
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Environmental transformation of natural and engineered carbon nanoparticles and implications for the fate of organic contaminants
Gabriel Sigmund
 ab,
Chuanjia Jiang
c,
Thilo Hofmann
*b and
Wei Chen
*c
ab,
Chuanjia Jiang
c,
Thilo Hofmann
*b and
Wei Chen
*c
aChair of Analytical Chemistry and Water Chemistry, Technical University of Munich, 9 Marchioninistrasse 17, 81377 Munich, Germany
bDepartment of Environmental Geosciences, University of Vienna, A-1090 Vienna, Austria. E-mail: thilo.hofmann@univie.ac.at
cCollege of Environmental Science and Engineering, Ministry of Education Key Laboratory of Pollution Processes and Environmental Criteria, Tianjin Key Laboratory of Environmental Remediation and Pollution Control, Nankai University, 38 Tongyan Rd., Tianjin 300350, China. E-mail: chenwei@nankai.edu.cn
First published on 1st October 2018
Abstract
Environmental transformation of carbon nanoparticles can significantly affect their transport, fate, and effects. The last decade of environmental nano-science has often focused on understanding the behavior of well-defined engineered carbon nanoparticles (eCNPs) in the natural environment. However, more complex pyrogenic/petrogenic carbon nanoparticles (pCNPs), including those derived from soot, fossil coal, wildfire charcoal, and biochar, are more than four orders of magnitude more abundant in the environment. This paper aims to review findings from investigations into eCNPs and to consider their transferability to pCNPs, in order to improve our understanding of pCNPs and identify gaps in our knowledge. Findings from previous investigations into the chemical, physical and biological transformation of larger carbonaceous particles, as well as of eCNPs, can help us to understand the transformation of pCNPs. The transformation of soot during atmospheric transport is relatively well documented, whereas the transformation of pCNPs in soil, sediment, and aqueous systems remains poorly understood. To bridge findings on particulate transport, contaminant binding, and contaminant transformation from eCNPs to pCNPs, the complex compositions of pCNPs need to be taken into account. We therefore suggest that future research on pCNP transformation should focus on changes in intrinsic porosity and on interactions with non-carbonized phases, tar phases, and mineral phases, as well as with organo-mineral complexes in soils, sediments and water bodies.
 Gabriel Sigmund | Gabriel Sigmund received his PhD in Environmental Sciences from the University of Vienna in 2017, and is currently working as post-doc at the Chair of Analytical Chemistry and Water Chemistry, Technical University of Munich. His research focuses on interactions of carbonaceous (nano)materials with organic contaminants and natural organic matter, as well as the characterisation of these materials. He is additionally in close collaboration with the Ithaka Institute for carbon strategies to translate his scientific findings into the development of practical applications of biochar in environmentally-friendly agriculture- and water treatment approaches. |
 Chuanjia Jiang | Chuanjia Jiang is an associate professor of environmental science and engineering at Nankai University, Tianjin, China. He received his B.S. and M.S. in Environmental Engineering from Tsinghua University, Beijing, in 2009 and 2011, respectively; his Ph.D. in Civil and Environmental Engineering from Duke University, Durham, NC, in 2016. His current research interests include environmental aquatic chemistry, and the environmental applications and implications of nanotechnology. |
 Thilo Hofmann | Thilo Hofmann received his Ph.D. in Aquatic Geochemistry from Bremen University in 1998. Since 2005 he has been a full professor and chair for Environmental Geosciences at the University of Vienna. His group works in nanogeosciences, environmental impact of nanotechnology, sorption of trace contaminants to carbonaceous materials and microplastics, and in hydrogeology, including vulnerability analysis. He is the director of the University of Vienna's Environmental Research Network including more than 130 scientists from natural sciences, social sciences, humanities, law, and economics. He is an Adjunct Full Professor at Duke University, U.S., and a Guest Professor of Nankai University, Tianjin, China. |
 Wei Chen | Wei Chen is a professor of Environmental Science and Engineering at Nankai University, Tianjin, China, and the director of Tianjin Key Laboratory of Environmental Remediation and Pollution Control. He received his B.S. in Environmental Chemistry from Nankai University in 1992, and M.S. and Ph.D. in Environmental Science and Engineering from Rice University, Houston, TX, USA, in 1997 and 2000. His current research interests are fate and transport of nanoparticles in the environment, and nanomaterial-based environmental technologies. Prof. Chen is an editor of Environmental Toxicology and Chemistry, and editorial advisory board members of Environmental Science & Technology and Environmental Science: Nano. |
Environmental significanceBoth natural and anthropogenic sources release large quantities of carbon nanoparticles (CNPs) into the environment. CNPs have strong adsorption affinities for organic contaminants and the accumulation of contaminants onto CNPs may significantly alter the effects of the CNPs, as well as the transport, reactivity, and bioavailability of environmental contaminants. Once released into the environment, CNPs can undergo physical, chemical and biological transformation, resulting in significant changes to their physicochemical properties, consequently altering their transport in natural environments and their interactions with contaminants through sorption and catalysis. Bridging findings from well-defined and well-studied engineered CNPs to the more complex, much more abundant and much less studied pyrogenic/petrogenic CNPs is critical for assessing the environmental impact and exploring applications of CNPs. |
1. Introduction
Carbon particles are released into the environment from both natural and anthropogenic sources.1,2 Forest fires, fossil fuel combustion and fossil coal transport all release large quantities of carbon into the environment2 including carbon nanoparticles (CNPs), which have at least one dimension between 1 and 100 nm. Those CNPs that derive from pyrogenic or petrogenic sources (pCNPs) have been released into the environment for millennia. Engineered CNPs (eCNPs), including carbon nanotubes, fullerenes and graphene (oxides), have also been commercialized over the last decade and are used in a range of consumer products and industry applications.3 Both eCNPs and pCNPs have strong adsorption affinities for a range of organic contaminants and are likely to be the predominant media with which contaminants are associated in many natural environments. The accumulation of organic contaminants onto CNPs may significantly alter the effects of the CNPs (e.g., their toxicity), as well as the transport, reactivity, and bioavailability of environmental contaminants.4–6Once released into the environment CNPs can undergo physical, chemical and biological transformation, resulting in significant changes to their physicochemical properties (e.g., their size, morphology, and surface functionality). The transformation of CNPs can have a marked effect on their surface charge, hydrophobicity, and reactivity, and may thus alter their aggregation and transport in natural environments and their interactions with contaminants through sorption and catalysis.
The formation and dispersal of carbonaceous particles,1,2,7 the transport of CNPs,7–10 and the sorption of organic contaminants by CNPs11–13 have all been extensively investigated and reviewed. The transformation of CNPs in natural environments has also recently been receiving increasing attention.14–17 However, the last decade of research has focused mainly on understanding the behavior and implications of well-defined eCNPs, whereas those of the more complex, naturally occurring pCNPs of pyrogenic and petrogenic origins, which comprise the dominant fraction of CNPs in natural environments, remain largely unexplored. A key aim of this review is therefore to compare findings from investigations into pristine eCNPs and consider their transferability to pCNPs, in order to improve our understanding of pCNPs as well as of how transformation of pCNPs can affect the fate of contaminants. This will allow identification of future research needs in the field of naturally-occurring CNPs. A good understanding of the sources, transport, and transformation of pCNPs and the associated implications for contaminant fate is critical for identifying risks associated with colloidal CNPs and may also shed light on possible uses of CNPs in remediation.
2. Sources of CNPs in the environment
Sources of CNPs found in the natural environment include fossil coal transportation (petrogenic origin from geological transformation of organic carbon), forest fires, fossil fuel combustion, biochar application for agricultural and remedial purposes (pyrogenic origin from incomplete combustion of organic carbon), the use of activated carbon for filtration and remediation purposes, and unintended releases of eCNPs from industrial processes or during the resulting products life-cycles.2,5,72.1 Fossil coal
Fossil coal is a major energy source in many industrial countries including the USA, China, and India, where fossil coal combustion remains a key factor for power production. Although there has been a recent decrease in global coal-based energy production, fossil coal production still amounts to approximately 6.5 Pg per year and no complete phase-out of coal-based power plants is foreseeable in the coming decades.18,19 The formation of pCNPs from fossil coal combustion residuals, which consist mainly of mineral phases, is negligible; pCNPs are, however, released during the mining and transport of fossil coal, where a mass loss of 1% has been reported.19 The decomposition of fossil coal depends on its rank (which is associated with its aromaticity) as well as on its volatile matter and O content. Coal stability decreases in the following order: anthracite > bituminous coal > subbituminous coal > lignite.20,21 Assuming that, on average, fossil coals disintegrate physically at a similar rate to biochar (see section 2.3) suggests a pCNP release of approximately 6.5 Tg per year from fossil coal.2.2 Soot
A very common type of pCNP is soot, which is the finest residual fraction from incomplete combustion; it results from natural processes such as forest fires as well as from anthropogenic processes such as fossil fuel combustion. Soot is formed through the re-condensation and nucleation of organic compounds such as acetylene and polycyclic aromatic hydrocarbons (PAHs) from a gas phase during combustion and is typically introduced into the environment via atmospheric transport.22 It can be found throughout all environmental compartments, including extreme examples such as in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 year-old Greenland ice core.23 Soot particles can vary greatly in their size distribution, surface chemistry, and morphology depending on the production conditions and other environmental factors. Soot generally contains large quantities of PAHs, which can become bioavailable.24,25 Estimates for the annual production of soot transportable via the atmosphere range from 6 to 28 Tg per year,26 with the most recent estimates being 17 Tg per year.2,27
000 year-old Greenland ice core.23 Soot particles can vary greatly in their size distribution, surface chemistry, and morphology depending on the production conditions and other environmental factors. Soot generally contains large quantities of PAHs, which can become bioavailable.24,25 Estimates for the annual production of soot transportable via the atmosphere range from 6 to 28 Tg per year,26 with the most recent estimates being 17 Tg per year.2,27
2.3 Biochar
Biochar is a product of biomass pyrolysis; it is used in agriculture to increase the nutrient- and water-holding capacity and pH of soils, and in remedial applications for sorption of contaminants. It also serves as a carbon sink for climate change mitigation.28 In contrast to soot or wildfire charcoals, biochar is produced under more controlled conditions, resulting in a “cleaner”, more carbonized material. For instance, when comparing biochars with wildfire charcoals from the same feedstock, PAHs are generally found to be present at lower concentrations and to be less bioavailable in biochar than in wildfire charcoal.29 Biochar particles produced by physical disintegration in soil are expected to be transported mainly by surface runoff and colloidal transport in pore water, rather than via the atmosphere.30 Understanding the release of pCNPs from biochar in the field remains challenging as the fraction of pCNPs varies greatly depending on the biochar feedstock, pyrolysis temperature, and soil/sediment conditions.30–32The application of biochar is currently limited globally to a number of test sites.28 However, if biochar was to become globally accepted as a means of carbon sequestration and soil improvement, Woolf et al.33 estimate that up to 220 Tg of biochar could be used every year, which would result in a considerable quantity of biochar-derived pCNPs being released into the environment. Spokas et al.30 have reported particle release from disintegration of ten different biochars ranging from 1 to 47%. A conservative estimate of 10% was assumed for Fig. 1, resulting in the release of up to 20 Tg per year of biochar-derived pCNPs. It is not currently possible to quantify more precisely the overall potential release of pCNPs from biochar due to the large differences in disintegration between different biochars.
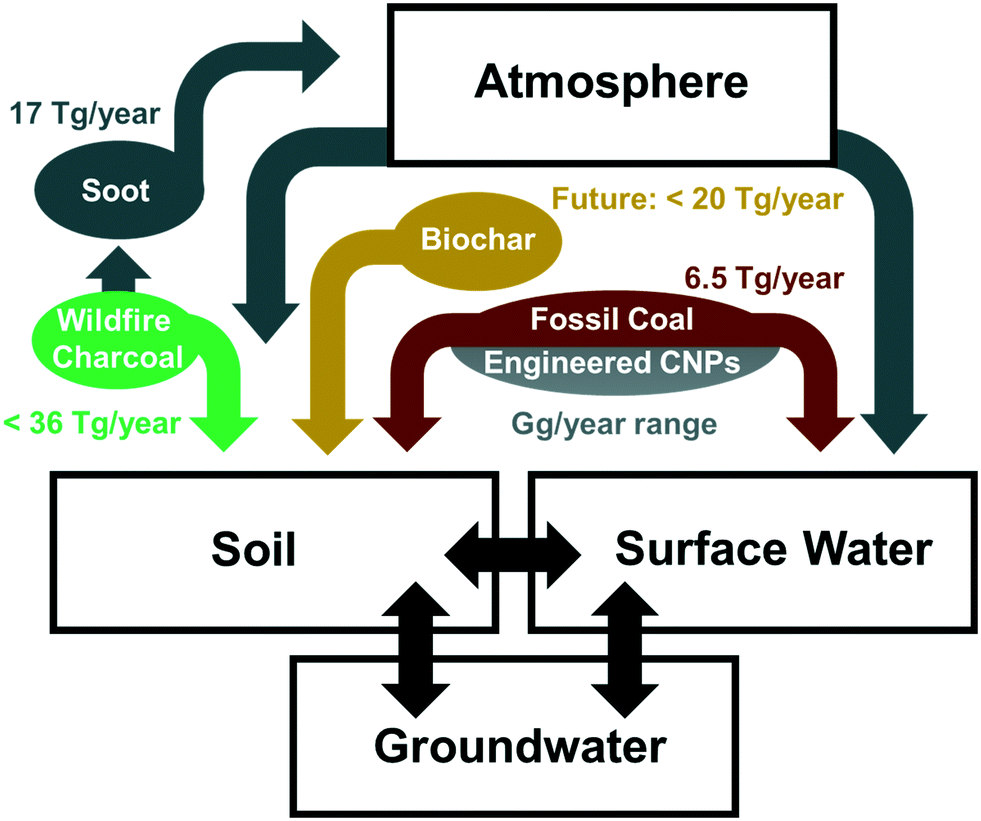 | ||
| Fig. 1 Major sources of CNPs in the natural environment with rough estimates of mass fluxes, as discussed in section 2. | ||
2.4 Wildfire charcoal
Wildfires burn 30–46 million km2 per year of the global land surface,34 producing up to 241 Tg per year of wildfire charcoal,26 the solid combustion residue of wildfires. Between 1 and 26% of the total carbon consumed during forest fires ends up as wildfire charcoal,35 with a generally lower proportion for grassland (<3%).26 The physicochemical properties of wildfire charcoal are strongly dependent on the feedstock, the maximum temperature of the fire, the heating duration, and the particle size of the feedstock.36,37 It has recently been noted that the physical location within a tree (e.g., base of main tree stem compared to the upper crown) also has a marked effect on the physicochemical properties of the wildfire charcoal produced.38 de Lafontaine and Asselin39 compared a large number of wildfire charcoals in different soils and found that soil type and climate only had a minor influence on their physical disintegration, indicating that disintegration mostly occurs soon after formation and is mainly driven by the physicochemical properties of the wildfire charcoal. Wildfire charcoals are typically less stable and more prone to transformation and physical disintegration than biochar.21,29 For a rough estimate of pCNPs produced a slightly larger proportion has therefore been assumed for wildfire charcoal (15%) than for biochar (10%), yielding an estimated pCNP release of up to 36 Tg per year. This is of the same order of magnitude as a recent estimate of global charcoal particle transport to the oceans (26.5 Tg per year).402.5 Engineered CNPs
eCNPs include carbon nanotubes (CNTs), fullerenes, graphene, and graphene oxide (GO); they can be released into the environment both intentionally and unintentionally.5,7,41 Global eCNP production was approximately 4.5 Gg in 20113 and is reported to have been approximately 20 Gg in 2016, with annual production predicted to increase by 20% per year until 2023.42 The use of activated carbon may also result in the release of a significant quantity of CNPs into the environment. Recent predictions suggest that activated carbon production will reach 3 Tg per year by 2024.43A significant fraction of eCNPs is incorporated into other matrices (e.g., CNTs may be part of a composite) and/or used under highly controlled conditions (e.g., activated carbon is used for water filtration) so that only a small proportion of eCNPs is expected to reach the natural environment, whether in wastewater streams or by direct introduction.
In summary, soot, fossil coal, and wildfire charcoal are important sources of naturally-occurring pCNPs (>99%; Tg per year range). In contrast, the better defined eCNPs, which have received the most attention over the last decade, make up a much smaller proportion of the CNPs in the natural environment (<1%; Gg per year range), as illustrated in Fig. 1. Although pCNPs and eCNPs share many molecular similarities such as their high aromaticity and high electron donor-acceptor capacity, they differ significantly in terms of structural irregularities, surface functionality, inorganic phases and porosity. For example, pCNPs generally possess more negatively charged surface groups and smaller aromatic sheets and thus have a lower sorption affinity for most organic contaminants compared to eCNPs. These structural and molecular differences are expected to play an important role in fate, transport, and effects (e.g., interactions with organic contaminants) of pCNPs. It therefore stands to reason that the scientific community needs to investigate in greater detail the often more complex and more abundant pCNPs described above. Although there is some current research into the transformation in natural environments of certain pCNPs, published literature on pCNPs and their transformation remains scarce. Therefore, findings for larger particles are taken into consideration in the subsequent discussion.
Recently, methods have been developed to identify and quantify some eCNPs in environmental matrices. For example, CNTs were quantified by measuring concentrations of characteristic metal catalyst impurities, laser fluorescence or microscopically by using the distinct shape of CNTs.44 Unfortunately, these techniques are not directly applicable for the identification of pCNPs, as pCNP physicochemical properties are less distinct than for CNTs. One promising strategy for future studies on pCNPs in the laboratory will be the use of isotopically labeled particles. However, further development on separation, extraction and quantification of pCNPs in environmental matrices is necessary for all currently existing methods.
3. Environmental transformation of CNPs
Following their formation and/or release into the natural environment, various processes including (photo)chemical (redox) reactions, biological interactions, and interactions with environmental constituents (e.g., natural organic matter, clay minerals) can alter the physicochemical properties of CNPs, in dependence of environmental conditions.31,45–47 These transformations can be categorized as chemical, physical, or biological transformations.453.1 Chemical transformation
Chemical transformation of CNPs refers to surface reactions (including direct and indirect photolysis and redox reactions), which can change the chemical structure of CNPs as well as the abundance and species of surface functionality.45 Research on eCNPs including CNTs,48 graphene,16 and GO,16,49 has shown that pre-existing functional groups, structural defects, mineral phases (e.g. residual metal catalysts), and the edges of the aromatic sheets forming the eCNPs, are sites that favor chemical transformation. These observations indicate that the pCNPs discussed above may be more prone to transformation than eCNPs as they generally contain smaller aromatic sheets, as well as more structural irregularities and mineral phases.28,50Chemical transformation of pCNPs can be influenced by environmental factors such as light,15,16,51 or the presence of oxidants (e.g., O2),15 reductants (e.g., Fe2+ and S2−),6,52–54 and natural organic matter (NOM).55,56 The factors driving chemical transformation differ markedly between (i) soot, which is mainly transported via the atmosphere and more susceptible to photochemical reactions involving reactive oxygen species (ROS), and (ii) other pCNPs, which are mainly transported by surface runoff and subsurface infiltration, where redox reactions with O2 or S2− are likely to be more important.
Over the last decade climate scientists have developed an increasing interest in the behavior of soot in the atmosphere, which has motivated investigations into its atmospheric transformation and transport.57–64 The chemical processes driving soot transformation are photochemical redox reactions with SOx, NOx, O3 and other ROS species in the atmosphere.57,58
In contrast, there has only been limited research into the chemical transformation of pCNPs released from fossil coal, biochar, and wildfire charcoal, either before or after transport by infiltration and surface runoff.65 Chemical transformation has been observed for the non-carbonized NOM, the mineral phase, and the carbonized graphene-like phase within large biochar particles.28 Transformation in soil is predominantly through oxidation and dissolution processes.28,66,67 Biochar transformation under reducing and anoxic conditions over a prolonged period of time has not been investigated in detail to date. Non-carbonized NOM phases and soluble constituents of the mineral phase within the biochar can be degraded or solubilized, thereby reducing the relative importance of NOM phases and soluble minerals over time. Furthermore, these processes can free up previously blocked pores thereby increasing the porosity of pCNPs. However, a general proportional increase in porosity and the stable carbonized phase following environmental transformation cannot be predicted due to the formation of organo-mineral complexes involving NOM, Ca2+, and redox-active Fe-particles,68,69 as well as interactions between pCNPs and NOM or minerals in the surrounding environment.28,70
Although the carbonized phase of pCNPs is generally very stable, transformations in the natural environment can increase the electron donating capacity and polarity of pCNPs due to the formation of oxygen-containing functional groups on the outer carbonized surface.31,70 In aqueous suspensions CNPs often carry a surface charge due to the presence of dissociable O- and occasionally N- or S-containing functional groups (including carboxyl, amine, and sulfonic groups), and to the adsorption of charged solutes from the aqueous media. The degree of oxidation thereby increases from the interior of the particle to the exterior surface, and also with the particle density.71 Under oxic conditions pCNPs therefore develop a more negative surface charge over time accompanied by a reduction in the release of positively charged ions from the soluble mineral phase. There have, however, been no systematic investigations into the transformation of pCNPs in natural environments, especially under reducing conditions.
The transformation of eCNPs such as CNTs and GO can provide important additional insights. For example, GO can be transformed in aquatic environments because of its abundant surface O-functional groups, while Fe2+ and S2− can reduce GO thereby increasing its hydrophobicity.6,52,54 However, Wang et al.53 found that GO reduction by Fe2+ resulted in a reduced content of alkoxy and hydroxy groups and an increased content of carboxy groups because of the hydrolysis reaction of acid anhydrides on GO. Chemical reduction can therefore also result in the formation of polar surface groups and the effect of redox reactions needs to be evaluated on a case-by-case basis.
While general trends and processes for chemical CNP transformation are reasonably well understood, the overall current state of knowledge remains incomplete for pCNPs derived from fossil coal, biochar, and wildfire charcoal. For instance, chemical transformation of the carbonized phases can be influenced by co-occurring mineral phases and non-carbonized NOM phases within the pCNPs, as well as by external factors such as soil type and pH; none of which are to date well understood.72,73 Future research therefore needs to build on the current understanding of the chemical transformation of soot during atmospheric transport based on work by other scientific communities, and to focus on closing the gaps in our understanding of the transformation of pCNPs in soil, sediment, and aquatic systems.
3.2 Physical transformation
Physical transformation of CNPs usually refers to changes in particle size and porosity, and to interactions with NOM and other particles. It can be caused by abrasion, by changes in pH, ionic strength, and intrinsic composition, or by the presence of NOM and minerals.45,47Physical disintegration is an important process in the transformation of pCNPs originating from fossil coal, biochar or wildfires (see section 2). Braadbaart et al.74 found that the physical exfoliation of carbonaceous materials can be chemically mediated by alkaline conditions in soils. Furthermore, the formation of cracks and fractures in biochar as a result of exposure to water and soil can lead to particle breakdown, with sandy soils resulting in higher disintegration rates than other soil types.30 Particles produced from high-lignin feedstocks (e.g., wood) disintegrate into smaller particles more readily than those from high cellulose feedstocks (e.g., grass). Additionally, the degree of disintegration and pCNP formation tends to decrease with an increase in carbonization and aromaticity.75 When carbonaceous materials and CNPs are exposed to soil they can interact with colloidal, dissolved, and particulate materials, which can fill exposed cavities thereby stabilizing particles and preventing further disintegration.30,71
In contrast to other pCNPs, soot particles are formed by nucleation of organic compounds such as acetylene and PAHs from the gas phase. Soot particles are oxidized by O2 and ROS both during and after their formation, which counteracts the growth of pCNPs. Following their initial formation, pCNPs coagulate to form larger particles that are initially spherical but can subsequently form chain-like fractal clusters.1,60 Both during soot particle formation and during atmospheric transport, interactions with other constituents of the gas phase can have a marked effect on particle size and shape.59–63 Over time, soot particle fractal dimensions tend to increase because they become either coated or entirely embedded within other aerosol components including sulfates, nitrates, sea salt, mineral dust, fly ash, and NOM.59,60,63 In aqueous suspensions all CNPs can interact with the surrounding environmental matrix, including NOM and mineral particles contained therein, which can affect the size, surface charge, and aggregation of particles (see section 4.2).
Naturally-occurring pCNPs such as wildfire charcoal and biochar contain a complex network of micropores and mesopores, as well as differently bound mineral phases including organo-mineral complexes. The chemical processes described in section 3.1 can increase the accessibility of CNP pores by dissolution, or block pores through the formation of precipitates and/or the binding of large NOM molecules onto the biochar surface (“fouling”).70,76,77 Changes in temperature, physical stress, and humidity can also lead to the disintegration and collapse of pore-networks.30,75 Changes in the composition and porosity70 of individual particles therefore need to be taken into account when investigating pCNP transformation. For instance, Liu et al.75 found that pCNP derived from biochar disintegration contain more oxygen and less aromatic structures compared to the original biochar.
3.3 Biological transformation
Biological transformation of CNPs refers to changes in the physicochemical properties of CNPs due to interactions with organisms, individual cells, and enzymes.45 For instance, the role of fungal hyphae in the physical breakdown of large fossil coal and biochar particles and of fungal extracellular enzymes in their chemical decomposition is widely recognized,21 but they have to date not been investigated with regard to the transformation of pCNPs, even though certain microorganisms (e.g., fungi) are capable of using highly aromatic structures such as lignin as carbon sources.78 The extracellular enzymes produced by these fungi, as well as by certain bacteria, include Mn-peroxidase, lignin-peroxidase and laccase,20,79 all of which are expected to oxidize pCNPs. Specifically, Hilscher et al.80 found that O-alkyl/alkyl-C groups are microbially transformed to carboxyl/carbonyl-C groups. These authors observed a reduction in aromaticity for some carbonaceous materials through the transformation of aryl-groups and the associated opening and partial oxidation of aromatic ring structures. Saquing et al.81 recently demonstrated that quinone groups in the carbonized phase of biochar can act both as electron donors and as acceptors for microbiota, thereby being oxidized or reduced, respectively. The biological transformation of pCNPs through redox processes is generally expected to result in changes in surface chemistry similar to those in non-biological systems (see section 3.1).Enzymatic transformations of CNPs are unfortunately often investigated using only pure enzymes and under laboratory conditions; they are consequently not very representative of enzymatic transformations in the natural environment. For instance, further investigation is required for the transfer and possible cellular-uptake required for the CNPs to reach degrading enzymes within the cells, as well as factors that drive the production and the associated gene expression of CNP-degrading enzymes (both intracellular and extracellular). Other factors influencing CNP degradation (e.g., microbial community composition, nutrient availability) also remain poorly understood. The enzymatic transformation of pCNPs has hardly been investigated to date but investigations into the enzymatic transformation of eCNPs can help to understand key enzymatic transformation processes that are also relevant to pCNP transformation. For instance, the horseradish peroxidase enzyme is able to catalyze the formation of oxygen radicals that are capable of degrading a range of eCNPs, including CNTs82,83 and GO,84 and are thus expected to also be capable of degrading pCNPs. O-functionalized CNPs are generally more degradable than non-functionalized CNPs because the associated structural irregularities facilitate (bio)degradation.82 However, Zhang et al.85 found that manganese peroxidase was able to transform CNTs but not O-functionalized CNTs. This was explained as being possibly due to the bridging of Mn2+ with carboxyl groups on the surface of O-functionalized CNTs, which interfered with the Mn2+/Mn3+ catalytic cycle, thereby suppressing transformation. The generalization that “more O-functionalization will result in more biological transformation” is therefore not always valid.
Apart from (bio)chemical transformation, cells can also physically interact with CNPs including soot,86–88 biochar particles,89 and CNTs.90,91 In such cases the CNPs are immobilized by aggregation or ingestion, while at the same time inducing negative biological effects on the cells (such as cytotoxicity). These interactions of CNPs with cells can reduce their stability and lead to aggregation (see section 4), thus affecting their transport.
The formation of a biofilm on the surfaces of larger carbonaceous particles has previously been reported,68 but it remains unclear if biofilms would also form on pCNPs. However, pCNP aggregates may well be susceptible to biofilm formation, which can have a variety of effects on the physicochemical properties of the pCNPs including changing their surface chemistry, increasing the particle size, and blocking pores.
Investigation of the biological transformation of pCNPs is challenging and a large number of factors need to be taken into account including their physicochemical properties, aggregation, and interactions with soil minerals, as well as the presence of NOM, microbial compositions, microbial activity, and nutrient accessibility. This may be the reason why systematic investigations into CNP transformations in conditions that are close to those found in natural environments remain so scarce. Such studies are, however, essential to achieving a better understanding of the effects that biological transformation processes have on the fate of, and possible risks associated with, CNPs. Ideally such investigations need to reflect the complexity of the investigated systems by combining the expertise of specialists from different fields, including microbiologists, environmental chemists, and colloidal scientists.
4. Effects of transformation on the colloidal stability and transport of CNPs
The colloidal stability, aggregation, and transport of CNPs are all significantly affected by their surface chemistries. Changes in surface chemistry are typically caused by chemical and biological transformation processes and by changes in the surrounding environment.4.1 Surface charge and surface potential of CNPs
The electric surface potential resulting from surface charges on CNPs affects the distribution of ions in aqueous solutions and is an important factor controlling the colloidal stability of CNPs in aqueous suspensions. Ions with opposite charges to the CNP surfaces (counterions) will accumulate around the CNPs and form an electrical double layer (EDL), which can be sub-divided into the Stern layer and the diffuse layer (see review by Wagner et al.92). The electric potential at the imaginary slipping plane between these two layers is referred to as the zeta potential (ζ-potential), which is indicative of the electric surface potential. The pH at which the measured ζ-potential approaches 0 mV is the isoelectric point (IEP) of a given CNP. In the absence of specific adsorption of charge-determining species onto the particle surface, the measured IEP is equal to the point of zero charge (PZC), i.e., the pH at which the net surface charge of the CNP is zero.The surface charge depends on the CNP functional groups and thus varies with the chemistry of the CNP surface, as well as with that of the solution. In natural environments most surfaces, including those of minerals, are negatively charged due to their crystallographic properties and/or the sorption of anionic polyelectrolytes such as NOM.47 Nevertheless, the PZC of pCNPs, at which particles tend to aggregate, can cover a wide range of pH values between 2 and 12.93,94 Both the surface functionalization and the composition of the mineral phases within the pCNPs can change upon environmental transformation (see section 3), which will in turn affect the ζ-potential, IEP and PZC of the pCNPs. For instance, the formation of O-functional groups and the leaching of positively charged mineral phases from pCNPs would render their surface charges more negative and reduce their ζ-potential, IEP and PZC.
4.2 Colloidal stability and aggregation of CNPs
The colloidal stability of CNPs in aqueous suspensions is typically controlled by their EDL and van der Waals interactions, as described by the classic Derjaguin–Landau–Verwey–Overbeek (DLVO) theory.10,92,95,96 Attractive van der Waals interactions cause the CNPs to aggregate, while the electrostatic repulsion between like-charged nanoparticles promotes stable colloidal systems. For example, Wang et al.32 found that biochars produced at 350 °C formed more stable pCNPs than biochars produced at 550 °C, which have lower negative surface charges. Further, the DLVO theory was extended to also include hydrogen bonding/electron donating–accepting capacity by van Oss et al., extending its predictive capability.97 Still, DLVO might not be able to predict environmental aggregation behavior in complex matrices.For CNPs with adsorbed organic molecules (such as, for example, NOM) steric repulsion is another important mechanism for colloidal stabilization. For instance, humic acid (a NOM fraction) has been found to increase the colloidal stability and mobility of biochar-derived pCNPs, which was explained as being due to an increase in electrostatic and steric repulsive interactions.98,99 The colloidal stability and aggregation of CNPs are not only influenced by surface functionality and water chemistry (e.g., pH, ionic strength and composition, NOM),95,96,100 but may also depend on morphological CNP properties. The effect that the physical properties of pCNPs have on their aggregation has not been investigated to date. However, the colloidal stability of GO in NOM-containing water has been found to depend on the morphology of the GO, with NOM stabilizing crumpled GO more effectively than flat GO, regardless of surface chemistry of these different GOs,100 indicating that the aggregation of pCNPs may also be influenced by their morphology.
CNPs with highly hydrophobic surfaces (e.g., pristine soot particles and reduced GO) are prone to aggregation in aqueous media due to the minimal electrostatic repulsion between the CNPs. Strong aggregation can even occur in CNPs with a higher surface charge (e.g., aged soot and GO) under certain conditions through one or more of the following mechanisms: (i) EDL compression induced by high ionic strength, (ii) adsorption and charge neutralization, where the surface charge is neutralized by adsorbed counterions within the Stern layer, (iii) bridging by NOM or other polymer chains and multivalent ions such as divalent Ca2+, and (iv) sweep flocculation, in which large particle aggregates enmesh individually dispersed nanoparticles.
The presence of a nonpolar hydrophobic organic contaminant (naphthalene) has been found to reduce pCNP mobility, probably due to the shielding of negative surface charges following naphthalene adsorption to the pCNPs.98,101 Bisiaux et al.65 found that pCNP pulses from wildfires and from urban runoff were rapidly attenuated in a river system, suggesting unexpected aggregation and/or disintegration of these particles. The presence of tar moieties formed during wildfires may partially explain high aggregation rates as the nonpolar tar components could shield the negative surface charges of the pCNPs, thereby reducing electrostatic repulsion between the wildfire-derived pCNPs.
In addition to interactions between similar CNPs (homo-aggregation), CNPs can also aggregate with other nanomaterials and larger colloids (hetero-aggregation) and this will generally be the dominant process in natural environments. Both NOM and mineral phases (mainly Al- and Fe-oxides) will interact with pCNP surfaces as well as filling their intra-particular cavities, thus changing the size of the CNP aggregate, its porosity, and its density.95,102–104 Yi et al.104 investigated the hetero-aggregation of biochar-derived pCNPs with CeO2 nanoparticles and found that at a pH where both particle groups are stable against homo-aggregation, hetero-aggregation occurred through a charge neutralization–charge reversal mechanism. In such cases primary hetero-aggregates are stable at both low and high pCNP concentrations due to electrostatic repulsion, but unstable at intermediate pCNP concentrations leading to secondary hetero-aggregation, which occurs through a charge-accumulation, core–shell stabilization mechanism. In this way pCNPs formed a negatively charged shell on the neutral surface of CeO2 nanoparticles, stabilizing the core–shell hetero-aggregate at a size that decreased with pCNP concentration.104 Similarly, the heteroaggregation of biochar-derived pCNP with goethite/hematite increased at low pCNP concentrations and formed more stable suspensions with increasing pCNP concentrations.75
4.3 Transport of CNPs in porous media
The transport of CNPs in porous media is an important process affecting their fate and effects in terrestrial environments. When CNPs are exposed to soil and sediment they are retained to some extent by physical sedimentation, straining, interception, or by attachment to collector particles through diffusion.105 The attachment of CNPs to porous media surfaces is affected by electrostatic interactions and also by non-DLVO interactions such as steric hindrance, hydrophobic effects, hydrogen bonding, and cation/anion bridging. The transformation of CNPs in natural environments can significantly affect the size, density, surface charge, and functionality of the particles, and consequently the interplay between CNPs and the porous media (Fig. 2).52,106,107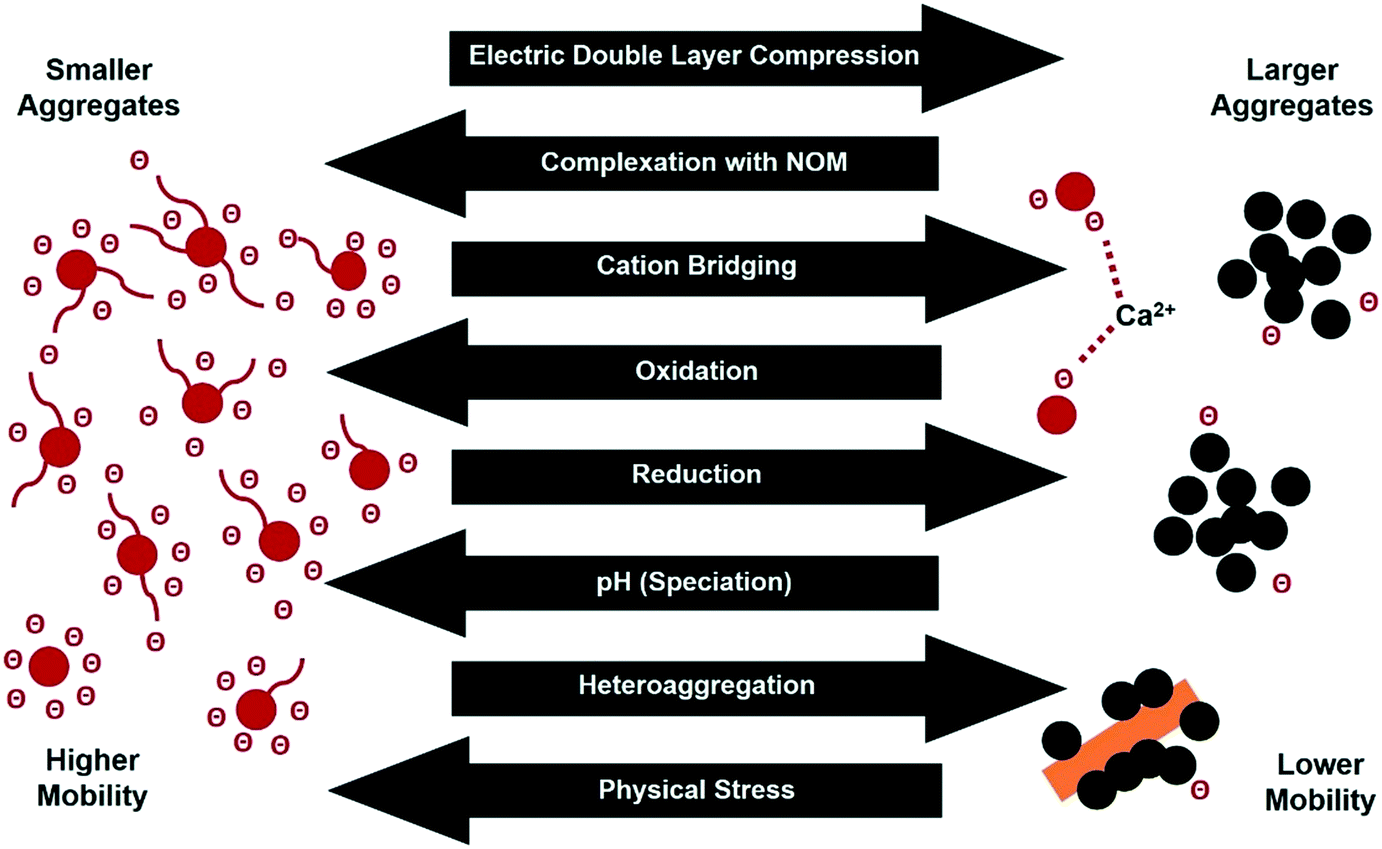 | ||
| Fig. 2 Transformation processes affecting CNP surface chemistry, aggregate size, and environmental behavior. | ||
CNP attachment generally decreases with increasing negative surface charge. Biochar-derived pCNPs, which contain the most O-functional groups and negative surface charge could therefore form more mobile pCNPs than less oxidized pCNPs.32 Any alteration of CNP surface charge (e.g., through an increase or decrease in surface O-functional groups, or adsorption of NOM) may significantly affect their transport. This is supported by Wang et al.,108 who found that pCNP interactions with humic acids increased pCNP transport due to increased electrosteric repulsion, whereas pCNP interactions with positively charged iron oxyhydroxide reduced pCNP transport due to electrostatic attraction.
Reduction of CNPs generally reduces the concentrations of surface O-functional groups, thus increasing the hydrophobicity of the particles and resulting in increased aggregation. Such reduction-induced effects can in turn significantly reduce particle transport, for example through physical straining. Conversely, oxidation of CNPs typically enhances their transport by increasing their surface hydrophilicity and inhibiting aggregation.52,106,109 It is important to note that the transformation of surface O-functional groups in natural environments is also critical to the mobility of CNPs. For instance, Xia et al.110 observed that the inhibition of GO transport in saturated porous media by metal ions followed the Hofmeister series (i.e., Na+ < K+ < Cs+ and Mg2+ < Ca2+ < Ba2+), and that the Hofmeister effect was more significant for reduced GO than for GO, probably due to the different quantities of metal complexing groups (including carboxyl and phenolic groups) in these different eCNPs. Furthermore, it has been reported that chloramine-treated GO (an oxidized form compared to pristine GO) exhibited greater mobility than pristine GO when Na+ was the dominant cation, whereas in the presence of Ca2+ its mobility was significantly lower than that of GO.106 This surprising lower mobility was attributed to the stronger cation-bridging effect associated with this material, in that the chloramine treatment increased the concentration of surface carboxyl groups (a strong metal-binding moiety).109,111
When extrapolating the results reported for eCNPs to the transport of pCNPs, the particle shape, density, and complex composition (including mineral phases) also need to be taken into account, but these have received little attention to date. The mineral components of pCNPs may, for example, impart heterogeneous surface charges (e.g., localized positive surface charges) to particles, which can in turn enhance particle deposition onto negatively charged sand grains. Moreover, most of the studies on transport of CNPs have been conducted with column experiments using mainly saturated quartz sand as the porous media. A few studies carried out column experiments using soils or under unsaturated conditions indicated, as shown much earlier for natural colloids,112 that heterogeneous soil components (e.g., iron oxides and clay minerals) and soil water content are important factors affecting transport of CNPs.99,113–115 For example, it was reported that increasing ionic strength and switching background solution from NaCl to CaCl2 had more significant effects on the deposition of fullerenes in a sandy soil than in purified quartz sand, likely because the clay minerals (and possibly soil organic matter) in soil responded to changes of ionic strength and species differently than quartz.113 In the natural subsurface environment (in particular, vadose zone) the effects of transformation on transport of CNPs are expected to be more complex compared to column tests using (purified) quartz sand, due to the non-stationary flow regimes and heterogeneity in porous media.
5. Effects of transformation on accumulation and release of organic contaminants by CNPs
Hydrophobic organic contaminants, as well as many ionizable organic contaminants, can sorb either reversibly or irreversibly onto CNPs.11,116 The transformation of CNPs in natural environments leads to changes in their physicochemical properties and aggregation, thereby affecting the sorption of organic contaminants.11,116,117 CNP transformation can therefore affect contaminant sequestration, enrichment, or release, depending on the properties of the particular contaminant involved, the properties of the CNPs, and the environmental conditions. Such alteration to the binding of organic contaminants to CNPs can in turn affect the transport of these contaminants, which is affected by colloidal CNPs.5.1 Effect of CNP surface chemistry on contaminant binding
The transformation of CNPs in natural environments can change their surface chemistry, and hence their hydrophobicity which has a major effect on the sorption of hydrophobic organic contaminants such as PAHs.31,116,118,119 For instance, the number of O-functional groups on the surfaces of CNPs will either increase with oxidation, or decrease with reduction. If the number of O-functional groups increases, the CNP surfaces become more hydrophilic and hence thermodynamically less attractive to hydrophobic contaminants. Furthermore, the formation of water clusters around negatively charged O-functional groups has been proposed as an explanation for the observed reduction in sorption of organic contaminants onto CNPs.120–122 Conversely, if the content of O-functional groups decreases, the CNPs become more hydrophobic and less negatively charged.The effects that changes in surface functional groups and surface charge have are more complex for polar ionic and ionizable contaminants, where the speciation of the contaminant, the pH, and the ionic strength are all important factors affecting their interactions with CNPs (see review by Kah et al.11). For instance, oxidation of CNPs can result in negatively charged contaminants being repulsed electrostatically from the negatively charged CNP surfaces.11,94 However, polar compounds sorbed to CNPs by hydrogen bonds will become more strongly bound with an increase in surface O-functional groups (e.g., following oxidation of the CNPs)107 and sorption can decrease with the disappearance of these functional groups (e.g., following reduction of the CNPs).11 Such enhanced or inhibited polar interactions between polar contaminants (e.g., 1-naphthol) and CNPs can then determine the extent of facilitated transport of contaminants by CNPs.107 Aromatic organic contaminants are strongly bound to CNPs, mainly through π–π electron-donor–acceptor interactions that occur between π-electrons of the aromatic surface of CNPs and π-electrons of the organic contaminants.123 Depending on the region on the CNP and the functional groups of the contaminant, both can act as either π-electron acceptors or donors. The reduction of oxidized surfaces during CNP transformation in natural environments can increase the electron density on CNP surfaces, which can in turn enhance the π–π interactions between CNPs and organic contaminants.6,124,125 In addition, Wang et al.6 found that reductions by sulfide can open the epoxy rings on GO surface to form hydroxyl and carbonyl groups. Because these two functional groups have the ability to form hydrogen bonds, the reduction could increase the sorption of a polar contaminant (1-naphthol). Again, such changes may affect the capability of CNPs as contaminant vectors of certain contaminants. Wang et al.117 also found that reducing GO through Fe2+ enhanced their capacity for 1-naphthol adsorption by increasing the hydrophobicity of, and π–π interactions with GO. However, reduction through Fe2+ reduced the adsorption capacity of hydroxylated CNTs for 1-naphthol. This can be explained by the ability of Fe2+ to reduce the surface O-functional groups of the CNTs, which would also reduce hydrogen bonding and cation bridging. The adsorption capacity would consequently be reduced, even though hydrophobicity and π–π interactions increased. These examples show that the importance of transformation-induced changes in contaminant binding depends on the relative contributions of different mechanisms to the overall sorption.
Similar effects can be expected for pCNPs derived from biochar and wildfire charcoal to those for eCNPs. Biochar transformation under oxidative conditions generally results in an increase in the number of oxygen-containing functional groups and the adsorption of partially oxidized organic matter and clay components, all of which reduce the number of accessible sorption sites resulting in a relative reduction in contaminant sorption with ageing.126–129 In contrast, reductive processes have to date not been investigated in this regard for biochar, wildfire charcoal, and the associated pCNPs. Nevertheless, the general trends illustrated in Fig. 3 can be expected to apply for all CNPs.
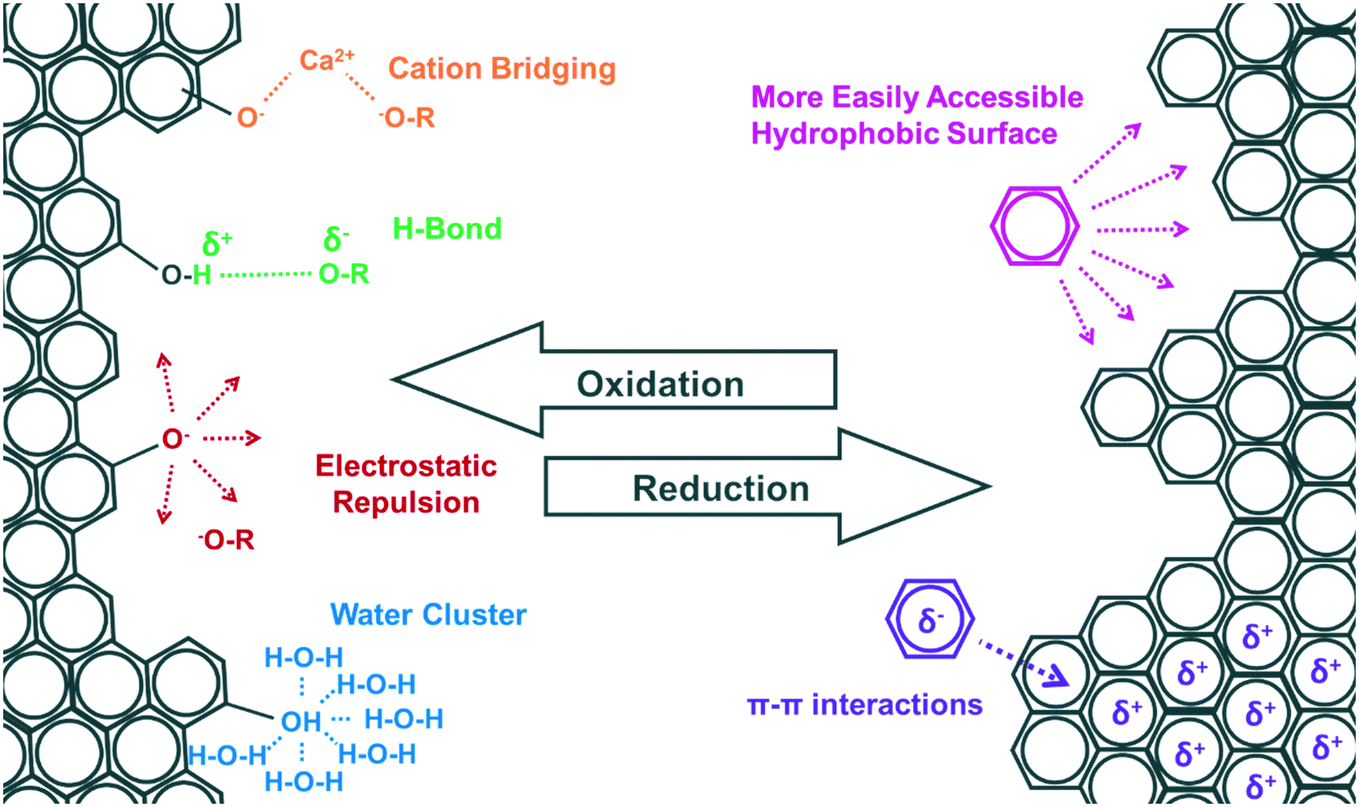 | ||
| Fig. 3 Effect of chemical/biological transformation of CNPs on binding of organic contaminants. | ||
5.2 Effect of CNP aggregation and porosity on contaminant binding
The effects of chemical and physical transformation of CNPs are interrelated. While changes in the surface properties of CNPs can affect their aggregation and adsorption potential, the aggregation of CNPs can in turn affect the kinetics of chemical transformation and the surface chemistry of the particles (e.g., decreasing negative charge as a result of cation bridging between surface O-functional groups of CNPs).130,131 Changes in aggregation of CNPs and in the pore structures of aggregates therefore affect their ability to sorb organic contaminants.132,133The aggregation of CNPs can reduce the number of accessible sorption sites134 and thereby reduce the binding capacity of CNPs.107 The aggregation of CNPs can, however, also form micropores and mesopores, which can become important sorption sites for organic contaminants.135,136 The structure formed by aggregation of tubular CNTs is relatively open, which means that organic molecules entering open spaces within the aggregate can interact with multiple CNTs at the same time, resulting in enhanced adsorption.130 In contrast, aggregation of spherical fullerenes can form a closed inner space that is not accessible to organic contaminants.134 Changes in the intrinsic pore size distributions of pCNPs as a result of transformation and leaching of soluble phases can also affect the pCNP-contaminant binding potential.137
Changes in water chemistry, surface functionality, and NOM loading all affect the aggregation of CNPs.15,47,107 Changes in CNP aggregation as a result of transformation in natural environments can alter the pore structure and pore volume, thus affecting the sorption and desorption of contaminants as well as their co-transport with the CNPs.130,134,138,139 The adsorption of organic contaminants onto the outer surfaces of CNPs is generally reversible. However, due to the capillary condensation effect or aggregate structure rearrangement in the process of adsorption, contaminants can become trapped in the internal pore spaces of CNP aggregates and thus be more strongly bound.132,138 For example, Wang et al.138 found that fullerene aggregates that had been modified by NOM and surfactants had markedly different pore geometries, and possibly also greater pore volumes, than unmodified fullerenes. The sorption of polychlorinated biphenyl (PCB) in these physically transformed fullerene aggregates consequently exhibited a much higher level of desorption resistance and, as a result, enhanced co-transport with fullerene (Fig. 4).
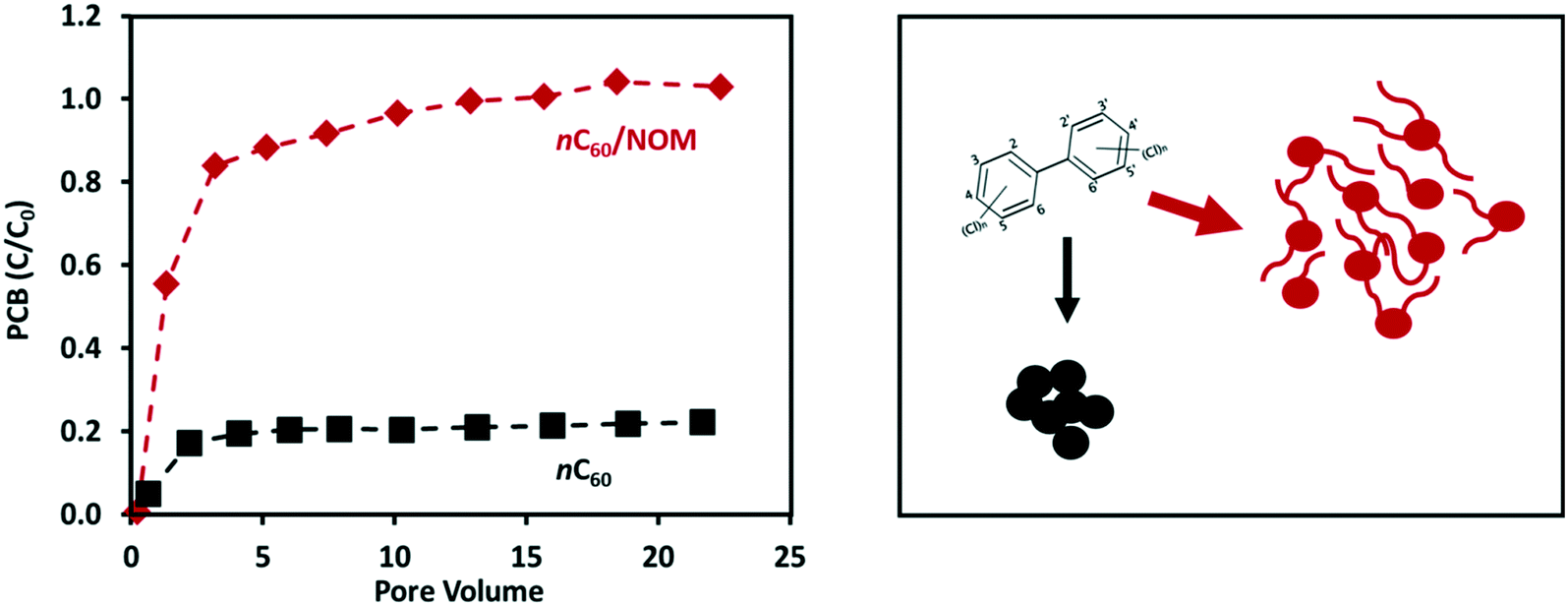 | ||
| Fig. 4 NOM loading can affect the pore structure and pore volume of fullerene (nC60) aggregates, thereby enhancing their ability to mobilize polychlorinated biphenyl (PCB) in porous media. Data adapted from Wang et al.138 | ||
The influence of NOM on sorption by CNPs is ambiguous. On the one hand NOM can improve the dispersion of CNPs, thereby increasing the number of available sorption sites, while on the other hand NOM can also bind to CNP sorption sites, thereby competing with organic contaminants for sorption sites. NOM can also block micropores in CNPs and their aggregates, with adverse effects on their binding capacity.130,140–143 In addition, interactions between CNPs and NOM can result in the blockage of micropores and mesopores, thereby trapping contaminants within these pore spaces.132,137
Organic contaminants sorbed by CNTs, fullerenes, fossil coal, and biochar are generally not very bioaccessible because of their high contaminant-binding capacities.116,144,145 However, when comparing these materials, which are generally produced under controlled conditions, with their counterparts from unintentional emissions (e.g., soot,24,25 biochar produced under poorly controlled conditions,146 or wildfire charcoal29), the sorption and release of organic contaminants can differ considerably. To derive conclusions for contaminant release from soot, biochar, and wildfire charcoal on the basis of findings for eCNPs is therefore not advisable, as additional factors need to be taken into account. For example, the intrinsic porosity of these materials has been identified as a key factor affecting the sorption and desorption of organic contaminants. Many pCNPs -especially wildfire charcoal and biochars- contain intrinsic micropore networks which can result in strong sorption hysteresis exceeding that of eCNPs, making these pCNPs potentially more effective contaminant vectors than eCNPs. Although preliminary results indicate that the micropore and mesopore size distributions of certain types of biochar may remain similar after transformation in the field,70,146 this observation cannot be extrapolated to other types of biochar, soot, or wildfire charcoal, which may be transformed under different conditions in the natural environment (e.g., under different temperatures, water saturation, or pH).
It is therefore important to complement findings from eCNPs with investigations into those additional factors that can affect contaminant binding and release by pCNPs in the natural environment. Additional factors to be considered include the intrinsic porosity, physical disintegration, non-carbonized phases, tar phases, and mineral phases of pCNPs.
6. Effects of transformation on catalytic efficiency of CNPs
The involvement of activated carbon in catalytic reactions under elevated temperatures has been well documented.147 The involvement of pyrogenic carbons including soot, biochar, and wildfire charcoal in catalytic reactions under conditions encountered in natural environments has also more recently been discussed.116 Pignatello et al.116 subdivided reactions that can be catalyzed by CNPs into two groups: (i) reactions involving surface-mediated electron transfer (including surface-mediated hydrolysis), and (ii) reactions involving the generation of ROS.6.1 Surface-mediated electron transfer
pCNPs typically contain both non-carbonized and mineral phases, in addition to the carbonized phase that reassembles eCNPs. Both the mineral phase and the non-carbonized NOM-like phase can interact with the carbonized phase, and can also participate in catalytic reactions. For instance, redox reactions and the hydrolysis of organic contaminants on mineral surfaces (e.g. Mn-oxides and Fe-rich minerals), which have been widely investigated, can both be affected by the presence of NOM. NOM can participate in and/or accelerate mineral dissolution and metal complexation, which in turn affects the catalytic capacity of minerals (see review by Polubesova and Chefetz56). All the phases of pCNPs, including carbonized, non-carbonized, and mineral phases, can be dissolved, exchanged, and transformed, which will affect their influences on the catalytic properties of the pCNPs. The catalytic properties of these phases in pCNPs and their transformation products have, to date, received little attention. However, results obtained for eCNPs may help understand the catalytic properties of the carbonized phase of pCNPs.The catalytic efficiencies of CNPs in hydrolysis reactions are largely affected by the acidity/basicity of their surface O-functional groups. For example, deprotonated surface O-functional groups of CNTs and GO have been shown to act as bases to catalyze the dehydrochlorination of chlorinated solvents.148,149 Even though the total number of surface functional groups on the GO decreased following photochemical transformation, the number of carboxyl groups increased,148,150 resulting in an increased catalytic efficiency for the dehydrochlorination of 1,1,2,2-tetrachloroethane compared with that of pristine GO.148 Furthermore, Fu and Zhu54 found that GO was reduced in the presence of Na2S and could therefore catalyze the reduction of nitrobenzene with Na2S. The edges of the reduced GO acted as the catalytic sites, while the basal plane of the reduced GO served as a conductor for the electron transfer during the catalytic process.
In addition to surface functionality, the aggregation of CNPs can also affect their catalytic efficiency. For example, oxidation increases the amount of O-functional groups on CNP surfaces, which can improve their catalytic efficiency for hydrolysis of organic contaminants. However, O-functional groups can form cation bridges with divalent cations (e.g. Ca2+) so that an abundance of O-functional groups can encourage aggregation of CNPs, thereby decreasing their catalytic efficiency. Furthermore, NOM can enhance the dispersion of CNPs leading to more surface functional groups becoming available as active catalytic sites.151,152 However, NOM may also block a proportion of the sorption and catalytic sites on CNP surfaces by sorbing to the CNPs.148,151,153 The effect of NOM on the catalytic properties of CNPs therefore needs to be carefully evaluated on a case by case basis.
6.2 Generation of reactive oxygen species
During pyrolytic pCNP formation unshared electrons and electron pairs can form free radicals that can persist within the pCNP structures for prolonged periods of time. The abundance of these persistent free radicals within pCNPs depends on the pyrolysis conditions, the composition of the original feedstock, the abundance of oxygen groups in the pCNPs, and the presence of external metals (see review by Qin et al.154). The concentration of persistent free radicals in pCNPs can reach up to 1019 spins per gram,116 which is two orders of magnitude above the concentrations commonly found in humic acids.155 When pCNPs are introduced into water these radicals can form H2O2 and ROS, but most of these reactions typically take place within the first few hours after contact with water, as was recently observed for biomass-based chars where H2O2 production peaked after approximately 1 hour and then declined.156CNP aggregation and pore blockage can decrease the release of free radicals and their products, similar to the reduced release of organic contaminants discussed in section 5.2. ROS deriving from pCNPs are capable of inducing the transformation of a range of organic contaminants including sulfadimidine,157 1,3-dichloropropene,158,159p-nitrophenol,160 trifluralin,161 pendimethalin,161 diethyl phthalate,162 nitrobenzene,163 and 2-chlorobiphenyl.164 Some compounds can react directly with carbonaceous materials, as has been observed for chloropicrin165 and p-nitrophenol.156 Pignatello et al.116 suggested that the intrinsic reactivity of carbonaceous materials could be due to their redox behavior. The transformation of pCNPs is expected to have a strong effect on H2O2 production, ROS release, and pCNP redox behavior. These aspects of pCNP-induced contaminant transformation remain largely unexplored and require further investigation.
Finally, it is important to note that the sorption of organic contaminants to CNPs can affect contaminant transformation. For example, sorption of nitroaromatics to CNPs favors electron transfer for redox transformation of these contaminants.166 Thus, the effects of CNP transformation on contaminant sorption discussed in section 5 can also affect the catalytic efficiency of CNPs for contaminant transformation.
7. Further research required
Our review indicates that engineered carbon nanoparticles (eCNPs) comprise a very small proportion (<1%, Gg per year range) of the CNPs in the natural environment and are therefore largely of academic interest compared to the generally more complex pyrogenic or petrogenic carbon nanoparticles (pCNPs) that derive from forest fires, fossil fuel combustion, and biochar application (>99%, Tg per year range).The translation of exiting findings on eCNPs to understand the behavior of pCNPs needs to carefully consider molecular differences between these materials. Generally, pCNP consists of smaller aromatic clusters and contains more structural irregularities and mineral phases than eCNPs, resulting in significant differences in surface chemistry, density, porosity, reactivity, and stability. Therefore, building on the knowledge obtained for eCNPs and soot, a special focus should be on changes in intrinsic porosity and on the transformation of the non-carbonized phases, the mineral phases, and the organo-mineral complexes of pCNPs. The complexity of these transformation processes calls for increased exchanges between different scientific communities, including atmospheric chemists, microbiologists, soil scientists, water chemists, and colloidal scientists.
Among the different pCNPs, soot and its transformation within the atmosphere are well understood because of a large body of research in atmospheric chemistry. In contrast, pCNPs deriving from fossil coal, wildfire charcoal, and biochar are less well understood. A strategy for future research could be to build on existing knowledge concerning the transformation of soot during atmospheric transport and focus on closing the existing knowledge gaps on the transformation of pCNPs in soil, sediment, and aquatic systems. Most of the reported findings have been obtained under oxidative conditions, while investigations under anoxic and reducing conditions are underrepresented in the scientific literature.
Investigations into the effects of transformation on CNP colloidal transport should take into account the geometry, porosity, and composition of pCNPs deriving from soot, fossil coal, wildfire charcoal, and biochar. Differences in aggregation compared to commonly investigated eCNPs (including GO and CNTs) can be expected and will require further investigation. Thereby, the effects of the pCNP phases that differ from those of eCNPs are of special interest. For instance, non-carbonized phases, Al- and Fe-oxides, organo-mineral complexes, and tar can bind to pCNP surfaces and affect their surface chemistry, porosity, and aggregation. Incorporating these factors into systematic prediction tools for pCNP transformation and aggregation that can take into account chemical, physical, and biological transformation processes would be of great assistance in obtaining a better assessment of naturally-occurring pCNPs.
Contaminant binding and release can be affected by CNP transformation, however, the importance of transformation-induced changes in contaminant binding depends on the relative contributions of particular mechanisms to the overall sorption, which needs to be carefully considered. Future research needs to complement the findings for pristine eCNPs with investigations into additional factors that might affect the binding and release of contaminants by pCNPs. The additional factors to be considered include changes in the intrinsic porosity, physical decomposition, non-carbonized phases, tar phases, and mineral phases of pCNPs.
The catalytic properties of pCNPs are expected to be affected by all of the different phases of pCNPs including carbonized, non-carbonized, and mineral phases, which can all be dissolved, exchanged, and transformed in the natural environment. The influence that these phases and their transformation have on the catalytic activity of pCNPs has received little attention to date and warrants further investigation.
Systematic investigations are required into the relative contributions of specific mechanisms to the overall sorption in order to better predict transformation and transformation-induced changes to contaminant binding. The development of generalized approaches such as the use of poly-parameter linear free-energy relationships (ppLFER), while taking into account pCNP properties and transformation processes, will be required to predict the sorption of both neutral and polar organic contaminants to CNPs in the natural environment. However, such comprehensive predictive tools are unlikely to be developed in the near future as more fundamental open questions, such as those discussed in this review, will first need to be addressed.
Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
This project was supported by the National Natural Science Foundation of China (Grant 21425729), the Ministry of Science and Technology of China (Grant 2014CB932001), the Fundamental Research Funds for the Central Universities, and the 111 Program of the Ministry of Education of China (T2017002).References
- M. Ni, J. Huang, S. Lu, X. Li, J. Yan and K. Cen, A Review on Black Carbon Emissions, Worldwide and in China, Chemosphere, 2014, 107, 83–93 CrossRef CAS PubMed.
- M. I. Bird, J. G. Wynn, G. Saiz, C. M. Wurster and A. McBeath, The Pyrogenic Carbon Cycle, Annu. Rev. Earth Planet. Sci., 2015, 43(1), 273–298 CrossRef CAS.
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Carbon Nanotubes: Present and Future Commercial Applications, Science, 2013, 339, 535–539 CrossRef CAS PubMed.
- M. R. Wiesner, G. V. Lowry, P. Alvarez, D. Dionysiou and P. Biswas, Assessing the Risks of Manufactured Nanomaterials, Environ. Sci. Technol., 2006, 40(14), 4336–4345 CrossRef CAS PubMed.
- B. Nowack and T. D. Bucheli, Occurrence, Behavior and Effects of Nanoparticles in the Environment, Environ. Pollut., 2007, 150(1), 5–22 CrossRef CAS PubMed.
- F. Wang, F. Wang, D. Zhu and W. Chen, Effects of Sulfide Reduction on Adsorption Affinities of Colloidal Graphene Oxide Nanoparticles for Phenanthrene and 1-Naphthol, Environ. Pollut., 2015, 196, 371–378 CrossRef CAS PubMed.
- E. J. Petersen, L. Zhang, N. T. Mattison, D. M. O'Carroll, A. J. Whelton, N. Uddin, T. Nguyen, Q. Huang, T. B. Henry and R. D. Holbrook, et al. Potential Release Pathways, Environmental Fate, and Ecological Risks of Carbon Nanotubes, Environ. Sci. Technol., 2011, 45(23), 9837–9856 CrossRef CAS PubMed.
- H. Weinberg, A. Galyean and M. Leopold, Evaluating Engineered Nanoparticles in Natural Waters, TrAC, Trends Anal. Chem., 2011, 30(1), 72–83 CrossRef CAS.
- C. M. Park, K. H. Chu, J. Heo, N. Her, M. Jang, A. Son and Y. Yoon, Environmental Behavior of Engineered Nanomaterials in Porous Media: A Review, J. Hazard. Mater., 2016, 309, 133–150 CrossRef CAS PubMed.
- W. J. G. M. Peijnenburg, M. Baalousha, J. Chen, Q. Chaudry, F. von der Kammer, T. A. J. Kuhlbusch, J. Lead, C. Nickel, J. T. K. Quik and M. Renker, et al. A Review of the Properties and Processes Determining the Fate of Engineered Nanomaterials in the Aquatic Environment, Crit. Rev. Environ. Sci. Technol., 2015, 45(19), 2084–2134 CrossRef CAS.
- M. Kah, G. Sigmund, F. Xiao and T. Hofmann, Sorption of Ionizable and Ionic Organic Compounds to Biochar, Activated Carbon and Other Carbonaceous Materials, Water Res., 2017, 124, 673–692 CrossRef CAS PubMed.
- V. K. Gupta and T. A. Saleh, Sorption of Pollutants by Porous Carbon, Carbon Nanotubes and Fullerene- An Overview, Environ. Sci. Pollut. Res., 2013, 20(5), 2828–2843 CrossRef CAS PubMed.
- J. G. Yu, X. H. Zhao, H. Yang, X. Q. H. Chen, Q. Yang, L. Y. Yu, J. H. Jiang and X. Q. H. Chen, Aqueous Adsorption and Removal of Organic Contaminants by Carbon Nanotubes, Sci. Total Environ., 2014, 482–483(1), 241–251 CrossRef CAS PubMed.
- G. D. Moon, S. Ko, Y. Min, J. Zeng, Y. Xia and U. Jeong, Chemical Transformations of Nanostructured Materials, Nano Today, 2011, 6(2), 186–203 CrossRef CAS.
- D. M. Mitrano, S. Motellier, S. Clavaguera and B. Nowack, Review of Nanomaterial Aging and Transformations through the Life Cycle of Nano-Enhanced Products, Environ. Int., 2015, 77, 132–147 CrossRef CAS PubMed.
- J. Zhao, Z. Wang, J. C. White and B. Xing, Graphene in the Aquatic Environment: Adsorption, Dispersion, Toxicity and Transformation, Environ. Sci. Technol., 2014, 48(17), 9995–10009 CrossRef CAS PubMed.
- S. M. Louie, R. Ma and G. V. Lowry, Transformations of Nanomaterials in the Environment, Front. Nanosci., 2014, 7, 55–87 Search PubMed.
- IEA International Energy Agency, World Energy Outlook 2017, 2017 Search PubMed.
- IEA Clean Coal Centre, Losses in the Coal Supply Chain, 2012 Search PubMed.
- L. Han, K. Sun, J. Jin and B. Xing, Some Concepts of Soil Organic Carbon Characteristics and Mineral Interaction from a Review of Literature, Soil Biol. Biochem., 2016, 94, 107–121 CrossRef CAS.
- K. A. Spokas, Review of the Stability of Biochar in Soils: Predictability of O:C Molar Ratios, Carbon Manage., 2010, 1(2), 289–303 CrossRef CAS.
- H. Wang, Formation of Nascent Soot and Other Condensed-Phase Materials in Flames, Proc. Combust. Inst., 2011, 33(1), 41–67 CrossRef CAS.
- L. E. Murr, E. V. Esquivel, J. J. Bang, G. de la Rosa and J. L. Gardea-Torresdey, Chemistry and Nanoparticulate Compositions of a 10,000 Year-Old Ice Core Melt Water, Water Res., 2004, 38(19), 4282–4296 CrossRef CAS PubMed.
- H. Richter and J. B. Howard, Formation of Polycyclic Aromatic Hydrocarbons and Their Growth to Soot—a Review of Chemical Reaction Pathways, Prog. Energy Combust. Sci., 2000, 26(4), 565–608 CrossRef CAS.
- M. T. O. Jonker and A. A. Koelmans, Sorption of Polycyclic Aromatic Hydrocarbons and Polychlorinated Biphenyls to Soot and Soot-like Materials in the Aqueous Environment: Mechanistic Considerations, Environ. Sci. Technol., 2002, 36(17), 3725–3734 CrossRef CAS PubMed.
- M. S. Forbes, R. J. Raison and J. O. Skjemstad, Formation, Transformation and Transport of Black Carbon (Charcoal) in Terrestrial and Aquatic Ecosystems, Sci. Total Environ., 2006, 370(1), 190–206 CrossRef CAS PubMed.
- T. C. Bond, S. J. Doherty, D. W. Fahey, P. M. Forster, T. Berntsen, B. J. Deangelo, M. G. Flanner, S. Ghan, B. Kärcher and D. Koch, et al. Bounding the Role of Black Carbon in the Climate System: A Scientific Assessment, J. Geophys. Res.: Atmos., 2013, 118(11), 5380–5552 CAS.
- J. Lehmann and S. Joseph, Biochar for Environmental Management - Science, Technology and Implementation, ed. J. Lehmann and S. Joseph, Routledge, London Sterling VA, 2nd ed., 2015 Search PubMed.
- C. Santín, S. H. Doerr, A. Merino, T. D. Bucheli, R. Bryant, P. Ascough, X. Gao and C. A. Masiello, Carbon Sequestration Potential and Physicochemical Properties Differ between Wildfire Charcoals and Slow-Pyrolysis Biochars, Sci. Rep., 2017, 7(1), 1–11 CrossRef PubMed.
- K. A. Spokas, J. M. Novak, C. A. Masiello, M. G. Johnson, E. C. Colosky, J. A. Ippolito and C. Trigo, Physical Disintegration of Biochar: An Overlooked Process, Environ. Sci. Technol. Lett., 2014, 1(8), 326–332 CrossRef CAS.
- F. Lian and B. Xing, Black Carbon (Biochar) in Water/Soil Environments: Molecular Structure, Sorption, Stability, and Potential Risk, Environ. Sci. Technol., 2017, 51(23), 13517–13532 CrossRef CAS PubMed.
- D. Wang, W. Zhang, X. Hao and D. Zhou, Transport of Biochar Particles in Saturated Granular Media: Effects of Pyrolysis Temperature and Particle Size, Environ. Sci. Technol., 2013, 47(2), 821–828 CrossRef CAS PubMed.
- D. Woolf, J. E. Amonette, F. A. Street-Perrott, J. Lehmann and S. Joseph, Sustainable Biochar to Mitigate Global Climate Change, Nat. Commun., 2010, 1, 56 Search PubMed.
- J. T. Randerson, Y. Chen, G. R. van der Werf, B. M. Rogers and D. C. Morton, Global Burned Area and Biomass Burning Emissions from Small Fires, J. Geophys. Res.: Biogeosci., 2012, 117(4), 1–23 Search PubMed.
- M. Reisser, R. S. Purves, M. W. I. Schmidt and S. Abiven, Pyrogenic Carbon in Soils: A Literature-Based Inventory and a Global Estimation of Its Content in Soil Organic Carbon and Stocks, Front. Earth Sci., 2016, 4(August), 1–14 Search PubMed.
- H. Knicker, A. Hilscher, F. J. González-Vila and G. Almendros, A New Conceptual Model for the Structural Properties of Char Produced during Vegetation Fires, Org. Geochem., 2008, 39(8), 935–939 CrossRef CAS.
- C. Nocentini, G. Certini, H. Knicker, O. Francioso and C. Rumpel, Nature and Reactivity of Charcoal Produced and Added to Soil during Wildfire Are Particle-Size Dependent, Org. Geochem., 2010, 41(7), 682–689 CrossRef CAS.
- D. Uhelski and J. R. Miesel, Physical Location in the Tree during Forest Fire Influences Element Concentrations of Bark-Derived Pyrogenic Carbon from Charred Jack Pines (Pinus Banksiana Lamb.), Org. Geochem., 2017, 110, 87–91 CrossRef CAS.
- G. de Lafontaine and H. Asselin, Soil Charcoal Stability over the Holocene across Boreal Northeastern North America, Quat. Res., 2011, 76(2), 196–200 CrossRef.
- R. Jaffé, Y. Ding, J. Niggemann, A. V. Vähätalo, A. Stubbins, R. G. M. Spencer, J. Campbell and T. Dittmar, Global Charcoal Mobilization from Soils via Dissolution and Riverine Transport to the Oceans, Science, 2013, 340(6130), 345–347 CrossRef PubMed.
- I. Bhatt and B. N. Tripathi, Interaction of Engineered Nanoparticles with Various Components of the Environment and Possible Strategies for Their Risk Assessment, Chemosphere, 2011, 82(3), 308–317 CrossRef CAS PubMed.
- Kuick Research, Global Carbon Nanotubes Market & Patent Insight 2023, Delhi, 2017 Search PubMed.
- Global Industry Analysts Inc., Stringent Environmental Regulations to Boost in the Global Activated Carbon Market, San Jose, 2017 Search PubMed.
- E. J. Petersen, D. X. Flores-Cervantes, T. D. Bucheli, L. C. C. Elliott, J. A. Fagan, A. Gogos, S. Hanna, R. Kägi, E. Mansfield and A. R. M. Bustos, et al. Quantification of Carbon Nanotubes in Environmental Matrices: Current Capabilities, Case Studies, and Future Prospects, Environ. Sci. Technol., 2016, 4587–4605 CrossRef CAS PubMed.
- G. V. Lowry, K. B. Gregory, S. C. Apte and J. R. Lead, Transformations of Nanomaterials in the Environment, Environ. Sci. Technol., 2012, 46(13), 6893–6899 CrossRef CAS PubMed.
- A. D. Dwivedi, S. P. Dubey, M. Sillanpää, Y. N. Kwon, C. Lee and R. S. Varma, Fate of Engineered Nanoparticles: Implications in the Environment, Coord. Chem. Rev., 2015, 287, 64–78 CrossRef CAS.
- P. Christian, F. von der Kammer, M. Baalousha and T. Hofmann, Nanoparticles: Structure, Properties, Preparation and Behaviour in Environmental Media, Ecotoxicology, 2008, 17(5), 326–343 CrossRef CAS PubMed.
- S. Banerjee, M. G. C. Kahn and S. S. Wong, Rational Chemical Strategies for Carbon Nanotube Functionalization, Chem. – A Eur. J., 2003, 9(9), 1898–1908 CrossRef CAS PubMed.
- Y. Li, N. Yang, T. Du, X. Wang and W. Chen, Transformation of Graphene Oxide by Chlorination and Chloramination: Implications for Environmental Transport and Fate, Water Res., 2016, 103, 416–423 CrossRef CAS PubMed.
- X. Xiao and B. Chen, A Direct Observation of the Fine Aromatic Clusters and Molecular Structures of Biochars, Environ. Sci. Technol., 2017, 51(10), 5473–5482 CrossRef CAS PubMed.
- I. Chowdhury, W.-C. Hou, D. Goodwin, M. Henderson, R. G. Zepp and D. Bouchard, Sunlight Affects Aggregation and Deposition of Graphene Oxide in the Aquatic Environment, Water Res., 2015, 78, 37–46 CrossRef CAS PubMed.
- T. Xia, J. D. Fortner, D. Zhu, Z. Qi and W. Chen, Transport of Sulfide-Reduced Graphene Oxide in Saturated Quartz Sand: Cation-Dependent Retention Mechanisms, Environ. Sci. Technol., 2015, 49(19), 11468–11475 CrossRef CAS PubMed.
- F. Wang, F. Wang, G. Gao and W. Chen, Transformation of Graphene Oxide by Ferrous Iron: Environmental Implications, Environ. Toxicol. Chem., 2015, 34(9), 1975–1982 CrossRef CAS PubMed.
- H. Fu and D. Zhu, Graphene Oxide-Facilitated Reduction of Nitrobenzene in Sulfide-Containing Aqueous Solutions, Environ. Sci. Technol., 2013, 47(9), 4204–4210 CrossRef CAS PubMed.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Reduction of Graphene Oxide Vial-Ascorbic Acid, Chem. Commun., 2010, 46(7), 1112–1114 RSC.
- T. Polubesova and B. Chefetz, DOM-Affected Transformation of Contaminants on Mineral Surfaces: A Review, Crit. Rev. Environ. Sci. Technol., 2014, 44(3), 223–254 CrossRef CAS.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, Wiley, 2016 Search PubMed.
- N. Riemer, H. Vogel and B. Vogel, Soot Aging Time Scales in Polluted Regions during Day and Night, Atmos. Chem. Phys., 2004, 4(7), 1885–1893 CrossRef CAS.
- R. Zhang, A. F. Khalizov, J. Pagels, D. Zhang, H. Xue and P. H. McMurry, Variability in Morphology, Hygroscopicity, and Optical Properties of Soot Aerosols during Atmospheric Processing, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(30), 10291–10296 CrossRef CAS PubMed.
- Y. Wang, F. Liu, C. He, L. Bi, T. Cheng, Z. Wang, H. Zhang, X. Zhang, Z. Shi and W. Li, Fractal Dimensions and Mixing Structures of Soot Particles during Atmospheric Processing, Environ. Sci. Technol. Lett., 2017, 4(11), 487–493 CrossRef CAS.
- J. A. S. Lighty, J. M. Veranth and A. F. Sarofim, Combustion Aerosols: Factors Governing Their Size and Composition and Implications to Human Health, J. Air Waste Manage. Assoc., 2000, 50(9), 1565–1618 CrossRef CAS.
- A. D'Anna, Combustion-Formed Nanoparticles, Proc. Combust. Inst., 2009, 32 I(1), 593–613 CrossRef.
- W. Li, J. Sun, L. Xu, Z. Shi, N. Riemer, Y. Sun, P. Fu, J. Zhang, Y. Lin and X. Wang, et al. A Conceptual Framework for Mixing Structures in Individual Aerosol Particles, J. Geophys. Res.: Atmos., 2016, 121(22), 13784–13798 Search PubMed.
- H. A. Michelsen, Probing Soot Formation, Chemical and Physical Evolution, and Oxidation: A Review of in Situ Diagnostic Techniques and Needs, Proc. Combust. Inst., 2017, 36(1), 717–735 CrossRef CAS.
- M. M. Bisiaux, R. Edwards, A. C. Heyvaert, J. M. Thomas, B. Fitzgerald, R. B. Susfalk, S. G. Schladow and M. Thaw, Stormwater and Fire as Sources of Black Carbon Nanoparticles to Lake Tahoe, Environ. Sci. Technol., 2011, 45(6), 2065–2071 CrossRef CAS PubMed.
- H. Fu, H. Liu, J. Mao, W. Chu, Q. Li, P. J. J. Alvarez, X. Qu and D. Zhu, Photochemistry of Dissolved Black Carbon Released from Biochar: Reactive Oxygen Species Generation and Phototransformation, Environ. Sci. Technol., 2016, 50(3), 1218–1226 CrossRef CAS PubMed.
- X. Qu, H. Fu, J. Mao, Y. Ran, D. Zhang and D. Zhu, Chemical and Structural Properties of Dissolved Black Carbon Released from Biochars, Carbon, 2016, 96, 759–767 CrossRef CAS.
- N. Hagemann, S. Joseph, H. P. Schmidt, C. I. Kammann, J. Harter, T. Borch, R. B. Young, K. Varga, S. Taherymoosavi and K. W. Elliott, et al. Organic Coating on Biochar Explains Its Nutrient Retention and Stimulation of Soil Fertility, Nat. Commun., 2017, 8(1), 1–11 CrossRef CAS PubMed.
- B. S. Archanjo, M. E. Mendoza, M. Albu, D. R. G. Mitchell, N. Hagemann, C. Mayrhofer, T. L. A. Mai, Z. Weng, A. Kappler and S. Behrens, et al. Nanoscale Analyses of the Surface Structure and Composition of Biochars Extracted from Field Trials or after Co-Composting Using Advanced Analytical Electron Microscopy, Geoderma, 2017, 294, 70–79 CrossRef CAS.
- G. Sorrenti, C. A. Masiello, B. Dugan and M. Toselli, Biochar Physico-Chemical Properties as Affected by Environmental Exposure, Sci. Total Environ., 2016, 563–564, 237–246 CrossRef CAS PubMed.
- S. Brodowski, W. Amelung, L. Haumaier, C. Abetz and W. Zech, Morphological and Chemical Properties of Black Carbon in Physical Soil Fractions as Revealed by Scanning Electron Microscopy and Energy-Dispersive X-Ray Spectroscopy, Geoderma, 2005, 128(1), 116–129 CrossRef CAS.
- F. Yang, L. Zhao, B. Gao, X. Xu and X. Cao, The Interfacial Behavior between Biochar and Soil Minerals and Its Effect on Biochar Stability, Environ. Sci. Technol., 2016, 50(5), 2264–2271 CrossRef CAS PubMed.
- Y. Yang, K. Sun, L. Han, J. Jin, H. Sun, Y. Yang and B. Xing, Effect of Minerals on the Stability of Biochar, Chemosphere, 2018, 204, 310–317 CrossRef CAS PubMed.
- F. Braadbaart, I. Poole and A. A. van Brussel, Preservation Potential of Charcoal in Alkaline Environments: An Experimental Approach and Implications for the Archaeological Record, J. Archaeol. Sci., 2009, 36(8), 1672–1679 CrossRef.
- G. Liu, H. Zheng, Z. Jiang, J. Zhao, Z. Wang, B. Pan and B. Xing, Formation and Physicochemical Characteristics of Nano Biochar: Insight into Chemical and Colloidal Stability, Environ. Sci. Technol., 2018, 52(18), 10369–10379 CrossRef CAS PubMed.
- A. M. P. Oen, B. Beckingham, U. Ghosh, M. E. Kruså, R. G. Luthy, T. Hartnik, T. Henriksen and G. Cornelissen, Sorption of Organic Compounds to Fresh and Field-Aged Activated Carbons in Soils and Sediments, Environ. Sci. Technol., 2012, 46(2), 810–817 CrossRef CAS PubMed.
- S. D. Joseph, M. Camps-Arbestain, Y. Lin, P. Munroe, C. Chia, J. Hook, L. van Zwieten, S. Kimber, A. Cowie and B. P. Singh, et al. An Investigation into the Reactions of Biochar in Soil, Soil Res., 2010, 501–515 CrossRef CAS.
- F. J. Ruiz-Dueñas and Á. T. Martínez, Microbial Degradation of Lignin: How a Bulky Recalcitrant Polymer Is Efficiently Recycled in Nature and How We Can Take Advantage of This, Microb. Biotechnol., 2009, 2(2 SPEC. ISS), 164–177 CrossRef PubMed.
- C. I. Czimczik and C. A. Masiello, Controls on Black Carbon Storage in Soils, Global Biogeochem. Cycles, 2007, 21(3), 1–8 CrossRef.
- A. Hilscher, K. Heister, C. Siewert and H. Knicker, Mineralisation and Structural Changes during the Initial Phase of Microbial Degradation of Pyrogenic Plant Residues in Soil, Org. Geochem., 2009, 40(3), 332–342 CrossRef CAS.
- J. M. Saquing, Y.-H. Yu and P. C. Chiu, Wood-Derived Black Carbon (Biochar) as a Microbial Electron Donor and Acceptor, Environ. Sci. Technol. Lett., 2016, 3(2), 62–66 CrossRef CAS.
- D. X. Flores-Cervantes, H. M. Maes, A. Schäffer, J. Hollender and H. P. E. Kohler, Slow Biotransformation of Carbon Nanotubes by Horseradish Peroxidase, Environ. Sci. Technol., 2014, 48(9), 4826–4834 CrossRef CAS PubMed.
- B. L. Allen, G. P. Kotchey, Y. Chen, N. V. K. Yanamala, J. Klein-Seetharaman, V. E. Kagan and A. Star, Mechanistic Investigations of Horseradish Peroxidase-Catalyzed Degradation of Single-Walled Carbon Nanotubes, J. Am. Chem. Soc., 2009, 131(47), 17194–17205 CrossRef CAS PubMed.
- G. P. Kotchey, B. L. Allen, H. Vedala, N. Yanamala, A. A. Kapralov, Y. Y. Tyurina, J. Klein-Seetharaman, V. E. Kagan and A. Star, The Enzymatic Oxidation of Graphene Oxide, ACS Nano, 2011, 5(3), 2098–2108 CrossRef CAS PubMed.
- C. Zhang, W. Chen and P. J. J. Alvarez, Manganese Peroxidase Degrades Pristine but Not Surface-Oxidized (Carboxylated) Single-Walled Carbon Nanotubes, Environ. Sci. Technol., 2014, 48(14), 7918–7923 CrossRef CAS PubMed.
- L. A. Sgro, A. Simonelli, L. Pascarella, P. Minutolo, D. Guarnieri, N. Sannolo, P. Netti and A. D'Anna, Toxicological Properties of Nanoparticles of Organic Compounds (NOC) from Flames and Vehicle Exhausts, Environ. Sci. Technol., 2009, 43(7), 2608–2613 CrossRef CAS PubMed.
- H. Kong, Y. Zhang, Y. Li, Z. Cui, K. Xia, Y. Sun, Q. Zhao and Y. Zhu, Size-Dependent Cytotoxicity of Nanocarbon Blacks, Int. J. Mol. Sci., 2013, 14(11), 22529–22543 CrossRef PubMed.
- K. M. Garza, K. F. Soto and L. E. Murr, Cytotoxicity and Reactive Oxygen Species Generation from Aggregated Carbon and Carbonaceous Nanoparticulate Materials, Int. J. Nanomed., 2008, 3(1), 83–94 CrossRef CAS PubMed.
- G. Sigmund, D. Huber, T. D. Bucheli, M. Baumann, N. Borth, G. M. Guebitz and T. Hofmann, Cytotoxicity of Biochar: A Workplace Safety Concern?, Environ. Sci. Technol. Lett., 2017, 4(9), 362–366 CrossRef CAS.
- A. Magrez, S. Kasas, V. Salicio, N. Pasquier, J. W. Seo, M. Celio, S. Catsicas, B. Schwaller and L. Forró, Cellular Toxicity of Carbon-Based Nanomaterials, Nano Lett., 2006, 6(6), 1121–1125 CrossRef CAS PubMed.
- P. Wick, P. Manser, L. K. Limbach, U. Dettlaff-Weglikowska, F. Krumeich, S. Roth, W. J. Stark and A. Bruinink, The Degree and Kind of Agglomeration Affect Carbon Nanotube Cytotoxicity, Toxicol. Lett., 2007, 168(2), 121–131 CrossRef CAS PubMed.
- S. Wagner, A. Gondikas, E. Neubauer, T. Hofmann and F. von der Kammer, Spot the Difference: Engineered and Natural Nanoparticles in the Environment-Release, Behavior, and Fate, Angew. Chem., Int. Ed., 2014, 53(46), 12398–12419 CAS.
- M. Kosmulski, PH-Dependent Surface Charging and Points of Zero Charge IV. Update and New Approach, J. Colloid Interface Sci., 2009, 337(2), 439–448 CrossRef CAS PubMed.
- G. Sigmund, H. Sun, T. Hofmann and M. Kah, Predicting the Sorption of Aromatic Acids to Noncarbonized and Carbonized Sorbents, Environ. Sci. Technol., 2016, 50(7), 3641–3648 CrossRef CAS PubMed.
- I. Chowdhury, M. C. Duch, N. D. Mansukhani, M. C. Hersam and D. Bouchard, Colloidal Properties and Stability of Graphene Oxide Nanomaterials in the Aquatic Environment, Environ. Sci. Technol., 2013, 47(12), 6288–6296 CrossRef CAS PubMed.
- K. L. Chen, B. A. Smith, W. P. Ball and D. H. Fairbrother, Assessing the Colloidal Properties of Engineered Nanoparticles in Water : Case Studies from Fullerene C60 Nanoparticles and Carbon Nanotubes, Environ. Chem., 2010, 7, 10–27 CrossRef CAS.
- C. J. van Oss, R. J. Good and M. K. Chaudhury, The Role of van Der Waals Forces and Hydrogen Bonds in “Hydrophobic Interactions” between Biopolymers and Low Energy Surfaces, J. Colloid Interface Sci., 1986, 111(2), 378–390 CrossRef CAS.
- W. Yang, Y. Wang, J. Shang, K. Liu, P. Sharma, J. Liu and B. Li, Antagonistic Effect of Humic Acid and Naphthalene on Biochar Colloid Transport in Saturated Porous Media, Chemosphere, 2017, 189, 556–564 CrossRef CAS PubMed.
- M. Chen, D. Wang, F. Yang, X. Xu, N. Xu and X. Cao, Transport and Retention of Biochar Nanoparticles in a Paddy Soil under Environmentally-Relevant Solution Chemistry Conditions, Environ. Pollut., 2017, 230, 540–549 CrossRef CAS PubMed.
- Y. Jiang, R. Raliya, P. Liao, P. Biswas and J. D. Fortner, Graphene Oxides in Water: Assessing Stability as a Function of Material and Natural Organic Matter Properties, Environ. Sci.: Nano, 2017, 4(7), 1484–1493 RSC.
- W. Yang, Y. Wang, P. Sharma, B. Li, K. Liu, J. Liu, M. Flury and J. Shang, Effect of Naphthalene on Transport and Retention of Biochar Colloids through Saturated Porous Media, Colloids Surf., A, 2017, 530(July), 146–154 CrossRef CAS.
- J. Zhao, F. Liu, Z. Wang, X. Cao and B. Xing, Heteroaggregation of Graphene Oxide with Minerals in Aqueous Phase, Environ. Sci. Technol., 2015, 49(5), 2849–2857 CrossRef CAS PubMed.
- K. A. Huynh, J. M. McCaffery and K. L. Chen, Heteroaggregation of Multiwalled Carbon Nanotubes and Hematite Nanoparticles: Rates and Mechanisms, Environ. Sci. Technol., 2012, 46(11), 5912–5920 CrossRef CAS PubMed.
- P. Yi, J. J. Pignatello, M. Uchimiya and J. C. White, Heteroaggregation of Cerium Oxide Nanoparticles and Nanoparticles of Pyrolyzed Biomass, Environ. Sci. Technol., 2015, 49(22), 13294–13303 CrossRef CAS PubMed.
- N. Tufenkji and M. Elimelech, Deviation from the Classical Colloid Filtration Theory in the Presence of Repulsive DLVO Interactions, Langmuir, 2004, 20(25), 10818–10828 CrossRef CAS PubMed.
- Y. Li, N. Yang, T. Du, T. Xia, C. Zhang and W. Chen, Chloramination of Graphene Oxide Significantly Affects Its Transport Properties in Saturated Porous Media, NanoImpact, 2016, 3–4, 90–95 CrossRef.
- Z. Qi, L. Hou, D. Zhu, R. Ji and W. Chen, Enhanced Transport of Phenanthrene and 1-Naphthol by Colloidal Graphene Oxide Nanoparticles in Saturated Soil, Environ. Sci. Technol., 2014, 48(17), 10136–10144 CrossRef CAS PubMed.
- D. Wang, W. Zhang and D. Zhou, Antagonistic Effects of Humic Acid and Iron Oxyhydroxide Grain-Coating on Biochar Nanoparticle Transport in Saturated Sand, Environ. Sci. Technol., 2013, 47(10), 5154–5161 CrossRef CAS PubMed.
- S. Park, K.-S. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff, Graphene Oxide Papers Modified by Divalent Ions—Enhancing Mechanical Properties via Chemical Cross-Linking, ACS Nano, 2008, 2(3), 572–578 CrossRef CAS PubMed.
- T. Xia, Y. Qi, J. Liu, Z. Qi, W. Chen and M. R. Wiesner, Cation-Inhibited Transport of Graphene Oxide Nanomaterials in Saturated Porous Media: The Hofmeister Effects, Environ. Sci. Technol., 2017, 51(2), 828–837 CrossRef CAS PubMed.
- K. L. Chen, S. E. Mylon and M. Elimelech, Aggregation Kinetics of Alginate-Coated Hematite Nanoparticles in Monovalent and Divalent Electrolytes, Environ. Sci. Technol., 2006, 40(5), 1516–1523 CrossRef CAS PubMed.
- R. Kretzschmar, M. Borkovec, D. Grolimund and M. Elimelech, Mobile Subsurface Colloids and Their Role in Contaminant Transport, Adv. Agron., 1999, 66, 121–193 CAS.
- L. Zhang, L. Hou, L. Wang, A. T. Kan, W. Chen and M. B. Tomson, Transport of Fullerene Nanoparticles (nC60) in Saturated Sand and Sandy Soil: Controlling Factors and Modeling, Environ. Sci. Technol., 2012, 46(13), 7230–7238 CrossRef CAS PubMed.
- W. Zhang, J. Niu, V. L. Morales, X. Chen, A. G. Hay, J. Lehmann and T. S. Steenhuis, Transport and Retention of Biochar Particles in Porous Media: Effect of pH, Ionic Strength and Particle Size, Ecohydrology, 2010, 3(4), 497–508 CrossRef CAS.
- W. Zhang, C. W. Isaacson, U. Rattanaudompol, T. B. Powell and D. Bouchard, Fullerene Nanoparticles Exhibit Greater Retention in Freshwater Sediment than in Model Porous Media, Water Res., 2012, 46(9), 2992–3004 CrossRef CAS PubMed.
- J. J. Pignatello, W. A. Mitch and W. Xu, Activity and Reactivity of Pyrogenic Carbonaceous Matter toward Organic Compounds, Environ. Sci. Technol., 2017, 51(16), 8893–8908 CrossRef CAS PubMed.
- F. Wang, L. Duan, F. Wang and W. Chen, Environmental Reduction of Carbon Nanomaterials Affects Their Capabilities to Accumulate Aromatic Compounds, NanoImpact, 2016, 1, 21–28 CrossRef.
- K. Yang, W. Wu, Q. Jing and L. Zhu, Aqueous Adsorption of Aniline, Phenol, and Their Substitutes by Multi-Walled Carbon Nanotubes, Environ. Sci. Technol., 2008, 42(21), 7931–7936 CrossRef CAS PubMed.
- S. Gotovac, C.-M. Yang, Y. Hattori, K. Takahashi, H. Kanoh and K. Kaneko, Adsorption of Polyaromatic Hydrocarbons on Single Wall Carbon Nanotubes of Different Functionalities and Diameters, J. Colloid Interface Sci., 2007, 314(1), 18–24 CrossRef CAS PubMed.
- S. Zhang, T. Shao, S. S. K. Bekaroglu and T. Karanfil, Adsorption of Synthetic Organic Chemicals by Carbon Nanotubes: Effects of Background Solution Chemistry, Water Res., 2010, 44(6), 2067–2074 CrossRef CAS PubMed.
- M. Franz, H. A. Arafat and N. G. Pinto, Effect of Chemical Surface Heterogeneity on the Adsorption Mechanism of Dissolved Aromatics on Activated Carbon, Carbon, 2000, 38, 1807–1819 CrossRef CAS.
- B. Pan and B. Xing, Adsorption Mechanisms of Organic Chemicals on Carbon Nanotubes, Environ. Sci. Technol., 2008, 42(24), 9005–9013 CrossRef CAS PubMed.
- D. Zhu and J. J. Pignatello, Characterization of Aromatic Compound Sorptive Interactions with Black Carbon (Charcoal) Assisted by Graphite as a Model, Environ. Sci. Technol., 2005, 39(7), 2033–2041 CrossRef CAS PubMed.
- W. Chen, L. Duan, L. Wang and D. Zhu, Adsorption of Hydroxyl- and Amino-Substituted Aromatics to Carbon Nanotubes, Environ. Sci. Technol., 2008, 42(18), 6862–6868 CrossRef CAS PubMed.
- W. Chen, L. Duan and D. Zhu, Adsorption of Polar and Nonpolar Organic Chemicals to Carbon Nanotubes, Environ. Sci. Technol., 2007, 41(24), 8295–8300 CrossRef CAS PubMed.
- S. M. Martin, R. S. Kookana, L. Van Zwieten and E. Krull, Marked Changes in Herbicide Sorption-Desorption upon Ageing of Biochars in Soil, J. Hazard. Mater., 2012, 231–232, 70–78 CrossRef CAS PubMed.
- S. Hale, K. Hanley, J. Lehmann, A. Zimmerman and G. Cornelissen, Effects of Chemical, Biological, and Physical Aging as Well as Soil Addition on the Sorption of Pyrene to Activated Carbon and Biochar, Environ. Sci. Technol., 2011, 45(24), 10445–10453 CrossRef CAS PubMed.
- C.-H. Cheng, T.-P. Lin, J. Lehmann, L.-J. Fang, Y.-W. Yang, O. V. Menyailo, K.-H. K.-H. Chang, J.-S. Lai and L. Lai, Sorption Properties for Black Carbon (Wood Char) after Long-Term Exposure in Soils, Org. Geochem., 2014, 70, 53–61 CrossRef CAS.
- K. Shi, Y. Xie and Y. Qiu, Natural Oxidation of a Temperature Series of Biochars: Opposite Effect on the Sorption of Aromatic Cationic Herbicides, Ecotoxicol. Environ. Saf., 2015, 114, 102–108 CrossRef CAS PubMed.
- L. Hou, D. Zhu, X. Wang, L. Wang, C. Zhang and W. Chen, Adsorption of Phenanthrene, 2-Naphthol, and 1-Naphthylamine to Colloidal Oxidized Multiwalled Carbon Nanotubes: Effects of Humic Acid and Surfactant Modification, Environ. Toxicol. Chem., 2013, 32(3), 493–500 CrossRef CAS PubMed.
- G.-C. Chen, X.-Q. Shan, Y.-S. Wang, B. Wen, Z.-G. Pei, Y.-N. Xie, T. Liu and J. J. Pignatello, Adsorption of 2,4,6-Trichlorophenol by Multi-Walled Carbon Nanotubes as Affected by Cu(II), Water Res., 2009, 43(9), 2409–2418 CrossRef CAS PubMed.
- K. Yang and B. Xing, Desorption of Polycyclic Aromatic Hydrocarbons from Carbon Nanomaterials in Water, Environ. Pollut., 2007, 145(2), 529–537 CrossRef CAS PubMed.
- B. Pan, D. Lin, H. Mashayekhi and B. Xing, Adsorption and Hysteresis of Bisphenol A and 17α-Ethinyl Estradiol on Carbon Nanomaterials, Environ. Sci. Technol., 2008, 42(15), 5480–5485 CrossRef CAS PubMed.
- L. Hou, J. D. Fortner, X. Wang, C. Zhang, L. Wang and W. Chen, Complex Interplay between Formation Routes and Natural Organic Matter Modification Controls Capabilities of C60 nanoparticles (nC60) to Accumulate Organic Contaminants, J. Environ. Sci., 2017, 51, 315–323 CrossRef PubMed.
- F. Wang, J. J.-H. Haftka, T. L. Sinnige, J. L. M. Hermens and W. Chen, Adsorption of Polar, Nonpolar, and Substituted Aromatics to Colloidal Graphene Oxide Nanoparticles, Environ. Pollut., 2014, 186, 226–233 CrossRef CAS PubMed.
- X. Zhang, M. Kah, M. T. O. Jonker and T. Hofmann, Dispersion State and Humic Acids Concentration-Dependent Sorption of Pyrene to Carbon Nanotubes, Environ. Sci. Technol., 2012, 46(13), 7166–7173 CrossRef CAS PubMed.
- B. Wang, W. Zhang, H. Li, H. Fu, X. Qu and D. Zhu, Micropore Clogging by Leachable Pyrogenic Organic Carbon: A New Perspective on Sorption Irreversibility and Kinetics of Hydrophobic Organic Contaminants to Black Carbon, Environ. Pollut., 2017, 220, 1349–1358 CrossRef CAS PubMed.
- L. Wang, Y. Huang, A. T. Kan, M. B. Tomson and W. Chen, Enhanced Transport of 2,2′,5,5′-Polychlorinated Biphenyl by Natural Organic Matter (NOM) and Surfactant-Modified Fullerene Nanoparticles (nC60), Environ. Sci. Technol., 2012, 46(10), 5422–5429 CrossRef CAS PubMed.
- T. Hofmann and F. von der Kammer, Estimating the Relevance of Engineered Carbonaceous Nanoparticle Facilitated Transport of Hydrophobic Organic Contaminants in Porous Media, Environ. Pollut., 2009, 157(4), 1117–1126 CrossRef CAS PubMed.
- J. Y. Chen, W. Chen and D. Zhu, Adsorption of Nonionic Aromatic Compounds to Single-Walled Carbon Nanotubes: Effects of Aqueous Solution Chemistry, Environ. Sci. Technol., 2008, 42(19), 7225–7230 CrossRef CAS PubMed.
- M. Kah, X. Zhang and T. Hofmann, Sorption Behavior of Carbon Nanotubes: Changes Induced by Functionalization, Sonication and Natural Organic Matter, Sci. Total Environ., 2014, 497–498, 133–138 CrossRef CAS PubMed.
- Q. Li, V. L. Snoeyink, B. J. Mariñas and C. Campos, Pore Blockage Effect of NOM on Atrazine Adsorption Kinetics of PAC: The Roles of PAC Pore Size Distribution and NOM Molecular Weight, Water Res., 2003, 37(20), 4863–4872 CrossRef CAS PubMed.
- Y. Qiu, X. Xiao, H. Cheng, Z. Zhou and G. D. Sheng, Influence of Environmental Factors on Pesticide Adsorption by Black Carbon: pH and Model Dissolved Organic Matter, Environ. Sci. Technol., 2009, 43(13), 4973–4978 CrossRef CAS PubMed.
- I. Hilber, P. Mayer, V. Gouliarmou, S. E. Hale, G. Cornelissen, H. P. Schmidt and T. D. Bucheli, Bioavailability and Bioaccessibility of Polycyclic Aromatic Hydrocarbons from (Post-Pyrolytically Treated) Biochars, Chemosphere, 2017, 174, 700–707 CrossRef CAS PubMed.
- Y. Yang, T. Hofmann, C. Pies and P. Grathwohl, Sorption of Polycyclic Aromatic Hydrocarbons (PAHs) to Carbonaceous Materials in a River Floodplain Soil, Environ. Pollut., 2008, 156(3), 1357–1363 CrossRef CAS PubMed.
- G. Sigmund, T. D. Bucheli, I. Hilber, V. Micić, M. Kah and T. Hofmann, Effect of Ageing on the Properties and Polycyclic Aromatic Hydrocarbon Composition of Biochar, Environ. Sci.: Processes Impacts, 2017, 19(5), 768–774 RSC.
- H. Marsh and F. Rodriguez-Reinoso, Activated Carbon, Elsevier Science & Technology Books, 2006 Search PubMed.
- X. Li, W. Chen, C. Zhang, Y. Li, F. Wang and W. Chen, Enhanced Dehydrochlorination of 1,1,2,2-Tetrachloroethane by Graphene-Based Nanomaterials, Environ. Pollut., 2016, 214, 341–348 CrossRef CAS PubMed.
- W. Chen, D. Zhu, S. Zheng and W. Chen, Catalytic Effects of Functionalized Carbon Nanotubes on Dehydrochlorination of 1,1,2,2-Tetrachloroethane, Environ. Sci. Technol., 2014, 48(7), 3856–3863 CrossRef CAS PubMed.
- W.-C. Hou, I. Chowdhury, D. G. Goodwin, W. M. Henderson, D. H. Fairbrother, D. Bouchard and R. G. Zepp, Photochemical Transformation of Graphene Oxide in Sunlight, Environ. Sci. Technol., 2015, 49(6), 3435–3443 CrossRef CAS PubMed.
- H. Hyung, J. D. Fortner, J. B. Hughes and J.-H. Kim, Natural Organic Matter Stabilizes Carbon Nanotubes in the Aqueous Phase, Environ. Sci. Technol., 2007, 41(1), 179–184 CrossRef CAS PubMed.
- X. Zhou, L. Shu, H. Zhao, X. Guo, X. Wang, S. Tao and B. Xing, Suspending Multi-Walled Carbon Nanotubes by Humic Acids from a Peat Soil, Environ. Sci. Technol., 2012, 46(7), 3891–3897 CrossRef CAS PubMed.
- Z. Chen, B. Chen, D. Zhou and W. Chen, Bisolute Sorption and Thermodynamic Behavior of Organic Pollutants to Biomass-Derived Biochars at Two Pyrolytic Temperatures, Environ. Sci. Technol., 2012, 46(22), 12476–12483 CrossRef CAS PubMed.
- Y. Qin, G. Li, Y. Gao, L. Zhang, Y. S. Ok and T. An, Persistent Free Radicals in Carbon-Based Materials on Transformation of Refractory Organic Contaminants (ROCs) in Water: A Critical Review, Water Res., 2018, 137, 130–143 CrossRef CAS PubMed.
- L. Shirshova, Redox Properties of Humic Acids, Geochim. Cosmochim. Acta, 1997, 61, 4599–4604 CrossRef.
- J. Yang, J. J. Pignatello, B. Pan and B. Xing, Degradation of p-Nitrophenol by Lignin and Cellulose Chars: H2O2-Mediated Reaction and Direct Reaction with the Char, Environ. Sci. Technol., 2017, 51(16), 8972–8980 CrossRef CAS PubMed.
- N. Chen, Y. Huang, X. Hou, Z. Ai and L. Zhang, Photochemistry of Hydrochar: Reactive Oxygen Species Generation and Sulfadimidine Degradation, Environ. Sci. Technol., 2017, 51(19), 11278–11287 CrossRef CAS PubMed.
- J. Qin, Q. Chen, M. Sun, P. Sun and G. Shen, Pyrolysis Temperature-Induced Changes in the Catalytic Characteristics of Rice Husk-Derived Biochar during 1,3-Dichloropropene Degradation, Chem. Eng. J., 2017, 330, 804–812 CrossRef CAS.
- J. Qin, Y. Cheng, M. Sun, L. Yan and G. Shen, Catalytic Degradation of the Soil Fumigant 1,3-Dichloropropene in Aqueous Biochar Slurry, Sci. Total Environ., 2016, 569–570, 1–8 CrossRef CAS PubMed.
- J. Yang, B. Pan, H. Li, S. Liao, D. Zhang, M. Wu and B. Xing, Degradation of p-Nitrophenol on Biochars: Role of Persistent Free Radicals, Environ. Sci. Technol., 2016, 50(2), 694–700 CrossRef CAS PubMed.
- W. Gong, X. Liu, S. Xia, B. Liang and W. Zhang, Abiotic Reduction of Trifluralin and Pendimethalin by Sulfides in Black-Carbon-Amended Coastal Sediments, J. Hazard. Mater., 2016, 310, 125–134 CrossRef CAS PubMed.
- G. Fang, C. Zhu, D. D. Dionysiou, J. Gao and D. Zhou, Mechanism of Hydroxyl Radical Generation from Biochar Suspensions: Implications to Diethyl Phthalate Degradation, Bioresour. Technol., 2015, 176, 210–217 CrossRef CAS PubMed.
- W. Gong, X. Liu, L. Tao, W. Xue, W. Fu and D. Cheng, Reduction of Nitrobenzene with Sulfides Catalyzed by the Black Carbons from Crop-Residue Ashes, Environ. Sci. Pollut. Res., 2014, 21(9), 6162–6169 CrossRef CAS PubMed.
- G. Fang, J. Gao, C. Liu, D. D. Dionysiou, Y. Wang and D. Zhou, Key Role of Persistent Free Radicals in Hydrogen Peroxide Activation by Biochar: Implications to Organic Contaminant Degradation, Environ. Sci. Technol., 2014, 48(3), 1902–1910 CrossRef CAS PubMed.
- Y. Li, J. M. Kemper, G. Datuin, A. Akey, W. A. Mitch and R. G. Luthy, Reductive Dehalogenation of Disinfection Byproducts by an Activated Carbon-Based Electrode System, Water Res., 2016, 98, 354–362 CrossRef CAS PubMed.
- W. Xu, J. J. Pignatello and W. A. Mitch, Reduction of Nitroaromatics Sorbed to Black Carbon by Direct Reaction with Sorbed Sulfides, Environ. Sci. Technol., 2015, 49(6), 3419–3426 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |