
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermination of polar organic micropollutants in surface and pore water by high-resolution sampling-direct injection-ultra high performance liquid chromatography-tandem mass spectrometry†
Malte
Posselt
 *a,
Anna
Jaeger
bc,
Jonas L.
Schaper
bd,
Michael
Radke
e and
Jonathan P.
Benskin
a
*a,
Anna
Jaeger
bc,
Jonas L.
Schaper
bd,
Michael
Radke
e and
Jonathan P.
Benskin
a
aDepartment of Environmental Science and Analytical Chemistry (ACES), Stockholm University, Stockholm, Sweden. E-mail: malte.posselt@aces.su.se
bLeibniz-Institute of Freshwater Ecology and Inland Fisheries, Ecohydrology Department, Berlin, Germany
cHumboldt University Berlin, Geography Department, Berlin, Germany
dTechnical University Berlin, Chair of Water Quality Engineering, Berlin, Germany
eInstitute for Hygiene and Environment, Free and Hanseatic City of Hamburg, Germany
First published on 12th October 2018
Abstract
Hyporheic zones (HZs) are dynamic and complex transition regions between rivers and aquifers which are thought to play an important role in the attenuation of environmental micropollutants. Non-steady state and small-scale hyporheic processes which affect micropollutants in the HZ are poorly characterized due to limitations in existing analytical methodologies. In this work we developed a method for high spatio-temporal resolution analysis of polar organic micropollutants (POMs) in hyporheic pore- and surface waters by combining (semi-) automatic low volume sampling techniques with direct-injection ultra-high performance liquid chromatography tandem mass spectrometry. The method is capable of quantifying 25 parent compounds and 18 transformation products (TPs) using only 0.4 mL of water and few preparation steps. Application of the method to both surface and pore water revealed significant (i.e. > an order of magnitude) differences in POM concentrations over small time and spatial scales (i.e. < a few hours and tens of cm, respectively). Guanylurea, a TP of the antidiabetic drug metformin was detected at unprecedentedly high concentrations. Collectively, this method is suitable for in situ characterization of POMs at high spatial and temporal resolution and with minimal disturbance of natural flow paths and infiltration of surface water.
Environmental significanceHyporheic zones are dynamic and complex transition regions between rivers and aquifers which serve a key role in the functioning of aquatic ecosystems, including the turnover and degradation of organic pollutants. Multi-dimensional sampling without disturbing the small scale hyporheic flow paths in the field is challenging and few methods exist. We therefore developed an efficient workflow for studying polar organic micropollutants (POMs) and their transformation products in the hyporheic zone at high spatio-temporal resolution. This was achieved by improving an existing sampling device and coupling it to a newly developed high throughput-direct injection-UHPLC-MS/MS method. Application of the method in the field revealed significant differences in POM concentrations that varied over small time- and spatial scales. These data shed new light on the behavior of POMs in the hyporheic zone. |
Introduction
Polar organic micropollutants (POMs) encompass a wide range of pharmaceuticals and personal care products, pesticides, performance chemicals, and transformation products which are widespread in the aquatic environment. Increasing evidence suggests that a large number of these substances pass through or are formed in wastewater treatment plants (WWTPs),1,2 making effluent a major source of POMs in water.3 Many POMs are pharmaceutically active, and may have adverse health effects on wildlife.4–12Increased awareness of POMs has resulted in major recent developments surrounding their regulation on the European level. In 2013, the European Commission for the first time added pharmaceutically active compounds to a Watch List13 under the environmental quality standard directive14 naming them as a potential risk to surface waters and effectively making them candidates for the Water Framework Directive priority list. A later addition to the priority list would demand management action and a progressive reduction of the emission of these compounds by member states.15 This represents a major change in how pharmaceutically active compounds are treated legally, and highlights ongoing efforts to characterize and reduce unintentional exposure to both humans and wildlife. The German Association of Energy and Water Industries, representing 1800 companies, called for a strategic action plan for the protection of water resources in the wake of increasing consumption of pharmaceuticals.16
The risks associated with POMs has led to a growing need to understand their fate and behavior in aquatic ecosystems. Within this environment, hyporheic zones (HZs) are thought to play a particularly important role in micropollutant attenuation. HZs are dynamic and complex transition regions between rivers and aquifers characterized by the simultaneous occurrence of multiple physical, biological and chemical processes.17 Fischer et al.18 described HZs as serving the “liver function” of a river with regards to cycling of nutrients and natural organic matter but also degradation of micropollutants.19,20 Nevertheless, contaminant attenuation in the HZ is poorly understood and there are few methods suitable for investigating advection and biogeochemical conditions in the field or simply for sampling along the complex, small-scale flow paths within the HZ. Studying this transition region is important for understanding micropollutant degradation processes, improving knowledge on the self-purification capacity of rivers and lakes, and ultimately facilitating a more comprehensive assessment of risks associated with release of POMs into the aquatic environment.
Traditionally, sampling of hyporheic pore water for both nutrient and micropollutant analysis involves either ex situ techniques such as squeezing21 and centrifugation22 or in situ techniques such as gel samplers,23 equilibrium passive-24 or dialysis samplers or suction filtration.25Ex situ techniques are highly invasive since they require the removal of sediment; therefore, they are of limited use for time-series sampling or long term observations. Passive, gel and dialysis samplers are less invasive but tend to provide time-weighted average concentrations, typically over periods of days or weeks. While these techniques can be used to estimate the overall contribution of the HZ to the micropollutant attenuation potential of a river system,26 they provide little information about the underlying small-scale processes occurring throughout the HZ, such as the influence of bedforms (i.e. advection), biogeochemical processes, or the abundance of microorganisms. Rivers may also undergo rapid changes (i.e. hours–days) for instance as a result of anthropogenic activities or storm events,27 which can alter the composition and concentration of POMs directly through remobilization28 but also indirectly e.g. through combined sewer overflows.29 To study non-steady state conditions and small-scale hyporheic processes, high spatial and temporal resolution, achieved through low-volume direct sampling of water from the HZ, is critical.
The objective of the present work was to develop a method for high spatio-temporal resolution sampling (i.e. few centimeter and minute scales, respectively) and analysis of POMs in hyporheic pore water (HPW) and surface water (SW). Previously, single- or multi-level suction filtration techniques have allowed sampling at higher spatial and temporal resolution.30 The present work built upon these initial efforts by developing and validating an in situ, minimally invasive sampling device based on the USGS minipoint sampler.31 Spatial and temporal resolution was achieved through low-volume (1 mL min−1) HPW sampling and the installation of autosamplers for collecting SW. When paired with a highly sensitive, low-volume-direct-injection ultra-high performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS) platform, the entire method was capable of quantitative analysis of 43 substances (25 parent compounds and 18 transformation products) with typical detection limits of ∼100 ng L−1 or lower using only 0.4 mL of water and few preparation steps. Collectively, this method facilitates high resolution sampling and analysis of POMs, suitable for in situ characterization of time and spatial profiles without disturbing natural flow paths or inducing SW infiltration.
Material and methods
Target compound selection
The 43 POMs studied were selected based on literature review,32,33 initial screening experiments, and standard availability. The selected compounds cover a wide range of physico-chemical properties (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kow between −2.03 and 5.39) and compound classes (Table S4 in the ESI†), including 6 analgesics (4 NSAIDs, acetaminophen, and the opioid analgesic tramadol), 3 β-blockers, 3 lipid regulators, 2 angiotensin II receptor antagonists, 2 antihyperglycemics, 2 diuretics, 2 corrosion inhibitors, 1 antianxiety agent, 1 antibiotic, 1 anticonvulsant, 1 stimulant, 1 antidepressant, 1 artificial sweetener and 18 TPs.
Kow between −2.03 and 5.39) and compound classes (Table S4 in the ESI†), including 6 analgesics (4 NSAIDs, acetaminophen, and the opioid analgesic tramadol), 3 β-blockers, 3 lipid regulators, 2 angiotensin II receptor antagonists, 2 antihyperglycemics, 2 diuretics, 2 corrosion inhibitors, 1 antianxiety agent, 1 antibiotic, 1 anticonvulsant, 1 stimulant, 1 antidepressant, 1 artificial sweetener and 18 TPs.
Standards and reagents
LC/MS grade water was purchased from VWR (Darmstadt, Germany), methanol (MeOH) from Merck KGaA (Darmstadt, Germany) and acetic acid (HAc, ≥99.7%) from Sigma-Aldrich (Darmstadt, Germany). Native and isotope-substituted internal standards (Table S5†) were purchased from Toronto Research Chemicals Inc., (North York, Canada) and Sigma-Aldrich. Stock solutions (c = 1 g L−1 in MeOH) were used to prepare two working solutions, one containing internal standards (c = 10 mg L−1 in MeOH) and a second containing native standards (c = 0.94–2.4 mg L−1 in MeOH, depending on the expected environmental concentrations of each compound). Working solution two was used to prepare calibration standards with LC/MS grade water (for concentration ranges see Table S6†).Study site and experimental overview
Sampling was carried out on the fourth order, eutrophic lowland river Erpe19 at the eastern edge of Berlin, Germany. The Erpe receives effluent water (EW) from several smaller WWTPs and one large WWTP (Muenchehofe) which has a dry weather capacity of 42500 m3 d−1 treating approximately 15% of Berlin's wastewater. The EW content of the stream underlies strong diurnal fluctuations and can reach more than 80% of the total discharge. Water samples were collected during field campaigns in April (24 h time series HPW + SW) and June (48 h time series SW) 2016. SW time series samples were collected hourly using an autosampler (ISCO 3700 portable sampler, Teledyne Isco, Lincoln NE, USA, equipped with 24 glass bottles that were cooled with ice and exchanged every 24 h) which was installed 700 m downstream of the EW inflow (uniform mixing was assumed at this point). Grab samples of surface water were collected upstream (USW) of the EW inflow to test for background POM concentrations. Discharge at the SW sampling site was determined by Q–h rating curves, using a handheld electromagnetic current meter (MF pro, OTT Hydromet, Kempten, Germany) and a water level logger (CTD-Diver, VanEssen Instruments, Delft, the Netherlands). Average discharge was 0.46 m3 s−1 (max: 0.68 m3 s−1, min: 0.26 m3 s−1) during sampling in April and 0.73 m3 s−1 (max: 1.38 m3 s−1, min: 0.26 m3 s−1) during sampling in June. Sediment characteristics were determined from one, 40 cm long sediment core taken directly at the HPW sampling location after the sampling event in June 2016 using a hand auger (9.0 cm ID). Porosity (determined from oven dried samples) decreased with depth, ranging from 0.6 in the top 8 cm of the streambed to 0.4 in the lower sections of the sediment profile (Table S1†). Saturated hydraulic conductivity, measured using a KSAT instrument (METER, Germany) ranged from 3.6 × 10−5 m s−1 to 1.7 × 10−4 m s−1. Grain size sieving indicated that streambed sediment predominantly consisted of medium and fine sand (Table S2†). Sediment characteristics are in close agreement with values reported previously for the same site.34
Modified minipoint samplers
Collection of HPW at high spatial and temporal resolution was achieved using modified USGS minipoint samplers (Fig. 1). Sampler dimensions are summarized in Table 1 along with a comparison to the original device described by Duff et al.31 The major modifications included increasing the sampler length, reducing the dead volume, and the use of both individual samplers and semi-automated sampling using a syringe pump. These changes had the net effect of increasing the sampling depth (down to 1.5 m), improving resolution, and facilitating automated collection of samples over 24 h. We also evaluated the temporal resolution/sampling rate and effective radius of the sediment sphere, which were not previously characterized in Duff et al.31 | ||
| Fig. 1 Schematic illustration of the minipoint samplers as they were assembled and applied in this study. Optodes for oxygen measurement attached to each sampler were used to control for potential surface water infiltration. | ||
| Parameter | Duff et al. | Present study |
|---|---|---|
| a n.a. – information not available. | ||
| Number of minipoint samplers | 6 | 3 (6 per syringe pump) |
| Orientation/spatial arrangement | Circular arrangement, 10 cm diameter | Row (flexible, individual samplers) |
| Outer diameter of minipoint | 0.318 cm | 0.3 cm |
| Number of slits | 3 | 4 |
| Dimension of slits | 0.8 × 0.04 cm | 1 × 0.05 cm |
| Maximum sampling depth evaluated (maximum feasible) | 15 cm (n.a.) | 30 cm (150 cm) |
| Maximum spatial resolution (maximum feasible) | 2.5 cm | 5 cm horizontal and 10 cm vertical distance (1.5 cm) |
| Maximum temporal resolution | n.a. | 30 min (June sampling) |
| Sampling rate (feasible) | n.a. | 1 mL min−1 (0.452 μL h−1 to 1451 mL h−1) |
| Effective radius of sediment sphere | n.a. | >0.5 cm (depending on sample volume) |
| Pump type | Peristaltic | Syringe pump |
| Dead volume | 2 mL | 1.4 mL |
Each sampler consisted of a 1.5 m steel tube (3 mm outer diameter) which was filter screened 5 mm from the closed tip (1 cm long laser cut slits, width ≈ 0.5 mm). Since sediment grain size was dominated by medium to fine sands, clogging of the minipoint sampler did not occur. The inner dead volume of the sampler was simultaneously reduced and fixed to just 1.4 cm3 using PEEK tubing (0.762 mm internal diameter × 3.05 m length, Sigma Aldrich) which was run through the length of the sampler to the perforated tip. Each sampler could be moved and positioned individually facilitating 3-dimensional sampling in the river bed. This offers the possibility to sample along hyporheic flow paths (e.g. sand dunes and ripples), which is an area of ongoing investigation in our labs. In addition, the low cross-sectional area of the individual tubes and the low form drag allowed sampling over several weeks without the need to adjust the sampler position due to hydrodynamic scour and the entanglement of flotsam.
Samplers were pushed vertically into the river sediment and then maintained at the desired depth using a rubber stopper (∼2.5 cm diameter, Fig. 1). The stopper sat flat on the sediment in order to prevent vertical infiltration of sediment and water along the tube. The PEEK tubing was connected to a sampling syringe (20 mL volume) via Swagelok fittings (Swagelok, Solon, USA) and the syringes were operated automatically using NE-1600 6-channel syringe pumps (New Era Pump Systems, Inc, Farmingdale, USA) and emptied after each sampling event. A total volume of 13 ± 0.5 mL pore water was sampled at a rate of 1 mL min−1. The first 2 mL (inner PEEK tube volume multiplied by 1.4) of the sampler were discarded to remove old water in the tubings.
The volume of pore water that can be sequentially sampled within a given time interval without considerably disturbing the hyporheic flow field (i.e. the temporal resolution of the sampler) is a function of both sediment porosity and pore water velocity. Vertical pore water velocity was calculated from temperature depth profiles, measured using a Multi-Level Temperature Stick (MLTS, UIT, Dresden), which is described in detail elsewhere.35 Temperature time series were measured in 5, 10, 15, 20, 30 and 40 cm depths from one week before until one week after the sampling event. Vertical Darcy velocities were calculated using the combined amplitude ratio/phase shift method presented by McCallum et al.,35 embedded in the VFLUX 2.0 code36 as described elsewhere.34 Modelled Darcy velocities were corrected for sediment porosity to obtain pore water velocities. Mean pore water velocities (positive indicating downward flow) calculated during the sampling events were 4.3 cm h−1 during the April sampling event and 7.5 cm h−1 during the June sampling event (Fig. S1†) which is in line with literature data37 and with flow rates previously calculated at the same site.34 With observed pore water velocities and sediment porosities, 50 min was identified as the minimum sampling interval under the conditions encountered during the April sampling campaign. A detailed discussion on the calculation of the minimum temporal resolution of the method is provided in the ESI.†
The minipoint HPW sampling apparatus was installed 800 m downstream of the EW outfall at a site where downwelling conditions prevailed (MLTS data; Fig. S1†). The sampling site was located in the middle of a relatively homogeneous section of the stream channel (a more detailed description of the site can be found in Schaper et al. 2018).34 Depending on the variability of the respective stream the sampling site identification as well as an estimation of the minimal sampling resolution can take place before to the beginning of a sampling campaign. The temporal resolution of the sampler is a function of both sediment porosity and pore water velocity, both of which can be determined prior to collection of samples for POM analysis. Vertical pore water velocity can be calculated from temperature depth profiles while sediment porosity can be obtained from sediment cores.
Water was sampled simultaneously at sediment depths of 10, 20, and 30 cm. Three samplers were positioned in a line with 5 cm gaps transversally to the river flow direction. At this distance no overlapping of sampling spheres was expected. A detailed calculation of the minimum spatial resolution of the method is provided in the ESI.† The sampling rate was set to 1 mL min−1 and one 13 ± 0.5 mL sample was taken per h for 24 h and split for different analyses. O2, conductivity and pH were monitored for signs of surface water infiltration. O2 content was measured using optodes attached to the samplers (Fig. 1; Fibox 4 O2 Meter with Oxygen Dipping Probe; Presense Precision Sensing GmbH, Regensburg, Germany). HPW for the inter-laboratory comparison experiment was sampled using a 1D dialysis sampler38 equipped with a polysulfone membrane (pore size 0.2 μm). All samples collected in the field were immediately frozen on dry ice and stored at −20 °C until further processing. Repeated measurements of a frozen sample and standards over 12 months revealed no significant changes in POM concentrations, therefore freezing was deemed to be an appropriate means of sample preservation in the present work.
Instrumental analysis
Prior to analysis, samples were defrosted in a 25 °C water bath and vortexed, after which a sample volume of 400 μL was combined with 97.5 μL MeOH and 2.5 μL internal standard mix. The samples were vortexed again and filtered (Filtropur S 0.45 μm, PES membrane, Sarstedt AG&Co, Nuembrecht, Germany) into microvials (2 mL; Thermo Scientific, Dreieich, Germany). Samples were either analyzed directly or stored at −20 °C prior to analysis.Samples were chromatographed on a Thermo Scientific Ultimate 3000 UHPLC system equipped with a Waters (Manchester, UK) Acquity UPLC HSS T3 column (1.8 μm, 2.1 mm × 100 mm). The mobile phase consisted of 10 mM acetic acid in deionized water (solvent A) and 10 mM acetic acid in methanol (solvent B). Starting conditions were 97% A/3% B, which were ramped to 40% B in 2.7 min, and finally to 97% B in 3.3 min. The system was maintained at isocratic conditions for 1.2 min after which it was returned to starting conditions and equilibrated for 4 min. The flow rate was set to 500 μL min−1 during the gradient and 1000 μL min−1 for equilibration. The column oven temperature was set to 45 °C.
MS/MS analysis was carried out using a Thermo Scientific Quantiva triple-quadrupole mass spectrometer equipped with a heated electrospray ionization source. The mass spectrometer was operated in polarity switching, selected reaction monitoring mode. The positive spray voltage was set to 3.7 kV, the negative spray voltage to 3.5 kV, the sheath gas to 38 arbitrary units and the auxiliary gas to 10 arbitrary units. The ion transfer tube temperature was set to 305 °C and the vaporizer temperature to 350 °C. Compound specific MS parameters were optimized using standard solutions (1 mg L−1 in 80% deionized water/20% MeOH) of each analyte which were infused directly into the mass spectrometer. Fine tuning of source gas pressures and selection of optimal product ions was carried out using samples (spiked samples if concentrations were low) from the river Erpe. Individual precursor/product ion transitions are provided in Table S5.† Multiple ions for a given target were monitored and their ratio was examined for signs of a co-eluting interference.
A series of calibration standards (prepared in 80% LC/MS grade water/20% MeOH) containing all target compounds and internal standards was measured in the beginning, middle and end of each series of samples (typically comprising 100–150 samples). The linearity (expressed as correlation coefficient, r2; detailed information in Table S6†) of the calibration curve was greater than 0.992 for all target compounds except for alpha-hydroxymetoprolol (0.987). In addition, one standard was measured every 15 samples for quality control. MS data were processed using Thermo Scientific Xcalibur 3.1.66.10 instrument software and quantified using the internal standards method. The chromatographic peaks of 2-chlorobenzoic acid and 4-chlorobenzoic acid as well as of 2-hydroxyibuprofen and 3-hydroxyibuprofen, respectively, were not fully separated. Therefore peak areas were integrated together and the sum of both compounds, 2/4-chlorobenzoic acid and 2/3-hydroxyibuprofen respectively, was processed further.
Matrix-specific LODs and LOQs were defined as the concentration producing a signal-to-noise ratio (s/n) of 3:1 and 10:1, respectively. Exceptions were carbamazepine, 4-hydroxydiclofenac, valsartan and venlafaxine, in which calculated LODs were negative due to the calibration curves having larger, positive y-intercepts. In these cases the LOD was set to the LOQ. For the calculation of means, measured concentrations above the LOD but below the LOQ were replaced by LOQ × 2−0.5; values below LOD were replaced by LOD × 2−0.5.
Method validation
Recovery experiments using spiked samples were carried out in 5 different matrices—SW, USW, HPW, tap water (TW) and Milli-Q-water (MQ)—at fortification levels of ∼0.5 μg L−1 and ∼5 μg L−1 (see Table S6† for exact concentrations). Accuracy was determined by comparing measured values with fortified concentrations while precision was assessed by calculating the relative standard deviation of replicates. Instrumental repeatability and reproducibility were also assessed through replicate measurements of a standard over the course of a day (N = 4) or week (N = 5), respectively. Matrix-induced ionization effects were evaluated by comparing the area of each isotopically-labelled compound in spiked samples to a standard in LC/MS grade water (20% MeOH).As a final validation of the method, two interlaboratory comparisons were carried out on a subset of parent compounds. First, samples collected by a dialysis sampler from the Erpe river sediment (N = 25) were split and analyzed in parallel using the current method and a method (method B) at the Institute for Hygiene and Environment (Hamburg, Germany) based on the German standard method DIN 38407-47:2017-07.39 Second, Erpe surface water samples (N = 46) were split and analysed in parallel using the current method and a method (method C) established previously at the Chair of Water Quality Engineering (Technical University Berlin, Berlin, Germany).40,41
Statistical analysis
Patterns in the fluctuation of individual POMs in both the 24 h time series were investigated graphically and statistically using MetaboAnalyst 3.0.42 Target compounds with more than 65% missing values were not included in the analysis. For the remaining targets, missing values were estimated using Bayesian principal component analysis, after which the entire dataset was cube root transformed or autoscaled (mean centering and dividing by the standard deviation of each variable). Two statistical analyses were carried out: first, hierarchical clustering was performed using Euclidean distance as a measure of similarity and clustering using Ward's linkage (which minimizes the sum of squares of any two clusters). The resulting cluster was then plotted as a heat map. A correlation matrix (presented in the ESI†) was also created to visualize overall correlations between different POMs using Spearman's rank correlation. A Shapiro–Wilk-test was used to control for normality and a Levene's test for homogeneity of group variances. Differences in group means were evaluated using ANOVA in cases where variances were equal and Welch's ANOVA when variances were unequal.Results and discussion
Method performance and validation
Results of spike/recovery experiments are summarized in Fig. S3† and Tables S7 and S8.† Accuracy and precision were excellent for most compounds at both fortification levels, with >75% of target compounds displaying recoveries between 75–125% and relative standard deviations (RSDs) of <25% in all 5 matrices at both fortification levels. Instrumental repeatability and reproducibility were also good, with RSDs of <24% and <18%, respectively, for all compounds (Tables S6 and S12†). Substances displaying sub-optimal performance included diclofenac amide, acridone and acridine (20–50% recovery in SW, USW, and HPW), 2/4-chlorobenzoic acid (∼135% recovery in SW, USW, and HPW, both fortification levels), and diclofenac, ibuprofen, and their respective transformation products, which could not be observed in samples of MQ or TW. One possible explanation was that the declustering potential (optimized in the presence of riverwater matrix) was too high for MQ samples and resulted in in-source fragmentation, leading to an apparent drop in signal. In most cases, precision remained excellent for these substances despite their poor recoveries. Given the focus on temporal and spatial changes in the present work (i.e. relative measurements), lower accuracy for these substances was acceptable, and the method was deemed fit-for-purpose.A comparison of matrix-induced ionization effects revealed moderate signal suppression in SW, USW and HPW matrices for most compounds (<50%), while signal intensities of acesulfame, acetaminophen, metoprolol, metoprolol acid and sotalol were enhanced (Fig. S4†). Matrix-specific method LODs and LOQs (Tables S10 and S11†) were comparable to other direct injection methods43,44 however we reach these limits over a challenging range of matrix parameters. For example, in the high organic carbon content (DOC: up to 11.6 mg L−1) and salinity (conductivity: ∼800–1200 mS cm−1) river investigated here, we achieve LODs below 100 ng L−1 for 35 of 43 targets. A limitation of the method was higher detection limits for ibuprofen and its TPs. We suggest using correction factors to handle the observed lower recoveries of some compounds. Overall, these data indicated excellent method performance across a diverse range of concentrations and matrices.
The exact sampling parameters of the minipoint apparatus depend on the respective sampling environment with sediment properties and pore water velocity being limiting factors. Obviously, the system works best in sandy sediments and cannot be applied in clayey sediment layers (see also ESI†). Pore water velocities need to be considered when defining sampling volume, frequency and extraction speed. The minipoints should therefore be combined with methods to determine flow.
As a final validation of the method, we compared concentrations of 12 compounds measured by the current method with concentrations generated by two other labs using similar methods. These data are summarized in Fig. S5.† The average relative deviation from our results ranged from 74 and 136% for all but two targets: higher levels of 1H-benzotriazole (194 ± 28%) were reported using the method B relative to the current method and higher levels of diclofenac (165 ± 36%) were reported using the method C relative to the current method. An explanation for the differences between labs remains unclear at this time but may be due to inconsistencies in sample preservation and/or sample handling during shipment.
POM profiles in the river Erpe
An overview of POM profiles in USW, HPW, and SW is provided in Fig. 2 and Table S9.† A total of 35 out of 43 targets (including 14 TPs) were detectable at concentrations up to 222 μg L−1 (guanylurea). Four substances were above LOQ in USW, possibly due to inputs from smaller WWTPs or diffuse discharges further upstream. Hence, the largest proportion of POMs in HPW and SW originates from the WWTP Muenchehofe. POM concentrations of frequently detected compounds in SW and HPW were mostly similar or higher in SW (Fig. 3 cluster 1). Significantly higher concentrations in SW relative to HPW (p < 0.05; ANOVA) were found for 17 compounds (2/4 chlorobenzoic acid, 4-hydroxydiclofenac, acesulfame, bezafibrate, carbamazepine, carbamazepine-10,11-epoxide, diclofenac, furosemide, guanylurea, irbesartan, metformin, metoprolol, O-desmethylvenlafaxine, sitagliptin, sotalol, valsartan and alpha-hydroxy-metoprolol), possibly due to transformation in the HZ. The dilution with groundwater can be excluded as a cause due to prevailing downwelling conditions. 1H-benzotriazole, chlorothiazide and valsartan acid were unusual in that they were the only targets present at significantly (p < 0.05; ANOVA) higher concentrations in HPW relative to SW (Fig. 3 cluster 2), suggesting formation or accumulation in the HZ. Higher concentrations of the TP valsartan acid fit the observation of lower concentrations of the respective parent compound valsartan in the HZ. | ||
| Fig. 2 Average Erpe surface water (SW, N = 24), hyporheic pore water (HPW, N = 72, all depths) ±SD (only + SD for diclofenac amide) and upstream surface water (USW, data > LOQ) concentrations measured over 24 h in April. * Indicates that SW and HPW concentrations were significantly different (p < 0.05; ANOVA). ♦ Indicates concentrations < LOQ. | ||
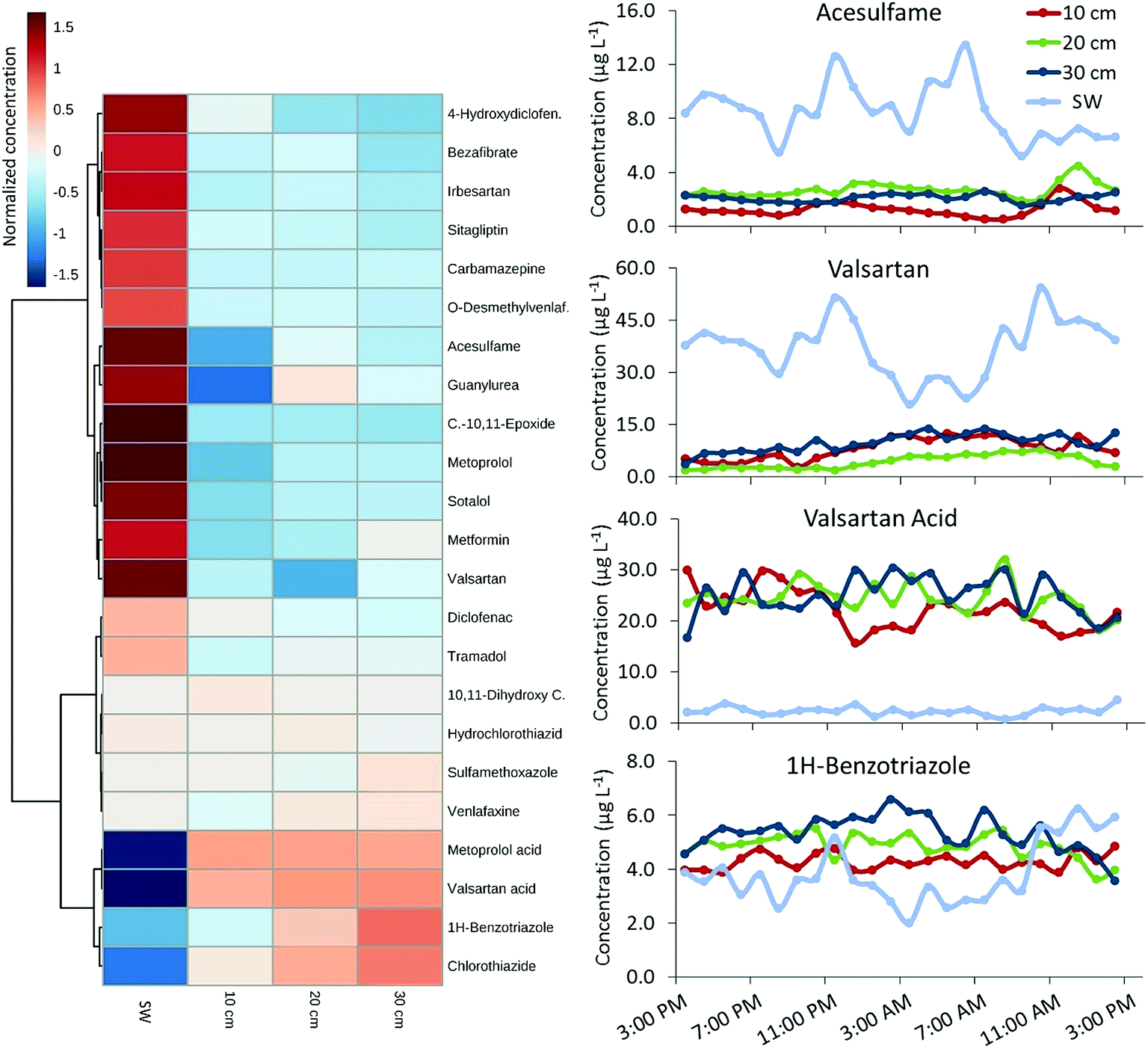 | ||
| Fig. 3 24 h time-series for micropollutants in Erpe surface water (SW) and hyporheic pore water extracted from three different depths (10, 20 and 30 cm below surface): clustered heat map with class averages of cube root transformed concentration data and exemplary time series plots (measured real concentrations) for acesulfame and valsartan (cluster 1), valsartan acid and 1H-benzotriazole (cluster 2). Samples were taken in April. | ||
Concentrations of parent compounds reported here are considerably higher than those found previously in similar environments,45,46 possibly due to the high EW ratio in this river. In fact, levels of diclofenac, bezafibrate, 1H-benzotriazole and ketoprofen reached or exceeded reported maximum concentrations from a comprehensive European survey that included data from 122 sampling stations.47 Concentrations of carbamazepine, hydrochlorothiazide, metoprolol and bezafibrate exceeded levels found in comprehensive WWTP effluent studies.45,48 Acetaminophen, gemfibrozil and ibuprofen were not detected in any samples. Considering the widespread use of these compounds their absence suggests efficient removal by the WWTP, which is supported by literature data.49–51 Naproxen was detected in SW only which could be due to efficient bacterial degradation in the HZ.52
Elevated concentrations of the TPs guanylurea, O-desmethylvenlafaxine and valsartan acid were also observed, in some cases exceeding their respective parent compound concentrations by over an order of magnitude. In fact, to our knowledge maximum concentrations of guanylurea reported here (222 μg L−1) are among the highest reported in the aquatic environment. In comparison, a literature review from 2014 reported that concentrations of guanylurea in small streams with high effluent load reached up to 30 μg L−1,53 while concentrations in EW were up to 100 μg L−1.54 To confirm whether our measurements were realistic, we compared our data with release estimates derived from metformin consumption data (the parent compound of guanylurea) in Germany. Details of these calculations can be found in the ESI.† Overall, the estimated maximum quantity of metformin released from the WWTP Muenchehofe on a daily basis (58.9 mol) is highly consistent with the daily sum of guanylurea and metformin measured empirically in the Erpe during two days in April 2016 (54.9 mol). These data confirm that the high concentrations of guanylurea reported here are realistic considering the consumption of metformin. This finding is of concern, considering that 40 μg L−1 of metformin was reported to induce significant adverse effects in male fish.55 To our knowledge there are no data on the ecotoxicity of guanylurea.
Temporal and spatial variability
A comparison of POM concentrations at various depths in HPW and SW (hourly sampling in April; Fig. 3) revealed considerable fluctuation over 24 h. For example, the range of HPW concentrations of sitagliptin and 4-hydroxydiclofenac varied by an order of magnitude (Table S9†), while SW concentrations of valsartan acid varied by a factor of 6 (i.e. 0.7–4.5 μg L−1; mean 2.3 μg L−1; Fig. 3 and Table S9†). SW concentrations over 48 h (hourly sampling in June) are provided in Fig. 4. For 12 of the 35 detected POMs, peak SW concentrations were observed between noon and 18:00 (Fig. 4: clusters 2a and 2b) while a drop in almost all concentrations was observed between 04:00 and 10:00. These local maxima and minima, respectively, are largely in line with the diurnal fluctuation of EW release from the WWTP which follows the water consumption of the local urban population with a temporal shift reflecting the duration of wastewater transport and treatment. This is particularly true for parent compounds, but for TPs the fluctuations were less pronounced (Fig. 3). While the source of parent compounds is primarily WWTP discharge, TPs are at least partially formed within the river. Possible explanations for the lower diurnal fluctuations for some substances may be back diffusion from the HZ or biofilms, but these hypotheses require further investigation.
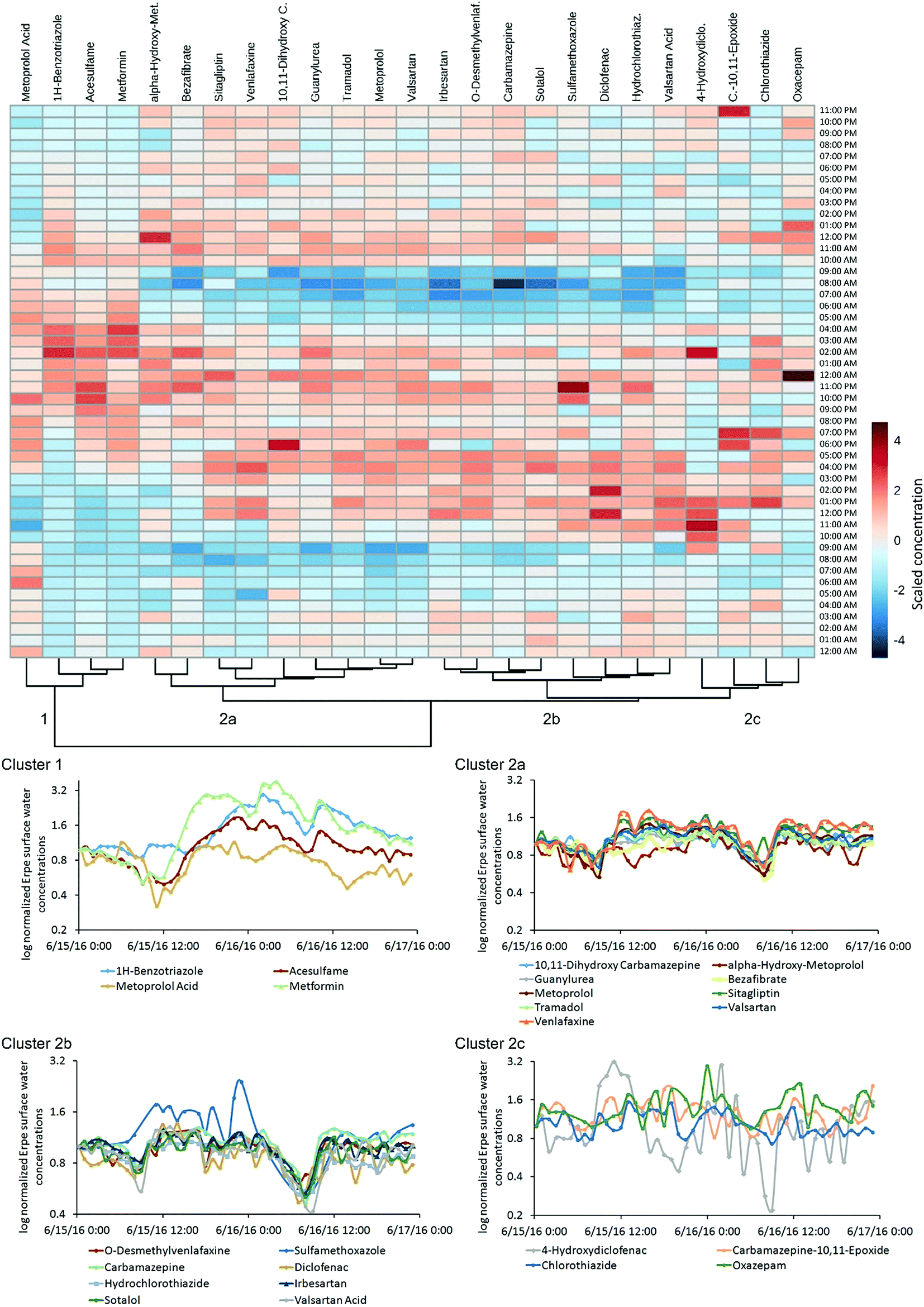 | ||
| Fig. 4 48 h time-series for micropollutants in river Erpe surface water in June 2016: clustered heat map of scaled concentration data and time series plots of log normalized concentration data for each cluster. Alpha-hydroxy-met. = alpha-hydroxy-metoprolol, C.-10,11-epoxide = carbamazepine-10,11-epoxide, 10,11-dihydroxy C. = 10,11-dihydroxy carbamazepine, 4-hydroxydiclo. = 4-hydroxydiclofenac, O-desmethylvenlaf. = O-desmethylvenlafaxine, 1-methyl-1H-BZ = 1-methyl-1H-benzotriazole, hydrochlorothiaz. = hydrochlorothiazide. 12:00 AM = 5/15/16 (MEZ + 1). | ||
Some substances had distinctly dissimilar temporal concentration trends than the majority of target compounds. The concentration of 1H-benzotriazole for instance was higher on the second day of sampling. Apart from its application as a corrosion inhibitor in dishwashing agents this compound is used in various industries and emissions may follow a different pattern. Clustering revealed unique behavior for 1H-benzotriazole, acesulfame, metformin and metoprolol acid relative to other substances, with a less distinct decrease in concentration in the early morning (cluster 1, Fig. 4). These substances might follow different consumption patterns (acesulfame) or be the subject of exceptional transformation processes, both of which are areas for further investigation.
Alongside the temporal variations, a comparison of 24 h POM depth profiles revealed remarkable and significant differences with depth, both within the HZ as well as between SW and HPW (Fig. 3). Differences were highly compound-specific; concentrations of substances such as carbamazepine or metoprolol acid were similar at different HPW depths, while those of sotalol, acesulfame and metoprolol were between 2- and 10- fold higher in SW compared to HPW. Significantly lower concentrations of these compounds were detected in HPW from 10 cm depth compared to the deeper sampling depths. In contrast, TPs such as valsartan acid appeared to form in the HZ (Fig. 3 and Table S9†), resulting in dramatic changes in parent compound (valsartan)/TP (valsartan acid) ratios between SW and HPW and over a 10 cm change in depth (p < 0.05; ANOVA). These differences can only be captured using a highly spatially resolved sampling procedure such as the one employed here. Most POMs occurred at lower concentrations in the upper HZ compared to the other HPW and SW sampling points. The concurrent observation of lower parent compound concentrations and higher TP concentrations in the upper HZ layer (10 cm depth) for metoprolol/metoprolol acid and metformin/guanylurea (Fig. 3) points to increased transformation rates in this region. The entire HZ but especially its upper boundary layer with the benthic zone presents a highly dynamic transition region; high productivity and occurrence of physical, biological and chemical processes seem to encourage multiple simultaneous micropollutant degradation mechanisms. For 1H-benzotriazole the HZ appears to act as a sink (highest concentrations were found in the deepest sampled layer) while the TPs valsartan acid and chlorothiazide seem to form and—at least temporarily—accumulate in deeper layers at concentrations above 30 μg L−1 and 1 μg L−1 respectively (Fig. 3 and S9†). Thus, changing hydraulic conditions—for instance following storm events—could turn the HZ into a source leading to TP concentration peaks in SW. A similar dynamic functioning of the HZ has been described for nutrients.56
The exfiltration of river water to groundwater in the sampling area was previously estimated at ∼800 m3 d−1,57 representing a mere 2% (maximum) of SW discharge. However, hyporheic exchange (i.e. infiltration and subsequent re-exfiltration to the SW) may vary along the river, depending on sediment characteristics. This heterogeneity makes quantification of reach-scale hyporheic exchange, and ultimately it's impact on SW quality, challenging, and beyond the scope of this study.58 Nevertheless the method presented here provides a means of investigating this in future work. Furthermore, POM data generated here together with data on reach-scale flow and hyporheic exchange could be used for a more quantitative understanding of polluted river systems (i.e. via calculation of removal rates). In addition, our sampling approach allows the coupling of online HPW monitoring with online flow investigations e.g. heat pulse sensing,59 opening up even more areas for potential application.
Conclusion
Tools for high temporal and spatial resolution analysis of POMs are important for characterizing their behavior in the environment. Until now, equilibrium samplers like peepers or passive samplers which provide time weighted average concentrations would not resolve such short term concentration peaks. Our method significantly increased the possibilities regarding active HPW sampling in the field: with a sampling volume of 13 ± 0.5 mL which is sufficient for a range of different analyses and the observed pore water velocity, a minimum sampling interval of 50 min and a minimum spatial resolution of ca. 4 cm (distance between samplers) was achievable under conditions observed during our river Erpe sampling campaign in April. In sediments with higher velocities and extracting only the theoretical minimum volume of 1.8 mL (dead volume + 400 μL minimum sample volume for UHPLC-MS/MS) our method is capable of reaching higher spatio-temporal resolutions (realistically 2 centimeter and minute scales, respectively).High-frequency sampling with a high spatial and temporal resolution enabled us to detect significant small-scale differences in micropollutant and TP concentrations that vary within short periods of time while estimations based on measured sediment porosity and pore water velocities showed that our sampling method lead to minimal (if any) disturbances of natural, hyporheic flow paths. In the future, we aim to apply this method in further comprehensive investigations on the fate of micropollutants in the aquatic environment and the role of the HZ in rivers. The exact understanding of the processes controlling micropollutant fate in rivers is further required to improve the efficacy of river engineering or restoration measures on micropollutant transformation/degradation but also to enable predictions of how for instance future changes of local climate conditions might affect self-purification capacities.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the European Union's Horizon2020 research and innovation programme under Marie–Skłodowska–Curie grant agreement No. 641939. Furthermore, this project has also received funding from the Research Training Group ‘Urban Water Interfaces (UWI)’ (Project N6 “Retention of chemical compounds in hyporheic reactors of urban freshwater systems”, GRK 2032/1), which is funded by the German Research Foundation (DFG).References
- C. D. Metcalfe, B. G. Koenig, D. T. Bennie, M. Servos, T. A. Ternes and R. Hirsch, Occurrence of neutral and acidic drugs in the effluents of Canadian sewage treatment plants, Environ. Toxicol. Chem., 2003, 22, 2872–2880 CrossRef CAS PubMed.
- R. Loos, R. Carvalho, D. C. António, S. Comero, G. Locoro, S. Tavazzi, B. Paracchini, M. Ghiani, T. Lettieri and L. Blaha, EU-wide monitoring survey on emerging polar organic contaminants in wastewater treatment plant effluents, Water Res., 2013, 47, 6475–6487 CrossRef CAS PubMed.
- T. A. Ternes, Analytical methods for the determination of pharmaceuticals in aqueous environmental samples, TrAC, Trends Anal. Chem., 2001, 20, 419–434 CrossRef CAS.
- S. Kusari, D. Prabhakaran, M. Lamshöft and M. Spiteller, In vitro residual anti-bacterial activity of difloxacin, sarafloxacin and their photoproducts after photolysis in water, Environ. Pollut., 2009, 157, 2722–2730 CrossRef CAS PubMed.
- J. Sunderland, C. M. Tobin, A. J. Hedges, A. P. MacGowan and L. O. White, Antimicrobial activity of fluoroquinolone photodegradation products determined by parallel-line bioassay and high performance liquid chromatography, J. Antimicrob. Chemother., 2001, 47, 271–275 CrossRef CAS PubMed.
- K. Fent, A. A. Weston and D. Caminada, Ecotoxicology of human pharmaceuticals, Aquat. Toxicol., 2006, 76, 122–159 CrossRef CAS PubMed.
- S. Jiao, S. Zheng, D. Yin, L. Wang and L. Chen, Aqueous photolysis of tetracycline and toxicity of photolytic products to luminescent bacteria, Chemosphere, 2008, 73, 377–382 CrossRef CAS PubMed.
- C. J. Sinclair and A. B. Boxall, Assessing the ecotoxicity of pesticide transformation products, Environ. Sci. Technol., 2003, 37, 4617–4625 CrossRef CAS PubMed.
- R. F. Dantas, O. Rossiter, A. K. R. Teixeira, A. S. Simões and V. L. da Silva, Direct UV photolysis of propranolol and metronidazole in aqueous solution, Chem. Eng. J., 2010, 158, 143–147 CrossRef CAS.
- M. DellaGreca, M. Brigante, M. Isidori, A. Nardelli, L. Previtera, M. Rubino and F. Temussi, Phototransformation and ecotoxicity of the drug naproxen-Na, Environ. Chem. Lett., 2003, 1, 237–241 CrossRef.
- M. l. Farré, S. Pérez, L. Kantiani and D. Barceló, Fate and toxicity of emerging pollutants, their metabolites and transformation products in the aquatic environment, TrAC, Trends Anal. Chem., 2008, 27, 991–1007 CrossRef.
- V. S. Thomaidi, A. S. Stasinakis, V. L. Borova and N. S. Thomaidis, Is there a risk for the aquatic environment due to the existence of emerging organic contaminants in treated domestic wastewater? Greece as a case-study, J. Hazard. Mater., 2015, 283, 740–747 CrossRef CAS PubMed.
- The European Parliament and the Council of the European Union, Directive 2013/39/EU of the European Parliament and of the Council of 12 August 2013 amending Directives 2000/60/EC and 2008/105/EC as regards priority substances in the field of water policy, Off. J. Eur. Union, 2013, 226, 1–17 Search PubMed.
- The European Parliament and the Council of the European Union, Directive 2008/105/EC of the European Parliament and of the Council of 16 December 2008 on environmental quality standards in the field of water policy, amending and subsequently repealing Council Directives 82/176/EEC, 83/513/EEC, 84/156/EEC, 84/491/EEC, 86/280/EEC and amending Directive 2000/60/EC of the European Parliament and of the Council, Off. J. Eur. Union, 2008, 348, 84–97 Search PubMed.
- The European Parliament and the Council of the European Union, Directive 2000/60/EC of the European Parliament and of the Council of 23 October 2000 establishing a framework for Community action in the field of water policy, Official Journal of the European Communities 2000, L327, 1–72 Search PubMed.
- German Association of Energy and Water Industries BDEW, Steigender Medikamentenverbrauch erfordert Maβnahmenpaket zum Schutz der Gewässer, https://www.bdew.de/presse/presseinformationen/steigender-medikamentenverbrauch-erfordert-ma%C3%9Fnahmenpaket-zum-gewaesserschutz/, (accessed Feb 26, 2018).
- A. J. Boulton, S. Findlay, P. Marmonier, E. H. Stanley and H. M. Valett, The functional significance of the hyporheic zone in streams and rivers, Annu. Rev. Ecol. Syst., 1998, 29, 59–81 CrossRef.
- H. Fischer, F. Kloep, S. Wilzcek and M. T. Pusch, A river's liver–microbial processes within the hyporheic zone of a large lowland river, Biogeochemistry, 2005, 76, 349–371 CrossRef CAS.
- J. Lewandowski, A. Putschew, D. Schwesig, C. Neumann and M. Radke, Fate of organic micropollutants in the hyporheic zone of a eutrophic lowland stream: results of a preliminary field study, Sci. Total Environ., 2011, 409, 1824–1835 CrossRef CAS PubMed.
- F. Jüttner, Efficacy of bank filtration for the removal of fragrance compounds and aromatic hydrocarbons, Water Sci. Technol., 1999, 40, 123–128 CrossRef.
- M. Bender, W. Martin, J. Hess, F. Sayles, L. Ball and C. Lambert, A whole-core squeezer for interfacial pore-water sampling, Limnol. Oceanogr., 1987, 32, 1214–1225 CrossRef CAS.
- G. Ankley and M. Schubauer-Berigan, Comparison of techniques for the isolation of sediment pore water for toxicity testing, Arch. Environ. Contam. Toxicol., 1994, 27, 507–512 CrossRef CAS.
- M. Krom, P. Davison, H. Zhang and W. Davison, High-resolution pore-water sampling with a gel sampler, Limnol. Oceanogr., 1994, 39, 1967–1972 CrossRef.
- G. Cornelissen, A. Pettersen, D. Broman, P. Mayer and G. D. Breedveld, Field testing of equilibrium passive samplers to determine freely dissolved native polycyclic aromatic hydrocarbon concentrations, Environ. Toxicol. Chem., 2008, 27, 499–508 CrossRef CAS PubMed.
- S. E. Bufflap and H. E. Allen, Sediment pore water collection methods for trace metal analysis: a review, Water Res., 1995, 29, 165–177 CrossRef CAS.
- U. Kunkel and M. Radke, Reactive tracer test to evaluate the fate of pharmaceuticals in rivers, Environ. Sci. Technol., 2011, 45, 6296–6302 CrossRef CAS PubMed.
- M. A. Zimmer and L. K. Lautz, Temporal and spatial response of hyporheic zone geochemistry to a storm event, Hydrol. Processes, 2014, 28, 2324–2337 CrossRef CAS.
- J. Eggleton and K. V. Thomas, A review of factors affecting the release and bioavailability of contaminants during sediment disturbance events, Environ. Int., 2004, 30, 973–980 CrossRef CAS PubMed.
- P. Phillips, A. Chalmers, J. Gray, D. Kolpin, W. Foreman and G. Wall, Combined sewer overflows: an environmental source of hormones and wastewater micropollutants, Environ. Sci. Technol., 2012, 46, 5336–5343 CrossRef CAS PubMed.
- M. Beck, O. Dellwig, K. Kolditz, H. Freund, G. Liebezeit, B. Schnetger and H. J. Brumsack, In situ pore water sampling in deep intertidal flat sediments, Limnol. Oceanogr.: Methods, 2007, 5, 136–144 CrossRef CAS.
- J. H. Duff, F. Murphy, C. C. Fuller, F. J. Triska, J. W. Harvey and A. P. Jackman, A mini drivepoint sampler for measuring pore water solute concentrations in the hyporheic zone of sand-bottom streams, Limnol. Oceanogr., 1998, 43, 1378–1383 CrossRef CAS.
- Z. Li, A. Sobek and M. Radke, Fate of pharmaceuticals and their transformation products in four small European rivers receiving treated wastewater, Environ. Sci. Technol., 2016, 50, 5614–5621 CrossRef CAS PubMed.
- Z. Li, A. Sobek and M. Radke, Flume experiments to investigate the environmental fate of pharmaceuticals and their transformation products in streams, Environ. Sci. Technol., 2015, 49, 6009–6017 CrossRef CAS PubMed.
- J. L. Schaper, W. Seher, G. Nützmann, A. Putschew, M. Jekel and J. Lewandowski, The fate of polar trace organic compounds in the hyporheic zone, Water Res., 2018, 140, 158–166 CrossRef CAS PubMed.
- A. McCallum, M. Andersen, G. Rau and R. Acworth, A 1-D analytical method for estimating surface water–groundwater interactions and effective thermal diffusivity using temperature time series, Water Resour. Res., 2012, 48, W11532 CrossRef.
- R. P. Gordon, L. K. Lautz, M. A. Briggs and J. M. McKenzie, Automated calculation of vertical pore-water flux from field temperature time series using the VFLUX method and computer program, J. Hydrol., 2012, 420, 142–158 CrossRef.
- S. B. Grant, K. Stolzenbach, M. Azizian, M. J. Stewardson, F. Boano and L. Bardini, First-order contaminant removal in the hyporheic zone of streams: physical insights from a simple analytical model, Environ. Sci. Technol., 2014, 48, 11369–11378 CrossRef CAS PubMed.
- J. Lewandowski, K. Rüter and M. Hupfer, Two-dimensional small-scale variability of pore water phosphate in freshwater lakes: results from a novel dialysis sampler, Environ. Sci. Technol., 2002, 36, 2039–2047 CrossRef CAS PubMed.
- Z. Li, M. P. Maier and M. Radke, Screening for pharmaceutical transformation products formed in river sediment by combining ultrahigh performance liquid chromatography/high resolution mass spectrometry with a rapid data-processing method, Anal. Chim. Acta, 2014, 810, 61–70 CrossRef CAS PubMed.
- F. Zietzschmann, G. Aschermann and M. Jekel, Comparing and modeling organic micro-pollutant adsorption onto powdered activated carbon in different drinking waters and WWTP effluents, Water Res., 2016, 102, 190–201 CrossRef CAS PubMed.
- F. Zietzschmann, R.-L. Mitchell and M. Jekel, Impacts of ozonation on the competition between organic micro-pollutants and effluent organic matter in powdered activated carbon adsorption, Water Res., 2015, 84, 153–160 CrossRef CAS PubMed.
- J. Xia, I. V. Sinelnikov, B. Han and D. S. Wishart, MetaboAnalyst 3.0—making metabolomics more meaningful, Nucleic Acids Res., 2015, 43, W251–W257 CrossRef CAS PubMed.
- T. Reemtsma, L. Alder and U. Banasiak, A multimethod for the determination of 150 pesticide metabolites in surface water and groundwater using direct injection liquid chromatography–mass spectrometry, J. Chromatogr. A, 2013, 1271, 95–104 CrossRef CAS PubMed.
- T. S. Oliveira, M. Murphy, N. Mendola, V. Wong, D. Carlson and L. Waring, Characterization of pharmaceuticals and personal care products in hospital effluent and waste water influent/effluent by direct-injection LC-MS-MS, Sci. Total Environ., 2015, 518, 459–478 CrossRef PubMed.
- B. Kasprzyk-Hordern, R. M. Dinsdale and A. J. Guwy, The removal of pharmaceuticals, personal care products, endocrine disruptors and illicit drugs during wastewater treatment and its impact on the quality of receiving waters, Water Res., 2009, 43, 363–380 CrossRef CAS PubMed.
- D. Bendz, N. A. Paxeus, T. R. Ginn and F. J. Loge, Occurrence and fate of pharmaceutically active compounds in the environment, a case study: Höje river in Sweden, J. Hazard. Mater., 2005, 122, 195–204 CrossRef CAS PubMed.
- R. Loos, B. M. Gawlik, G. Locoro, E. Rimaviciute, S. Contini and G. Bidoglio, EU-wide survey of polar organic persistent pollutants in European river waters, Environ. Pollut., 2009, 157, 561–568 CrossRef CAS PubMed.
- M. S. Kostich, A. L. Batt and J. M. Lazorchak, Concentrations of prioritized pharmaceuticals in effluents from 50 large wastewater treatment plants in the US and implications for risk estimation, Environ. Pollut., 2014, 184, 354–359 CrossRef CAS PubMed.
- A. Joss, E. Keller, A. C. Alder, A. Göbel, C. S. McArdell, T. Ternes and H. Siegrist, Removal of pharmaceuticals and fragrances in biological wastewater treatment, Water Res., 2005, 39, 3139–3152 CrossRef CAS PubMed.
- Z. Li, E. Undeman, E. Papa and M. S. McLachlan, High-throughput evaluation of organic contaminant removal efficiency in a wastewater treatment plant using direct injection UHPLC-Orbitrap-MS/MS, Environ. Sci.: Processes Impacts, 2018, 20, 561–571 RSC.
- N. Paxeus, Removal of selected non-steroidal anti-inflammatory drugs (NSAIDs), gemfibrozil, carbamazepine, b-blockers, trimethoprim and triclosan in conventional wastewater treatment plants in five EU countries and their discharge to the aquatic environment, Water Sci. Technol., 2004, 50, 253–260 CrossRef CAS PubMed.
- D. Wojcieszyńska, D. Domaradzka, K. Hupert-Kocurek and U. Guzik, Bacterial degradation of naproxen–undisclosed pollutant in the environment, J. Environ. Manage., 2014, 145, 157–161 CrossRef PubMed.
- T. Ter Laak and K. Baken, The occurrence, fate and ecological and human health risks of metformin and guanylurea in the water cycle-A literature review, The Water Research Institute, KWR, 2014, p. 24 Search PubMed.
- C. I. Kosma, D. A. Lambropoulou and T. A. Albanis, Comprehensive study of the antidiabetic drug metformin and its transformation product guanylurea in Greek wastewaters, Water Res., 2015, 70, 436–448 CrossRef CAS PubMed.
- N. J. Niemuth, R. Jordan, J. Crago, C. Blanksma, R. Johnson and R. D. Klaper, Metformin exposure at environmentally relevant concentrations causes potential endocrine disruption in adult male fish, Environ. Toxicol. Chem., 2015, 34, 291–296 CrossRef CAS PubMed.
- C. Maazouzi, C. Claret, M.-J. Dole-Olivier and P. Marmonier, Nutrient dynamics in river bed sediments: effects of hydrological disturbances using experimental flow manipulations, J. Soils Sediments, 2013, 13, 207–219 CrossRef CAS.
- H. Verleger and F. Schumacher, Auswirkungen von Maßnahmen zur Gewässerentwicklung und zur Verbesserung des Hochwasserschutzes in der Erpe auf den Grundwasserstand, Erläuterungsbericht Staatsverwaltung für Stadtentwicklung und Umwelt, Berlin, 2012 Search PubMed.
- M. C. Westhoff, M. N. Gooseff, T. A. Bogaard and H. H. G. Savenije, Quantifying hyporheic exchange at high spatial resolution using natural temperature variations along a first-order stream, Water Resour. Res., 2011, 47, W10508 CrossRef.
- J. Lewandowski, L. Angermann, G. Nützmann and J. H. Fleckenstein, A heat pulse technique for the determination of small-scale flow directions and flow velocities in the streambed of sand-bed streams, Hydrol. Processes, 2011, 25, 3244–3255 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8em00390d |
| This journal is © The Royal Society of Chemistry 2018 |