
Graphene analogues in aquatic environments and porous media: dispersion, aggregation, deposition and transformation
Xuemei
Ren
 ab,
Jie
Li
c,
Changlun
Chen
*abd,
Yang
Gao
a,
Diyun
Chen
e,
Mianhua
Su
e,
Ahmed
Alsaedi
d and
Tasawar
Hayat
d
ab,
Jie
Li
c,
Changlun
Chen
*abd,
Yang
Gao
a,
Diyun
Chen
e,
Mianhua
Su
e,
Ahmed
Alsaedi
d and
Tasawar
Hayat
d
aCAS Key Laboratory of Photovoltaic and Energy Conservation Materials, Institute of Plasma Physics, Chinese Academy of Sciences, P.O. Box 1126, Hefei 230031, P.R. China. E-mail: clchen@ipp.ac.cn; Fax: +86 551 65591310; Tel: +86 551 65592788
bCollaborative Innovation Center of Radiation Medicine of Jiangsu Higher Education Institutions, Soochow University, Suzhou 215123, P.R. China
cSchool of Resources and Environment, Anhui Agricultural University, Hefei 230036, P.R. China
dNAAM Research Group, Faculty of Science, King Abdulaziz University, Jeddah 21589, Saudi Arabia
eGuangdong Province Key Laboratory of Radioactive Contamination Control and Resources, Guangzhou University, Guangzhou 510006, P.R. China
First published on 19th April 2018
Abstract
The potential extensive application of graphene analogues (GAs), such as graphene, graphene oxide and reduced graphene oxide, in various fields results in the possibility of their release into the natural environment with negative impacts on humans and the ecosystem. The aggregation and transformation of GAs in aqueous solutions as well as the deposition of GAs on model environmental surfaces and in porous media control their fate, transport, and bio-toxicity in aquatic environments, aquatic-terrestrial transition zones and subsurface environments. The change in solution chemistry, physicochemical properties of GAs and interaction of GAs with water or a solid surface will alter the aggregation, deposition and transformation of GAs. The present review aims to provide a comprehensive overview of current knowledge concerning the aggregation, deposition and transformation of GAs with particular attention paid to the key factors governing the behavior of GAs in natural environments, which is a critical component of their life-cycle analysis, risk assessment, and waste management. We expect that this review can provide useful insights into some current knowledge gaps and can also reveal clues about needed future developments.
 Xuemei Ren | Xuemei Ren is an associate Professor at the Institute of Plasma Physics, Chinese Academy of Sciences. She received her Ph.D. (2013) from Hefei Institutes of Physical Sciences, Chinese Academy of Sciences. After that, she became a research assistant at the Institute of Plasma Physics. Currently, her research interests focus on the fate and toxicity of nanomaterials before and after they are used as contaminant carriers. She, as the first author and the first corresponding author, has published more than 20 peer-reviewed journal publications. |
 Jie Li | Jie Li is an associate professor at the School of Resources and Environment, Anhui Agricultural University. She received her PhD in 2017 from the University of Science and Technology of China under the supervision of Prof. Xiangke Wang. During her Ph.D., she joined Prof. Zhiyong Tang's research group at the National Center for Nanoscience and Technology as a joint trained student for one and a half years, working on the synthesis of nanomaterials and their application in the fields of toxic metal ion decontamination and nuclear waste management. |
 Changlun Chen | Changlun Chen is an associate Professor at the Institute of Plasma Physics, Chinese Academy of Sciences. He received his Ph.D. (2008) from Hefei Institutes of Physical Sciences, Chinese Academy of Sciences. In 2008–2010, he worked in Shizuoka University, Japan, and then returned to the institute. Currently, his research interests focus on nanomaterial preparation and functionalization, application of nanostructured materials and nanotechnology for environmental pollution treatment. He has published more than 120 peer-reviewed journal publications with >9600 citations. http://www.researcherid.com/rid/H-9177-2012 |
Environmental significanceGraphene analogues (GAs) have a negative impact on the environment and ecology. The fate, transport, and bioavailability of GAs are determined by their aggregation, deposition and transformation. The present review aims to provide a comprehensive overview of current knowledge concerning the aggregation, deposition and transformation of GAs with particular attention paid to the key factors governing the behavior of GAs in the environment. This is valuable for the life-cycle analysis, risk assessment, and waste management of GAs. |
Introduction
Graphene, one layer of sp2-hybridized carbon atoms arranged in a two-dimensional honeycomb lattice of six-membered rings, can be considered as the mother of carbonaceous materials. It can be wrapped up into zero-dimensional fullerenes, rolled into one-dimensional carbon nanotubes or stacked into three-dimensional graphite. Graphene oxide (GO), the oxidized form of graphene synthesized by oxidative exfoliation of graphite, can serve as a substrate for mass fabrication and manipulation of graphene and graphene-based nanomaterials. Reduced graphene oxide (rGO) is produced by thermal annealing or chemical treatment to eliminate oxygen-containing functional groups on GO. These three graphene analogues (GAs) exhibit unique structure and remarkable physicochemical properties, resulting in a wide range of applications in various industries and customer products.1–4 With their large production and widespread use, GAs have the potential to be released into the environment incidentally or intentionally during their whole life-cycle including manufacture, transport, use and eventual disposal of GAs and GA-containing products. Recent studies have highlighted that GAs are toxic to living organisms. For example, graphene has shown toxic effects on algal cells,5 bacteria,6 mammalian fibroblasts7 and Daphnia magna.8 Both GO and rGO are toxic to human umbilical vein endothelial cells9 and bacteria.10 The introduction of GO results in severe and persistent lung injuries.11 Therefore, GAs may pose potential environmental and human health risks if they are released into soil and groundwater systems.The colloidal stability of GAs plays an important role in determining their use in industrial applications, their environmental fate and transport, their risk assessment, their bioavailability, their reactive oxygen species production, their toxicity to live beings, their removal during wastewater treatment, and their adsorption capacity for other contaminants (e.g., organic pollutants and heavy metal ions). The colloidal properties of GAs depend on their physicochemical properties within a given aqueous medium and are ultimately reflected in their aggregation and deposition behaviors. Therefore, a comprehensive understanding of the aggregation and deposition behaviors of GAs is important not only for promoting their industrial applications but also for more accurately evaluating their environmental fate and mobility and consequently assessing their environmental impact and risks.
Several reviews have summarized the recent developments in graphene-based materials.3,12,13 Zhao et al.12 provided a nice overview of four aspects of graphene-family nanomaterials in aquatic environments including adsorption, dispersion, toxicity and transformation. A recent review by He et al.13 summarized available studies occurring before 2017 on the behavior of graphene nanomaterials under various aquatic environmental factors, their transport in soil and porous media, and their effects on microbial communities. This present thorough review aims to provide more detailed information on the fate of GAs in three kinds of environments: aquatic environments, aquatic-terrestrial transition zones, and subsurface environments, in which the latest studies occurring in 2017 on this issue are included. In aquatic environments, GAs and aqueous media components such as natural organic matter (NOM) and inorganic ions exist simultaneously. Therefore, the colloidal behavior of GAs will be changed by the aqueous media components and the environmental factors. In aquatic-terrestrial transition zones, natural colloidal particles are ubiquitous, and the interactions of GAs with these natural colloidal particles are likely to occur besides the interactions of GAs with themselves and the interaction of GAs with aqueous media components, which play an important role in the removal of GAs from aquatic environments. When they are transported from surface environments to subsurface environments, the interaction of GAs with the porous media will affect their capacity to be released into subsurface environments. The corresponding phenomena, factors, and potential mechanisms involved in the fate and transport processes are also summarized thoroughly, mainly in order to provide a theoretical basis and technical guidance for accurate assessment of the fate and toxicity of GAs in diverse environments. Additionally, this review is expected to identify key knowledge gaps and offer future perspectives. Fig. 1 presents the different processes controlling the fate of GAs in aquatic environments, aquatic-terrestrial transition zones, and subsurface environments. Surface transformation (process 1 in Fig. 1) only occurs under special conditions. Homoaggregation is the aggregation process of GAs with each other (process 2 in Fig. 1). Heteroaggregation is the aggregation of GAs with natural colloids or other nanoparticles (process 3 in Fig. 1). The deposition refers to the attachment of GAs to a pore wall/a much larger colloidal particle by Brownian diffusion or direct interception and/or by gravitational sedimentation (process 4 in Fig. 1). The homoaggregation and heteroaggregation of GAs are more common and control the fate of GAs in the whole environmental zone. The deposition of GAs contributes to the fate of GAs in aquatic-terrestrial transition zones and subsurface environments. The transport of GAs is only found in subsurface environments.
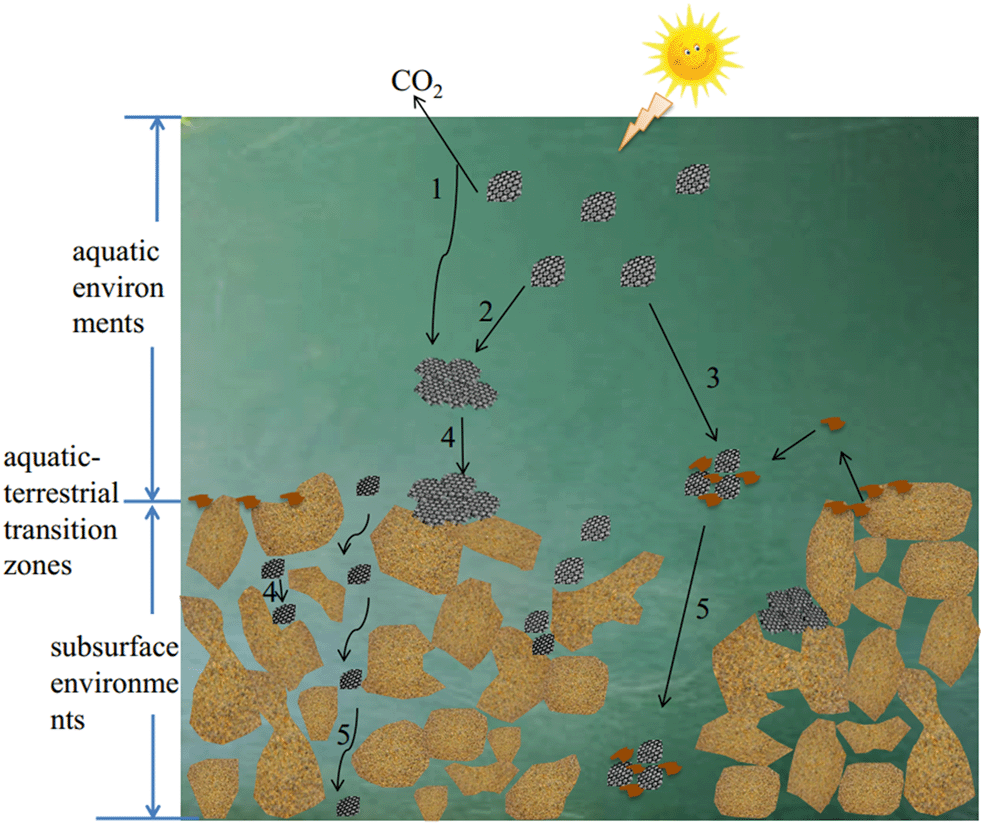 | ||
| Fig. 1 Schematic overview of the main processes controlling the fate of graphene analogues (GAs). 1. Transformation; 2. homoaggregation; 3. heteroaggregation; 4. deposition; 5. transport. | ||
Material properties of GAs relevant to their colloidal behavior
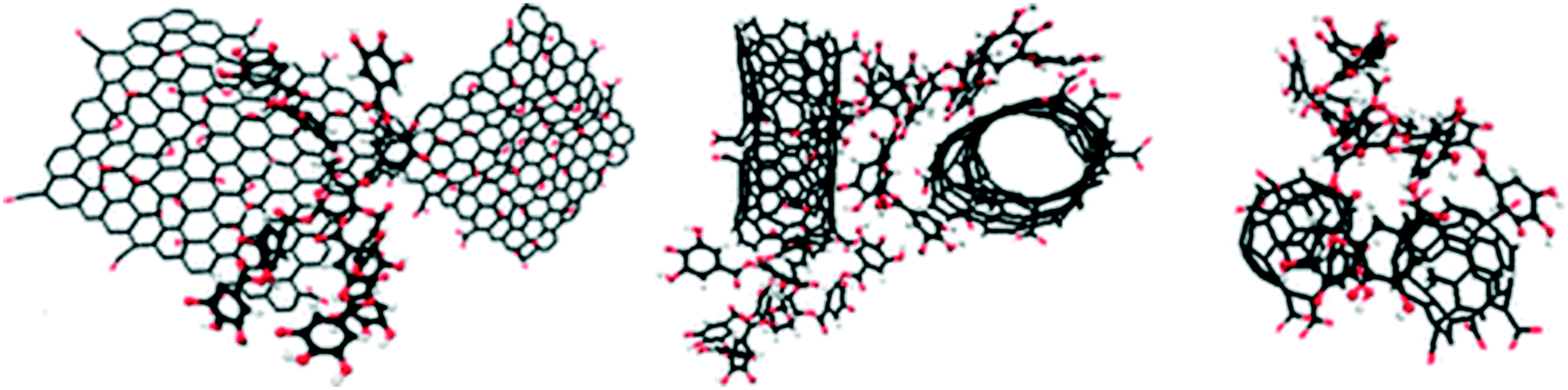
The different physicochemical properties (e.g., morphology, size, specific surface area, functional groups, surface charge, polarity, and so on) of GAs result in different interactions of graphene substrates with themselves or with guest media (e.g., aqueous media or porous media) and consequently variation in aggregation and deposition behaviors. Fig. 2 shows the most important GAs and some structural features relevant for their colloidal behavior. The section below summarizes the material properties of GAs relevant to their colloidal behavior in detail.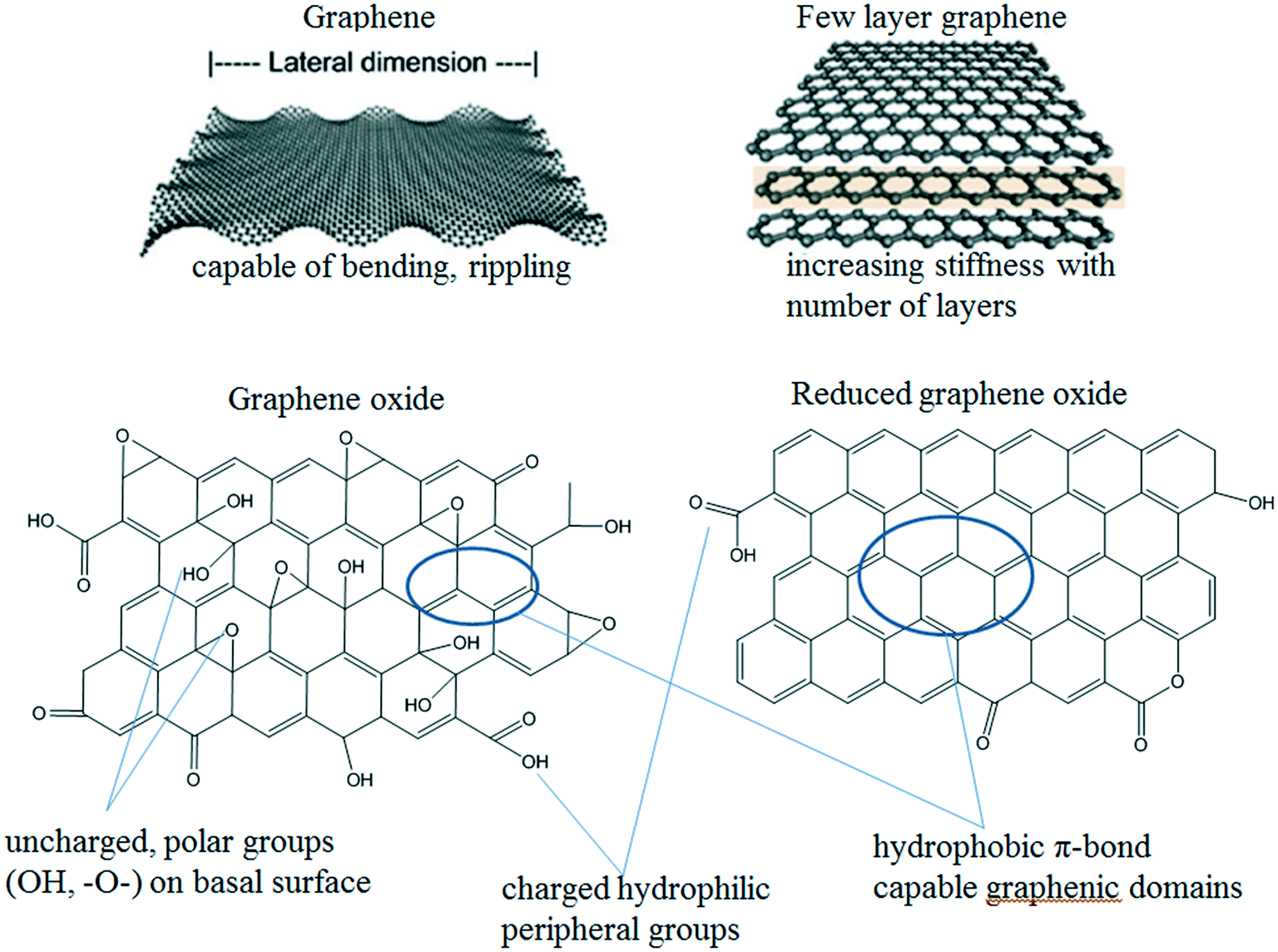 | ||
| Fig. 2 Examples of GAs and selected properties relevant to their colloidal behavior.12,14 Reprinted with permission from ref. 12. Copyright 2014, American Chemical Society. Reprinted with permission from ref. 14. Copyright 2012, American Chemical Society. | ||
Pristine graphene is composed of C and H atoms. An ideal monolayer graphene consists of a one-atom thick sheet of sp2-hybridized carbon atoms arrayed in a honeycomb pattern. In this extreme case, every carbon atom lies on the surface and is exposed to the surrounding medium on two sides, giving a theoretical maximum surface area as high as 2600 m2 g−1 and a theoretical thickness of 0.34 nm.14,15 The reported thickness of monolayer graphene is usually higher than 0.34 nm due to the existence of functional groups across the graphene surface.16 Corrugation and scrolling are part of the intrinsic nature of monolayer graphene, since the 2D membrane structure becomes thermodynamically stable via bending.17 The increased thickness will enhance the stiffness and rigidity of graphene. The large delocalized π-electron system of graphene sheets results in the formation of multilayers via π–π stacking interaction between graphene sheets. Therefore, graphene used in numerous reports usually contains several layers. Graphene exhibits hydrophobicity and easily forms agglomerates irreversibly or even restacks to form graphite via van der Waals interactions in aqueous solutions, so it is difficult to disperse graphene in water. It is suggested that the original size and crystallographic face/lattice of graphene that control the packing also play a role in its aggregation.18 The initial aggregation rate of graphene is fast. However, it has been shown recently that graphene sheets obtained from chemical reduction of GO can readily form stable aqueous colloids via electrostatic stabilization over a restricted range of pH values (e.g., above a pH value of 6.1).19
GO consists of C, O and H atoms. It is commonly synthesized by modified Hummers' methods and can be considered as a precursor for graphene/rGO synthesis by either chemical or thermal reduction processes. The obtained GO is presented as irregularly shaped flakes composed of one or more layers of graphene sheets with abundant oxygen-containing functional groups. Regardless of the preparation method, all the GO samples have the same set of oxygen-containing functional groups. A high concentration of basal plane epoxides is a remarkable feature of GO. However, the different synthetic protocols and the extent of the reaction result in different C/O ratios. The C/O ratios of GO prepared by modified Hummers' methods are usually in the range of 2–3.20 As an indication of the oxidation degree, C/O ratios are negatively correlated with the values of the critical coagulation concentration (CCC, determined from the intersection of extrapolated lines through the diffusion and reaction limited regimes, is a threshold concentration of the electrolyte necessary to cause the rapid coagulation of GAs) of GAs.21,22 At the highest oxidation level, the GO's π-systems are heavily interrupted, giving a stable bright yellow solution.20 The oxidization level also affects the thickness of GO. The increase in the thickness of GO is predicted to have a profound effect on its stability.23 GO shows characteristic ultraviolet (UV)-vis absorbance peaks at 230 nm (λmax) due to the π–π* transition and at 305 nm due to the n–π* transition. The OH-rich GO red-shifts the π–π* transition to 250 nm and decreases the n–π* transition strength; meanwhile, the COOH-rich GO blue-shifts the π–π* transition to 200 nm and increases the n–π* transition strength.24 As the λmax is in direct proportion to the ratio of sp2-hybridized to sp3-hybridized carbon in the GO sheets,25 the oxidation and reduction of GO are accompanied by the blue shift and red shift of λmax, respectively. A modest decrease in the size of the π conjugated domains also causes the blue shift of λmax (230 nm) to a shorter wavelength (225 nm).26 GO contains a wide range of hydrophilic oxygen-containing functional groups such as carboxyl, carbonyl, hydroxyl, and phenol that cover a range of acid dissociation coefficients (pKa). It was reported that the pKa value of carboxylic and phenolic hydroxyl groups on GO sheets is 6.6 and 9.8, respectively. The presence of phenolic hydroxyl groups in proximity to the carboxylic groups in GO lowers the pKa value of the carboxylic group to 4.3 by intramolecular hydrogen bonding, which allows GO to form stable aqueous dispersions with long term stability over a wide range of pH values.27 The different ionization degrees of the acidic oxygen-containing functional groups on the GO surface affect the stability of GO in water, which is pH dependent. It is reported that the isoelectric point of GO is lower than pH 2.28 In an aqueous solution with the same pH value, smaller GO sheets should have higher solubility compared to larger ones, since the decrease in the lateral dimension of GO results in the increase of the edge-to-area ratio of a GO sheet and a higher density of ionized carboxylic groups.26 GO is an amphiphilic material with a hydrophobic basal plane and hydrophilic edges. On the one hand, the hydrophobic basal plane has a large intact sp2-hybridized carbon backbone structure, allowing GO to maintain the potential of strong π–π interactions between the conjugated sp2 network structures of GO nanosheets.29 On the other hand, electrostatic repulsion between the negatively charged ionized edges associated with carboxylate groups makes GO soluble in water.
rGO is the product of treating GO with high temperature, UV irradiation or reducing chemical agents. The reduction of GO removes most of the oxygen-containing functional groups and restores the majority of the conjugated graphene networks, altering a number of GO properties such as hydrophobicity, holes/defects in the carbon lattice, surface charge and water dispersibility. There is only one type of acidic group, namely the carboxylic group with pKa = 7.9, on rGO sheets. In some cases, the reduction results in the transformation of epoxy groups into hydroxyl groups.21,30 The removal of oxygen-containing functional groups decreases the repulsive forces between the graphene sheets and the restoration of the conjugated graphene networks strengthens the attractive ones, decreasing the water dispersibility of rGO. Photo-, chemical-, and thermal-reduction are accompanied by a red shift of λmax (230 nm) to a longer wavelength (∼270 nm). Upon reduction, a brown to black color change of GO aqueous dispersions can be observed. So far, the largest C/O ratio of rGO reported is >246.31 As the reduction depth increases, the rGO sheets become more aromatic and more prone to interparticle stacking due to the strong van der Waals forces among rGO sheets, facilitating the aggregation of rGO sheets. Thus, plenty of rGO sheets precipitate after the rGO dispersion is left to stand for 2 h.10 Taken together, the origin of the instability of rGO is attributed to the following reasons: higher hydrophobicity, lower water solubility, π–π stacking of rGO sheets, the increase in attractive forces (van der Waals forces) and the decrease in repulsive forces (electrostatic repulsion).23,32,33
In a word, GAs have a similar structure and all of them include graphene domains, defects, and oxygen-containing functional groups. Considering the potential acid/base reactions of functional groups on the GA surface, GAs obtain a negative surface charge via deprotonation of carboxylic, enolic, and phenolic groups (i.e., –C–COOH + OH− ↔ –COO− + H2O, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–OH + OH− ↔ –CC–O− + H2O).34 Meanwhile, via proton complexation of the π-electron system of graphene planes (Cπ + H2O ↔ CπH+ + OH−) and various Brønsted basic oxygen species (ethers, carbonyl groups), GAs obtain a positive surface charge. On both GO and rGO sheets, the ionization of the carboxylic groups is primarily responsible for the build-up of the surface charge, but on GO sheets, the presence of phenolic and hydroxyl groups in close proximity to the carboxylic groups lowers the pKa value by stabilizing the carboxylate anion, resulting in superior water dispersibility.27,35 GO sheets form stable dispersions at pH > 4, while rGO sheets develop a sufficient negative charge to form stable dispersions only at pH > 8.27 Gudarzi23 stated that the sulphate groups in GO also contribute to the negative surface charge of GO at low pH. Dimiev et al.36 stated that the number of carboxylic acid moieties situated on the edges of the GO flake is small and cannot account for the acidic properties of GO. They deemed that the generation of protons during the reaction of GO with water is the main factor contributing to GO acidity. Table 1 attempts to highlight the main physicochemical characteristics of GAs relevant to their colloidal behaviour, offering a qualitative comparison of their differences.
C–OH + OH− ↔ –CC–O− + H2O).34 Meanwhile, via proton complexation of the π-electron system of graphene planes (Cπ + H2O ↔ CπH+ + OH−) and various Brønsted basic oxygen species (ethers, carbonyl groups), GAs obtain a positive surface charge. On both GO and rGO sheets, the ionization of the carboxylic groups is primarily responsible for the build-up of the surface charge, but on GO sheets, the presence of phenolic and hydroxyl groups in close proximity to the carboxylic groups lowers the pKa value by stabilizing the carboxylate anion, resulting in superior water dispersibility.27,35 GO sheets form stable dispersions at pH > 4, while rGO sheets develop a sufficient negative charge to form stable dispersions only at pH > 8.27 Gudarzi23 stated that the sulphate groups in GO also contribute to the negative surface charge of GO at low pH. Dimiev et al.36 stated that the number of carboxylic acid moieties situated on the edges of the GO flake is small and cannot account for the acidic properties of GO. They deemed that the generation of protons during the reaction of GO with water is the main factor contributing to GO acidity. Table 1 attempts to highlight the main physicochemical characteristics of GAs relevant to their colloidal behaviour, offering a qualitative comparison of their differences.
| Physicochemical characteristics | Graphene | rGO | GO |
|---|---|---|---|
| Shape (similar) | Flake-like stacks of 2–10 graphene layers14 | Several layers stacked on top of one another like sheets of paper, with silky, wrinkle and flower-like curling morphology37 | Multi-layered, wavy, folded flakes37 |
| Surface area (different) | 400–700 m2 g−1 (ref. 38) | 466 m2 g−1 for hydrazine mediated rGO32 | 130.8 m2 g−1 (ref. 39) |
| Lateral sizes (similar) | 60–590 nm (ref. 40) | 0.5–3 μm (ref. 41) | 100–5000 nm (ref. 42) |
| Relationship with colloidal behavior: few layer graphene (FLG) nanoparticles with smaller lateral sizes are shown to agglomerate more slowly than larger FLG;40 the smaller particle size intensifies the hydrophilicity43 | |||
| Color (different) | Dark | Dark-brown44 | Slight-brown45 |
| Thickness (similar) | 1.05–4.05 nm (ref. 46) | ∼4.23 nm for bacterially reduced graphene oxide47 | 0.3–1.4 nm (ref. 28) |
| Relationship with colloidal behavior: the increase in the thickness of GO is predicted to have a profound effect on its stability23 | |||
C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O ratio (different) :O ratio (different) |
89:6 (ref. 46) |
8–246 (ref. 48) | 2–4 (ref. 48) |
| Relationship with colloidal behavior: as an indication of the oxidation degree, C/O ratios are negatively correlated with CCC values21 | |||
| UV characteristic peak (different) | No obvious UV-vis absorption region46 | 260–270 nm depending on the reduction extent49 | A maximum absorption peak at 232 nm which was attributed to the π–π* transition of the aromatic CC bonds and a weak shoulder at 300 nm due to the n–π* transition of CO bonds45 |
| Relationship with colloidal behavior: the absorption peak of the GO dispersion at 231 nm gradually redshifts to 270 nm and the absorption in the whole spectral region (231 nm) increases with reaction time, suggesting that the electronic conjugation within the graphene sheets is restored upon hydrazine reduction19 | |||
| Zeta potential (ZP) pH at the point of zero charge (pHPZC) (different) | pHPZC = 5.5;50 a net negative electrophoretic mobility (EPM) at pH 7.0 (ref. 51) | ZP of rGO is below −30 mV when the pH is greater than 6.1;19 the rGO sheets develop a sufficient negative charge to form stable dispersions above a pH value of 8 (ref. 27) | GO remains negatively charged over the pH range from 2 to 10, and the isoelectric point of GO is <pH 2 (ref. 28) |
| Relationship with colloidal behavior: ZP values higher than −30 mV are usually considered as the threshold for colloidal stability in water21 | |||
|
I
D:IG |
1.08 (ref. 5) | ∼1.1–1.5 for chemically or thermally reduced GO52 | 1.8 (ref. 37) |
| d-Spacing (different) | 0.39 nm (ref. 5) | 0.356 nm (ref. 52) | More than 0.625 nm (ref. 53) |
| 0.344 nm (ref. 46) | |||
| Relationship with colloidal behavior: the introduction of water molecules and modification of functional groups lead to expansion of the d-spacing of the structure5 | |||
| Colloidal behaviour (different) | Agglomerated46 | Aggregated54 | Stable28 |
| Acidic groups (different) | Hydroxyl groups51 | pKa of oxygenated groups in rGO is around 8;23 covalently bonded sulfate groups are perhaps responsible for the stability of rGO at low pH values23 | Carboxylic (pKa, 4.3), phenolic hydroxyl (pKa, 9.8) and sulfate groups23 |
| Relationship with colloidal behavior: the deprotonation of carboxyl groups with low acid dissociation constants is the main contributor to ZP and thus material aqueous stability21 | |||
Aggregation behavior of GO in aquatic environments
The surface of GO sheets is functionalized with a set of oxygen containing functional groups, which allows GO sheets to form relatively stable suspensions. Upon release into aquatic environments, it is inevitable for GO sheets to come into contact with natural system constituents such as inorganic ions, surface active molecules, NOM, colloidal particles and bio-colloids, which can affect the colloidal behavior of GO and can further dominate the fate of GO in aquatic environments. It has been reported that pH, divalent cations, and NOM can play complex roles in the fate of GO and rGO (Fig. 3).55 Although GO is not a spherical colloid, the aggregation of GO follows colloidal theory including Derjaguin–Landau–Verwey–Overbeek (DLVO) theory and the Schulze–Hardy rule.28,56,57 This section mainly covers the colloidal behavior of GO as a function of pH, ionic strength (IS), salt type, NOM, natural colloidal particles, and toxic heavy metal ions combined with natural colloidal particles under various aquatic environmental conditions.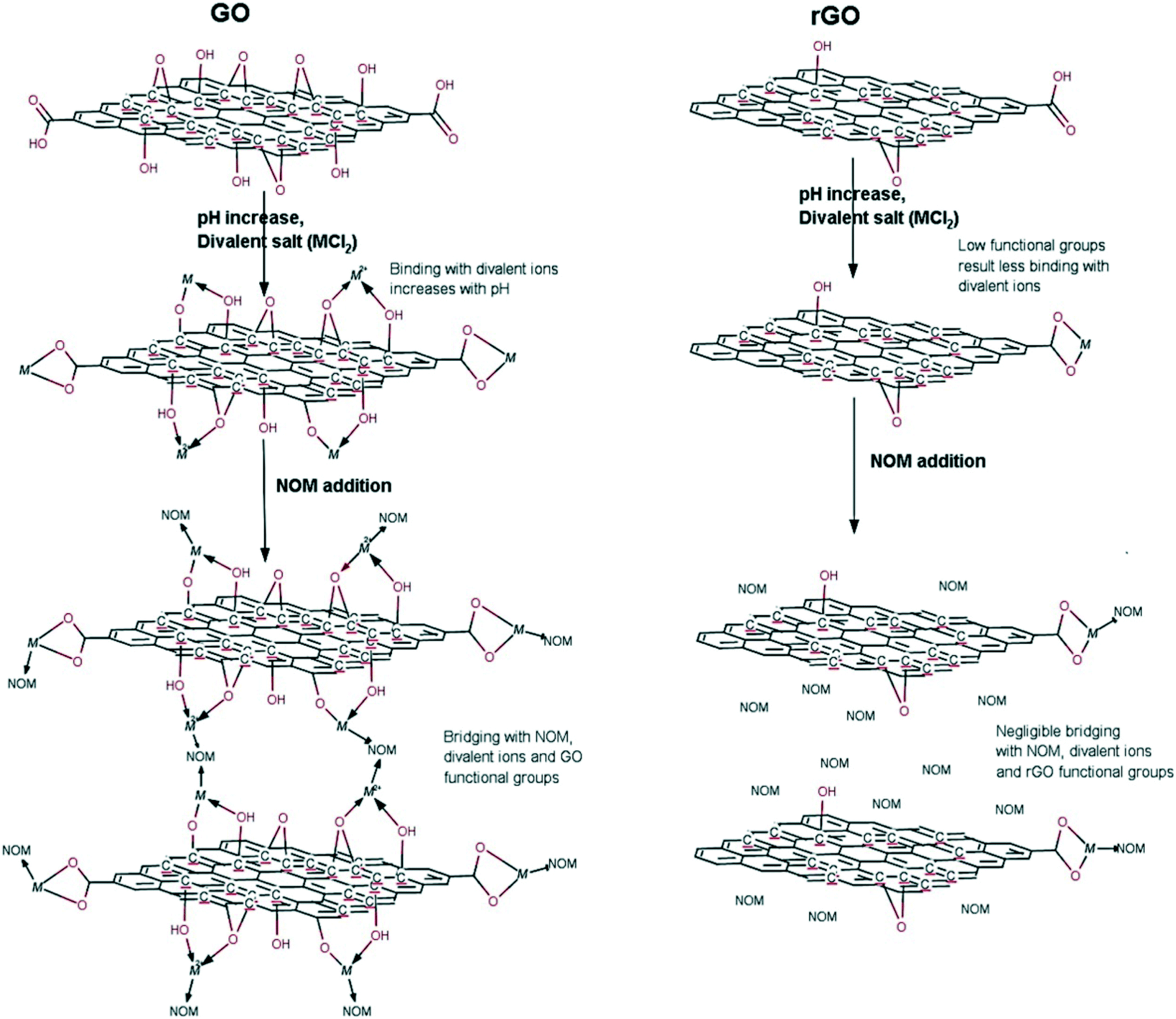 | ||
| Fig. 3 Interaction mechanisms of pH, divalent cations (M: Ca2+, Mg2+) and natural organic matter (NOM) with GO and rGO.55 Reprinted with permission from ref. 55. Copyright 2015, American Chemical Society. | ||
Effect of pH
The solution pH value is one of the most important factors controlling the stability of GO in water. The pH-dependent reactions, phenomena, and application are listed in Table 2. pH values affect the physicochemical properties of GO, especially the degree of ionization of the oxygen-containing functional groups on the surfaces of GO, so the stability of GO in water is pH dependent. The GO suspension exhibits very different appearances at extremely acidic pH values (pH 1–3), intermediate pH values (pH 3–12), and extremely alkaline pH values (pH 12–14). GO precipitates at pH < 3 and is stably suspended in the range of pH 3–10. Shih and coworkers58 observed that the GO suspension was visually homogeneous with a dark-brown color at pH 14. Taniguchi and coworkers59 observed that the color of the GO suspension changes from slight-brown to dark-brown when the pH value increases from 3 to 12. They attributed this to the pH-driven reversible epoxide ring opening/closing on the GO basal plane (Fig. 4).| pH dependent reactions | pH dependent phenomena | pH dependent application |
|---|---|---|
| Note: Reprinted with permission from ref. 59. Copyright 2014, Elsevier. Reprinted with permission from ref. 23. Copyright 2016, American Chemical Society. | ||
| Protonation or deprotonation of a high density of oxygen-containing functional groups13 | Largely reversible color changes from slight brown to dark brown59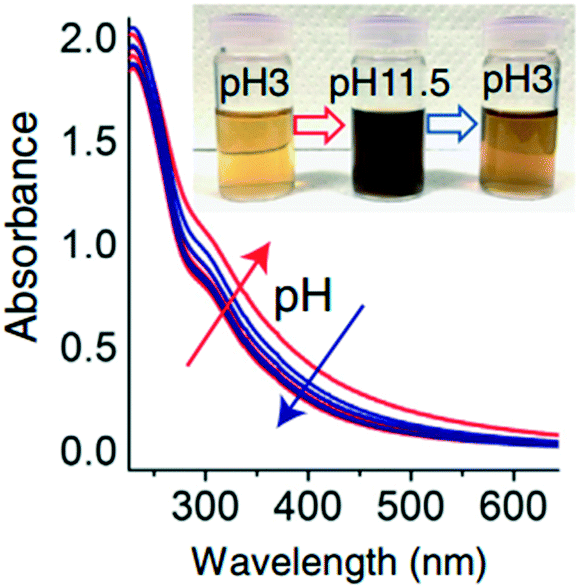 |
By pH adjustment, separation of larger size GO from smaller size GO60 |
| Deoxygenation of GO under alkaline conditions36 | pH-Dependent photoluminescence efficiency: photoluminescence quenching of GO in alkaline solutions59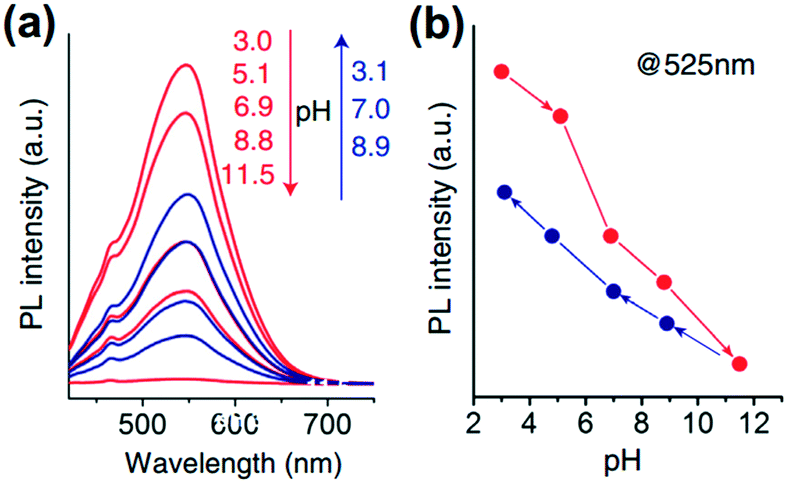 |
|
| Reversible epoxy ring opening and closing based on epoxy–hydroxyl conversion59 | pH driven conformational changes: at low pH, numerous sheets fold and networks of sheets are observed to agglomerate. At high pH, each sheet undergoes extensive collapse, condensing against neighboring sheets into larger macro-scale agglomerates61 | |
pH dependent CCC23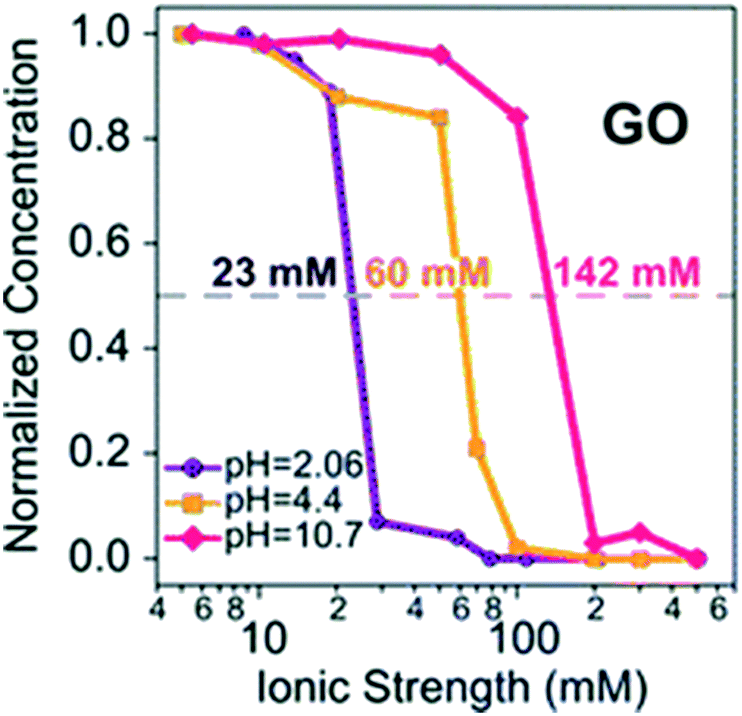 |
||
| The water contact angle of GO decreases sharply from 42.3° to 11.6° when the pH value increases from 3.4 to 10.5 (ref. 43) | ||
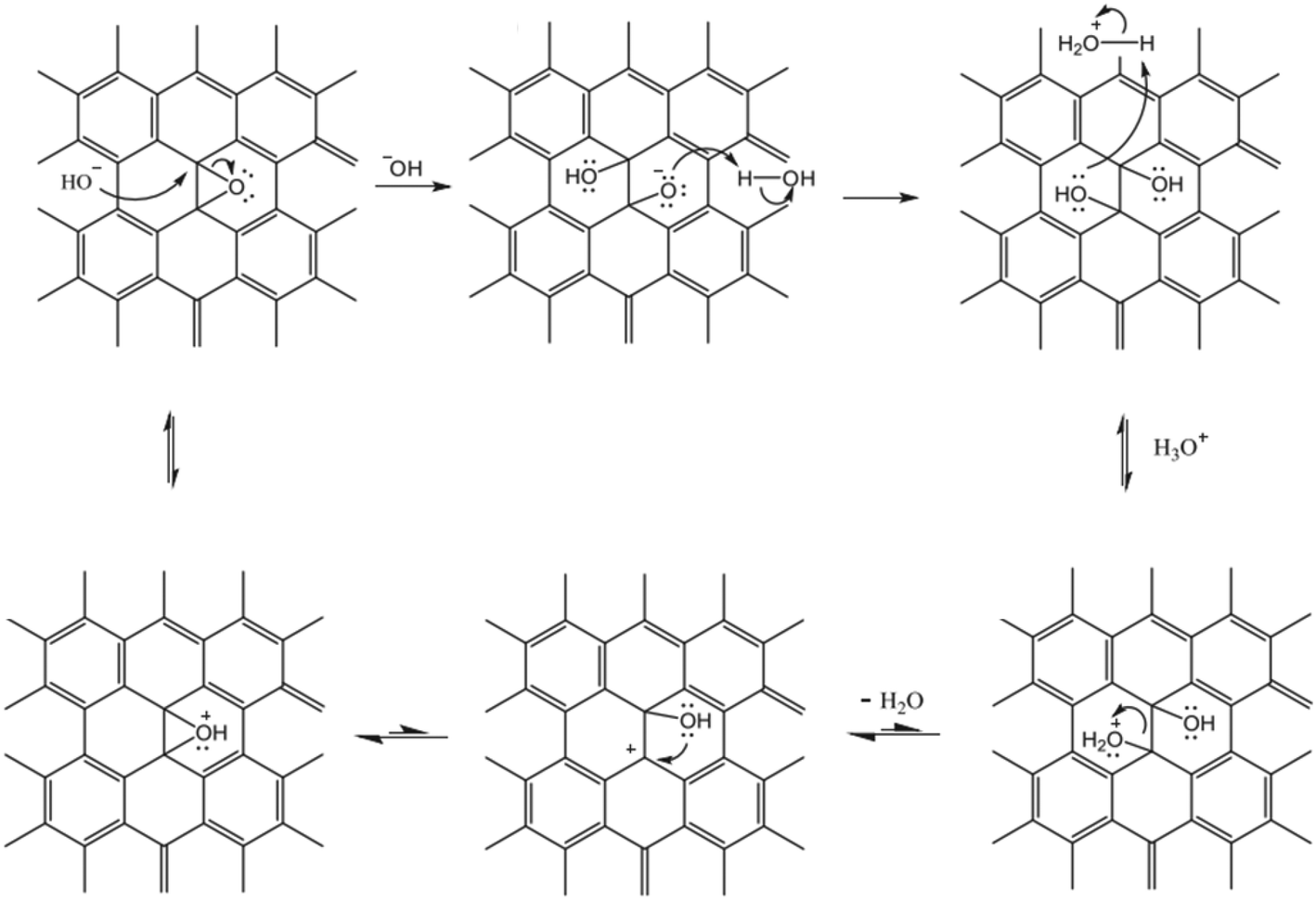 | ||
| Fig. 4 Proposed pH-dependent epoxy ring opening/closing reactions.59 Reprinted with permission from ref. 59. Copyright 2014, Elsevier. | ||
As an amphoteric substance, positively-, neutral-, and negatively-charged surface species all coexist in the GO colloidal form. Their balance in the GO colloidal structure can be easily destroyed and restructured when pH changes. Thus, the response of GO stability to varying pH value mainly results from the pH-induced charge variation (i.e., protonation or deprotonation of a high density of oxygen-containing functional groups). At low solution pH values, the presence of a large amount of protons may suppress the deprotonation of the carboxyl groups and reduces the hydrophilicity at the edge. Meanwhile, the intact carboxyl groups readily form carboxylic dimers through strong intermolecular hydrogen bonding.62 As a result, GO may aggregate instead of staying in the bulk aqueous solution. At even lower pH values, the carboxyl groups at the edge of GO are gradually protonated due to the decrease in pH, which makes GO sheets more hydrophobic, forming aggregates. In these cases, van der Waals interaction should dominate over electrostatic repulsion. In addition, the residual π-conjugation in the basal plane of GO may also contribute to the aggregation via π–π stacking. Both cases would favor GO aggregation via the face-to-face pattern. By employing molecular dynamics (MD) simulation, Shih and coworkers58 stated that the GO aggregates may exhibit a GO/water/GO sandwich-like structure (Fig. 5). In a word, the instability of GO occurring at low pH values can stem from the increased hydrophobicity, decreased electrostatic repulsion, increased intermolecular hydrogen bonding, increased van der Waals interaction and possible π–π stacking. By employing MD simulations, Tang et al.63 stated that hydrophobic interaction played a dominant role in the aggregation of GO at low pH values.
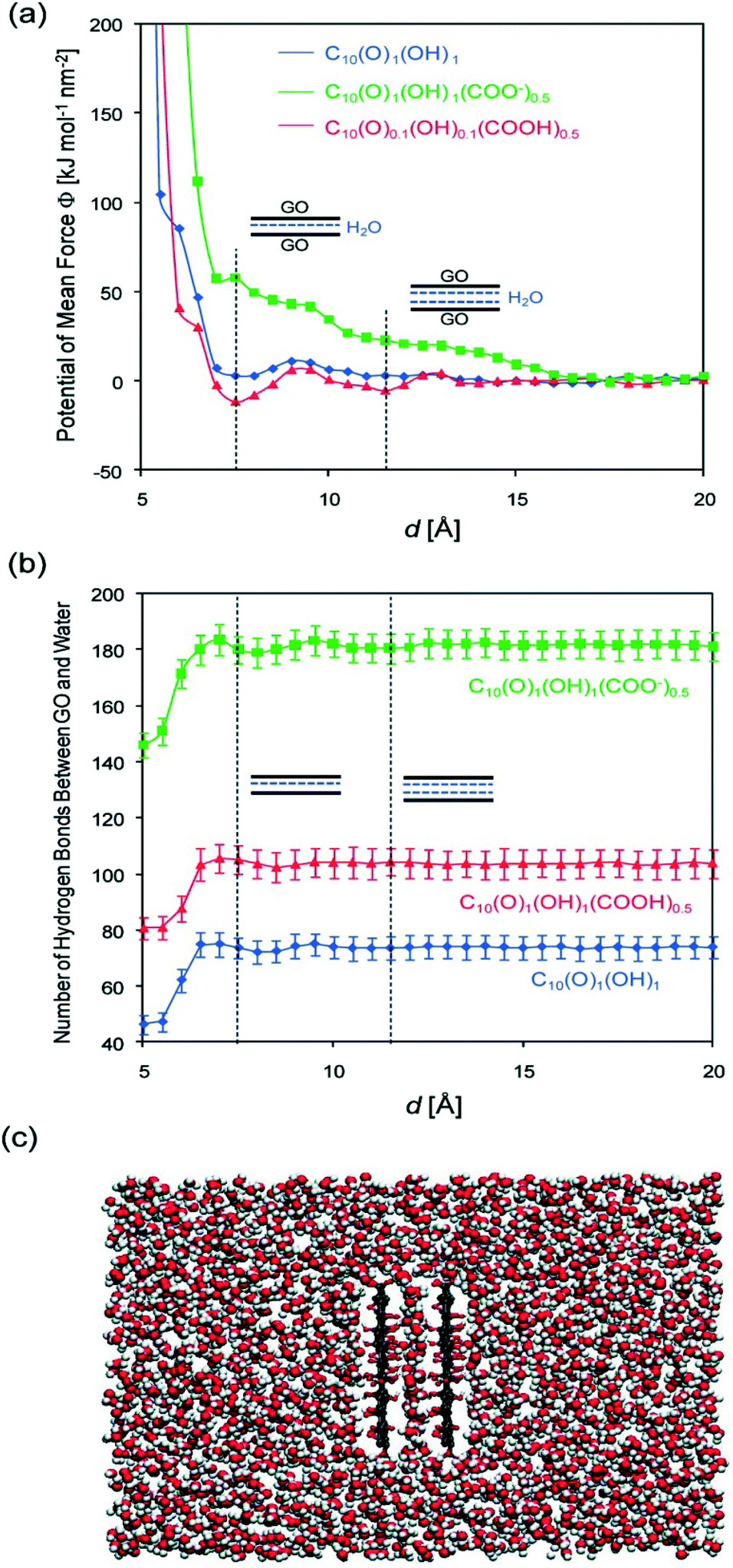 | ||
| Fig. 5 Simulated (a) potential of the mean force between two parallel, fixed GO sheets, and (b) the number of H-bonds formed between the two sheets and the surrounding water molecules as a function of the intersheet separation, d. Three forms of GO [C10(O)1(OH)1, C10(O)1(OH)1(COOH)0.5, and C10(O)1(OH)1(COO)0.5] are considered. The vertical dashed lines correspond to the energy-minimized configurations of GO/single-layer water/GO and GO/two-layer water/GO sandwich structures. (c) Post equilibrium MD simulation snapshot of two parallel C10O1(OH)1 (COOH)0.5 sheets solvated in water at d = 7.5 Å showing a single layer of water molecules being confined between the two GO sheets. Color code: red, oxygen; white, hydrogen; and gray, carbon.58 Reprinted with permission from ref. 58. Copyright 2012, American Chemical Society. | ||
As the pH values increase, the degree of deprotonation of carboxylic and phenolic groups increases in water. As a result, electrostatic stabilization of GO in aqueous solutions can be achieved when the suspension pH is higher than the pKa of carboxyls or phenolic hydroxyls. Compared with carboxyls, the deprotonation of hydroxyls is much weaker and may not be important to the surface charge acquisition of GO.34,61,64 Since GO is both hydrophobic and hydrophilic, the deprotonation of carboxyl groups will maximize the difference in the extent of hydrophobicity between the edge and the basal plane.58 According to the principle that “like dissolves like”, the strong hydrophilicity of carboxylic groups at the edge may pull GO into bulk water, which results in its easy dispersion in water. Besides, at higher pH, more H-bonds formed between GO sheets and water, making GO more hydrophilic to dissolve in water.65 Therefore, it can be concluded that the degree of deprotonation of the edge carboxylic groups, the formation of H-bonds and the electrostatic repulsion between the negatively charged deprotonated carboxylic groups are the driving forces for the increased stability of GO at higher pH values. At extremely high pH values, an excessively large volume of NaOH is added to adjust the pH values, so the “salting out” effect should be considered. Whitby et al.61 observed that single layer GO aggregation occurred at pH 14 due to the salting out effect, while Shih et al.58 observed that GO was stably suspended at pH 14. The inconsistency of the results in these two studies may result from variations in the physiochemical properties of the examined GO sheets. The conformational changes in single layer GO may be easier to occur. The phenomenon of the decrease in solubility at high pH values was also observed for oxidized carbon nanotubes (CNTs). Shieh and coworkers62 reported that the solubility of oxidized CNTs at pH 12 was lower than that at pH 10. They attributed this to the larger amount of sodium cations in the aqueous solution of pH 12 causing the coagulation of some carboxylate anions.
The macroscopic effects of solution pH on the surface charge, size, and suspended mass concentration of GO have been systematically studied. Broadly, previous studies monitor the surface charge, size, and suspended mass concentration as a function of pH mainly by using a Zeta Sizer Nano ZS instrument, time-resolved dynamic light scattering, and UV-vis spectroscopy, respectively.28,66 The ZP values of GO are reported to be lower than −30 mV at pH > 4.0.27,60,67 Generally speaking, negatively charged colloids with ZP more than −30 mV are considered to be electrostatically stable and well dispersed in water.22,35,68 Therefore, GO sheets are stable at pH > 4. The change in ZP (or EPM) and hydrodynamic diameters (HD) of GO as a function of pH shows a similar trend.28,67 The values of ZP and HD increase significantly with decreasing pH at low pH values (pH 2–4), while they remain very similar at high pH values (pH 4–10).28 Huang et al.,69 Chowdhury et al.,28 Lanphere et al.,70 Wu et al.,64 Ren et al.66 and Yang et al.71 separately reported that pH did not play a significant role in altering the surface potential and HD of GO over the pH range from 3 to 12. It is worth noting that all of the above-mentioned studies were carried out under conditions with the concentration of the monovalent electrolyte lower than the CCC value of GO. The pH of a natural aquatic environment is usually observed in the range of 5–9. Across these pH values, the pH will have no obvious effect on the fate of GO without considering the effect of the coexisting metal cations (Fig. 6). Meanwhile, in the presence of Ca2+ and Mg2+, Hua et al.72 reported that the values of EPM and HD increased with increasing pH values across pH 5–9. The CCCNa values of GO are very sensitive to pH. Gudarzi et al.23 reported that GO sheets began to aggregate at IS higher than 20 mM for pH 2.06, 50 mM for pH 4.4, and 100 mM for pH 10.7. These phenomena are due to the effect of the cation nature, which will be discussed in the next part.
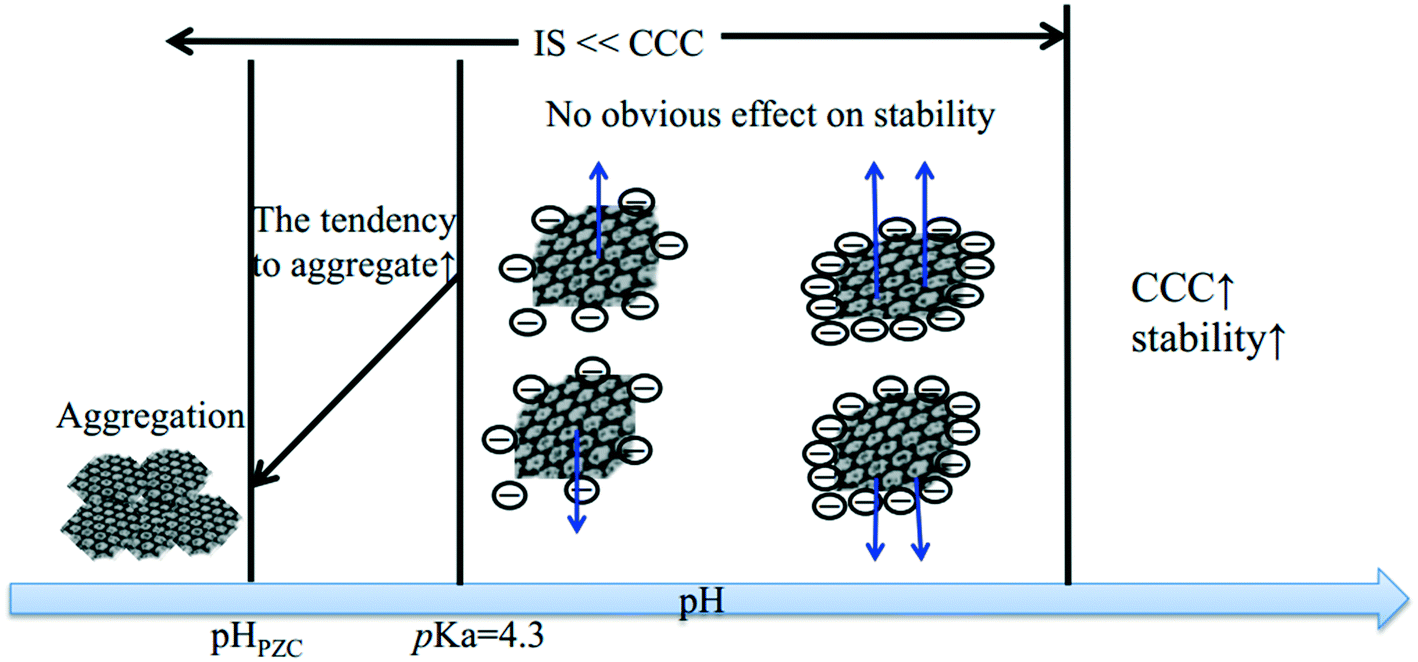 | ||
| Fig. 6 Diagrams of the impact of pH on GO stability. | ||
Effect of IS and salt type
Surface water and groundwater environments contain a complex mixture of inorganic ions such as Na+, K+, Mg2+, Ca2+, Cl−, HCO3−, SO42−, etc., which change with location and can have a significant impact on the electro-kinetic behavior of colloids and hence their colloidal properties.73 Ions with an opposite charge to the surface charge of colloids, namely counter-ions, tend to accumulate at the charged interface and can strongly compress the electrostatic double layer (EDL) and effectively reduce the energy barrier between colloids by neutralizing the particle surface charge. This results in the attractive van der Waals interactions becoming dominant and the enhancement of particle aggregation.74 Charge neutralization can also occur as a result of specific adsorption of counter-ions on the charged surface. The degree of charge neutralization is dependent on the electrolyte concentration, valence of counter-ions, and interaction of the electrolyte with colloids. Surface charge screening is directly reflected in the EPM and ZP of the colloids. With an increase in the counter-ion concentration, the absolute values of EPM and ZP approach zero, indicating a favorable aggregation condition. Also, the counter-ion concentration approaches and exceeds the CCC, indicating a favorable aggregation condition, known as the diffusion-limited regime (the second stage of colloid aggregation), where the aggregation kinetics of the colloids are controlled by diffusion and independent of the counter-ion concentration.75 However, in the initial stage of colloid aggregation, known as the reaction-limited regime, the aggregation kinetics of the colloid increase linearly with increasing counter-ion concentration up to the CCC.76 The values of CCC of GAs are gathered in Table 3. One can see that the CCC values of GO are significantly different depending on the surface chemistry of GO and solution chemistries. Table 3 also shows that the multivalent CCC values are significantly lower than monovalent CCC values, which is expected due to the bridging/crosslinking behavior of multivalent ions. The ratio of CCCMg/Ca to CCCNa for GO is almost approximated as z−6, indicating that although GO is not spherical in shape, the aggregation and stability of GO in aquatic environments follow the Schulze–Hardy rule.| GAs | Conditions | CCC | CCC ratio of Mg2+ to Na+ | CCC ratio of Ca2+ to Na+ | Conclusions | ||
|---|---|---|---|---|---|---|---|
| GO–COOH | 25 mg L−1 | 175 mM Na+ | 4.2 mM Ca2+ | 2–5.42 | The introduction of carboxylic groups increases the stability of GO in water63 | ||
| GO | 25 mg L−1 | 110 mM Na+ | 2.66 mM Ca2+ | 2–5.73 | |||
| GO | pH 5.5, 20 mg L−1 GO | 50 mM Na+ | 28 mM Ca2+ | The fate of GO in aquatic-terrestrial transition zones will be influenced by its interaction with hydrous oxides and clay minerals69 | |||
| pH 5.5, 20 mg L−1 GO, 2 g L−1 kaolinite | 20 mM Na+ | ||||||
| pH 5.5, 20 mg L−1 GO, 2 g L−1 kaolinite–goethite associations | <1 mM Na+ | ||||||
| pH 9, 20 mg L−1 GO, 2 g L−1 kaolinite–goethite associations | 5 mM Na+ | ||||||
| GO | 44 mM Na+ | 1.3 mM K+ | 0.9 mM Mg2+ | 2–5.61 | 2–5.08 | The presence of HA improves the stability of GO significantly28 | |
| Suwannee River humic acid (SRHA) | 125 mM Na+ | 3.9 mM K+ | 2.2 mM Mg2+ | 2–5.82 | 2–5.00 | ||
| GO | pH 5.2 ± 0.3, 10 mg L−1 GO | 200 mM Na+ | ∼0.7 mM Mg2+ | The stability of GO in water decreases with successive reduction of functional groups55 | |||
| rGO-1h | pH 5.2 ± 0.3, 10 mg L−1 rGO | ∼35 mM Na+ | ∼0.7 mM Mg2+ | ||||
| rGO-2h | pH 5.2 ± 0.3, 10 mg L−1 rGO | ∼30 mM Na+ | ∼0.7 mM Mg2+ | ||||
| GO | pH 5.2, 12.4 mg TOC L−1 GO | ∼222 mM Na+ | Heteroaggregation of GO with hematite influences the fate and transport of GO77 | ||||
| GO–HemNP nanohybrids | pH 5.2, 12.4 mg TOC L−1 GO and 1.24 mg L−1 HemNPs | ∼308 mM Na+ | |||||
| GO | pH 2.06, 20 mg L−1 GO | 20 mM Na+ | Both GO and rGO are less prone to aggregation in basic media as the CCC increases with increasing pH23 | ||||
| pH 4.4, 20 mg L−1 GO | 50 mM Na+ | ||||||
| pH 10.7, 20 mg L−1 GO | 100 mM Na+ | ||||||
| rGO | pH 2.06, 10 mg L−1 rGO | 14 mM Na+ | |||||
| pH 10.6, 10 mg L−1 rGO | 42 mM Na+ | ||||||
| GO | pH 6.0 ± 0.3, 40 mg L−1 GO | 68.7 mM Na+ | 1.91 mM Mg2+ | 1.57 mM Ca2+ | 2–5.17 | 2–5.45 | CCC values are correlated with the physicochemical properties of the material including the shape, zeta potentials, C/O ratios, carboxyl group concentrations, and C–C fractions21 |
| CGO-200 | pH 6.0 ± 0.3, 40 mg L−1 CGO | 81.7 mM Na+ | 3.09 mM Mg2+ | 2.5 mM Ca2+ | 2–4.72 | 2–5.03 | |
| CGO-400 | pH 6.0 ± 0.3, 40 mg L−1 CGO | 73.9 mM Na+ | 2.77 mM Mg2+ | 2.24 mM Ca2+ | 2–4.74 | 2–5.04 | |
| CGO-500 | pH 6.0 ± 0.3, 40 mg L−1 CGO | 59.3 mM Na+ | 2.05 mM Mg2+ | 1.86 mM Ca2+ | 2–4.86 | 2–5.00 | |
| CGO-600 | pH 6.0 ± 0.3, 40 mg L−1 CGO | 13.4 mM Na+ | 1.06 mM Mg2+ | 0.92 mM Ca2+ | 2–3.66 | 2–3.86 | |
| CGO-800 | pH 6.0 ± 0.3, 40 mg L−1 CGO | 9.9 mM Na+ | 0.94 mM Mg2+ | 0.82 mM Ca2+ | 2–3.40 | 2–3.59 | |
| GO | pH 6.0–7.0, 20 mg L−1 GO, | 67 mM Na+ | 1.4 mM Ca2+ | NOM type has a significant impact on material stability, particularly in the presence of monovalent cations30 | |||
| pH 6.0–7.0, 20 mg L−1 GO, 2.7 mg C L−1 SRHA | 145 mM Na+ | 2.5 mM Ca2+ | |||||
| pH 6.0–7.0, 20 mg L−1 GO, 2.7 mg C L−1 SRFA | 109 mM Na+ | 1.8 mM Ca2+ | |||||
| pH 6.0–7.0, 20 mg L−1 GO, 2.7 mg C L−1 AHA | 267 mM Na+ | 2.1 mM Ca2+ | |||||
| CGO-400 | pH 6.0–7.0, 20 mg L−1 CGO | 80 mM Na+ | 2.0 mM Ca2+ | ||||
| pH 6.0–7.0, 20 mg L−1 CGO 2.7 mg C L−1 SRHA | 1177 mM Na+ | 5.4 mM Ca2+ | |||||
| pH 6.0–7.0, 20 mg L−1 CGO 2.7 mg C L−1 SRFA | 744 mM Na+ | 5.2 mM Ca2+ | |||||
| pH 6.0–7.0, 20 mg L−1 CGO 2.7 mg C L−1 AHA | 1572 mM Na+ | 5.5 mM Ca2+ | |||||
| CGO-800 | pH 6.0–7.0, 20 mg L−1 CGO | 17 mM Na+ | 1.3 mM Ca2+ | ||||
| pH 6.0–7.0, 20 mg L−1 CGO, 2.7 mg C L−1 SRHA | 772 mM Na+ | 5.5 mM Ca2+ | |||||
| pH 6.0–7.0, 20 mg L−1 CGO, 2.7 mg C L−1 SRFA | 439 mM Na+ | 5.6 mM Ca2+ | |||||
| pH 6.0–7.0, 20 mg L−1 CGO, 2.7 mg C L−1 AHA | 845 mM Na+ | 5.4 mM Ca2+ | |||||
| GO | 10 mgL−1 GO, pH 5.6 ± 0.2 | 210 mM Na+ | The order of CCC values correlates reasonably with the degree of reduction of rGO, as indicated by the C/O ratios22 | ||||
| VC–rGO | 10 mg L−1 rGO, pH 5.5 ± 0.2 | 113 mM Na+ | |||||
| NaBH4–rGO | 10 mg L−1 rGO pH 6.0 ± 0.2 | 78 mM Na+ | |||||
| N2H4–rGO | 10 mg L−1 rGO pH 6.0 ± 0.2 | 41 mM Na+ | |||||
| GO | pH 3 | >1000 mM Na+ | 7 mM Ca2+ | 0.05 mM Eu3+ | Coagulation is a sorption driven mechanism78 | ||
| pH 7 | 400 mM Na+ | 1 mM Ca2+ | 0.05 mM Eu3+ | ||||
| GO | 5 mg L−1 GO, pH 7 | 184 mM Na+ | The presence of KGa-1b destabilizes the GO suspension slightly79 | ||||
| GO-(KGa-1b) | 5 mg L−1 GO, 50 mg L−1 KGa-1b, pH 7 | 152 mM Na+ | |||||
| GO | pH 6.5, 18 mg L−1 GO | 50 mM Na+ | 0.5 mM Mg2+ | 0.018 mM Fe3+ | 2–6.64 | Multivalent cations are more effective in destabilizing the GO suspension68 | |
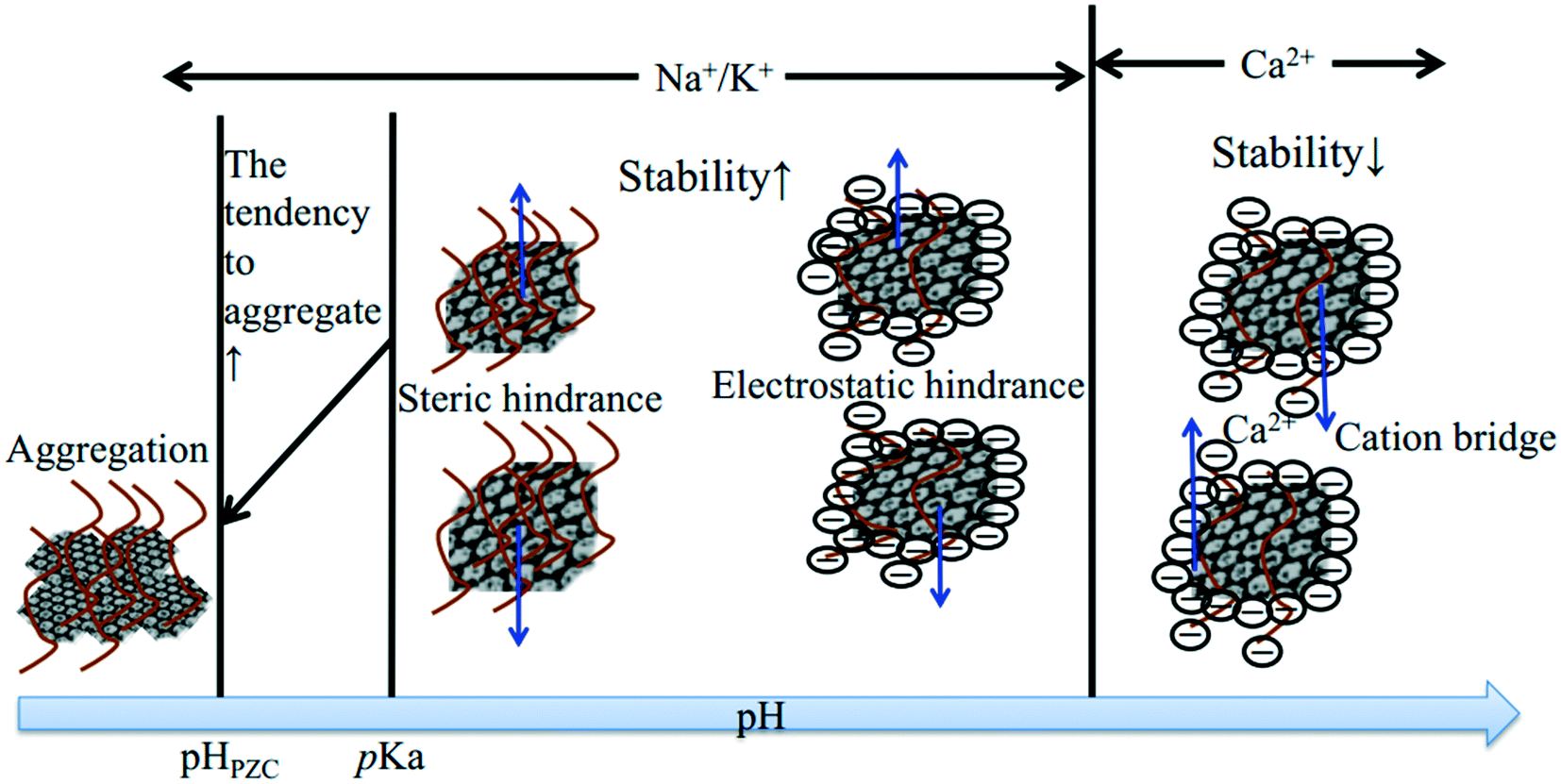
Even though no clear standard relates ZP to colloidal stability, the colloid system is considered to be stable at an absolute ZP of >30 mV.80,81 Lots of studies have showed that the absolute ZP of a colloid decreases with increasing IS at specific pH, in accordance with classical electrostatic theory.80,82 The representative EPM of GO as a function of IS is shown in Fig. 7. For all electrolytes, the EPM values increase marginally at low IS and more substantially at high IS. This increase obviously depends not only on IS but also on the nature of the added counter-ion (e.g., cation valence, cation radius/hydrated radius), and can be explained by the suppression of the EDL, surface charge neutralization, preferential adsorption, or a combination of them by the increased IS. When cations are not chemically adsorbed onto GO, the capacity of cations to neutralize the surface charge of GO is related to the cation valence, to the ability of cations to approach the charged interface, and to the state of hydration of cations.83 Cations with a higher valence are more efficient in destabilizing GO. For counter-ions with the same valence, the smaller hydrated radius results in more effective screening of the surface charge of GO. The effects of the common environmental cations including Na+, K+, Mg2+ and Ca2+ on the surface potential of GO are well established. The cations increase the surface charge of colloids in the order K+ > Na+ for monovalent counter-ions22,74,83 and Ca2+ > Ba2+ > Mg2+ for bivalent counter-ions.83 Qi et al.22 attributed the greater aggregation effect of K+ than Na+ to the less dense hydration of K+, and the higher aggregation effect of Ca2+ than Mg2+ to the less dense hydration and the bridging effect of Ca2+.
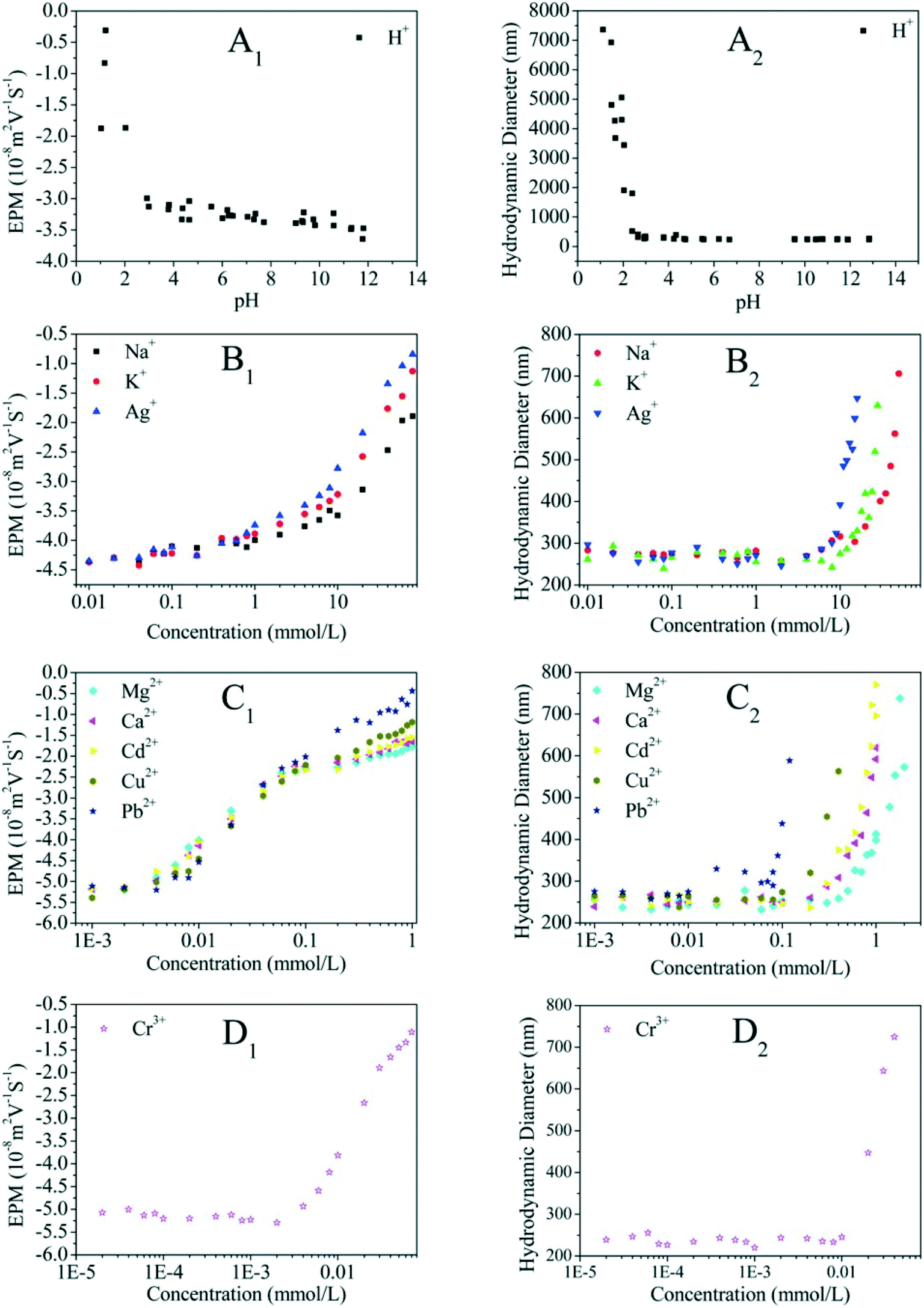 | ||
| Fig. 7 Electrophoretic mobility (EPM) and hydrodynamic diameter of GO as a function of the cation concentration of H+ (A1 and A2), monovalent metal cations (B1 and B2), divalent metal cations (C1 and C2), and trivalent metal cations (D1 and D2).71 Reprinted with permission from ref. 71. Copyright 2016, American Chemical Society. | ||
Besides neutralizing surface charges, bivalent electrolytes which can specifically interact with GO to cross-link them are much more effective in destabilizing GO suspensions. Wu and coworkers64 proposed that Ca2+ and Mg2+ induced the aggregation of GO sheets through the following three types of cross-linking interactions (Fig. 8): (1) bridging the edges of the GO sheets through chelating carboxylate groups, (2) intercalating between the basal planes through either weak alkoxide or dative bonds from carbonyl and hydroxyl groups, and (3) cross-linking of the H-bonds formed among the oxygen-containing functional groups on GO surfaces and the interlamellar water molecules. Specially, preferential adsorption plays an important role in the destabilization of GO by Ca2+. Chowdhury and coworkers28 observed that the ZP values of GO as a function of IS were quite similar for both CaCl2 and MgCl2, but the hydrodynamic sizes of GO as a function of IS were significantly different for CaCl2 and MgCl2. They attributed the higher effectiveness of Ca2+ in destabilizing GO than Mg2+ to the preferential adsorption of Ca2+ through the binding of Ca2+ with oxygen containing functional groups available on GO surfaces. Similarly, Wu and coworkers64 attributed the lower CCC of GO in Ca2+ than that in Mg2+ to the higher propensity of Ca2+ to form complexes with carboxylated groups.
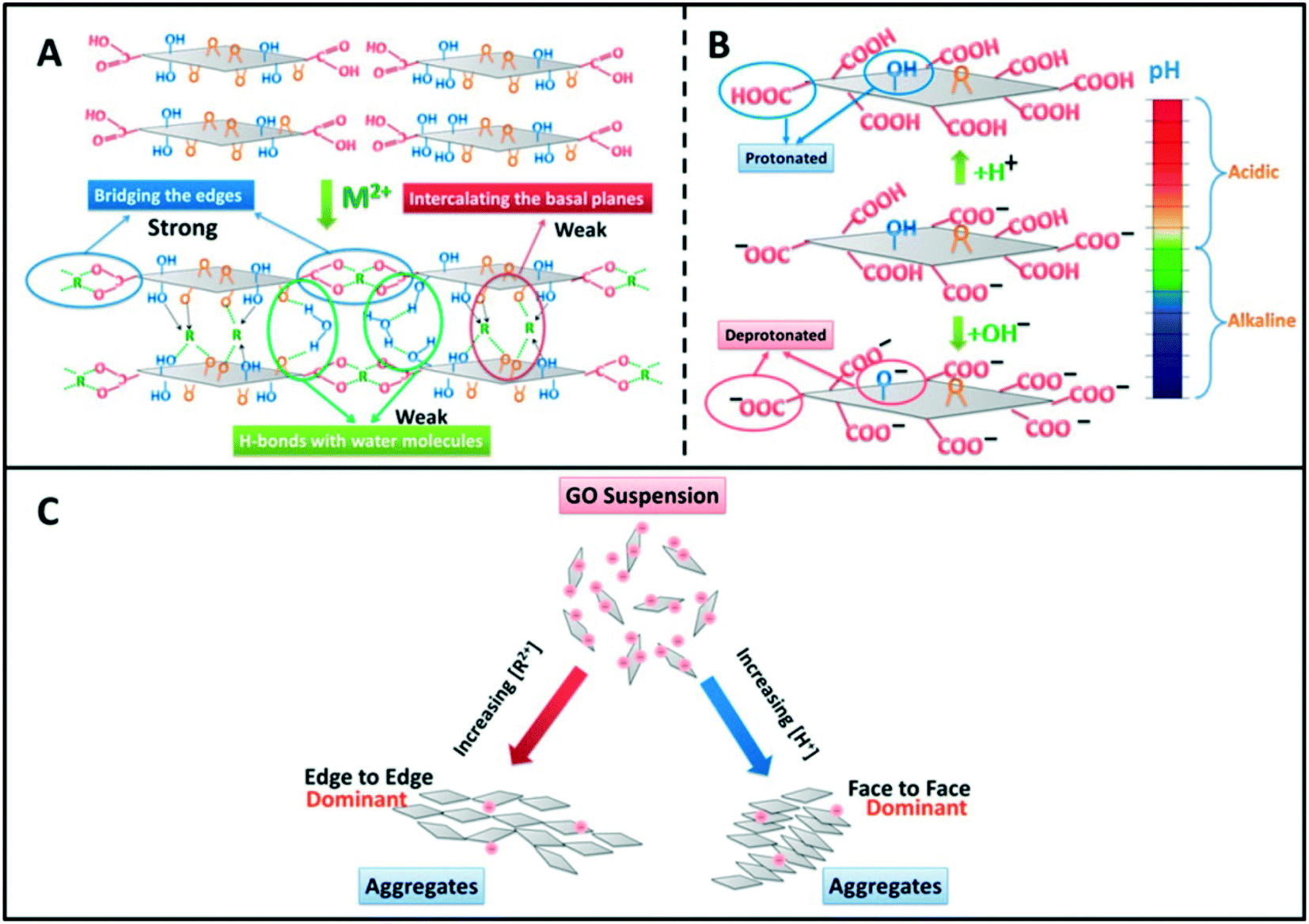 | ||
| Fig. 8 GO aggregation mechanisms: (A) cross-linking by divalent cations (Mg2+ and Ca2+), (B) deprotonation of the carboxyl groups at the edges when pH increases, and (C) summary of aggregation modes of GO nanosheets.64 Reprinted with permission from ref. 64. Copyright 2013, American Chemical Society. | ||
Besides the common environmental cations Na+, K+, Mg2+ and Ca2+, Yang and coworkers71 investigated for the first time the effect of heavy metal cations (Ag+, Pb2+, Cu2+, Cd2+ and Cr3+) on the aggregation of GO. Based on the CCC values (Table 4), they found that the destabilizing ability of cations follows the order Cr3+ ≫ Pb2+ > Cu2+ > Cd2+ > Ca2+ > Mg2+ ≫ Ag+ > K+ > Na+. The discrepancy in destabilizing ability between different valence cations follows the classic Schulze–Hardy rule, but the difference in the destabilizing ability of equivalent cations does not. This is because in addition to the suppression of the EDL, surface binding, which is determined by the electronegativity and hydration shell thickness of metal cations, contributes to the aggregation of GO. By employing the element mapping of transmission electron microscopy (TEM), we observed that the monovalent cation (i.e., Na+) was distributed on the GO surface inhomogeneously and intensively located at the boundary of GO, while multi-valent cations (i.e., Mg2+, Al3+) were distributed on the GO surface homogeneously (Fig. 9).67 These results evidence that it is hard for monovalent cations to cross the EDL and induce GO aggregation via EDL suppression; in contrast, multivalent cations can easily cross the EDL, bind to the oxygen containing functional groups on the GO surface, and then induce GO aggregation via EDL suppression and surface binding. By adding polyacrylic acid (PAA), the GO aggregates induced by Na+, K+, Ca2+ or Al3+ can be re-dispersed partly in solution, while those induced by Mg2+ can be re-dispersed completely (Fig. 10). This suggests that the aggregation processes of GO induced by EDL suppression and strong complexing are not fully reversible and only those induced by weak complexing are fully reversible.
| Metal cations | Ionic radius (Å) | Hydration shell thickness (Å) | Electronegativity | CCC (mM) | Ratio of CCC to CCCNa |
|---|---|---|---|---|---|
| Na+ | 0.95 | 2.63 | 0.93 | 36 | 1 |
| K+ | 1.33 | 1.98 | 0.82 | 28 | 0.778 |
| Ag+ | 1.26 | 2.15 | 1.93 | 19 | 0.528 |
| Mg2+ | 0.65 | 3.63 | 1.31 | 1.5 | 2−4.585 |
| Ca2+ | 0.99 | 3.13 | 1.01 | 1.35 | 2−4.737 |
| Cd2+ | 0.97 | 3.29 | 1.69 | 1.3 | 2−4.791 |
| Cu2+ | 0.72 | 3.47 | 1.90 | 0.725 | 2−5.634 |
| Pb2+ | 1.32 | 2.69 | 2.33 | 0.3 | 2−6.907 |
| Cr3+ | 0.64 | 3.48 | 1.66 | 0.085 | 3−5.506 |
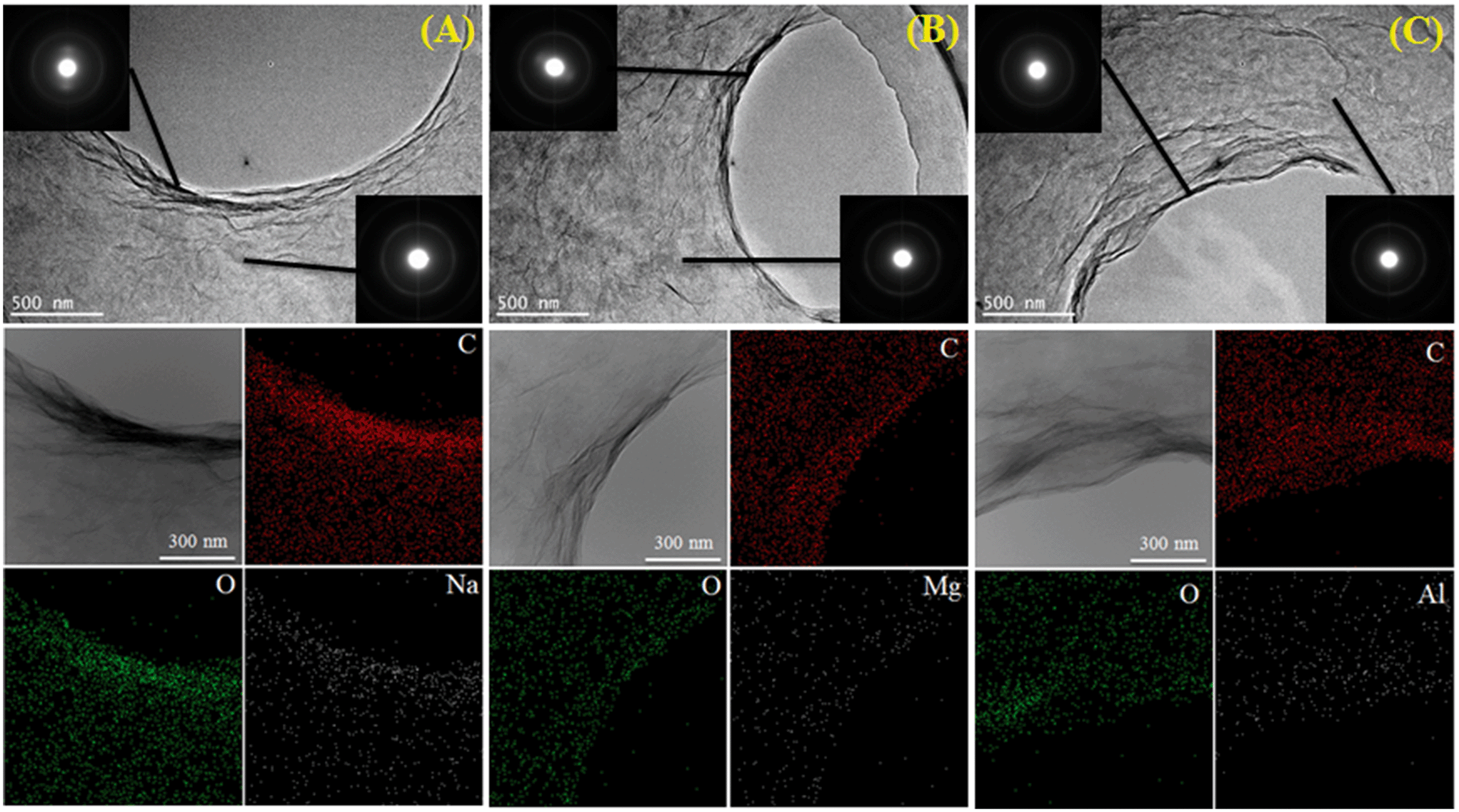 | ||
| Fig. 9 TEM, selected area electron diffraction (SAED) and element mapping pattern of GO aggregates in the presence of Na+ (A), Mg2+ (B) or Al3+ (C) electrolyte (Na+: 50 mM, Mg2+: 1 mM, Al3+: 0.1 mM and GO: 20 mg L−1).67 Reprinted with permission from ref. 67. Copyright 2017, Elsevier. | ||
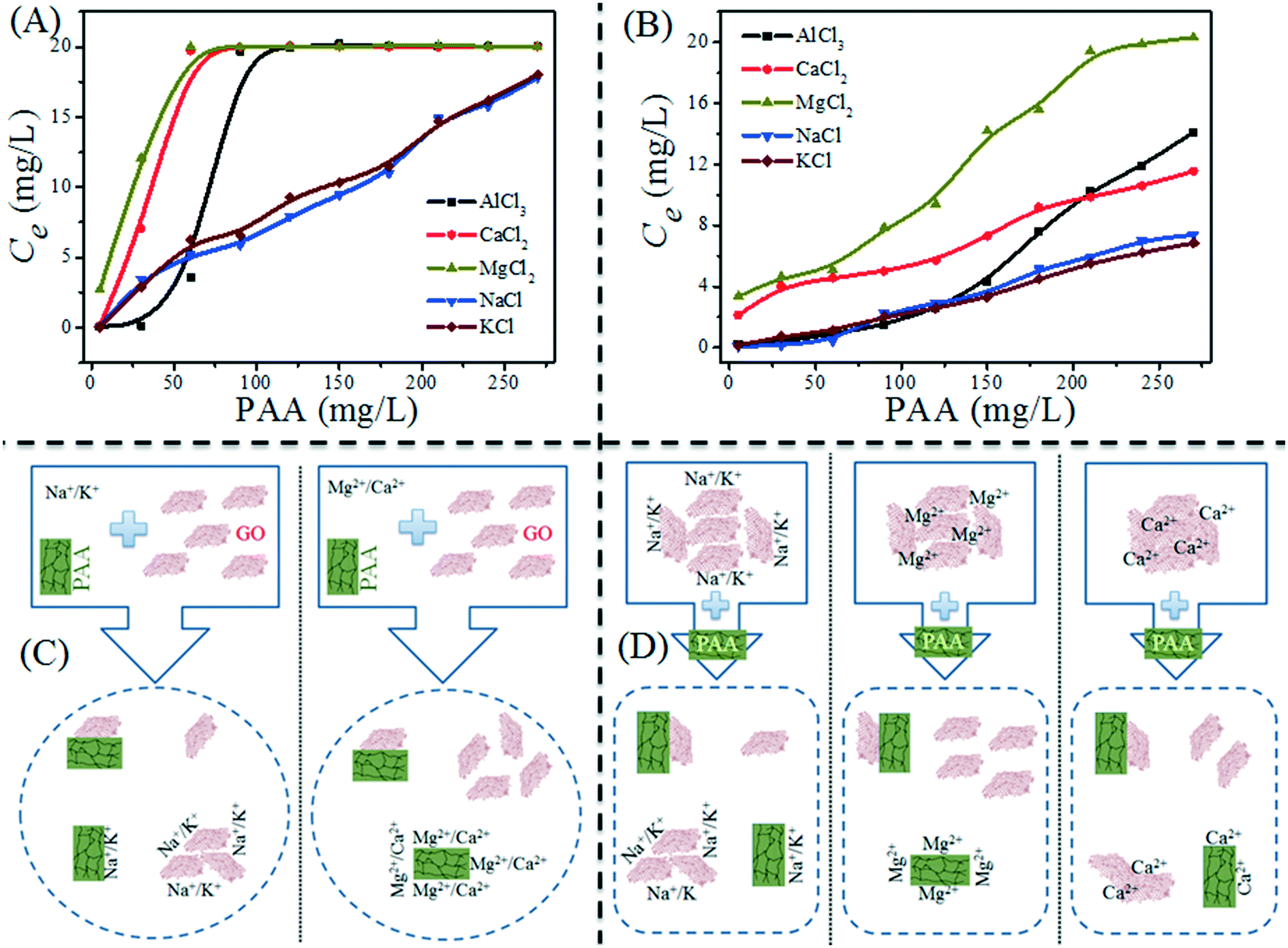 | ||
| Fig. 10 (A) Effect of polyacrylic acid (PAA) concentration on GO sediment: adding PAA, GO and background electrolyte at the same time; (B) PAA-mediated re-dispersion of GO aggregates: adding PAA after background electrolyte inducing GO sediment. C(GO)initial = 20 mg L−1, pH = 5.0 ± 0.5. The concentrations of cations used to induce GO aggregation are 50 mM NaCl, 50 mM KCl, 1 mM MgCl2, 1 mM CaCl2 and 0.1 mM AlCl3. (C) Schematic diagram of the PAA effect on GO sediment; (D) schematic diagram of PAA-mediated re-dispersion of GO aggregates.67 Reprinted with permission from ref. 67. Copyright 2017, Elsevier. | ||
Although cations play a dominant role in the aggregation of GO, their effectiveness in destabilizing GO is affected by the coexisting anion (co-ion) type. Depending on the complexation ability of anions with cations, the coexisting anions will affect the concentrations of the free cations and their ability to destabilize GO. Typically, our group84 has characterized the coexisting anion effect and found that the effect of the electrolytes on GO aggregation decreased in the following order: Na2SO4 > NaCl > NaH2PO4. In another study, our group studied the colloidal behavior of GO in the coexistence of Cd(II) and phosphate (P(V)).66 We found that when Cd(II), P(V) and GO were in contact simultaneously, the presence of P(V) enhanced the tendency of GO to agglomerate in a Cd(II) aqueous solution but reduced its sedimentation rate, which makes GO aggregates transport over a relatively long distance. In a very recent study, we found that the anions affected the threshold values of cations destabilizing GO and that the effectiveness of the electrolytes in destabilizing GO followed the order Na2SO4 > Na2HPO4 > NaCl > NaHCO3.67
Most of the above-mentioned investigations are mainly focused on single-salt solutions. The information on the aggregation behavior of GO in mixed-cation solutions is limited. Chowdhury et al.55 characterized the EPM and HD of GO in mixed Na–Ca and Na–Mg electrolyte systems. They found that a small amount of background CaCl2 (0.1 mM) can significantly affect the stability of GO while a small amount of background MgCl2 (0.1 mM) cannot. Based on their findings, GO cannot remain suspended for a long time in natural aquatic environments, where the concentration of CaCl2 often exceeds 0.1 mM. Similarly, our group67 investigated the sedimentation kinetics of GO in mixed Na–Mg electrolyte systems. We found that the settling tendency of GO was dependent on the ratio of Na to Mg. The prevailing electrolyte played a dominant role in the sedimentation processes.
Effect of NOM
Macromolecular NOM with various carbon functionalities (e.g., carbonyl, carboxyl, acetal, aromatic, heteroaliphatic, and aliphatic carbons), ubiquitous in natural aquatic systems, is known to function as a stabilizing agent for colloidal suspensions. Typically, the concentration of NOM is in the range from 0.1 to 2 mg L−1 in ground waters and as high as 20 mg L−1 in surface water.30 Humic substances, as typical NOM analogues, can be operationally classified into fulvic acid (FA, which is soluble at all pH levels) and humic acid (HA, which is soluble in alkaline aqueous solutions and insoluble in acidic aqueous solutions) depending on water solubility.NOM and its analogues can be adsorbed on GAs via hydrophobic interaction, π–π interaction, Lewis acid–base interaction, hydrogen bonding, and/or covalent and electrostatic interaction.56,85,86 The exact interaction mechanism of GAs with NOM will depend on both GA characteristics and functional derivatization as well as NOM characteristics including size, aromaticity, and charge density. The surface adsorption of NOM will alter the size, shape, morphology and surface chemistry of GO, especially the charge acquisition forms of GO, influencing its aqueous colloidal behavior and consequently more or less affecting its mobility and contaminant-mobilizing capabilities. For example, NOM consists of hydrophilic side chains (carboxylic and hydroxyl groups), a hydrophobic backbone, and a large number of cross-linked aromatic rings. They show a high tendency to pack parallel to the surface of GO by strong π–π interactions or to be readily adsorbed onto the basal plane of GO through hydrophobic interaction. The abundant carboxyl and hydroxyl groups from the adsorbed NOM protrude into the water and make the GO surface more hydrophilic. These structural characteristics not only create a thermodynamically suitable surface in water but also provide steric or electrostatic repulsion among dispersed GO, providing them with excellent capacities to disperse the hydrophobic graphene analogue.87 Electrostatic repulsion can be judged by comparing the changes in surface charge (ZP and EPM) of GO in the absence and presence of NOM, since the surface charge of NOM is less negative than that of GO. Generally speaking, ZP or EPM values in the presence of NOM remain unchanged, indicating that the enhanced stability can be due to steric stabilization, not electrostatic repulsion.88,89 If the changes in surface charge do not correlate well with the changes in particle size, the electrostatic interaction is not the only mechanism for particle stability.90,91 The effect of NOM on the colloidal behavior of GAs and the corresponding mechanisms in the literature are listed in Table 5.
| NOM | Conditions | EMP/ZP | HD | GA stability | Mechanism and ref. | CCC |
|---|---|---|---|---|---|---|
| SRHA | No effect | ↓ | ↑ | Steric repulsion28 | 125 mM NaCl | |
| SRHA | pH 7, 40 mg L−1 GO, 10 mg L−1 | ↑ | ↓ | ↑ | Electrostatic repulsion, steric repulsion (predominant)72 | |
| SRHA | 25 mg L−1 GO, 31.6 mM KCl, 0.1–10 mg L−1 SRHA | Negligible effect | ↓ | ↑ | Steric repulsion73 | |
| SRHA | 5 mg L−1 SRHA, 40 mg L−1 GO, pH 5.5 | No significant effect | ↑ | Steric repulsion28 | 125 mM NaCl, 2.2 mM CaCl2, 3.9 mM MgCl2 | |
| SRHA | Synthetic surface water, 5 mg L−1 SRHA, 10 mg L−1 rGO | Short term stability of GAs ↑; long term stability of GO and fully rGO ↓ | First, SRHA can bind with GO which provides steric repulsion and increases stability. Second, SRHA can facilitate binding with GO functional groups in the presence of divalent (Ca2+, Mg2+) cations, which can increase aggregation of GO flakes and reduce stability55 | |||
| SRNOM | 0.57 mg TOC L−1, pH 7.0, 1 mg L−1 FLG | ↓ | ↑ | Steric and electrostatic repulsion forces40 | 95 mM NaCl | |
| Low-molecular-weight organic acids | No significant effect | ↓ | ↑ | Steric repulsion92 |
SRHA is widely employed as a model to investigate the impact of NOM on GO stability. It is well established that the addition of SRHA reduces the aggregation of GO in NaCl, MgCl2 and CaCl2 solutions. Jiang et al.30 reported that the addition of 1.7 mg L−1 Aldrich humic acid (AHA) made the CCC value of GO increase from 67 to 220 mM for NaCl and from 1.4 to 2.2 mM for CaCl2. Chowdhury et al.28 reported that the addition of 5 mg L−1 SRHA increased the CCC of GO from 44 to 125 mM for NaCl, from 1.2 to 3.9 mM for MgCl2, and from 0.8 to 2.2 mM for CaCl2. Accordingly, the significance of stability enhancement by HA is largely dependent on the concentration and the type of background electrolyte. Generally speaking, when the background cation is Na+/K+ with a low concentration, the stability enhancement effect of NOM is not significant. In this case, NOM has a negligible effect on the EPM and HD of GO. When the background cation is Na+/K+ with a high concentration, the stability enhancement effect of NOM is very significant. In this case, NOM has no obvious effect on the EPM of GO while it decreases the HD of GO, so NOM stabilizes GO via steric repulsion. When the background cation is Na+/K+ with an extremely high concentration, NOM may fail to inhibit the aggregation of GO. In a solution containing 31.6 mM KCl, the presence of 1 mg L−1 SRHA was found to significantly decrease the HD of GO from 1598.1 ± 105.1 to 573.7 ± 56.1 nm.73 At 20 mM NaCl, the presence of tannic acid (TA) was reported to suppress the GO aggregation; meanwhile at 200 mM NaCl, the adsorption bridging effect of TA was more significant than its steric effect, which leads to TA failing to inhibit the aggregation of GO via the formation of GO–TA–GO aggregates (Fig. 11).63 When the background cation is Ca2+/Mg2+, NOM shows lower effectiveness in stabilizing GO or even facilitates the aggregation of GO. In this case, two opposing processes govern the interactions of NOM with GO (Fig. 3). First, NOM can bind with GO, which provides steric repulsion and increases the stability of GO. Second, NOM can facilitate binding with GO functional groups in the presence of divalent (Ca2+, Mg2+) cations, which can increase the aggregation of GO and reduce its stability. Either of the above-mentioned two opposing processes plays the dominant role depending on the experimental conditions. Lanphere et al.73 reported that although the presence of 1 mg L−1 SRHA decreased the HD of GO in a solution containing 1 mM CaCl2 from 2797.8 ± 211.6 to 1556.4 ± 182.2 nm, it cannot prevent the aggregation of GO. Chowdhury et al.55 found that in synthetic surface water containing Mg2+ and Ca2+, the addition of 5 mg L−1 SRHA improved the short-term stability of GO but did not improve the long-term stability of GO. Two modalities of GO aggregation and breakage in the presence of Mg2+/Ca2+ with NOM (Fig. 12) were reported.72
 | ||
| Fig. 11 Configurations of the graphene–tannic acid–graphene (GN–TA–GN) aggregate.65 Reprinted with permission from ref. 65. Copyright 2017, American Chemical Society. | ||
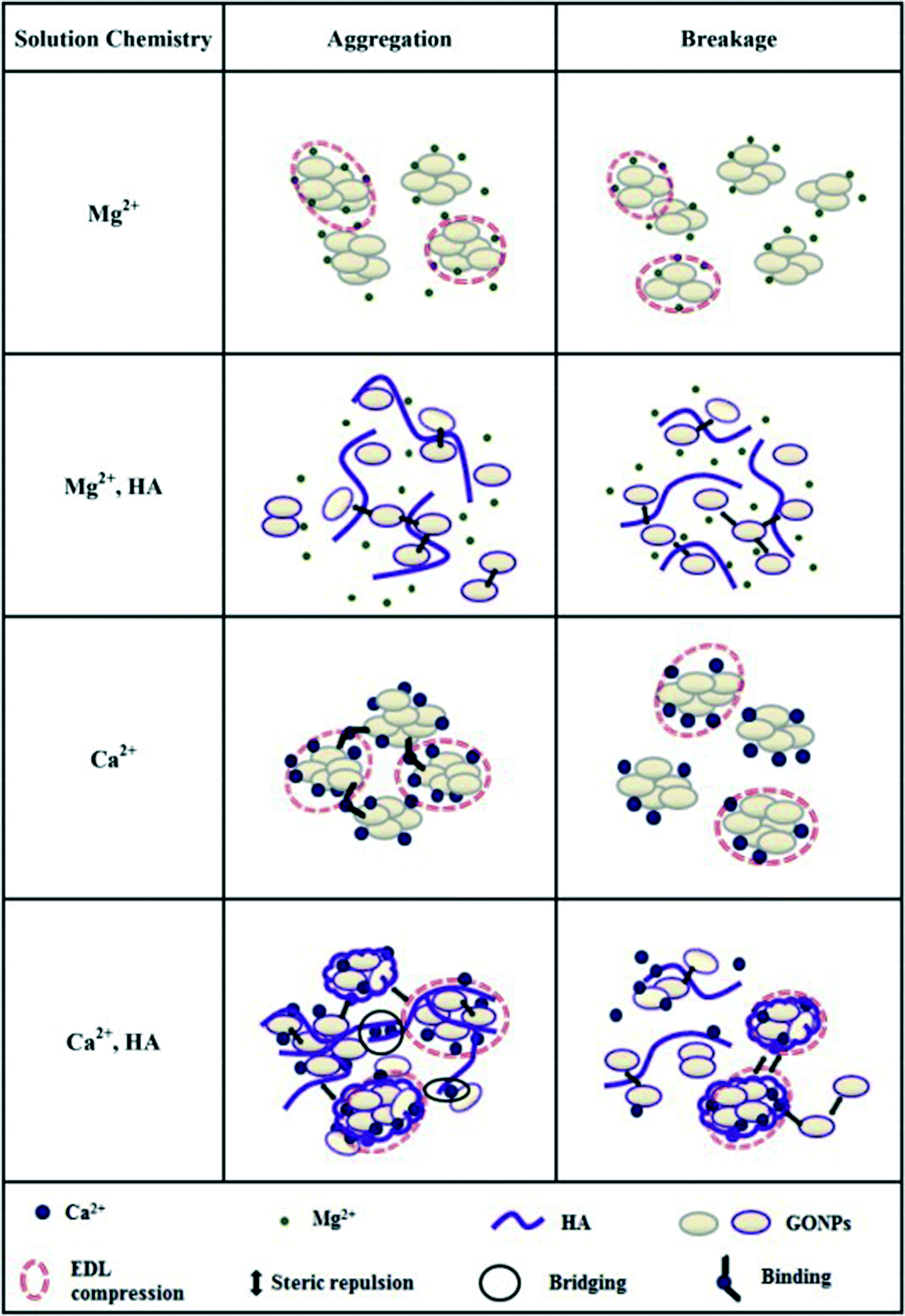 | ||
| Fig. 12 Models of aggregation and breakage of the GO structure. The Lifshitz–van der Waals interaction is not additionally displayed due to its ubiquity.72 Reprinted with permission from ref. 72. Copyright 2015, Elsevier. | ||
Besides the concentration and type of cations, the solution pH value also affects the stabilizing efficiency of SRHA. Hua et al.72 observed that in the presence of SRHA with Mg2+, both the EPM and HD of GO decreased as the pH value increased from 5 to 7 and increased as the pH value further increased from 7 to 9. Accordingly, in the presence of Mg2+, SRHA can enhance the GO stability in an acidic solution and reduce the GO stability in an alkaline solution. The enhancement is due to the steric hindrance of HA surpassing the EDL compression induced by Mg2+. The reduction is due to the desorption of HA which weakens the steric hindrance and electrostatic repulsion. Meanwhile, SRHA cannot stabilize GO in a solution containing Ca2+ at pH 5–9.72 This is because a higher pH would accelerate HA deprotonation and facilitate the bridging effect of Ca2+ on the HA adsorbed on the GO surface.
The NOM character especially aromaticity also has a significant impact on the stabilizing efficiency of NOM. A recent study suggested that the stabilization power of NOM followed the order AHA > SRHA > SRFA in the presence of NaCl.30 The differential stabilization capacity of NOM could be explained by the conformational and structural characteristics of the adsorbed NOM layers by inducing steric hindrance, as determined by both the NOM affinity to GO and the adsorbed layer thickness.93 Hyung and Kim94 and Kennedy et al.95 attributed the greater stabilization effectiveness of HA than FA to its higher aromaticity and molecular mass. Additionally, the stability capacities of NOM vary with the NOM concentration. The high(er) concentrations of NOM in aqueous environments will likely further enhance the stability of GO sheets. As the concentration of AHA increases from 1.7 mg L−1 to 2.7 mg L−1, Qi et al.22 reported that the CCC value of GO for NaCl increases from 1354 to 1572 mM, and Jiang et al.30 reported that the CCC value of GO for NaCl increases from 220 to 267 mM. The aggregation of GO in 0.5 mM Ca2+ can be completely inhibited by 10 mg L−1 SRHA.22
In a word, the stability efficiencies of NOM on GAs vary with the solution chemistries (e.g., solution pH, IS, and complexing cations), since the structure and conformation of NOM are pH-, IS-, and cation-dependent. NOM is more flexible and expanded under higher pH and/or lower IS, and becomes more rigid and compact at lower pH and/or higher IS. Multivalent complexing cations (e.g., Ca2+, Mg2+) could also cause intra-molecular contraction or intermolecular aggregation of NOM due to charge neutralization and cation bridging.96 Besides, the solution pH, IS and complexing cations will affect the interaction of NOM with GO. For example, the adsorption of NOM on GO decreases with increasing pH. Ca2+, as a bridge, could connect the HA polymers to the surface of GO by interacting with the carboxylic and hydroxyl groups on HA and GO (Fig. 13). When attempting to predict the fate of GAs as a function of NOM in a natural aquatic environment, the change of salinity cannot be ignored. The cations (Mg2+, Ca2+, Fe3+, etc.), which can specifically bind with the acidic functional groups of NOM and GAs, will influence the adsorbed NOM layer characteristics and the NOM–NOM, NOM–GA, GA–GA interaction, and thus affect the stability capacity of NOM. In most of the above-mentioned studies, the concentration of NOM varies from 1 to 10 mg L−1 total organic carbon (TOC), which captures the typical range of NOM concentrations in ground and surface waters. The information on the stabilization capacity of NOM with a concentration beyond this range is not available.
 | ||
| Fig. 13 Diagrams of the impact of NOM on GO stability. | ||
Effect of natural colloidal particles
In natural systems, the concentrations of naturally occurring colloids are typically several orders of magnitude higher than those of GO, ranging from 1 to 20 mg L−1 in freshwaters, marginally lower in seawater, and higher in soil solutions.97 Upon release into waters, sediments, and soils, GAs may interact with fine natural colloidal particles via electrostatic interaction, hydrogen bonding, Lewis acid–base interaction, van der Waals forces and hydrophobic interaction.68 Hydrophobic interaction may not be the dominant force for GO-colloid association due to the high hydrophilicity of GO. Heteroaggregation with natural colloids is therefore likely to control the fate, transport, and bioavailability of GO sheets. In principle, heteroaggregation processes of GO with environmentally relevant natural colloidal particles of nano- or micro-size may be of more environmental importance as compared to homoaggregation between GO sheets.98 However, due to the lack of analytical techniques or strategies to investigate heteroaggregation processes, there are only a few studies investigating heteroaggregation processes in the laboratory. In the current situation, the heteroaggregation between GO and nanometer-sized colloids can be quantified by dynamic light scattering, while the heteroaggregation between GO and micrometer-sized colloids can be characterized through batch adsorption and sedimentation experiments. Overall, the hetero-mixtures in such studies show lower stability compared to their homogeneous suspensions under similar electrolyte conditions. The heteroaggregation of GO with natural colloids would reduce the mobility of GO in aquatic environments. Indeed, Wang's group found that the presence of Al2O3, layered double oxides, TiO2, MgO, ZnO, and Mg/Al layered double hydroxides (LDHs) significantly decreases the residual GO concentration in the supernatant.84,99–104 By employing plane-wave-based density functional theory calculations, Wang's group99 stated that hydrogen bonds and electrostatic interactions are the dominant heteroaggregation mechanisms of GO with LDHs (a special positively charged mineral clay) from aqueous solutions (Fig. 14).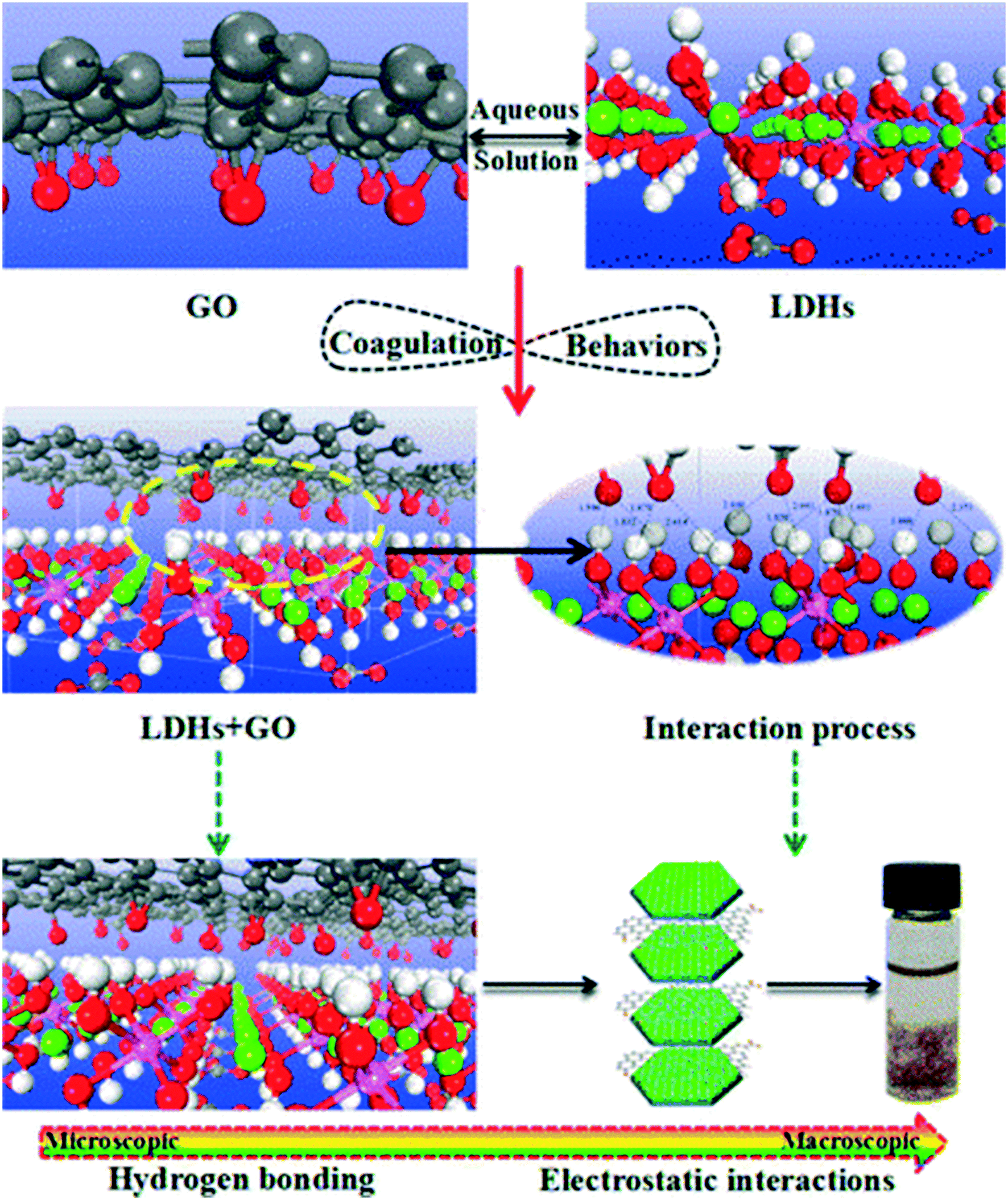 | ||
| Fig. 14 Optimized geometrical structures and coagulation processes of GO on layered double hydroxides (LDHs).99 Reprinted with permission from ref. 99. Copyright 2016, American Chemical Society. | ||
In addition, heteroaggregation of GO with several other mineral clays including montmorillonite, goethite, kaolinite, and hematite has been investigated recently.68,77,79,105 For positively charged goethite and hematite, the heteroaggregation with GO is electrostatically favourable and occurs through electrostatic attraction. In these cases, the heteroaggregation processes are governed by an electrostatic patching effect, resulting in a flat configuration of heteroaggregates. For negatively charged montmorillonite and kaolinite, the heteroaggregation with GO is electrostatically unfavourable due to the strong electrostatic repulsion. Zhao et al.68 observed no adsorption of GO on montmorillonite and kaolinite, indicating the absence of heteroaggregation between GO and montmorillonite/kaolinite. Meanwhile, Yang et al.105 observed the heteroaggregation of GO with kaolinite. They attributed this to charge repulsion being overcome by Lewis acid–base and hydrogen bonding interactions. Huang et al.69 demonstrated that heteroaggregates between GO and kaolinite can be formed at pH below pHPZC of kaolinite. The discrepancy may be due to the inconsistent experimental conditions especially the different sizes and surface charges of the employed kaolinites. Feng et al.77 investigated the heteroaggregation of GO with nanometer- and micrometer-sized hematite colloids and found that the size of hematite affects the stability of GO–hematite heteroaggregates. Accordingly, the physicochemical properties of natural colloids play an important role in their heteroaggregation with GO. In addition, recent studies demonstrated that the concentration ratio of the negatively charged colloid to the positively charged colloid (e.g., silver and hematite,106 carbon nanotubes and hematite,107 and n-CeO2 and pyrogenic carbonaceous materials108) played an appreciable role in their heteroaggregation rates and the conformation of heteroaggregates. Indeed, Feng and co-workers77 established that the trends in the variation of the heteroaggregate growth rate were dependent on the GO/hematite ratio. Generally, the heteroaggregate growth rate increases with an initial increase in the GO/hematite ratio, reaches a maximum at an optimal GO/hematite ratio, and then decreases as the GO/hematite ratio is further increased. At a low GO/hematite ratio, multiple hematite nanoparticles attached to the surface of GO, forming a small amount of large heteroaggregates; at an intermediate GO/hematite ratio, GO served as a bridge between hematite nanoparticles, forming primary heteroaggregates; at a high GO/hematite ratio, hematite was wrapped by GO, forming stable GO–hematite nanohybrids. Therefore, the concentrations of both GO and natural colloids need to be accounted for during the assessment of the mobility of GO. In contrast to the heteroaggregation of GO with colloids, the dissolution of colloids at environmentally relevant pH values has no obvious effect on GO stability and can be neglected.68
The deposition of GO occurs when GO sheets collide with and are sorbed onto an immobile surface in a complex subsurface environment. Relative to the GO sheets, these mineral and soil surfaces can be considered infinitely large particles and therefore this kind of deposition can be studied as a form of heteroaggregation. Several studies have monitored the deposition of GO using a quartz crystal microbalance with dissipation monitoring. Typically, Chowdhury and coworkers56,109 have monitored the deposition and release of GO onto environmental surfaces including silica-, poly-L-lysine-, SRHA-, SRFA-, alginate-, and aluminium oxide-coated surfaces. They found that the deposition and the release of GO onto these surfaces were dependent on the GO concentration, IS, cation nature, and physicochemical properties of the surface. Due to multiple orientation deposition and multilayer formation, a significantly higher mass of GO was deposited on NOM-coated surfaces than aluminum oxide-coated surfaces.56 The deposition of GO on these environmental surfaces is highly reversible, so it is possible to release and re-mobilize the deposited GO from these surfaces in a natural aquatic environment. Photo-transformation caused by sunlight will decrease the deposition of GO on many environmental surfaces and reduce remobilization of GO in aquatic environments.110
Most of the studies on heteroaggregation were carried out in simple aqueous–solid systems. However, in a real natural environment, more than one kind of colloid is usually associated with another. There is only one study investigating the heteroaggregation of GO in a more complex system containing clay minerals (kaolinite) and metal (hydr)oxides (goethite).69 The kaolinite–goethite associations show higher ability to aggregate GO than kaolinite (Fig. 15A). The increase in the content of goethite in kaolinite–goethite associations decreases the stability of GO. Similar to the heteroaggregation of GO with kaolinite, the possible heteroaggregation between GO and kaolinite–goethite associations occurs via kaolinite–goethite associations interacting with the edge of GO sheets and then wrapped by GO at pH < pHPZC of kaolinite–goethite associations (Fig. 15B and C). The heteroaggregation of GO with kaolinite–goethite associations is dependent on IS, pH and GO concentration. The increase in IS and the decrease in pH will reduce the energy barrier between GO and kaolinite–goethite associations, facilitating the occurrence of heteroaggregation. With increasing GO concentration, more large-size GO can efficiently wrap kaolinite–goethite associations, hindering the occurrence of heteroaggregation. These findings are valuable in understanding and predicting the fate of GO in relatively complicated aquatic and soil environments.
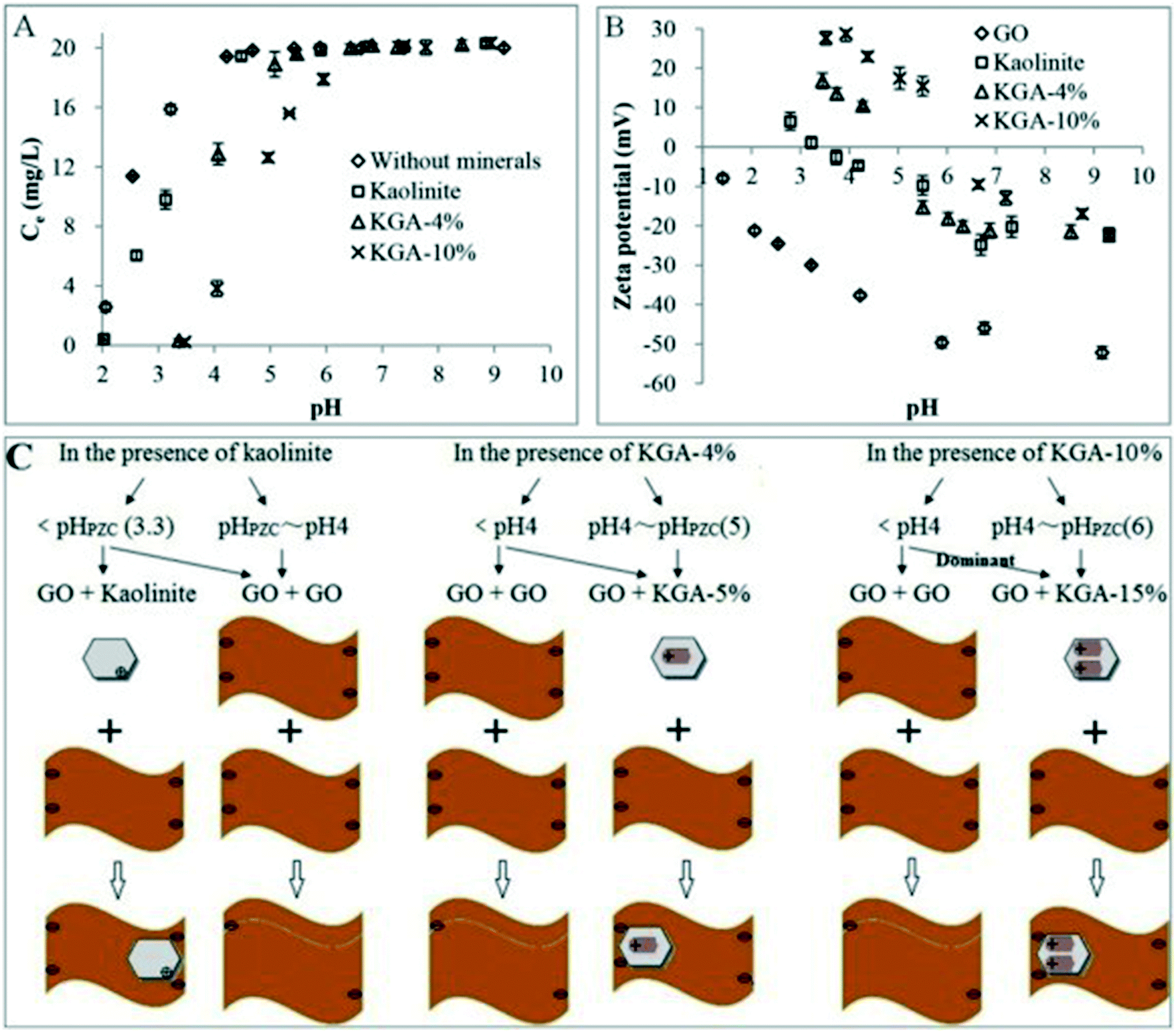 | ||
| Fig. 15 (A) Concentrations of GO in the supernatant as a function of pH in the absence and presence of minerals with 20 mg L−1 of initial GO (without background electrolyte). (B) Zeta potentials of GO and minerals as a function of pH (GO concentration = 20 mg L−1, minerals = 2 g L−1, without background electrolyte). (C) Illustrative diagram of aggregation modes of GO in the presence of minerals as a function of pH.69 KGA-4%, KGA-5%, KGA-10%, KGA-15% mean 4%, 5%, 10%, and 15% of goethite in the kaolinite–goethite association (KGA). Reprinted with permission from ref. 69. Copyright 2016, Elsevier. | ||
Effect of both toxic heavy metal ions and natural colloidal particles
Due to its special physicochemical properties including an extremely large surface area to mass ratio, abundant oxygen-containing functional groups, high negative charge density, and stable stability in aqueous solutions, GO is found to work effectively in removing a range of heavy metal ions and radionuclides.111–113 When GO is employed as an adsorbent for heavy metal ion or radionuclide removal, heavy metal ions or radionuclides may come into contact with GO in a common occurrence. The adsorbed heavy metal ions or radionuclides will have an important influence on the fate and mobility of GO. Recently, several groups have studied the effect of coexisting heavy metal ions or radionuclides on the heteroaggregation behavior of GO with natural colloidal particles. Wang's group114 studied the heteroaggregation behavior of GO with TiO2 in the coexistence of Cu(II) and found that the presence of Cu(II) enhanced GO deposition onto TiO2. Recently, Sheng's group115,116 carried out a series of studies about GO deposition onto LDHs in the presence of a radionuclide (e.g., Eu(III)) or a heavy metal ion (e.g., Cu(II)). They found that the metal ion or radionuclide could enhance the deposition of GO onto LDHs by the formation of LDH–cation–GO ternary surface complexes. Collectively, these studies might facilitate the evaluation of the potential physicochemical behavior of GO after being used as a contaminant carrier in natural aquatic environments.Aggregation behavior of graphene and rGO in aquatic environments
Prevention of graphene aggregation
Due to their strong tendency to form irreversible agglomerates or even restack to form graphite, graphene sheets are unlikely to be stable in water. In contrast to GO, only a few studies characterized graphene aggregation behavior in aquatic environments. Findings suggest that the aggregation behavior of graphene can be affected by the graphene concentration, particle size, ambient water, visible-light irradiation and low-molecular-weight organic acids.5,40,92 FLG agglomerates formed rapidly in deionized water at concentrations higher than 3 mg L−1, and smaller FLG sheets with lateral sizes of 25–75 nm agglomerated more slowly than larger ones.40 The hydration and irradiation weakened the aggregation of graphene.5 Graphene was found to be more stable in the presence of low-molecular-weight organic acids (benzoic acid, gallic acid).92Since the formation of stable aqueous graphene dispersions is important for many applications, many studies concentrated on the prevention of graphene aggregation. The liquid-phase exfoliation of more pristine forms of graphite in the presence of a surfactant is widely employed to prepare relatively defect-free graphene. A low concentration of graphene on the order of 0.01 mg mL−1 can be stabilized by ionic surfactants.117–122 Lin et al.123 investigated the dispersion stability of graphene in the presence of ionic surfactants by molecular simulation, theoretical modelling, and experimental measurements. The corresponding results disclosed that the steric hindrance was more crucial than electrostatic interactions in graphene dispersion. For a high concentration of graphene up to about 1 mg mL−1, Guardia et al.124 found that nonionic surfactants were more effective in comparison with ionic counterparts and pointed out that steric repulsion was more efficient than electrostatic repulsion. The stability of graphene dispersed in an aqueous surfactant solution is influenced by the amount, charge, and morphology of the adsorbed surfactant. Hsieh et al.125 systematically explored the relation between surfactant adsorption and the dispersion stability of graphene. They found the adsorption of sodium dodecyl sulfate (SDS) on graphene can be divided into four stages, including isolated monomer adsorption, monolayer adsorption, surface micelle formation, and bulk micelle formation (Fig. 16). SDS concentrations, above the onset of monolayer adsorption on graphene (i.e., ≥40 μM), are sufficient to achieve colloidal stability in aqueous graphene dispersions. Besides surfactants, polymers are also used to assist aqueous-phase exfoliation of graphite for the scalable production of graphene without oxygen containing groups and defects.126 Bourlinos et al.127 reported that the aqueous-phase exfoliation of graphite in the presence of polyvinylpyrrolidone resulted in a stable aqueous dispersion containing solubilized graphene (Fig. 17). Additionally, a few studies have focused on the role of macromolecules in graphene stability. Recently, it was found that DNA not only enhanced the water solubility and the dispersion of graphene by the formation of a DNA/graphene hybrid (Fig. 18) but it was also used for specific DNA–DNA hybridization, which is an essential requirement for biological applications of graphene.128 By employing MD simulations, it was found that oligodeoxynucleotides could prevent graphene aggregation by the disruption of interlayer van der Waals force.129 Without the aid of either polymeric or surfactant stabilizers, Li et al.19 obtained stable aqueous graphene dispersions successfully by controlling the chemical conversion of GO.
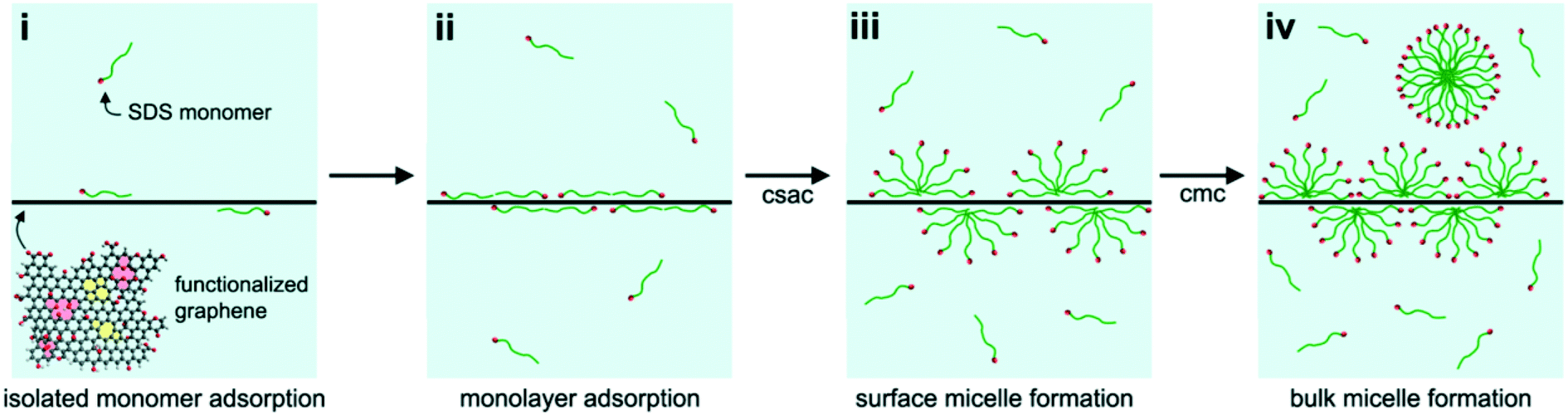 | ||
| Fig. 16 Schematic representation of the four stages of sodium dodecyl sulfate (SDS) adsorption onto functionalized graphene: (i) adsorption of isolated surfactant monomers, (ii) adsorption of a surfactant monolayer, (iii) formation of hemi-cylindrical surface micelles, and (iv) formation of micelles in bulk solution. Also indicated are the critical surface aggregation concentration and the critical micelle concentration. The functionalized graphene schematic shows oxygen functionalities (red), 5–8–5 and 5–7–7–5 topological defects (yellow), and lattice vacancies (pink).125 Reprinted with permission from ref. 125. Copyright 2013, American Chemical Society. | ||
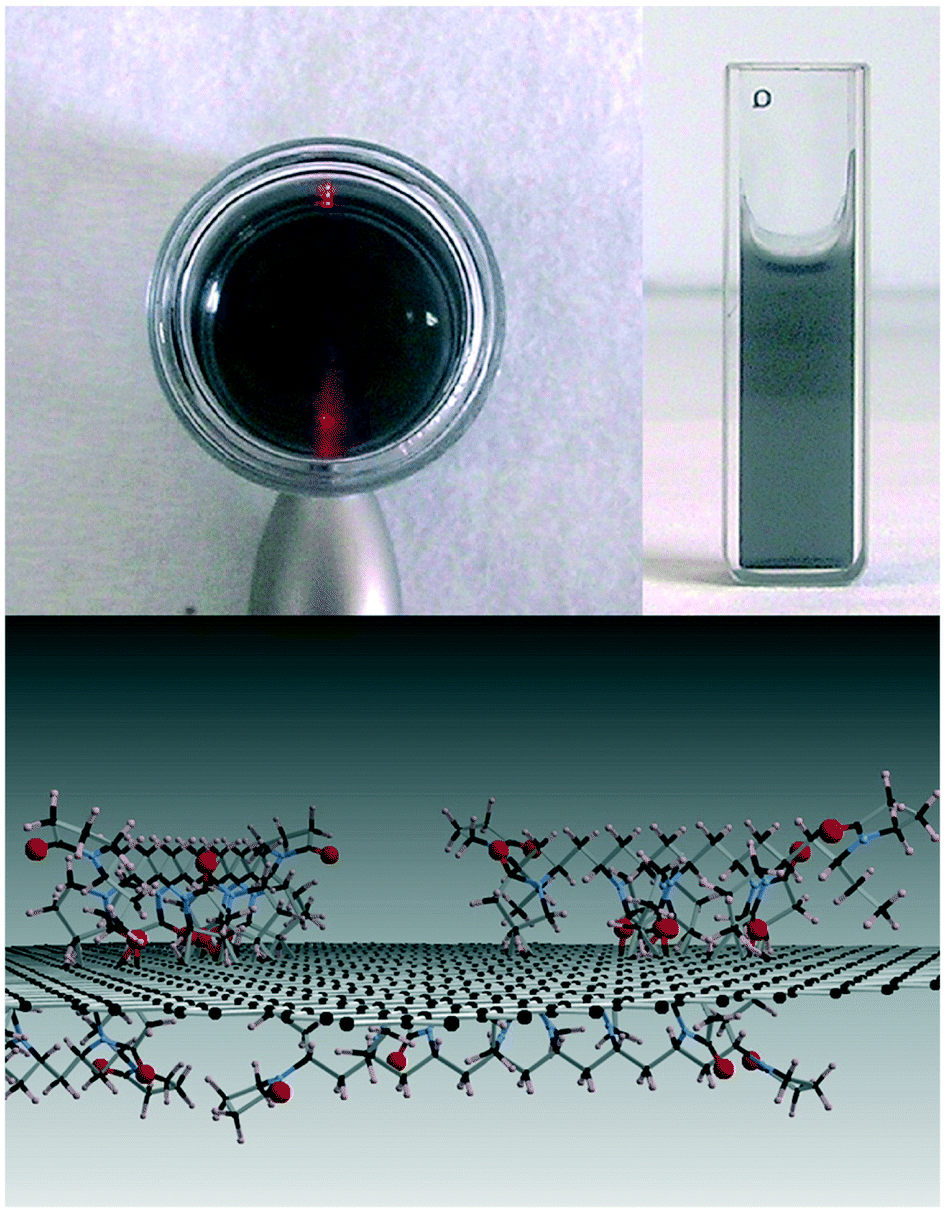 | ||
| Fig. 17 Top: The Tyndall scattering effect is seen in the left image for the aqueous colloidal dispersion of graphite after settling and centrifugation (0.1 mg ml−1). The cuvette on the right shows the optical transparency of the same dispersion. Bottom: Schematic model of polyvinylpyrrolidone-coated graphene.127 Reprinted with permission from ref. 127. Copyright 2009, Elsevier. | ||
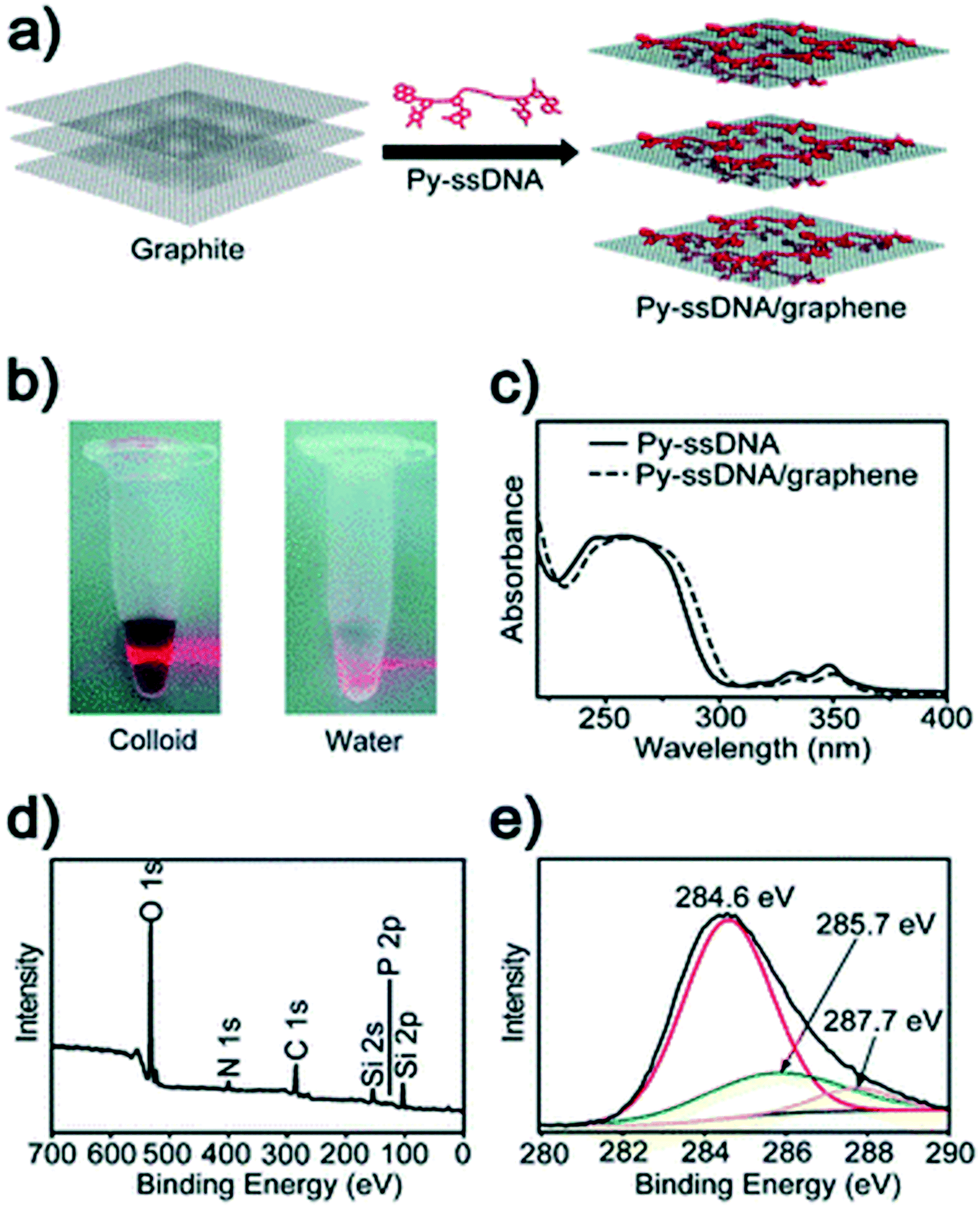 | ||
| Fig. 18 (a) Schematics of the pyrene-labeled single stranded DNA (Py-ssDNA)/graphene hybrid. (b) Tyndall effect of a Py-ssDNA/graphene solution. (c) UV-vis absorption spectra of Py-ssDNA and Py-ssDNA/graphene. XPS data for (d) Py-ssDNA/graphene hybrids and (e) core C 1s level.128 Reprinted with permission from ref. 128. Copyright 2010, Royal Society of Chemistry. | ||
It is reported that external forces such as sonication, mechanical shear force, and milling could provide the activation energy to weaken layer-to-layer van der Waals force between the layered graphene sheets in the presence of a suitable stabilizing agent, thus resulting in a well-dispersed graphene suspension.38 Knieke et al.130 reported that 25 g L−1 of mono- and multi-layer graphene sheets can be produced by stirred media milling. Paton et al.131 reported that large quantities of defect-free graphene suspensions can be produced by shear mixing. Yang et al.132 showed that stirring could induce graphene aggregation in solvents such as benzene, toluene and xylene, which led to reversible agglomeration and folding of graphene sheets.
Aggregation behavior of rGO
Due to the specific reduction approaches used, commercially available rGO will vary remarkably in the concentration, type and distribution of surface oxygen functionalities. The reduction results in four possible consequences: (1) considerable losses of surface oxygen functionalities; (2) a significant increase in the hydrophobicity of GO; (3) partial restoration of the graphitic structure of GO, enhancing the π–π stacking of GO sheets; (4) the presence of cationic groups in hydrazine-reduced GO. All of these increase the aggregation tendency of rGO. Gudarzi23 attributed the origin of the low stability of rGO to the high van der Waals forces among rGO sheets, and particularly, to the removal of negatively charged groups, and possibly the formation of some cationic groups during reduction. Chen's group22,133 mainly attributed the increased aggregation tendency of rGO to the increased hydrophobicity, not to the decreased surface charge negativity. The discrepancy may be due to different study protocols.The reduction degree and the solution chemistry have a significant impact on the aggregation tendency of rGO. In systems dominated by monovalent cations, the aggregation tendency of rGO correlates well with the degree of reduction. Chowdhury et al.55 reported that the aggregation rate of rGO increased in the order rGO-5h (fully reduced) > rGO-2h (intermediately reduced) > rGO-1h (partially reduced). Meanwhile, in systems dominated by multivalent cations capable of forming complexes with surface oxygen functional groups of GO (e.g., Ca2+), the aggregation tendency of rGO can correlate poorly with the degree of reduction but strongly with the type and amount of surface oxygen functionalities, which determine the nature and strength of interactions between rGO and multivalent cations.
Due to the significant decrease in the concentration of surface oxygen functional groups, the findings from the studies on the aggregation and stability of GO as a function of solution chemistry including pH, IS, cation valence, and NOM will be not applicable to rGO. In contrast to GO, pH values have a great influence on rGO stability. In aqueous media, rGO sheets are dispersible only above a pH value of 8.27,35 At a matching pH, rGO sheets aggregate at a much lower IS compared to GO. The aggregation of rGO induced by Ca2+ ions is not as significant as that observed for GO. In the presence of Ca2+, NOM shows greater effectiveness in inhibiting the aggregation of rGO, since rGO has fewer surface functional groups than GO, binding with Ca2+ in the presence of NOM.55 The findings further underline the stability of nanomaterials being controlled by the complex interplay between nanomaterial surface properties and solution chemistry factors.
Aggregation behavior of GAs in natural and synthetic waters
The conditions in natural aquatic environmental systems are very complex. Surface water and groundwater environments include a complex mixture of ions (e.g., Na+, K+, Mg2+, Ca2+, Cl−, HCO3−, and SO42−) and NOM, which can have a significant impact on the stability of GAs.28,55,84 Currently there are several studies that investigate the effects of complex solution chemistry found in surface water and groundwater environments on the stability and behavior of GAs. For example, Chowdhury et al.28 studied the initial aggregation kinetics of GO for 60 min and the long-term stability of GO over 28 days in natural and synthetic waters including Call's Creek water, Oconee wastewater, synthetic surface water and synthetic wastewater. They found that the stability of GO both over very short time frames and over longer time scales depends on the water type. They pointed out that the initial aggregation kinetics data cannot predict the long-term stability of GAs in natural aquatic environments. In another study, they investigated the long-term stability of rGO in Call's Creek water, synthetic surface water, synthetic groundwater, wastewater treatment plant effluent, and synthetic wastewater and showed that the combination of NOM and divalent ions played major roles in the stability of rGO.55 Similarly, Lanphere et al.73 found that there was a significant difference between the behavior of GO in groundwater and surface water systems, since groundwater and surface water systems had different concentrations of hardness (e.g., Ca2+ and Mg2+) and NOM. Recently, Su et al.40 characterized the sedimentation kinetics of FLG in eight ambient waters (i.e., East China Sea, Yangtze River, Beihai Lake, Qinghai Lake, Bapai Spring, Daijia Deng Spring, wastewater treatment plant influent and wastewater treatment plant effluent) (Fig. 19) and found that besides the water type (relating to IS and NOM), the concentration of FLG had a notable influence on the stability of FLG. Our group67 systematically studied the sedimentation kinetics of GO in three natural surface waters collected from Dongpu Lake, Nanfei River and Chaohu Lake in Hefei City (Fig. 20). We found that water hardness (i.e., the concentration of Ca2+ and Mg2+) played a key role in the sedimentation processes and that NOM in the three natural surface waters was not high enough to stabilize GO. As a result, GO may be much less stable and mobile in Dongpu Lake, Nanfei River and Chaohu Lake than expected. In another study,84 we observed that when GO sheets were in Dongpu Lake water, synthetic groundwater, synthetic surface water, and tap water, they settled down to the bottom of the bottle (Fig. 21). In a word, the fate of GO in natural aquatic environments are a complex function of many factors. In order to more accurately assess the environmental impact and the ecological risk of GAs, the colloidal properties of GAs should be investigated under environmentally relevant conditions.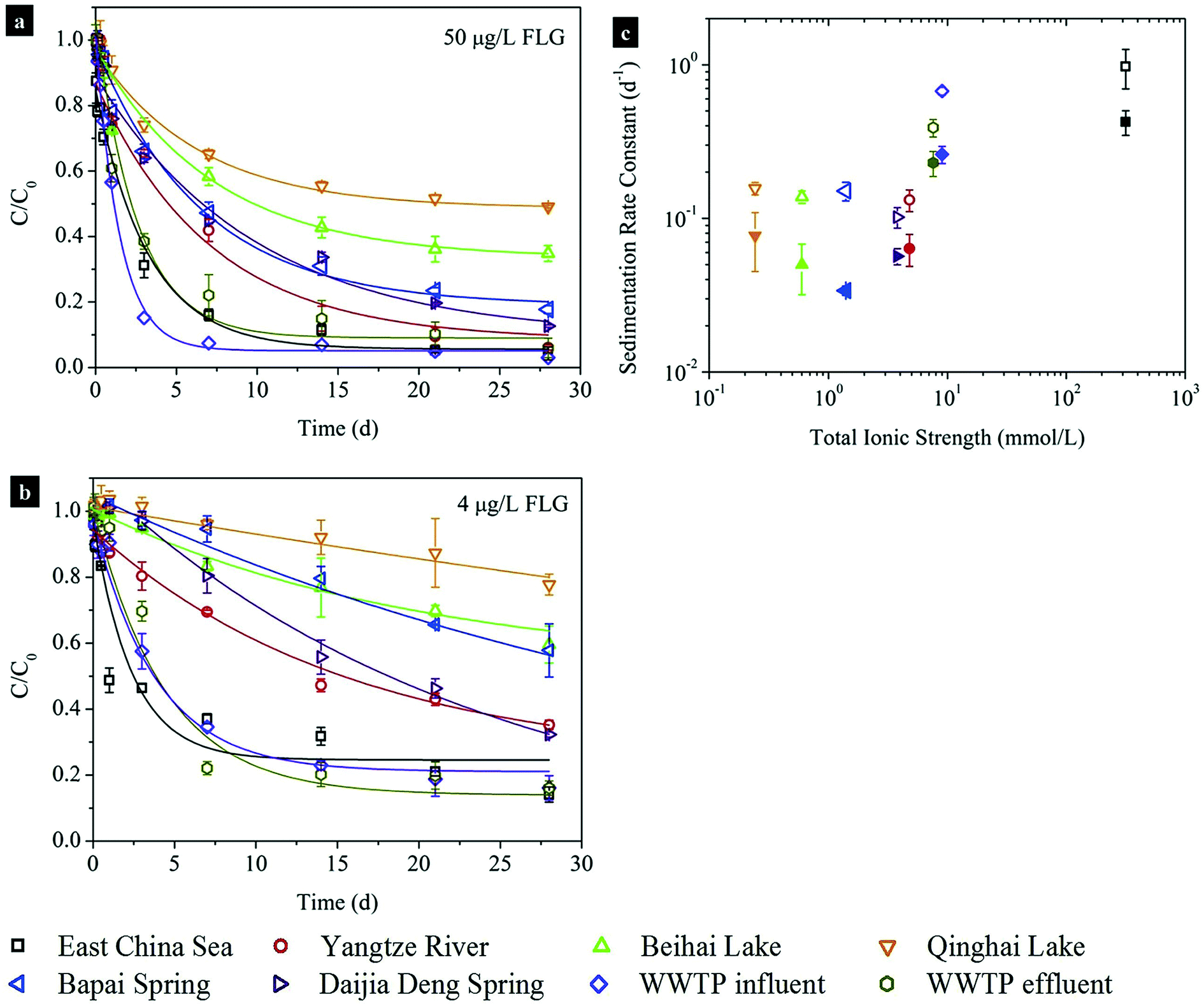 | ||
| Fig. 19 Long-term stability of few layer graphene (FLG) in ambient waters and wastewater samples from a municipal wastewater treatment plant (WWTP). Normalized FLG concentrations in different types of water; the initial FLG concentration was (50 ± 2) μg L−1 (a) and (3.90 ± 0.03) μg L−1 (b), respectively (n = 3; data points are mean and standard deviation values calculated from triplicate samples). Sedimentation rate constants (c) of FLG at initial concentrations of 50 μg L−1 (open symbols) and 4 μg L−1 (solid symbols) as a function of total ionic strength of the water samples.40 Reprinted with permission from ref. 40. Copyright 2016, Elsevier. | ||
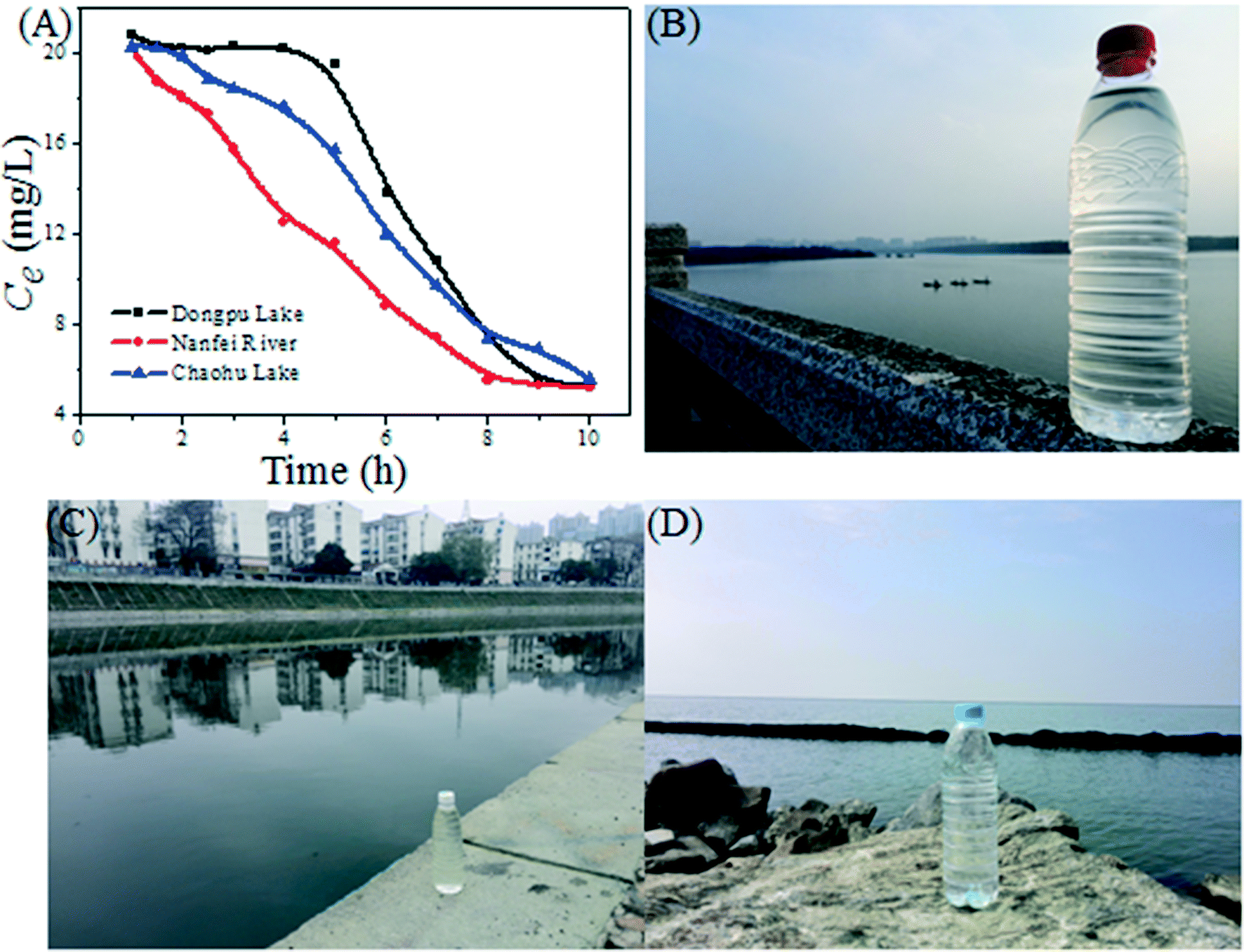 | ||
| Fig. 20 (A) Sedimentation kinetics of GO sheets in the three natural surface waters collected from Dongpu Lake (B), Nanfei River (C), and Chaohu Lake (D).67 Reprinted with permission from ref. 67. Copyright 2017, Elsevier. | ||
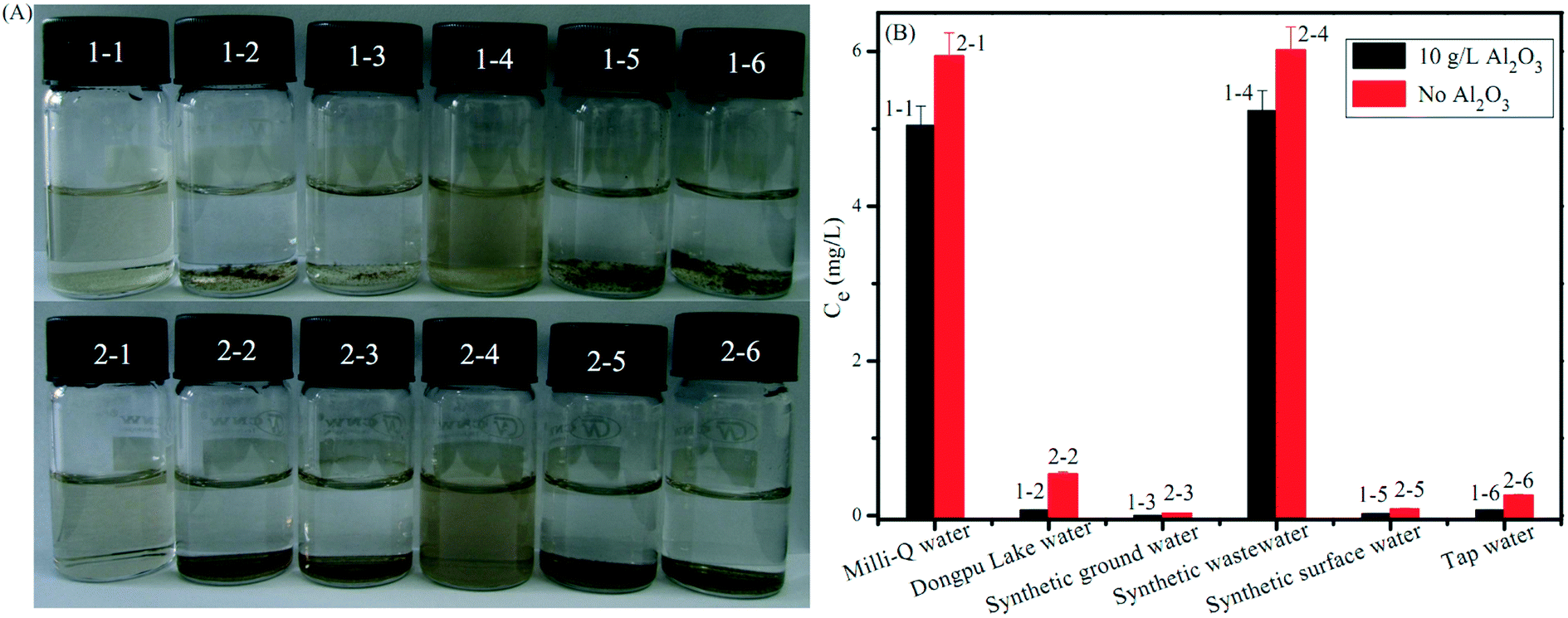 | ||
| Fig. 21 (A) Visual images of GO in the absence and presence of Al2O3 in different waters. (B) Concentrations of the residual GO sheets in the supernatant as a function of cation types and concentrations in the absence and presence of Al2O3 in natural and synthetic waters. C(GO)initial = 6 mg L−1, m/V = 10 g L−1 (i.e., Al2O3 concentration).84 Reprinted with permission from ref. 84. Copyright 2014, American Chemical Society. | ||
GAs in comparison with other carbonaceous nanomaterials
Carbonaceous nanomaterials mainly include three types of stable forms, namely fullerene (C60), carbon nanotubes (CNTs), and graphene (or GO). Different extents of sp3 (aliphatic) or sp2 (aromatic) hybridization of the carbon atoms in the carbonaceous nanomaterials lead to different bonding and ring structures. The defect-free C60 in enclosed cage-like structures is composed of twelve 5-member rings (sp3) and an unspecified number of 6-member rings (sp2). As the 2D counterpart of naturally occurring 3D graphite, graphene is a hypothetical infinite aromatic sheet with sp2 hybridization of the carbon atoms. The CNT structure can be considered to stem from the folding of one or several graphene sheets (sp2-hybridized carbon) aligned in a concentric manner. GO can be regarded as the oxidized graphene with the basal planes decorated mostly by epoxide and hydroxyl groups and the edges by carbonyl and carboxyl groups. This topological variation results in different physicochemical properties (e.g., morphology, size, specific surface area, functional groups, surface charge, polarity, and so on) among carbon nanomaterials and different interactions of the carbon nanomaterial substrates with themselves or with guest molecule media (e.g., aqueous media, porous media) and consequently variation in aggregation and deposition behavior. The huge difference in physicochemical properties of C60, CNTs, and GO results in fundamental differences in the interfacial-interaction characteristics of these carbon nanomaterials in aquatic environments. Therefore, the factors controlling the colloidal properties of carbon nanomaterials can be classified into peculiar factors and common factors. The common factors include pH, IS, cation nature, NOM and sunlight (or UV) exposure.28,75,110,134 The peculiar factors for C60 include preparation methods,135 size (hydroxylation degree),136 and ozone.137 The peculiar factors for CNTs include synthesis methods,138 chiral literature,139 diameter,140 surface oxidization degrees,141 and purification.142 The peculiar factors for GO include the reduction degree,22 lateral size,40 and layer.123The distinct differences in the physical shape and surface chemistry of GO compared to those of C60 and CNTs lead to the colloidal properties of GO, in response to common factors, differing distinctly from those of other carbonaceous nanomaterials. GO is more resilient than C60 and CNTs to changes in solution chemistry (e.g., pH, IS) that favor aggregation and deposition of C60/CNTs. Specially, unlike C60 having to undergo solvent exchange or sonication and CNTs requiring the addition of stabilizing reagents, GO can possess negative surface charges and is stable in relatively wide ranges of environmental conditions. Its relatively hydrophilic nature makes GO more mobile than C60 and CNTs, even the surface oxidized ones. In the case of GAs, the stability of GO is higher than that of rGO in aqueous solutions. Although the shapes of CNTs and GO are not spherical, their aggregation and stability in aquatic environments are found to follow colloidal theory, including DLVO theory and the Schulze–Hardy rule, as C60 does.28,88,143
It is reported that the aggregation of CNTs could be decreased in the presence of NOM (SRHA), polysaccharides (alginate), proteins (bovine serum albumin), and cell culture medium (Luria–Bertani broth).144 TA also shows the ability to stabilize the CNT suspension via steric repulsion.145 It is likely that a stable graphene or rGO suspension might be obtained in the presence of the above-mentioned macromolecules due to the structural similarity of graphene or rGO with CNTs. Additionally, Zinchenko et al.146 found that long-chain DNA–chitosan complexes can be employed to entrap C60 and CNTs and to remove C60 and CNTs from water by centrifugation, filtration, or decantation; similarly, decontamination from GO can be achieved by other combinations of low-cost natural (e.g., alginic acid, chondroitin sulfate, etc.) or synthetic (e.g., poly(acrylic acid), carboxymethyl cellulose, etc.) polyelectrolytes with DNA. Besides, due to their great water solubility and hydrophobic central plane, the GO sheets can disperse carbonaceous nanomaterials including C60, graphene and CNTs to form stable suspensions via the mechanism of strong π–π interaction.26,29
Transport and deposition behavior of GAs in porous media
A key process of nanomaterial exposure is infiltration from the point of nanomaterial release to the subsurface system. A number of laboratory studies have reported that the mobility of GAs in porous media is controlled by several subsurface environmental factors, including solution chemistry (e.g., IS, cation type, NOM, and pH), hydrodynamic factors (e.g., flow rate, moisture content), particle concentration and surface properties, and media characters (e.g., grain size, surface potential). In the following subsections, we will elaborate the factors in the column system affecting the transport and retention of GAs. It is possible that a combination of several processes, such as dissolution, homoaggregation, heteroaggregation, straining, and deposition, is responsible for the high retention of nanoparticles.Effect of pH
Under unfavorable attachment conditions, pH affects transport mainly by masking the heterogeneities of grain surfaces (e.g., metal oxides). DLVO profiles show that solution pH has very small effects on the depth of secondary minimum for rGO transport in quartz sand,133 little effect on the interaction force/energy between the carboxyl-functionalized graphene and the two types of sand (i.e., acid-cleaned and natural sand),147 negligible effects on the depth of the secondary minimum energy well and only small effects on the repulsive energy barrier between particles and collectors for the transport of GO in quartz sand.148 By comparing the breakthrough curves, Qi et al.148,149 observed that increasing the pH resulted in slightly enhanced transport of GO in Lula soil, had a negligible effect on pre-cleaned Sigma sand, and had a small effect on the transport of GO in quartz sand. The author attributed the small enhancement in transport to the effects of pH on soil grains and concluded that the major mechanism affecting the transport of GO in quartz sand is the deposition in the secondary minimum. Similarly, Liu et al.147 reported that the increase in solution pH can enhance the mobility of carboxyl-functionalized graphene in natural sand media. They attributed the lower deposition to the influence of solution pH on the surface charges of media. As for the transport of rGO in saturated quartz sand, the pH effects are largely dependent on the background cation type. Xia et al.133 reported that when the background electrolyte was Na+, increasing the pH value from 5 to 9 had a drastic effect on the transport of rGO mainly by affecting the EDL properties of sand grains; meanwhile when the background electrolyte was Ca2+, the pH effect was negligible due to Ca2+ bridging. Findings show that mathematic models based on the advection–dispersion–reaction equation could be used as a monitoring tool to predict the fate and transport of GAs in soil and groundwater systems in some cases.Effect of IS and ion types
Natural aquatic environments contain many different ions. These ions, varying significantly in charge density, size and complexing capability, can affect the transport of nanoparticles very differently. DLVO calculations suggest that the particle–surface interaction energy profiles are sensitive to changes in IS, and the energy barrier gradually decreases with increasing IS. The laboratory tests show that increasing the IS reduces the transport of GAs in saturated sand packs.70,150–152 Cations can enhance the deposition of GAs in the following ways. First, cations affect the size of GA aggregates by decreasing the electrostatic repulsion between GA–GA sheets, which in turn may affect the transport properties of GAs through physical straining. Second, cations affect the surface charges of GAs and collectors, facilitating the interaction of GAs with collectors. Third, cations deepen the secondary minimum energy well. Fourth, accumulation of cations on the surface of collectors may interfere with GA deposition through steric hindrance. Fifth, some cations can serve as a bridging agent, strengthening the interaction of GAs with collectors. As IS increases, GAs have the capacity to aggregate more, to interact with the quartz sand collectors increasingly and consequently to deposit more. Previous studies have shown that a divalent cation (e.g., Ca2+) is more effective in inhibiting the transport of GAs than a monovalent cation (e.g., Na+),133,153 not only because a divalent cation is more effective in compressing the EDL than a monovalent cation but also because it can increase the deposition of GAs via cation bridging.Interestingly, cations with the same valence have markedly different effects on the transport of GO and rGO. Xia et al.153 found that the effects of cations on the transport of GAs in saturated quartz sand obeyed the Hofmeister series, decreasing in the order Na+ < K+ < Cs+ for alkali metal ions and Mg2+ < Ca2+ < Ba2+ for alkaline earth metal ions. When Na+ and Ca2+ coexist, the molar ratio of Ca2+ to Na+ plays an important role in the transport of GO in saturated porous media especially at high IS.154 However, unlike aqueous solutions, studies on the effect of coexisting heavy metal ions on the transport of GO in soil are far fewer and need further investigation.
Effect of NOM and surfactants
NOM is ubiquitous in natural surface water environments. Under unfavourable deposition conditions, NOM likely enhances the transport of GAs via the following mechanisms. First, the adsorption of NOM on the surfaces of GAs and collectors enhances the steric repulsion between GAs and collectors, thus inhibiting the deposition of GAs. Second, the adsorption of NOM on GAs reduces the size of GA aggregates, affecting the deposition of GAs via straining. The effectiveness of NOM in enhancing the transport of GAs is dependent on the cation type, cation concentration and collector nature. Xia et al.133 reported that SRHA was much less effective in mitigating the transport-inhibiting effects of divalent cations (i.e., Ca2+) than those of monovalent cations (i.e., Na+). Qi et al.148 found that the transport enhancement effect of SRHA was more profound at a higher IS (35 mM NaCl) compared to that at a lower IS (10 mM NaCl). In another study, Qi et al.149 observed that SRHA showed weaker effects on the transport of GO in Lula soil than in Sigma sand. Surfactant modification is a common practice to disperse graphene. Although it is mainly used as a dispersion agent, the surfactant also plays important roles in controlling the environmental fate and transport of GAs. Liu et al.155 found that the surfactant type and concentration strongly affected the retention and transport of graphene in porous media.Effect of collector properties
Sand with a negative charge is widely used as a collector to investigate the transport and deposition of GAs in porous media. Negatively charged GAs are expected to deposit on negatively charged sand surfaces due to the existing strong primary energy barriers. Instead, GO sheets may interact with sand in a secondary minimum. The sand grain size has a strong influence on the amount of GO transport and retention.51,156 Sun et al.157 investigated the transport of GO in laboratory columns packed with quartz sand of three grain sizes. They found that the retention of GO at a given initial concentration tended to increase with decreasing sand size (e.g., fine > medium > coarse).Very recently, researchers have also noted that different surface coatings on sand collectors show different influences on the retention of GAs. He et al.158 reported that extracellular polymeric substances exhibited a negligible influence on GO transport due to the hydration effect and steric repulsion. In another study, they reported that biofilms significantly enhanced the retention of GO due to the following reasons: (1) GO specially interacts with biofilms via H-bonds, Lewis acid–base, and π–π interactions; (2) the inhomogeneous coverage of biofilms increases the physical roughness of the sand collector, creating a low flow velocity region; (3) biofilms decrease the absolute surface charge of the collectors and increase the degree of charge heterogeneity of the collectors; (4) biofilms grown on sand surfaces reduce the porosity and narrow the pore sizes of packed columns, thereby greatly inducing physical straining.151 Wang et al.159 reported that hematite coating reduced the mobility of GO in sand due to surface charge heterogeneity. In addition, surface impurities also affect the deposition and transport behaviors of GAs in saturated porous media via surface charge heterogeneity. Liu et al.147 reported that the mobility of carboxyl-functionalized graphene was lower in natural sand than in acid cleaned sand. A similar phenomenon was observed by Wang et al.160 They attributed the higher retention of GO in natural sand columns than in acid-cleaned sand to the presence of additional attachment sites and metal impurities.
Effect of other factors
The decrease in the flow velocity greatly enhances the retention of graphene.51 Meanwhile the effect of flow velocity on the transport of GO depends on the type of collector and IS. Qi et al.149 observed that the flow velocity had noticeable effects on the transport of GO in Lula soil and had essentially no effects on the transport of GO in Sigma sand. In another study, Qi et al.148 observed that the varying flow velocity had little effect on the transport of GO at low IS (e.g., 10 mM) and a notable effect on the transport of GO at high IS (e.g., 35 mM). Furthermore, the moisture content also plays an important role in the retention of GO in porous media.152,156 Reducing the moisture content increases the retention of GO in porous media. The mobility of GAs is lower in unsaturated porous media than in saturated porous media due to film straining.152 The input concentration of GAs shows different influences on the transport of GO and graphene in saturated quartz sand. Sun et al.157 reported a decreasing trend of GO retention with increasing input concentration and attributed this effect to fast blocking of a limited number of deposition sites by a higher particle concentration. In contrast, Sun et al.51 reported that an increase in the particle concentration caused greater retention of graphene via straining. The discrepancy is attributed to the different surface properties of graphene from those of GO. In addition, the transport of GAs is also affected by the temperature. A higher temperature results in less mobile GO sheets.160 However, the mechanisms remain unclear. Recently, Lu et al.161 studied the effects of three typical clay minerals (kaolinite, montmorillonite, and illite) on the transport of GO in porous media and found that the transport of GO was inhibited remarkably in the presence of clay minerals, following the order kaolinite > montmorillonite > illite. Zhou et al.162 found that the addition of a minute quantity of GO can cause a significant change in the physical and mechanical properties and microstructure of clayey sand. These findings disclose that the transport of GAs in actual subsurface systems is very complex.Transformation and degradation of GAs in aquatic environments
Once GAs enter the natural environment, they can undergo physical and/or chemical transformations. Possible chemical transformations include (1) degradation of surface coatings when in contact with sunlight, (2) oxidation/reduction as a result of exposure to concomitant chemicals such as hydroxyl radicals, ozone, Fe(II), sulfides, chlorine or chloramine, (3) bio-transformations when GAs are exposed to microorganisms or plants. All of these transformation processes could modify the environmental behavior (e.g., transport and aggregation state) and ecological effects (e.g., toxicity) of GAs. So far, the numerous studies on the transformation of GAs have been carried out under laboratory conditions, not in real natural environments.Contact with sunlight and UV irradiation
Sunlight photolysis is one of the primary routes by which GAs react in natural waters. The photo-transformation shows different effects on graphene and GO. Generally speaking, depending on the original state of the carbon surface (oxidized or not), the overall effect of photolysis appears to fall into one of the two categories, oxidation or reduction. It has been shown that photolysis often results in the introduction of oxygen containing functional groups onto the graphene surface. For example, Hu et al.5 reported that graphene was gradually oxygenated after visible-light irradiation under an ambient atmosphere. They also found that the irradiated graphene was more stable and less toxic than graphene.In contrast, photo-reduction appears to be the dominant transformation route of GO. Irradiation time has an important effect on the photo-chemical transformation of GO. Andryushina et al.25 found that the hydrodynamic size of GO increased sharply after UV light exposure for 30 min due to the elimination of certain functional groups of GO and the breakup of H-bonds between the fragments of GO particles. After UV light exposure for 90 min, the hydrodynamic size of GO decreased due to the π–π stacking interaction between the aromatic areas of GO and crumpling of the GO sheets (Fig. 22). After 1 h of irradiation, Abraham et al.163 observed the formation of nanopores during the photo-reaction processes of GO in the presence of O2 (Fig. 23). After 11 h of irradiation, Hou et al.164 found that GO was photo-chemically converted into CO2, low molecular-weight species, and fragmented photo-products similar to rGO (Fig. 24). Lastly, they predicted that photo-transformed products will show different transport behaviours and eco-toxicological effects from the parent GO. Indeed, Chowdhury et al.110 found that the irradiation of simulated sunlight increased the aggregation of GO and decreased the deposition of GO on SRHA coated silicon with lower possibility of remobilization. Namely, the photo-transformation can reduce the stability of GO in aquatic environments, increase the mobility of GO and reduce the remobilization of GO in aquatic-terrestrial transition zones, where NOM-coated surfaces commonly exist. Recently, Hou et al.165 reported that the bacterial toxicity of directly photo-transformed GO (without H2O2) increased due to greater cell membrane disruption and oxidative stress, while that of indirectly photo-transformed GO (with H2O2) decreased due to the loss of the TOC. During water and wastewater treatment, GO may come into contact with other chemicals used in water treatment (e.g., chlorine) in addition to sunlight irradiation. Du et al.166 observed that simulated sunlight irradiation enhanced the chlorination of GO depending on the chlorine concentration. Finally, the colloidal stability and the antibacterial effect of the photo-chlorinated nanomaterials decreases and increases, respectively.
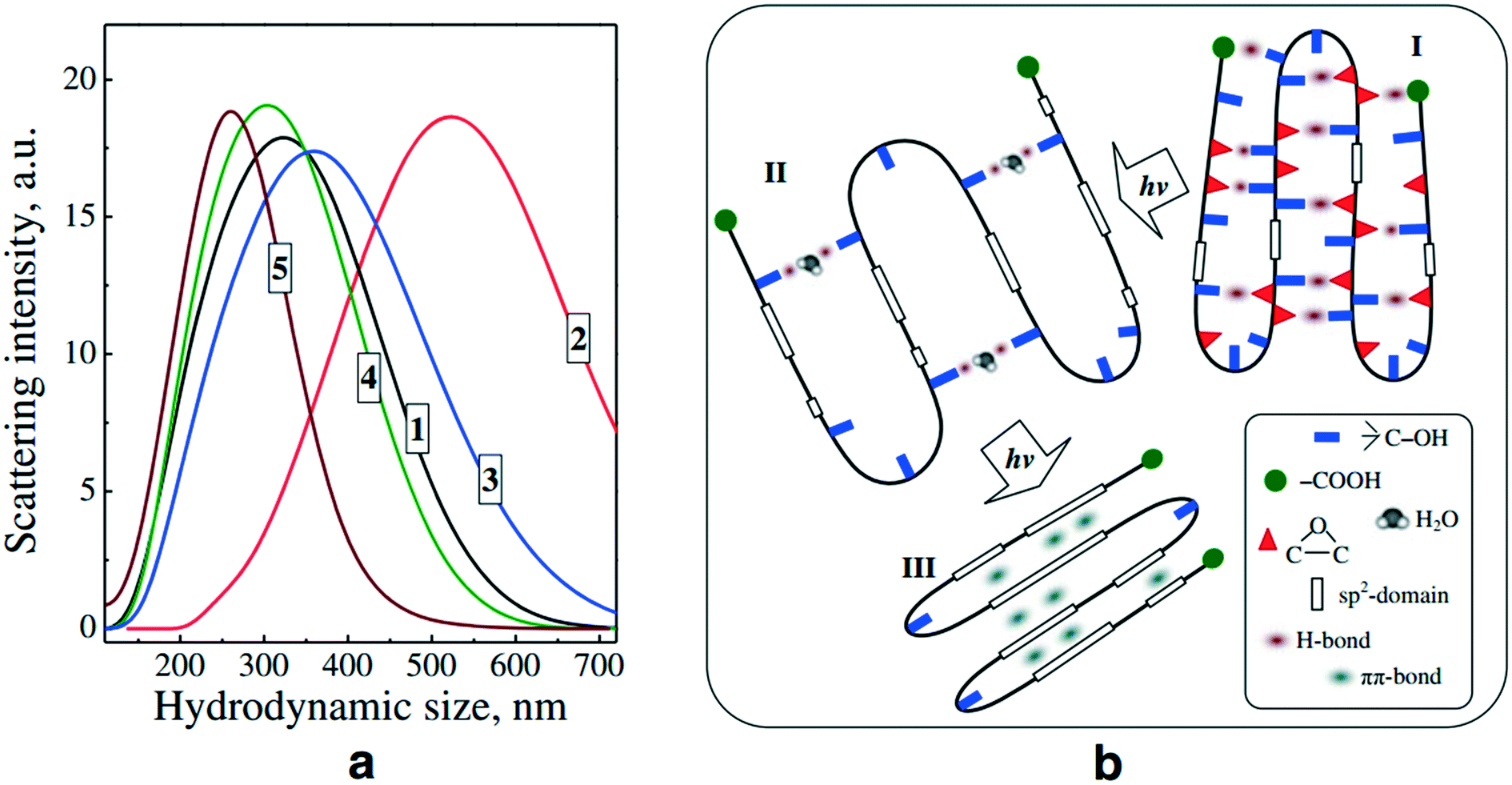 | ||
| Fig. 22 (a) Hydrodynamic size distribution of colloidal GO at pH 6 before photo-reduction (curve 1) and after illumination with UV light for 30 min (curve 2), 60 min (curve 3), 90 min (curve 4), and 180 min (curve 5). GO = 0.025 mg mL−1. (b) Photo-induced changes in shape and structure of colloidal GO particles.25 Reprinted with permission from ref. 25. Copyright 2013, Springer Nature. | ||
 | ||
| Fig. 23 (a) Models of the pore production by the photoreaction of GO in N2 and O2. (b) Model of proton conduction at epoxide groups.163 Reprinted with permission from ref. 163. Copyright 2012, American Chemical Society. | ||
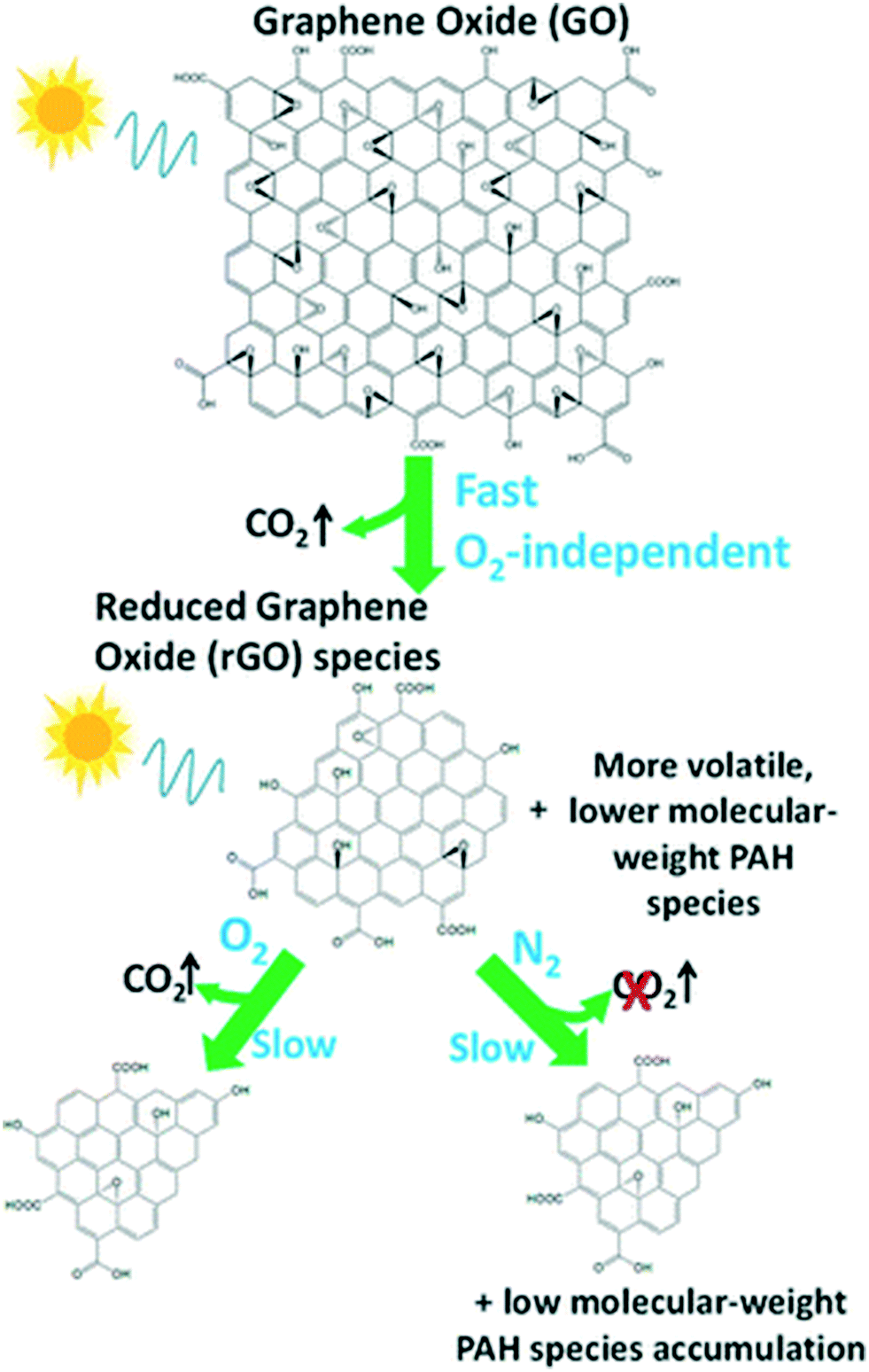 | ||
| Fig. 24 Pathways of GO photo-transformation in sunlight.164 Reprinted with permission from ref. 164. Copyright 2015, American Chemical Society. | ||
Contact with microorganisms
Our recent study showed that GO would coat the bacteria and cause cell membrane damage,167,168 but the fate of GO after being in contact with GO resistant microorganisms needs to receive much attention. It is well established that after being in contact with GO resistant microorganisms, GO sheets would be reduced or degraded by some microbes (Fig. 25), which may further cause changes in the fate and toxicity of GO in aquatic environments. For the first time, Salas et al.169 reported that GO could be reduced by the metal-reducing and environmental microbe from Shewanella strains under strictly anaerobic conditions, which was confirmed by Jiao et al.170 Wang et al.112 also confirmed similar observations and further found that the microbial reduction of GO by Shewanella can occur in a normal aerobic cultural setup. Afterwards, GO reduced by Escherichia coli, one of the most widely present stains in aquatic environments, was observed under anaerobic conditions.171 In addition, it has been found that many other microbial species including Escherichia fergusonii,172Halomonas strains,173Bacillus marisflavi,47Azotobacter chroococcum,174Bacillus subtilis,175Gluconacetobacter xylinus,176Fontibacillus aquaticus isolated from nanomaterial contaminated pond soil,177 aerobic microorganisms isolated from river sediments,178 entophytic microorganisms present in the carrot root,179 and even baker's yeast180 are capable of reducing GO to rGO. After being in contact with bacteria, the transformation of GO occurs via the mechanism of bacterial respiration and glycolysis. Several groups found that besides the reduction potential, the biomass could also act as a stabilizing agent, resulting in the good stability of synthesized graphene in water.47,181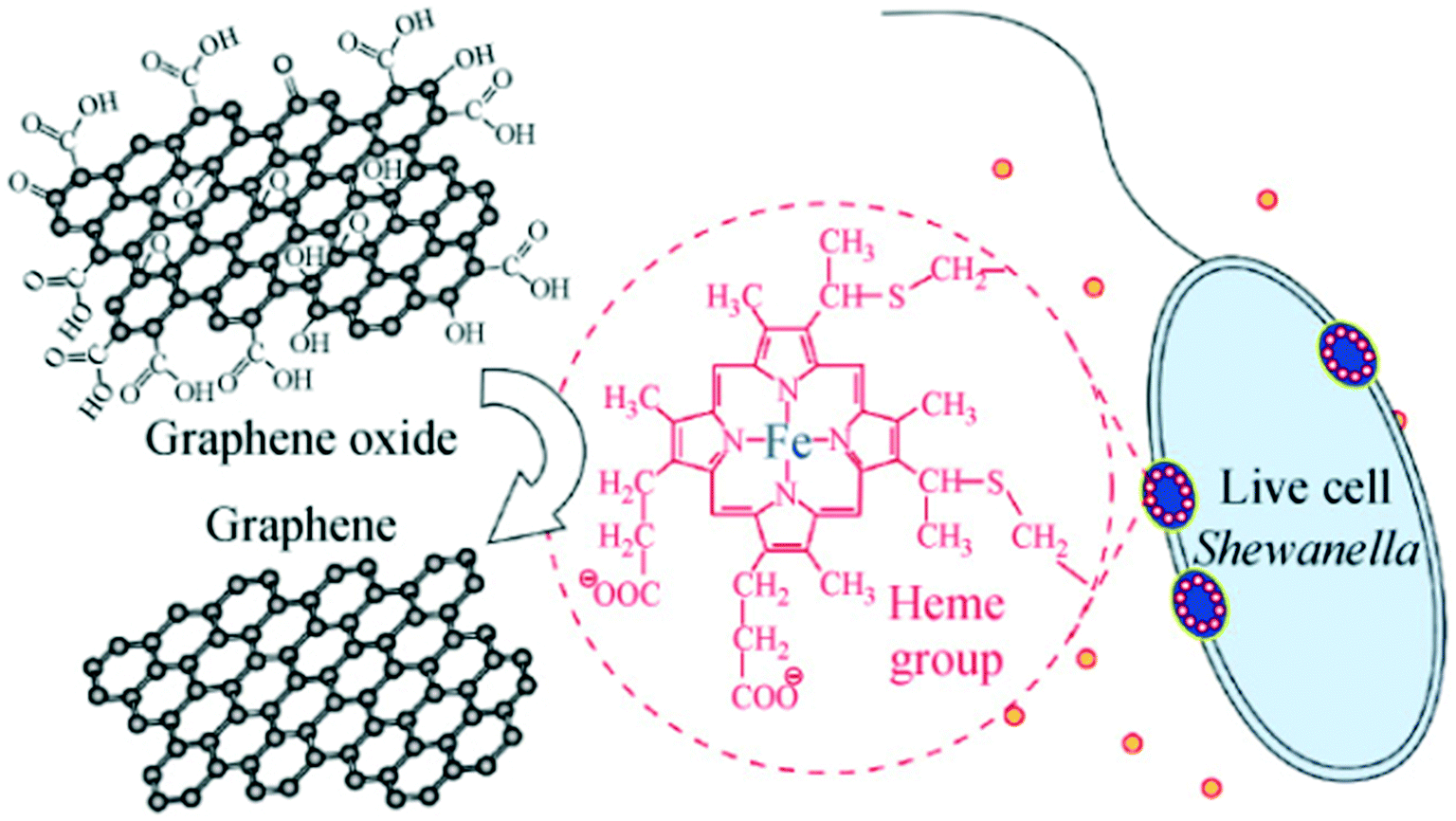 | ||
| Fig. 25 Proposed mechanism of microbial reduction of GO. Orange dots represent self-secreted electron mediators; blue circles with white dots represent multiheme-containing outermembrane c-type cytochromes; the molecular structure of the heme group is shown in the dashed circle.44 Reprinted with permission from ref. 44. Copyright 2011, Springer Nature. | ||
A few works have reported that bacteria show the ability not only to reduce GO but also to oxidize graphitic materials (graphite and rGO). Liu et al.182 reported that naphthalene degrading Pseudomonas bacteria isolated from a graphite mine could degrade GO and oxidize rGO and graphite. Recently, Guo et al.183 reported that GO was reduced while rGO was oxidized after incubation with Escherichia coli and Staphylococcus aureus. They also found that the formation and development of biofilms attenuated the toxicity of rGO. The organisms used in graphene synthesis vary from simple prokaryotic systems to complex eukaryotes. In a word, after being in contact with some special microbes, GO sheets would be transformed, which would cause changes in the inherent properties, stability and fate of GO in aquatic environments. On the other hand, understanding the abilities and mechanisms of GO transformation by microbes will broaden our knowledge of the microbe-mediated green synthesis of high performance nanomaterials.
Contact with plant extracts
The reduction of GO can also occur after accumulation within a plant. Many reports have demonstrated that GO transformation could be stimulated by plant extracts from leaves, fruits and flowers of plants. In general, plant extracts contain various types of phytochemicals, such as anthocyanins, glycosides, terpenoids, polyphenols, pectins, vitamins, flavonoids, ascorbic acid, apigenin, luteolin and various flavones.45,49,184,185 These phytochemicals have a high tendency to get oxidized and play an important role in the bio-reduction of GO and stabilization of graphene (Fig. 26). Phytoextracts have tremendous potential to be used as reducing agents for the reduction of GO, such as those derived from Ganoderma spp. mushroom,37Ginkgo biloba leaf,49Hibiscus sabdariffa L.,184Pulicaria glutinosa,186Terminalia chebula seeds,187Colocasia esculenta leaf,45Mesua ferrea Linn leaf,45 orange peel,45 pollen grains of Peltophorum pterocarpum,188 tea,185Rosa damascene,189etc. Phytoextracts derived from different plants show different abilities in reducing GO. Lee and Kim190 compared the ability of seven leaf extracts from cherry, magnolia, platanus, persimmon, pine, maple, and ginkgo to reduce GO. They found that among them, cherry showed the best performance in reducing GO. The coexisting metal ions may influence the reducing ability of plant extracts. Akhavan et al.191 reported that the presence of iron can increase the activity of green tea polyphenols in reducing GO due to the formation of a Fe2+-polyphenol complex, resulting in better recovery of the graphitic structure of the GO sheets and obtaining lasting and homogenous dispersion of the reduced GO sheets in water. In most cases, extracts from plants can act both as a reducing and stabilizing agent, resulting in the formation of water-soluble and cyto-compatible rGO.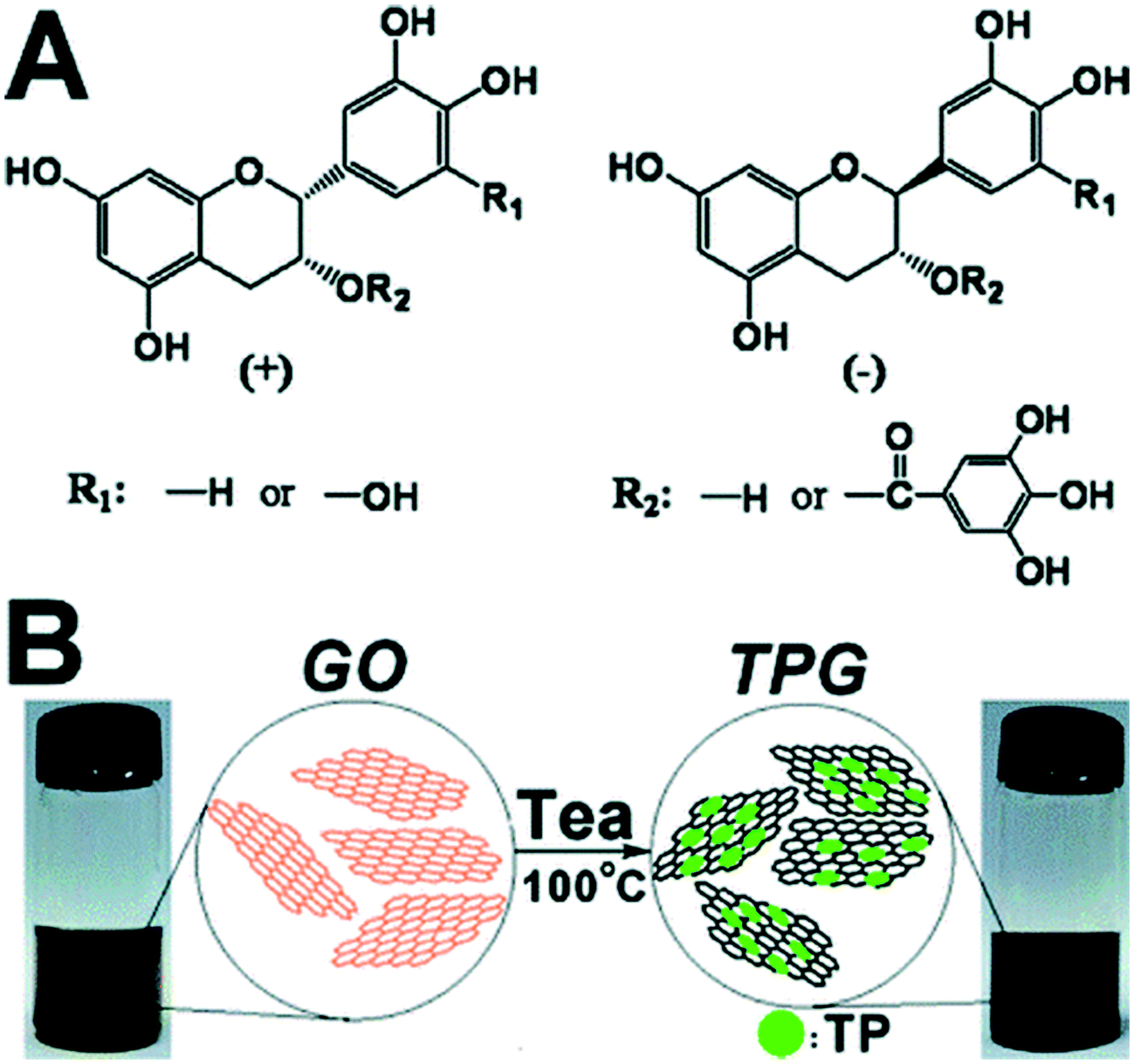 | ||
| Fig. 26 (A) Chemical structure of tea polyphenols (TPs). (B) Schematic illustration of the preparation of TP reduced graphene.185 Reprinted with permission from ref. 185. Copyright 2011, American Chemical Society. | ||
As compared with the chemical method for the reduction of GO, plant-based methods can not only avoid the use of harsh, toxic, and expensive chemicals such as hydrazine hydrate hydrazine, sodium citrate, and sodium borohydride, but they can also avoid the irreversible aggregation of rGO. Meanwhile as compared with microorganism-based methods for the reduction of GO, plant-based methods eliminate the need for high maintenance cell cultures and can be readily adapted for large-scale production of graphene. Because the plant extracts are green and abundant in nature, GO degradation to rGO by plant-based methods is promising. The toxicity of the obtained extract-rGO depends on the extract. Ginkgo biloba extract-rGO is less toxic than GO, while Ganoderma spp. mushroom extract-rGO is more toxic to cancer cells than GO.37
Contact with biomolecules
After their release into aquatic environments, GAs can enter the human body, thus understanding the biotransformation of GAs once they come into contact with biomolecules is a fundamental issue for assessing the potential environmental health risks of GAs. The human body contains many proteins, enzymes, small organic molecules and other active components. The effects of a single enzyme or protein on the biodegradation of GO have been widely studied. It is reported that human myeloperoxidase, eosinophil peroxidase, lactoperoxidase, hemoglobin and xanthine oxidase might be effective in oxidative biodegradation of GO.192,193 Initially, Kotchey et al.194 demonstrated the biodegradation of GO via the catalysis of horseradish peroxidase. Later, Kurapati et al.195 reported that human myeloperoxidase could catalyse the degradation of GO in the presence of H2O2 and found that its biodegradation capability depended on the hydrophilicity, negative surface charge, and colloidal stability of the aqueous GO.195 The biodegradability of GO in vivo was investigated by Girish et al.,196 who found that macrophages played an important role in the biodegradation. Very recently, the biotransformation of GO in real blood plasma was investigated by Hu et al.,193 who found that free radicals and biological molecules in human blood plasma simultaneously drive the biotransformation of GO sheets. Importantly, the bio-transformed GO induced lower levels of cell ultrastructure damage than did pristine GO. Finally, the biotransformation of GAs will alter their uptake and bioavailability in the biological microenvironment compared with pristine GAs. On the other hand, the protein in the biological microenvironment can be adsorbed on GAs via hydrophobic interaction and strong π–π stacking interaction.41 Protein adsorption on GAs will influence the cellular recognition and uptake of GAs, changing the biological response toward these foreign entities. Chong et al.41 found that coating GAs with bovine serum albumin, bovine fibrinogen, immunoglobulin, and transferrin can effectively reduce the cytotoxicity of GO. Along with the alleviation of GO cytotoxicity, Li et al.197 found that coating GO with bovine serum albumin inhibited the oxidative biodegradation of GO catalysed by horseradish peroxidase. In a word, these findings can provide information on the environmental health risks of GAs, thereby avoiding overestimating their relevant biological risks.Contact with inorganic oxidants and reductants
Fenton's reagent (Fe2+/Fe3+/H2O2) is well known as a strong oxidizing agent with the production of the highly reactive hydroxyl radical (˙OH), which plays an important role in oxidative degradation of GAs.198 UV irradiation will accelerate the production of ˙OH and improve the degradation rate.199 Fenton's reagent has been applied not only to decompose aromatic organic pollutants in water,200,201 but also to understand the degradation mechanism of carbon nanomaterials in enzymatic-catalyzed systems with ferric heme iron (Fe3+) in the catalytically active center.202,203 Therefore, naturally occurring Fenton's reagent may play a significant role in controlling the environmental fate and biological effect of GAs. Initially, Zhou et al.204 demonstrated that the photo-Fenton reaction can degrade GO into graphene quantum dots (Fig. 27). After that, Bai et al.205 identified the degradation products of GO driven by the photo-Fenton reaction. They demonstrated that the early stage of photo-Fenton degradation of GO yields oxidized polycyclic aromatic hydrocarbons. After a longer reaction time, the formation of holes on the basal plane of the GO sheets was observed, and the GO flakes began to break apart into graphene quantum dots. By employing 14C-labeling, Feng et al.46 quantitatively measured the Fenton degradation of a low concentration of FLG under environmentally relevant conditions. They found that the degradation products of FLG became more stable in water compared to the unreacted one, and were less easily accumulated in Daphnia magna. Recently, Zhang et al.206 reported that GO can be degraded completely by the photo-Fenton method to CO2 after 28 days. Based on the above-mentioned statement, naturally occurring Fenton-like reactions may drive the transformation of GAs, and the resulting change in the morphology, properties and degradation intermediates of GAs should be taken into account when assessing their potential ecological risks. | ||
| Fig. 27 (A) Schematic diagram demonstrating the degradation of graphene oxide (GO) via the photo-Fenton reaction, which results in the formation of oxidized polycyclic aromatic hydrocarbon (o-PAH) intermediates and graphene quantum dot (GQDs) products with time. (B) AFM images of as-received GO after reaction with Fenton's reagent under UV irradiation for 0, 18, 36, and 54 h. All images were obtained in tapping mode, and the scale bars are 500 nm.204 Reprinted with permission from ref. 204. Copyright 2012, American Chemical Society. | ||
Another possible and highly environmentally relevant route for the abiotic reduction of GO is the reaction with a naturally occurring reductant. Chen's group has examined the effects of abiotic transformation of GO by two environmentally relevant reducing agents including one strong reductant (i.e., S2−) and one rather mild reductant (i.e., ferrous iron, Fe(II)) on the fate and transport of GO systematically.57,133,207,208 They found that both sulfide and Fe(II) reduction resulted in significant changes in the distribution of surface oxygen functionality. After being in contact with S2−, GO is destabilized due to the increase in the surface hydrophobicity of rGO. Besides the increase in the surface hydrophobicity of rGO, the π–π interaction between the graphitic structures and the stacking of GO sheets through Fe2+/Fe3+ cation bridging also contribute to the settlement of GO after being in contact with Fe2+. Moreover, a mild reductant of Fe2+ only caused partial reduction of GO, so even the GO samples treated with high doses of Fe2+ still possessed considerable colloidal stability. Accordingly, mild reductants might exert even greater effects on the environmental risks of GO than strong reductants such as sulfur-containing compounds, whose strong reduction can result in a significant loss of GO oxygen functionalities and consequently much inhibited the mobility of GO. This aspect should be given a consideration in the risk assessment of GO.
Chen's group209,210 also characterized the effect of the water and wastewater treatment especially chlorination/chloramination on the physicochemical properties of GAs and the subsequent effects on their fate and transport. They found that GO can undergo further oxidation upon chlorine or chloramine treatment. When the background cation was Na+ (soft water), chlorination or chloramination of GO may enhance the mobility and transport of GO by making the material more negatively charged and less prone to agglomeration. At 35 mM NaCl, the decrease in the mobility followed the order chlorine-treated GO > chloramine-treated GO > pristine GO.210 When the background cation was Ca2+ (hard water), chloramination of GO could reduce the mobility and transport of GO due to the increase in surface carboxyl groups from chloramination, which induced the deposition of GO through cation-bridging between GO and surface hydroxyl groups of quartz sand.209 One can see that even relatively mild treatments such as chloramination can result in profound changes in GO transport properties which calls for the need to fully understand the significant impact of wastewater treatment processes on the environmental behavior of GAs.
Challenges and perspectives
GAs, as advanced nanomaterials, have driven widespread development in various research and engineering fields. Research relating to GAs is increasing at an incredible rate in a variety of disciplines. During the development processes of GAs, it is inevitable to induce positive and negative impacts on humans and the ecosystem. This report focuses on describing the environmental behavior of GAs in both water and subsurface environments. Current batch and column studies generally examined various influencing factors, and came to different conclusions due to inconsistent experimental conditions. Therefore, it is necessary to study the effects of the various influencing factors under a uniform experimental condition. Nevertheless, a number of scientific “blind spots” and knowledge gaps in GA research still exist.(1) Study the environmental behavior and the toxicity of GAs after being used as a contaminant carrier. Due to its special physicochemical properties, especially an extremely large surface area to mass ratio, the hydrophobicity of the basal plane, the hydrophilicity of the edge, and the stable stability in aqueous solutions, GO shows potential for adsorption of both heavy metal ions and organic contaminants in environmental pollution management. By serving as a contaminant carrier, GO can significantly enhance the transport and risks of contaminants. However, studies on the effect of the adsorbed contaminants on GA's colloidal stability and toxicity are still scarce.
(2) Investigate the effect of the contaminant mixtures on the aggregation and deposition behaviors of GAs. The effect of other contaminant mixtures including organic–organic mixtures, metal–metal mixtures, and organic–metal mixtures on the aggregation and deposition behaviors of GAs needs to be investigated.
(3) Find the correlation between adsorption, colloidal behavior and the combined toxicity of GO and other contaminants. In most studies, the adsorption, colloidal behavior and the combined toxicity of GO and heavy metal ions were investigated separately. To develop effective in situ or ex situ bioremediation technologies and assess the ecological risk of GO after being used as a contaminant carrier, there must be a better understanding of the correlation between adsorption, colloidal behavior and the combined toxicity of GO and contaminants.
(4) Determine the colloidal behavior of GAs in natural waters. A complex mixture of cations (e.g., Na+, K+, Mg2+, and Ca2+), anions (e.g., Cl−, HCO3−, and SO42−) and NOM in surface water environments has a complicated impact on the GO stability. Meanwhile, there are huge differences in the chemical composition of natural surface waters. There is still a great knowledge gap between the simplified laboratory results and the actual behavior of GO in natural waters. Sufficient information on the colloidal properties of GAs in natural waters from all over the world is needed to determine the actual behavior of GO in real worldwide aquatic environments.
(5) Research on the heteroaggregation, cotransport, and combined toxicity of GAs and other nanoparticles. With the rapid development of the nanotechnology industry, it will be inevitable that different types of nanoparticles will enter the natural environment simultaneously. Accordingly, the fate, transport, and toxic potential of nanomaterial mixtures are likely to be distinct from those of individual materials. Ag, zero-valent iron, CuO, ZnO, TiO2, and Al2O3 nanoparticles, which have been widely applied in a wide variety of fields, show a high possibility to come into contact with GAs. However, information on the heteroaggregation, cotransport, and combined toxicity of GAs and these metal/metal oxide nanoparticles is still unavailable.
(6) Explore the environmental behavior and toxicity of the functionalized GAs. To improve the special properties of GAs such as adsorption capacity, biocompatibility, and stability for a wide range of applications, other functional groups have been grafted onto GAs through various chemical reactions, forming functionalized GAs. The physiochemical properties of the functionalized GAs are distinctly different from those of the parent GAs, which may result in the aggregation and deposition properties of the functionalized GAs, in response to the different solution chemistry conditions and hydrodynamic factors, differing significantly from those of the parent GAs. Therefore, the environmental behavior and toxicity of the functionalized GAs cannot be extrapolated from the available findings.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (21477133), Anhui Provincial Natural Science Foundation (1608085QB44), CAS Key Laboratory of Photovoltaic and Energy Conservation Materials, Jiangsu Provincial Key Laboratory of Radiation Medicine and Protection, and Priority Academic Program Development of Jiangsu Higher Education Institutions is acknowledged.Notes and references
- B.-T. Zhang, X. Zheng, H.-F. Li and J.-M. Lin, Application of carbon-based nanomaterials in sample preparation: A review, Anal. Chim. Acta, 2013, 784, 1–17 CrossRef CAS PubMed.
- K. Scida, P. W. Stege, G. Haby, G. A. Messina and C. D. Garcia, Recent applications of carbon-based nanomaterials in analytical chemistry: Critical review, Anal. Chim. Acta, 2011, 691, 6–17 CrossRef CAS PubMed.
- Y. Shen, Q. Fang and B. Chen, Environmental applications of three-dimensional graphene-based macrostructures: Adsorption, transformation, and detection, Environ. Sci. Technol., 2014, 49, 67–84 CrossRef PubMed.
- M. S. Mauter and M. Elimelech, Environmental applications of carbon-based nanomaterials, Environ. Sci. Technol., 2008, 42, 5843–5859 CrossRef CAS PubMed.
- X. G. Hu, M. Zhou and Q. X. Zhou, Ambient water and visible-light irradiation drive changes in graphene morphology, structure, surface chemistry, aggregation, and toxicity, Environ. Sci. Technol., 2015, 49, 3410–3418 CrossRef CAS PubMed.
- O. Akhavan and E. Ghaderi, Toxicity of graphene and graphene oxide nanowalls against bacteria, ACS Nano, 2010, 4, 5731–5736 CrossRef CAS PubMed.
- K.-H. Liao, Y.-S. Lin, C. W. Macosko and C. L. Haynes, Cytotoxicity of graphene oxide and graphene in human erythrocytes and skin fibroblasts, ACS Appl. Mater. Interfaces, 2011, 3, 2607–2615 CAS.
- K. Lu, Q. Huang, P. Wang and L. Mao, Physicochemical Changes of Few-Layer Graphene in Peroxidase-Catalyzed Reactions: Characterization and Potential Ecological Effects, Environ. Sci. Technol., 2015, 49, 8558–8565 CrossRef CAS PubMed.
- S. Das, S. Singh, V. Singh, D. Joung, J. M. Dowding, D. Reid, J. Anderson, L. Zhai, S. I. Khondaker, W. T. Self and S. Seal, Oxygenated functional group density on graphene oxide: Its effect on cell toxicity, Part. Part. Syst. Charact., 2013, 30, 148–157 CrossRef CAS.
- S. B. Liu, T. H. Zeng, M. Hofmann, E. Burcombe, J. Wei, R. R. Jiang, J. Kong and Y. Chen, Antibacterial activity of graphite, graphite oxide, graphene oxide, and reduced graphene oxide: Membrane and oxidative stress, ACS Nano, 2011, 5, 6971–6980 CrossRef CAS PubMed.
- M. C. Duch, G. R. S. Budinger, Y. T. Liang, S. Soberanes, D. Urich, S. E. Chiarella, L. A. Campochiaro, A. Gonzalez, N. S. Chandel, M. C. Hersam and G. M. Mutlu, Minimizing oxidation and stable nanoscale dispersion improves the biocompatibility of graphene in the lung, Nano Lett., 2011, 11, 5201–5207 CrossRef PubMed.
- J. Zhao, Z. Wang, J. C. White and B. Xing, Graphene in the aquatic environment: Adsorption, dispersion, toxicity and transformation, Environ. Sci. Technol., 2014, 48, 9995–10009 CrossRef CAS PubMed.
- K. He, G. Chen, G. Zeng, M. Peng, Z. Huang, J. Shi and T. Huang, Stability, transport and ecosystem effects of graphene in water and soil environments, Nanoscale, 2017, 9, 5370–5388 RSC.
- V. C. Sanchez, A. Jachak, R. H. Hurt and A. B. Kane, Biological interactions of graphene-family nanomaterials: An interdisciplinary review, Chem. Res. Toxicol., 2012, 25, 15–34 CrossRef CAS PubMed.
- X. J. Deng, L. L. Lu, H. W. Li and F. Luo, The adsorption properties of Pb(II) and Cd(II) on functionalized graphene prepared by electrolysis method, J. Hazard. Mater., 2010, 183, 923–930 CrossRef CAS PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Functionalization of graphene: Covalent and non-covalent approaches, derivatives and applications, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- Z.-J. Fan, W. Kai, J. Yan, T. Wei, L.-J. Zhi, J. Feng, Y.-m. Ren, L.-P. Song and F. Wei, Facile synthesis of graphene nanosheets via Fe reduction of exfoliated graphite oxide, ACS Nano, 2011, 5, 191–198 CrossRef CAS PubMed.
- A. R. Petosa, D. P. Jaisi, I. R. Quevedo, M. Elimelech and N. Tufenkji, Aggregation and deposition of engineered nanomaterials in aquatic environments: role of physicochemical interactions, Environ. Sci. Technol., 2010, 44, 6532–6549 CrossRef CAS PubMed.
- D. Li, M. B. Mueller, S. Gilje, R. B. Kaner and G. G. Wallace, Processable aqueous dispersions of graphene nanosheets, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- T. M. Swager, Functional graphene: Top-down chemistry of the π-Surface, ACS Macro Lett., 2012, 1, 3–5 CrossRef CAS.
- Y. Jiang, R. Raliya, J. D. Fortner and P. Biswas, Graphene oxides in water: Correlating morphology and surface chemistry with aggregation behavior, Environ. Sci. Technol., 2016, 50, 6964–6973 CrossRef CAS PubMed.
- Y. Qi, T. Xia, Y. Li, L. Duan and W. Chen, Colloidal stability of reduced graphene oxide materials prepared using different reducing agents, Environ. Sci.: Nano, 2016, 3, 1062–1071 RSC.
- M. M. Gudarzi, Colloidal stability of graphene oxide: Aggregation in two dimensions, Langmuir, 2016, 32, 5058–5068 CrossRef CAS PubMed.
- S. K. Cushing, M. Li, F. Huang and N. Wu, Origin of strong excitation wavelength dependent fluorescence of graphene oxide, ACS Nano, 2014, 8, 1002–1013 CrossRef CAS PubMed.
- N. S. Andryushina, O. L. Stroyuk, I. B. Yanchuk and A. V. Yefanov, A dynamic light scattering study of photochemically reduced colloidal graphene oxide, Colloid Polym. Sci., 2014, 292, 539–546 CAS.
- J. Luo, L. J. Cote, V. C. Tung, A. T. L. Tan, P. E. Goins, J. Wu and J. Huang, Graphene oxide nanocolloids, J. Am. Chem. Soc., 2010, 132, 17667–17669 CrossRef CAS PubMed.
- B. Konkena and S. Vasudevan, Understanding aqueous dispersibility of graphene oxide and reduced graphene oxide through pKa measurements, J. Phys. Chem. Lett., 2012, 3, 867–872 CrossRef CAS PubMed.
- I. Chowdhury, M. C. Duch, N. D. Manuskhani, M. C. Hersam and D. Bouchard, Colloidal properties and stability of graphene oxide nanomaterials in the aquatic environment, Environ. Sci. Technol., 2013, 47, 6288–6296 CrossRef CAS PubMed.
- Y. Li, J. Yang, Q. Zhao and Y. Li, Dispersing carbon-based nanomaterials in aqueous phase by graphene oxides, Langmuir, 2013, 29, 13527–13534 CrossRef CAS PubMed.
- Y. Jiang, R. Raliya, P. Liao, P. Biswas and J. D. Fortner, Graphene oxides in water: assessing stability as a function of material and natural organic matter properties, Environ. Sci.: Nano, 2017, 4, 1484–1493 RSC.
- S. Mao, H. H. Pu and J. H. Chen, Graphene oxide and its reduction: Modeling and experimental progress, RSC Adv., 2012, 2, 2643–2662 RSC.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- C. K. Chua and M. Pumera, Chemical reduction of graphene oxide: a synthetic chemistry viewpoint, Chem. Soc. Rev., 2014, 43, 291–312 RSC.
- T. Szabo, E. Tombacz, E. Illes and I. Dekany, Enhanced acidity and pH-dependent surface charge characterization of successively oxidized graphite oxides, Carbon, 2006, 44, 537–545 CrossRef CAS.
- B. Konkena and S. Vasudevan, Covalently linked, water-dispersible, cyciodextrin: Reduced-graphene oxide sheets, Langmuir, 2012, 28, 12432–12437 CrossRef CAS PubMed.
- A. M. Dimiev, L. B. Alemany and J. M. Tour, Graphene oxide. Origin of acidity, its instability in water, and a new dynamic structural model, ACS Nano, 2013, 7, 576–588 CrossRef CAS PubMed.
- S. Gurunathan, J. Han, J. H. Park and J. H. Kim, An in vitro evaluation of graphene oxide reduced by Ganoderma spp. in human breast cancer cells (MDA-MB-231), Int. J. Nanomed., 2014, 9, 1783–1797 CrossRef PubMed.
- A. Ciesielski and P. Samori, Graphene via sonication assisted liquid-phase exfoliation, Chem. Soc. Rev., 2014, 43, 381–398 RSC.
- W. Cheng, C. Ding, Q. Wu, X. Wang, Y. Sun, W. Shi, T. Hayat, A. Alsaedi, Z. Chai and X. Wang, Mutual effect of U(VI) and Sr(II) on graphene oxides: evidence from EXAFS and theoretical calculations, Environ. Sci.: Nano, 2017, 4, 1124–1131 RSC.
- Y. Su, G. Yang, K. Lu, E. J. Petersen and L. Mao, Colloidal properties and stability of aqueous suspensions of few-layer graphene: Importance of graphene concentration, Environ. Pollut., 2017, 220(Part A), 469–477 CrossRef CAS PubMed.
- Y. Chong, C. C. Ge, Z. X. Yang, J. A. Garate, Z. L. Gu, J. K. Weber, J. J. Liu and R. H. Zhou, Reduced cytotoxicity of graphene nanosheets mediated by blood-protein coating, ACS Nano, 2015, 9, 5713–5724 CrossRef CAS PubMed.
- M. Hu and B. X. Mi, Enabling graphene oxide nanosheets as water separation membranes, Environ. Sci. Technol., 2013, 47, 3715–3723 CrossRef CAS PubMed.
- X. B. Hu, Y. Yu, W. M. Hou, J. E. Zhou and L. X. Song, Effects of particle size and pH value on the hydrophilicity of graphene oxide, Appl. Surf. Sci., 2013, 273, 118–121 CrossRef CAS.
- G. Wang, F. Qian, C. W. Saltikov, Y. Jiao and Y. Li, Microbial reduction of graphene oxide by Shewanella, Nano Res., 2011, 4, 563–570 CrossRef CAS.
- S. Thakur and N. Karak, Green reduction of graphene oxide by aqueous phytoextracts, Carbon, 2012, 50, 5331–5339 CrossRef CAS.
- Y. Feng, K. Lu, L. Mao, X. Guo, S. Gao and E. J. Petersen, Degradation of 14C-labeled few layer graphene via Fenton reaction: Reaction rates, characterization of reaction products, and potential ecological effects, Water Res., 2015, 84, 49–57 CrossRef CAS PubMed.
- S. Gurunathan, J. W. Han, V. Eppakayala and J.-H. Kim, Green synthesis of graphene and its cytotoxic effects in human breast cancer cells, Int. J. Nanomed., 2013, 8, 1015–1027 CrossRef PubMed.
- F. Perreault, A. F. de Faria and M. Elimelech, Environmental applications of graphene-based nanomaterials, Chem. Soc. Rev., 2015, 44, 5861–5896 RSC.
- S. Gurunathan, J. W. Han, J. H. Park, V. Eppakayala and J.-H. Kim, Ginkgo biloba: a natural reducing agent for the synthesis of cytocompatible graphene, Int. J. Nanomed., 2014, 9, 363–377 CrossRef PubMed.
- J. Lakshmi and S. Vasudevan, Graphene-a promising material for removal of perchlorate (ClO4−) from water, Environ. Sci. Pollut. Res., 2013, 20, 5114–5124 CrossRef CAS PubMed.
- Y. Su, B. Gao and L. Mao, Concurrent agglomeration and straining govern the transport of 14C-labeled few-layer graphene in saturated porous media, Water Res., 2017, 115, 84–93 CrossRef CAS PubMed.
- K. Parvez, Z.-S. Wu, R. Li, X. Liu, R. Graf, X. Feng and K. Müllen, Exfoliation of graphite into graphene in aqueous solutions of inorganic salts, J. Am. Chem. Soc., 2014, 136, 6083–6091 CrossRef CAS PubMed.
- R. Sitko, E. Turek, B. Zawisza, E. Malicka, E. Talik, J. Heimann, A. Gagor, B. Feist and R. Wrzalik, Adsorption of divalent metal ions from aqueous solutions using graphene oxide, Dalton Trans., 2013, 42, 5682–5689 RSC.
- Y. Matsumoto, M. Koinuma, S. Ida, S. Hayami, T. Taniguchi, K. Hatakeyama, H. Tateishi, Y. Watanabe and S. Amano, Photoreaction of graphene oxide nanosheets in water, J. Phys. Chem. C, 2011, 115, 19280–19286 CAS.
- I. Chowdhury, N. D. Mansukhani, L. M. Guiney, M. C. Hersam and D. Bouchard, Aggregation and stability of reduced graphene oxide: Complex roles of divalent cations, pH, and natural organic matter, Environ. Sci. Technol., 2015, 49, 10886–10893 CrossRef CAS PubMed.
- I. Chowdhury, M. C. Duch, N. D. Mansukhani, M. C. Hersam and D. Bouchard, Interactions of graphene oxide nanomaterials with natural organic matter and metal oxide surfaces, Environ. Sci. Technol., 2014, 48, 9382–9390 CrossRef CAS PubMed.
- H. Y. Fu, X. L. Qu, W. Chen and D. Q. Zhu, Transformation and destabilization of graphene oxide in reducing aqueous solutions containing sulfide, Environ. Toxicol. Chem., 2014, 33, 2647–2653 CrossRef CAS PubMed.
- C. J. Shih, S. C. Lin, R. Sharma, M. S. Strano and D. Blankschtein, Understanding the pH-dependent behavior of graphene oxide aqueous solutions: A comparative experimental and molecular dynamics simulation study, Langmuir, 2012, 28, 235–241 CrossRef CAS PubMed.
- T. Taniguchi, S. Kurihara, H. Tateishi, K. Hatakeyama, M. Koinuma, H. Yokoi, M. Hara, H. Ishikawa and Y. Matsumoto, pH-driven, reversible epoxy ring opening/closing in graphene oxide, Carbon, 2015, 84, 560–566 CrossRef CAS.
- X. Wang, H. Bai and G. Shi, Size fractionation of graphene oxide sheets by pH-assisted selective sedimentation, J. Am. Chem. Soc., 2011, 133, 6338–6342 CrossRef CAS PubMed.
- R. L. D. Whitby, A. Korobeinyk, V. M. Gun'ko, R. Busquets, A. B. Cundy, K. Laszlo, J. Skubiszewska-Zieba, R. Leboda, E. Tombacz, I. Y. Toth, K. Kovacs and S. V. Mikhalovsky, pH-driven physicochemical conformational changes of single-layer graphene oxide, Chem. Commun., 2011, 47, 9645–9647 RSC.
- Y. T. Shieh, G. L. Liu, H. H. Wu and C. C. Lee, Effects of polarity and pH on the solubility of acid-treated carbon nanotubes in different media, Carbon, 2007, 45, 1880–1890 CrossRef CAS.
- H. Tang, Y. Zhao, X. Yang, D. Liu, P. Shao, Z. Zhu, S. Shan, F. Cui and B. Xing, New insight into the aggregation of graphene oxide using molecular dynamics simulations and extended Derjaguin–Landau–Verwey–Overbeek theory, Environ. Sci. Technol., 2017, 51, 9674–9682 CrossRef CAS PubMed.
- L. Wu, L. Liu, B. Gao, R. Muñoz-Carpena, M. Zhang, H. Chen, Z. Zhou and H. Wang, Aggregation kinetics of graphene oxides in aqueous solutions: Experiments, mechanisms, and modeling, Langmuir, 2013, 29, 15174–15181 CrossRef CAS PubMed.
- H. Tang, Y. Zhao, X. Yang, D. Liu, S. Shan and F. Cui, Understanding the roles of solution chemistries and functionalization on the aggregation of graphene-based nanomaterials using molecular dynamic simulations, J. Phys. Chem. C, 2017, 121, 13888–13897 CAS.
- X. Ren, Q. Wu, H. Xu, D. Shao, X. Tan, W. Shi, C. Chen, J. Li, Z. Chai, T. Hayat and X. Wang, New insight into GO, cadmium(II), phosphate interaction and its role in GO colloidal behavior, Environ. Sci. Technol., 2016, 50, 9361–9369 CrossRef CAS PubMed.
- Y. Gao, X. Ren, X. Tan, T. Hayat, A. Alsaedi and C. Chen, Insights into key factors controlling GO stability in natural surface waters, J. Hazard. Mater., 2017, 335, 56–65 CrossRef CAS PubMed.
- J. Zhao, F. Liu, Z. Wang, X. Cao and B. Xing, Heteroaggregation of graphene oxide with minerals in aqueous phase, Environ. Sci. Technol., 2015, 49, 2849–2857 CrossRef CAS PubMed.
- G. Huang, H. Guo, J. Zhao, Y. Liu and B. Xing, Effect of co-existing kaolinite and goethite on the aggregation of graphene oxide in the aquatic environment, Water Res., 2016, 102, 313–320 CrossRef CAS PubMed.
- J. D. Lanphere, C. J. Luth and S. L. Walker, Effects of solution chemistry on the transport of graphene oxide in saturated porous media, Environ. Sci. Technol., 2013, 47, 4255–4261 CrossRef CAS PubMed.
- K. Yang, B. Chen, X. Zhu and B. Xing, Aggregation, adsorption and morphological transformation of graphene oxide in aqueous solutions containing different metal cations, Environ. Sci. Technol., 2016, 50, 11066–11075 CrossRef CAS PubMed.
- Z. Hua, Z. Tang, X. Bai, J. Zhang, L. Yu and H. Cheng, Aggregation and resuspension of graphene oxide in simulated natural surface aquatic environments, Environ. Pollut., 2015, 205, 161–169 CrossRef CAS PubMed.
- J. D. Lanphere, B. Rogers, C. Luth, C. H. Bolster and S. L. Walker, Stability and transport of graphene oxide nanoparticles in groundwater and surface Water, Environ. Eng. Sci., 2014, 31, 350–359 CrossRef CAS PubMed.
- B. Mukherjee and J. W. Weaver, Aggregation and charge behavior of metallic and nonmetallic nanoparticles in the presence of competing similarly-charged inorganic ions, Environ. Sci. Technol., 2010, 44, 3332–3338 CrossRef CAS PubMed.
- K. L. Chen and M. Elimelech, Influence of humic acid on the aggregation kinetics of fullerene (C60) nanoparticles in monovalent and divalent electrolyte solutions, J. Colloid Interface Sci., 2007, 309, 126–134 CrossRef CAS PubMed.
- H. Mashayekhi, S. Ghosh, P. Du and B. S. Xing, Effect of natural organic matter on aggregation behavior of C60 fullerene in water, J. Colloid Interface Sci., 2012, 374, 111–117 CrossRef CAS PubMed.
- Y. Feng, X. Liu, K. A. Huynh, J. M. McCaffery, L. Mao, S. Gao and K. L. Chen, Heteroaggregation of graphene oxide with nanometer- and micrometer-sized hematite colloids: Influence on nanohybrid aggregation and microparticle sedimentation, Environ. Sci. Technol., 2017, 51, 6821–6828 CrossRef CAS PubMed.
- A. Y. Romanchuk, A. S. Slesarev, S. N. Kalmykov, D. V. Kosynkin and J. M. Tour, Graphene oxide for effective radionuclide removal, Phys. Chem. Chem. Phys., 2013, 15, 2321–2327 RSC.
- N. P. Sotirelis and C. V. Chrysikopoulos, Heteroaggregation of graphene oxide nanoparticles and kaolinite colloids, Sci. Total Environ., 2017, 579, 736–744 CrossRef CAS PubMed.
- Y. K. Yang, N. Nakada, R. Nakajima, M. Yasojima, C. Wang and H. Tanaka, pH, ionic strength and dissolved organic matter alter aggregation of fullerene C60 nanoparticles suspensions in wastewater, J. Hazard. Mater., 2013, 244, 582–587 CrossRef PubMed.
- A. Schierz and H. Zänker, Aqueous suspensions of carbon nanotubes: Surface oxidation, colloidal stability and uranium sorption, Environ. Pollut., 2009, 157, 1088–1094 CrossRef CAS PubMed.
- K. L. Chen and M. Elimelech, Relating colloidal stability of fullerene (C60) nanoparticles to nanoparticle charge and electrokinetic properties, Environ. Sci. Technol., 2009, 43, 7270–7276 CrossRef CAS PubMed.
- J. Brant, H. Lecoanet, M. Hotze and M. Wiesner, Comparison of electrokinetic properties of colloidal fullerenes (n-C60) formed using two procedures, Environ. Sci. Technol., 2005, 39, 6343–6351 CrossRef CAS PubMed.
- X. M. Ren, J. X. Li, X. L. Tan, W. Q. Shi, C. L. Chen, D. D. Shao, T. Wen, L. F. Wang, G. X. Zhao, G. P. Sheng and X. K. Wang, Impact of Al2O3 on the aggregation and deposition of graphene oxide, Environ. Sci. Technol., 2014, 48, 5493–5500 CrossRef CAS PubMed.
- T. Hartono, S. B. Wang, Q. Ma and Z. H. Zhu, Layer structured graphite oxide as a novel adsorbent for humic acid removal from aqueous solution, J. Colloid Interface Sci., 2009, 333, 114–119 CrossRef CAS PubMed.
- S. B. Wang, H. Q. Sun, H. M. Ang and M. O. Tade, Adsorptive remediation of environmental pollutants using novel graphene-based nanomaterials, Chem. Eng. J., 2013, 226, 336–347 CrossRef CAS.
- T. T. Li, D. H. Lin, L. Li, Z. Y. Wang and F. C. Wu, The kinetic and thermodynamic sorption and stabilization of multiwalled carbon nanotubes in natural organic matter surrogate solutions: The effect of surrogate molecular weight, Environ. Pollut., 2014, 186, 43–49 CrossRef CAS PubMed.
- N. B. Saleh, L. D. Pfefferle and M. Elimelech, Aggregation kinetics of multiwalled carbon nanotubes in aquatic systems: measurements and Environmental implications, Environ. Sci. Technol., 2008, 42, 7963–7969 CrossRef CAS PubMed.
- L. K. Duncan, J. R. Jinschek and P. J. Vikesland, C60 colloid formation in aqueous systems: Effects of preparation method on size, structure, and surface charge, Environ. Sci. Technol., 2008, 42, 173–178 CrossRef CAS PubMed.
- B. Xie, Z. Xu, W. Guo and Q. Li, Impact of natural organic matter on the physicochemical properties of aqueous C60 nanoparticles, Environ. Sci. Technol., 2008, 42, 2853–2859 CrossRef CAS PubMed.
- Q. L. Li, B. Xie, Y. S. Hwang and Y. J. Xu, Kinetics of C60 fullerene dispersion in water enhanced by natural organic matter and sunlight, Environ. Sci. Technol., 2009, 43, 3574–3579 CrossRef CAS PubMed.
- Z. Wang, Y. Gao, S. Wang, H. Fang, D. Xu and F. Zhang, Impacts of low-molecular-weight organic acids on aquatic behavior of graphene nanoplatelets and their induced algal toxicity and antioxidant capacity, Environ. Sci. Pollut. Res., 2016, 23, 10938–10945 CrossRef CAS PubMed.
- W. Zhang, U. s. Rattanaudompol, H. Li and D. Bouchard, Effects of humic and fulvic acids on aggregation of aqu/nC60 nanoparticles, Water Res., 2013, 47, 1793–1802 CrossRef CAS PubMed.
- H. Hyung and J. H. Kim, Natural organic matter (NOM) adsorption to multi-walled carbon nanotubes: Effect of NOM characteristics and water quality parameters, Environ. Sci. Technol., 2008, 42, 4416–4421 CrossRef CAS PubMed.
- A. J. Kennedy, J. C. Gunter, M. A. Chappell, J. D. Goss, M. S. Hull, R. A. Kirgan and J. A. Steevens, Influence of nanotube preparation in aquatic bioassays, Environ. Toxicol. Chem., 2009, 28, 1930–1938 CrossRef CAS PubMed.
- L.-F. Wang, L.-L. Wang, X.-D. Ye, W.-W. Li, X.-M. Ren, G.-P. Sheng, H.-Q. Yu and X.-K. Wang, Coagulation kinetics of humic aggregates in mono- and di-valent electrolyte solutions, Environ. Sci. Technol., 2013, 47, 5042–5049 CrossRef CAS PubMed.
- G. E. Batley, J. K. Kirby and M. J. McLaughlin, Fate and risks of nanomaterials in aquatic and terrestrial environments, Acc. Chem. Res., 2013, 46, 854–862 CrossRef CAS PubMed.
- J. Liu, S. Legros, F. Von Der Kammer and T. Hofmann, Natural organic matter concentration and hydrochemistry influence aggregation kinetics of functionalized engineered nanoparticles, Environ. Sci. Technol., 2013, 47, 4113–4120 CrossRef CAS PubMed.
- Y. Zou, X. Wang, Y. Ai, Y.-H. Liu, J.-X. Li, Y. Ji and X. Wang, Coagulation behavior of graphene oxide on nanocrystallined Mg/Al layered double hydroxides: Batch experimental and theoretical calculation study, Environ. Sci. Technol., 2016, 50, 3658–3667 CrossRef CAS PubMed.
- J. Wang, Y. Li, W. Chen, J. Peng, J. Hu, Z. Chen, T. Wen, S. Lu, Y. Chen, T. Hayat, B. Ahmad and X. Wang, The rapid coagulation of graphene oxide on La-doped layered double hydroxides, Chem. Eng. J., 2017, 309, 445–453 CrossRef CAS.
- J. Wang, X. Wang, L. Tan, Y. Chen, T. Hayat, J. Hu, A. Alsaedi, B. Ahmad, W. Guo and X. Wang, Performances and mechanisms of Mg/Al and Ca/Al layered double hydroxides for graphene oxide removal from aqueous solution, Chem. Eng. J., 2016, 297, 106–115 CrossRef CAS.
- J. Wang, S. Yu, Y. Zhao, X. Wang, T. Wen, T. Yang, Y. Ai, Y. Chen, T. Hayat, A. Alsaedi and X. Wang, Experimental and theoretical studies of ZnO and MgO for the rapid coagulation of graphene oxide from aqueous solutions, Sep. Purif. Technol., 2017, 184, 88–96 CrossRef CAS.
- S. Yu, X. Wang, R. Zhang, T. Yang, Y. Ai, T. Wen, W. Huang, T. Hayat, A. Alsaedi and X. Wang, Complex roles of solution chemistry on graphene oxide coagulation onto titanium dioxide: Batch experiments, spectroscopy analysis and theoretical calculation, Sci. Rep., 2017, 7, 39625 CrossRef CAS PubMed.
- Y. Zou, X. Wang, Z. Chen, W. Yao, Y. Ai, Y. Liu, T. Hayat, A. Alsaedi, N. S. Alharbi and X. Wang, Superior coagulation of graphene oxides on nanoscale layered double hydroxides and layered double oxides, Environ. Pollut., 2016, 219, 107–117 CrossRef CAS PubMed.
- Z. Yang, H. Yan, H. Yang, H. Li, A. Li and R. Cheng, Flocculation performance and mechanism of graphene oxide for removal of various contaminants from water, Water Res., 2013, 47, 3037–3046 CrossRef CAS PubMed.
- H. Khanh An, J. M. McCaffery and K. L. Chen, Heteroaggregation reduces antimicrobial activity of silver nanoparticles: Evidence for nanoparticle-cell proximity effects, Environ. Sci. Technol. Lett., 2014, 1, 361–366 CrossRef.
- K. A. Huynh, J. M. McCaffery and K. L. Chen, Heteroaggregation of multiwalled carbon nanotubes and hematite nanoparticles: Rates and mechanisms, Environ. Sci. Technol., 2012, 46, 5912–5920 CrossRef CAS PubMed.
- P. Yi, J. J. Pignatello, M. Uchimiya and J. C. White, Heteroaggregation of cerium oxide nanoparticles and nanoparticles of pyrolyzed biomass, Environ. Sci. Technol., 2015, 49, 13294–13303 CrossRef CAS PubMed.
- I. Chowdhury, M. C. Duch, N. D. Mansukhani, M. C. Hersam and D. Bouchard, Deposition and release of graphene oxide nanomaterials using a quartz crystal microbalance, Environ. Sci. Technol., 2014, 48, 961–969 CrossRef CAS PubMed.
- I. Chowdhury, W.-C. Hou, D. Goodwin, M. Henderson, R. G. Zepp and D. Bouchard, Sunlight affects aggregation and deposition of graphene oxide in the aquatic environment, Water Res., 2015, 78, 37–46 CrossRef CAS PubMed.
- G. Zhao, T. Wen, X. Yang, S. Yang, J. Liao, J. Hu, D. Shao and X. Wang, Preconcentration of U(VI) ions on few-layered graphene oxide nanosheets from aqueous solutions, Dalton Trans., 2012, 41, 6182–6188 RSC.
- G. X. Zhao, J. X. Li, X. M. Ren, C. L. Chen and X. K. Wang, Few-layered graphene oxide nanosheets as superior sorbents for heavy metal ion pollution management, Environ. Sci. Technol., 2011, 45, 10454–10462 CrossRef CAS PubMed.
- G. X. Zhao, X. M. Ren, X. Gao, X. L. Tan, J. X. Li, C. L. Chen, Y. Y. Huang and X. K. Wang, Removal of Pb(II) ions from aqueous solutions on few-layered graphene oxide nanosheets, Dalton Trans., 2011, 40, 10945–10952 RSC.
- S. Yu, X. Wang, Y. Ai, Y. Liang, Y. Ji, J. Li, T. Hayat, A. Alsaedi and X. Wang, Spectroscopic and theoretical studies on the counterion effect of Cu(II) ion and graphene oxide interaction with titanium dioxide, Environ. Sci.: Nano, 2016, 3, 1361–1368 RSC.
- K. Chang, Y. Sun, F. Ye, X. Li, G. Sheng, D. Zhao, W. Linghu, H. Li and J. Liu, Macroscopic and molecular study of the sorption and co-sorption of graphene oxide and Eu(III) onto layered double hydroxides, Chem. Eng. J., 2017, 325, 665–671 CrossRef CAS.
- B. Hu, C. Huang, X. Li, G. Sheng, H. Li, X. Ren, J. Ma, J. Wang and Y. Huang, Macroscopic and spectroscopic insights into the mutual interaction of graphene oxide, Cu(II), and Mg/Al layered double hydroxides, Chem. Eng. J., 2017, 313, 527–534 CrossRef CAS.
- J. M. Englert, J. Roehrl, C. D. Schmidt, R. Graupner, M. Hundhausen, F. Hauke and A. Hirsch, Soluble graphene: Generation of aqueous graphene solutions aided by a perylenebisimide-based bolaamphiphile, Adv. Mater., 2009, 21, 4265–4269 CrossRef CAS.
- A. A. Green and M. C. Hersam, Solution phase production of graphene with controlled thickness via density differentiation, Nano Lett., 2009, 9, 4031–4036 CrossRef CAS PubMed.
- R. Hao, W. Qian, L. Zhang and Y. Hou, Aqueous dispersions of TCNQ-anion-stabilized graphene sheets, Chem. Commun., 2008, 6576–6578, 10.1039/b816971c.
- J. H. Lee, D. W. Shin, V. G. Makotchenko, A. S. Nazarov, V. E. Fedorov, J. H. Yoo, S. M. Yu, J.-Y. Choi, J. M. Kim and J.-B. Yoo, The superior dispersion of easily soluble graphite, Small, 2010, 6, 58–62 CrossRef CAS PubMed.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, Liquid phase production of graphene by exfoliation of graphite in surfactant/water solutions, J. Am. Chem. Soc., 2009, 131, 3611–3620 CrossRef CAS PubMed.
- M. Zhang, R. R. Parajuli, D. Mastrogiovanni, B. Dai, P. Lo, W. Cheung, R. Brukh, P. L. Chiu, T. Zhou, Z. Liu, E. Garfunkel and H. He, Production of graphene sheets by direct dispersion with aromatic healing agents, Small, 2010, 6, 1100–1107 CrossRef CAS PubMed.
- S. Lin, C.-J. Shih, M. S. Strano and D. Blankschtein, Molecular insights into the surface morphology, layering structure, and aggregation kinetics of surfactant-stabilized graphene dispersions, J. Am. Chem. Soc., 2011, 133, 12810–12823 CrossRef CAS PubMed.
- L. Guardia, M. J. Fernández-Merino, J. I. Paredes, P. Solís-Fernández, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, High-throughput production of pristine graphene in an aqueous dispersion assisted by non-ionic surfactants, Carbon, 2011, 49, 1653–1662 CrossRef CAS.
- A. G. Hsieh, S. Korkut, C. Punckt and I. A. Aksay, Dispersion stability of functionalized graphene in aqueous sodium dodecyl sulfate solutions, Langmuir, 2013, 29, 14831–14838 CrossRef CAS PubMed.
- T. Hasan, F. Torrisi, Z. Sun, D. Popa, V. Nicolosi, G. Privitera, F. Bonaccorso and A. C. Ferrari, Solution-phase exfoliation of graphite for ultrafast photonics, Phys. Status Solidi B, 2010, 247, 2953–2957 CrossRef CAS.
- A. B. Bourlinos, V. Georgakilas, R. Zboril, T. A. Steriotis, A. K. Stubos and C. Trapalis, Aqueous-phase exfoliation of graphite in the presence of polyvinylpyrrolidone for the production of water-soluble graphenes, Solid State Commun., 2009, 149, 2172–2176 CrossRef CAS.
- F. Liu, J. Y. Choi and T. S. Seo, DNA mediated water-dispersible graphene fabrication and gold nanoparticle-graphene hybrid, Chem. Commun., 2010, 46, 2844–2846 RSC.
- L. Liang, T. Wu, Y. Kang and Q. Wang, Dispersion of Graphene Sheets in Aqueous Solution by Oligodeoxynucleotides, ChemPhysChem, 2013, 14, 1626–1632 CrossRef CAS PubMed.
- C. Knieke, A. Berger, M. Voigt, R. N. K. Taylor, J. Rohrl and W. Peukert, Scalable production of graphene sheets by mechanical delamination, Carbon, 2010, 48, 3196–3204 CrossRef CAS.
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O'Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O'Brien, E. K. McGuire, B. M. Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Scalable production of large quantities of defect-free few-layer graphene by shear exfoliation in liquids, Nat. Mater., 2014, 13, 624–630 CrossRef CAS PubMed.
- W. Yang, E. Widenkvist, U. Jansson and H. Grennberg, Stirring-induced aggregation of graphene in suspension, New J. Chem., 2011, 35, 780–783 RSC.
- T. Xia, J. D. Fortner, D. Zhu, Z. Qi and W. Chen, Transport of sulfide-reduced graphene oxide in saturated quartz sand: Cation-dependent retention mechanisms, Environ. Sci. Technol., 2015, 49, 11468–11475 CrossRef CAS PubMed.
- S. R. Chae, Y. Xiao, S. H. Lin, T. Noeiaghaei, J. O. Kim and M. R. Wiesner, Effects of humic acid and electrolytes on photocatalytic reactivity and transport of carbon nanoparticle aggregates in water, Water Res., 2012, 46, 4053–4062 CrossRef CAS PubMed.
- L. Wang, L. Hou, X. Wang and W. Chen, Effects of the preparation method and humic-acid modification on the mobility and contaminant-mobilizing capability of fullerene nanoparticles (nC60), Environ. Sci.: Processes Impacts, 2014, 16, 1282–1289 CAS.
- S. R. Chae, A. R. Badireddy, J. F. Budarz, S. H. Lin, Y. Xiao, M. Therezien and M. R. Wiesner, Heterogeneities in fullerene nanoparticle aggregates affecting reactivity, bioactivity, and transport, ACS Nano, 2010, 4, 5011–5018 CrossRef CAS PubMed.
- J. D. Fortner, D. I. Kim, A. M. Boyd, J. C. Falkner, S. Moran, V. L. Colvin, J. B. Hughes and J. H. Kim, Reaction of water-stable C60 aggregates with ozone, Environ. Sci. Technol., 2007, 41, 7497–7502 CrossRef CAS PubMed.
- I. Chowdhury, M. C. Duch, C. C. Gits, M. C. Hersam and S. L. Walker, Impact of synthesis methods on the transport of single walled carbon nanotubes in the aquatic environment, Environ. Sci. Technol., 2012, 46, 11752–11760 CrossRef CAS PubMed.
- I. A. Khan, A. Afrooz, J. R. V. Flora, P. A. Schierz, P. L. Ferguson, T. Sabo-Attwood and N. B. Saleh, Chirality affects aggregation kinetics of single-walled carbon nanotubes, Environ. Sci. Technol., 2013, 47, 1844–1852 CrossRef CAS PubMed.
- J. G. Duque, A. N. G. Parra-Vasquez, N. Behabtu, M. J. Green, A. L. Higginbotham, B. K. Price, A. D. Leonard, H. K. Schmidt, B. Lounis, J. M. Tour, S. K. Doorn, L. Cognet and M. Pasquali, Diameter-dependent solubility of single-walled carbon nanotubes, ACS Nano, 2010, 4, 3063–3072 CrossRef CAS PubMed.
- Y. C. Bai, F. C. Wu, D. H. Lin and B. S. Xing, Aqueous stabilization of carbon nanotubes: effects of surface oxidization and solution chemistry, Environ. Sci. Pollut. Res., 2014, 21, 4358–4365 CrossRef CAS PubMed.
- H. Hu, A. P. Yu, E. Kim, B. Zhao, M. E. Itkis, E. Bekyarova and R. C. Haddon, Influence of the zeta potential on the dispersability and purification of single-walled carbon nanotubes, J. Phys. Chem. B, 2005, 109, 11520–11524 CrossRef CAS PubMed.
- X. L. Qu, Y. S. Hwang, P. J. J. Alvarez, D. Bouchard and Q. L. Li, UV irradiation and humic acid mediate aggregation of aqueous fullerene (nC60) nanoparticles, Environ. Sci. Technol., 2010, 44, 7821–7826 CrossRef CAS PubMed.
- N. B. Saleh, L. D. Pfefferle and M. Elimelech, Influence of Biomacromolecules and Humic Acid on the Aggregation Kinetics of Single-Walled Carbon Nanotubes, Environ. Sci. Technol., 2010, 44, 2412–2418 CrossRef CAS PubMed.
- D. Lin and B. Xing, Tannic Acid Adsorption and Its Role for Stabilizing Carbon Nanotube Suspensions, Environ. Sci. Technol., 2008, 42, 5917–5923 CrossRef CAS PubMed.
- A. A. Zinchenko, N. Maeda, S. Pu and S. Murata, Entrapping of fullerenes, nanotubes, and inorganic nanoparticles by a DNA–chitosan complex: A method for nanomaterials removal, Environ. Sci. Technol., 2013, 47, 4489–4496 CrossRef CAS PubMed.
- L. Liu, B. Gao, L. Wu, L. Y. Yang, Z. H. Zhou and H. Wang, Effects of pH and surface metal oxyhydroxides on deposition and transport of carboxyl-functionalized graphene in saturated porous media, J. Nanopart. Res., 2013, 15, 2079 CrossRef.
- Z. C. Qi, L. L. Zhang, F. Wang, L. Hou and W. Chen, Factors controlling transport of graphene oxide nanoparticles in saturated sand columns, Environ. Toxicol. Chem., 2014, 33, 998–1004 CrossRef CAS PubMed.
- Z. C. Qi, L. L. Zhang and W. Chen, Transport of graphene oxide nanoparticles in saturated sandy soil, Environ. Sci.: Processes Impacts, 2014, 16, 2268–2277 CAS.
- L. Feriancikova and S. Xu, Deposition and remobilization of graphene oxide within saturated sand packs, J. Hazard. Mater., 2012, 235, 194–200 CrossRef PubMed.
- J.-Z. He, D.-J. Wang, H. Fang, Q.-L. Fu and D.-M. Zhou, Inhibited transport of graphene oxide nanoparticles in granular quartz sand coated with Bacillus subtilis and Pseudomonas putida biofilms, Chemosphere, 2017, 169, 1–8 CrossRef CAS PubMed.
- L. Liu, B. Gao, L. Wu, V. L. Morales, L. Y. Yang, Z. H. Zhou and H. Wang, Deposition and transport of graphene oxide in saturated and unsaturated porous media, Chem. Eng. J., 2013, 229, 444–449 CrossRef CAS.
- T. Xia, Y. Qi, J. Liu, Z. Qi, W. Chen and M. R. Wiesner, Cation-inhibited transport of graphene oxide nanomaterials in saturated porous media: The Hofmeister effects, Environ. Sci. Technol., 2017, 51, 828–837 CrossRef CAS PubMed.
- W. Fan, X. Jiang, W. Yang, Z. Geng, M. Huo, Z. Liu and H. Zhou, Transport of graphene oxide in saturated porous media: Effect of cation composition in mixed Na–Ca electrolyte systems, Sci. Total Environ., 2015, 511, 509–515 CrossRef CAS PubMed.
- L. Liu, B. Gao, L. Wu, Y. Sun and Z. Zhou, Effects of surfactant type and concentration on graphene retention and transport in saturated porous media, Chem. Eng. J., 2015, 262, 1187–1191 CrossRef CAS.
- S. Dong, X. Shi, B. Gao, J. Wu, Y. Sun, H. Guo, H. Xu and J. Wu, Retention and release of graphene oxide in structured heterogeneous porous media under saturated and unsaturated conditions, Environ. Sci. Technol., 2016, 50, 10397–10405 CrossRef CAS PubMed.
- Y. Y. Sun, B. Gao, S. A. Bradford, L. Wu, H. Chen, X. Q. Shi and J. C. Wu, Transport, retention, and size perturbation of graphene oxide in saturated porous media: Effects of input concentration and grain size, Water Res., 2015, 68, 24–33 CrossRef CAS PubMed.
- J. Z. He, C. C. Li, D.-J. Wang and D.-M. Zhou, Biofilms and extracellular polymeric substances mediate the transport of graphene oxide nanoparticles in saturated porous media, J. Hazard. Mater., 2015, 300, 467–474 CrossRef CAS PubMed.
- D. Wang, C. Shen, Y. Jin, C. Su, L. Chu and D. Zhou, Role of solution chemistry in the retention and release of graphene oxide nanomaterials in uncoated and iron oxide-coated sand, Sci. Total Environ., 2017, 579, 776–785 CrossRef CAS PubMed.
- M. Wang, B. Gao, D. Tang, H. Sun, X. Yin and C. Yu, Effects of temperature on graphene oxide deposition and transport in saturated porous media, J. Hazard. Mater., 2017, 331, 28–35 CrossRef CAS PubMed.
- T. Lu, T. Xia, Y. Qi, C. Zhang and W. Chen, Effects of clay minerals on transport of graphene oxide in saturated porous media, Environ. Toxicol. Chem., 2017, 36, 655–660 CrossRef CAS PubMed.
- G. Zhou, J. Zhong, H. Zhang, X. Hu, J. Wu, N. Koratkar and X. Shi, Influence of releasing graphene oxide into a clayey sand: physical and mechanical properties, RSC Adv., 2017, 7, 18060–18067 RSC.
- M. Koinuma, C. Ogata, Y. Kamei, K. Hatakeyama, H. Tateishi, Y. Watanabe, T. Taniguchi, K. Gezuhara, S. Hayami, A. Funatsu, M. Sakata, Y. Kuwahara, S. Kurihara and Y. Matsumoto, Photochemical engineering of graphene oxide nanosheets, J. Phys. Chem. C, 2012, 116, 19822–19827 CAS.
- W.-C. Hou, I. Chowdhury, D. G. Goodwin, W. M. Henderson, D. H. Fairbrother, D. Bouchard and R. G. Zepp, Photochemical transformation of graphene oxide in sunlight, Environ. Sci. Technol., 2015, 49, 3435–3443 CrossRef CAS PubMed.
- W.-C. Hou, P.-L. Lee, Y.-C. Chou and Y.-S. Wang, Antibacterial property of graphene oxide: The role of phototransformation, Environ. Sci.: Nano, 2017, 4, 647–657 RSC.
- T. Du, A. S. Adeleye, A. A. Keller, Z. Wu, W. Han, Y. Wang, C. Zhang and Y. Li, Photochlorination-induced transformation of graphene oxide: Mechanism and environmental fate, Water Res., 2017, 124, 372–380 CrossRef CAS PubMed.
- Y. Gao, X. Ren, J. Wu, T. Hayat, A. Alsaedi, C. Cheng and C. Chen, Graphene oxide interactions with co-existing heavy metal cations: adsorption, colloidal properties and joint toxicity, Environ. Sci.: Nano, 2018, 5, 362–371 RSC.
- Y. Gao, J. Wu, X. Ren, X. Tan, T. Hayat, A. Alsaedi, C. Cheng and C. Chen, Impact of graphene oxide on the antibacterial activity of antibiotics against bacteria, Environ. Sci.: Nano, 2017, 4, 1016–1024 RSC.
- E. C. Salas, Z. Sun, A. Lüttge and J. M. Tour, Reduction of graphene oxide via bacterial respiration, ACS Nano, 2010, 4, 4852–4856 CrossRef CAS PubMed.
- Y. Jiao, F. Qian, Y. Li, G. Wang, C. W. Saltikov and J. A. Gralnick, Deciphering the electron transport pathway for graphene oxide reduction by Shewanella oneidensis MR-1, J. Bacteriol., 2011, 193, 3662–3665 CrossRef CAS PubMed.
- O. Akhavan and E. Ghaderi, Escherichia coli bacteria reduce graphene oxide to bactericidal graphene in a self-limiting manner, Carbon, 2012, 50, 1853–1860 CrossRef CAS.
- S. Gurunathan, J. W. Han, V. Eppakayala, M. Jeyaraj and J.-H. Kim, An environmentally friendly approach to the reduction of graphene oxide by Escherichia fergusoni, J. Nanosci. Nanotechnol., 2013, 13, 2091–2098 CrossRef CAS PubMed.
- S. Raveendran, N. Chauhan, Y. Nakajima, H. Toshiaki, S. Kurosu, Y. Tanizawa, R. Tero, Y. Yoshida, T. Hanajiri, T. Maekawa, P. M. Ajayan, A. Sandhu and D. S. Kumar, Ecofriendly route for the synthesis of highly conductive graphene using extremophiles for green electronics and bioscience, Part. Part. Syst. Charact., 2013, 30, 573–578 CrossRef CAS.
- Y. Chen, Y. Niu, T. Tian, J. Zhang, Y. Wang, Y. Li and L. Qin, Microbial reduction of graphene oxide by Azotobacter chroococcum, Chem. Phys. Lett., 2017, 677, 143–147 CrossRef CAS.
- H. Zhang, X. Yu, D. Guo, B. Qu, M. Zhang, Q. Li and T. Wang, Synthesis of bacteria promoted reduced graphene oxide-nickel sulfide networks for advanced supercapacitors, ACS Appl. Mater. Interfaces, 2013, 5, 7335–7340 CAS.
- A. G. Nandgaonkar, Q. Wang, K. Fu, W. E. Krause, Q. Wei, R. Gorga and L. A. Lucia, A one-pot biosynthesis of reduced graphene oxide (RGO)/bacterial cellulose (BC) nanocomposites, Green Chem., 2014, 16, 3195–3201 RSC.
- R. S. Chouhan, A. Pandey, A. Qureshi, V. Ozguz and J. H. Niazi, Nanomaterial resistant microorganism mediated reduction of graphene oxide, Colloids Surf., B, 2016, 146, 39–46 CrossRef CAS PubMed.
- Y. Tanizawa, Y. Okamoto, K. Tsuzuki, Y. Nagao, N. Yoshida, R. Tero, S. Iwasa, A. Hiraishi, Y. Suda, H. Takikawa, R. Numano, H. Okada, R. Ishikawa and A. Sandhu, in Asia-Pacific Interdisciplinary Research Conference 2011, ed. A. Sandhu, 2012, vol. 352 Search PubMed.
- T. Kuila, S. Bose, P. Khanra, A. K. Mishra, N. H. Kim and J. H. Lee, A green approach for the reduction of graphene oxide by wild carrot root, Carbon, 2012, 50, 914–921 CrossRef CAS.
- P. Khanra, T. Kuila, N. H. Kim, S. H. Bae, D.-s. Yu and J. H. Lee, Simultaneous bio-functionalization and reduction of graphene oxide by baker's yeast, Chem. Eng. J., 2012, 183, 526–533 CrossRef CAS.
- S. Gurunathan, J. W. Han, V. Eppakayala and J.-H. Kim, Microbial reduction of graphene oxide by Escherichia coli: A green chemistry approach, Colloids Surf., B, 2013, 102, 772–777 CrossRef CAS PubMed.
- L. Liu, C. Zhu, M. Fan, C. Chen, Y. Huang, Q. Hao, J. Yang, H. Wang and D. Sun, Oxidation and degradation of graphitic materials by naphthalene-degrading bacteria, Nanoscale, 2015, 7, 13619–13628 RSC.
- Z. Guo, C. Xie, P. Zhang, J. Zhang, G. Wang, X. He, Y. Ma, B. Zhao and Z. Zhang, Toxicity and transformation of graphene oxide and reduced graphene oxide in bacteria biofilm, Sci. Total Environ., 2017, 580, 1300–1308 CrossRef CAS PubMed.
- H.-J. Chu, C.-Y. Lee and N.-H. Tai, Green reduction of graphene oxide by Hibiscus sabdariffa L. to fabricate flexible graphene electrode, Carbon, 2014, 80, 725–733 CrossRef CAS.
- Y. Wang, Z. Shi and J. Yin, Facile synthesis of soluble graphene via a green reduction of graphene oxide in tea solution and its biocomposites, ACS Appl. Mater. Interfaces, 2011, 3, 1127–1133 CAS.
- M. Khan, A. H. Al-Marri, M. Khan, N. Mohri, S. F. Adil, A. Al-Warthan, M. R. H. Siddiqui, H. Z. Alkhathlan, R. Berger, W. Tremel and M. N. Tahir, Pulicaria glutinosa plant extract: a green and eco-friendly reducing agent for the preparation of highly reduced graphene oxide, RSC Adv., 2014, 4, 24119–24125 RSC.
- S. B. Maddinedi, B. K. Mandal, R. Vankayala, P. Kalluru and S. R. Pamanji, Bioinspired reduced graphene oxide nanosheets using Terminalia chebula seeds extract, Spectrochim. Acta, Part A, 2015, 145, 117–124 CrossRef CAS PubMed.
- O. S. A. Rahman, V. Chellasamy, N. Ponpandian, S. Amirthapandian, B. K. Panigrahi and P. Thangadurai, A facile green synthesis of reduced graphene oxide by using pollen grains of Peltophorum pterocarpum and study of its electrochemical behavior, RSC Adv., 2014, 4, 56910–56917 RSC.
- B. Haghighi and M. A. Tabrizi, Green-synthesis of reduced graphene oxide nanosheets using rose water and a survey on their characteristics and applications, RSC Adv., 2013, 3, 13365–13371 RSC.
- G. Lee and B. S. Kim, Biological reduction of graphene oxide using plant leaf extracts, Biotechnol. Prog., 2014, 30, 463–469 CrossRef CAS PubMed.
- O. Akhavan, M. Kalaee, Z. S. Alavi, S. M. A. Ghiasi and A. Esfandiar, Increasing the antioxidant activity of green tea polyphenols in the presence of iron for the reduction of graphene oxide, Carbon, 2012, 50, 3015–3025 CrossRef CAS.
- H. Kim, J. Kim, M. Lee, H. C. Choi and W. J. Kim, Stimuli-regulated enzymatically degradable smart graphene-oxide-polymer nanocarrier facilitating photothermal gene delivery, Adv. Healthcare Mater., 2016, 5, 1918–1930 CrossRef CAS PubMed.
- X. Hu, D. Li and L. Mu, Biotransformation of graphene oxide nanosheets in blood plasma affects their interactions with cells, Environ. Sci.: Nano, 2017, 4, 1569–1578 RSC.
- G. P. Kotchey, B. L. Allen, H. Vedala, N. Yanamala, A. A. Kapralov, Y. Y. Tyurina, J. Klein-Seetharaman, V. E. Kagan and A. Star, The enzymatic oxidation of graphene oxide, ACS Nano, 2011, 5, 2098–2108 CrossRef CAS PubMed.
- R. Kurapati, J. Russier, M. A. Squillaci, E. Treossi, C. Menard-Moyon, A. E. Del Rio-Castillo, E. Vazquez, P. Samori, V. Palermo and A. Bianco, Dispersibility-dependent biodegradation of graphene oxide by myeloperoxidase, Small, 2015, 11, 3985–3994 CrossRef CAS PubMed.
- C. M. Girish, A. Sasidharan, G. S. Gowd, S. Nair and M. Koyakutty, Confocal raman imaging study showing macrophage mediated biodegradation of graphene In vivo, Adv. Healthcare Mater., 2013, 2, 1489–1500 CrossRef CAS PubMed.
- Y. Li, L. Feng, X. Shi, X. Wang, Y. Yang, K. Yang, T. Liu, G. Yang and Z. Liu, Surface coating-dependent cytotoxicity and degradation of graphene derivatives: Towards the design of non-toxic, degradable nano-graphene, Small, 2014, 10, 1544–1554 CrossRef CAS PubMed.
- R. V. Lloyd, P. M. Hanna and R. P. Mason, The origin of the hydroxyl radical oxygen in the Fenton reaction, Free Radical Biol. Med., 1997, 22, 885–888 CrossRef CAS PubMed.
- R. Bauer and H. Fallmann, The Photo-Fenton oxidation - A cheap and efficient wastewater treatment method, Res. Chem. Intermed., 1997, 23, 341–354 CrossRef CAS.
- C. Sirtori, A. Zapata, I. Oller, W. Gernjak, A. Agueera and S. Malato, Decontamination industrial pharmaceutical wastewater by combining solar photo-Fenton and biological treatment, Water Res., 2009, 43, 661–668 CrossRef CAS PubMed.
- M. M. Cheng, W. H. Ma, J. Li, Y. P. Huang and J. C. Zhao, Visible-light-assisted degradation of dye pollutants over Fe(III)-loaded resin in the presence of H2O2 at neutral pH values, Environ. Sci. Technol., 2004, 38, 1569–1575 CrossRef CAS PubMed.
- B. L. Allen, G. P. Kotchey, Y. Chen, N. V. K. Yanamala, J. Klein-Seetharaman, V. E. Kagan and A. Star, Mechanistic investigations of horseradish peroxidase-catalyzed degradation of single-walled carbon nanotubes, J. Am. Chem. Soc., 2009, 131, 17194–17205 CrossRef CAS PubMed.
- G. P. Kotchey, S. A. Hasan, A. A. Kapralov, S. H. Ha, K. Kim, A. A. Shvedova, V. E. Kagan and A. Star, A natural vanishing act: The enzyme-catalyzed degradation of carbon nanomaterials, Acc. Chem. Res., 2012, 45, 1770–1781 CrossRef CAS PubMed.
- X. J. Zhou, Y. Zhang, C. Wang, X. C. Wu, Y. Q. Yang, B. Zheng, H. X. Wu, S. W. Guo and J. Y. Zhang, Photo-Fenton reaction of graphene oxide: A new strategy to prepare graphene quantum dots for DNA cleavage, ACS Nano, 2012, 6, 6592–6599 CrossRef CAS PubMed.
- H. Bai, W. T. Jiang, G. P. Kotchey, W. A. Saidi, B. J. Bythell, J. M. Jarvis, A. G. Marshall, R. A. S. Robinson and A. Star, Insight into the mechanism of graphene oxide degradation via the photo-Fenton reaction, J. Phys. Chem. C, 2014, 118, 10519–10529 CAS.
- C.-Z. Zhang, T. Li, Y. Yuan and J. Xu, An efficient and environment-friendly method of removing graphene oxide in wastewater and its degradation mechanisms, Chemosphere, 2016, 153, 531–540 CrossRef CAS PubMed.
- F. F. Wang, F. Wang, G. D. Gao and W. Chen, Transformation of graphene oxide by ferrous iron: Environmental implications, Environ. Toxicol. Chem., 2015, 34, 1975–1982 CrossRef CAS PubMed.
- F. Wang, F. Wang, D. Zhu and W. Chen, Effects of sulfide reduction on adsorption affinities of colloidal graphene oxide nanoparticles for phenanthrene and 1-naphthol, Environ. Pollut., 2015, 196, 371–378 CrossRef CAS PubMed.
- Y. Li, N. Yang, T. Du, T. Xia, C. Zhang and W. Chen, Chloramination of graphene oxide significantly affects its transport properties in saturated porous media, NanoImpact, 2016, 3–4, 90–95 CrossRef.
- Y. Li, N. Yang, T. Du, X. Wang and W. Chen, Transformation of graphene oxide by chlorination and chloramination: Implications for environmental transport and fate, Water Res., 2016, 103, 416–423 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |