, S. F. Bent and J. K. Nørskov, J. Am. Chem. Soc., 2016, 138, 3705–3714 CrossRef PubMed.
- R. Burch and M. I. Petch, Appl. Catal., A, 1992, 88, 39–60 CrossRef.
- M. M. Bhasin, W. J. Bartley, P. C. Ellgen and T. P. Wilson, J. Catal., 1978, 54, 120–128 CrossRef.
- T. P. Wilson, P. H. Kasai and P. C. Ellgen, J. Catal., 1981, 69, 193–201 CrossRef.
- M. Ichikawa and T. Fukushima, J. Phys. Chem., 1985, 89, 1564–1567 CrossRef.
- M. A. Haider, M. R. Gogate and R. J. Davis, J. Catal., 2009, 261, 9–16 CrossRef.
- J. Wang, Q. Zhang and Y. Wang, Catal. Today, 2011, 171, 257–265 CrossRef.
- J. Yu, D. Mao, L. Han, Q. Guo and G. Lu, J. Ind. Eng. Chem., 2013, 19, 806–812 CrossRef.
- F. Xue, W. Chen, X. Song, X. Cheng and Y. Ding, RSC Adv., 2016, 6, 35348–35353 RSC.
- Y. Liu, F. Göeltl, I. Ro, M. R. Ball, C. Sener, I. B. Aragão, D. Zanchet, G. W. Huber, M. Mavrikakis and J. A. Dumesic, ACS Catal., 2017, 7, 4550–4563 CrossRef.
- R. M. Palomino, J. W. Magee, J. Llorca, S. D. Senanayake and M. G. White, J. Catal., 2015, 329, 87–94 CrossRef.
- F. G. A. van den Berg, J. H. E. Glezer and W. M. H. Sachtler, J. Catal., 1985, 93, 340–352 CrossRef.
- W. M. H. Sachtler and M. Ichikawa, J. Phys. Chem., 1986, 90, 4752–4758 CrossRef.
- A. S. Lisitsyn, S. A. Stevenson and H. Knözinger, J. Mol. Catal., 1990, 63, 201–211 CrossRef.
- D. Mei, R. Rousseau, S. M. Kathmann, V.-A. Glezakou, M. H. Engelhard, W. Jiang, C. Wang, M. A. Gerber, J. F. White and D. J. Stevens, J. Catal., 2010, 271, 325–342 CrossRef.
- W. Mao, J. Su, Z. Zhang, X.-C. Xu, W. Dai, D. Fu, J. Xu, X. Zhou and Y.-F. Han, Chem. Eng. Sci., 2015, 135, 312–322 CrossRef.
- W. Mao, J. Su, Z. Zhang, X.-C. Xu, D. Fu, W. Dai, J. Xu, X. Zhou and Y.-F. Han, Chem. Eng. Sci., 2015, 135, 301–311 CrossRef.
- N. Yang, J. S. Yoo, J. Schumann, P. Bothra, J. A. Singh, E. Valle, F. Abild-Pedersen, J. K. Nørskov and S. F. Bent, ACS Catal., 2017, 5746–5757, DOI:10.1021/acscatal.7b01851.
- I. A. W. Filot, R. J. P. Broos, J. P. M. van Rijn, G. J. H. A. van Heugten, R. A. van Santen and E. J. M. Hensen, ACS Catal., 2015, 5, 5453–5467 CrossRef.
- J. Wang, Z. Liu, R. Zhang and B. Wang, J. Phys. Chem. C, 2014, 118, 22691–22701 Search PubMed.
- I. A. W. Filot, S. G. Shetty, E. J. M. Hensen and R. A. van Santen, J. Phys. Chem. C, 2011, 115, 14204–14212 Search PubMed.
- N. Kapur, J. Hyun, B. Shan, J. B. Nicholas and K. Cho, J. Phys. Chem. C, 2010, 114, 10171–10182 Search PubMed.
- M. Bwoker, Catal. Today, 1992, 15, 77–100 CrossRef.
- L. E. Y. Nonneman, A. G. T. M. Bastein, V. Ponec and R. Burch, Appl. Catal., 1990, 62, L23–L28 CrossRef.
- M. R. Gogate and R. J. Davis, ChemCatChem, 2009, 1, 295–303 CrossRef.
- P. R. Watson and G. A. Somorjai, J. Catal., 1982, 74, 282–295 CrossRef.
- J. W. Magee, R. M. Palomino and M. G. White, Catal. Lett., 2016, 146, 1771–1779 CrossRef.
- H.-Y. Luo, P.-Z. Lin, S.-B. Xie, H.-W. Zhou, C.-H. Xu, S.-Y. Huang, L.-W. Lin, D.-B. Liang, P.-L. Yin and Q. Xin, J. Mol. Catal. A: Chem., 1997, 122, 115–123 CrossRef.
- Y. Wang, Z. Song, D. Ma, H. Luo, D. Liang and X. Bao, J. Mol. Catal. A: Chem., 1999, 149, 51–61 CrossRef.
- M. Ojeda, M. L. Granados, S. Rojas, P. Terreros, F. J. García-García and J. L. G. Fierro, Appl. Catal., A, 2004, 261, 47–55 CrossRef.
- D. Jiang, Y. Ding, Z. Pan, W. Chen and H. Luo, Catal. Lett., 2008, 121, 241–246 CrossRef.
- W. M. H. Sachtler, D. F. Shriver, W. B. Hollenberg and A. F. Lang, J. Catal., 1985, 92, 429–431 CrossRef.
- P. R. Watson and G. A. Somorjai, J. Catal., 1981, 72, 347–363 CrossRef.
- S. S. C. Chuang and S. I. Pien, J. Catal., 1992, 135, 618–634 CrossRef.
- F. Li and W. Qian, Appl. Petrochem. Res., 2017, 7, 161–167 CrossRef.
- J. Yu, D. Mao, L. Han, Q. Guo and G. Lu, Catal. Commun., 2012, 27, 1–4 CrossRef.
- H. Yin, Y. Ding, H. Luo, H. Zhu, D. He, J. Xiong and L. Lin, Appl. Catal., A, 2003, 243, 155–164 CrossRef.
- C. Schlesiger, L. Anklamm, H. Stiel, W. Malzer and B. Kanngiesser, J. Anal. At. Spectrom., 2015, 30, 1080–1085 RSC.
- XAFS Spectra library, http://cars.uchicago.edu/xaslib/search.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef PubMed.
- C. H. M. van Oversteeg, H. Q. Doan, F. M. F. de Groot and T. Cuk, Chem. Soc. Rev., 2017, 46, 102–125 RSC.
- V. Schwartz, A. Campos, A. Egbebi, J. J. Spivey and S. H. Overbury, ACS Catal., 2011, 1, 1298–1306 CrossRef.
- S. K. Kim, Y. Tian, F. Jona and P. M. Marcus, Phys. Rev. B, 1997, 56, 9858–9863 CrossRef.
- G. A. Somorjai, Chem. Rev., 1996, 96, 1223–1236 CrossRef PubMed.
- G. J. P. Abreu, A. Pancotti, L. H. de Lima, R. Landers and A. de Siervo, J. Nanopart. Res., 2013, 15, 1510 CrossRef.
- J. L. Huang, Z. Li, H. H. Duan, Z. Y. Cheng, Y. D. Li, J. Zhu and R. Yu, J. Am. Chem. Soc., 2017, 139, 575–578 CrossRef PubMed.
- X. Wang and L. Andrews, J. Phys. Chem. A, 2002, 106, 3706–3713 CrossRef.
- X. Wang and L. Andrews, J. Phys. Chem. A, 2003, 107, 4081–4091 CrossRef.
- V. Schunemann, H. Trevino, G. D. Lei, D. C. Tomczak, W. M. H. Sachtler, K. Fogash and J. A. Dumesic, J. Catal., 1995, 153, 144–157 CrossRef.
- I. M. Hamadeh and P. R. Griffiths, Appl. Spectrosc., 1987, 41, 682–688 CrossRef.
- P. Basu, D. Panayotov and J. T. Yates, J. Am. Chem. Soc., 1988, 110, 2074–2081 CrossRef.
- C. Yang and C. W. Garl, J. Phys. Chem., 1957, 61, 1504–1512 CrossRef.
- J. T. Yates, T. M. Duncan, S. D. Worley and R. W. Vaughan, J. Chem. Phys., 1979, 70, 1219–1224 CrossRef.
- S. Trautmann and M. Baerns, J. Catal., 1994, 150, 335–344 CrossRef.
- Y. Koretaka, K. Yukinori, K. Akihide and T. Isao, J. Phys.: Condens. Matter, 2014, 26, 355006 CrossRef PubMed.
- G. Axel, J. Phys.: Condens. Matter, 2009, 21, 084205 CrossRef PubMed.
- H. Kusama, K. Sayama, K. Okabe and H. Arakawa, Nippon Kagaku Kaishi, 1995, 1995, 875–880 CrossRef.
- H. Kusama, K. Okabe, K. Sayama and H. Arakawa, Catal. Today, 1996, 28, 261–266 CrossRef.
- S. A. Stevenson, A. Lisitsyn and H. Knoezinger, J. Phys. Chem., 1990, 94, 1576–1581 CrossRef.
- T. P. Beebe, P. Gelin and J. T. Yates Jr, Surf. Sci., 1984, 148, 526–550 CrossRef.
- M. I. Zaki and H. Knözinger, Mater. Chem. Phys., 1987, 17, 201–215 CrossRef.
- A. F. H. Wielers, A. J. H. M. Kock, C. E. C. A. Hop, J. W. Geus and A. M. van Der Kraan, J. Catal., 1989, 117, 1–18 CrossRef.
- M. Kantcheva, M. U. Kucukkal and S. Suzer, J. Catal., 2000, 190, 144–156 CrossRef.
- M. Jiang, N. Koizumi and M. Yamada, J. Phys. Chem. B, 2000, 104, 7636–7643 CrossRef.
- K. I. Hadjiivanov and G. N. Vayssilov, in Advances in Catalysis, Academic Press Inc, San Diego, 2002, vol. 47, pp. 307–511, Vol 47 Search PubMed.
- H. Y. Luo, A. G. T. M. Bastein, A. A. J. P. Mulder and V. Ponec, Appl. Catal., 1988, 38, 241–253 CrossRef.
- T. Mineva, N. Russo and H.-J. Freund, J. Phys. Chem. A, 2001, 105, 10723–10730 CrossRef.
- T. Iizuka and Y. Tanaka, J. Catal., 1981, 70, 449–450 CrossRef.
- F. Solymosi and M. Lancz, J. Chem. Soc., Faraday Trans. 1, 1986, 82, 883–897 RSC.
- S. D. Worley, G. A. Mattson and R. Caudill, J. Phys. Chem., 1983, 87, 1671–1673 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: XPS and additional TEM and FTIR. See DOI: 10.1039/c7fd00215g |
| This journal is © The Royal Society of Chemistry 2018 |