
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Whole metagenome sequencing of chlorinated drinking water distribution systems
Isabel
Douterelo
 *a,
Carolina
Calero-Preciado
a,
Victor
Soria-Carrasco
b and
Joby B.
Boxall
a
*a,
Carolina
Calero-Preciado
a,
Victor
Soria-Carrasco
b and
Joby B.
Boxall
a
aPennine Water Group, Department of Civil and Structural Engineering, University of Sheffield, Mappin Street, Sheffield S1 3JD, UK. E-mail: i.douterelo@sheffield.ac.uk
bDepartment of Animal and Plant Sciences, University of Sheffield, Sheffield S10 2TN, UK
First published on 17th October 2018
Abstract
There has been an explosion of research into the microorganisms present within drinking water distribution systems (DWDS). However, previous studies have focused mainly on the taxonomic composition and little is known about the actual genes composing the metagenomes of DWDS and their function or whether such information could be used for genetic profiling and monitoring processes taking place in DWDS. We use here for the first time whole metagenome shotgun sequencing to characterise microbial communities from both biofilm and bulk water samples from operational, chlorinated DWDS. Gene content analysis revealed habitat-specific (biofilm vs. water) differences in terms of organisms as well as gene functions, suggesting adaptation to specific environments. In addition, several resistance mechanisms were identified preferentially within biofilms, including genes associated with the prevention and repair of disinfectant radical-induced damage and antibiotic resistance. This research highlights the potential of such information to help protect drinking water quality and safety in the future following further research and wider application.
Water impactWe have used whole metagenome sequencing to assess the taxonomic affiliation and functional potential of chlorinated drinking water distribution systems (DWDS). This study illustrates how high throughput sequencing of environmental samples can inform on essential processes occurring in DWDS. This information can help on the development of faster and cost-effective monitoring strategies to indicate infrastructure or treatment failures and have the potential to be used in water risk assessment to protect water quality and safety. |
Introduction
The importance of microbial communities within DWDS is widely recognised, and recent research has illuminated the diversity and complexity of these communities.1–3 Microorganisms in DWDS, particularly those inhabiting biofilms, govern key processes including; transformation of metals involved in corrosion,4–6 degradation of disinfectant residual,7,8 changes in water organoleptic characteristics,9 discolouration,10 sheltering pathogens11 and production of toxins and virulence factors.12,13 However, there is virtually no understanding of how microorganisms work individually or as a community, and the impacts on the quality and safety of the drinking water or on the performance of the ageing pipe infrastructure. Prokaryotes are considered as simple organisms at genetic level when compared with eukaryotes, but they do have complex mechanisms to regulate gene expression. Prokaryotes have genomes that contain thousands of different genes which encode proteins with roles in different processes such as metabolism, defense mechanisms against other organisms, maintenance of cell structure, etc. All these genes are not expressed all the time and there are various ways of gene regulation to control which genes are expressed and at what levels.14 The aim of this study was to obtain a genetic fingerprint (gene presence) of microbial habitats in DWDS (biofilm vs. water) as a first step to understand genetic diversity. This initial knowledge will guide further steps in understanding specific mechanisms that can affect water infrastructure and quality such as biofilm formation, discolouration, corrosion, etc. Thus, our approach consists in understanding first what is there (all genes) to then focus further studies and monitoring strategies in specific functions and regulatory processes. To date, microbial ecological and metabolic interactions in drinking water-related systems have been studied using culturing methods, with a combination of two or three isolated microorganisms and under controlled laboratory conditions.15,16 The information presented by these laboratory studies is limited because: i) the laboratory growth conditions used are far from those experienced in real DWDS, and ii) interactions between a few selected microorganisms do not reflect highly diverse communities such as those present in biofilms. Currently, cultivation-independent methods based on targeted sequencing of specific genetic markers, such as the 16S rRNA gene, have provided a greater understanding of the bacterial diversity in DWDS17–20 and can be used to establish microbial networks and co-occurrence pattern.21 However, they do not yield information about prokaryotes and eukaryotes interactions and on the functional or genetic components of microbial communities. A step further in the use of molecular methods to understand DWDS is shotgun metagenomics, the use of DNA-based whole metagenomic analyses can define both taxonomy and potential function (presence of gene pools and expression when sequencing RNA) of complex microbial communities, providing greater insights into the processes and pathways of whole communities and their ecology.22 The whole metagenome sequencing analysis presented here includes the study of eukaryotes, organisms that despite their proven presence in these systems23 have been long ignored in most microbial studies in DWDS, with the exception of amoebas24,25 and the protozoa Cryptosporidium.26 In order to understand dynamics in DWDS and how they work as an ecosystem, we need to consider all interactions including those between eukaryotes and prokaryotes which are important for the maintenance of ecological processes.Shotgun sequencing has been used in microbial ecology to identify environment-specific genes and interpreting environments27 and to study functional adaptation of planktonic prokaryotes in oceans.28 Environmental metagenome shotgun sequencing can provide new information on rapid diagnostic and monitoring tools29 that can be applied to analyse failures in DWDS, thus useful for public health protection. This research was conducted to assess the potential of whole metagenome sequencing to genetically characterise the ecology of DWDS, in particular aimed at uncovering habitat-specific genetic profiles that can provide further insights into the life styles of organisms living in DWDS. This information can be then used to inform next-generation management strategies that focus on the promotion of “friendly microorganisms” that outcompete pathogenic or undesirable ones. The proposed new monitoring strategies have the potential to be more sustainable, strengthening infrastructure performance while minimizing costly interventions by water utilities.
Material and methods
Distribution systems (ground water vs. surface water)
We studied two operational DWDS by sampling two different sites located in the Southwest of UK one of them supplied by surface water and another one by groundwater. The groundwater site is supplied with a mixture of water from boreholes and treated by marginal chlorination using sodium hypochlorite solution as disinfection residual. The surface water site is supplied with water from local springs, river abstraction and treated by coagulation with aluminium sulphate, flocculation and removal of floc particles by dissolved air flotation. Subsequently the water is filtered using sand filters and granular activated carbon in order to absorb and remove organics. Chlorine is used for disinfection and as a residual in the system. Biofilm sampling devices (Fig. 1) replacing 1 m high density polyethylene (HDPE) pipe in the system were installed at these two sites as described in Douterelo et al.3 These devices have coupons inserted that allowed for analysing compositional and functional characteristics of biofilms developed for a year under realistic conditions. Importantly, these coupons fit flush with the curved inner pipe surface thus maintaining boundary hydraulic conditions, such as shear stress for mobilising force and turbulence for exchange with the bulk water.30,31 Water that supplied both the ground and the surface water site were used to characterise planktonic communities and functional patterns under two different water supplies. | ||
| Fig. 1 A) Biofilm sampling devices installed in two chlorinated distribution systems in the south of the UK. The devices have 10 coupons (B) that were inserted in the system and left biofilm develop for a whole year, after this period coupons were collected for biofilm analysis (C). | ||
Sampling biofilm and water samples
On the day of coupon collection, after one year of biofilm development in situ, samples of the bulk water that supplied the systems were collected using designated containers for physico-chemical and microbiological characterization via sampling taps located immediately upstream of the devices.Temperature and pH were measured in situ using a Hanna portable meter and probe HI 991003. All the other parameters were obtained by analysis of discrete water samples by an UK-accredited drinking water laboratory. Flow was measured by magnetic flow meters upstream of the biofilm. Coupons were installed and biofilm left to develop for a period of one year (2014–2015).
DNA was isolated from 24 samples, 6-bulk water and 6 biofilm samples at each site. To obtain DNA from biofilm samples, the whole area of the coupons was brushed and suspended in phosphate buffered saline (PBS), and then biofilm suspensions were concentrated by filtration in 0.22 μm nitrocellulose membrane (Millipore, Corp.). Bulk water samples (6 replicates of 3 L per site) were filtered through 0.22 μm membrane filters for subsequent DNA analysis. Biofilm and bulk water samples were then preserved in the dark at −80 °C until DNA was extracted. For the extractions we used a method based on proteinase K digestion followed by a standard chloroform/isoamyl alcohol method.32 The quantity and purity of the extracted DNA were assessed using Nanodrop ND-1000 spectrophotometer (NanoDrop, Wilmington, USA). In order to obtain enough DNA for whole metagenome sequencing it was necessary to pool DNA from the 6 coupons and the 6 water samples at each site, giving a total of 4 samples. However, because low DNA concentration of the pooled samples (<2 ng ul−1), whole genome amplification was carried out by the sequencing facility Research & Testing Laboratories. After amplification, DNA from the pooled biofilm samples from the surface water supply system was not of enough quantity and quality to proceed with whole metagenome sequencing. Thus, 2 water pooled samples (surface vs. ground) and 1 biofilm final pooled sample (groundwater) were used to obtain sequencing libraries and a description of the gene content of each habitat.
Metagenomics analysis
Sequencing libraries of the three pooled samples were prepared by Research and Testing Laboratory (Texas, USA) using a Kapa HyperPlus Single-Index Adapter Kit with TruSeq adapters (Illumina, San Diego, CA, USA), with sample-specific multiplex adaptor according to the manufacturer's instructions. The libraries were sequenced using a single lane in a HiSeq2500 System (Illumina).MG-RAST Metagenomics Analysis Server v.4.0 (http://metagenomics.anl.gov)33 was used for shotgun metagenomics sequence analysis. We followed the standard steps: pre-processing and quality control, feature identification, feature annotation and profile generation. MG-RAST quality control included the removal of artificial replicates according to the method of Gomez-Alvarez et al.34 and removal of low quality sequences using a modified Dynamic trim35 for sequences with 5 bp below a 15 Phred score 8–23% of the reads were retained after this step (Table 1). Taxonomic profiling was carried out using searches against the SEED Subsystems database, which contains all publicly available genome sequences36 with maximum E-value of 10–5, minimum identity of 60%, and minimum alignment length of 15 amino acids.37 To identify genes and their functions, the reads were annotated using the clusters of orthologous groups of proteins (COGs) database,38 using the same parameters for the searches.
| Water_surface | Water_ground | Biofilm_ground | |
|---|---|---|---|
| Total sequences | 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 653827 653827 |
50098184 |
29554050 |
| Basepairs | 7719069672 |
8275671991 |
4832305234 |
| Average length | 173 ± 42 bp | 165 ± 35 bp | 164 ± 34 bp |
| No pass QC | 39957241 (89.48%) |
42688313 (85.21%) |
21780435 (73.70%) |
| Artificial duplicate reads (dereplication) | 36039642 |
34371110 |
18127888 |
| Passed QC | 3754681 (8.41%) |
6145584 (12.27%) |
21780435 (73.70%) |
| Sequences contain ribosomal RNA genes | 4981 (0.13%) | 458519 (7%) |
16527 (0.25%) |
| Sequences contain predicted proteins with known functions | 3098757 (82.53%) |
3360956 (54.69%) |
5567919 (83.40%) |
| Sequences contain predicted proteins with unknown function | 650943 (17.34%) |
2326109 (37.85%) |
1091397 (16.35%) |
| Unknown | 941905 (2.11%) |
1264287 (2.52%) |
1097772 (3.71%) |
Results
Physico-chemical analysis
The flow in the groundwater site was low (<5 ML s−1) during the biofilm developmental period when compared with the surface supplied site which fluctuated between 2.5 to 12.5 ML s−1 (data not shown). Table 2 shows the results from the analysis of water samples collected on the day of coupon sampling, after one year of biofilm development. Results from the physico-chemical analysis showed that conductivity and the levels of the nitrous compounds were higher in the groundwater samples compared to the surface water ones. The other analysed parameters were similar at both sites. Very low levels of metals (≤0.01 mg l−1) and turbidity (0.11–≤0.1 NTU) were measured at both sites. Free chlorine concentrations were higher at the surface water site (0.62 mg l−1) than at the groundwater site (0.32 mg l−1).| Water_surface | Water_ground | |
|---|---|---|
| Turbidity (NTU) | 0.11 | <0.1 |
| Conduct 20 °C (μS cm−1) | 144 | 446 |
| pH | 7.9 | 7.5 |
| Ammonia as N (mg N per L) | <0.02 | 0.14 |
| Tot oxid N (mg N per L) | 1.4 | 8.8 |
| Nitrite as N (mg N per L) | <0.003 | <0.003 |
| Nitrate as N (mg N per L) | 1.37 | 8.81 |
| Ammonia (mg NH3 per L) | 0.023 | 0.182 |
| Nitrite (mg NO2 per L) | <0.01 | <0.01 |
| Nitrate (mg NO3 per L) | 3.06 | 39 |
| Orthophos (mg P per L) | <0.03 | <0.03 |
| Sulphate (mg SO4 per L) | 20 | 18 |
| Chloride (mg Cl per L) | 15 | 18 |
| Free Cl2 ATS (mg Cl per L) | 0.62 | 0.32 |
| Total Cl2 ATS (mg Cl per L) | 0.71 | 0.37 |
| Al mg L−1 | 0.01 | <0.01 |
| Mn mg L−1 | 0.002 | <0.001 |
| Fe mg L−1 | 0.01 | <0.01 |
Differences in taxonomic composition
The structural composition of biofilm and bulk water samples was characterised at a domain level (Fig. 2). Bacteria were clearly dominating the composition of all the studied samples, with a relative abundance of >85% in all samples. However, eukaryotes (4–5%) and viruses (5–6%) were slightly more represented in the structure of the planktonic communities than in biofilms. Among the source water type, the patterns were similar for both type of water, only archaea were slightly more abundant (0.3%) in surface water samples.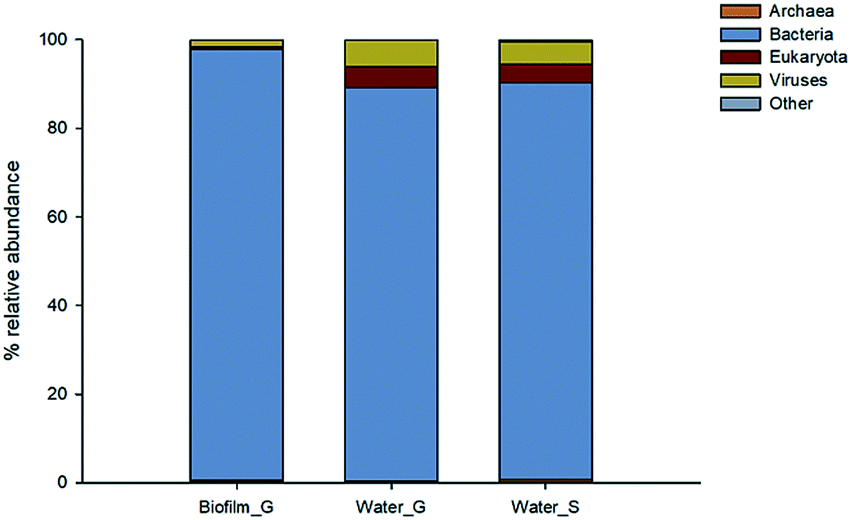 | ||
| Fig. 2 Taxonomic analysis at domain level of water and biofilm samples. | ||
Fig. 3 shows the eukaryotic profiles from biofilm and water samples at the groundwater site. Biofilms harboured a relatively high abundance of Chordata (28%), Ascomycota (23%), Streptophyta (15%) and Arthropoda (15%) (Fig. 3A). However, in bulk water diversity was dominated by fungal Ascomycota taxa representing 70% of all the sequences analysed, followed by Chordata (13%) and Streptophyta (3%) (Fig. 3B). Although found in relatively high abundance, animal and plant taxa are likely to be external contributions captured from the water sources. However, fungi are known to be the main eukaryotic components of biofilms in DWDS32,39 and were highly abundant in our samples.
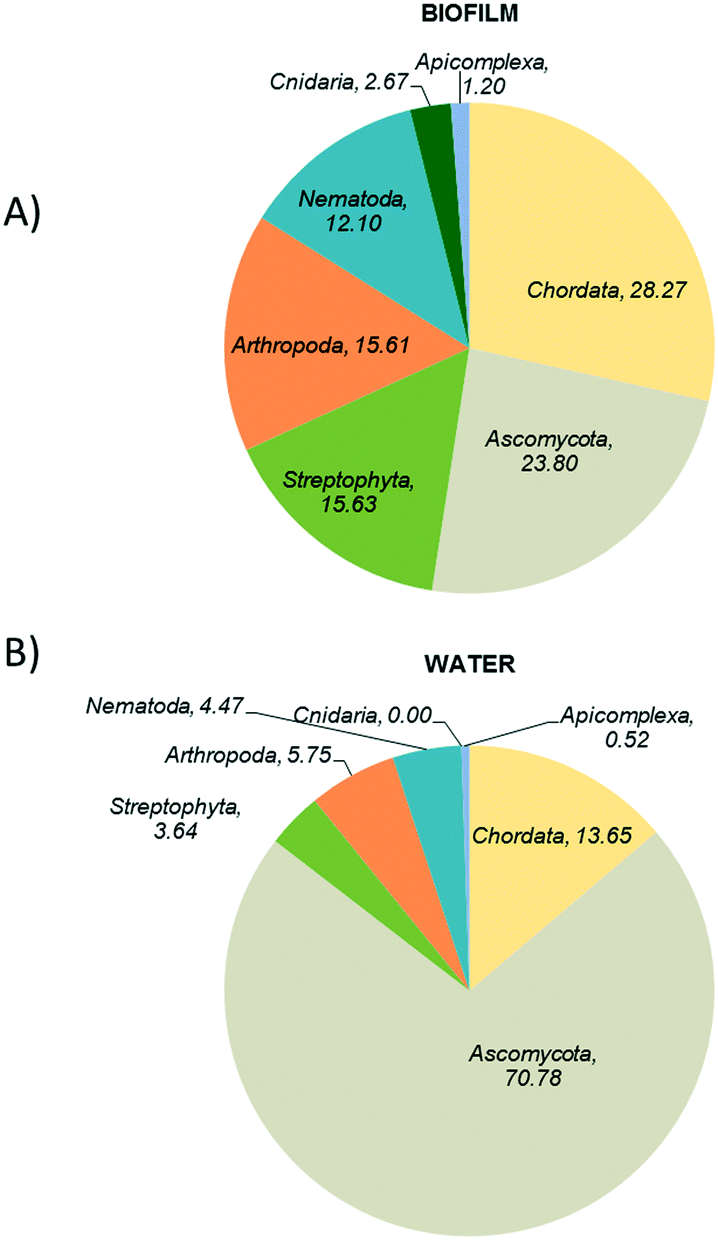 | ||
| Fig. 3 Taxonomic analysis of eukaryotes in the ground water supplied systems A) biofilm and B) water. | ||
Fungal diversity (Fig. 4) was mostly composed by the phylum Ascomycota followed by Basidiomycota and Chytridiomicota. Within Ascomycota, the relative abundance of the genus Gibberella was high in both biofilm (37%) and bulk water (ground = 32% and surface = 24%). Schizosaccaromyces was highly abundant in biofilms (26%) whilst other fungal genera samples such as Neurospora (ground = 19% and surface = 13%) and Magnaporthe (ground = 12% and surface = 6%) were mainly present in bulk water.
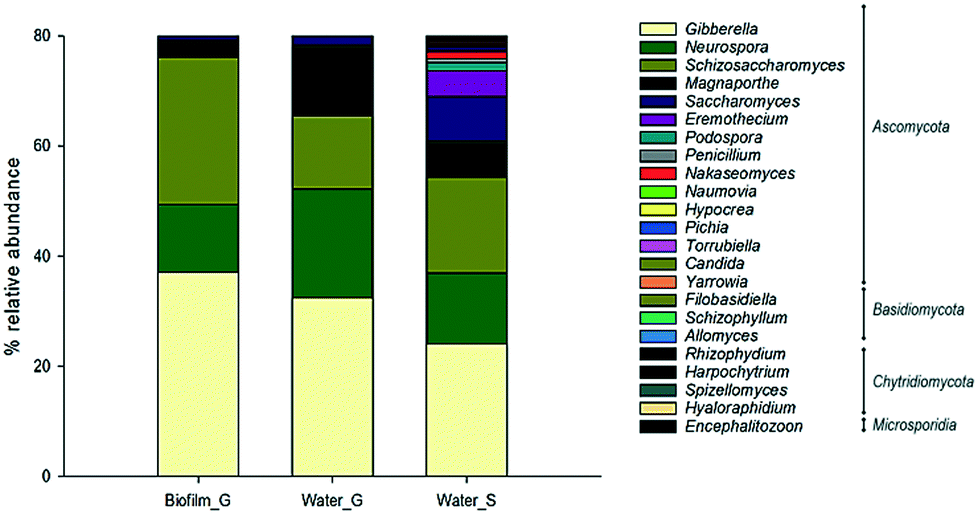 | ||
| Fig. 4 Most abundant fungi phylum and genera in the analysed samples in ground (G) and surface (S) supplied systems. | ||
Regarding bacteria, the most abundant phylum in the two habitats were Bacteroidetes, Firmicutes and Proteobacteria (Fig. 5). Within biofilms Bacterioidetes was clearly dominating the composition of the community, with high relative abundance of genera such as Chitinophaga (19%) and Pedobacter (7%). In bulk water samples, Firmicutes was the main phylum represented in the samples, mainly the genera Staphylococus (11–13%) and Bacillus, which clearly dominated the composition of the surface water samples (25%).
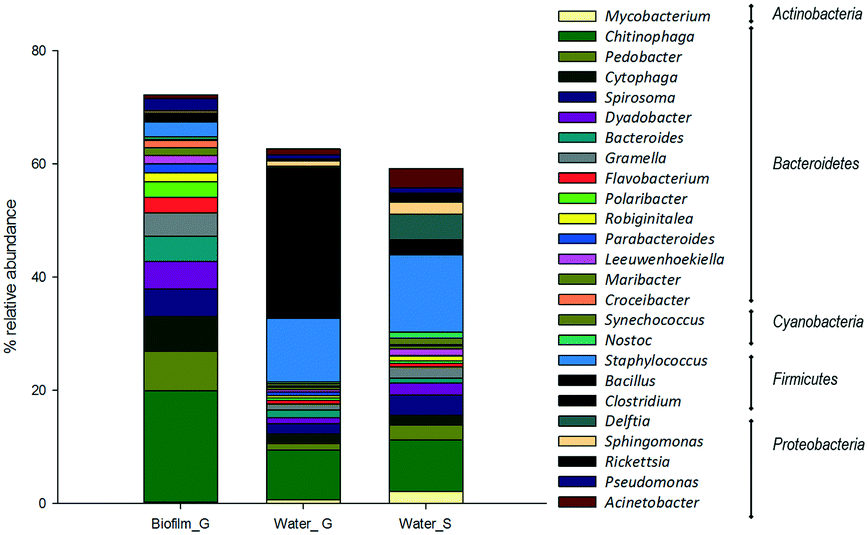 | ||
| Fig. 5 Most abundant bacteria phylum and genera in the analysed samples in ground (G) and surface (S) supplied systems. | ||
Differences in annotated genes and their functions
Fig. 6 shows a heatmap with the relative abundance of annotated genes in the whole metagenome of each of the sequenced samples. MG-RAST analyses of biofilm and bulk water samples showed similitudes in the relative abundance of the most relevant level 1 (highest COGs functional level) functional categories, which are grouped in 4 categories; 1) cellular processes, 2) information storage and processing, 3) metabolism and 4) poorly characterized. When the genetic profile of biofilm and water samples for the groundwater site was characterised, the most relatively abundant level 1 functional categories were metabolism (biofilm average 50% and water 46%), followed by information storage and processing (biofilm and water average 18%) and cellular process and signaling (biofilm average 19% and water 20%). The fingerprint for both types of waters was similar.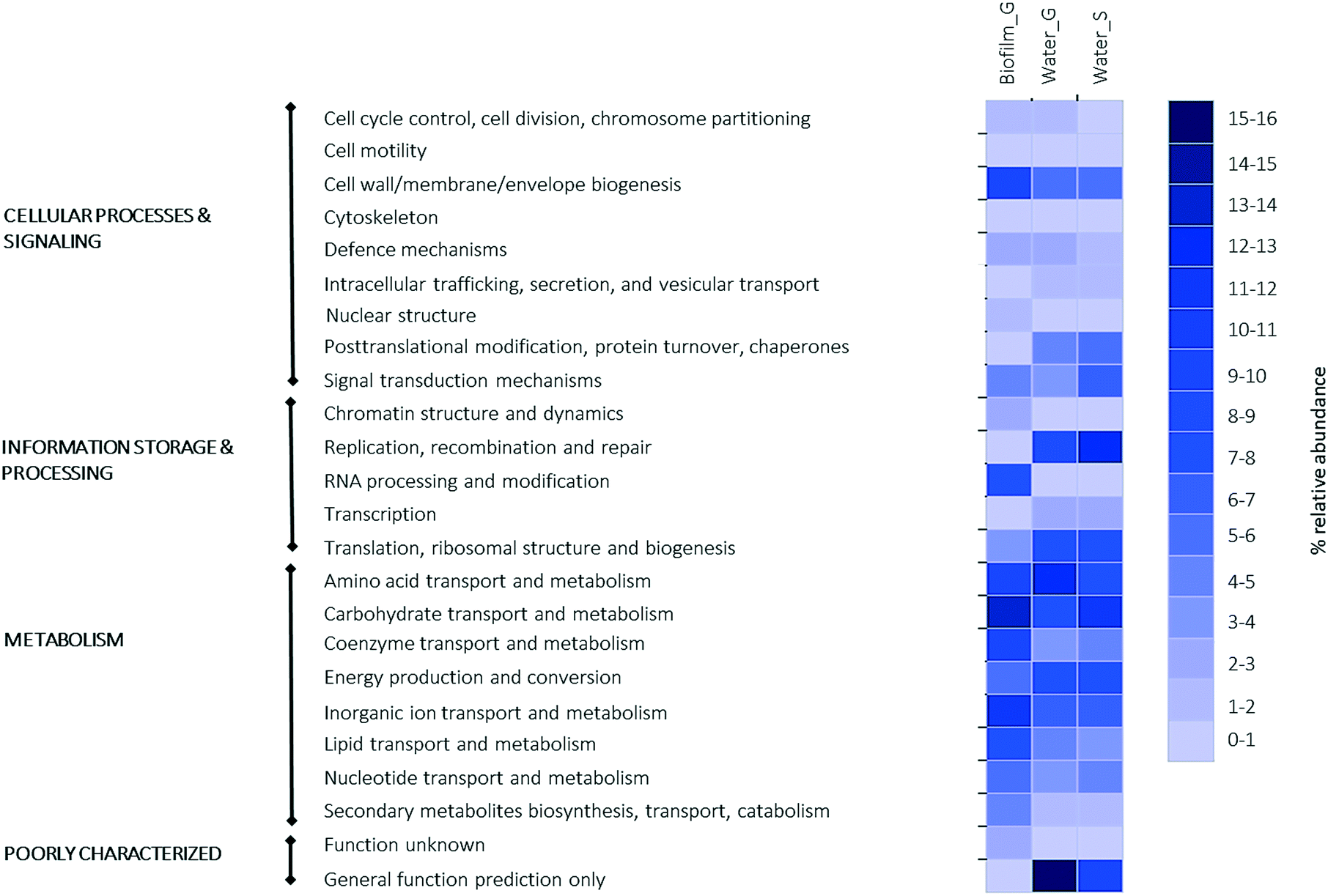 | ||
| Fig. 6 Heatmap of the relative abundance of annotated genes in the whole metagenome of each of the sequenced samples. COGs functional levels 1 and 2. | ||
Within biofilm and analysing the genetic data at functional level 2, most of the genes involved in metabolism were related to aminoacid transport and metabolism (12%), with genes related to the expression of peptidases (enzymes that hydrolase proteins) in particular dipeptidyl aminopeptidases/acylaminoacyl-peptidases. Within metabolism, most genes were also related with energy production (9%) and the most abundant gene in this category was the aerobic-type carbon monoxide dehydrogenase. Carbon monoxide dehydrogenase is an enzyme that plays an important role in the carbon cycle and is involved in the metabolism of methanogenic, aerobic carboxidotrophic, acetogenic, sulfate reducing, and hydrogenogenic bacteria. Regarding the functional category of information storage and processing, most of the reads (8%) matched genes involved in translation, ribosomal structure and biogenesis and in particular the Asp-tRNAAsn/Glu-tRNAGln amidotransferase gene.
Gene functions related to biofilm formation and resistance mechanisms
Table 3 shows the results for the specific functional annotated categories related to stress and resistance mechanisms and biofilm formation in samples from the groundwater supplied site. Different gene functions related to cell resistance and protection mechanisms were observed in biofilm and bulk water samples. Specific examples included extracellular polymeric matrix production and degradation (biofilm formation), glutathione protection, SoxRS system, the OyxR system and the RpoS regulated gene (all involved in cell protection), and antibiotic resistance. As it was expected, extracellular polymeric substances (EPS) production and degradation genes were almost exclusively identified in biofilms. The most abundant genes in the samples related to biofilm formation were sialic acid synthesis (0.09%) and dTDP-rhamanose synthesis (0.06%). Both of these genes are related to the synthesis of carbohydrates which enhances biofilm formation and influences motility and pathogenicity of bacteria. Gene categories related to cellular protection mechanisms were predominantly present in biofilms, such as catalase (0.13%) within the OxyR system and superoxide dismutase (0.14%) and aconitase A (0.28%) in the SoxRS system. Antibiotic related resistance genes were clearly more abundant in biofilms, represented mainly by beta-lactamase resistance genes (0.21%).| Relative distribution (%) | |||
|---|---|---|---|
| Water | Biofilm | ||
| Glutathione protection | Glutathione peroxidase | 0.10 | 0.03 |
| Glutathione S-transferase | 0.02 | 0.02 | |
| Glutathione synthetase/ribosomal protein S6 modification enzyme (glutaminyl transferase) | 0.00 | 0.01 | |
| OxyR system | Arsenate reductase and related proteins, glutaredoxin family | 0.14 | 0.02 |
| Bacterial nucleoid DNA-binding protein | 0.01 | 0.04 | |
| Catalase (peroxidase I) | 0.01 | 0.13 | |
| Thioredoxin reductase | 0.09 | 0.10 | |
| SoxRS system | Superoxide dismutase | 0.02 | 0.14 |
| Aconitase A | 0.09 | 0.28 | |
| Fumarase | 0.04 | 0.08 | |
| Glucose-6-phosphate 1-dehydrogenase | 0.04 | 0.01 | |
| Glucose-6-phosphate isomerase | 0.03 | 0.09 | |
| Endonuclease IV | 0.02 | 0.00 | |
| RpoS regulated genes | RNA polymerase sigma factor (predicted RNA polymerase sigma factor containing a TPR repeat domain) | 0.00 | 0.04 |
| EPS production and degradation (biofilms) | Sialic acid synthase | 0.01 | 0.09 |
| dTDP-rhamnose synthesis (dTDP-4-dehydrorhamnose reductase) | 0.01 | 0.04 | |
| dTDP-rhamnose synthesis (dTDP-4-dehydrorhamnose 3,5-epimerase and related enzymes) | 0.00 | 0.06 | |
| Antibiotic resistance | Beta-lactamase (beta-lactamase class C and other penicillin binding proteins) | 0.05 | 0.21 |
| Negative regulator of beta-lactamase expression | 0.00 | 0.04 | |
| Sulfonamides, dihydropteroate synthase (dihydropteroate synthase and related enzymes) | 0.01 | 0.04 | |
| Multidrug efflux pump and transporter | Multidrug efflux transporter (ABC-type multidrug transport system, ATPase and permease components) | 0.54 | 0.37 |
| ABC-type multidrug transport system, ATPase component | 0.33 | 0.40 | |
| ABC-type multidrug transport system, permease component | 0.01 | 0.02 | |
| Cation/multidrug efflux pump | 0.77 | 0.68 | |
| Na+-driven multidrug efflux pump | 0.09 | 0.08 | |
| Multidrug resistance efflux pump | 0.00 | 0.01 | |
| Potential degradation genes | Arylsulfatase A and related enzymes | 0.05 | 0.34 |
| Beta-glucosidase (related glycosidases) | 0.15 | 0.13 | |
| Beta-galactosidase | 0.00 | 0.01 | |
| Beta-glucosidase/6-phospho-beta-glucosidase/beta-galactosidase | 0.11 | 0.04 | |
| Beta-galactosidase/beta-glucuronidase | 0.02 | 0.04 | |
Genes related to multidrug efflux pump and transport mechanisms were similarly represented in both habitats (biofilm and bulk water), with multidrug efflux transport genes (biofilm = 0.37% and water = 0.54%) and cation/multidrug efflux pump (biofilm = 0.68% and water = 0.77%). Within the potential degradation genes, arylsulfatase A and related enzymes were highly represented in biofilms (0.34%) and these enzymes are related to the decomposition of organic matter and aromatic compounds. Beta-glucosidase activity related to the breakdown of carbohydrates was equally represented in both habitats (biofilm = 0.13% and water = 0.15%).
Discussion
Eukaryotes and prokaryotes: diversity and interactions
Our results show that not only prokaryotes are relevant in DWDS. Other taxonomic domains, including viruses, eukaryote and archaea were identified in the analysed samples, mostly in the bulk water samples at both sites. Eukaryotes are part of the ecological network of these engineered systems and consequently they can affect the quality and safety of the water. There are few studies based on eukaryotes in DWDS and those have been mainly focused in the parasitic protozoan Cryptosporidium and Giardia, or in planktonic amoeba studies24,40 and to some extent fungi, mainly Ascomycota.41,42 One of the few drinking water studies that reports the mixed presence of different eukaryotes in water from a desalination plant is that of Belila et al.39 The authors reported that uncultured fungi were the major group of eukaryotes, followed by Chordata and Arthropoda. We can confirm the taxonomic identification of different type of eukaryotes in chlorinated DWDS (water and biofilm); nematodes, amphipods, insect and copepods larvae or most likely fragments of those, these can overcome water treatment and enter in DWDS. Of particular interest in this study is the presence of a high percentage of Chordata, Arthropoda and Nematoda in the analysed biofilm samples that is different from bulk water samples mainly dominated by the fungal group Ascomycota. It must be noted that the identification of metazoan DNA does not necessarily implies that the actual organisms are in the samples. The identification of such organisms could be explained by the presence of free DNA released from animals or plants into the original source water. Alternatively, there may be potential sources of living organisms and biomass in DWDS other than the original treated water. Various interactions and sources through the DWDS, including service reservoirs used for breaks and repairs, ingress due to transients, back siphoning, or cross connections can influence and enrich biofilm communities.43 Thus, external organic matter contributions may be a key source of nutrients for biofilm. If that was the case, a potential good mechanism to control their development in DWDS would be to limit the entry of these sources at treatment work level. Similarly, monitoring for specific members of eukaryotic communities can offer an alternative way of assessing external organic matter entries in DWDS.The importance of eukaryote–prokaryote relationships has been established in other environments and these interactions can be either beneficial or disadvantageous. There are cross-kingdom cell-to-cell signalling mechanisms that involve small molecules, mainly hormones, that are produced by eukaryotes and hormone-like chemicals produced by bacteria.44 Interactions between algae and bacteria are common in roots of higher plants for example, where microalgae serve as firm matrix to help create biofilms.45 Similarly, fungi hyphae can serve as building blocks and/or provide biotic support for the establishment and colonization of surfaces by bacteria. Cross-kingdom interactions such as those of bacterial-fungal associations are involved also in contact, adhesion and colonization of surfaces, thus in the process of mixed species biofilm formation.46 This study suggests that the contribution of fungi, particularly Ascomycota, to the microbial ecology of real DWDS is important, as has been previously reported in previous studies.41 Ascomycota form spores to propagate and the small size of these together with their resistant to disinfection can explain why they were the main representatives of eukaryotes in water samples. Bioremediation studies have proved the potential of non-ligninolytic fungi such as Ascomycota to degrade chlorinated and polycyclic aromatic hydrocarbons.47 Besides fungal–bacterial interactions, other cross-kingdom interactions reported in DWDS are predation of bacteria and viruses by amoeba or protozoa.48 The main concern related to the presence of amoeba and protozoa in DWDS, is the association of these with the transport of pathogens such as Legionella or viruses.49 Thus, based on this and our previous results in the same DWDS,41 it can be concluded that bacteria are the main inhabitants of these ecosystems, this can indicate that interactions between bacteria with many other domains are probably non beneficial for these organisms or that bacteria are the main organisms with the necessary mechanisms to thrive in this type of environment.
Biofilm formation
Knowing the mechanisms of biofilm formation will open the door to predicting the consequences of environmental changes and to engineering them to fulfil particular functions in DWDS. For instance, by directing biofilm formation we could favour “friendly” microbial communities that produce infrastructure-protective EPSs or neutralise pathogens.EPS production comprises metabolic processes where microorganisms produce polysaccharides, proteins and other extracellular compounds such as lipid and extracellular DNA.50 Gene functional categories documented in this study suggest that the main metabolic pathways associated with biofilm formation are polysaccharide biosynthesis and sialic acid synthesis. Sialic acids are sugars found as the terminal units on carbohydrate chains linked to proteins or lipids and they help to create a barrier when microorganisms are inhabiting or invading a surface. Sialic acids are a potential source of carbon, nitrogen and cell wall metabolites, that are necessary for bacterial colonization, persistence, and growth. In fact, it has been also shown that the presence of sialic acid can enhance biofilm formation by certain microorganisms.51
Consequently, we suggest that by understanding of the metabolic use of a group of prevalent carbohydrates, such as sialic acids, it may be possible to partially identify factors controlling bacterial colonization of pipe surfaces and the probability of biofilm formation thus helping to predict it.
Following on our discussion on biofilm formation and the central role of bacteria on this, we observed that species belonging to the genus Pseudomonas were more abundant in biofilms. This is not surprising, since the involvement of these microorganisms in the initial steps of biofilm formation has been previously observed in this habitat.3 Our study proposes the potential use of other bacterial groups as alternative bioindicators, of biofilm formation such as Bacteroidetes. Lin et al.52 studying bacterial communities in a drinking water treatment plant proposed Bacterioidetes as a rapid indicator of faecal pollution since its presence can indicate failures in the system earlier than other commonly used faecal indicators such as E. coli. As has been indicated before, such taxonomic information is of practical relevance to applications such as design of new bioindicators. These two are examples of how the information obtained at genetic level can help to improve risk assessment, monitoring and management strategies.
Most genes in biofilms were related to the metabolism of amino acids and carbohydrates. This suggests that microbial communities in biofilms play a key role in carbon and nitrogen cycling through for example organic matter decomposition. Previous observation that biofilms may be self-sufficient in the generation of biomass brings into question the use of nutrient management strategies in DWDS.53 Therefore, rather than intervening at the water treatment works to control the incoming water quality, it might be better or also necessary to invest in network interventions, such as cleaning strategies. However, as highlighted by Husband et al.10 such operations are not a one off, biofilms and associated discolouration risk will re-accumulate/grow, thus an ongoing maintenance strategy is required.
Regarding the functional category of information storage and processing, most of the reads matched genes involved in translation, ribosomal structure and biogenesis and in particular the Asp-tRNAAsn/Glu-tRNAGln amidotransferase gene. This enzyme is fundamental for protein biosynthesis and is involved in the process of RNA translation into proteins. Our observations at a genetic level are complementary to previous studies at a structural level (i.e. physical structure) in DWDS biofilm. Fish et al.,54 showed by performing experimental tests in a chlorinated DWDS, that the main structural components of biofilms were proteins and carbohydrates. The authors demonstrated that the ratio of these two components varied with the flow rate in the system and was associated to the strength/mobilisation of biofilm and hence discolouration risk. Since the production of these structural components is regulated at a genetic level, we can suggest that hydraulic regimes might trigger or suppress the expression and level of expression of genes involved in EPS production such as those characterised in this study. These genetic changes can be also attributed to an evolutionary selection process, as long as the selective pressure (i.e. hydraulic regime) is kept long enough in the system. Future studies focused on linking genetic information and expression of these genes (e.g. amidotransferase gene), can certainly yield better insights into the mechanisms of biofilm formation and on how to improve biofilm control strategies in DWDS.
Resistance to oxidative stress and antibiotics
This study has characterised genes related to microbial resistance and cellular protection, that aid biofilm to be successful in DWDS, whilst affecting water quality and safety. Microbial cells can encounter reactive oxygen species (ROS), which are toxic due to their ability to damage molecules such as DNA, proteins, and lipids.55 Prokaryotes use defence mechanisms based on antioxidant scavenging enzymes, such as superoxide dismutase (SOD), peroxiredoxin, and catalase, to protect cells from ROS damage.56 In most gram-negative bacteria, the main defence systems induced under oxidative stress are: i) the OxyR system (responds to hydrogen peroxide); ii) the SoxRS system (respond to redox-active compound) and ii) several genes regulated by RpoS.57 Oxidative stress can stimulate OxyR and induce the transcription of several antioxidants, including glutaredoxin and thioredoxin reductases. Several genes related to the SoxRS system were found in this study, included superoxidase dismutase, acotinase A and glucose-6-phosphate isomerase. Another protection mechanisms characterised in this study is based on the activity of the enzyme glutathione reductase. Glutathione reductase reduces glutathione disulphide to the sulfhydryl form glutathione (GSH), which plays a key role in maintaining the reducing environment of the cells and resisting oxidative stress.58 GSH genes have been previously associated with enhanced resistance to chlorine by bacteria.59 Chao et al.,60 using drinking water biofilms grown on annular reactors suggested that oligotrophic conditions and chlorination can stimulate GSH synthesis, supporting previous observations on the limited effect of chlorine residual in controlling microbial growth and biofilm formation in DWDS.32 Gomez-Alvarez et al.,61 analysing drinking water receiving different disinfection treatments found that sequences related to glutathione reductase (gorA) and thioredoxin reductase (trxB) were highly present in water treated with chlorine and suggested that the gorA reductase serves as a repair mechanism against damage caused by oxygen radicals. Several studies have been able to associate the response of certain microorganism with specific genetic functions related to chlorine resistance. For example, Gomez-Alvarez et al.,61 in the study mentioned above observed that Gram-negative bacteria related to the families Caulobacteraceae and Rhodobacteraceae were responsible of encoding gorA reductase genes. Similarly, Chao et al.,60 noted that bacteria belonging to Alphaproteobacteria and Sphingomonaceae, which are resistance to chlorination, commonly carry GSH related genes. These are examples of how the specific association between a process (i.e. chlorine/chloramine resistance) and a certain type of microorganism can help to determine the type of treatment needed in DWDS and equally the inefficiency of it. This study succeed at characterising genes involved in processes used by microorganisms to respond to oxidative and chemical stress in water and biofilm, suggesting that both habitats experience oxidative stress. Although, the presence of these genes was higher in biofilms, indicating once again the protective habitat that biofilms offer to its inhabitants, and the limited effect that chlorine impose to control their development in DWDS.This study reported high relative abundance of genes related to β-lactamase resistance, a common antibiotic, supporting the idea that attached communities on pipes have greater protection to environmental stressors. It has been documented that chlorination increases resistance to antibiotics62,63 and that the higher abundance of chlorine-resistant bacteria (e.g. Pseudomonas and Acidovorax) can contribute to the net increase in antibiotic resistance genes.64 However, it must be noted that antibiotic production and resistance are processes expected to be found in natural communities as a result of the competition between microorganisms.65 Distinguishing between human-driven and natural processes will require identifying and analysing the concentration of the antibiotics in bulk water in future studies. Genes that confer resistance can be transferred from one bacterium to another by mobile genetic elements, and they can be used by bacteria as a natural defence mechanism to avoid competition.66 Further research is also needed to disentangle the role of particular microbial species in biofilms in relation to the exchange of resistance mechanisms, as well as which factors favour the occurrence of such mechanisms.
Future research and steps for the development of new monitoring strategies in DWDS
This is the first ‘proof of concept’ and documented evidence of a whole metagenome sequencing study carried out in operational DWDS. As a consequence, we identified a number of procedural challenges and concerns while carrying out this study. One of such challenges is the difficulty to access biofilm samples in real DWDS, which in this case, this resulted in the need to pool samples prior whole metagenome sequencing. The amount of DNA recommended for whole metagenome sequencing using Illumina is 50–100 ng.67 Our coupons design had yielded enough DNA for our previous 16S-based analyses. However, it did not allow to obtain the larger amounts of DNA required for biological replication and whole metagenome sequencing. Future studies should utilise improved protocols that ensure that sufficient biofilm and bulk water is sampled to enable replicate results. Larger biofilm samples could be obtained by scratching a larger area of the internal pipe surface. However, the coupon design to enable this is difficult, as it would also have to follow the pipe curvature to maintain critical boundary effects that include shear stress and nutrient transfer. The use of removable pipe sections would be more feasible, but there are various complexities that would need to be addressed to ensure lack of contamination during removal and to replacement effects if multiple time points were to be studied. Another potential sampling strategy is to flushing sections of pipes by creating excess hydraulic forces and hence mobilise biofilm into the bulk water. However, it is well documented that this does not completely remove the attached biofilm and only the weakly adhered material would be sampled.68 Obtaining large water samples is relatively easier. For example, instead of filtering by vertical pressure, tangential flow filtration69 could be used that allows for the concentration of a higher volumes of water and consequently higher concentration of cells for subsequent DNA extraction.Another area that could be improved is the efficiency of DNA extractions. As part of the preliminary analyses of the samples included this study, we tried several DNA extraction methods including Mobio biofilm and water extraction kits. However, the method that yielded more DNA and of better quality was the one based on using lysozyme/SDS/proteinase K, followed by CTAB and a chloroform step that was eventually used. Additionally, the use of a combination of different DNA extractions methods could be useful to recover metagenome sequences of rare and uncultured bacteria as recommended by Albertsen et al.70
Another challenge associated with the low amounts of DNA we isolated is that they are particularly sensitive to any contamination, especially when whole metagenome amplification is used. Contamination controls are used for NGS as a matter of routine, but additional controls when collecting samples and processing them in the laboratory might be advisable. For example, although we were careful in our procedures, we cannot completely rule out that the presence of metazoan DNA in our samples is not due to contamination instead of the most likely interpretation that it is DNA derived from organisms present in the original water source.
Despite the operational issues found, this study has shown for the first time that the use of whole metagenome shotgun sequencing is a promising powerful tool to better understanding the biological processes taking place in DWDS. Future steps will likely involve the selection of genetic markers as indicators of specific processes in DWDS, for example; corrosion, biofilm growth and virulence factors and ultimately test their use for monitoring purposes in real drinking water networks. With this aim, it will be necessary to establish a baseline of the presence (i.e. relative abundance) of these genes under optimal operation conditions. Once the baseline is established and known, significant changes in the relative abundance of these marker genes can indicate when a failure in the system occurs. This will yield data to inform a new risk assessment framework for DWDS that the water utilities can use to better manage these systems. Moreover, we have shown that it is possible to obtain additional information about the genes and their function, which could potentially be used to develop new and more sophisticated monitoring strategies. Thanks to this study, the follow-up research on microbial communities on DWDS will have a more solid methodological background. This will allow future studies to focus on linking genetic taxonomic information and functional analysis of genes, and on how to use our knowledge of the mechanisms of biofilm formation and bacterial resistance for improving the biofilm control strategies in DWDS.
Conclusion
We have used whole metagenome shotgun metagenomics to assess the taxonomic affiliation and functional potential of chlorinated DWDS for the first time. The information obtained in this study not only reinforces previous observations that DWDS hold a diverse community of prokaryotes and eukaryotes, but also provides new insight on the environmental genetic pool and the function of the genes involved. In particular, we have detected, mainly in biofilms, genes related to biofilm formation as well as mechanisms of resistance and damage-repair to external stressors such as chlorine and antibiotics. This study illustrates how high throughput sequencing of genetic data can inform on essential processes occurring in DWDS. This information could inform and help on the development of new indicators of infrastructure or treatment failures and has the potential to be used to protect and promote water quality and safety.Research data management
The sequencing data used for this project was deposited and is openly accessible in the MG-RAST server under project number ID MGP80824.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research reported here was supported by the UK Engineering and Physical Sciences Research Council (EPSRC). EPSRC-LWEC Challenge Fellowship EP/N02950X/1, the Challenging Engineering Grant EP/G029946/1 and the EPSRC Platform Grant Funding EP/1029346/1. Victor Soria Carrasco is supported by a Leverhulme Trust Early Career Fellowship. We would like to recognise and thank Wessex Water Services Ltd for sampling sites, field work and physico-chemical sample analysis.References
- G. Liu, J. Q. J. C. Verberk and J. C. Van Dijk, Bacteriology of drinking water distribution systems: an integral and multidimensional review, Appl. Microbiol. Biotechnol., 2013, 97, 9265–9276 CrossRef CAS PubMed.
- S. Liu, C. Gunawan, N. Barraud, S. A. Rice, E. J. Harry and R. Amal, Understanding, Monitoring, and Controlling Biofilm Growth in Drinking Water Distribution Systems, Environ. Sci. Technol., 2016, 50, 8954–8976 CrossRef CAS PubMed.
- I. Douterelo, J. B. Boxall, P. Deines, R. Sekar, K. E. Fish and C. A. Biggs, Methodological approaches for studying the microbial ecology of drinking water distribution systems, Water Res., 2014, 65, 134–156 CrossRef CAS PubMed.
- X. Li, H. Wang, C. Hu, M. Yang, H. Hu and J. Niu, Characteristics of biofilms and iron corrosion scales with ground and surface waters in drinking water distribution systems, Corros. Sci., 2015, 90, 331–339 CrossRef CAS.
- H. Sun, B. Shi, F. Yang and D. Wang, Effects of sulfate on heavy metal release from iron corrosion scales in drinking water distribution system, Water Res., 2017, 114, 69–77 CrossRef CAS PubMed.
- Y. Zhu, H. Wang, X. Li, C. Hu, M. Yang and J. Qu, Characterization of biofilm and corrosion of cast iron pipes in drinking water distribution system with UV/Cl2 disinfection, Water Res., 2014, 60, 174–181 CrossRef CAS PubMed.
- C. Montoya-Pachongo, I. Douterelo, C. Noakes, M. A. Camargo-Valero, A. Sleigh, J.-C. Escobar-Rivera and P. Torres-Lozada, Field assessment of bacterial communities and total trihalomethanes: Implications for drinking water networks, Sci. Total Environ., 2018, 616–617, 345–354 CrossRef CAS PubMed.
- H. Wang, Y. Zhu and C. Hu, Impacts of bacteria and corrosion on removal of natural organic matter and disinfection byproducts in different drinking water distribution systems, Int. Biodeterior. Biodegrad., 2017, 117, 52–59 CrossRef CAS.
- X. Zhou, K. Zhang, T. Zhang, C. Li and X. Mao, An ignored and potential source of taste and odor (T&O) issues—biofilms in drinking water distribution system (DWDS), Appl. Microbiol. Biotechnol., 2017, 101, 3537–3550 CrossRef CAS PubMed.
- S. Husband, K. E. Fish, I. Douterelo and J. Boxall, Linking discolouration modelling and biofilm behaviour within drinking water distribution systems, Water Sci. Technol.: Water Supply, 2016, 16, 942–950 CAS.
- A. Nocker, M. Burr and A. Camper, in Microbiology of Waterborne Diseases, ed. M. V. Yates, D. W. Williams, R. M. Chalmers and N. F. Gray, 2nd edn, Academic Press, London, 2014, pp. 3–32 Search PubMed.
- M. Pablos, J. M. Rodríguez-Calleja, J. A. Santos, A. Otero and M.-L. García-López, Occurrence of motile Aeromonas in municipal drinking water and distribution of genes encoding virulence factors, Int. J. Food Microbiol., 2009, 135, 158–164 CrossRef CAS PubMed.
- X. He, Y.-L. Liu, A. Conklin, J. Westrick, L. K. Weavers, D. D. Dionysiou, J. J. Lenhart, P. J. Mouser, D. Szlag and H. W. Walker, Toxic cyanobacteria and drinking water: Impacts, detection and treatment, Harmful Algae, 2016, 54, 174–193 CrossRef CAS PubMed.
- T. Brock, M. Madigan, J. Martinko and J. Parker, Biology of microorganisms, 7th ed., 1994 Search PubMed.
- J. Vornhagen, M. Stevens, D. McCormick, S. E. Dowd, J. N. S. Eisenberg, B. R. Boles and A. H. Rickard, Coaggregation occurs amongst bacteria within and between domestic showerhead biofilms, Biofouling, 2013, 29, 53–68 CrossRef PubMed.
- B. Ramalingam, R. Sekar, J. Boxall and C. Biggs, Aggregation and biofilm formation of bacteria isolated from domestic drinking water, Water Sci. Technol.: Water Supply, 2013, 13, 1016–1023 Search PubMed.
- J. L. A. Shaw, et al. Using amplicon sequencing to characterize and monitor bacterial diversity in drinking water distribution systems, Appl. Environ. Microbiol., 2015, 81(18), 6463–6473 CrossRef CAS PubMed.
- J. El-Chakhtoura, E. Prest, P. Saikaly, M. van Loosdrecht, F. Hammes and H. Vrouwenvelder, Dynamics of bacterial communities before and after distribution in a full-scale drinking water network, Water Res., 2015, 74, 180–190 CrossRef CAS PubMed.
- V. Gomez-Alvarez, S. Pfaller and R. P. Revetta, Draft Genome Sequence of Two Sphingopyxis sp. Strains, Dominant Members of the Bacterial Community Associated with a Drinking Water Distribution System Simulator, Genome Announc., 2016, 4(2), 1–2 Search PubMed.
- N. M. Hull, E. P. Holinger, K. A. Ross, C. E. Robertson, J. K. Harris, M. J. Stevens and N. R. Pace, Longitudinal and Source-to-Tap New Orleans, LA, U.S.A. Drinking Water Microbiology, Environ. Sci. Technol., 2017, 51, 4220–4229 CrossRef CAS PubMed.
- K. Faust, G. Lima-Mendez, J.-S. Lerat, J. F. Sathirapongsasuti, R. Knight, C. Huttenhower, T. Lenaerts and J. Raes, Cross-biome comparison of microbial association networks, Front Microbiol., 2015, 6, 1200 Search PubMed.
- A. Escobar-Zepeda, A. Vera-Ponce de León and A. Sanchez-Flores, The Road to Metagenomics: From Microbiology to DNA Sequencing Technologies and Bioinformatics, Front. Genet., 2015, 6, 348 Search PubMed.
- H. Y. Buse, J. Lu, I. T. Struewing and N. J. Ashbolt, Eukaryotic diversity in premise drinking water using 18S rDNA sequencing: implications for health risks, Environ. Sci. Pollut. Res., 2013, 20, 6351–6366 CrossRef CAS PubMed.
- G. J. Puzon, J. T. Wylie, T. Walsh, K. Braun and M. J. Morgan, Comparison of biofilm ecology supporting growth of individual Naegleria species in a drinking water distribution system, FEMS Microbiol. Ecol., 2017, 93(4), 1–8 CrossRef PubMed.
- H. C. Miller, M. J. Morgan, J. T. Wylie, A. H. Kaksonen, D. Sutton, K. Braun and G. J. Puzon, Elimination of Naegleria fowleri from bulk water and biofilm in an operational drinking water distribution system, Water Res., 2017, 110, 15–26 CrossRef CAS PubMed.
- M. I. Z. Sato, A. T. Galvani, J. A. Padula, A. C. Nardocci, M. d. S. Lauretto, M. T. P. Razzolini and E. M. Hachich, Assessing the infection risk of Giardia and Cryptosporidium in public drinking water delivered by surface water systems in Sao Paulo State, Brazil, Environ. Sci. Pollut. Res., 2013, 442, 389–396 CAS.
- S. G. Tringe, C. von Mering, A. Kobayashi, A. A. Salamov, K. Chen, H. W. Chang, M. Podar, J. M. Short, E. J. Mathur, J. C. Detter, P. Bork, P. Hugenholtz and E. M. Rubin, Comparative Metagenomics of Microbial Communities, Science, 2005, 308, 554–557 CrossRef CAS PubMed.
- J. C. Venter, K. Remington, J. F. Heidelberg, A. L. Halpern, D. Rusch, J. A. Eisen, D. Wu, I. Paulsen, K. E. Nelson, W. Nelson, D. E. Fouts, S. Levy, A. H. Knap, M. W. Lomas, K. Nealson, O. White, J. Peterson, J. Hoffman, R. Parsons, H. Baden-Tillson, C. Pfannkoch, Y.-H. Rogers and H. O. Smith, Environmental Genome Shotgun Sequencing of the Sargasso Sea, Science, 2004, 304, 66–74 CrossRef CAS PubMed.
- E. Afshinnekoo, C. Chou, N. Alexander, S. Ahsanuddin, A. N. Schuetz and C. E. Mason, Precision Metagenomics: Rapid Metagenomic Analyses for Infectious Disease Diagnostics and Public Health Surveillance, J. Biomol. Tech., 2017, 28, 40–45 CrossRef PubMed.
- H. Fang, Y. Chen, L. Huang and G. He, Analysis of biofilm bacterial communities under different shear stresses using size-fractionated sediment, Sci. Rep., 2017, 7, 1299 CrossRef PubMed.
- E. Tsagkari and W. T. Sloan, Turbulence accelerates the growth of drinking water biofilms, Bioprocess Biosyst. Eng., 2018, 41(6), 757–770 CrossRef CAS PubMed.
- I. Douterelo, M. Jackson, C. Solomon and J. Boxall, Spatial and temporal analogies in microbial communities in natural drinking water biofilms, Sci. Total Environ., 2017, 581, 277–288 CrossRef PubMed.
- K. P. Keegan, E. M. Glass and F. Meyer, MG-RAST, a Metagenomics Service for Analysis of Microbial Community Structure and Function, Methods Mol. Biol., 2016, 44, 590–594 Search PubMed.
- V. Gomez-Alvarez, T. K. Teal and T. M. Schmidt, Systematic artifacts in metagenomes from complex microbial communities, ISME J., 2009, 3, 1314–1317 CrossRef PubMed.
- M. P. Cox, D. A. Peterson and P. J. Biggs, SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data, BMC Bioinf., 2010, 11, 1471–2105 CrossRef PubMed.
- T. Disz, S. Akhter, D. Cuevas, R. Olson, R. Overbeek, V. Vonstein, R. Stevens and R. A. Edwards, Accessing the SEED Genome Databases via Web Services API: Tools for Programmers, BMC Bioinf., 2010, 11, 319 CrossRef PubMed.
- R. Overbeek, T. Begley, R. M. Butler, J. V. Choudhuri, H. Y. Chuang, M. Cohoon, V. de Crecy-Lagard, N. Diaz, T. Disz, R. Edwards, M. Fonstein, E. D. Frank, S. Gerdes, E. M. Glass, A. Goesmann, A. Hanson, D. Iwata-Reuyl, R. Jensen, N. Jamshidi, L. Krause, M. Kubal, N. Larsen, B. Linke, A. C. McHardy, F. Meyer, H. Neuweger, G. Olsen, R. Olson, A. Osterman, V. Portnoy, G. D. Pusch, D. A. Rodionov, C. Ruckert, J. Steiner, R. Stevens, I. Thiele, O. Vassieva, Y. Ye, O. Zagnitko and V. Vonstein, The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes, Nucleic Acids Res., 2005, 33, 5691–5702 CrossRef CAS PubMed.
- L. J. Jensen, P. Julien, M. Kuhn, C. von Mering, J. Muller, T. Doerks and P. Bork, eggNOG: automated construction and annotation of orthologous groups of genes, Nucleic Acids Res., 2008, 36, 16 CrossRef PubMed.
- A. Belila, J. El-Chakhtoura, P. E. Saikaly, M. C. M. van Loosdrecht and J. S. Vrouwenvelder, Eukaryotic community diversity and spatial variation during drinking water production (by seawater desalination) and distribution in a full-scale network, Environ. Sci.: Water Res. Technol., 2017, 3, 92–105 RSC.
- H. C. Miller, M. J. Morgan and J. T. Wylie, et al. Elimination of Naegleria fowleri from bulk water and biofilm in an operational drinking water distribution system, Water Res., 2017, 110, 15–26 CrossRef CAS PubMed.
- I. Douterelo, M. Jackson, C. Solomon and J. Boxall, Microbial analysis of in situ biofilm formation in drinking water distribution systems: implications for monitoring and control of drinking water quality, Appl. Microbiol. Biotechnol., 2016, 100, 3301–3311 CrossRef CAS PubMed.
- V. M. Siqueira, H. M. B. Oliveira, C. Santos, R. R. M. Paterson, N. B. Gusmão and N. Lima, Biofilms from a Brazilian water distribution system include filamentous fungi, Can. J. Microbiol., 2012, 59, 183–188 CrossRef PubMed.
- V. Goncharuk, Drinking Water: Physics, Chemistry and Biology, Cham: Springer International Publishing, 2014 Search PubMed.
- D. T. Hughes and V. Sperandio, Inter-kingdom signalling: communication between bacteria and their hosts, Nat. Rev. Microbiol., 2008, 6, 111–120 CrossRef CAS PubMed.
- L. E. de-Bashan, M. Schmid, M. Rothballer, A. Hartmann and Y. Bashan, Cell-cell interation in the eukaryote-prokaryote model of the microalgae Chlorella vulgaris and the bacterium Azospirillum brasilense inmobilized in polymer beads, J. Phycol., 2011, 47, 1350–1359 CrossRef PubMed.
- P. Frey-Klett, P. Burlinson, A. Deveau, M. Barret, M. Tarkka and A. Sarniguet, Bacterial-Fungal Interactions: Hyphens between Agricultural, Clinical, Environmental, and Food Microbiologists, Microbiol. Mol. Biol. Rev., 2011, 75, 583–609 CrossRef CAS PubMed.
- E. Marco-Urrea, I. García-Romera and E. Aranda, Potential of non-ligninolytic fungi in bioremediation of chlorinated and polycyclic aromatic hydrocarbons, New Biotechnol., 2015, 32, 620–628 CrossRef CAS PubMed.
- N. Ashbolt, Environmental (Saprozoic) Pathogens of Engineered Water Systems: Understanding Their Ecology for Risk Assessment and Management, Pathogens, 2015, 4, 390 CrossRef PubMed.
- M. Taylor, K. Ross and R. Bentham, Legionella, Protozoa, and Biofilms: Interactions Within Complex Microbial Systems, Microb. Ecol., 2009, 58, 538–547 CrossRef PubMed.
- H.-C. Flemming and J. Wingender, The biofilm matrix, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS PubMed.
- C. Trappetti, L. Gualdi, L. Di Meola, P. Jain, C. C. Korir, P. Edmonds, F. Iannelli, S. Ricci, G. Pozzi and M. R. Oggioni, The impact of the competence quorum sensing system on Streptococcus pneumoniae biofilms varies depending on the experimental model, BMC Microbiol., 2011, 11(75), 1–12 Search PubMed.
- H. Lin, S. Zhang, S. Zhang, W. Lin and X. Yu, The function of advanced treatment process in a drinking water treatment plant with organic matter-polluted source water, Environ. Sci. Pollut. Res., 2017, 24, 8924–8932 CrossRef CAS PubMed.
- T. J. Battin, K. Besemer, M. M. Bengtsson, A. M. Romani and A. I. Packmann, The ecology and biogeochemistry of stream biofilms, Nat. Rev. Microbiol., 2016, 14, 251 CrossRef CAS PubMed.
- K. Fish, A. M. Osborn and J. B. Boxall, Biofilm structures (EPS and bacterial communities) in drinking water distribution systems are conditioned by hydraulics and influence discolouration, Sci. Total Environ., 2017, 593–594, 571–580 CrossRef CAS PubMed.
- B. Halliwell, Reactive Species and Antioxidants. Redox Biology Is a Fundamental Theme of Aerobic Life, Plant Physiol., 2006, 141, 312–322 CrossRef CAS PubMed.
- S. W. Seo, D. Kim, R. Szubin and B. O. Palsson, Genome-wide Reconstruction of OxyR and SoxRS Transcriptional Regulatory Networks under Oxidative Stress in Escherichia coli K-12 MG1655, Cell Rep., 2015, 12, 1289–1299 CrossRef CAS PubMed.
- A. R. Krapp, M. V. Humbert and N. Carrillo, The soxRS response of Escherichia coli can be induced in the absence of oxidative stress and oxygen by modulation of NADPH content, Microbiology, 2011, 157, 957–965 CrossRef CAS PubMed.
- J. P. Kehrer, J. D. Robertson and C. V. Smith, 1.14 - Free Radicals and Reactive Oxygen Species, Compr. Toxicol., 2010, 1, 277–307 CrossRef.
- J. A. Chesney, J. W. Eaton and J. R. Mahoney, Bacterial glutathione: a sacrificial defense against chlorine compounds, J. Bacteriol., 1996, 178, 2131–2135 CrossRef CAS PubMed.
- Y. Chao, Y. Mao, Z. Wang and T. Zhang, Diversity and functions of bacterial community in drinking water biofilms revealed by high-throughput sequencing, Sci. Rep., 2015, 5, 10044 CrossRef CAS PubMed.
- V. Gomez-Alvarez, R. P. Revetta and J. W. S. Domingo, Metagenomic Analyses of Drinking Water Receiving Different Disinfection Treatments, Appl. Environ. Microbiol., 2012, 78, 6095–6102 CrossRef CAS PubMed.
- M. R. Templeton, F. Oddy, W.-k. Leung and M. Rogers, Chlorine and UV disinfection of ampicillin-resistant and trimethoprim-resistant Escherichia coli, Journal of Environmental Engineering and Science, Can. J. Civ. Eng., 2009, 36, 889–894 CrossRef CAS.
- Y. Chao, L. Ma, Y. Yang, F. Ju, X.-X. Zhang, W.-M. Wu and T. Zhang, Metagenomic analysis reveals significant changes of microbial compositions and protective functions during drinking water treatment, Sci. Rep., 2013, 3, 3550 CrossRef PubMed.
- S. Jia, P. Shi, Q. Hu, B. Li, T. Zhang and X.-X. Zhang, Bacterial Community Shift Drives Antibiotic Resistance Promotion during Drinking Water Chlorination, Environ. Sci. Technol., 2015, 49, 12271–12279 CrossRef CAS PubMed.
- M. E. Hibbing, C. Fugua, M. R. Parsek and S. B. Peterson, Bacterial competition: surviving and thriving in the microbial jungle, Nat. Rev. Microbiol., 2010, 8(1), 15–25 CrossRef CAS PubMed.
- G. D. Wright, Antibiotic resistance in the environment: a link to the clinic?, Curr. Opin. Microbiol., 2010, 13, 589–594 CrossRef CAS PubMed.
- M. Schirmer, R. D'Amore, U. Z. Ijaz, N. Hall and C. Quince, Illumina error profiles: resolving fine-scale variation in metagenomic sequencing data, BMC Bioinf., 2016, 17, 125 CrossRef PubMed.
- I. Douterelo, R. L. Sharpe and J. B. Boxall, Influence of hydraulic regimes on bacterial community structure and composition in an experimental drinking water distribution system, Water Res., 2013, 47, 503–516 CrossRef CAS PubMed.
- B. Petru
![[s with combining breve]](https://www.rsc.org/images/entities/char_0073_0306.gif) evski, G. Bolier, A. N. Van Breemen and G. J. Alaerts, Tangential flow filtration: A method to concentrate freshwater algae, Water Res., 1995, 29, 1419–1424 CrossRef.
evski, G. Bolier, A. N. Van Breemen and G. J. Alaerts, Tangential flow filtration: A method to concentrate freshwater algae, Water Res., 1995, 29, 1419–1424 CrossRef. - M. Albertsen, P. Hugenholtz, A. Skarshewski, K. L. Nielse, G. W. Tysson and P. H. Nielsen, Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes, Nat. Biotechnol., 2013, 31, 533–538 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |