
Influence of light wavelength on the photoactivity, physicochemical transformation, and fate of graphene oxide in aqueous media†
Tingting
Du
a,
Adeyemi S.
Adeleye‡
 b,
Tong
Zhang
a,
Chuanjia
Jiang
a,
Min
Zhang
a,
Huihui
Wang
a,
Yao
Li
*a,
Arturo A.
Keller
*b and
Wei
Chen
a
b,
Tong
Zhang
a,
Chuanjia
Jiang
a,
Min
Zhang
a,
Huihui
Wang
a,
Yao
Li
*a,
Arturo A.
Keller
*b and
Wei
Chen
a
aCollege of Environmental Science and Engineering, Ministry of Education Key Laboratory of Pollution Processes and Environmental Criteria, Tianjin Key Laboratory of Environmental Remediation and Pollution Control, Nankai University, 38 Tong Yan Road, Tianjin 300350, China. E-mail: hkliyao@nankai.edu.cn; Fax: 86 22 2350 1117; Tel: 86 22 2350 1117
bBren School of Environmental Science and Management, University of California, Santa Barbara, California 93106, USA. E-mail: keller@bren.ucsb.edu; Fax: 86 22 2350 1117; Tel: 86 22 2350 1117
First published on 11th September 2018
Abstract
The rapidly increasing production and application of graphene oxide (GO) calls for much-needed attention to aspects related to the environmental implications of this unique nanomaterial. To date, very little is known about the transformation of GO when exposed to electromagnetic radiation at different wavelengths, which can occur in natural water systems. This study explored the changes in the physicochemical properties of GO when exposed to the UV and visible wavelength ranges of simulated solar light. Irradiation, especially under UV light, led to remarkable changes in the surface oxygen (O)-functionalities of GO, and cleavage of GO's graphitic bond. The carbon/oxygen (C/O) ratio of GO increased by 41% and 31% after irradiation with simulated solar light and UV irradiation, respectively. Visible light also changed the morphology of GO, but the C/O ratio only increased by 5.5%. The formation of hydroxyl radicals (˙OH), superoxide (O2˙−) and singlet oxygen (1O2) by GO was observed during irradiation with both UV and visible lights, but the proportions were different. 1O2 was primarily formed during visible light irradiation, while O2˙− and ˙OH were mostly formed under UV light. ˙OH hydroxylated GO, which mainly resulted in the decomposition of GO. The changes in O-containing functional groups on GO by irradiation influenced the colloidal stability of GO in aqueous media, but the physical wrinkle on GO surface may strongly affect its interactions with other organic materials.
Environmental significanceThe applications of graphene oxide (GO) are leading to increasing environmental release of these novel materials. As a reactive nanomaterial, it is unlikely that GO will maintain its pristine form in the environment; instead, GO can undergo substantial transformation, e.g. under the irradiation of sunlight. However, the full band of solar light will be filtered or partially minimized in natural water systems, e.g. UV light could be filtered out with increasing water depth. It is unclear how the physicochemical properties of GO will change in natural waters when exposed to lights with different wavelength ranges (i.e., UV or visible light). Thus, knowledge on the wavelength-dependent photo-transformation of GO is necessary to better understand the fate and transport of GO nanomaterials in the environment. |
1 Introduction
Graphene oxide (GO) is a structural analog of graphene, and it possesses abundant oxygen (O)-containing functionalities such as epoxide (C–O–C), hydroxyl (C–OH), carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and carboxyl groups (O–CO) covalently bound to either its basal planes (C–O–C and C–OH groups) or edges (CO and O–CO groups).1–3 GO can be stabilized in aqueous media as colloidal agglomerates because of its functionalization, and it is more reactive in water than other carbon nanomaterials, such as C60 and carbon nanotubes.4,5 GO is increasingly being proposed for commercial products, such as nanopaints, polymer composites, functional coatings, and water treatment nanohybrids, which could lead to release of the nanomaterial into the environment.6–9 Thus, it is expected that wastes containing GO will be generated and released into the environment, and may accumulate in landfills or surface waters.
O), and carboxyl groups (O–CO) covalently bound to either its basal planes (C–O–C and C–OH groups) or edges (CO and O–CO groups).1–3 GO can be stabilized in aqueous media as colloidal agglomerates because of its functionalization, and it is more reactive in water than other carbon nanomaterials, such as C60 and carbon nanotubes.4,5 GO is increasingly being proposed for commercial products, such as nanopaints, polymer composites, functional coatings, and water treatment nanohybrids, which could lead to release of the nanomaterial into the environment.6–9 Thus, it is expected that wastes containing GO will be generated and released into the environment, and may accumulate in landfills or surface waters.
As a reactive nanoparticle, it is unlikely that GO will maintain its pristine form in the environment. The physicochemical characteristics of GO can change substantially when it is exposed to the environment via several natural or engineered processes.10–16 In the environment, solar irradiation is the most common factor that may induce or affect the transformation of GO, which can change the chemical characteristics as well as the morphology of pristine GO. Several studies indicated that GO could be reduced and decomposed in the presence of simulated sunlight irradiation,17–19 producing CO2 and other small sized particles, such as polycyclic aromatic hydrocarbons (PAHs). However, most of these studies only considered the transformation of GO under full-spectrum simulated solar light, while no study has focused on the transformation of GO under light of different wavelengths. In natural waters, the UV fraction of solar light will be significantly filtered with increasing water depth,20–22 and the remaining visible light will be mainly responsible for the transformation of GO. In addition, UV light, which is frequently used in wastewater treatment systems,23 can directly change GO's physicochemical characteristics within wastewater treatment processes. The above-mentioned light conditions may induce different patterns of GO transformation and possibly change the environmental fate of GO transformation products.17 Besides, GO may undergo photocatalytic reactions in water to produce different types of reactive oxygen species (ROS). In a recent study where GO was exposed to solar irradiation, we demonstrated the production of hydroxyl radicals (˙OH), superoxide radical anions (O2˙−) and singlet oxygen (1O2) by GO.24 However, the specific effect of each distinct fraction (of the solar spectrum) is still unknown, and some ROS, such as ˙OH, have high reactivity and can generate highly oxidizing organic radicals.25 The mechanism and effects of the produced ROS on the transformation of GO are also unclear.
In summary, the objectives of the study were to (1) investigate the physicochemical transformation of GO under the UV (290–420 nm) and visible wavelength range (>420 nm), (2) explore the ratio of the ROS formed under different light wavelengths and their impacts on GO transformation, and (3) determine the influence of GO transformation under UV and visible light on its environmental fate and interactions.
2 Experimental
2.1 Materials and stock preparation
Graphene oxide (GO) nanoparticles (>99%) were purchased from Plannano Materials Tech. Co. (Tianjin, China) and used as received. The nanomaterials were synthesized using a modified Hummers method, according to the information provided by the manufacturer.26 To prepare GO stock suspensions, 90 mg of pristine GO nanosheets was added into 300 mL of deionized (DI) water in a 500 mL Erlenmeyer flask and then ultrasonicated at 100 W for 4 h using a Vibra-Cell VCX800 sonicator (Sonics & Materials, USA). A uniform dark-brown suspension was obtained and kept in the dark at 4 °C until use. The GO in the suspension had an average hydrodynamic parameter of 129.5 ± 23.2 nm, according to dynamic light scattering (DLS) analysis (Zetasizer Nano ZS90, Malvern Instruments, Worcestershire, UK).Methanol, acetonitrile, toluene, acetone, chloroform, furfuryl alcohol (FFA), terephthalic acid (TA), phenanthrene, and 1-naphthol were purchased from Sigma-Aldrich (St. Louis, USA).
2.2 Set-up of photochemistry experiments
The photochemistry set-up was similar to the one used previously.24 In summary, a photochemical reaction apparatus (XPA-7, Xu Jiang Machine Plant, Nanjing, China) connected to an 800 W xenon lamp was used as the light source, and a 290 nm cutoff filter was used to adjust the light irradiation to simulate the solar spectrum typical of the real environment (λ > 290 nm). A 420 nm cutoff filter was used to allow only visible light (λ > 420 nm), and another filter was used to allow only UV light (λ = 290–420 nm). Fig. S1† shows a comparison of the spectra for these different wavelengths and natural solar irradiation (collected on May 28, 2015).The concentration of GO used for the experiment was 10 mg L−1. The photochemistry experiment was conducted in DI water in order to simplify the system enough for us to clearly decipher the role of the different light wavelengths on the photo-transformation of GO. The reaction was carried out in a 60 mL grinding-mouth transparent quartz tube, and a 1.5 cm magnetic stirrer was used for mixing. The quartz tubes were submerged in a thermostat-controlled water bath (25 °C) during irradiation experiments. Samples were irradiated for 8 h per day for 3 days, making a total of 24 h. All the samples, including GO (the pristine nanomaterial not exposed to any irradiation), GOfull (GO with full-spectrum simulated solar irradiation, λ > 290 nm), GOUV (GO with only UV light irradiation, λ = 290–420 nm) and GOvis (GO with visible light irradiation, λ > 420 nm), were prepared for characterization.
2.3 Graphene oxide characterization
GO was characterized as pristine particles and after irradiation treatments. For post-irradiation characterization, dry GO particles were obtained from the treated GO suspensions via freeze-drying. The chemical composition and functional groups of pristine and photo-treated GO (GOfull, GOUV and GOvis) were characterized via X-ray photoelectron spectroscopy (XPS, using a PHI 5000 VersaProbe, Japan), Fourier transform infrared (FTIR) transmission spectroscopy (110 Bruker TENSOR 27 apparatus, Germany), and Raman spectroscopy (Renishaw InVia Raman spectrometer, England). The UV-visible absorbance spectra of the GO suspensions were obtained with a UV-2401 spectrophotometer (Shimadzu Scientific Instruments, USA). The morphology of GO was characterized by atomic force microscopy (AFM, MMAFM/STM, D3100M, Digital Ltd., USA), scanning electron microscopy (SEM, FEI Nova NanoSEM 430, The Netherlands) and transmission electron microscopy (TEM, FEI Tecnai G2100 F-20, The Netherlands).2.4 Reactive oxygen species (ROS) detection
The steady-state concentrations of 1O2 were determined indirectly by monitoring the concentrations of FFA, as explained in a previous study24 and summarized here (eqn (1) and (2)). The concentration of the GO suspension used was 10 mg L−1, and a known amount of FFA standard water stock solution was injected into the quartz container with a micro-syringe to achieve a FFA concentration of 50 μM. Samples were collected from the quartz container (after irradiation for 0, 1, 2, 4, 6, 16, and 24 h) using a 1 mL glass syringe and passed through 0.22 μm filters to remove GO particles prior to high-performance liquid chromatography (HPLC) analysis. [1O2]ss was determined by dividing the observed FFA degradation rate by the reaction rate constant of FFA with 1O2 and the residual concentration of FFA (which was determined via HPLC using a Waters e2695 (Waters Technology, USA) equipped with a symmetry reversed phase C18 column (4.6 × 150 mm)). FFA was detected with a Waters 2489 UV/visible detector at a wavelength of 219 nm; the mobile phase used was 40% acetonitrile and 60% H2O with a flow rate of 1.0 mL min−1.FFA + 1O2 → reaction![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) products products | (1) |
 | (2) |
The steady-state concentrations of O2˙− were measured with a chemiluminescent probe (2-methyl-6-[p-methoxyphenyl]-3,7-dihydroimidazo[1,2-a]pyrazin-3-one (MCLA)), which can selectively react with O2˙−. The detailed procedure was described by Adeleye et al. (2018).24 A 200 μM MCLA stock solution was prepared and subsequently frozen at 4 °C. The analytical reagent contained 1.0 μM MCLA buffered with 0.05 M sodium acetate, and the final pH of this reagent was adjusted to 5.5 using quartz-distilled HCl. During the measurement, the analytical reagent and GO/GOfull/GOUV/GOvis (10 mg L−1) were pumped at a 1:1 (v/v) ratio into a purpose-built flow injection analysis (FIA) system (Waterville Analytical, Waterville, ME, US). The reaction between O2˙− and MCLA resulted in a chemiluminescence signal at 455 nm, which was detected using a photomultiplier.
Terephthalic acid (TA) was used to measure the steady-state concentrations of ˙OH. 2-Hydroxy-terephthalic acid (2HTA) is generated during the reaction of TA with ˙OH, and the reaction is unaffected by the presence of other reactive species such as O2˙−, HO2˙, and H2O2−.28 2HTA was analyzed via HPLC and detected with a Waters 2475 fluorescence detector at an excitation wavelength of 315 nm and an emission wavelength of 425 nm; the mobile phase consisted of 65% trifluoroacetic acid buffer (0.05% v/v) and 35% acetonitrile (v/v) at a flow rate of 1.0 mL min−1.
Tert-butanol (t-BuOH) was used to eliminate ˙OH radicals in the suspension. 10 mM t-BuOH was mixed into the GO samples before irradiation and then the irradiated GO samples were collected for further characterization.29,30
2.5 Influence of photo-treatment on GO fate
To understand how the exposure of GO to different light wavelengths will influence the environmental fate of the nanomaterial, additional studies were carried out to determine changes in the hydrodynamic diameter (Dh), agglomeration kinetics, and zeta (ζ) potential of the photo-treated GO suspensions at 25 °C using the Zetasizer. Furthermore, changes in the adsorption properties of GO were also characterized.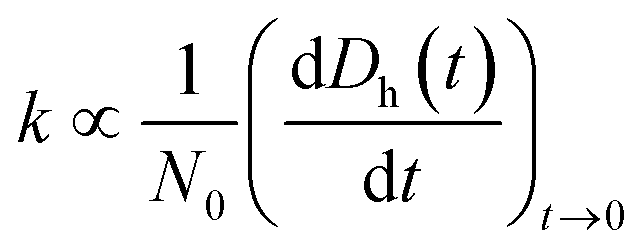 | (3) |
The particle attachment efficiency (α) was used to quantify the GO agglomeration kinetics, and it was calculated by normalizing the measured k at a given ionic strength (with or without HA) with the agglomeration rate constant measured under diffusion limited (fast) conditions, as shown in eqn (4):
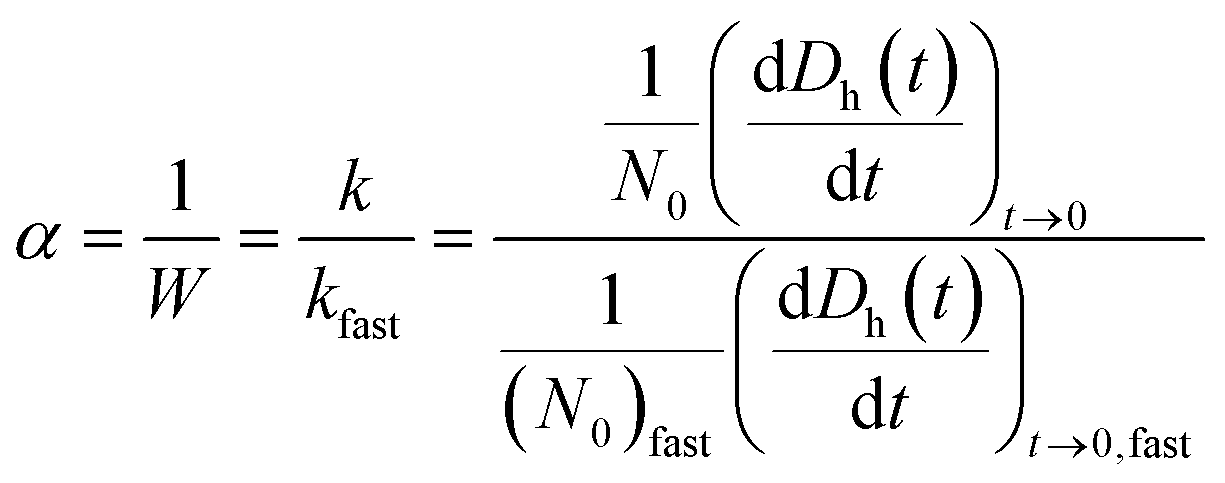 | (4) |
The diffusion-limited (DLCA) and reaction-limited (RLCA) clustering agglomeration regimes, and thus, the critical coagulation concentrations (CCCs), can be identified in a plot of log10[NaCl] versus log10α. DLCA occurs when the attachment efficiency between particles is close to unity, whereas RLCA dominates at very low attachment efficiencies.36 CCC is the point of intersection between DLCA and RLCA.
3 Results and discussion
3.1 Characterization of pristine and photo-treated GO
The chemical transformation of GO upon irradiation under different light wavelengths was evaluated. The XPS spectra of pristine GO, GOfull, GOUV and GOvis are provided in Fig. 1a–d and the associated quantitative data are summarized in Table 1. The binding energy level and FWHM values of GOs are summarized in Table S1.† The carbon/oxygen (C/O) ratio increased by 41% from 1.80 for GO to 2.54 for GOfull, which indicates that reduction of GO occurred during the 24 h irradiation under full-spectrum simulated solar light. After irradiation by UV and visible light, the C/O ratio increased by 31% and 5.5%, respectively, which also confirmed the loss of O-functional groups (i.e., reduction of GO) when GO was exposed to irradiation in the UV and visible light regions. In addition, the relative abundance of the different O-functional groups on the surface of GO changed remarkably upon irradiation at the different irradiation wavelengths, as shown by the deconvolution of the XPS C 1s spectra (summarized in Table 1).38 The relative abundance of GO's aromatic C–C/CC group (binding energy [BE] = 284.6 eV) increased from 45.16% (in pristine GO) to 62.42%, 57.13% and 49.15% after irradiation with full-spectrum solar, UV and visible light, respectively. Irradiation, especially with full-spectrum solar and UV light, resulted in a considerable decrease in the C–O–C/C–OH (BE = 286.6 eV) and CO (BE = 287.8 eV) groups, while the relative abundance of the O–CO group (BE = 288.8 eV) increased. The changes in GO's functional groups were minimal when irradiated by visible light alone.
 | ||
| Fig. 1 Photo-transformation of GO. High resolution C 1s XPS spectra of (a) pristine GO, (b) GO exposed to the full solar spectrum (λ > 290 nm), (c) GO exposed to UV light (λ = 290–420 nm), and (d) GO exposed to visible light (λ > 420 nm). Changes in GO after exposure to light of different wavelengths were also determined via (e) FTIR spectroscopy and (f) UV-vis absorbance spectroscopy. | ||
| Sample ID | C (atom %) | Total C (atom %) | Total O (atom %) | C/O ratio | |||
|---|---|---|---|---|---|---|---|
| C–C/CC |
C–OH/C–O–C | CO |
O–CO |
||||
| GO | 45.16 | 43.71 | 4.77 | 6.36 | 64.29 | 35.72 | 1.80 |
| GOfull | 62.42 | 25.14 | 3.14 | 9.30 | 71.76 | 28.24 | 2.54 |
| GOUV | 57.13 | 32.62 | 3.06 | 7.19 | 70.25 | 29.74 | 2.36 |
| GOvis | 49.15 | 40.26 | 4.30 | 6.29 | 65.47 | 34.53 | 1.90 |
Changes in the surface chemistry of GO upon irradiation with different electromagnetic wavelengths were further characterized by other spectroscopic techniques including FTIR and UV-vis absorption spectroscopy (Fig. 1e and f). The FTIR spectrum of pristine GO exhibited four characteristic peaks located at 1055, 1383, 1628 and 1735 cm−1, corresponding to C–O–C stretching, a C–O group, O–CO stretching, and CO stretching, respectively.39–41 The C–O, C–O–C and CO groups decreased slightly after irradiation with the full solar spectrum and UV light. The characteristic absorbance peak of GO at 230 nm was observed for pristine GO, which can be ascribed to π–π* transitions in small electronically conjugated domains present in the material.42 The characteristic absorbance peak however shifted to 240 nm in GOUV and 250 nm in GOfull (after 24 h exposure), which indicates an increase in the electronically conjugated domain, resulting from the reduction of GO. The time-series UV-vis spectra of irradiated GO showed progressive red-shifting of the GO peak at 230 nm when exposed to the full solar spectrum and UV irradiation but no substantial shift under visible light irradiation for up to 32 h (Fig. S2†). In addition, the peak at around 300 nm, originating from the n–π* transitions of the O-functionalities, was clearly observed in pristine GO.3 The intensity of the peak decreased in GOvis and was not observed in GOUV and GOfull, indicating reduction of GO under those conditions. The UV-vis absorbance includes the absorbance of unreacted GO, light-transformed GO and any other photoreaction products. The increase in absorbance (especially when GO was exposed to the full spectrum and UV) thus shows that light-absorbing photoproducts were formed upon irradiation of GO. In addition, the increased peak intensity at around 254 nm, which reflects aromaticity, may also indicate an increasing degree of graphitization of GO after its reduction by irradiation.
Changes in the morphology of GO after exposure to light of different irradiation wavelengths were also examined. Untreated GO nanosheets appeared smooth, with some minimal wrinkles under the TEM (Fig. 2a) and SEM (Fig. 2e). The nanomaterials, however, appeared broken up into smaller-sized sheets upon exposure to full-spectrum solar (Fig. 2b and f) and UV lights (Fig. 2c and g). The GO samples exposed to visible light were not remarkably broken up, but more wrinkles were observed in the TEM micrographs (Fig. 2d and h). Breaking up of GO sheets into smaller pieces upon photo-treatment with full-spectrum solar and UV light was confirmed by AFM analysis, which also showed no remarkable difference in size distribution between pristine GO and GO exposed to visible light. The Raman spectra of the GO samples (Fig. S3†) indicated an increase in the ID/IG ratio (the ratio of the intensity of the D band (around 1350 cm−1) to that of the G band (around 1580 cm−1)) after 24 h irradiation for all the samples, especially GOUV. Since ID/IG is a measure of the average size of the in-plane sp2 domains and molecular defects,43 the results showed that more defect sites were formed on the graphenic sheets upon irradiation for all light sources. The physical appearance of the GO suspension also changed when it was exposed to different irradiation wavelengths for 24 h (Fig. S4†). Specifically, the color of GO darkened strongly upon irradiation with the full solar spectrum and UV light. The GO suspension was only slightly darker upon its exposure to visible light for 24 h. Time series experiments showed that the darkening of the GO suspension became stronger over time upon exposure to all three irradiation types (Fig. S5†). In general, the intensity of the GO darkening observed was GOfull > GOUV ≫ GOvis (Fig. S5†). Darkening of GO is mainly due to the reduction of GO functional groups by the irradiation, which may destroy some chromophore units (such as C–OH and CO groups on the graphitic carbon).17,44
 | ||
| Fig. 2 TEM micrographs of (a) pristine GO, (b) GOfull, (c) GOUV, and (d) GOvis. SEM micrographs of (e) pristine GO, (f) GOfull, (g) GOUV, and (h) GOvis. AFM images of (i) pristine GO, (j) GOfull, (k) GOUV, and (l) GOvis. The irradiation time was 24 h for all the samples. | ||
In general, characterization of GO, GOfull, GOUV, and GOvis showed that irradiation strongly changed the physicochemical properties of the nanomaterials and the changes were correlated with the wavelength, and thus, the photon energy, of the irradiation. The characterization data showed that UV and full spectrum light cause remarkable physicochemical changes in GO, which is mainly because the energy of the photons of UV exceeds the bandgap of the GO domains. In contrast, the photons of visible light do not have as much energy. Thus, more substantial changes in the physicochemical properties of GO were observed in GOfull (which included irradiation in the UV region) and GOUV compared to GOvis. The XPS and FTIR spectra indicated that the reduction of GO by irradiation was mostly due to UV photons, while visible light only generated modest changes in GO's functional groups. The TEM, SEM and AFM micrographs clearly showed a sharp decrease in the particle size of GOfull and GOUV, indicating that the graphenic sheets of GO were probably cleaved by the UV radiation but the effects of visible light were not as obvious.
3.2 Reactive oxygen species (ROS) formation
In this study, three major ROS, i.e., 1O2, O2˙− and ˙OH, were detected when GO was exposed to irradiation at different wavelengths. Fig. 3a shows the steady-state concentration of 1O2 generated by GO, a photosensitizer, under different classes of irradiation at pH 7. The results indicate that 1O2 could be generated by GO exposed to both UV and visible light. Surprisingly, more 1O2 was generated by GO under visible light than UV light, contrary to a previous study which showed a higher production of 1O2 by fullerol under UV irradiation compared to visible light.45 Generally, the energy transfer process leads to the formation of 1O2 from the interaction between the excited state of a substance (such as GO) with the ground state of O2. The energy required to promote ground-state O2 to 1O2 is 94 kJ mol−1.46 Our results indicated that UV and visible light can both excite GO to GO*, which promotes ground-state O2 to 1O2 (eqn (5) and (6)). The generation of a smaller amount of 1O2 when GO was irradiated with UV light, relative to irradiation with visible light, may be because UV rapidly reduced GO to reduced GO (rGO), resulting in the formation of less GO* and thus a lower production of 1O2.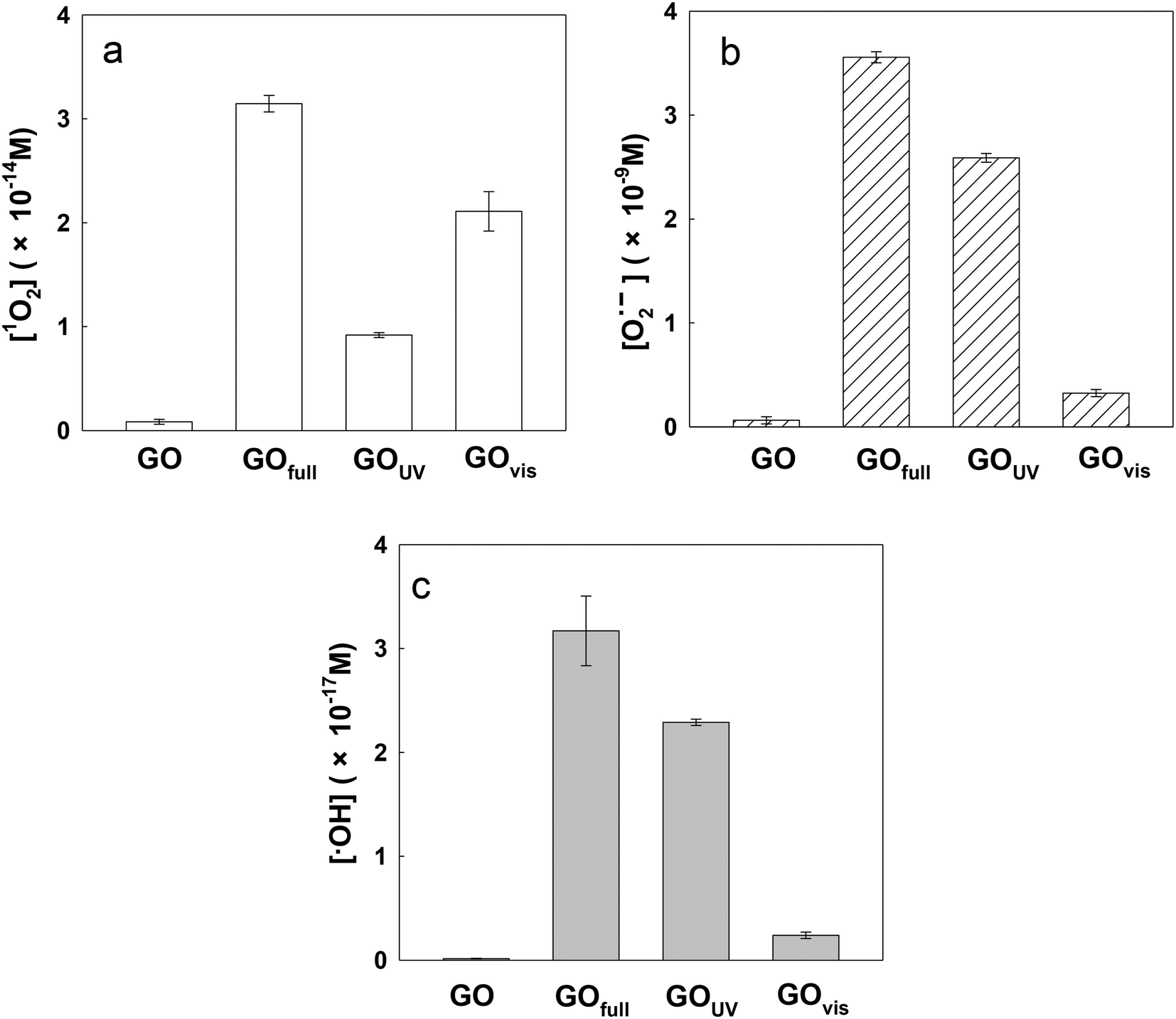 | ||
| Fig. 3 Steady-state concentrations of (a) singlet oxygen (1O2), (b) hydroxyl radicals (˙OH), and (c) superoxide radical anions (O2˙−) in GO suspensions in the dark (GO) and exposed to full-spectrum solar (GOfull), UV (GOUV), and visible light (GOvis). [GO] = 10 mg L−1; pH = 7. | ||
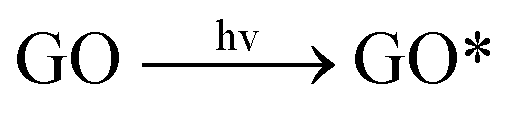
| (5) |
|
GO* + O2 → GO + 1O2
| (6) |
Fig. 3b and c show the steady-state concentrations of O2˙− and ˙OH generated by GO. Since O2˙− disproportionates rapidly in water to H2O2, which is an important precursor for ˙OH, the relative ratio between O2˙− and ˙OH may be similar for all light sources.25 As shown in Fig. 3b and c, the production of O2˙− and ˙OH was mainly under UV light. It has been reported that the threshold for GO reduction is 3.2 eV (λ < 390 nm).47 Based on the literature, the semiconducting domains of GO can act as photocatalysts when irradiated with UV light of energy exceeding the bandgap of the domains.19 Therefore, UV irradiation of GO results in the formation of electron–hole pairs; the co-occurrence of oxidative (valence band holes, hvb+) and reductive (conduction band electron, eaq−) transients agrees with concurrent GO oxidation and reduction (or disproportionation).17,48,49 Pickering and Wiesner (2005)45 hypothesized that irradiation of fullerol led to the formation of the fullerol radical anion, which acted as a precursor for the formation of O2˙−via a type I photochemical reaction, and a similar reaction may also occur on the surface of GO. As shown in eqn (7) and (8), the conduction band electron (eaq−) produced during irradiation of the GO suspension may act as an electron source for the generation of GO radical anions and subsequent formation of O2˙−.
| GO + eaq− → GO˙− | (7) |
| GO˙− + O2 → GO + O2˙− | (8) |
It should be noted that the amount of radicals formed by full spectrum irradiation is approximately equal to the amount of radicals formed under UV and visible light combined for all the three ROS determined. This demonstrates the additive effect of light wavelength on ROS production by GO.
3.3 Mechanisms controlling GO transformation from UV light, visible light and produced ROS
Of all the ROS, 1O2 and O2˙− appear to have little effects on GO transformation because of poor oxidation effects. However, ˙OH shows extremely high reactivity and can unselectively react with most organic substances.25,50 As ˙OH was formed on the surface of GO during the irradiation with UV light, it is possible that ˙OH can attack the surface of GO, similar to the oxidation of the unfunctionalized aromatic ring of small molecular compounds.51 To understand the effects of ˙OH radicals on the oxidation of GO, we used tert-butanol (t-BuOH), a scavenger that can selectively eliminate ˙OH radicals in suspension,52 to further clarify the specific role of ˙OH in the transformation of GO. Fig. 4a–c shows the FTIR spectra of GO under full-spectrum, UV and visible light with or without t-BuOH. As shown in the figure, the O-containing functional groups decreased significantly in GOfull and GOUV samples when t-BuOH was present, but there was no substantial change for GOvis. | ||
| Fig. 4 FTIR spectra of (a) GOfull, GOfull with quenching (using t-BuOH) (b) GOUV, GOUV with t-BuOH and (c) GOvis, GOvis with t-BuOH; XPS spectra of (d) GOfull, GOfull with quenching (t-BuOH) (e) GOUV, GOUV with t-BuOH and (f) GOvis, GOvis with t-BuOH. | ||
Compared with pristine GO (Fig. 1e), FTIR spectroscopy revealed that the O-functional groups changed substantially both without t-BuOH (i.e., when both UV light and ˙OH were active species) and when t-BuOH was present (i.e., in the absence of ˙OH) during UV irradiation, indicating that UV light and ˙OH both have effects on the transformation of GO. The XPS analysis (Fig. 4d–f) further illustrates that ˙OH radicals can hydroxylate GO. Changes in the morphology of GO under UV light with and without t-BuOH were compared by TEM, SEM and AFM (Fig. 5). The results clearly show that without ˙OH, GO could hardly be broken into small pieces. This result illustrates that ˙OH may cause the fragmentation of GO.
 | ||
| Fig. 5 TEM micrographs, SEM micrographs and AFM images of GO exposed to UV light (λ = 290–420 nm) (a–c) and exposed to UV light with t-BuOH (d–f). | ||
Based on the results in this study and in the literature,13,17 we believe that the physicochemical properties of GO are mainly modified by UV light. When GO was irradiated with UV (and full spectrum) light, electron–hole pairs (hvb+ and eaq−) were formed because the photon energy of the light exceeds the bandgap of the domains.17 Oxidation of O-functional groups on GO by hvb+ results in the loss of functional groups and the formation of nanoholes on the surface of GO. eaq− results in the formation of GO˙− and ROS such as O2˙− and ˙OH (Fig. S6†). Furthermore, ˙OH then attacks the surface of GO, leading to the addition of OH groups, as well as the oxidation of C–OH to CO and O–CO groups, opening the aromatic rings, fragmenting the GO nanosheets and forming lower molecular weight compounds, as determined from the MS spectra (Fig. S7†).
From these characterization results, we find that the O-containing functional groups on GO did not change significantly under visible light. However, an interesting observation (Fig. 2d and h) was that GO wrinkled significantly during visible light irradiation, and 1O2 formation was also higher than under UV light. Generally, wrinkling of nanostructures typically occurs under aggressive treatment, such as sonication, which may change the structure of the carbon nanosheets.53 In this study, light irradiation (both UV and visible light) may impact the structure of GO in a similar manner. The energy transfer during visible light irradiation may cause an unbalanced force on the basal plane of GO, leading to wrinkles in the nanomaterials.
In the natural environment, water chemistry factors (such as pH, ionic strength, and natural organic matter (NOM)) as well as turbidity may affect the transformation of GO under different light scenarios. For example, salts and NOM may adsorb onto the surface of GO and prohibit its reduction by light. Very turbid water will most likely absorb light and make it less accessible to the GO particles, which also weakens the reduction of GO. The roles of the physicochemical properties of water on GO transformation under different light conditions, thus, need to be studied further.
3.4 Influence of light on the environmental fate of GO
As we have shown, irradiation of GO led to the loss of O-functional groups on the surface of the nanomaterial, which may decrease the hydrophilicity of the nanomaterial in aqueous media. In addition, the morphology of GO was drastically changed, with much smaller sizes observed upon irradiation with full-spectrum solar and UV light and sheet wrinkling observed upon irradiation with visible light. All of these light-induced changes may affect the behavior of GO in the natural environment. We, therefore, examined the changes in the electrokinetic properties of GO, induced by different light sources, their effect on agglomeration in aqueous media, and the adsorption affinity of GO for common organic contaminants.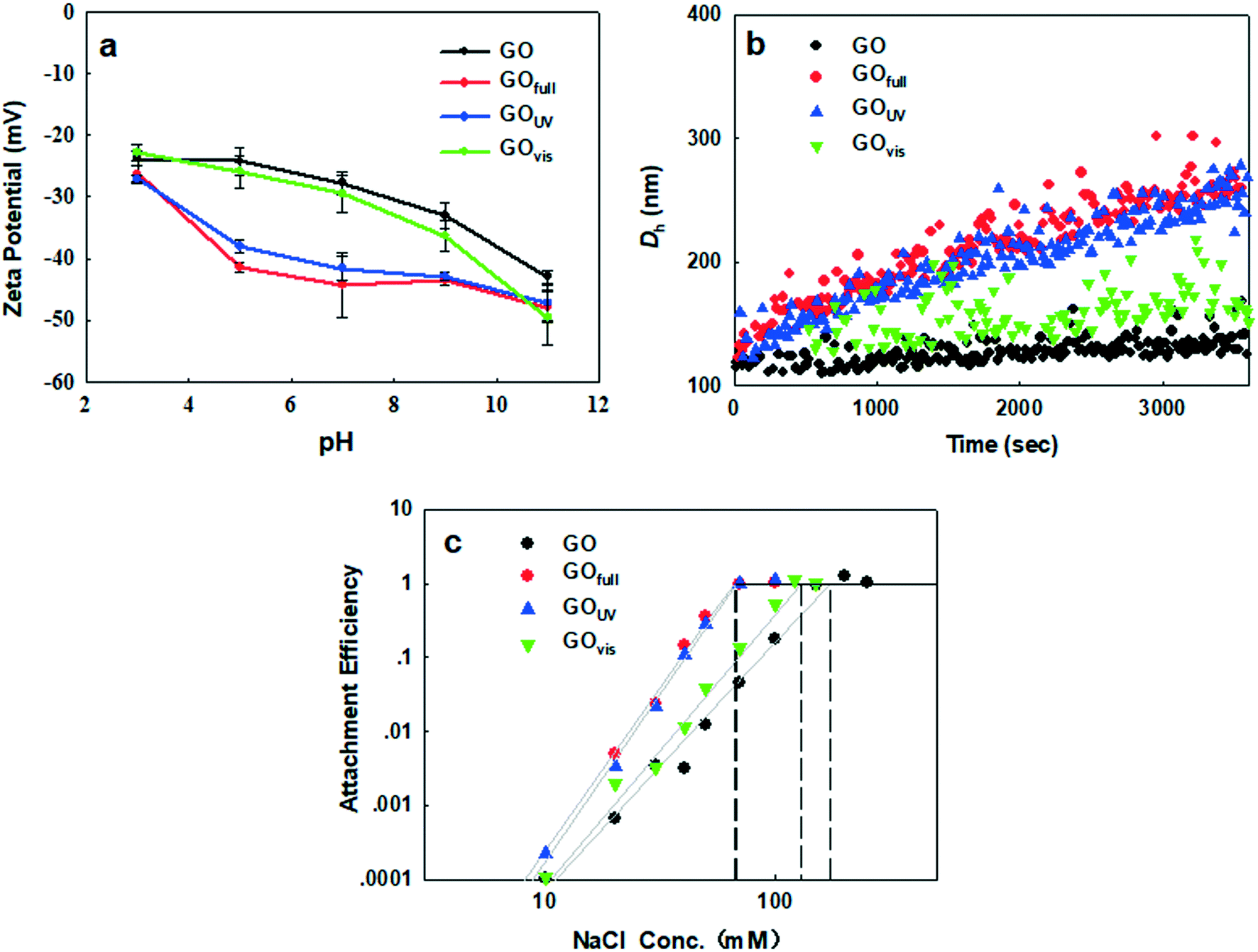 | ||
| Fig. 6 Influence of different irradiation types on the physicochemical properties of graphene oxide in water. (a) Zeta potential; (b) agglomeration kinetics. [Na+] = 30 mg L−1, pH = 7; (c) attachment efficiency as a function of NaCl concentration at pH 7. | ||
As the acidity of the aqueous medium increased, the ζ potential of GO decreased in magnitude, regardless of irradiation treatment, because of the protonation of the OH and COOH groups on GO. In contrast, as the basicity of the suspension media increased we observed an increased negativity due to the presence of negatively charged O− and COO− groups. While the negativity of GOfull and GOUV increased more rapidly (than that of pristine GO and GOvis) between pH 3 and pH 9, GO and GOvis demonstrated a more rapid increase in negativity above pH 9. All these trends show that light of different wavelengths has different impacts on the surface properties of GO in water, which will influence the colloidal behavior of the nanomaterials, as well as its surface interactions with other constituents (organic and inorganic) present in natural waters.
Since the particle size affects the agglomeration rate, we further determined and compared the CCC of the pristine and transformed GO in order to clearly determine how light-induced transformations influence the stability of GO in aqueous media. To achieve this, the changes in the attachment efficiency (α) as a function of the NaCl concentration were determined for each GO type. DLCA and RLCA regimes were observed for all four materials, i.e. pristine GO, GOvis, GOfull and GOUV (Fig. 6c). According to the Derjaguin–Landau–Verwey–Overbeek (DLVO) theory, increasing the ionic strength reduces the electrostatic energy barrier and deepens the secondary minimum well, which promotes agglomeration so that the attachment efficiency gradually increases.58 As shown in Fig. 6c, the observed CCC values were 171 mM NaCl for GO, 130 mM NaCl for GOvis, 67 mM NaCl for GOUV, and 66 mM NaCl for GOfull. This agrees very well with the trend observed in k, with GO becoming only less stable after visible light treatment but much more unstable when treated with UV and the full light spectrum. In addition, the observed trend in CCC correlates well with the degree of transformation of the GO materials (i.e. chemical reduction as indicated by the C/O ratios and size decrease as indicated by AFM) after their exposure to the different light sources. This strongly suggests that the colloidal stability of GO in natural waters is a function of its transformation in the natural environment.
Increased destabilization of GO after UV and full-spectrum solar irradiation could not have been predicted by the ζ potential, which, as reported earlier, increased in magnitude under these conditions. The observation of lower colloidal stability of GOfull and GOUV, despite having a higher negative charge on their surfaces compared to pristine GO and GOvis, suggests that factors other than electrostatic repulsion played important roles in the agglomeration kinetics of the nanomaterials. We hypothesize that a decrease in the particle size of GOfull and GOUV (as shown by electron microscopy and AFM in Fig. 2), a decrease in the van der Waals force, and an increase in the hydrophobic effect led to their increased agglomeration. A decrease in particle size changes the structure and surface energy characteristics of nanomaterials, which may impact the total potential energy of interaction.59 Meanwhile, van der Waals forces and hydrophobic effect may also have effects on the GO agglomeration.
Using the classical DLVO theory, He et al. (2008)59 showed that the interaction potential decreases as the particle size decreases, leading to increased agglomeration of smaller-sized nanomaterials. Also, smaller GO particles possess a higher fraction of their atoms on the edges, which may increase the density of active sites. A higher density of active sites and a larger specific surface area of smaller-sized GO can enhance the interaction with counterions in the surrounding media, leading to decreased colloidal stability. The van der Waals force between GOfull and GOUV samples increased due to the decrease in particle size. However, as the van der Waals force is weak, it probably has only slight effects on the interaction of the GO particles. On the other hand, the hydrophobic effect may be much more important. As O-functional groups decreased and the C/O ratio increased for GOfull and GOUV samples, the hydrophobic effect may be substantially increased, promoting the agglomeration of GO particles.
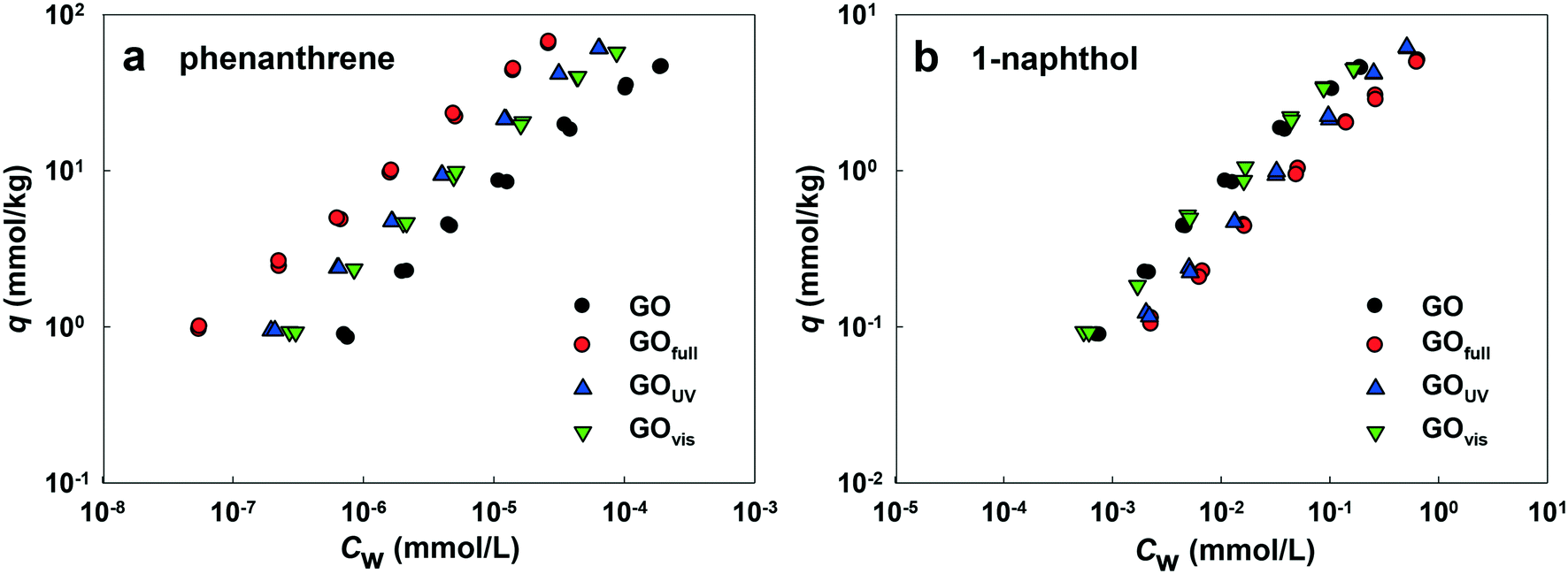 | ||
| Fig. 7 Isotherms of adsorption of phenanthrene (a) and 1-naphthol (b) onto the colloidal GO, GOfull, GOUV and GOvis suspensions; q (mmol kg−1) and CW (mmol L−1) are the equilibrium concentrations of an adsorbate on GO and in the solution, respectively. | ||
Fig. 7a shows the adsorption affinities of GO for phenanthrene. The results indicate that adsorption was enhanced with irradiation from all light sources. The adsorption isotherms of GOfull, GOUV and GOvis were substantially shifted compared with the isotherm obtained for pristine GO. This demonstrates that the adsorption of PAHs onto GO is mainly driven by hydrophobic effects and particle size.60,61 Thus, the reduction of the surface O-containing functional groups of GO by full-spectrum and UV light irradiation could affect the adsorption of phenanthrene due to increased surface hydrophobicity and decreased particle size (which may result in higher surface area). Surprisingly, the adsorption affinity of GOvis was also increased, even as much as GOUV. As no obvious chemical change was observed on the surface of GOvis, the increased adsorption of phenanthrene may be due to the wrinkles formed on the surface of GOvis, which created additional sites for phenanthrene attachment.
Fig. 7b shows that the adsorption affinity of GO for 1-naphthol was also enhanced after light irradiation, but the extent of adsorption enhancement was much less compared with phenanthrene. This is mainly because the logKOW value of 1-naphthol (2.85) is much less than that of phenanthrene (4.57), and the aqueous solubility of 1-naphthol (3.04 mmol L−1) is much greater than that of phenanthrene (6.31 × 10−3 mmol L−1).37 Hence, the mechanisms controlling the adsorption of 1-naphthol and phenanthrene onto GO are different. The results indicate that adsorption of 1-naphthol onto GO is strongly affected by hydrogen bonding between the OH group of 1-naphthol and the surface O-functionalities of GOs,62 as well as the π–π interactions. As such, the adsorption of 1-naphthol by GOUV and GOfull decreased because of the loss of OH groups upon irradiation.
4 Conclusions
Release of GO materials into surface water will facilitate some physicochemical transformations by light irradiation. As shown in this study, the O-containing functional groups of GO are decreased by UV irradiation (or the UV fraction of solar light). These chemical changes lead to physical variation in the nanosheets—mainly cleavage of the GO nanosheets. The ˙OH radicals, which are formed during UV irradiation, are also involved in the transformation of GO, hydroxylating the surface of GO, as well as breaking up the GO nanosheets into small pieces. Visible light (or the visible range of solar light) can hardly alter the chemical composition of GO but changes the appearance of GO and wrinkled the GO nanosheets. The transformation of GO by light may be affected by water chemistry. The chemical changes in GO, especially the loss of O-containing functional groups, is the main factor which affects the agglomeration of GO, while the changes in GO morphology and hydrophobicity strongly affect the adsorption of hydrophobic organic pollutants.Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
This research was supported by the National Basic Research Program of China (Grant 2014CB932001 and Grant 2015CB459000), the National Natural Science Foundation of China (Project No. 21677078), the 111 Program, Ministry of Education, China (Grant T2017002), and the Basic Scientific Research Program (Grant 7954).References
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, The chemistry of graphene oxide, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, New insights into the structure and reduction of graphite oxide, Nat. Chem., 2009, 1, 403–408 CrossRef CAS PubMed.
- D. G. Goodwin, Jr., A. S. Adeleye, L. Sung, K. T. Ho, R. M. Burgess and E. J. Petersen, Detection and quantification of graphene-family nanomaterials in the environment, Environ. Sci. Technol., 2018, 52, 4491–4513 CrossRef PubMed.
- I. Chowdhury, M. C. Duch, N. D. Mansukhani, M. C. Hersam and D. Bouchard, Colloidal properties and stability of graphene oxide nanomaterials in the aquatic environment, Environ. Sci. Technol., 2013, 47, 6288–6296 CrossRef CAS PubMed.
- Y. Li, J. Yang, Q. Zhao and Y. Li, Dispersing carbon-based nanomaterials in aqueous phase by graphene oxides, Langmuir, 2013, 29, 13527–13534 CrossRef CAS PubMed.
- A. S. Adeleye, J. R. Conway, K. Garner, Y. Huang, Y. Su and A. A. Keller, Engineered nanomaterials for water treatment and remediation: Costs, benefits, and applicability, Chem. Eng. J., 2016, 286, 640–662 CrossRef CAS.
- K. Krishnamoorthy, K. Jeyasubramanian, M. Premanathan, G. Subbiah, H. S. Shin and S. J. Kim, Graphene oxide nanopaint, Carbon, 2014, 72, 328–337 CrossRef CAS.
- R. Song, Y. Qin, S. Suh and A. A. Keller, Dynamic model for the stocks and release flows of engineered nanomaterials, Environ. Sci. Technol., 2017, 51, 12424–12433 CrossRef CAS PubMed.
- S. C. Ray, in Applications of Graphene and Graphene-Oxide Based Nanomaterials, William Andrew Publishing, Oxford, 2015, pp. 39–55, DOI:10.1016/B978-0-323-37521-4.00002-9.
- T. Du, Y. Wang, X. Yang, W. Wang, H. Guo, X. Xiong, R. Gao, X. Wuli, A. S. Adeleye and Y. Li, Mechanisms and kinetics study on the trihalomethanes formation with carbon nanoparticle precursors, Chemosphere, 2016, 154, 391–397 CrossRef CAS PubMed.
- T. Du, A. S. Adeleye, A. A. Keller, Z. Wu, W. Han, Y. Wang, C. Zhang and Y. Li, Photochlorination-induced transformation of graphene oxide: Mechanism and environmental fate, Water Res., 2017, 124, 372–380 CrossRef CAS PubMed.
- L. Duan, R. Hao, Z. Xu, X. He, A. S. Adeleye and Y. Li, Removal of graphene oxide nanomaterials from aqueous media via coagulation: Effects of water chemistry and natural organic matter, Chemosphere, 2017, 168, 1051–1057 CrossRef CAS PubMed.
- W.-C. Hou, W. M. Henderson, I. Chowdhury, D. G. Goodwin, Jr., X. Chang, S. Martin, D. H. Fairbrother, D. Bouchard and R. G. Zepp, The contribution of indirect photolysis to the degradation of graphene oxide in sunlight, Carbon, 2016, 110, 426–437 CrossRef CAS.
- Y. Li, N. Yang, T. Du, X. Wang and W. Chen, Transformation of graphene oxide by chlorination and chloramination: Implications for environmental transport and fate, Water Res., 2016, 103, 416–423 CrossRef CAS PubMed.
- F. Wang, F. Wang, G. Gao and W. Chen, Transformation of graphene oxide by ferrous iron: Environmental implications, Environ. Toxicol. Chem., 2015, 34, 1975–1982 CrossRef CAS PubMed.
- T. Xia, J. D. Fortner, D. Zhu, Z. Qi and W. Chen, Transport of sulfide-reduced graphene oxide in saturated quartz sand: Cation-dependent retention mechanisms, Environ. Sci. Technol., 2015, 49, 11468–11475 CrossRef CAS PubMed.
- W.-C. Hou, I. Chowdhury, D. G. Goodwin, Jr., W. M. Henderson, D. H. Fairbrother, D. Bouchard and R. G. Zepp, Photochemical transformation of graphene oxide in sunlight, Environ. Sci. Technol., 2015, 49, 3435–3443 CrossRef CAS PubMed.
- Y. Matsumoto, M. Koinuma, S. Y. Kim, Y. Watanabe, T. Taniguchi, K. Hatakeyama, H. Tateishi and S. Ida, Simple photoreduction of graphene oxide nanosheet under mild conditions, ACS Appl. Mater. Interfaces, 2010, 2, 3461–3466 CrossRef CAS PubMed.
- Y. Matsumoto, M. Koinuma, S. Ida, S. Hayami, T. Taniguchi, K. Hatakeyama, H. Tateishi, Y. Watanabe and S. Amano, Photoreaction of graphene oxide nanosheets in water, J. Phys. Chem. C, 2011, 115, 19280–19286 CrossRef CAS.
- M. Tedetti and R. Sempere, Penetration of ultraviolet radiation in the marine environment. A review, Photochem. Photobiol., 2006, 82, 389–397 CrossRef CAS PubMed.
- E. W. Helbling, K. S. Gao, R. J. Goncalves, H. Y. Wu and V. E. Villafane, Utilization of solar UV radiation by coastal phytoplankton assemblages off SE China when exposed to fast mixing, Mar. Ecol.: Prog. Ser., 2003, 259, 59–66 CrossRef CAS.
- H. Piazena, E. Perez-Rodrigues, D. P. Hader and F. Lopez-Figueroa, Penetration of solar radiation into the water column of the central subtropical Atlantic Ocean - optical properties and possible biological consequences, Deep Sea Res., Part II, 2002, 49, 3513–3528 CrossRef CAS.
- Q. Zhao, C. Shang, X. Zhang, G. Ding and X. Yang, Formation of halogenated organic byproducts during medium-pressure UV and chlorine coexposure of model compounds NOM and bromide, Water Res., 2011, 45, 6545–6554 CrossRef CAS PubMed.
- A. S. Adeleye, X. Wang, F. Wang, R. Hao, W. Song and Y. Li, Photoreactivity of graphene oxide in aqueous system: Reactive oxygen species formation and bisphenol A degradation, Chemosphere, 2018, 195, 344–350 CrossRef CAS PubMed.
- D. Zhang, S. Yan and W. Song, Photochemically induced formation of reactive oxygen species (ROS) from effluent organic matter, Environ. Sci. Technol., 2014, 48, 12645–12653 CrossRef CAS PubMed.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, High-throughput solution processing of large-scale graphene, Nat. Nanotechnol., 2009, 4, 25–29 CrossRef CAS PubMed.
- D. E. Latch, B. L. Stender, J. L. Packer, W. A. Arnold and K. McNeill, Photochemical fate of pharmaceuticals in the environment: Cimetidine and ranitidine, Environ. Sci. Technol., 2003, 37, 3342–3350 CrossRef CAS PubMed.
- M. Sahni and B. R. Locke, Quantification of hydroxyl radicals produced in aqueous phase pulsed electrical discharge reactors, Ind. Eng. Chem. Res., 2006, 45, 5819–5825 CrossRef CAS.
- J. D. Fortner, D.-I. L. Kim, A. M. Boyd, J. C. Falkner, S. Moran, V. L. Colvin, J. B. Hughes and J.-H. Kim, Reaction of Water-Stable C60 aggregates with ozone, Environ. Sci. Technol., 2007, 41, 7497–7502 CrossRef CAS PubMed.
- S. Kim and W. Choi, Kinetics and mechanisms of photocatalytic degradation of (CH3)nNH4-n+ (0 ≤ n ≤ 4) in TiO2 suspension: The role of OH radicals, Environ. Sci. Technol., 2002, 36, 2019–2025 CrossRef CAS PubMed.
- K. L. Chen and M. Elimelech, Influence of humic acid on the aggregation kinetics of fullerene (C60) nanoparticles in monovalent and divalent electrolyte solutions, J. Colloid Interface Sci., 2007, 309, 126–134 CrossRef CAS PubMed.
- A. S. Adeleye, J. R. Conway, T. Perez, P. Rutten and A. A. Keller, Influence of extracellular polymeric substances on the long-term fate, dissolution, and speciation of copper-based nanoparticles, Environ. Sci. Technol., 2014, 48, 12561–12568 CrossRef CAS PubMed.
- A. S. Adeleye and A. A. Keller, Interactions between algal extracellular polymeric substances and commercial TiO2 nanoparticles in aqueous media, Environ. Sci. Technol., 2016, 50, 12258–12265 CrossRef CAS PubMed.
- A. S. Adeleye and A. A. Keller, Long-term colloidal stability and metal leaching of single wall carbon nanotubes: Effect of temperature and extracellular polymeric substances, Water Res., 2014, 49, 236–250 CrossRef CAS PubMed.
- A. S. Adeleye, S. Pokhrel, L. Maedler and A. A. Keller, Influence of nanoparticle doping on the colloidal stability and toxicity of copper oxide nanoparticles in synthetic and natural waters, Water Res., 2018, 132, 12–22 CrossRef CAS PubMed.
- V. Runkana, P. Somasundaran and P. C. Kapur, Reaction-limited aggregation in presence of short-range structural forces, AIChE J., 2005, 51, 1233–1245 CrossRef CAS.
- F. Wang, F. Wang, D. Zhu and W. Chen, Effects of sulfide reduction on adsorption affinities of colloidal graphene oxide nanoparticles for phenanthrene and 1-naphthol, Environ. Pollut., 2015, 196, 371–378 CrossRef CAS PubMed.
- X. Li, W. Chen, C. Zhang, Y. Li, F. Wang and W. Chen, Enhanced dehydrochlorination of 1,1,2,2-tetrachloroethane by graphene-based nanomaterials, Environ. Pollut., 2016, 214, 341–348 CrossRef CAS PubMed.
- C. Nethravathi, T. Nisha, N. Ravishankar, C. Shivakumara and M. Rajamathi, Graphene-nanocrystalline metal sulphide composites produced by a one-pot reaction starting from graphite oxide, Carbon, 2009, 47, 2054–2059 CrossRef CAS.
- S. Park, K. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff, Graphene oxide papers modified by divalent ions - Enhancing mechanical properties via chemical cross-linking, ACS Nano, 2008, 2, 572–578 CrossRef CAS PubMed.
- C. Rodriguez-Gonzalez, A. L. Martinez-Hernandez, V. M. Castano, O. V. Kharissova, R. S. Ruoff and C. Velasco-Santos, Polysaccharide nanocomposites reinforced with graphene oxide and keratin-grafted graphene oxide, Ind. Eng. Chem. Res., 2012, 51, 3619–3629 CrossRef CAS.
- L. Guardia, S. Villar-Rodil, J. I. Paredes, R. Rozada, A. Martinez-Alonso and J. M. D. Tascon, UV light exposure of aqueous graphene oxide suspensions to promote their direct reduction, formation of graphene-metal nanoparticle hybrids and dye degradation, Carbon, 2012, 50, 1014–1024 CrossRef CAS.
- M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Perspectives on carbon nanotubes and graphene Raman spectroscopy, Nano Lett., 2010, 10, 751–758 CrossRef CAS PubMed.
- L. Szpyrkowicz, C. Juzzolino and S. N. Kaul, A comparative study on oxidation of disperse dyes by electrochemical process, ozone hypochlorite and Fenton reagent, Water Res., 2001, 35, 2129–2136 CrossRef CAS PubMed.
- K. D. Pickering and M. R. Wiesner, Fullerol-sensitized production of reactive oxygen species in aqueous solution, Environ. Sci. Technol., 2005, 39, 1359–1365 CrossRef CAS PubMed.
- F. Wilkinson, W. P. Helman and A. B. Ross, Quantum yields for the photosensitized formation of the lowest electronically excited singlet state of molecular oxygen in solution, J. Phys. Chem. Ref. Data, 1993, 22, 113–262 CrossRef CAS.
- V. A. Smirnov, A. A. Arbuzov, Y. M. Shul'ga, S. A. Baskakov, V. M. Martynenko, V. E. Muradyan and E. I. Kresova, Photoreduction of graphite oxide, High Energy Chem., 2011, 45, 57–61 CrossRef CAS.
- H. Xu, W. J. Cooper, J. Jung and W. Song, Photosensitized degradation of amoxicillin in natural organic matter isolate solutions, Water Res., 2011, 45, 632–638 CrossRef CAS PubMed.
- Y. Zhang, L. Guo, H. Xia, Q. Chen, J. Feng and H. Sun, Photoreduction of graphene oxides: Methods, properties, and applications, Adv. Opt. Mater., 2014, 2, 10–28 CrossRef.
- M. O. Buffle, S. Galli and U. von Gunten, Enhanced bromate control during ozonation: The chlorine-ammonia process, Environ. Sci. Technol., 2004, 38, 5187–5195 CrossRef CAS PubMed.
- J. Yao, Z. Huang and S. J. Masten, The ozonation of pyrene: Pathway and product identification, Water Res., 1998, 32, 3001–3012 CrossRef CAS.
- J. Staehelin and J. Hoigne, Decomposition of ozone in water in the presence of organic solutes acting as promoters and inhibitors of radical chain reactions, Environ. Sci. Technol., 1985, 19, 1206–1213 CrossRef CAS PubMed.
- M. V. Savoskin, V. N. Mochalin, A. P. Yaroshenko, N. I. Lazareva, T. E. Konstantinova, I. V. Barsukov and L. G. Prokofiev, Carbon nanoscrolls produced from acceptor-type graphite intercalation compounds, Carbon, 2007, 45, 2797–2800 CrossRef CAS.
- D. Li, M. B. Mueller, S. Gilje, R. B. Kaner and G. G. Wallace, Processable aqueous dispersions of graphene nanosheets, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- T. Szabo, O. Berkesi, P. Forgo, K. Josepovits, Y. Sanakis, D. Petridis and I. Dekany, Evolution of surface functional groups in a series of progressively oxidized graphite oxides, Chem. Mater., 2006, 18, 2740–2749 CrossRef CAS.
- B. Konkena and S. Vasudevan, Engineering a water-dispersible, conducting photoreduced graphene oxide, J. Phys. Chem. C, 2015, 119, 6356–6362 CrossRef CAS.
- B. Konkena and S. Vasudevan, Understanding aqueous dispersibility of graphene oxide and reduced graphene oxide through pKa measurements, J. Phys. Chem. Lett., 2012, 3, 867–872 CrossRef CAS PubMed.
- M. Elimelech, J. Gregory, X. Jia and R. A. Williams, in Particle Deposition and Aggregation, ed. M. Elimelech, J. Gregory, X. Jia and R. A. Williams, Butterworth-Heinemann, 1995, pp. 310–343, DOI:10.1016/B978-0-7506-0743-8.50015-7.
- Y. T. He, J. Wan and T. Tokunaga, Kinetic stability of hematite nanoparticles: The effect of particle sizes, J. Nanopart. Res., 2008, 10, 321–332 CrossRef CAS.
- J. Wang, Z. Chen and B. Chen, Adsorption of polycyclic aromatic hydrocarbons by graphene and graphene oxide nanosheets, Environ. Sci. Technol., 2014, 48, 4817–4825 CrossRef CAS PubMed.
- J. Xu, L. Wang and Y. Zhu, Decontamination of bisphenol A from aqueous solution by graphene adsorption, Langmuir, 2012, 28, 8418–8425 CrossRef CAS PubMed.
- Z. Pei, L. Li, L. Sun, S. Zhang, X. Shan, S. Yang and B. Wen, Adsorption characteristics of 1,2,4-trichlorobenzene, 2,4,6-trichlorophenol, 2-naphthol and naphthalene on graphene and graphene oxide, Carbon, 2013, 51, 156–163 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8en00593a |
| ‡ Current address: Department of Civil and Environmental Engineering, University of California, Irvine, CA 92697, United States. |
| This journal is © The Royal Society of Chemistry 2018 |