 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of SCILL catalysts for the S–S coupling of thiols to disulfides†
Octavian D.
Pavel
 a,
Iunia
Podolean
a,
Vasile I.
Parvulescu
*a,
S. F. Rebecca
Taylor
c,
Haresh G.
Manyar
b,
Kathryn
Ralphs
b,
Peter
Goodrich
b and
Christopher
Hardacre
*bc
a,
Iunia
Podolean
a,
Vasile I.
Parvulescu
*a,
S. F. Rebecca
Taylor
c,
Haresh G.
Manyar
b,
Kathryn
Ralphs
b,
Peter
Goodrich
b and
Christopher
Hardacre
*bc
aUniversity of Bucharest, Faculty of Chemistry, Department of Organic Chemistry, Biochemistry and Catalysis, 030018, Bucharest, Romania. E-mail: vasile.parvulescu@chimie.unibuc.ro
bQueen’s University Belfast, The QUILL Centre/School of Chemistry and Chemical Engineering, Belfast, Northern Ireland, UK. E-mail: c.hardacre@manchester.ac.uk
cThe University of Manchester, School of Chemical Engineering & Analytical Science, Manchester, UK
First published on 10th May 2017
Abstract
This study reports the behaviour of SCILL based catalysts in the oxidative S–S coupling of aliphatic and aromatic thiols, namely 1-butanethiol and thiophenol, to dibutyl disulfide and diphenyl disulfide. A range of ionic liquids (1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide, 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide, 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide) and metal supported catalysts (5% Pt/SiO2; 5% Ru/SiO2; 5% Ru/C; 5% Pt/OMS-2) were used to prepare the SCILL catalysts and all were found to be active for the reaction following the trend 5% Pt-OMS-2 > 5% Pt/SiO2 > 5% Ru/C > 5% Ru/SiO2. The presence of SCILL catalysts afforded high selectivity to the disulfide, and the activity of the SCILL catalyst was dependent on the ionic liquid used. A significant increase in the stability of all the supported metal catalysts was found in the presence of the ionic liquid, and there was no change in the selectivity towards disulfides. This demonstrated that the ionic liquids protect the active sites of the catalyst against sulfation, thus providing more stable and active catalysts.
Introduction
Disulfide bonds play an important role in the folding and stability of many proteins.1–4 For example, S–S bonds are formed following the oxidative folding process between the thiol groups of cysteine, which can also hold different subunits of larger protein complexes together such as antibodies.5 These are also found in keratin in hair.6 In the refining of crude oil, oxidative desulfurization (ODS) of thiols forms the corresponding disulfides7 which then undergo further oxidation to thiosulfinates, disulfoxides, sulfinyl sulfones or disulfones. Disulfides are also used as vulcanizing agents for rubber8 and elastomers,9 in drugs,10 and in the design of rechargeable lithium batteries,11 for example.Traditionally, disulfides are produced through the oxidation of thiols in the presence of homogeneous catalysts including tributylammonium chlorochromate,12 metal phthalocyanines,13 cobalt(II) phthalocyanine tetrasodium sulfonate attached to poly(vinylamine),14 porphyrazinatocobalt(II) complex15 and iron(III)-ethylenediaminetetraacetic acid.16 These reactions are commonly performed in the presence of environmentally unfriendly oxidising agents such as sodium perborate,17 nitric oxide or nitrogen dioxide,18 permanganate,19 ammonium persulfate20 or haloacid/dimethyl sulfoxide.21 Industrially, the thiols are converted in aqueous caustic soda in order to increase their reactivity to dioxygen facilitating the use of air as the oxidant.22 However, the use of homogeneous catalysts coupled with these oxidizing agents has a number of disadvantages including catalyst separation issues, over-oxidation of the disulphide or thiol to sulfoxides, long reaction times, heavy metal contamination, toxicity and high cost. The use of a heterogeneous catalyst system has the potential to circumvent these challenges leading to more environmentally friendly processes.
To date, several systems have been explored including gold catalysts with O2,23 cobalt porphyrin intercalated into a phosphatoantimonic acid with air,24 reduced graphene oxide supported silicotungstic acid with H2O2,25 Bi2WO6 nanoparticles with air,26 chlorinated silica with O2,27 or Schiff base complexes of Ni, Co, Cr, Cd and Zn supported on magnetic nanoparticles with H2O2.28 Among these Suib reported the behavior of the OMS-2 catalyst for the oxidation of thiols in organic solvents.29 The present investigation focuses on the development of new ionic liquid modified catalysts for disulfide production.
Ionic liquids have received increasing attention for oxidation reactions. For example, 1-butyl-3-methylimidazolium methylselenite30 and 1-butyl-3-methylimidazolium tetrafluoroborate ([Bmim][BF4]) with oxygen,31 pyridinium-based ionic liquids with H2O2,32 or cobalt(II) phthalocyanines in [Bmim][BF4] with pure O2,33 have all been examined. Ionic liquids have some advantages over organic solvents in oxidations including non-flammability and the capacity to dissolve in various organic, inorganic and organometallic compounds. Another advantage of these solvents is that they facilitate the synthesis of highly polar inorganic raw materials under ambient conditions or under anhydrous conditions with low levels of water. In this way, the formation of metal oxy-hydroxides or hydrates can be suppressed. Ionic liquids also provide a medium where nano-metallic catalysts may be activated.34 For example, palladium complexes such as [C3CNpy]2[PdCl4], [PdCl2(C3CNpy)2][PdCl4] and [Pd(C3CNpy)2Cl2][PF6]2 are stable in air and do not decompose by washing with water or alcohol at room temperature.
More recently, three concepts allowed processes to ameliorate bi- and tri-phasic reactions. The Supported Ionic Liquid Phase (SILP)35–38 concept consists of a metal complex dissolved in a thin IL layer that is immobilized on an inert support material. SILP catalyst systems combine the benefits of homogeneous catalysts such as high activity and selectivity with stability and easy separation of product and catalyst. The second concept (Supported Ionic Liquid Catalyst (SILC)) refers to the anchoring of an ionic liquid on the surface of a support via a covalent bond.39 The third is defined as Solid Catalyst with an Ionic Liquid Layer (SCILL) and consists of a heterogeneous catalyst that is coated by a thin layer of an ionic liquid.40
This paper reports on the preparation of several SCILL catalysts, where the ionic liquids contain the same anion (bis(trifluoromethylsulfonyl)imide) and a range of cations (1-butyl-3-methylimidazolium, 1-ethyl-3-methylimidazolium, 1-butyl-1-methylpyrrolidinium) and are deposited onto active solids: 5 wt% Pt/SiO2, 5 wt% Ru/SiO2, 5 wt% Ru/C, 5 wt% Pt/OMS-2. These new catalysts were investigated in the aerobic oxidative coupling of 1-butanethiol and thiophenol to dibutyl disulfide and diphenyl disulfide, respectively, under mild temperatures and pressures using air as the oxidation source.
Experimental
All the organic reagents were commercial products of analytical purity and were used without further purification. The silica and carbon based heterogeneous catalysts were obtained from Johnson Matthey (5% Pt/SiO2 Batch M04483, JM ref. 108070; 5% Ru/SiO2 Batch M04484, JM ref. 108070; 5% Ru/C Batch M02070, JM ref. 94890). The 5% Pt/OMS-2 (cryptomelane-type manganese oxide) was prepared as reported previously.41 1-Butanethiol (99%) and thiophenol (≥99%) were purchased from Sigma-Aldrich.Catalysts preparation
All the ionic liquids were prepared following standard procedures from the bromide salt of the parent cation.42 The ionic liquids were dried for 24 h under temperature and vacuum (333.15 K, 0.02 mbar). The resulting ionic liquids were characterized using 13C and 1H NMR spectroscopy and Inductively Coupled Plasma Optical Emission Spectrometry (ICP OES). The NMR data and the Li content for all the ionic liquids are presented in Fig. SI1–SI6† and are consistent with previously reported NMR data.The SCILLs were prepared by mixing a solution of the ionic liquid in dichloromethane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 vol/vol ratio) with each catalyst using a 1:1 (w/w) ratio of IL to catalyst. The SCILL suspension was stirred for 2 h followed by solvent removal under vacuum and drying for 24 h under temperature and vacuum (333.15 K, 0.02 mbar).
:3 vol/vol ratio) with each catalyst using a 1:1 (w/w) ratio of IL to catalyst. The SCILL suspension was stirred for 2 h followed by solvent removal under vacuum and drying for 24 h under temperature and vacuum (333.15 K, 0.02 mbar).
Catalytic tests
S–S coupling of thiols to disulfides, Scheme 1, was performed in a glass batch reactor equipped with a condenser system. Typically, 0.09 mmol of thiol (1-butanethiol or thiophenol) was stirred at 600 rpm with 10 cm3 of cyclohexane (the nonpolar solvent) and heated in a silicon oil bath to the reflux temperature. Once the reaction temperature was stabilized, the catalysts (0.18 g SCILL: 0.09 g pure ionic liquid and 0.09 g supported metal catalysts) were added and the reaction started. In order to follow the progress of the reaction, aliquots of 0.5 cm3 were periodically removed from the reactor, filtered and analyzed by GC-MS and GC where the retention times of the peaks were compared against authentic samples obtained from Aldrich.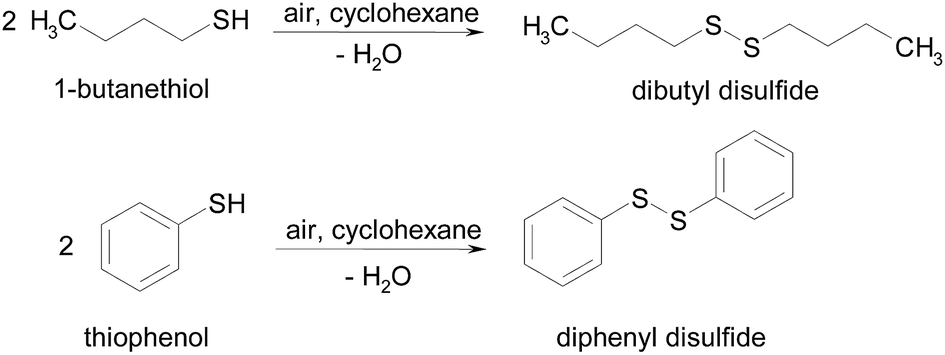 | ||
| Scheme 1 Pathway of the oxidation with air of both thiols under mild conditions. | ||
Catalyst recycling
After the reactions, the catalysts were removed by filtration, washed 3 times with 10 cm3 of cyclohexane followed by drying for 1 h at 80 °C. Then, they were re-introduced in the glass batch reactor after the reaction temperature had reached reflux conditions.Characterization techniques
1H NMR spectra were recorded on a Bruker UltraShield 500 MHz spectrometer. Powder X-ray diffraction patterns were recorded with a Shimadzu XRD 7000 diffractometer using Cu Kα radiation (λ = 1.5418 Å, 40 kV, 40 mA) at a scanning speed of 0.10° min−1 in the 5–80° 2θ range. The leaching of metals from the catalyst was monitored by induced coupled plasma analysis on an Agilent Technologies 715 ICP-OES apparatus. XPS analysis was carried out using Kratos AXIS Ultra DLD apparatus equipped with a monochromated Al Kα X-ray source, a charge neutralizer and a hemispherical electron energy analyzer with a pass energy of 160 eV. Background subtraction was performed using a Shirley background, and CasaXPS software was employed to treat the data. The XPS was referenced to the aliphatic C 1s feature at 284.8 eV.Results and discussion
Following deposition onto the catalyst surfaces, the ionic liquids (ILs) showed no discernible changes in the NMR spectra, Fig. SI7–SI10,† and no leaching of K was found in the case of OMS-2. In addition, no chemical interaction between the IL and the investigated solid surfaces was evidenced by XPS. For example (Table SI1†), on depositing [Bmim][NTf2] on 5 wt% Pt/OMS-2 no significant changes were observed in the binding energies of the Pt 4f features compared with the fresh sample41 or the C 1s features compared with the pure ionic liquid.43The XRD patterns of OMS-2 and 5 wt% Pt/OMS-2 present only the lines assigned to the support (Fig. 1), which are characteristic of 2 × 2 tunnel structure manganese oxide phases with a chemical composition of KMn8O16.44,45 No other manganese oxide forms were detected. No XRD features associated with Pt were observed, consistent with a high metal dispersion. However, Pt incorporation led to a decrease of the intensity of the diffraction lines of the OMS-2 phase, as compared to the Pt free support (Fig. SI11†). The XRD also confirmed that no significant changes in the structure of the support had occurred on deposition of the ILs. Similar observations were found for the carbon and silica based catalysts (Fig. 2 and 3).
 | ||
| Fig. 1 XRD patterns of SCILL materials with 5% Pt/OMS-2 as the support. | ||
 | ||
| Fig. 2 XRD patterns of SCILL samples with charcoal as the support. | ||
 | ||
| Fig. 3 XRD patterns of SCILL samples with silica as the support. | ||
The oxidation of the thiols was conducted using the SCILL catalysts, the catalyst without IL modification, and in the pure IL. In the latter, although some oxidation of thiols occurred, the conversions were found to be below 4% which is consistent with the background reaction in the absence of a radical initiator.46 Spontaneously, this reaction occurs at a very slow rate, whenever any thiol is exposed to atmospheric oxygen.47
The selectivity towards the corresponding disulfides was, however, higher than 99%, confirming the fact that under these conditions the oxidation is mild compared with the use of more aggressive conditions. For example, using hydrogen peroxide, thiols (RSH) are oxidized to sulfonic acid (RSO3H) via sulfenic acid (RSOH) and sulfinic acid (RSO2H).48
It should be noted that the selectivity was found to be quantitative to the disulfide for all the reactions investigated in this study, irrespective of the system used.
The catalytic activity for the oxidation of both 1-butanethiol and thiophenol was examined and the results are shown in Fig. 4 and 5, respectively, and summarized in Table 2. For 1-butanethiol the conversion varied in the following order: 5% Pt/OMS-2 > 5% Pt/SiO2 > 5% Ru/C > 5% Ru/SiO2 (Fig. 4).
 | ||
| Fig. 4 The activity of the catalysts in the conversion of 1-butanethiol (0.09 mmol thiol; 600 rpm; 10 cm3 cyclohexane; 0.09 g supported metal catalysts or 0.18 g SCILL; reflux; 5 h). | ||
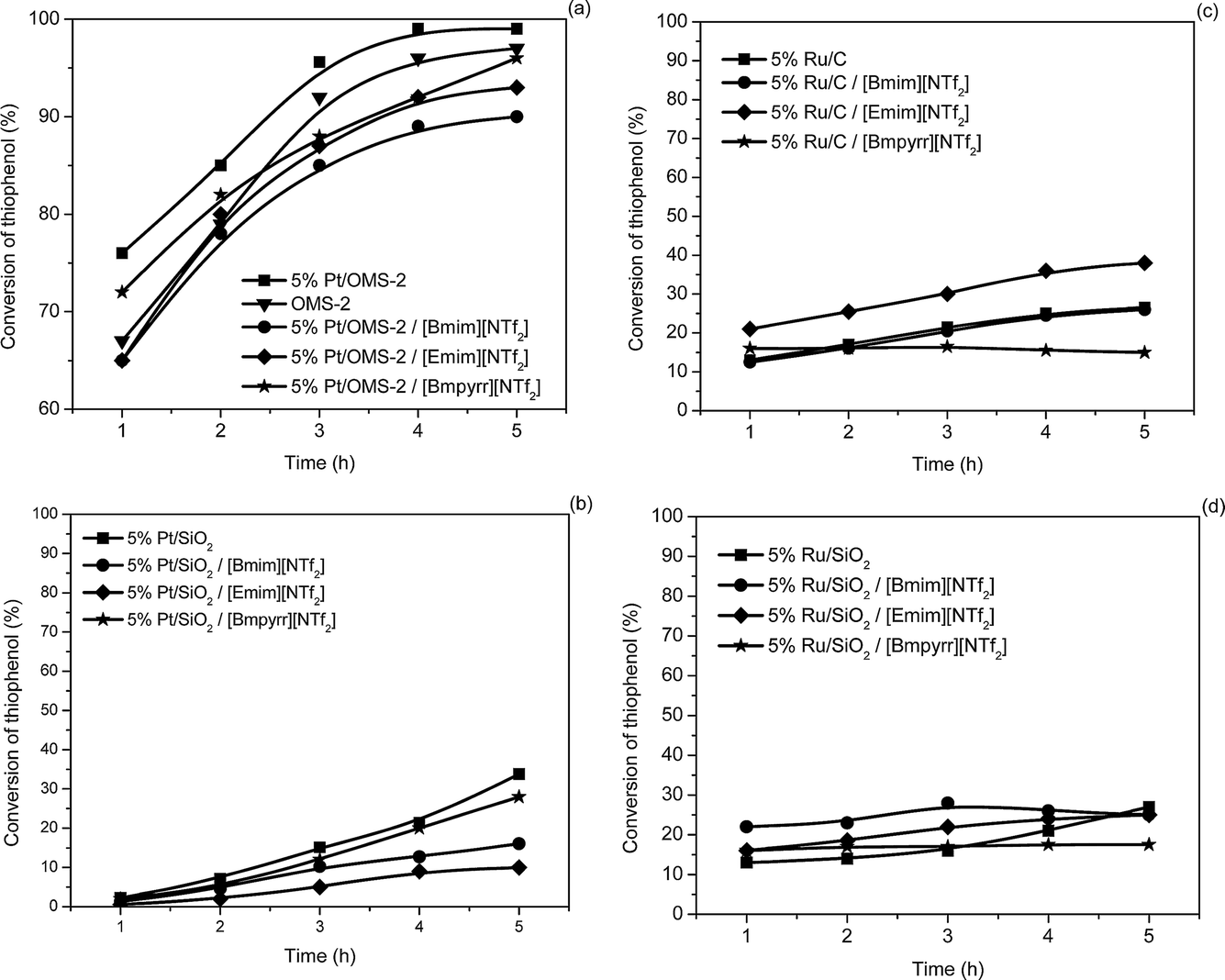 | ||
| Fig. 5 The activity of the catalysts in the conversion of thiophenol (0.09 mmol thiol; 600 rpm; 10 cm3 cyclohexane; 0.09 g supported metal catalysts or 0.18 g SCILL; reflux; 5 h). | ||
5% Pt/OMS-2 exhibited the highest catalytic activity, which may be associated with the increased basicity as a result of a synergistic effect of potassium associated to octahedral MnO6 and platinum. To date, manganese is one of the most used cations for the catalysts utilized in the Merox process,49 while K cation intermediates are observed in the production of disulfides via the formation of potassium mercaptide. Platinum also plays a clear role that is highlighted by the difference in the activity between 5% Pt/OMS-2 (99%) and OMS-2 (90%) after 1 h (Fig. 4a). After 5 h reaction time the conversion was 95% for OMS-2 and 99% for 5% Pt/OMS-2. The total available total surface oxygen atoms is of the order of 2.4 × 1022 atoms, while the number of reactant molecules present was ∼6.0 × 1022. Since not all the surface oxygen is likely to be active and for every 2 thiol molecules 1 oxygen atom is required, the process is likely to occur via a Mars van Krevelen process since a Wolkenstein mechanism will generate a total oxidation that has not been observed. This mechanism is in accordance with that proposed by Suib et al.29 and was confirmed by additional experiments carried out in nitrogen. Under the inert atmosphere the conversion on 5% Pt/OMS-2 catalyst was 17% after 5 h.
It is clear from Fig. 4 and Table 1 that, compared to reactions carried out with the pure supported metal catalysts, the SCILL catalysts resulted in slightly lower conversions after 5 h, in general. This effect is small, and may be associated with a slightly slower diffusion of the substrate or the oxygen through the IL layer and thus a reduction in the surface concentration. It is interesting to note that the conversions do not reach 100% in many of the cases for 1-butanethiol, which is thought to be indicative of product poisoning of the catalyst surface.
| Samples | N W–P × 106 | |
|---|---|---|
| 1-Butanethiol | Thiophenol | |
| OMS-2 | 1.83 | 1.73 |
| 5% Pt/OMS-2 | 1.90 | 1.91 |
| 5% Pt/OMS-2/[Bmim][NTf2] | 1.78 | 1.79 |
| 5% Pt/OMS-2/[Emim][NTf2] | 1.81 | 1.85 |
| 5% Pt/OMS-2/[Bmpyrr][NTf2] | 1.71 | 1.87 |
In order to estimate the pore diffusion influence upon the reaction rate, the Weisz–Prater criterion50 has been calculated for all the OMS-2-based catalysts used using eqn (1):
| NW–P = R × ρp × Rp2/Cs × Deff | (1) |
Table 1 summarises the calculated values which are of the order of 10−6. These are five orders smaller than the limit of 0.3, indicating the absence of the pore diffusion limitation. Importantly neither the size nor structure of the IL used has any significant influence on this effect.
The activity in the presence of 100% oxygen is far superior to air (Table 2). The activity of SCILL based on 5% Pt/OMS-2 and 5% Pt/SiO2 is >99% for all the ILs tested after 5 h using O2 as the oxidant. In comparison, lower conversions were obtained using air.
| Catalysts | Conversion of 1-butanethiol (%) | |
|---|---|---|
| With air | With O2 | |
| a Reaction conditions: 0.09 mmol of 1-butanethiol; 10 cm3 of cyclohexane; 0.18 g SCILL; reflux; 5 h. | ||
| 5% Pt/OMS-2/[Bmim][NTf2] | 92 | >99 |
| 5% Pt/OMS-2/[Emim][NTf2] | 94 | >99 |
| 5% Pt/OMS-2/[Bmpyrr][NTf2] | 89 | >99 |
| 5% Pt/SiO2/[Bmim][NTf2] | 90 | >99 |
| 5% Pt/SiO2/[Emim][NTf2] | 82 | >99 |
| 5% Pt/SiO2/[Bmpyrr][NTf2] | 92 | >99 |
| 5% Ru/C/[Bmim][NTf2] | 37 | 60 |
| 5% Ru/C/[Emim][NTf2] | 45 | 67 |
| 5% Ru/C/[Bmpyrr][NTf2] | 41 | 63 |
| 5% Ru/SiO2/[Bmim][NTf2] | 21 | 51 |
| 5% Ru/SiO2/[Emim][NTf2] | 22 | 56 |
| 5% Ru/SiO2/[Bmpyrr][NTf2] | 20 | 55 |
This is due to the lower partial pressure of O2 in the feed and, therefore, the reduced surface concentration. In addition, the competition between nitrogen and oxygen through ionic liquids is another controlling factor. This has been shown by changing the balance gas from nitrogen to argon. A small but significant increase in the conversion (95%) of 1-butanethiol using 5% Pt/OMS-2/[Bmim][NTf2] was obtained when working with 20% O2 in Ar compared to 92% in 20% O2 in N2. It should be noted that although this difference is small it is reproducible over 5 repeats. Thus, the solubility of oxygen, nitrogen and argon in pure ionic liquids expressed by Henry’s law constant at 1 atm and 70 °C is different [KH = 1854 (for O2), 3064 (for N2) and 2707 (for Ar)].52 This may correspond to more inert gas in the ionic liquid in the case of nitrogen than in the case of argon. As a direct consequence the concentration of oxygen in the reaction conditions is higher when it is mixed with argon. Also, the oxygen solubility in ionic liquids is higher compared to molecular solvents.52 Previous reports on the oxidation of n-dodecyl amine in the presence of SILP catalysis, with [Bmim]Cl as the ionic liquid, indicated that a decrease in the nitrogen content corresponded to a significant increase in the conversion with no effect on the selectivity.53 Previously, a positive effect of CO2 in terms of the increase of the solubility of oxygen (from air) in ionic liquid has been reported.52 However, under our experimental conditions the concentration of CO2 was very low and no such effect has been evidenced.
For the reaction with sulfides the stability of the noble metal against sulfur poisoning is also very important. The data presented in this study indicated better performance for Pt compared to Ru, in agreement with previous reports.54
Changing the aliphatic substrate with a thiophenol led, after 5 h, using 5% Pt/OMS-2-SCILL (Fig. 5a), to comparable conversions as for 1-butanethiol. Again, OMS-2 behaved as the best support in this reaction. The 5% Pt/SiO2 catalyst showed a lower activity (Fig. 5b) in the oxidative coupling of thiophenol irrespective of the reaction conditions, i.e. in the presence or absence of ILs. As was found for 1-butanethiol, the structure of the IL in the investigated series exhibits almost no influence.
Stability and catalyst recycling
In all cases, after S–S coupling, the catalyst could be easily separated by simple filtration and reused. Induced coupled plasma analysis confirmed that no metal was present in the filtrate (for SCILL and fresh solids), showing no significant leaching of the metal from the catalysts. No significant structural changes were observed in the XRD patterns of the 5% Pt/OMS-2 fresh and spent catalysts (Fig. SI12†). However, a continuous decay in the activity has been measured. The decrease, presented in Table 3, is attributed to the blockage of the active sites by sulfur.| Substrate | Catalyst | Cycle 1 | Cycle 2 | Cycle 3 |
|---|---|---|---|---|
| a IL = [Bmim][NTf2]. | ||||
| 1-Butanethiol | 5% Pt/OMS-2 | >99 | 70 | 32 |
| 5% Pt/OMS-2/ILa | 92 | 80 | 61 | |
| 5% Pt/SiO2 | 85 | 72 | 63 | |
| 5% Pt/SiO2/ILa | 90 | 79 | 72 | |
| 5% Ru/C | 54 | 39 | 24 | |
| 5% Ru/C/ILa | 37 | 32 | 27 | |
| Thiophenol | 5% Pt/OMS-2 | >99 | 75 | 51 |
| 5% Pt/OMS-2/ILa | 90 | 72 | 60 | |
| 5% Ru/C | 26 | 21 | 14 | |
| 5% Ru/C/ILa | 26 | 22 | 18 | |
The nature of the thiol influences this process. 1-Butanethiol led to a faster deactivation of unmodified 5% Pt/OMS-2 than thiophenol. In contrast, in the presence of the IL, the catalyst retained significantly more activity on recycling. In addition, the recyclability using the aromatic and aliphatic thiols was similar in the presence of the IL. This is likely to be associated with the IL reducing the adsorption strength of the disulfide or thiol, which allows the sites to be free for the oxidation.
As mentioned above the nature of the catalyst is also responsible for the deactivation. The presence of potassium in the catalyst may generate potassium mercaptides that reduce the interaction with the noble metals. However, it is noticeable that the SCILL catalyst delays these complexation processes and, as a consequence, preserves the catalytic activity on cycling. On this basis we may speculate that the ionic solvent may better extract sulfur species from the catalyst surface. Differences between Ru and Pt can be assigned to the strength of the Ru–S and Pt–S bond energies. The strong adsorption in the case of Ru led to a faster deactivation, and as a consequence lower conversions. Ionic liquids also ensure an increased solubility of the aromatic thiols compared to the aliphatic thiols due to the interaction of the aromatic ring with the cation of the ionic liquids thus increasing the effectiveness of the catalyst for aromatic disulfide formation.55
The support influences the solid recycling. Thus, for the oxidation of 1-butanethiol, SCILL deactivation is less significant for Pt/SiO2 with a decrease of the conversion of 20% from the initial value compared with 33% for the Pt/OMS-2 catalyst. This is in agreement with the fact that the reaction is not limited for the SiO2 catalyst compared with the OMS-2 system and may reflect the weaker binding of the disulphide, for example.
Conclusions
The investigated SCILL catalysts were easily prepared by depositing various ionic liquids (1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide, 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide, 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide) onto the prepared catalyst materials. The activity of the SCILL catalysts in the oxidation of the investigated thiols varied as follows: 5% Pt-OMS-2 > 5% Pt/SiO2 > 5% Ru/C > 5% Ru/SiO2. Although a small decrease in activity was observed on using the SCILL based catalysts compared with the ionic liquid-free systems, the latter showed a decreased stability for both aliphatic and aromatic thiols. This was attributed to the ability of the ionic liquid to weaken the adsorption strength of the sulfur species on the metal and decrease the issue of reactant/product poisoning.Acknowledgements
The UK Catalysis Hub is kindly thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC (Portfolio Grants EP/K014706/2, EP/K014668/1, EP/K014854/1, EP/K014714/1 and EP/I019693/1). Open access data can be found via the University of Manchester research portal.Notes and references
- T. E. Creighton, BioEssays, 1988, 8, 57 CrossRef CAS PubMed.
- S.-G. Chang, K.-D. Choi, S.-H. Jang and H.-C. Shin, Mol. Cells, 2003, 16, 323 CAS.
- W. J. Wedemeyer, E. Welker, M. Narayan and H. A. Scheraga, Biochemistry, 2000, 39, 4207 CrossRef CAS PubMed.
- G. Bulaj, Biotechnol. Adv., 2005, 23, 87 CrossRef CAS PubMed.
- R. Glockshuber, T. Schmidt and A. Pliickthun, Biochemistry, 1992, 31, 1270 CrossRef CAS PubMed.
- R. D. B. Fraser, T. P. MacRae and L. G. Sparrow, Int. J. Biol. Macromol., 1988, 10, 106 CrossRef CAS.
- R. Javadli and A. de Klerk, Appl. Petrochem. Res., 2012, 1, 3 CrossRef CAS.
- A. Das, N. Naskar and D. K. Basu, J. Appl. Polym. Sci., 2004, 91, 1913 CrossRef CAS.
- M. Akiba and A. S. Hashim, Prog. Polym. Sci., 1997, 22, 475 CrossRef CAS.
- G. Saito, J. A. Swanson and K.-D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199 CrossRef CAS PubMed.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742 CrossRef CAS.
- S. Ghammamy, Asian J. Chem., 2007, 19, 917 CAS.
- C. F. Cullis and D. L. Trimm, Discuss. Faraday Soc., 1968, 46, 144 RSC.
- W. M. Brouwer and A. L. German, J. Mol. Catal., 1984, 22, 297 CrossRef CAS.
- H. Karaca, M. Teker and A. Gül, Chem. J., 2016, 6, 55 Search PubMed.
- T. V. Rao, B. Sain, P. S. Murthy, T. S. R. Prasada Rao, A. K. Jain and G. C. Joshi, J. Chem. Res., 1997, 300 RSC.
- A. McKillop, D. K. Krief, W. Dumont, P. Renier and M. Trabelsi, Tetrahedron Lett., 1990, 31, 5007 CrossRef CAS.
- W. A. Pryor, D. F. Church, C. K. Govindan and G. Crank, J. Org. Chem., 1982, 47, 156 CrossRef CAS.
- A. Shaabani and D. G. Lee, Tetrahedron Lett., 2001, 42, 5833 CrossRef CAS.
- R. S. Varma, H. M. Meshramzt and R. Dahiyal, Synth. Commun., 2000, 30, 1249 CrossRef CAS.
- P. Natarajan, H. Sharma, M. Kaur and P. Sharma, Tetrahedron Lett., 2015, 56, 5578 CrossRef CAS.
- C. Marty, White product refining by sweetening, in Petroleum refining, Conversion processes, ed. P. Leprince, Editions Technip, Paris, 2001, vol. 3, pp. 503–532 Search PubMed.
- A. Corma, T. Ródenasa and M. J. Sabater, Chem. Sci., 2012, 3, 398 RSC.
- X. Hu, Z. Huang, G. Gu, L. Wang and B. Chen, J. Mol. Catal. A: Chem., 1998, 132, 171 CrossRef CAS.
- S. V. Nipane, M. G. Mali and G. S. Gokavi, Ind. Eng. Chem. Res., 2014, 53, 3924 CrossRef CAS.
- Y. P. Bhoi, D. P. Rout and B. G. Mishra, J. Cluster Sci., 2016, 27, 267 CrossRef CAS.
- M. Sathe, R. Ghorpade and M. P. Kaushik, Chem. Lett., 2006, 35, 1048 CrossRef CAS.
- A. Ghorbani-Choghamarani, Z. Darvishnejad and B. Tahmasbi, Inorg. Chim. Acta, 2015, 435, 223 CrossRef CAS.
- S. Dharmarathna, C. K. King’ondu, L. Pahalagedara, C.-H. Kuo, Y. Zhang and S. L. Suib, Appl. Catal., B, 2014, 147, 124 CrossRef CAS.
- S. Thurow, V. A. Pereira, D. M. Martinez, D. Alves, G. Perin, R. G. Jacob and E. J. Lenardão, Tetrahedron Lett., 2011, 52, 640 CrossRef CAS.
- D. Singh, F. Z. Galetto, L. C. Soares, O. E. Dorneles Rodrigues and A. L. Braga, Eur. J. Org. Chem., 2010, 2661 CrossRef CAS.
- D. Zhao, Y. Wang and E. Duan, Molecules, 2009, 14, 4351 CrossRef CAS PubMed.
- S. M. S. Chauhan, A. Kumar and K. A. Srinivas, Chem. Commun., 2003, 2348 RSC.
- V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615 CrossRef PubMed.
- C. P. Mehnert, E. J. Mozeleski and R. A. Cook, Chem. Commun., 2002, 3010 RSC.
- C. P. Mehnert, R. A. Cook, N. C. Dispenziere and M. Afeworki, J. Am. Chem. Soc., 2002, 124, 12932 CrossRef CAS PubMed.
- A. Riisager, K. M. Eriksen, P. Wasserscheid and R. Fehrmann, Catal. Lett., 2003, 90, 149 CrossRef CAS.
- A. Riisager, P. Wasserscheid, R. van Hal and R. Fehrmann, J. Catal., 2003, 219, 452 CrossRef CAS.
- T. Selvam, A. MacHoke and W. Schwieger, Appl. Catal., A, 2012, 445–446, 92 CrossRef CAS.
- U. Kernchen, B. Etzold, W. Korth and A. Jess, Chem. Eng. Technol., 2007, 30, 985 CrossRef CAS.
- I. J. McManus, H. Daly, H. G. Manyar, S. F. Rebecca Taylor, J. M. Thompson and C. Hardacre, Faraday Discuss., 2016, 188, 451 RSC.
- D. Zhao, Z. Fei, R. Scopelliti and P. Dyson, Inorg. Chem., 2004, 43, 2197 CrossRef CAS PubMed.
- O. Hofft, S. Bahr, M. Himmerlich, S. Krischok, J. A. Schaefer and V. Kempter, Langmuir, 2006, 22, 7120 CrossRef CAS PubMed.
- P. H. Ho, S. C. Lee, J. Kim, D. Lee and H. C. Woo, Fuel Process. Technol., 2015, 131, 238 CrossRef CAS.
- C. C. King’ondu, N. Opembe, C. Chen, K. Ngala, H. Huang, A. Iyer, H. F. Garcés and S. L. Suib, Adv. Funct. Mater., 2011, 21, 312 CrossRef.
- P. V. Ioannou, Main Group Chem., 2013, 12, 125 CAS.
- N. Kharasch, in Organic Sulfur Compounds, Pergamon Press, New York, Oxford, London, Paris, 1961 Search PubMed.
- L. A. H. van Bergen, G. Roos and F. De Proft, J. Phys. Chem. A, 2014, 118, 6078 CrossRef CAS PubMed.
- B. Basu, S. Satapathy and A. K. Bhatnagar, Catal. Rev. Sci. Eng., 1993, 35, 571 CAS.
- P. B. Weisz and C. D. Prater, Adv. Catal., 1954, 6, 143 CAS.
- O. Levenspiel, Chemical Reaction Engineering, John Wiley & Sons, 3rd edn, 1999, ch. 3 Search PubMed.
- Z. Lei, C. Dai and B. Chen, Chem. Rev., 2014, 114, 1289 CrossRef CAS PubMed; J. Jacquemin, M. F. Costa Gomes, P. Husson and V. Majer, J. Chem. Thermodyn., 2006, 38, 490 CrossRef.
- O. D. Pavel, P. Goodrich, L. Cristian, S. M. Coman, V. I. Pârvulescu and C. Hardacre, Catal. Sci. Technol., 2015, 5, 2696 CAS.
- C. H. Bartholomew, Adv. Catal., 1982, 31, 135 CAS.
- S. Zhang and Z. C. Zhang, Fuel Chemistry Division Preprints, 2002, 47, 449 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7fd00159b |
| This journal is © The Royal Society of Chemistry 2018 |