
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Integrated, multi-process approach to total nutrient recovery from stored urine†
Neha
Jagtap
 *ab and
Treavor H.
Boyer
b
*ab and
Treavor H.
Boyer
b
aDepartment of Environmental Engineering Sciences, Engineering School of Sustainable Infrastructure & Environment (ESSIE), University of Florida, P.O. Box 116450, Gainesville, Florida 32611-6450, USA. E-mail: njagtap@ufl.edu; Tel: +1 352 346 0057
bSchool of Sustainable Engineering and the Built Environment (SSEBE), Arizona State University, P.O. Box 873005, Tempe, Arizona 85287-3005, USA
First published on 28th August 2018
Abstract
This study investigated an integrated, multi-process approach of using struvite precipitation, ammonia stripping–acid absorption, and evaporation to recover phosphorus (P), nitrogen (N), and potassium (K), respectively, from stored urine. The process produces separate nutrient products that can then be recombined to produce customized fertilizers of any NPK ratio. Bench-scale experiments were conducted using three stored urine solutions: synthetic urine, synthetic urine with six endogenous metabolites, and real urine. For struvite precipitation, MgCl2·6H2O, MgCO3, and MgO were tested and dosed at a molar ratio of 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Mg:P. There was a statistically significant difference between total phosphate (TP) recovered by each magnesium (Mg) source and urine solution; MgCl2·6H2O (91–94% TP recovered) > MgCO3 (55–77%) > MgO (52–66%) and real urine > synthetic urine with six endogenous metabolites > synthetic urine. For ammonia stripping–acid absorption, there was a statistically significant difference between TAN recovery and experimental stripping conditions where increasing both the pH and temperature recovered a higher percent of TAN compared to solely increasing the pH or temperature of the solution. In real urine, consumed cost for stripping increased as follows: control condition of pH 9.2, 22 °C < elevated pH condition of pH 10.5, 22 °C < elevated temperature condition of pH 9.2, 70 °C. There was no statistically significant difference between the Mg source and TAN recovery in real urine and synthetic urine with metabolites but there was in synthetic urine. Furthermore, the amount of TAN recovered in real urine and synthetic urine with metabolites was consistently greater than or approximately equal to synthetic urine. This suggests that using synthetic urine as a proxy for real urine is not suitable for N recovery. For evaporation, there was a statistically significant difference between the urine solution and conditions for N recovery (i.e., temperature and/or pH) on K recovery and product purity. As the pH was increased, the purity of the final K product, potash, decreased due to sodium from NaOH. Results from this study show that an integrated, multi-process approach to urine treatment can achieve approximately 99% N, 91% P, and 80% K recovery as fertilizer products.
:1 Mg:P. There was a statistically significant difference between total phosphate (TP) recovered by each magnesium (Mg) source and urine solution; MgCl2·6H2O (91–94% TP recovered) > MgCO3 (55–77%) > MgO (52–66%) and real urine > synthetic urine with six endogenous metabolites > synthetic urine. For ammonia stripping–acid absorption, there was a statistically significant difference between TAN recovery and experimental stripping conditions where increasing both the pH and temperature recovered a higher percent of TAN compared to solely increasing the pH or temperature of the solution. In real urine, consumed cost for stripping increased as follows: control condition of pH 9.2, 22 °C < elevated pH condition of pH 10.5, 22 °C < elevated temperature condition of pH 9.2, 70 °C. There was no statistically significant difference between the Mg source and TAN recovery in real urine and synthetic urine with metabolites but there was in synthetic urine. Furthermore, the amount of TAN recovered in real urine and synthetic urine with metabolites was consistently greater than or approximately equal to synthetic urine. This suggests that using synthetic urine as a proxy for real urine is not suitable for N recovery. For evaporation, there was a statistically significant difference between the urine solution and conditions for N recovery (i.e., temperature and/or pH) on K recovery and product purity. As the pH was increased, the purity of the final K product, potash, decreased due to sodium from NaOH. Results from this study show that an integrated, multi-process approach to urine treatment can achieve approximately 99% N, 91% P, and 80% K recovery as fertilizer products.
Water impactUrine is a unique waste stream because it contains nutrients that are valuable in agriculture yet problematic in excess in aquatic environments. Unlike traditional fertilizer production that requires finite resources, urine is widely available. Separation and treatment of urine can recover nitrogen, phosphorus, and potassium as separate products of value, thereby reducing the negative impacts of nutrients on the environment. |
1. Introduction
The separation and treatment of urine has gained considerable attention in the past two decades as a viable process for producing fertilizer.1–5 Urine is a unique waste stream because it contains several plant nutrients—nitrogen (N), phosphorus (P), potassium (K), sulfur (S), chloride (Cl−), and magnesium (Mg2+).6,7 In agriculture, N, P, and K (also referred to as NPK) are key nutrients and are essential for soil and plant nutrition as they are needed for healthy and optimum growth.8 Conventional production of NPK fertilizers involves mining (from the ground or atmosphere), transporting, and processing of location-specific raw materials, which requires water, energy, and other finite resources.9 Urine, conversely, is a renewable resource that is produced everywhere, making it a favorable candidate for nutrient recovery. Accordingly, separation and treatment of urine has been proposed as an alternative approach to fertilizer production.7,10,11The chemical composition of human urine varies with diet, physiological factors, and further changes upon storage.12 When excreted from the human body, urine contains a high concentration of N in the form of urea, inorganic cations (Na+, Ca2+, Mg2+, K+), inorganic anions (Cl−, SO42−, PO43−), organic compounds (e.g., endogenous metabolites such as creatinine, taurine, hippurate) and possibly trace contaminants.13–15 This is referred to as fresh urine in the literature. When urine is stored, bacteria containing the urease enzyme hydrolyze urea to produce ammonia and bicarbonate.16 Urea hydrolysis raises the pH of urine from pH 6 to pH 9. Other impacts of storage and hydrolysis include the presence of a high concentration of ammonia, which can act as a biocide to reduce the number of pathogens, and create supersaturated conditions that favor the precipitation of magnesium phosphate and calcium phosphate minerals.17,18 This is referred to as stored urine in the literature. Although previous studies show low or no presence of citrate and hippurate in stored urine,19,20 there is limited published data on the extent of endogenous metabolite degradation during storage.6,20 Therefore, one important assumption made for this study was that the concentration of metabolites in fresh urine and stored urine were equal and no degradation of endogenous metabolites occurred.
Although urine contains numerous nutrients, direct application of urine as a liquid fertilizer has disadvantages. These include a predetermined NPK ratio that may be suitable for some plants but not others as well as labor and storage costs of collecting and transporting large volumes of urine.21–24 These disadvantages can be mitigated by producing separate N, P, and K products which will result in two main benefits: 1) the ability to customize the ratio of NPK in the fertilizer by recombining each individual product to fit the nutrient needs of any crop and 2) to concentrate nutrients in solid and liquid fertilizers that will have higher nutrient density than liquid urine.5,7,24
Struvite (MgNH4PO4·6H2O) precipitation has gained attention because it is an effective treatment process for P recovery. With the addition of a magnesium source (Mg source) 95% of P can be recovered within an hour in the form of an odorless, slow release fertilizer with little heavy metal contamination when compared with commercial fertilizers.21,24–26 Although struvite contains multiple key nutrients, N and P, the elemental composition of struvite and the composition of urine limits the amount of N that can be recovered from urine. Considering the composition of synthetic urine used in this study (Table 1) and the concentration of Mg2+ added for struvite precipitation, 15.4 mmol L−1, approximately 6.8 g L−1 N or 97% of N would remain in solution if all the P precipitated as struvite. The processes of struvite precipitation has also been investigated to recover potassium as K-struvite, KMgPO4·6H2O. However, the precipitation of K-struvite is not likely until N is depleted to concentrations below K from urine.27–30 Consequently, a significant portion of N and K remain in the solution after the struvite precipitation process.
| Chemicals | Synthetic urinea | Synthetic urine with metabolitesa | Real urinea | |||
|---|---|---|---|---|---|---|
| mmol L−1 | mg L−1 | mmol L−1 | mg L−1 | mmol L−1 | mg L−1 | |
| a Concentrations are measured except endogenous metabolites, which were calculated in synthetic urine based on recipe and not measured (nm) in real urine. b TAN = total ammonia nitrogen = NH3 (g) + NH4+ (aq) (mg L−1 N). c TP = total phosphate = H2PO4− + HPO42− (mg L−1 P). | ||||||
| SO42− | 15.14 | 1453 | 14.73 | 1414 | 12.74 | 1223 |
| Na+ | 98.26 | 2259 | 112.4 | 2585 | 57.66 | 1326 |
| K+ | 37.72 | 1471 | 43.21 | 1685 | 27.73 | 1081 |
| TANb | 453.8 | 6350 | 459.3 | 6430 | 359.8 | 5037 |
| Cl− | 96.40 | 3413 | 94.82 | 3356 | 73.15 | 2590 |
| TPc | 13.3 | 411 | 15.2 | 470 | 11.1 | 344 |
| Conductivity (μS cm−1) | 38.45 | 39.20 | 45.24 | |||
| Citrate | 2.486 | 2.486 | nm | — | ||
| Creatinine | 0.5633 | 0.5633 | nm | — | ||
| Glycine | 1.237 | 1.237 | nm | — | ||
| Hippurate | 2.804 | 2.804 | nm | — | ||
| L-Cysteine | 0.8058 | 0.8058 | nm | — | ||
| Taurine | 0.9919 | 0.9919 | nm | — | ||
| pH | 9.2 | 9.2 | 9.0 | |||
Due to the high concentration of total ammonia nitrogen (TAN), NH3(g) + NH4+(aq), in stored urine, many technologies have been investigated for the recovery of N, including air stripping–acid absorption (hereafter referred to as ammonia stripping–acid absorption) which has shown high nutrient recovery (>80% N recovery).31–34 The ammonia stripping–acid absorption process targets N recovery by removing ammonia from solution and concentrating it in a sulfuric acid solution. This is done by shifting the solution ammonia acid/base equilibrium towards NH3 (unionized ammonia) via a temperature and/or pH increase. Using a sufficient air to liquid flow ratio, NH3 is transferred from the liquid to gas phase and is subsequently ionized via absorption by sulfuric acid to produce (NH4)2SO4, ammonium sulfate, a liquid N fertilizer. Since all of the K is present in the soluble form, K should remain in solution and not be affected by the ammonia stripping–acid absorption process.
Precipitation in the form of K-struvite, KMgPO4·6H2O, has been researched as a treatment for the recovery of K from N-depleted urine. However, due to the elemental composition of K-struvite additional chemical input of Mg2+ and P is required for effective K recovery.28,30
TAN recovery by ammonia stripping–acid absorption32,34,35 and TP recovery by struvite precipitation21,25,27,36–50 have been studied extensively to recover a significant portion of N and P in urine. For example, major conclusions from previous research include >95% TP recovery using MgCl2 and >90% TAN recovery by increasing the pH and temperature of the solution. The gap in knowledge lies in the few studies that combine N and P treatment in series to produce separate N and P products,30,31,51–54 and minimal research on treatment processes to recover N, P, and K at significant concentrations.30,55,56 For example, major conclusions on treatment processes to recover N, P, and K include a lack of significant N and K recovery via precipitation without the equimolar addition of P and Mg2+ to N or K+ in solution.
NPK are all essential for plant nutrition yet the majority of studies done on nutrient recovery from urine focuses on N and P recovery. This is mainly because of the high concentration of N in urine and the limited availability of raw P sources which are location specific. Based on the urine composition shown in Table 1, the concentration of nutrients NPK increase from P < K < N where the concentration of N is approximately 4.5 times greater than K, which is approximately 3.5 times greater than P. Considering the approximate concentration of NPK in urine and the market value of each NPK fertilizer, the economic value of each nutrient ($ nutrient per 10000 L urine) was calculated for N, P, and K as shown in Table S1 in the ESI.† The economic value of NPK in urine decreases from N > K > P where the value of N is approximately 1.7 times greater than K, which is approximately 14 times greater than P. Results from this table show that K has comparable economic value to N and greater economic value compared to P, and should therefore be a focus for studies investigating nutrient recovery from urine. Due to the agricultural and economic value of separate N, P, and K products, identifying effective nutrient recovery treatment processes are necessary. Furthermore, understanding the effect of treatment processes on subsequent treatment operations is required for maximum nutrient recovery from urine. As a result, this study aims to fill this gap in knowledge by providing an integrated, multi-process approach of combining nutrient recovery processes into a single sequence to understand the recovery of N, P, and K as separate products of economic and agricultural value.
The goal of this study was to investigate the downstream impacts of each treatment process on NPK recovery and identify conditions that would yield high NPK recovery as agricultural products. The specific objectives of this study were to (i) compare the effect of urine solution chemistry on NPK recovery; (ii) compare the process of struvite precipitation using three Mg source inputs (MgCl2·6H2O, MgO, and MgCO3) in terms of TP recovery (%), struvite product purity, and urine supernatant solution chemistry (pH and ion concentration); (iii) compare TAN recovery (%), liquid ammonium sulfate product purity, and urine solution chemistry (ion concentration) via ammonia stripping–acid absorption, using urine supernatant with pH (chemical) and temperature (physical) adjustments, (iv) evaluate the composition of the final product, potash, compare K recovery (%), and compare the mass of product via evaporation, and (v) evaluate the economic value of each urine derived fertilizer product.
2. Materials and methods
2.1 Synthetic and real urine
Three types of stored urine were used in this study (Table 1): synthetic stored urine without endogenous metabolites (hereafter referred to as synthetic urine), synthetic stored urine with six endogenous metabolites (hereafter referred to as synthetic urine with metabolites), and real stored human urine (hereafter referred to as real urine). The synthetic urine recipe was adapted from previous studies (Table S2 in ESI†)45,52,57 and contained no calcium or magnesium to account for spontaneous precipitation that occurs during storage.17 Endogenous metabolites used in the recipe were selected based on prevalence and concentration in fresh urine.13 The assumption was that there was no degradation of added endogenous metabolites from fresh to stored urine. All reagents used were of ACS grade or equivalent.The University of Florida Institutional Review Board approved real urine collection as exempt. Informed consent was obtained for any experimentation with human subjects. Undiluted real human urine was collected from healthy males and females ages 16–50 and stored for four months at 20–30 °C. The extent of urea hydrolysis was 80% as determined by total nitrogen (TN) and TAN concentrations. Initial nutrient concentrations were established by samples taken after the storage period (pre struvite precipitation).
2.2 Struvite precipitation
Bench-scale struvite precipitation experiments were done in triplicate using 1 L of urine. Three Mg sources were used: MgCl2·6H2O, MgO, and MgCO3, and dosed at a molar ratio of 1.1:1 Mg:P to ensure maximum TP recovery.30,58,59 Urine was mixed for 60 min at 100 rpm via a jar tester apparatus and allowed to settle in Imhoff cones for 60 min.25,31,59 The P-depleted supernatant was decanted into 1 L collapsible polyethylene terephthalate containers, compressed to minimize head space, and stored at 4 °C for analysis and subsequent experiments. To maintain struvite crystal structure, the resulting precipitate was collected and dried at 30 °C to prevent losses of TAN.44,60,61
Based on the solubility (g/100 mL at 25 °C) of Mg sources in water (MgCl2·6H2O = 52.9, MgO = 0.0086, MgCO3 = 0.0139), preliminary Mg2+ solubility tests were conducted to determine the solubility of each Mg source in solutions of increasing ionic strength and the expected TP recovery for struvite precipitation experiments. Experiments were done in triplicate using the same parameters detailed for struvite precipitation experiments (Mg:P molar ratio, mixing speed and time, and settling time), 250 mL of deionized (DI) water, and a modified synthetic urine made without the addition of phosphate (referred to as synthetic urine with no P). Solubility was determined based on the concentration of Mg2+ in solution.
2.3 Ammonia stripping–acid absorption
Ammonia stripping–acid absorption experiments were conducted with P-depleted supernatant following struvite precipitation. Batch experiments were performed using two glass 125 mL fritted gas-washing bottles (standard taper 34/28 top joint and a coarse fritted glass tube for gas dispersion). The stripping column contained 100 mL of stored urine and the absorption column contained 30 mL of 1 M sulfuric acid solution (made from 98% w/w H2SO4 purchased from Merck (Rahway, NJ)). The airflow rate (N2 at 1 L min−1) and experimental time (3 h) were kept constant and taken from the study conducted by Basakcilardan-Kabakci et al. to maximize N recovery potential.35 Each sample had five experimental conditions: (i) pH 9.6, 55 °C; (ii) pH 10, 40 °C; (iii) pH 10.5, 22 °C; (iv) pH 9.2, 70 °C; (v) pH 9.2, 22 °C. Visual MINTEQ 3.1, a chemical equilibrium software, was used to predict experimental conditions that would yield approximately 90% TAN as NH3(g).62 Conditions that had a pH above 9.2 were adjusted with a 10 M sodium hydroxide (NaOH) solution (Fisher certified grade) and conditions that were above 22 °C were heated with a water bath. During the ammonia stripping–acid absorption process, foam production was observed when real urine was used and this resulted in cross-contamination of urine from the stripping column to the absorption column. To reduce the foam production and prevent cross contamination, 2 mL of vegetable oil was added to each urine sample in the stripping column. For condition pH 9.2, 70 °C, significant foam production was still observed after the addition of vegetable oil. Further discussion can be found in section 2.2 in the ESI.†2.4 Evaporation
Following ammonia stripping–acid absorption, 5 mL of urine from the stripping column was filtered and analyzed for Mg2+, K+, Na+, Cl−, SO42−, TAN, and TP concentration. The remaining 95 mL of urine was dried in an oven at 50 °C for 48 h and the solid was analyzed for mineral identification.2.5 Analytical methods
All stock solutions and synthetic urine were prepared using 18.2 MΩ cm ultra-pure water and ACS reagent grade purity chemicals. Liquid urine samples were taken at three points: pre struvite precipitation (initial), post struvite precipitation, and post ammonia stripping–acid absorption. Conductivity and pH were measured with Orion Star A215 conductivity meter and Accumet AB 15 pH meter, respectively. In real urine at pH 9, both N and P are present as two species: NH3/NH4+ for nitrogen and H2PO4−/HPO42− for phosphorus. Therefore, samples were analyzed for total ammonia nitrogen (TAN) and total phosphate (TP). TP (reported as mg P L−1) was measured following Standard Method 4500P ascorbic acid method (EPA 356.3) using a Hitachi U-2900 spectrophotometer at 880 nm and a 1 cm quartz cuvette. Total ammonia nitrogen (TAN) was measured using flow injection analysis (Lachat's QuikChem® 8500 Series 2 Flow Injection Analysis System). Samples were acidified to pH < 2 and reported as mg N L−1. Magnesium (Mg2+), sodium (Na+), and potassium (K+) concentrations were measured using inductively coupled plasma (ICP) optical emission spectrometer (Thermo Fisher iCap 6000 ICP-OES). Samples were acidified with 2% trace metal grade nitric acid. Chloride (Cl−) and sulfate (SO42−) concentrations were measured via ion chromatography (Dionex ICS-3000) following standard methods.63 Analytical methods are further explained in the ESI,† section 1.1.Solid samples were taken at two points: post struvite precipitation and post evaporation. Samples were ground to a powder using a mortar and pestle then examined by X-ray diffraction (XRD). XRD analysis was performed using a Siemens D5000 X-ray diffractometer with monochromic Cu Kα radiation. The scanning rate used was 2.0° min−1 with a 2θ range of 10–70. The quality of precipitate obtained was compared with standard XRD patterns from the International Centre for Diffraction Data (ICDD).
2.6 Statistical analysis
ANOVA: two way with replication was used to compare the variation (factors) in each treatment process on nutrient recovery. Examples of each factor include urine solutions (factor i) and magnesium sources (factor ii) for struvite precipitation. Table S3a–e in ESI† reports p-values for each factor. Two null hypotheses were tested with a significance level of 5%: (1) there was no statistically significant difference between nutrient recovery by factor (i) and (2) there was no statistically significant difference between nutrient recovery by factor (ii). The alternative hypotheses stated that there was a significant difference between nutrient recovery by factor (i) and factor (ii).3. Results and discussion
3.1 Effect of Mg source on P recovery by struvite precipitation and implications for N and K recovery
While TP recovery by struvite precipitation has been well-researched, the effect of struvite precipitation on downstream processes (e.g., ammonia stripping–acid absorption for TAN recovery and evaporation for K recovery) has not been extensively investigated. Therefore, understanding the effect of each chemical input (Mg source) on the pH and ion concentrations of the supernatant (post struvite precipitation) and the implications on subsequent N and K recovery was essential. MgCl2·6H2O was chosen because of its high solubility (52.9 g/100 mL water) and addition of Cl− to the solution. Theoretically, all the Mg added would dissolve and precipitate with P and N to form struvite and the addition of Cl− would increase the purity of the K product because Cl− is a component of potash. MgCO3 and MgO were chosen because a pH increase of the solution was desired for subsequent N recovery. If the pH of the solution increased from the initial pH of approximately 9.2, theoretically less NaOH would be needed to reach the desired pH value of each ammonia stripping–acid absorption condition. The amount of NaOH in solution would also affect the purity of the potash product because low concentrations of Na+ and high concentrations of K+, Cl−, and SO42− are needed.The percent of TP recovered by each Mg source, percent of TP in the product, and the crystal structure identified using XRD are shown in Table 2. The amount of TP recovered by each Mg source was MgCl2·6H2O > MgCO3 > MgO, where only MgCl2·6H2O followed trends reported in literature25,48 whereas MgO and MgCO3 did not. For example, MgO recovered <66% TP, unlike the study conducted by Lind et al. in which >99% TP recovery was observed using a ratio less than 1.1:1 Mg:P (experimental dose).51 Etter et al. observed up to 91% TP recovery via filtration when MgO was dosed at 1.1:1 Mg:P molar ratio and less than 50% via sedimentation.25 Several struvite precipitation studies were conducted with MgO, however, the dose was greater than 1.1:1 Mg:P molar ratio and therefore could not be used for direct comparison.25,31,64 Due to the low solubility of MgCO3 in water, MgCO3 has not been investigated for struvite precipitation, but rather it has been treated to produce soluble MgO.42,65 Krahenuhl et al. saw only 1% of the added Mg2+ had dissolved in solution after six hours of calcination at 400 °C, which is less than the >55% TP recovered by MgCO3 in synthetic urine in this study.
:P molar ratio of 1.1:1)
| Urine solution | Magnesium source | Ce,a mean (mmol L−1) | Ce,a std (mmol L−1) | TP recovered (%) | P in solidb (%) |
|---|---|---|---|---|---|
| Initial TP concentration of urine solution is as follows: synthetic urine with metabolites (15.2 mmol L−1), synthetic urine (13.3 mmol L−1), and real urine (11.1 mmol L−1). Reported values are the averages of triplicates. a Ce = effluent concentration. b XRD analysis identified struvite as the dominant mineral phase for all solids. | |||||
| Synthetic urine | MgCl2·6H2O | N/A | N/A | 92.7 | 12.0 |
| MgO | 6.93 | 0.544 | 47.8 | 10.1 | |
| MgCO3 | 5.99 | 0.478 | 54.9 | 10.1 | |
| Synthetic urine with metabolites | MgCl2·6H2O | N/A | N/A | 93.6 | 12.5 |
| MgO | 5.20 | 0.868 | 65.7 | 10.8 | |
| MgCO3 | 5.23 | 1.59 | 65.5 | 11.4 | |
| Real urine | MgCl2·6H2O | N/A | N/A | 91.3 | 12.6 |
| MgO | 3.74 | 0.535 | 66.4 | 10.7 | |
| MgCO3 | 2.58 | 0.520 | 76.8 | 10.5 | |
Based on preliminary Mg2+ solubility experiments (Table S4 in ESI†), the solubility of MgO and MgCO3 increased with ionic strength (i.e., synthetic urine with no P > DI water) and were expected to recover approximately 100% and 30% TP, respectively, in synthetic urine since all of the dissolved Mg2+ would precipitate with P and N species. However, MgO and MgCO3 did not follow this trend and recovered approximately 52% and 55% of TP during struvite precipitation experiments, respectively, in synthetic urine (Table 2). Conductivity measurements were taken as a surrogate of ionic strength (Table 1) and the increasing trend was synthetic urine < synthetic urine with metabolites < real urine. For struvite precipitation experiments, TP recovered when MgCl2·6H2O was used was approximately equal (91–94%) in all urine solutions. The specific results are further explained in the ESI,† section 1.2. However, TP recovery when MgO and MgCO3 was used as the Mg source for struvite precipitation was highest in real urine followed by synthetic urine with metabolites and finally synthetic urine. The TP results supported the idea that the solubility of MgO and MgCO3 increased with increasing ionic strength.
Regardless of the Mg source and urine solution, all solid materials obtained were crystalline and agreed well with the peak positions of the standard XRD pattern for struvite (Fig. S1 in ESI†). Based on the stoichiometry of MgNH4PO4·6H2O, the theoretical percent of P in struvite is 12.6%, which is approximately four times greater than the percent of P in liquid urine. Samples that contained MgCl2·6H2O consistently had values of approximately 12.6% (12.6% in real urine, 12.5% in synthetic urine with metabolites, and 12.0% in synthetic urine), while MgO and MgCO3 consistently had values below 12.6%, indicating that an approximately pure struvite was produced albeit with some difference. Previous struvite precipitation work reported MgO as a more effective source for struvite precipitation than MgCO3 because of lower costs, greater percent of dissolved Mg2+ in solution, higher TP recovery, and higher P content in the product.25,42,43 A simple economic cost analysis for the production of struvite from 10000 L of real urine was calculated as shown in Table S5 in the ESI.† Although MgO had the greatest net profit, MgCO3 had the highest cost per mol of P recovered. Furthermore, MgCO3 recovered a greater percent of TP compared with MgO and had comparable struvite purity (approximately 11% in real urine, 11% in synthetic urine with metabolites, and 10% in synthetic urine), demonstrating that it is an equally effective Mg source for struvite precipitation. To determine which Mg source would be the most effective (high nutrient recovery and low cost) the location of the treatment process must be considered. For example, the abundance of MgCO3 in Nepal could reduce consumed costs by using a locally available resource and make MgCO3 more cost effective than pretreating MgCO3 to produce MgO and/or buying MgO from the market.25
The purpose of using three urine solutions was to compare the effect of urine solution chemistry under identical experimental conditions on NPK recovery, e.g., the effect of endogenous metabolites on nutrient recovery. The amount of P in the struvite product and TP recovered from solution increased as the conductivity of the solution increased, suggesting that metabolites had a positive effect on TP recovery and struvite purity. When MgCO3 was used as the Mg source, the percent of TP recovered from solution was 52%, 62%, and 66% in synthetic urine, synthetic urine with metabolites, and real urine, respectively (Table 2). Previous literature observed the opposite effect where the endogenous metabolites either had no effect on the precipitation potential of struvite and could be neglected or acted as organic complexing agents and reduced the amount of free cations in solution and thus the extent of struvite precipitation. For example, Udert et al. 2003 investigated precipitation effects in a urine collecting system and concluded that endogenous metabolites (organic compounds), specifically citrate and oxalate, had a negative effect on the struvite precipitation process because the metabolites would adsorb to the crystals during the nucleation step and inhibit the rate of precipitation by a factor of 4.17 However, the difference in precipitation potential was mainly noticed during the hydrolysis process. Once at least 16% of the urea was hydrolyzed, the precipitation potential in solutions with organic complexing agents and without organic complexing agents only differed slightly until complete urea hydrolysis was reached.17,19 For this study, approximately 80% of urea was hydrolyzed in real urine and 100% of urea was hydrolyzed in the synthetic urine solutions. Therefore, the difference in urea hydrolysis from previous literature and the current study could explain for the variation observed in this study. However, since TP recovery in the presence of metabolites has not been observed in pervious literature, future research detailing the mechanisms of prevalent metabolites in urine is required to fully understand and compare this result from this study.
Since stored urine is buffered by ammonia and carbonate, there was no significant increase or decrease in pH due to the Mg source (as shown in Table S6 in the ESI†).
Results from the ANOVA two-way test with replication for TP recovery (Table S3.a in ESI†) show that p-values for factor (i) and (ii) are <0.05. Therefore, there is a statistically significant different between TP recovery and varying Mg sources, where MgCl2·6H2O recovered the greatest TP, and between TP recovery and varying urine solutions, where the greatest TP was recovered in real urine.
3.2 Effect of pH and temperature on N recovery by ammonia stripping–acid absorption and implication for K recovery
To test the effect of pH and temperature on N recovery by ammonia stripping–acid absorption, the treatment process was initially tested with urine at pH 9.2, 22 °C, which corresponds to the conditions of post struvite precipitation and ambient laboratory temperature. Under this condition, approximately 47% of TAN occurs as NH3 (g), while 53% occurs as NH4+ (aq). A combination of pH and temperature conditions were determined to yield approximately 90% TAN as NH3(g) using Visual MINTEQ 3.1. Since stored urine is buffered by ammonia and carbonate, there was no significant increase in pH due to the Mg source and approximately similar volumes of NaOH were added, regardless of the Mg source, to increase the pH of urine for each condition (Table S7 in the ESI†) (e.g., for pH 10, 40 °C in real urine, MgCl2·6H2O, MgO and MgCO3 required 2.4, 2.3, and 2.4 mL, respectively). Conditions that had only elevated pH, only elevated temperature, and both elevated pH and temperature were compared because of the variation in operating costs for each condition. For example, elevated pH is achieved by chemical addition (one type of operating cost), whereas elevated temperature is achieved by heat transfer (which is a very different type of operating cost). Experimental conditions for ammonia stripping–acid absorption were compared in terms of TAN recovery efficiency (eqn (S1) and section 2.1 in the ESI†), consumed costs (Table S8 in the ESI†), and percent N (NH4+–N) in the liquid ammonium sulfate ((NH4)2SO4) fertilizer product (Table 3).| Urine solution | Mg source | Ammonia stripping–acid absorption conditions | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pH 9.6, 55 °C | pH 10, 40 °C | pH 10.5, 22 °C | pH 9.2, 70 °C | pH 9.2, 22 °C | ||||||
| % Na | % Ka | % Na | % Ka | % Na | % Ka | % Na | % Na | % Ka | ||
| a Percent nutrient in the product calculated as w/w. | ||||||||||
| Real urine | MgCl2·6H2O | 1.5 | 4.4 | 1.4 | 5.1 | 1.2 | 2.6 | 2.4 | 0.83 | 6.6 |
| MgO | 1.4 | 4 | 1.3 | 5.1 | 1.3 | 3.4 | 2.1 | 0.96 | 6.6 | |
| MgCO3 | 1.6 | 3.7 | 1.4 | 4.9 | 1.3 | 3.5 | 2.2 | 0.62 | 6.6 | |
| Synthetic with metabolites | MgCl2·6H2O | 1.7 | 6.6 | 1.6 | 7.1 | 1.4 | 5.3 | 2.1 | 0.98 | 6.7 |
| MgO | 1.8 | 6.7 | 1.7 | 6.7 | 1.5 | 5.5 | 1.9 | 0.91 | 6.5 | |
| MgCO3 | 2.0 | 5.9 | 1.6 | 6.1 | 1.9 | 5.5 | 2.3 | 1.1 | 9.2 | |
| Synthetic | MgCl2·6H2O | 1.7 | 6.7 | 1.6 | 7.4 | 1.3 | 5.8 | 2.2 | 0.93 | 9.3 |
| MgO | 1.9 | 5.5 | 1.6 | 6.2 | 1.4 | 5.2 | 2.3 | 0.96 | 9.2 | |
| MgCO3 | 2.0 | 5.2 | 1.8 | 6.6 | 1.6 | 5.2 | 1.7 | 0.98 | 9.1 | |
Increasing both the pH and temperature of the solution recovered the greatest percent of TAN in real urine and synthetic urine with metabolites, regardless of the Mg source. The trend of TAN recovery in real urine for this study follows trends in literature where solely increasing the temperature, rather than solely increasing the pH, of the solution resulted in a higher percentage of TAN recovery, pH 9.2, 70 °C > pH 10.5, 22 °C > pH 9.2, 22 °C (Fig. 1).32,33,66 Although solely increasing the temperature resulted in a higher percent of TAN recovered, operational issues such as excess foaming, precipitation, and evaporation were observed. Specific details are further explained in the ESI,† section 2.2. Experimental conditions were designed to yield TAN recovery of approximately 90%, however, only temperature adjusted samples recovered >90% TAN while samples adjusted for only pH (condition pH 10.5, 22 °C) recovered <75% TAN in all urine solutions. During ammonia stripping–acid absorption experiments, temperature was maintained using a water bath while pH was not monitored throughout. For pH measurements, samples were adjusted (to the condition pH) and measured before and after the treatment process (Table S9 in the ESI†). Due to the slight variation in pH, it is difficult to conclude why samples only adjusted for pH recovered <75% TAN instead of >90% TAN.
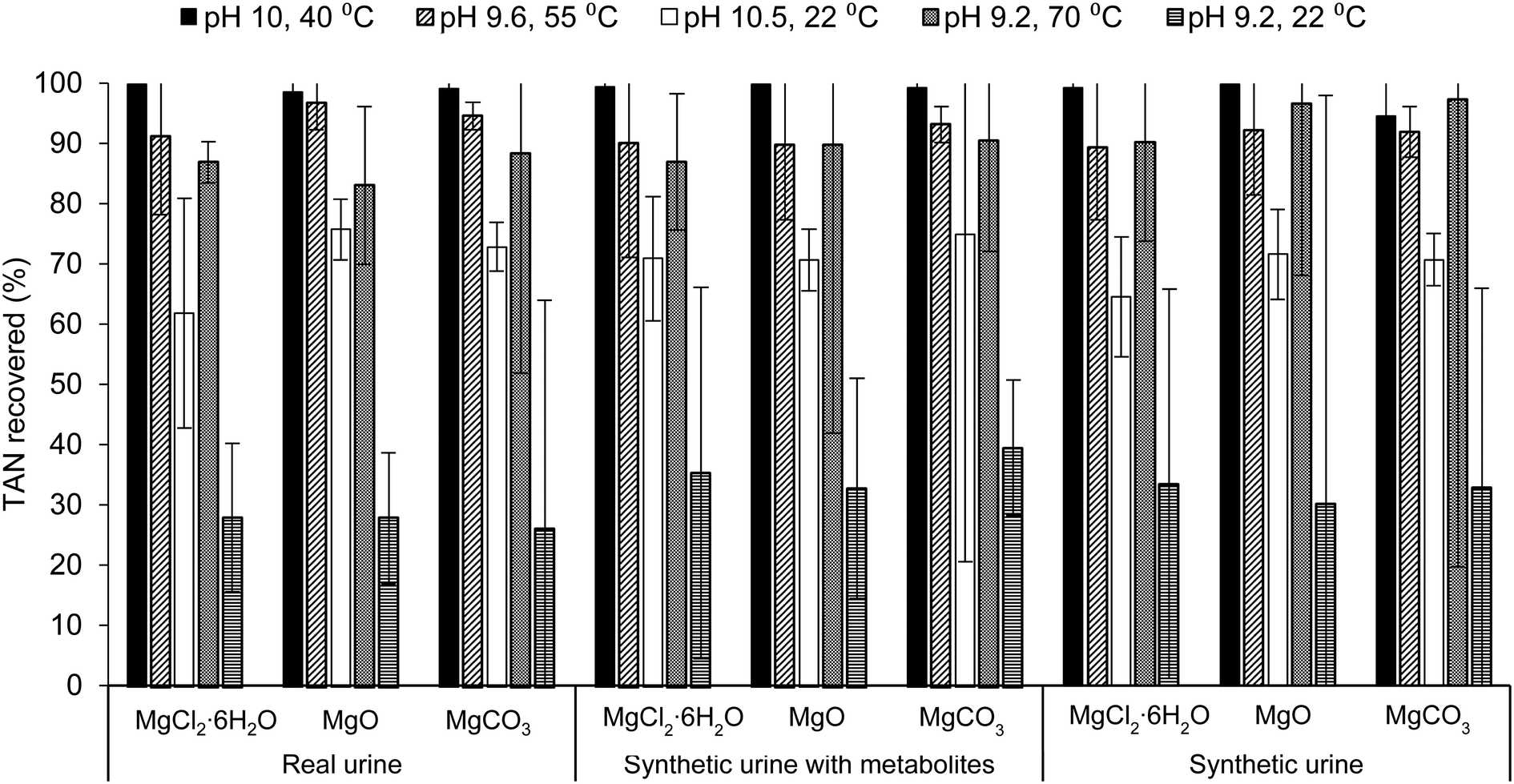 | ||
| Fig. 1 Effect of ammonia stripping–acid absorption conditions, Mg sources, and urine solution on TAN recovery via ammonia stripping–acid absorption after 3 h of operation. Urine composition and struvite precipitation conditions reported on x-axis labels. Ammonia stripping–acid absorption conditions reported in legend. All measured data are average values of triplicate samples with error bars showing one standard deviation. | ||
Results from the ANOVA two-way test with replication (Table S3.b and c in ESI†) for TAN recovery in samples containing MgCl2·6H2O, MgO, and MgCO3 indicate that there was a statistically significant difference between TAN recovery by varying ammonia stripping–acid absorption condition (p-value <0.05) and between TAN recovery in varying urine solutions. This suggests that the percent of TAN recovered by MgO, for example, in all urine solutions was significantly different, where a greater percent was recovered in real urine for all conditions aside from the baseline and pH 9.2, 70 °C. Furthermore, samples containing MgO and MgCO3 consistently had higher TAN recovery compared to samples containing MgCl2·6H2O for all conditions aside from the baseline and pH 9.2, 70 °C as well as in all urine solutions. For experiments done with real urine and synthetic urine with metabolites, there was no statically significant difference (p-value >0.05) between TAN recovery and varying Mg source but there was a statistically significant difference for experiments done with synthetic urine. This suggests that using synthetic urine as a proxy for real urine is not suitable for N recovery studies.
In post ammonia stripping–acid absorption, the (NH4)2SO4 product was kept in liquid form to minimize N loss and the percent N (w/w) in the product was calculated (Table 3).33,35 Calculation details for percent N (w/w) in the product are explained in the ESI,† eqn (S1)–(S6). The H2SO4 solution used in this study had the capability of producing a 2.67% N (w/w) product (eqn (S3) and (S4) in the ESI†). Condition pH 9.2, 70 °C produced the highest percent N (w/w) in the product, however due to operational issues detailed in the ESI,† this condition would not be ideal for treatment. The ammonium sulfate product derived from treating 100 mL of real urine contained approximately 1.18–1.47% N (w/w). Although this is less than the 8% N (w/w) in commercial liquid ammonium sulfate products on the market,67 it is 2–3 times more concentrated that the percent N (w/w) in urine. Furthermore, the concentration and volume of sulfuric acid (H2SO4) could be increased and reused to treat multiple batches of urine and produce a product with a higher N content (eqn (S5) and (S6) in the ESI†). Assuming the urination volume for each person is 1.4 L d−1, a single person can supply enough N to produce an 8% N ammonium sulfate liquid fertilizer in less than a month.
3.3 Effect of P and N recovery on K recovery by evaporation
To investigate the effect of P and N recovery on K recovery, the mass of K was tracked through each treatment process as shown in Fig. S3 in the ESI.† To summarize, the initial volume of 100 mL used for the ammonia stripping–acid absorption process was used to calculate the mass of K pre struvite precipitation and post struvite precipitation. The volume remaining in the stripping column after ammonia stripping–acid absorption was used to calculate the mass for the treatment process of ammonia stripping–acid absorption. The mass of K in samples post struvite precipitation remained approximately the same compared to samples pre-struvite precipitation. Post ammonia stripping–acid absorption, there was a decrease in K mass. Following the ammonia stripping–acid absorption treatment process, a liquid sample was taken from both the stripping and absorption column and analyzed for K concentration. The volume remaining in each column, the stripping and the absorption column, was used to calculate the mass of K in samples after the ammonia stripping–acid absorption treatment process. The volume remaining in the stripping column was evaporated and analyzed via XRD to identify the dominant mineral phase present in the product and possible precipitates and weighed to determine the purity of the potash product. XRD analysis was not sufficient to identify K mineral phases present, as shown in Fig. S2 in the ESI,† as only 50–60% of the peaks could be identified due to the addition of oil during the treatment process.The mass of K present in the stripping column post ammonia stripping–acid absorption treatment, as shown in Table S10 in the ESI,† and the percent K in the potash product, as shown in Table S11 in the ESI,† followed the same trend where K decreased from pH 9.2, 22 °C > pH 10, 40 °C > pH 9.6, 55 °C > pH 10.5, 22 °C in all urine samples. Due to the high solubility of K+ in solution, similar masses of K was expected before and after the ammonia stripping–acid absorption treatment process. However, this was not the case. Furthermore, all ammonia stripping–acid absorption conditions aside from the baseline condition, pH 9.2, 22 °C had over 15% of the initial mass of K that was not present in either the stripping or absorption column. Since XRD results were insufficient to draw conclusions and did not identify any insoluble K minerals, Visual MINTEQ 3.1 was used to predict possible K precipitates that could explain for the unaccounted K. None were identified. Although XRD and Visual MINTEQ 3.1 did not identify K precipitates or insoluble K minerals, both tools have limitations, and it may be possible that the elevated pH conditions resulted in precipitation of K. Since samples were filtered prior to potassium analysis, it is possible that the precipitates were excluded by the filter and therefore unaccounted for during the measurement.
Potash is a common term for nutrient forms of the element K such as KCl and K2SO4.68 Therefore, potash purity was determined for all samples by comparing potash nutrients, K, SO42−, and Cl− (% w/w) to Na, which is undesired by soils and plants because it inhibits the uptake of desired nutrients such as N and K which are essential to plant growth (Table S11 in the ESI†). Specific details are discussed in the ESI,† section 2.3. The percent of Na+ in solution was consistently greater than the percent of potash in all samples, and increased with pH, where condition pH 10.5, 22 °C had the greatest percent of Na by mass in the product followed by pH 10, 40 °C > pH 9.6, 55 °C > pH 9.2, 22 °C. Samples adjusted for pH contained approximately 30–50% by mass Na indicating that the use of NaOH as a pH adjustment would not be suitable for effective nutrient recovery. Marketable potash fertilizers range from 17–52% K,69 which is over 4 times greater than what was obtained in this work. One potential solution to reduce Na+ in solution and increase K+ in solution would be to substitute KOH ($480 per metric ton) in lieu of NaOH ($400 per metric ton) for pH adjustment during ammonia stripping–acid absorption (specific cost details located in section 2.5 in ESI†). However, the volume of KOH must be considered to draw concrete conclusions on the effect on potash purity. Furthermore, it is important to note that the presence of organic compounds such as endogenous metabolites and pharmaceuticals, could render the potash product unusable, and further treatment, such as the addition of acid and the use of biochar to reduce the odor and pharmaceutical concentration in urine should be explored.70,71
Results from the ANOVA two-way test with replication (Table S3.d and e in ESI†) indicate that there was a statistically significant difference between K recoveries for varying ammonia stripping–acid absorption conditions (p-value <0.05) as well as between K recoveries for varying urine solutions. Following the baseline condition (pH 9.2, 22 °C) which recovered the greatest percent K in all urine solutions, i.e., the mass of K present in the stripping column post ammonia stripping–acid absorption, increasing both pH and temperature (pH 10, 40 °C) recovered the second greatest percent K in all urine solutions. The trend for increasing K recovery under condition pH 10, 40 °C, for samples containing MgCl2·6H2O and MgO was real urine > synthetic urine with metabolites > synthetic urine, whereas the trend for samples containing MgCO3 was real urine > synthetic urine > synthetic urine with metabolites. These findings, which have not been previously reported in the literature, indicate that the urine solution and conditions for N recovery (i.e., temperature and/or pH) dictate the purity of K in the final product whereas conditions for P recovery (i.e., Mg source) have no significant effect on the purity of the K product.
3.4 Economic analysis of integrated, multi-process approach
To evaluate the integrated, multi-process approach for selective nutrient recovery of N, P, and K and the production of customizable fertilizers for any NPK ratio, a preliminary economic analysis was done. Using experimental data, the production of NPK fertilizer from 10000 L of real urine was calculated (Table 4). Following ammonia stripping–acid absorption, all samples were dried at 50 °C for 48 h. Therefore, energy costs during the evaporation process were neglected, since costs would be equivalent for all samples, and not considered in the cost analysis for this process. Out of the three nutrient recovery processes, only P and K recovery showed a positive net profit, and only condition pH 9.2, 22 °C, which required no pH and temperature adjustment, had a positive net profit from complete NPK recovery. The consumed costs of N recovery is detailed in Table S8 in the ESI.† In real urine, the control condition (pH 9.2, 22 °C) recovered approximately 28% TAN and cost $13.7 per 10000 L. Whereas increasing the pH (pH 10.5, 22 °C) recovered 60–75% N and cost approximately $94 per 10000 L and increasing the temperature (pH 9.2, 70 °C) recovered 83–96% N and cost approximately $100 per 10000 L. Therefore, changes must be made to the ammonia stripping–acid absorption process to make the combined treatment process profitable. Although increasing the pH and temperature of each sample resulted in additional consumed costs and subsequently lower net profits compared to the control condition, low-cost alternatives to increase the pH and temperature of urine can be used to minimize additional costs. These includes using air to strip CO2 in lieu of NaOH to increase the pH of urine,72 and using solar energy or waste heat in lieu of electricity as a source for transferring heat and increasing the temperature of urine.73
000 L of real urine using MgCl2·6H2O as the magnesium source for struvite precipitation
| P fertilizer ($) | N fertilizer ($) | P and N fertilizer ($) | K fertilizer ($) | N and K fertilizer ($) | NPK fertilizer ($) | |
|---|---|---|---|---|---|---|
| a Total nutrient recovery involving K fertilizer could not be calculated due to cross contamination of potassium during the ammonia stripping–acid absorption process. | ||||||
| pH 9.6, 55 °C | 4.99 | −75.89 | −70.89 | 13.12 | −62.77 | −57.77 |
| pH 10, 40 °C | 4.99 | −75.50 | −70.50 | 13.44 | −62.06 | −57.06 |
| pH 10.5, 22 °C | 4.99 | −75.88 | −70.88 | 12.09 | −63.79 | −58.79 |
| pH 9.2 70 °Ca | 4.99 | −73.78 | −68.78 | — | — | — |
| pH 9.2, 22 °C | 4.99 | −4.34 | 0.65 | 14.06 | 9.72 | 14.72 |
4. Conclusion
• An integrated, multi-process approach of struvite precipitation, ammonia stripping–acid absorption, and evaporation was able to recover separate P, N, and K products of agricultural and economic value.• To maximize N, P, and K recovery and purity of the corresponding products from real urine, MgCl2·6H2O is recommended as the input for struvite precipitation followed by an increase in pH and temperature to pH 10 and 40 °C. Under these conditions, over 91% of P, 99% of N, and 80% of K can be recovered.
• The Mg source used for P recovery did not impact subsequent N or K recovery in real urine or synthetic urine with metabolites.
• Conditions for N recovery affected subsequent K recovery where increasing both the pH and temperature of urine (pH 10, 40 °C) maximized K recovery and only increasing the pH decreased product purity.
• Replacing NaOH with KOH, another strong base to adjust the pH of urine, would increase the concentration of K+ (desired by soils and plants) in solution rather than Na+ (undesired by soils and plants) and create a K product of higher purity.
• TP, TAN, and K recovery in real urine was consistently higher than or equivalent to synthetic urine. Therefore, using synthetic solutions as a proxy is not suitable for robust conclusions regarding complete NPK recovery from real urine.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication is based upon work supported by the National Science Foundation, NSF CAREER grant CBET-1150790. Any opinions, findings, conclusions or recommendations expressed in this publication are those of the authors and do not necessarily reflect the views of NSF. This publication was improved by the thoughtful comments of three anonymous reviewers.References
- M. C. Chrispim, W. A. Tarpeh, D. T. Salinas and M. A. Nolasco, The sanitation and urban agriculture nexus: urine collection and application as fertilizer in São Paulo, Brazil, J. Water, Sanit. Hyg. Dev., 2017, 7, 455–465 CrossRef.
- P. J. Talboys, J. Heppell, T. Roose, J. R. Healey, D. L. Jones and P. J. Withers, Struvite: a slow-release fertiliser for sustainable phosphorus management?, Plant Soil, 2016, 401, 109–123 CrossRef PubMed.
- S. K. L. Ishii and T. H. Boyer, Life cycle comparison of centralized wastewater treatment and urine source separation with struvite precipitation: Focus on urine nutrient management, Water Res., 2015, 79, 88–103 CrossRef PubMed.
- T. Karak and P. Bhattacharyya, Human urine as a source of alternative natural fertilizer in agriculture: A flight of fancy or an achievable reality, Resour., Conserv. Recycl., 2011, 55, 400–408 CrossRef.
- H. Kichmann and S. Pettersson, Human urine–chemical composition and fertilizer efficiency, Fert. Res., 1995, 40, 149–154 CrossRef.
- K. Udert, T. Larsen and W. Gujer, Fate of major compounds in source-separated urine, Water Sci. Technol., 2006, 54, 413–420 CrossRef PubMed.
- T. A. Larsen and W. Gujer, Separate management of anthropogenic nutrient solutions (human urine), Water Sci. Technol., 1996, 34, 87–94 CrossRef.
- E. Simonne and G. Hochmuth, Soil and fertilizer management for vegetable production in Florida, Vegetable production handbook for Florida, 2005, vol. 2006, pp. 3–15 Search PubMed.
- C. W. Gellings and K. E. Parmenter, Energy efficiency in fertilizer production and use, Efficient use and conservation of energy, EOLSS Publications, 2016, pp. 123–131 Search PubMed.
- J. A. Wilsenach and M. C. van Loosdrecht, Integration of processes to treat wastewater and source-separated urine, J. Environ. Eng., 2006, 132, 331–341 CrossRef.
- J. Wilsenach and M. Van Loosdrecht, Impact of separate urine collection on wastewater treatment systems, Water Sci. Technol., 2003, 48, 103–110 CrossRef PubMed.
- C. Ricos, C. V. Jiménez, A. Hernández, M. Simón, C. Perich, V. Alvarez, J. Minchinela and M. Maciá, Biological variation in urine samples used for analyte measurements, Clin. Chem., 1994, 40, 472–477 Search PubMed.
- S. Bouatra, F. Aziat, R. Mandal, A. C. Guo, M. R. Wilson, C. Knox, T. C. Bjorndahl, R. Krishnamurthy, F. Saleem and P. Liu, The human urine metabolome, PLoS One, 2013, 8, e73076 CrossRef PubMed.
- H. Jönsson, A. R. Stintzing, B. Vinnerås and E. Salomon, Guidelines on the use of urine and faeces in crop production, EcoSanRes Programme, 2004 Search PubMed.
- P. Heitland and H. D. Köster, Fast, simple and reliable routine determination of 23 elements in urine by ICP-MS, J. Anal. At. Spectrom., 2004, 19, 1552–1558 RSC.
- H. Mobley and R. Hausinger, Microbial ureases: significance, regulation, and molecular characterization, Microbiol. Rev., 1989, 53, 85–108 Search PubMed.
- K. M. Udert, T. A. Larsen, M. Biebow and W. Gujer, Urea hydrolysis and precipitation dynamics in a urine-collecting system, Water Res., 2003, 37, 2571–2582 CrossRef PubMed.
- L. Decrey, S. Kazama, K. M. Udert and T. Kohn, Ammonia as an in situ sanitizer: inactivation kinetics and mechanisms of the ssRNA virus MS2 by NH3, Environ. Sci. Technol., 2014, 49, 1060–1067 CrossRef PubMed.
- K. M. Udert, T. A. Larsen and W. Gujer, Estimating the precipitation potential in urine-collecting systems, Water Res., 2003, 37, 2667–2677 CrossRef PubMed.
- B. D. Sykes, Urine stability for metabolomic studies: effects of preparation and storage, Metabolomics, 2007, 3, 19–27 CrossRef.
- M. Ronteltap, M. Maurer and W. Gujer, The behaviour of pharmaceuticals and heavy metals during struvite precipitation in urine, Water Res., 2007, 41, 1859–1868 CrossRef PubMed.
- H. Kirchmann and S. Pettersson, Human urine-chemical composition and fertilizer use efficiency, Nutr. Cycling Agroecosyst., 1994, 40, 149–154 Search PubMed.
- H. N. Bischel, B. D. Ö. Duygan, L. Strande, C. S. McArdell, K. M. Udert and T. Kohn, Pathogens and pharmaceuticals in source-separated urine in eThekwini, South Africa, Water Res., 2015, 85, 57–65 CrossRef PubMed.
- M. Maurer, W. Pronk and T. Larsen, Treatment processes for source-separated urine, Water Res., 2006, 40, 3151–3166 CrossRef PubMed.
- B. Etter, E. Tilley, R. Khadka and K. Udert, Low-cost struvite production using source-separated urine in Nepal, Water Res., 2011, 45, 852–862 CrossRef PubMed.
- M. Maurer, P. Schwegler and T. A. Larsen, Nutrients in urine: energetic aspects of removal and recovery, Water Sci. Technol., 2003, 48, 37–46 CrossRef PubMed.
- J. Wilsenach, C. Schuurbiers and M. Van Loosdrecht, Phosphate and potassium recovery from source separated urine through struvite precipitation, Water Res., 2007, 41, 458–466 CrossRef PubMed.
- K. Xu, C. Wang, H. Liu and Y. Qian, Simultaneous removal of phosphorus and potassium from synthetic urine through the precipitation of magnesium potassium phosphate hexahydrate, Chemosphere, 2011, 84, 207–212 CrossRef PubMed.
- K. Xu, C. Wang, X. Wang and Y. Qian, Laboratory experiments on simultaneous removal of K and P from synthetic and real urine for nutrient recycle by crystallization of magnesium–potassium–phosphate–hexahydrate in a draft tube and baffle reactor, Chemosphere, 2012, 88, 219–223 CrossRef PubMed.
- K. Xu, C. Zhang, J. Li, X. Cheng and C. Wang, Removal and recovery of N, P and K from urine via ammonia stripping and precipitations of struvite and struvite-K, Water Sci. Technol., 2017, 75, 155–164 CrossRef PubMed.
- S. Antonini, S. Paris, T. Eichert and J. Clemens, Nitrogen and phosphorus recovery from human urine by struvite precipitation and air stripping in Vietnam, Clean: Soil, Air, Water, 2012, 39, 1099–1104 Search PubMed.
- B. Liu, A. Giannis, J. Zhang, V. W. C. Chang and J. Y. Wang, Air stripping process for ammonia recovery from source-separated urine: modeling and optimization, J. Chem. Technol. Biotechnol., 2015, 90, 2208–2217 CrossRef.
- S. K. Pradhan, A. Mikola and R. Vahala, Nitrogen and phosphorus harvesting from human urine using a stripping, absorption, and precipitation process, Environ. Sci. Technol., 2017, 5165–5171 CrossRef PubMed.
- H. Gulyas, S. Zhang and R. Otterpohl, Pretreating Stored Human Urine for Solar Evaporation by Low-Technology Ammonia Stripping, J. Environ. Prot., 2014, 5, 962–969 CrossRef.
- S. Basakcilardan-Kabakci, A. N. Ipekoglu and I. Talinli, Recovery of ammonia from human urine by stripping and absorption, Environ. Eng. Sci., 2007, 24, 615–624 CrossRef.
- S. Gadekar and P. Pullammanappallil, Validation and applications of a chemical equilibrium model for struvite precipitation, Environ. Model Assess., 2010, 15, 201–209 CrossRef.
- Z. Ganrot, A. Slivka and G. Dave, Nutrient Recovery from Human Urine Using Pretreated Zeolite and Struvite Precipitation in Combination with Freezing-Thawing and Plant Availability Tests on Common Wheat, Clean: Soil, Air, Water, 2008, 36, 45–52 Search PubMed.
- M. G. Grau, S. L. Rhoton, C. J. Brouckaert and C. A. Buckley, Development of a fully automated struvite reactor to recover phosphorus from source separated urine collected at urine diversion toilets in eThekwini, Proceedings of the Water Environment Federation, 2013, vol. 2013, pp. 74–83 Search PubMed.
- H. Harada, Y. Shimizu, Y. Miyagoshi, S. Matsui, T. Matsuda and T. Nagasaka, Predicting struvite formation for phosphorus recovery from human urine using an equilibrium model, Water Sci. Technol., 2006, 54, 247–255 CrossRef PubMed.
- P. Kemacheevakul, S. Chuangchote, S. Otani, T. Matsuda and Y. Shimizu, Effect of magnesium dose on amount of pharmaceuticals in struvite recovered from urine, Water Sci. Technol., 2015, 72, 1102–1110 CrossRef PubMed.
- E. Kirinovic, A. R. Leichtfuss, C. Navizaga, H. Zhang, J. D. Schuttlefield Christus and J. Baltrusaitis, Spectroscopic and Microscopic Identification of the Reaction Products and Intermediates during the Struvite (MgNH4PO4·6H2O) Formation from Magnesium Oxide (MgO) and Magnesium Carbonate (MgCO3) Microparticles, ACS Sustainable Chem. Eng., 2017, 1567–1577 CrossRef.
- M. Krähenbühl, B. Etter and K. M. Udert, Pretreated magnesite as a source of low-cost magnesium for producing struvite from urine in Nepal, Sci. Total Environ., 2016, 542, 1155–1161 CrossRef PubMed.
- B. Liu, A. Giannis, J. Zhang, V. W.-C. Chang and J.-Y. Wang, Characterization of induced struvite formation from source-separated urine using seawater and brine as magnesium sources, Chemosphere, 2013, 93, 2738–2747 CrossRef PubMed.
- M. Prabhu and S. Mutnuri, Cow urine as a potential source for struvite production, Int. J. Recycl. Org. Waste Agric., 2014, 3, 49 CrossRef.
- M. Ronteltap, M. Maurer and W. Gujer, Struvite precipitation thermodynamics in source-separated urine, Water Res., 2007, 41, 977–984 CrossRef PubMed.
- M. Ronteltap, M. Maurer, R. Hausherr and W. Gujer, Struvite precipitation from urine–influencing factors on particle size, Water Res., 2010, 44, 2038–2046 CrossRef PubMed.
- S. R. Sakthivel, E. Tilley and K. M. Udert, Wood ash as a magnesium source for phosphorus recovery from source-separated urine, Sci. Total Environ., 2012, 419, 68–75 CrossRef PubMed.
- E. Tilley, J. Atwater and D. Mavinic, Recovery of struvite from stored human urine, Environ. Technol., 2008, 29, 797–806 CrossRef PubMed.
- F. Abbona, H. L. Madsen and R. Boistelle, Crystallization of two magnesium phosphates, struvite and newberyite: effect of pH and concentration, J. Cryst. Growth, 1982, 57, 6–14 CrossRef.
- P. Zamora, T. Georgieva, I. Salcedo, N. Elzinga, P. Kuntke and C. J. Buisman, Long-term operation of a pilot-scale reactor for phosphorus recovery as struvite from source-separated urine, J. Chem. Technol. Biotechnol., 2017, 92, 1035–1045 CrossRef.
- B.-B. Lind, Z. Ban and S. Bydén, Nutrient recovery from human urine by struvite crystallization with ammonia adsorption on zeolite and wollastonite, Bioresour. Technol., 2000, 73, 169–174 CrossRef.
- K. Udert and M. Wächter, Complete nutrient recovery from source-separated urine by nitrification and distillation, Water Res., 2012, 46, 453–464 CrossRef PubMed.
- A. Fumasoli, B. Etter, B. Sterkele, E. Morgenroth and K. M. Udert, Operating a pilot-scale nitrification/distillation plant for complete nutrient recovery from urine, Water Sci. Technol., 2016, 73, 215–222 CrossRef PubMed.
- S. Yao, L. Chen, D. Guan, Z. Zhang, X. Tian, A. Wang, G. Wang, Q. Yao, D. Peng and J. Li, On-site nutrient recovery and removal from source-separated urine by phosphorus precipitation and short-cut nitrification-denitrification, Chemosphere, 2017, 210–218 CrossRef PubMed.
- Z. G. Liu, Q. L. Zhao, K. Wang, W. Qiu, W. Li and J. F. Wang, Comparison between complete and partial recovery of N and P from stale human urine with MAP crystallization, J. Environ. Eng. Sci., 2008, 7, 223–228 CrossRef.
- M. A. Rahman and V. M. Chariar, Process Optimization for Sequential Recovery of N, P and K from Human Urine, South Asian J. Exp. Biol., 2016, 5, 205–221 Search PubMed.
- K. A. Landry and T. H. Boyer, Diclofenac removal in urine using strong-base anion exchange polymer resins, Water Res., 2013, 47, 6432–6444 CrossRef PubMed.
- K. M. Udert, C. A. Buckley, M. Wächter, C. S. McArdell, T. Kohn, L. Strande, H. Zöllig, A. Fumasoli, A. Oberson and B. Etter, Technologies for the treatment of source-separated urine in the eThekwini Municipality, Water SA, 2015, 41, 212–221 CrossRef.
- S. Başakçılardan-Kabakcı, A. N. İpekoğlu and I. Talınlı, Precipitation of urinary phosphate, Environ. Eng. Sci., 2007, 24, 1399–1408 CrossRef.
- R. L. Frost, M. L. Weier and K. L. Erickson, Thermal decomposition of struvite, J. Therm. Anal. Calorim., 2004, 76, 1025–1033 CrossRef.
- M. I. H. Bhuiyan, D. S. Mavinic and F. A. Koch, Thermal decomposition of struvite and its phase transition, Chemosphere, 2008, 70, 1347–1356 CrossRef PubMed.
- J. P. Gustafsson, Visual MINTEQ (version 3.1), 2014 Search PubMed.
- J. D. Pfaff, Method 300.0 Determination of inorganic anions by ion chromatography, 1993 Search PubMed.
- C. K. Abegglen, PhD, Swiss Federal Institute of Technology Zurich, 2008 Search PubMed.
- A. Gunay, D. Karadag, I. Tosun and M. Ozturk, Use of magnesit as a magnesium source for ammonium removal from leachate, J. Hazard. Mater., 2008, 156, 619–623 CrossRef PubMed.
- R. L. Siegrist, in Decentralized Water Reclamation Engineering, Springer, 2017, pp. 141–180 Search PubMed.
- T. B. I. John Weiss, M. Hunter, J. L. Karl Czymmek and Q. Ketterings, Nitrogen Fertilizers for Field Crops, 2009 Search PubMed.
- H. J. S. Finch, A. M. Samuel and G. P. F. Lane, in Lockhart & Wiseman's Crop Husbandry Including Grassland, Woodhead Publishing, 9th edn, 2014, pp. 63–91 Search PubMed.
- D. W. M. M. Stewart, Potash terminology and facts, 1996, p. 7 Search PubMed.
- J. V. Bergen, Industrial Odor Control, J. Air Pollut. Control Assoc., 1958, 8, 101–111 CrossRef.
- A. Solanki and T. H. Boyer, Pharmaceutical removal in synthetic human urine using biochar, Environ. Sci.: Water Res. Technol., 2017, 553–565 RSC.
- M. H. Winkler, F. Rossum, N. Oskam, L. Dijk and G. J. van de Pol, presented in part at the WEF/IWA Nutrient Removal and Recovery: Trends in Resource Recovery and Use, Vancouver, BC, Canada, 2013 Search PubMed.
- H. Siegrist, M. Laureni, K. M. Udert, T. Larsen, K. Udert and J. Lienert, Transfer into the gas phase: ammonia stripping, Source Separation and Decentralization for Wastewater Management, 2013, pp. 337–350 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ew00004b |
| This journal is © The Royal Society of Chemistry 2018 |