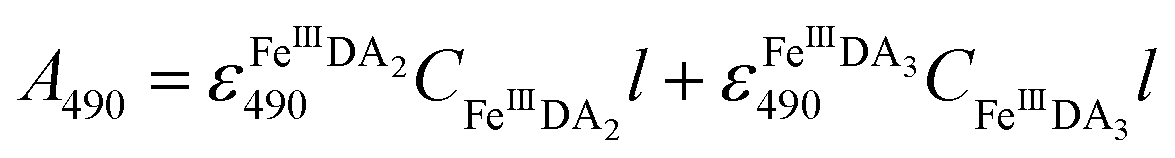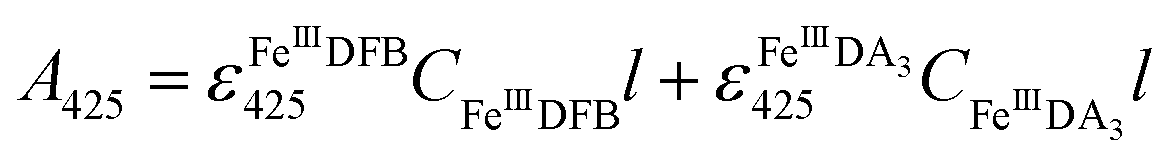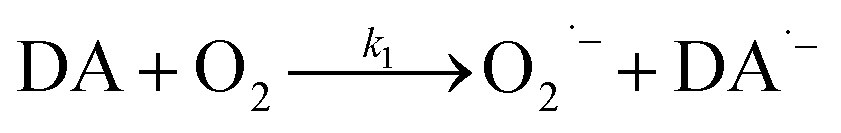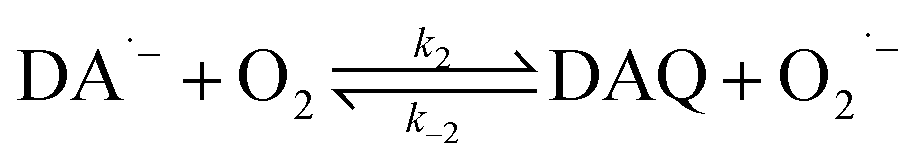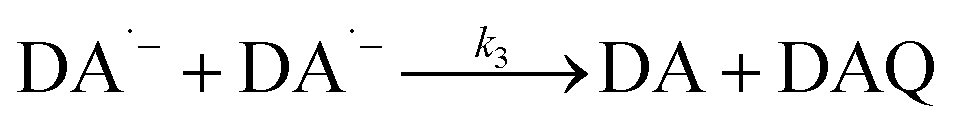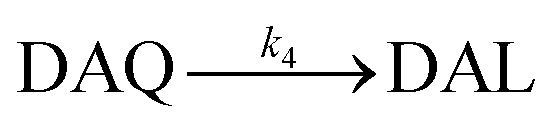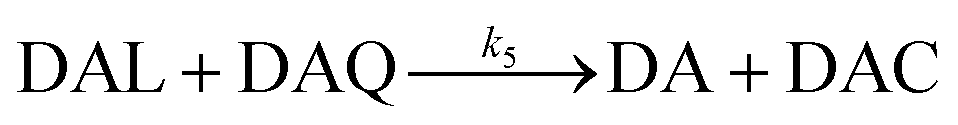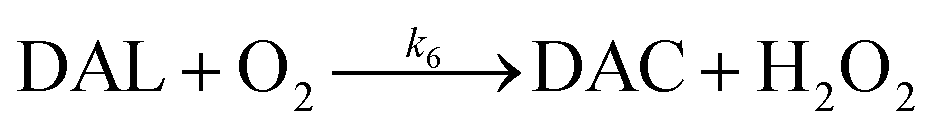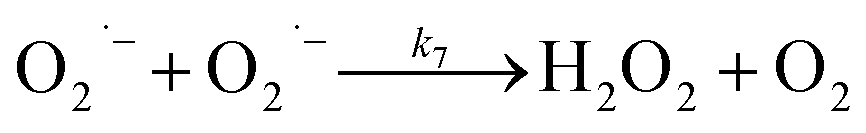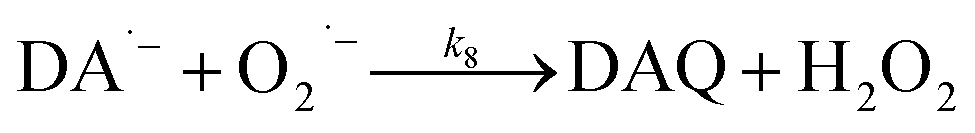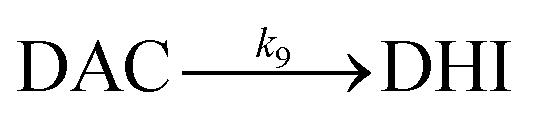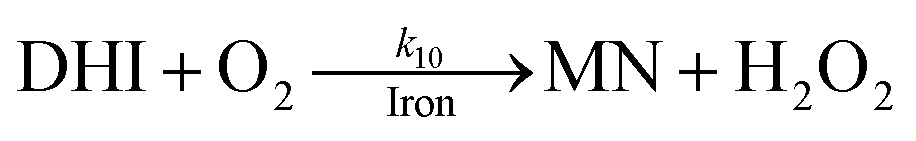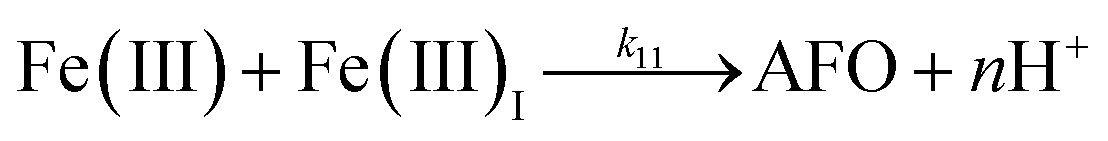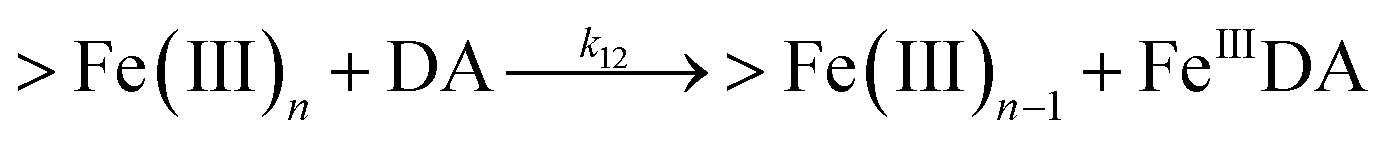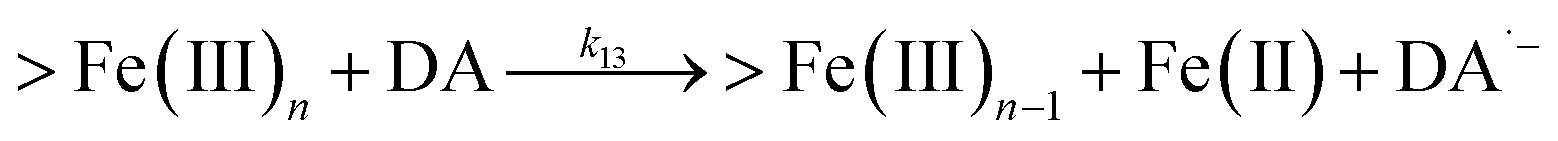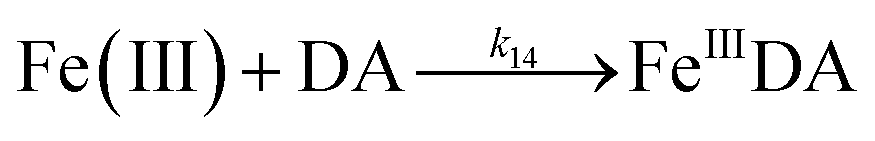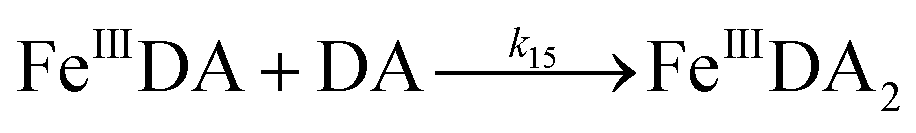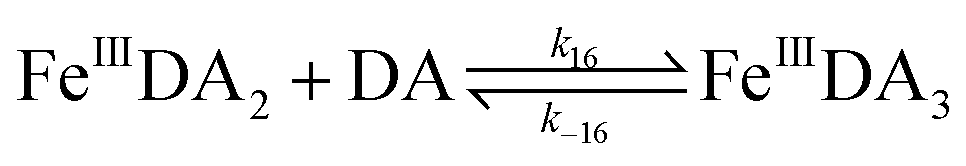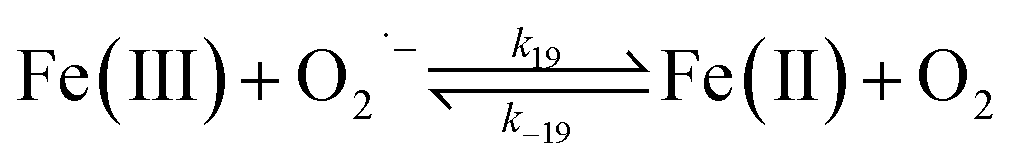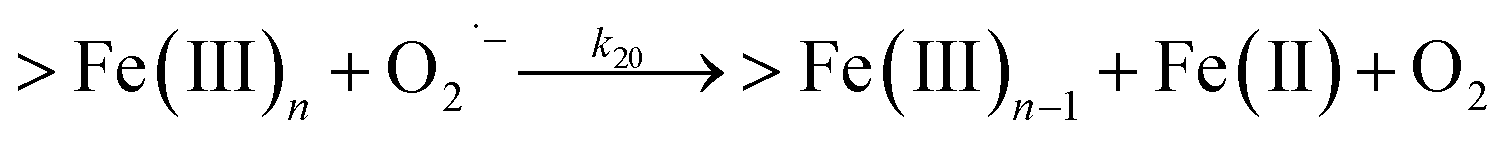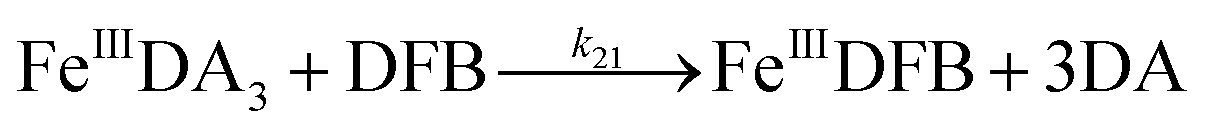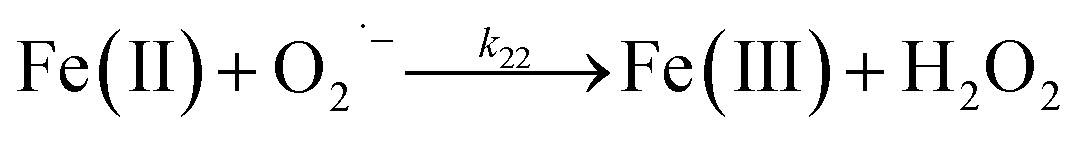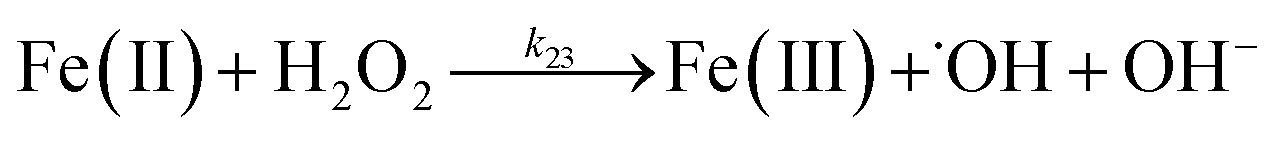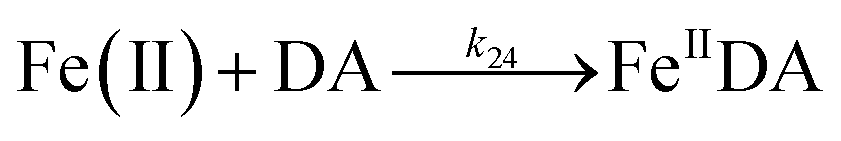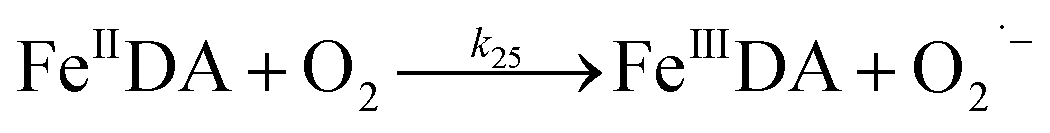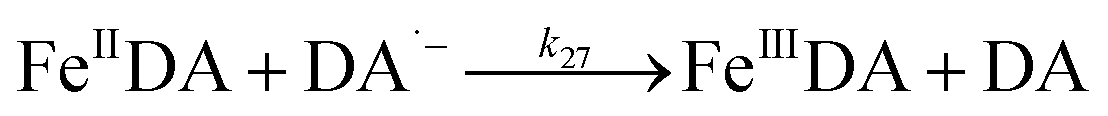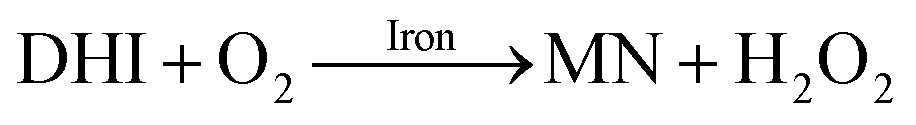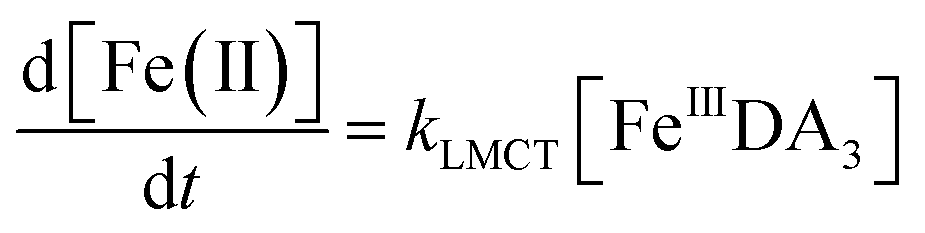Effect of release of dopamine on iron transformations and reactive oxygen species (ROS) generation under conditions typical of coastal waters†
Received
22nd October 2017
, Accepted 11th December 2017
First published on 11th December 2017
Abstract
Seasonally persistent blooms of Ulvaria obscura var. blyttii, the prominent species present in green tides in the northern Pacific and Atlantic, have been well documented in recent decades. The synthesis and release of dopamine (DA) by Ulvaria obscura var. blyttii has been proposed to be associated with the suppression and inhibition of the growth of other organisms competing for limited resources. To better understand the potential benefits obtained from the release of DA, the transformation of DA as well its concomitant impact on the local seawater environment are investigated in this study. The results show that, despite several toxic quinones being produced during the oxidation of DA, aminochrome (DAC) is likely to be the only quinone playing an allelopathic role in view of its expected accumulation in the surrounding environment. As a consequence of the direct oxidation of DA and DA induced generation of 5,6-dihydroxyindole (DHI), high concentrations of H2O2 accumulate over time, especially in the presence of elements including iron, calcium and magnesium. The oxidative stress to other organisms induced by the release of DA may be particularly detrimental as a result of H2O2 induced reduction in photosynthesis, inactivation of antioxidant systems or even the generation of ˙OH. DA induced iron mobilization may benefit the continuously persistent blooms of Ulvaria obscura var. blyttii or even the whole community via alleviation in iron deficiency within the bloom region.
Environmental significance
The occurrence of Ulvaria obscura var. blyttii in the northern Pacific Ocean has increased in recent years, most likely as a result, at least in part, of anthropogenic influences. Despite the absence of solid evidence, release of dopamine (DA) has been purported to be an element of the allelopathic strategy developed by Ulvaria obscura var. blyttii. The results from this work reveal that, of all the oxidative metabolites, H2O2 rather than DA related quinones is likely to be most detrimental in view of its extended half life and its critical role in oxidative stress. Additional ecological benefits may also be obtained by Ulvaria obscura var. blyttii as a result of the DA related alleviation in iron stress through inducement of reduction of iron to the relatively soluble Fe(II) form and the formation of relatively stable DA bound iron complexes.
|
Introduction
Increasing input of nutrients as a result of anthropogenic interference1 has resulted in the frequent occurrence of green tides in the northern Pacific Ocean in the past few decades with the seaweed Ulvaria obscura var. blyttii being a prominent component.2,3 Seaweeds such as Ulvaria may significantly impact the local environment including the alteration of dissolved gases,4 depletion and oversupply of essential nutrients5 and the release of allelochemicals.6 Even though the effects of tidal dilution and the strong buffering capacity of the marine environment can largely minimize the local influence induced by green tides, severe chemical modifications have been reported. Field measurements around Washington State suggest that fluctuation in pH as a result of the photosynthesis and respiration of macroalgae can be up to 1.8 units.7 Among all the modifications, the release of allelochemicals, a suite of organic compounds produced by marine macroalgae to mediate interactions with other organisms, is strongly related to the occurrence of blooms of the prominent species present. For Ulvaria obscura var. blyttii, it has been reported that dopamine (DA) is synthesized in chloroplasts' stromal regions and stored in vesicles with the primary goal of functioning as a feeding deterrent.7,8 Laboratory simulations indicate that more than 500 μM DA can be released into surrounding environments as a result of desiccation induced tissue damage.9 In a manner similar to that of dimethylsulfoniopropionate (DMSP), another abundant allelochemical produced by algae,10 the oxidative metabolites of DA can result in severe toxicity to nearby organisms. Inhibition of the germination of seaweeds within the bloom area has been frequently reported.11 Interestingly, while it has been reported that the release of DA can result in the mortality of some of the Ulvaria obscura var. blyttii as well, continuous growth is still observed.12 The survival of Ulvaria obscura var. blyttii suggests that benefits arising from the DA induced chemical modifications to the bloom region may possibly exist.
In general, besides the metabolites generated via the oxidative degradation of DA, the presence of large amounts of DA may also exert a significant influence on the fate of trace elements such as iron via complexation and possibly reduction processes.13,14 Biologically, availability of iron is critical for the growth and metabolism of phytoplankton and seagrasses including photosynthesis and respiration.15,16 While the optimal concentration of iron for biological growth is around 100 nM, the concentration of dissolved iron in oceanic waters typically ranges from 0.02–2 nM with these low concentrations rendering iron one of the key limiting factors for the growth of seagrasses.16–18 As such, DA induced iron mobilization may contribute to the continued survival of Ulvaria obscura var. blyttii. However, the interaction between DA and iron can also dramatically accelerate the oxidative transformation of DA19 with these transformations potentially aggravating the environmental modifications induced by the release of DA. Therefore, to better understand both the benefits and disadvantages associated with the production and storage of DA by Ulvaria obscura var. blyttii, it is imperative to clarify and quantify the mutual influence between DA and iron, especially in view of the toxicity induced by the oxidative metabolites of DA and bioavailability of iron.
While numerous studies having been conducted on the interaction between DA and iron, most previous work has been undertaken at the typical pH of extracellular fluid (ECF) in view of the particular interest in the role played by these two substances in the progression of Parkinson's disease.20,21 As such, limited information with regard to the interaction between DA and iron has been provided under seawater conditions. As pH-sensitive substances, variation in pH will change the speciation of both DA and iron, thereby potentially exerting a significant influence on the reactivity of DA and iron. In addition, while the oxidative metabolites of DA have long been proposed to be associated with the allelopathic effects induced by DA, most previous studies focused on the in situ influence on marine organisms.11 Thus, quantitative description and the relative importance of the role played by different toxic metabolites are far from well understood. Even though enhancement in the concentration of dissolved iron is evident as a result of DA induced iron mobilization,19,22 the fate and bioavailability of the DA bound iron have not been investigated.
To address the uncertainties mentioned above, we focus in this study on the generation of aminochrome (DAC) and H2O2via the oxidative degradation of DA both in the absence and presence of iron under marine conditions. Given the unique role of iron in the metabolism of seagrasses, the influence of DA on the fate of iron as well as the subsequent acquisition of DA bound iron via the naturally produced siderophore, desferrioxamine B (DFB), is also investigated. To further assist our understanding of the mutual influence of iron and DA, a kinetic model is developed herein based on the experimental data collected in this study.
Materials and methods
A complete description of the materials and methods used in this paper is presented in ESI 1.†
All experiments were conducted at pH 8.0 and under dark conditions in order to avoid the possible effects of light on iron transformations. While light effects are certainly relevant to natural systems, detailed studies of photochemical processes will be the subject of future investigations. The temperature of all the experiments was controlled at 22 ± 0.6 °C with this temperature within the range from 5 °C to 29 °C over which Ulvaria obscura is able to survive.23 Both Ca2+ and Mg2+ were found to significantly modify the UV-Vis spectrum of the iron complex formed with DA. As such, most of the experiments described were conducted in 0.1 M NaCl, 2 mM NaHCO3 and 10 mM 3-(N-morpholino)propanesulfonic acid (MOPS) solutions (referred to as buffer solutions hereafter) in order to enable comparison with previous work. However, in order to further examine the effect of Ca2+ and Mg2+ on the generation of DA derived toxicants, typical concentrations of these cations in seawater (i.e., 54.4 mM MgCl2 and 10.5 mM CaCl2) were added in several experiments to solutions containing 0.7 M NaCl and 10 mM MOPS (referred to herein as synthetic seawater) to mimic the marine environment.24
Experimental measurements
DAC measurement.
The concentration of DAC was quantified spectrophotometrically at 475 nm in a 10 cm cuvette using a Cary 60 spectrophotometer with baseline correction at 850 nm.25,26 To investigate the decay of DAC, a 1 mM DAC working solution was prepared by adding 1 mM DA and 2 mM NaIO4 into buffer solutions containing 20 mM HCl before each experiment with the actual substance initially present in the solution being DA o-quinone (DAQ).27 The calibration curves of DAC were developed at different DAC concentrations. Details of the spectrum can be found in Fig. S1.† Briefly, to accelerate the cyclization rate of DAQ initially formed in the acidic DAC working solution and prevent the significant decay of DAC at high pH, the calibration curves were conducted by adding different concentrations of the freshly prepared DAC working solution into buffer solutions at pH 7.0. Even though DAC is the intermediate formed during the oxidation of DA, the interference arising from its precursors or its degradation product on the measurement should be negligible either as a result of their extremely short half-life (as detailed below) or their non-absorbance above 350 nm. Specifically, most phenol type organics only have absorbance around 280 nm, including DA (shown in Fig. S2†) and 5,6-dihydroxyindole (DHI).28
H2O2 measurement.
The H2O2 formed during the course of DA oxidation (both in the absence and presence of iron) was measured using the modified DPD method.19,29 The measurement was performed in a 10 cm cuvette by a Cary 60 spectrophotometer at 551 nm with baseline correction at 690 nm. 1 mM DTPA was added to halt the iron catalyzed continuous generation of H2O2 during the measurements.19 The detection limit of this method is ∼ 10 nM in aqueous solutions when 10 cm cuvettes are used.
FeIIIDA2 and FeIIIDA3 measurement.
In general, DA is expected to form three different complexes with Fe(III). Under marine condition, the bis-complex (denoted hereafter as FeIIIDA2, ε580 nm = 3330 M−1 cm−1) and the tris-complex (denoted hereafter as FeIIIDA3, ε490 nm = 4190 M−1 cm−1) are generally considered to be formed with the dominant species being pH and concentration dependent.30,31 To avoid interference arising from the oxidative metabolites of DA on the spectrum of these complexes, the distribution of DA bound iron complexes was measured in deoxygenated solutions. The concentrations of the FeIIIDA2 and FeIIIDA3 complexes were determined by measuring the absorbance at 580 nm and 490 nm with baseline correction undertaken at 850 nm.30,32 The measurements were conducted in a 10 cm cuvette using a Cary 60 spectrophotometer. Theoretically, the absorbance at a particular wavelength is the sum of the contributions from different species as a result of the overlap of the spectrum. Given the coexistence of the bis- and tris-complexes at pH 8.0, the concentrations of each species can be determined by solving a system of two linear equations as previously reported.33| | 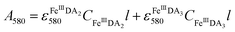 | (1) |
| | 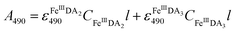 | (2) |
where A is the overall absorbance at a specific wavelength, εij is the molar absorptivity of component j at wavelength i, Cj is the concentration of component j and l is length of the cuvette, which is 10 cm in this study.
FeIIIDFB measurement.
Considering the extremely close peak absorbance of DAC and FeIIIDFB, ligand exchange between DA bound Fe(III) and DFB was performed in deoxygenated solutions. Briefly, the solutions were sparged for 1 h using a special gas mixture of 297 ± 6 ppm CO2 in argon (BOC) prior to the addition of DA. The solution was then bubbled for another 10 min before the addition of Fe(III). To guarantee the complete formation of FeIIIDA3 complex, the DA and Fe(III) containing solution was bubbled for at least one hour before the addition of DFB. The concentration of FeIIIDFB was determined spectrophotometrically at 425 nm with baseline correction at 800 nm.34,35 The measurements were conducted in a 10 cm cuvette using a Cary 60 spectrophotometer. Given the significant overlap of the spectrum of FeIIIDA3 and FeIIIDFB,32,35 instead of using the peak absorbance at 490 nm, the concentration of FeIIIDA3 was calculated first by using the linear regression curves at 650 nm with baseline correction at 850 nm. At this wavelength, the influence of FeIIIDFB on the measurement of FeIIIDA3 is negligible. According to eqn (3):| | 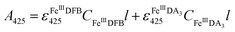 | (3) |
the concentration of FeIIIDFB in the presence of DA can be calculated by deducting the interference arising from FeIIIDA3 at 425 nm.
Fe(II) measurement.
The generation and accumulation of Fe(II) as a result of the interaction between FeIIIDFB and ascorbate was measured spectrophotometrically by using 2,2′-bipyridyl in a 10 cm cuvette.36 The measurements were performed using a Cary 60 spectrophotometer at 522 nm with baseline correction at 650 nm. The detection limit of this method is ∼ 20 nM in aqueous solutions when 10 cm cuvettes are used. To avoid the rapid oxidation of Fe(II) at pH 8.0, 2,2′-bipyridyl was directly added into the buffer solutions containing FeIIIDFB and ascorbate. The FeIIIDFB complex used in this work was prepared by adding 0.5 mM Fe(III) and 5 mM DFB into 10 mM HCl.
Speciation modeling
Knowledge of both ferric and ferrous iron speciation in the presence of various DA concentrations was utilized to determine the major DA bound iron species at particular conditions. Speciation calculations were undertaken using the program Visual Minteq37 with the equilibrium reactions and stability constants provided in Table S1.† Details of the distribution of Fe(III)/Fe(II)–DA species are provided in Fig. S3.†
Kinetic modeling
A kinetic model constituting a set of reactions describing the key processes in this study coupled with appropriate rate constants for each reaction was used to describe the experimental data over a range of conditions via the utilization of the kinetic program Kintek Explorer.38 The outputs of this model represent predictions based on the hypothesized reaction set and associated rate constants that are considered most appropriate for the conditions being examined. Rate constants used were generally based on either literature or experimentally determined values though in some instances a value for a particular reaction was determined from goodness of model description to the complete set of experimental data over all conditions considered.
It is important to note that an R2 value representing agreement of model prediction to a particular set of data is inappropriate for the model developed here as good description of the complete data ensemble represents a more appropriate fitting parameter though this “goodness of fit” is not obtained by curve fitting. Rather, “goodness of fit” is judged by the ability of the reaction set used (and the associated set of coupled differential equations representing the rate expressions for each reaction) to describe the time dependent concentrations of reactants, intermediates and products. If the model provides a poor description of the data, there is more than likely either a flaw in the reaction set or rate constant(s) used. Details of the model developed in this study and justification for the various reactions used in the model can be found in Tables 1–3 and ESI 3,† respectively. The sensitivity of the model to the changes in individual rate constant values was analysed by using the program Kintecus39 combined with a Visual Basic for Applications (VBA) program.
Table 1 Modelled reactions and rate constants for the autoxidation of DAa
| No. reactions |
Rate constants (M−1 s−1 or s−1) |
Reference |
|
DA, dopamine; DA˙−, semiquinone radical; DAQ, dopamine-o-quinone; DAC, dopaminochrome; DAL, leukoaminochrome and O2˙−, superoxide.
|
| 1 |
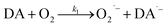
|
k
1 = 1.36 × 10−2 |
This study |
| 2 |
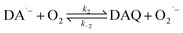
|
k
2 = 2.95 × 103 |
Ref. 26
|
|
k
−2 = 1 × 109 |
Ref. 26
|
| 3 |

|
k
3 = 2.35 × 108 |
Ref. 40
|
| 4 |

|
k
4 = 18.3 |
This study |
| 5 |

|
k
5 = 5.3 × 106 |
Ref. 41
|
| 6 |
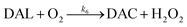
|
k
6 = 22.7 |
This study |
| 7 |

|
k
7 = 6.0 × 104 |
Ref. 42
|
| 8 |
|
k
8 = 8.27 × 109 |
Ref. 19
|
| 9 |

|
k
9 = 2.42 × 10−4 |
This study |
Table 2 Modelled reactions and rate constants for Fe(III)-catalyzed oxidation of DAa
| No. reactions |
Rate constants (M−1 s−1 or s−1) |
Reference |
|
DA, dopamine; DA˙−, semiquinone radical; DAQ, dopamine-o-quinone; O2˙−, superoxide; Fe(III), inorganic ferric ion; Fe(III)I, total inorganic Fe(III); AFO, ferrihydrite and Fe(II), inorganic ferrous iron.
|
| 10 |
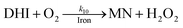
|
k
10 = 29 |
This study |
| 11 |
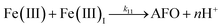
|
k
11 = 1.61 × 107 |
Ref. 43
|
| 12 |
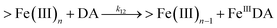
|
k
12 = 11.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
This study |
| 13 |
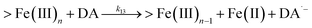
|
k
13 = 2.64 |
This study |
| 14 |
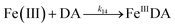
|
k
14 = 1.1 × 106 |
This study |
| 15 |
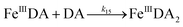
|
k
15 = 4.5 × 105 |
Ref. 44
|
| 16 |
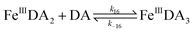
|
k
16 = 4.5 × 105 |
Ref. 44
|
|
k
−16 = 43.5 |
This study |
| 17 |
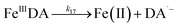
|
k
17 = 0.23 |
Ref. 45
|
| 18 |
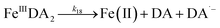
|
k
18 = 7.26 × 10−5 |
Ref. 19
|
| 19 |
|
k
19 = 1.5 × 108 |
Ref. 46
|
|
k
−19 = 5.79 |
This study |
| 20 |
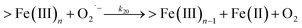
|
k
20 = 3.7 × 105 |
Ref. 19
|
| 21 |

|
k
21 = 5.43 × 102 |
This study |
Table 3 Modelled reactions and rate constants for Fe(II)-catalyzed oxidation of DAa
| No. reactions |
Rate constants (M−1 s−1 or s−1) |
Reference |
|
DA, dopamine; DA˙−, semiquinone radical; O2˙−, superoxide; Fe(III), inorganic ferric ion; Fe(II), inorganic ferrous ion; H2O2, peroxide and ˙OH, hydroxyl radicals.
|
| 22 |
|
k
22 = 1 × 107 |
Ref. 46
|
| 23 |
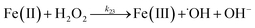
|
k
23 = 3.72 × 104 |
Ref. 47
|
| 24 |
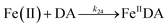
|
k
24 = 5.06 × 103 |
Ref. 19
|
| 25 |
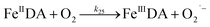
|
k
25 = 3.47 × 102 |
Ref. 19
|
| 26 |
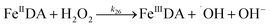
|
k
26 = 3.72 × 104 |
Ref. 47
|
| 27 |
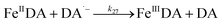
|
k
27 = 1.92 × 105 |
Ref. 19
|
Results and discussion
Formation and transformation of DA related quinones
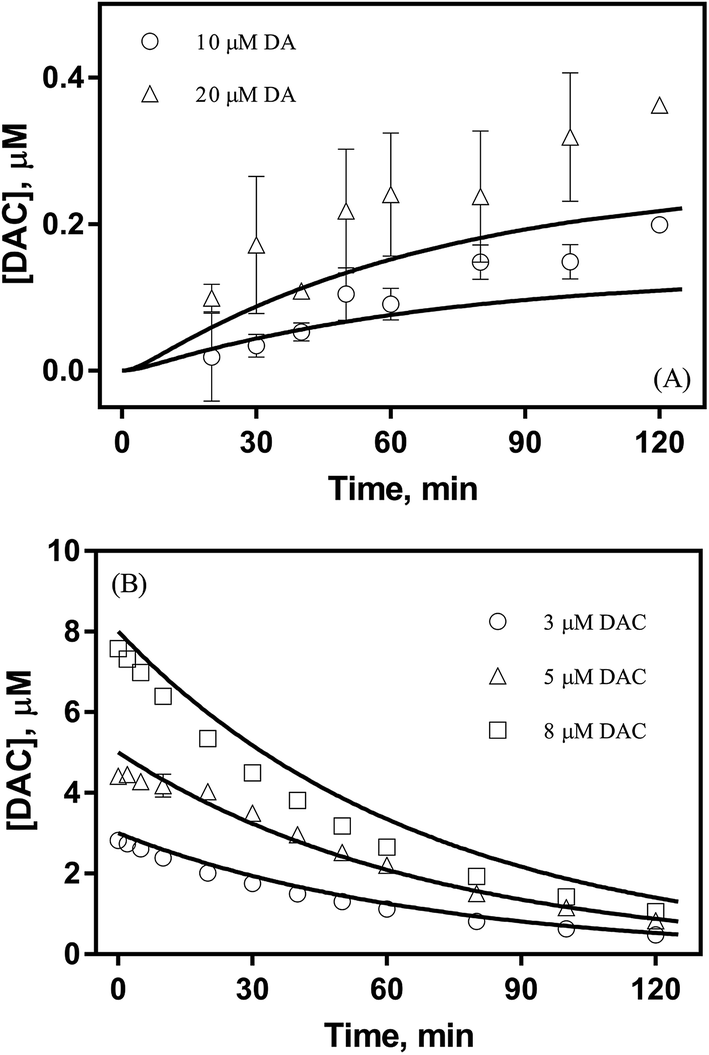
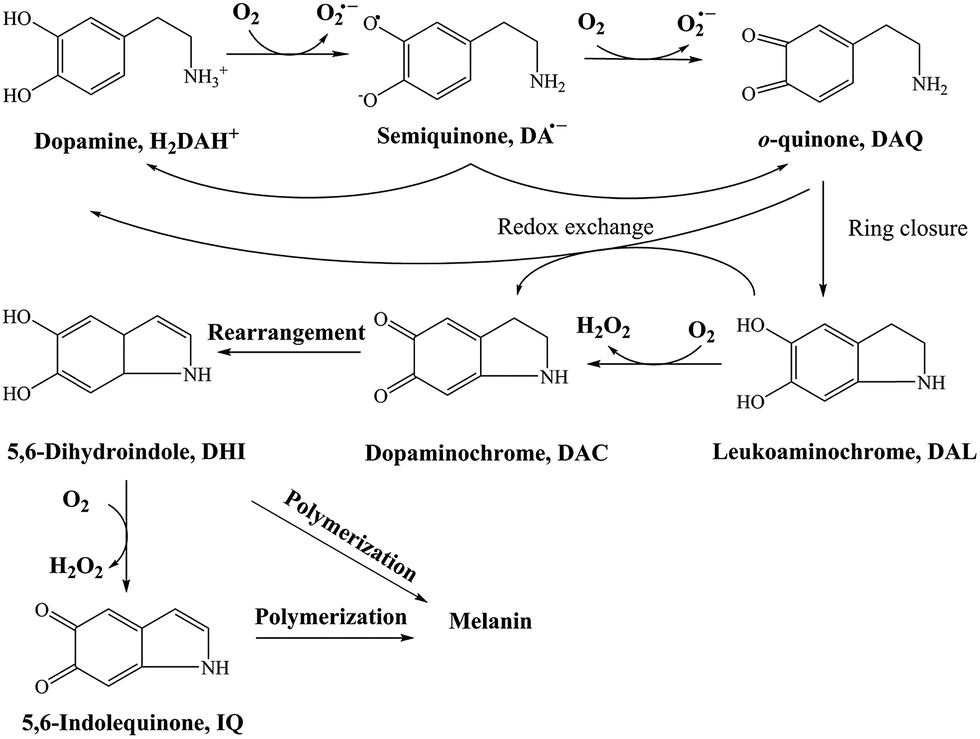
DA, of its own accord, is relatively non-toxic and, indeed, is a well-known functional neurotransmitter.48 The toxicity induced by DA mainly arises from its oxidative metabolites, such as DA derived quinones, reactive oxygen species (ROS) or even the 6-hydroxydopamine (6-OHDA). In general, there are several DA derived quinones that can be formed during the oxidative transformation of DA, including DAQ, DAC and 5,6-indolequinone (IQ). 26,49 While one or more of these metabolites have been associated with the allelopathic effects of DA, the relative importance of each quinone with regard to toxicity is far from clear. Based on the results from previous studies, the accumulation and transformation of DAC was measured herein with particular attention given to this metabolite for three reasons: (i) the higher molar absorptivity (εDAC = 3245 M−1 cm−1 and εDAQ = 1228 M−1 cm−1, which is calculated from the calibration curves shown in Fig. S5†), (ii) the relatively extended half-life in comparison with DAQ and IQ (as discussed below) and (iii) the well-recognized toxicity of this metabolite.50,51
As shown in Fig. 1A, despite a gradually increasing trend, the accumulation of DAC resulting from the autoxidation of 10 μM and 20 μM DA was only around 0.2 μM and 0.3 μM respectively at the conclusion of the two-hour experiment. As shown in Fig. 1B, the half-life observed for the decay of pure DAC was around 60 min at pH 8.0. Accompanied by the rapid decay of DAC, a concomitant significant increase in the baseline of the spectrum was evident (shown in Fig. S6†) with this increase suggestive of the formation of the end-product melanin via the oxidation and polymerization of the rearrangement product DHI.52 This phenomenon is also consistent with the black color observed in previous studies.9
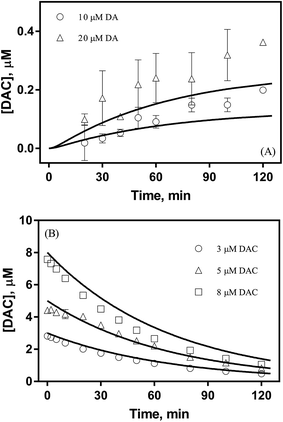 |
| | Fig. 1 Formation of DAC in the presence (○) 10 μM and (△) 20 μM DA (panel A) and decay of (○) 3 μM, (△) 5 μM and (□) 8 μM DAC (panel B) in air-saturated 0.1 M NaCl solutions at pH 8.0. Error bars are standard errors from triplicate measurements and solid lines represent the model fit. | |
As a direct oxidative metabolite of DA, DAQ will be generated either through two one-electron transfer steps (eqn (4) and (5)) or as a result of the disproportionation of DA semiquinone (DA˙−) (eqn (6)) (Scheme 1):
| | | DA˙− + O2 → DAQ + O2˙− | (5) |
| | | DA˙− + DA˙− → DA + DAQ | (6) |
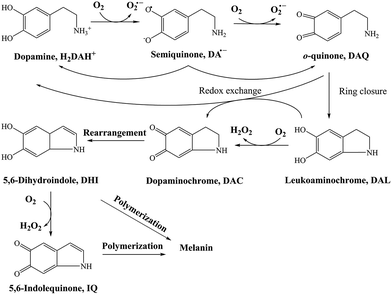 |
| | Scheme 1 Transformation of DA in the presence of O2.26 | |
In the absence of direct measurement, the concentration of DAQ can be predicted using the model developed in this study. As shown in Fig. S7,† in the presence of fixed O2 concentration (243 μM), a typical value for air-saturated solutions, the steady-state concentrations of DAQ achieved in the presence of fixed concentrations of DA are only around 10−2 nM to 10−1 nM both in the absence and presence of fixed concentrations of AFO, the most active iron oxide in natural environments. Even though a 2 nM steady-state concentration of DAQ is predicted under these conditions, the concentrations of DAQ decreases to 10−2 nM level as a result of the associated consumption of O2. Indeed, the negligible accumulation of DAQ at pH 8.0 is reasonable given its pH-dependent transformation behaviour 53,54 with the half-life of DAQ reported to be around 2.7 min, even under mild acidic conditions (pH 5.5).55 In addition, the reasonable molar absorptivity derived from the calibration curves at pH 7 indirectly supports the conclusion that any DAQ formed via the oxidation of DA at pH 8.0 should be converted to DAC immediately. These observations and deductions are consistent with previous NMR spectra obtained at the typical extracellular fluid (ECF) pH of 7.4, which indicates that DAQ may only be a transient oxidative product during the formation of melanin.27 As such, any toxicity induced by DAQ to marine organisms should be negligible.
Due to the rapid cyclization of DAQ, the orange-yellow colored DAC can be formed via either the redox exchange between DAQ and DAL or as a result of the direct oxidation of DAL:
Despite accumulation of DAC being observed, the rapid decay of DAC shown in Fig. 1B would result in significant amounts of the oxidized DA being converted into DHI and its corresponding quinone, IQ. The rapid rearrangement of DAC and the subsequent oxidation of DHI has been confirmed by the reddish-purple color observed in situ by Nelson and co-workers.2 In the presence of O2, DHI can be oxidized in a manner similar to that of DA with this process accompanied by the generation of H2O2 (as discussed in detail below) and IQ.56 Even though the actual existence of IQ has been confirmed indirectly via nucleophilic trapping,57,58 details of the characterization of IQ is elusive since its lability results in the absence of NMR signals.59 As the most unstable quinone formed during the oxidation of DA, it has been proposed that IQ will be incorporated into melanin instantaneously once formed.56 The extreme reactivity of IQ has also been demonstrated by its complete adduction with α-synuclein and resultant absence of formation of melanin.27 Indeed, it has been frequently documented that IQ together with its adducts are one of the major precursors of eumelanin.28,60 Therefore, it is reasonable to deduce that, despite several quinones being formed during the oxidative transformation of DA, DAC is likely the only one involved in quinone-related allelopathic effects. Even though DAC is less active than IQ, the ability of DAC to induce toxicity is well-recognized, which includes formation of adducts with proteins and binding to DNA with resultant dysfunction of these macromolecule.61 As such, the extended lifetime of DAC coupled with its toxicity result in its likely association with the inhibition in germination of seaweeds and survival of zoeae. To mimic the typical marine environment, the decay of DAC in synthetic seawater was further investigated. As shown in Fig. S8,† in addition to the obvious shoulder around 580 nm, a substantially greater increase in the baseline was evident. This phenomenon indicates that the presence of Ca2+ and Mg2+ may enhance the transformation of DAC, possibly via the formation of complexes with its rearrangement product DHI. From this perspective, under marine condition, the toxicity arising from the formation of DAC might be expected to be alleviated as a result of the presence of Ca2+ and Mg2+ cations and their role in facilitation of the formation of melanin.
Production of H2O2via the oxidation of DA
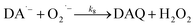
In the presence of O2, the oxidation of DA is accompanied by the generation of ROS, such as O2˙− and H2O2 either via two one-electron oxidation steps (shown in eqn (4) and (5)) or as a result of the disproportionation of the O2˙− formed on DA oxygenation; i.e.| | | O2˙− + O2˙− → H2O2 + O2 | (9) |
To better understand the oxidative stress induced by the release of DA into coastal waters, the concentration of H2O2 formed was measured herein. As shown in Fig. 2A, the concentration of H2O2 (0.52 μM) was almost three fold greater than that of DAC (0.19 μM) under the same conditions (in the presence of 10 μM DA). More particularly, the generation of H2O2via the autoxidation process was strongly concentration-dependent with around 0.5, 1 and 1.4 μM H2O2 being produced at the conclusion of the two-hour experiment in the presence of 10 μM, 20 μM and 30 μM DA.
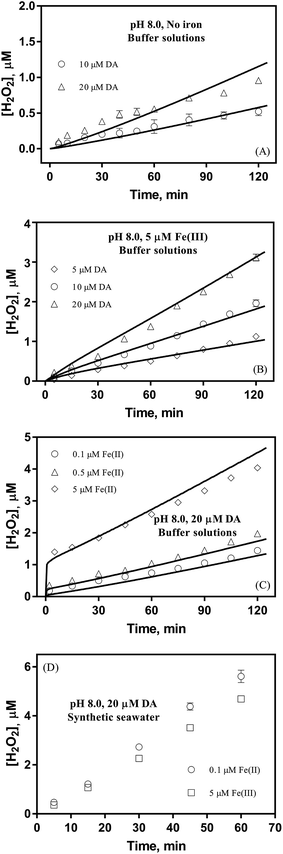 |
| | Fig. 2 Formation of H2O2 in buffer solutions in the absence of iron (panel A), in the presence of 5 μM Fe(III) (panel B), in the presence of 20 μM DA and Fe(II) (panel C) and in synthetic seawater containing 20 μM DA and iron (panel D) at pH 8.0. Error bars are standard errors from triplicate measurements and solid lines represent the model fit. | |
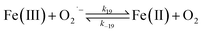
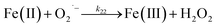
Despite high concentrations of H2O2 being produced via the autoxidation process, any redox active metals present could further aggravate the resultant oxidative stress by accelerating the oxidation of DA via ligation of the d orbitals between DA and O2.62 Given our particular interest in the mutual influence between iron and DA, the effects of ferric iron on the production of H2O2 were investigated at micromolar concentrations in view of the wide distribution of iron oxides, while the effects of ferrous iron were examined at nanomolar concentrations as this is typical of coastal marine environments. As shown in Fig. 2B and C, as expected, considerably higher concentrations of H2O2 were generated in the presence of both Fe(III) and Fe(II). Specifically, around 3 μM and 4 μM H2O2 were generated in the presence of 20 μM DA and 5 μM ferric and ferrous iron, respectively, with these concentrations 3 or 4 times greater than was the case in the absence of iron. Obviously, Fe(II) was much more effective in H2O2 generation as almost similar amounts of H2O2 (around 2 μM) was generated at the conclusion of the two hour experiment in the presence of 0.5 μM Fe(II) and 5 μM Fe(III). This phenomenon may contribute to the extensive hydroxylation and precipitation of the ferric iron that occurs at oceanic pH.43 In contrast to the gradual production of H2O2 in the presence of Fe(III), considerably higher concentrations of H2O2 were generated initially in the presence of Fe(II). The elevated accumulation of H2O2 may be attributed to the enhanced oxidation of Fe(II) as a result of the decrease in the reduction potential of the dominant iron couple via the formation of the FeIIDA complex.19
| | | FeIIDA + O2 → FeIIIDA + O2˙− | (11) |
Therefore, in addition to the production of H2O2, the presence of large amounts of DA will significantly deplete the most bioavailable iron species, Fe(II), with resultant formation of DA bound Fe(III) (shown in Fig. S9†) as follows:
| | | FeIIIDA + DA → FeIIIDA2 | (12) |
| | | FeIIIDA2 + DA → FeIIIDA3 | (13) |
As a result of the catalytic effects of iron, the presence of iron (both Fe(III) and Fe(II)) would be expected to accelerate the oxidative transformation of DA with resultant enhanced generation of intermediates, such as DHI. Despite the fact that we do not have direct measurement, the estimated half-life of DHI from previous work is only around 10 min at pH 7.4.27 As a pH-dependent process, the enhanced oxidation of DHI in coastal waters should significantly enhance the production of H2O2 and the concomitant formation of melanin with this process being dramatically accelerated by iron:
| | 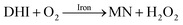 | (14) |
Theoretical analysis coupled with the agreement of the model to experimental data (shown in Fig. S10†) suggest that the iron induced generation coupled with the iron catalyzed oxidation of DHI play a pivotal role in the accumulation of H2O2 in waters containing high concentrations of DA, especially in the later stages of the reaction.
The production of H2O2 was also investigated in synthetic seawater in order to examine the possible role of Ca2+ and Mg2+ in the transformation of DA. In comparison with the results shown in Fig. 2B and C, the enhanced production of H2O2 shown in Fig. 2D may be attributed to the formation of DA bound Ca2+ and Mg2+ as a result of the evident absorbance, even in the presence of high concentrations of DTPA (shown in Fig. S11†). While the presence of Ca2+ and Mg2+ resulted in a 7 fold increase in concentration of H2O2 (to 5.6 μM) in the presence of 0.1 μM of Fe(II), only around 3.5 times increase in the concentration of H2O2 (to 4.6 μM) was evident in the presence of 5 μM Fe(III). This obvious difference indicates that iron may possess a higher affinity towards DA with this process strongly inhibiting the competition of Ca2+ and Mg2+ for DA even though the concentrations of these two cations are more than 2000 times greater than that of iron. As such, it is reasonable to deduce that, despite the presence of high concentrations of competing cations, DA may still exert a significant influence on the fate of iron in the marine environment as a result of its higher affinity for DA.
Dry and wet deposition, photochemistry and passive diffusion of excess ROS produced by marine organisms have long been proposed to be the main processes contributing to the influx of H2O2 in marine environments with the maximum concentration of H2O2 being around 200 nM.63,64 However, as shown in this study, the seasonal and spatial generation of long-lived H2O2 from biotic sources may well be more significant than previously expected. In contrast to short-lived O2˙− and ˙OH, the long half-life of H2O2 (10 to 30 hours) may extend the range in toxicity induced by H2O2 both temporally and spatially,65 with the high concentrations of extracellular H2O2 potentially being related to the allelopathic effects.7 On one hand, for predators of Ulvaria, the presence of high concentrations of H2O2 may be partially responsible for the change in development of invertebrate larvae with this alteration possibly benefiting the blooms of Ulvaria.8,66,67 On the other hand, as a result of membrane permeability, both surface and intracellular damage can be caused by H2O2 for co-occurring seagrasses.68 The reported H2O2 induced reduction in the efficiency of photosynthesis for Alaria esculenta is more than 80% and bleaching has also been observed for the algae thalli.69 The ongoing presence of high concentrations of H2O2 may cause severe damage to endogenous antioxidant systems with this process further aggravating oxidative stress,69–71 especially in the presence of transition metals.72 While generally detrimental, the presence of extracellular and intracellular ROS may benefit Ulvaria obscura var. blyttii and, indeed, the whole community, especially when it is recognized that these oxidants may stimulate the synthesis by Ulvaria of phenolase, an enzyme that acts to induce the polymerization of dopamine to melanin and reduce the steady state concentration of toxic quinone intermediates.73 Indeed, it has been reported that the presence of extracellular O2˙− may contribute to pathogen resistance while intracellular ROS may function as systemic signaling molecules in developing a generic defence response.74,75 As such, while highly speculative, the production of O2˙− and H2O2 accompanied by the rapid formation and decay of DAC in the presence of tyrosinase, a readily available analogue of phenolase (see Fig. S12†), may benefit algal survival. From this perspective, Ulvaria may obtain an ecological advantage as a result of the ROS-induced synthesis of phenolase which, in turn, acts to scavenge toxic quinones and induce the formation of a protective coating of melanin.
Effect of DA on the transformation of iron
In view of the potential for significant environmental modification, the influence exerted by DA on the transformation of iron was investigated from the perspective that iron limitation has been suggested as a factor limiting nutrient uptake by seagrasses.16
Effect of DA on transformation of ferric iron
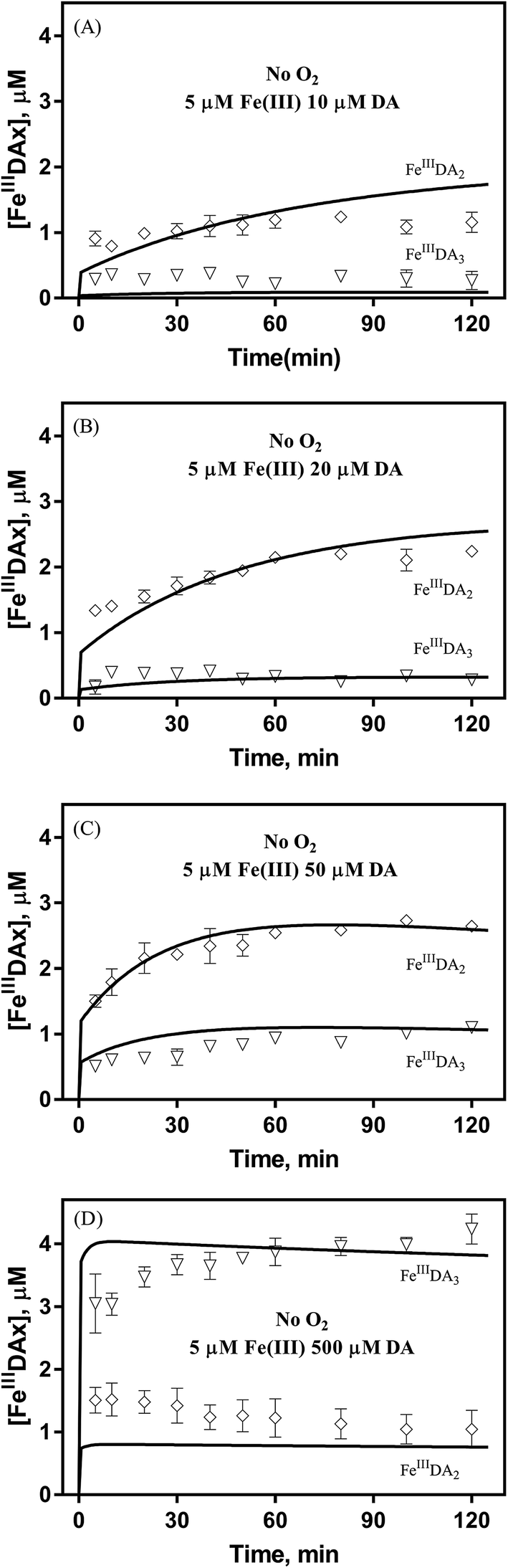
It is to be expected that in the presence of O2 and iron, the oxidative transformation of DA would result in a significant spectrum overlap of different species, including FeIIIDA2, FeIIIDA3 and DAC. To avoid interference arising from the presence of the oxidative metabolites and improve the accuracy of data analysis, measurements of the distribution of DA bound iron species were undertaken under deoxygenated conditions. As shown in Fig. 3, in the absence of O2, significant amounts of DA bound iron were rapidly formed on addition of Fe(III) into DA containing solutions, the total concentrations of which are considerably DA concentration-dependent. For instance, when the concentration of DA was greater than 500 μM, almost all the added Fe(III) was present as dissolved species. In general, as a bidentate chelator, DA can form three different complexes with Fe(III) with the dominant species being concentration and pH-dependent (Fig. S3†). Under marine conditions (pH approximately 8.0), both FeIIIDA2 and FeIIIDA3 were formed with the proportion of the tris-complex increasing dramatically on increase in DA concentration (Fig. 3). Interestingly, compared with the results obtained at lower pH, despite the precipitation rate of iron at pH 8 being more than tenfold greater than that at pH 6.5 (kpH 8.0 = 16.1 × 106 M−1 s−1 and kpH 6.5 = 1 × 106 M−1 s−1), substantially higher proportions of DA bound iron were formed at the same reactant concentrations at pH 8.0.43 The favoured formation of DA bound iron at pH 8.0 indicates that DA induced complexation is extremely competitive with the precipitation process under the conditions investigated herein.
 |
| | Fig. 3 Distribution of FeIIIDA2 and FeIIIDA3 complex in the presence of 5 μM Fe(III) and 10 μM DA (panel A), 20 μM DA (panel B), 50 μM DA (panel C) and 500 μM DA (panel D) in deoxygenated 0.1 M NaCl solutions at pH 8.0. Error bars are standard errors from triplicate measurements and solid lines represent the model fit. | |
The formation of DA bound Fe(III) is initiated by the formation of the rate limiting mono-complex, FeIIIDA, followed by the subsequent replacement of coordinated water by additional molecules of DA.19
| | | Fe(III) + DA → FeIIIDA | (15) |
| | | FeIIIDA + DA → FeIIIDA2 | (16) |
| | | FeIIIDA2 + DA → FeIIIDA3 | (17) |
In general, the above processes can be facilitated by the deprotonation of DA since increase in pH reduces the extent of dissociation (details can be found in ESI 5†) and enhances the electrostatic attraction between positively charged ferric species and the DA anion. Specifically, the proportional increase in HDA− and DA2− content for a certain DA concentration at pH 8.0 is one and three orders of magnitudes greater than that at pH 6.5 (Fig. S3†). Therefore, it is not unexpected that substantially higher initial concentrations of dissolved iron were measured at pH 8.0.
Following the rapid initial formation of Fe(III)–DA complexes, ongoing gradual increase in the concentration of DA bound iron is evident within the first 60 minutes with this increase most likely attributable to DA induced iron mobilization from any iron oxyhydroxides that do form; i.e.
| | | >Fe(III)n + DA → >Fe(III)n−1 + FeIIIDA | (18) |
| | | >Fe(III)n + DA → >Fe(III)n−1 + Fe(II) + DA˙− | (19) |
In addition to direct ligand-mediated dissolution of iron oxyhydroxide particles, reductive dissolution as a result of the ligand-to-metal charge transfer (LMCT) might also be expected, particularly in view of the ability of DA to reduce Fe(III) to Fe(II).19 The release of both Fe(II) and the quinone precursor, DA˙−, via this reductive dissolution pathway may play a pivotal role in the allelopathic effects of DA in view of both the formation of toxic quinones and the Fe(II)-mediated generation of H2O2 which can, in turn, react with Fe(II) resulting in production of powerful oxidants via the Fenton reaction. As such, it is reasonable to deduce that, in addition to the increase in the content of dissolved iron, the iron mobilization induced by DA may also contribute to DA related allelopathic effects.
Effect of DA on iron acquisition
Even though endocytosis may result in direct uptake of iron oxides,76 in general, soluble organically bound iron is more bioavailable with uptake occurring as a result of either assimilation of siderophore-bound Fe(III) or via reduction of Fe(III) to the much more soluble Fe(II).77–80 As such, the DA induced increase in dissolved iron concentration may be critical to the bioavailability of iron within the bloom region, even for competitor organisms. However, the assimilation of organically bound iron has been, and remains, controversial.81–83 Given the convergence of the DA bound Fe(III) and Fe(II), the possibility of the acquisition of DA bound iron is investigated by using the fully complexed FeIIIDA3 and DFB as typical compounds. On one hand, despite both FeIIIDA2 and FeIIIDA3 tending to be formed in coastal waters, iron within the FeIIIDA3 complex is much more difficult to be assimilated as a result of steric hindrance. On the other hand, even though DFB is commonly produced by terrestrial bacteria, evidence exists revealing that a structurally similar compound, desferrioxamine G (DFG), is produced by marine phytoplankton and heterotrophic bacteria and utilized for iron assimilation by using the same outer membrane receptor protein as that used in the case of DFB.84–87
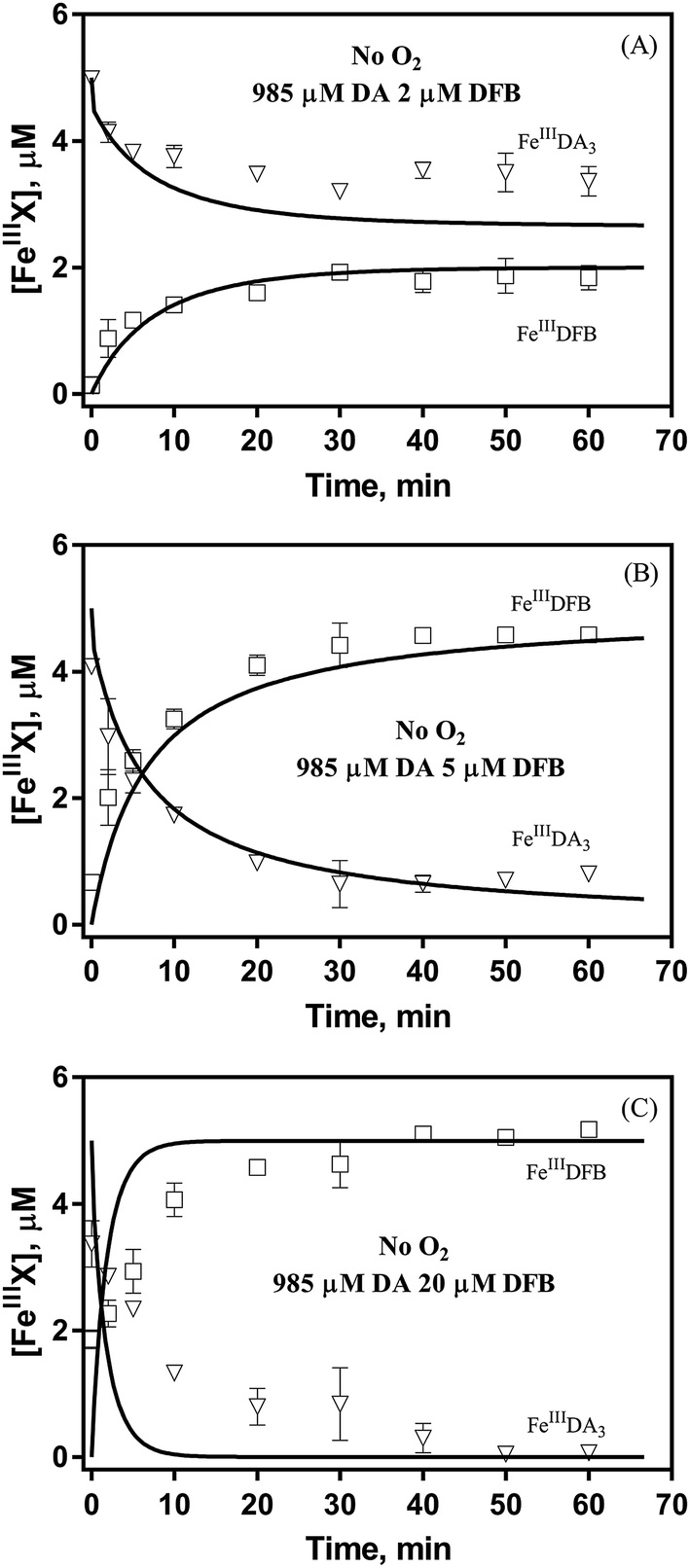
Theoretically, O2 is critical to the redox cycling of iron. However, in view of the transformation of ferric iron between different iron chelators, the role played by O2 should be negligible. Therefore, to avoid the interference arising from the oxidative transformation of DA, the possibility of the chelation of DA bound Fe(III) by DFB was investigated in the absence of O2. As shown in Fig. 4, in the absence of O2, despite the presence of high concentrations of excess DA (985 μM), addition of DFB into solutions containing 5 μM FeIIIDA3 resulted in the gradual formation of FeIIIDFB and, accordingly, a concomitant decrease in the concentrations of FeIIIDA3. In general, an equivalent concentration of iron was transformed to a certain concentration of DFB from DA complexes with the time consumed by this process decreasing dramatically on increase in the concentrations of DFB. Theoretically, the transformation of Fe(III) is unexpected since all the coordination sites of the central iron are fully occupied by DA. In the presence of DFB, two pathways may exist for the formation of the FeIIIDFB complex: (i) chelation of ferrous iron formed as a result of possible LMCT within the FeIIIDA3 complex followed by the subsequent oxidation of DFB bound Fe(II) by the trace amount of O2 present in the sparging solution, or (ii) direct chelation of ferric iron as a result of dissociation of the FeIIIDA3 complex. If chelation of ferrous iron is the dominant pathway, the LMCT process should be the rate-limiting process. Therefore, according to the rate law:
| | 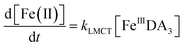 | (20) |
the release of Fe(
II) can be roughly estimated by the term
kLMCT[Fe
IIIDA
3]
t. As shown in
Fig. 4, under all conditions, the chelation process occurs within 30 min. In view of the reduction in the LMCT rate on increase in the number of coordinated ligands, the upper limit of iron release can be estimated by using the previously reported internal electron transfer rate (7.26 × 10
−5 M
−1 s
−1) for Fe
IIIDA
2. As such, at the conclusion of 30 min, the concentration of Fe(
II) released from Fe
IIIDA
3 is calculated to be around 0.65 μM which is much lower than the concentration of iron trapped in the DFB complex as measured experimentally. In addition, if the LMCT process occurred to a significant extent, a decrease in the concentration of Fe
IIIDA
3 in the absence of O
2 should be evident over time. However, this was not the case as evident from Fig. S13.
† As such, chelation of Fe(
II) released from Fe
IIIDA
3 by DFB is unlikely to occur to any significant extent. While unlikely to be the dominant pathway, the dissociation process would enable the dynamic formation of vacant coordination sites
88 with the presence of these sites enhancing the likelihood of ligand exchange. In addition, under the conditions investigated herein, the presence of high concentrations of mono-deprotonated DFB (p
Ka1 = 8.32) would result in an enhanced electrostatic attraction between Fe(
III) and DFB, which may further facilitate the dissociation of the coordinated DA.
89 As a hexadentate chelator, the formation of fully DFB complexed iron will minimize the influence of the dissociation process, resulting in a much more stable complex than that formed with DA. Based on the observations in this study, we conclude that the formation of DA bound Fe(
III) as a result of the release of high concentrations of DA would increase the dissolved iron content within the bloom region as long as a stronger siderophore can be utilized by the marine organisms.
 |
| | Fig. 4 Formation of FeIIIDFB and concomitant loss of FeIIIDA3 followed by the addition of 2 μM DFB (panel A), 5 μM DFB (panel B) and 20 μM DFB (panel C) to deoxygenated 0.1 M NaCl solutions containing 5 μM FeIIIDA3 and 985 μM DA at pH 8.0. Error bars are standard errors from triplicate measurements and solid lines represent the model fit. | |
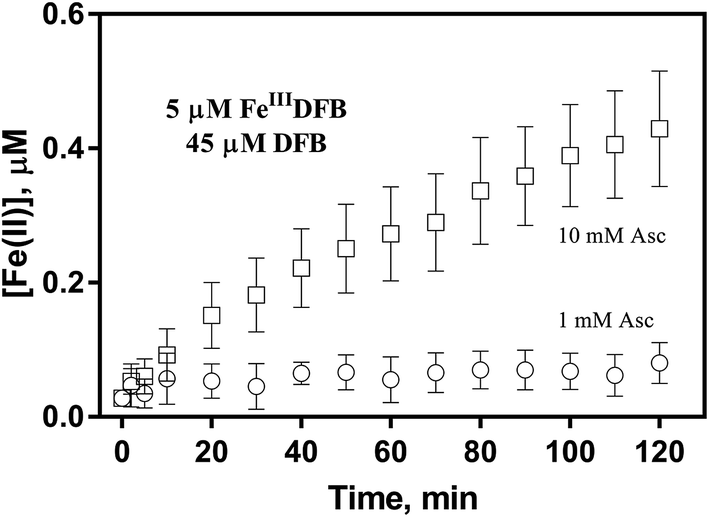
In order to examine if the DFB bound Fe(III) can also be utilized via the reductive pathway, the generation of Fe(II) in the presence of ascorbate has also been investigated herein. As shown in Fig. 5, while the concentration of Fe(II) generated in the presence of 1 mM ascorbate is only around 0.1 μM, continuous generation of Fe(II) in the presence of 10 mM ascorbate is evident with the concentration of Fe(II) up to more than 0.4 μM at the conclusion of the two-hour experiment. As such, it can be concluded that, despite the absence of a specific outer membrane receptor protein for DFB bound Fe(III), this source of dissolved iron can also be assimilated via the reductive pathway if a high concentration of reductant is present in the vicinity of the outer membrane of marine organisms.
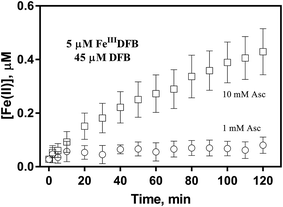 |
| | Fig. 5 Formation and accumulation of Fe(II) from the ascorbate induced reduction of FeIIIDFB at pH 8.0 in 0.1 M NaCl. Error bars are standard errors from triplicate measurements. | |
It should be noted that the pH within the bloom area may fluctuate as a result of photosynthesis and respiration with generally higher pH expected at noon and lower pH expected at night and early morning. Previous studies of iron–DA interactions at physiological pH (7.4)19 reveal somewhat lower rates of production of H2O2 and reduced extent of Fe(III)–DA complexation than at pH 8 with these effects due to the increased protonation of DA at lower pH. As such, the environmental impact of the release of DA may be expected to be magnified somewhat as a result of photosynthesis.
Conclusions and implications
The results of this study indicate that, in the presence of O2, substantial release of DA by Ulvaria obscura var. blyttii gives rise to the generation of DA related toxic quinones with DAC likely to be the major toxic component as a result of the extended lifetime and well-recognized toxicity of this compound. Compared with the generation of DAC, considerably higher concentrations of H2O2 are produced during the autoxidation of DA with this process being significantly accelerated in the presence of iron. In view of the catechol type functional group, the presence of DA can result in the formation of various DA bound iron species with the concentration of the tris-complex increasing significantly on increase in the concentration of DA. While the presence of DA can deplete any bioavailable Fe(II) present, the total concentrations of dissolved iron may still be significantly enhanced as a result of DA mediated iron transformations.
Our results suggest that the production of H2O2 resulting from the oxidation of DA may be much more detrimental to other organisms than DAC given the quantity and extended lifetime of this reactive oxygen species. In addition to inhibition in growth of other organisms as a result of the production of toxic DA metabolites, the alleviation in iron stress as a result of the formation of DA bound iron may contribute to the net fitness to Ulvaria obscura var. blyttii and its long-term persistence. While an ecological advantage may be obtained by Ulvaria obscura var. blyttii as a result of the DA induced production of ROS, the DA induced enhancement in dissolved iron concentration may benefit the surrounding community.
Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgements
We gratefully acknowledge the China Scholarship Council and the University of New South Wales for scholarship support to Yingying Sun. Support provided to Professor Waite by the Australian Research Council through Discovery Project DP150102248 is gratefully acknowledged.
References
- D. M. Anderson, P. M. Glibert and J. M. Burkholder, Harmful algal blooms and eutrophication: nutrient sources, composition, and consequences, Estuaries, 2002, 25(4), 704–726 CrossRef.
- T. A. Nelson, D. J. Lee and B. C. Smith, Are “green tides” harmful algal blooms? Toxic properties of water-soluble extracts from two bloom-forming macroalgae, Ulva Fenestrata and Ulvaria obscura (Ulvophyceae), J. Phycol., 2003, 39(5), 874–879 CrossRef CAS.
-
R. M. O'Clair, S. C. Lindstrom and S. Olsen, North Pacific Seaweeds, Plant Press, 2000 Search PubMed.
- L. Axelsson, C. Larsson and H. Ryberg, Affinity, capacity and oxygen sensitivity of two different mechanisms for bicarbonate utilization in Ulva lactuca L. (Chlorophyta), Plant, Cell Environ., 1999, 22(8), 969–978 CrossRef CAS.
- A. Tyler, K. McGlathery and I. Anderson, Macroalgae mediation of dissolved organic nitrogen fluxes in a temperate coastal lagoon, Estuarine, Coastal Shelf Sci., 2001, 53(2), 155–168 CrossRef CAS.
-
C. D. Amsler, Algal Chem. Ecol., Springer, 2008, vol. 468 Search PubMed.
- K. L. Van Alstyne, T. A. Nelson and R. L. Ridgway, Environmental chemistry and chemical ecology of “green tide” seaweed blooms, Integr. Comp. Biol., 2015, 55(3), 518–532 CrossRef PubMed.
- K. L. Van Alstyne, A. V. Nelson, J. R. Vyvyan and D. A. Cancilla, Dopamine functions as an antiherbivore defense in the temperate green alga Ulvaria obscura, Oecologia, 2006, 148(2), 304–311 CrossRef PubMed.
- K. L. Van Alstyne, K. J. Anderson, A. K. Winans and S.-A. Gifford, Dopamine release by the green alga Ulvaria obscura after simulated immersion by incoming tides, Mar. Biol., 2011, 158(9), 2087–2094 CrossRef.
- K. L. Van Alstyne and M. P. Puglisi, DMSP in marine macroalgae and macroinvertebrates: distribution, function, and ecological impacts, Aquat. Sci., 2007, 69(3), 394–402 CrossRef CAS.
- K. L. Van Alstyne, E. L. Harvey and M. Cataldo, Effects of dopamine, a compound released by the green-tide macroalga Ulvaria obscura (Chlorophyta), on marine algae and invertebrate larvae and juveniles, Phycologia, 2014, 53(2), 195–202 CrossRef CAS.
- K. L. Van Alstyne, K. J. Anderson, D. H. Hees and S. A. Gifford, Dopamine release by Ulvaria obscura (Chlorophyta): environmental triggers and impacts on photosynthesis, growth, and survival of the releaser, J. Phycol., 2013, 49(4), 719–727 CrossRef CAS PubMed.
- K. Kuma, J. Nishioka and K. Matsunaga, Controls on iron(III) hydroxide solubility in seawater: the influence of pH and natural organic chelators, Limnol. Oceanogr., 1996, 41(3), 396–407 CrossRef CAS.
- K. S. Johnson, R. M. Gordon and K. H. Coale, What controls dissolved iron concentrations in the world ocean? Authors' closing comments, Mar. Chem., 1997, 57(3–4), 181–186 CrossRef CAS.
- W. G. Sunda and S. A. Huntsman, Iron uptake and growth limitation in oceanic and coastal phytoplankton, Mar. Chem., 1995, 50(1), 189–206 CrossRef CAS.
- P. Viaroli, M. Bartoli, R. Azzoni, G. Giordani, C. Mucchino, M. Naldi, D. Nizzoli and L. Tajé, Nutrient and iron limitation to Ulva blooms in a eutrophic coastal lagoon (Sacca di Goro, Italy), Hydrobiologia, 2005, 550(1), 57–71 CrossRef CAS.
- C. M. Duarte, M. Martín and G. Margarita, Evidence of iron deficiency in seagrasses growing above carbonate sediments, Limnol. Oceanogr., 1995, 40(6), 1153–1158 CrossRef CAS.
- P. M. Vitousek and R. W. Howarth, Nitrogen limitation on land and in the sea: how can it occur?, Biogeochemistry, 1991, 13(2), 87–115 CrossRef.
- Y. Sun, A. N. Pham and T. D. Waite, Elucidation of the interplay between Fe(II), Fe(III), and dopamine with relevance to iron solubilization and reactive oxygen species generation by catecholamines, J. Neurochem., 2016, 137(6), 955–968 CrossRef CAS PubMed.
- D. Jiang, S. Shi, L. Zhang, L. Liu, B. Ding, B. Zhao, G. Yagnik and F. Zhou, Inhibition of the Fe(III)-catalyzed dopamine oxidation by ATP and its relevance to oxidative stress in Parkinson's disease, ACS Chem. Neurosci., 2013, 4(9), 1305–1313 CrossRef CAS PubMed.
- J. Smythies, The neurotoxicity of glutamate, dopamine, iron and reactive oxygen species: functional interrelationships in health and disease: a review—discussion, Neurotoxic. Res., 1999, 1(1), 27–39 CrossRef CAS.
- J. M. Santana-Casiano, M. González-Dávila, A. G. González and F. J. Millero, Fe(III) Reduction in the Presence of Catechol in Seawater, Aquat. Geochem., 2010, 16(3), 467–482 CrossRef CAS.
- Z. Guo and A. Mathieson, Physiological ecology of four ulvoid green algae, Bot. Mar., 1992, 35(6), 523–534 Search PubMed.
-
M. J. Kennish, Practical handbook of marine science, crc press, 2000 Search PubMed.
- E. Herlinger, R. F. Jameson and W. Linert, Spontaneous autoxidation of dopamine, J. Chem. Soc., Perkin Trans. 2, 1995, 2, 259–263 RSC.
- A. N. Pham and T. D. Waite, Cu(II)-catalyzed oxidation of dopamine in aqueous solutions: mechanism and kinetics, J. Inorg. Biochem., 2014, 137, 74–84 CrossRef CAS PubMed.
- M. Bisaglia, S. Mammi and L. Bubacco, Kinetic and structural analysis of the early oxidation products of dopamine analysis of the interactions with α-synuclein, J. Biol. Chem., 2007, 282(21), 15597–15605 CrossRef CAS PubMed.
- Y. V. Il'ichev and J. D. Simon, Building blocks of eumelanin: relative stability and excitation energies of tautomers of 5,6-dihydroxyindole and 5,6-indolequinone, J. Phys. Chem. B, 2003, 107(29), 7162–7171 CrossRef.
- H. Bader, V. Sturzenegger and J. Hoigne, Photometric method for the determination of low concentrations of hydrogen peroxide by the peroxidase catalyzed oxidation of N,N-diethyl-p-phenylenediamine (DPD), Water Res., 1988, 22(9), 1109–1115 CrossRef CAS.
- A. Avdeef, S. R. Sofen, T. L. Bregante and K. N. Raymond, Coordination chemistry of microbial iron transport compounds. 9. Stability constants for catechol models of enterobactin, J. Am. Chem. Soc., 1978, 100(17), 5362–5370 CrossRef CAS.
- L. K. Charkoudian and K. J. Franz, Fe(III)-coordination properties of neuromelanin components: 5,6-dihydroxyindole and 5,6-dihydroxyindole-2-carboxylic acid, Inorg. Chem., 2006, 45(9), 3657–3664 CrossRef CAS PubMed.
- M. J. Sever and J. J. Wilker, Visible absorption spectra of metal–catecholate and metal–tironate complexes, J. Chem. Soc., Dalton Trans., 2004, 7, 1061–1072 RSC.
- X. Yuan, C. J. Miller, A. N. Pham and T. D. Waite, Kinetics and mechanism of auto-and copper-catalyzed oxidation of 1,4-naphthohydroquinone, Free Radical Biol. Med., 2014, 71, 291–302 CrossRef CAS PubMed.
- B. Monzyk and A. L. Crumbliss, Kinetics and mechanism of the stepwise dissociation of iron(III) from ferrioxamine B in aqueous acid, J. Am. Chem. Soc., 1982, 104(18), 4921–4929 CrossRef CAS.
- B. Faller and H. Nick, Kinetics and mechanism of iron(III) removal from citrate by desferrioxamine B and 3-hydroxy-1, 2-dimethyl-4-pyridone, J. Am. Chem. Soc., 1994, 116(9), 3860–3865 CrossRef CAS.
- B. M. Voelker and B. Sulzberger, Effects of fulvic acid on Fe(II) oxidation by hydrogen peroxide, Environ. Sci. Technol., 1996, 30(4), 1106–1114 CrossRef CAS.
-
J. P. Gustafsson, Visual MINTEQ 3.0 user guide, Dep. of Land and Water Resour. Eng., KTH Royal Inst. of Technol., Stockholm, Sweden, 2005 Search PubMed.
- K. A. Johnson, Z. B. Simpson and T. Blom, Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data, Anal. Biochem., 2009, 387(1), 20–29 CrossRef CAS PubMed.
-
J. C. Ianni, A comparison of the Bader-Deuflhard and the Cash-Karp Runge-Kutta integrators for the GRI-MECH 3.0 model based on the chemical kinetics code Kintecus, Comput. Fluid. Solid. Mech., 2003, pp. 1368–1372 Search PubMed.
- J. Borovansky, R. Edge, E. J. Land, S. Navaratnam, S. Pavel, C. A. Ramsden, P. A. Riley and N. P. Smit, Mechanistic studies of melanogenesis: the influence
of N-substitution on dopamine quinone cyclization, Pigm. Cell Res., 2006, 19(2), 170–178 CrossRef CAS PubMed.
- E. Land, S. Ito, K. Wakamatsu and P. Riley, Rate constants for the first two chemical steps of eumelanogenesis, Pigm. Cell Res., 2003, 16(5), 487–493 CrossRef CAS.
- O. C. Zafiriou, Chemistry of superoxide ion-radical (O2−) in seawater. I. pK*aswb (HOO) and uncatalyzed dismutation kinetics studied by pulse radiolysis, Mar. Chem., 1990, 30, 31–43 CrossRef CAS.
- A. N. Pham, A. L. Rose, A. J. Feitz and T. D. Waite, Kinetics of Fe(III) precipitation in aqueous solutions at pH 6.0–9.5 and 25 °C, Geochim. Cosmochim. Acta, 2006, 70(3), 640–650 CrossRef CAS.
- M. A. Blesa and E. Matijević, Phase transformations of iron oxides, oxohydroxides, and hydrous oxides in aqueous media, Adv. Colloid Interface Sci., 1989, 29(3), 173–221 CrossRef CAS.
- U. El-Avaan, E. Herlinger, R. Jameson and W. Linert, Anaerobic oxidation of dopamine by iron(III), J. Chem. Soc., Dalton Trans., 1997, 16, 2813–2818 RSC.
- J. D. Rush and B. Bielski, Pulse radiolytic studies of the reactions of HO2/O2− with Fe(II)/Fe(III) ions. The reactivity of HO2/O2− with ferric ions and its implication on the occurrence of the Haber–Weiss reaction, J. Phys. Chem., 1985, 89(23), 5062–5066 CrossRef CAS.
- M. González-Davila, J. M. Santana-Casiano and F. J. Millero, Oxidation of iron(II) nanomolar with H2O2 in seawater, Geochim. Cosmochim. Acta, 2005, 69(1), 83–93 CrossRef.
- O. Hornykiewicz, Dopamine (3-hydroxytyramine) and brain function, Pharmacol. Rev., 1966, 18(2), 925–964 CAS.
- E. Herlinger, R. F. Jameson and W. Linert, Spontaneous autoxidation of dopamine, J. Chem. Soc., Dalton Trans., 1995, 2, 259–263 Search PubMed.
- A. Herrera, P. Muñoz, I. Paris, G. Díaz-Veliz, S. Mora, J. Inzunza, K. Hultenby, C. Cardenas, F. Jaña and R. Raisman-Vozari, Aminochrome induces dopaminergic neuronal dysfunction: a new animal model for Parkinson's disease, Cell. Mol. Life Sci., 2016, 1–15 Search PubMed.
- I. Paris, S. Cardenas, J. Lozano, C. Perez-Pastene, R. Graumann, A. Riveros, P. Caviedes and J. Segura-Aguilar, Aminochrome as a preclinical experimental model to study degeneration of dopaminergic neurons in Parkinson's disease, Neurotoxic. Res., 2007, 12(2), 125–134 CrossRef CAS.
- H. S. Mason, The chemistry of melanin III. Mechanism of the oxidation of dihydroxyphenylalanine by tyrosinase, J. Biol. Chem., 1948, 172(1), 83–99 CAS.
- T. E. Young and B. W. Babbitt, Electrochemical study of the oxidation of α-methyldopamine, α-methylnoradrenaline and dopamine, J. Org. Chem., 1983, 48(4), 562–566 CrossRef CAS.
- M. Hawley, S. Tatawawadi, S. Piekarski and R. Adams, Electrochemical studies of the oxidation pathways of catecholamines, J. Am. Chem. Soc., 1967, 89(2), 447–450 CrossRef CAS PubMed.
- J. Li and B. M. Christensen, Effect of pH on the oxidation pathway of dopamine and dopa, J. Electroanal. Chem., 1994, 375(1–2), 219–231 CrossRef CAS.
- J. Segura-Aguilar, I. Paris, P. Muñoz, E. Ferrari, L. Zecca and F. A. Zucca, Protective and toxic roles of dopamine in Parkinson's disease, J. Neurochem., 2014, 129, 898–915 CrossRef CAS PubMed.
- M. d'Ischia, A. Napolitano and G. Prota, Sulphydryl compounds in melanogenesis: part I. Reaction of cysteine and glutathione with 5,6-dihydroxyindoles, Tetrahedron, 1987, 43(22), 5351–5356 CrossRef.
- A. Napolitano, A. Palumbo, M. d'Ischia and G. Prota, Mechanism of selective incorporation of the melanoma seeker 2-thiouracil into growing melanin, J. Med. Chem., 1996, 39(26), 5192–5201 CrossRef CAS PubMed.
- A. Pezzella, O. Crescenzi, A. Natangelo, L. Panzella, A. Napolitano, S. Navaratnam, R. Edge, E. J. Land, V. Barone and M. d'Ischia, Chemical, pulse radiolysis and density functional studies of a new, labile 5,6-indolequinone and its semiquinone, J. Org. Chem., 2007, 72(5), 1595–1603 CrossRef CAS PubMed.
- R. L. Schroeder, K. L. Double and J. P. Gerber, Using Sepia melanin as a PD model to describe the binding characteristics of neuromelanin–A critical review, J. Chem. Neuroanat., 2015, 64, 20–32 CrossRef PubMed.
- A. H. Stokes, T. G. Hastings and K. E. Vrana, Cytotoxic and genotoxic potential of dopamine, J. Neurosci. Res., 1999, 55(6), 659–665 CrossRef CAS PubMed.
- D. M. Miller, G. R. Buettner and S. D. Aust, Transition metals as catalysts of “autoxidation” reactions, Free Radical Biol. Med., 1990, 8(1), 95–108 CrossRef CAS PubMed.
- R. G. Zika, J. W. Moffett, R. G. Petasne, W. J. Cooper and E. S. Saltzman, Spatial and temporal variations of hydrogen peroxide in Gulf of Mexico waters, Geochim. Cosmochim. Acta, 1985, 49(5), 1173–1184 CrossRef CAS.
- D. H. Van Hees and K. L. Van Alstyne, Effects of emersion, temperature, dopamine, and hypoxia on the accumulation of extracellular oxidants surrounding the bloom-forming seaweeds Ulva lactuca and Ulvaria obscura, J. Exp. Mar. Biol. Ecol., 2013, 448, 207–213 CrossRef CAS.
- R. G. Petasne and R. G. Zika, Hydrogen peroxide lifetimes in south Florida coastal and offshore waters, Mar. Chem., 1997, 56(3), 215–225 CrossRef CAS.
- Y. R. Vázquez, K. L. Van Alstyne and B. L. Bingham, Exudates of the green alga Ulvaria obscura (Kützing) affect larval development of the sand dollar Dendraster excentricus (Eschscholtz) and the Pacific oyster Crassostrea gigas (Thunberg), Mar. Biol., 2017, 164(10), 194 CrossRef.
- D. K. Adams, M. A. Sewell, R. C. Angerer and L. M. Angerer, Rapid adaptation to food availability by a dopamine-mediated morphogenetic response, Nat. Commun., 2011, 2, 592 CrossRef PubMed.
- G. P. Bienert, A. L. Møller, K. A. Kristiansen, A. Schulz, I. M. Møller, J. K. Schjoerring and T. P. Jahn, Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes, J. Biol. Chem., 2007, 282(2), 1183–1192 CrossRef CAS PubMed.
- A. Dummermuth, U. Karsten, K. Fisch, G. König and C. Wiencke, Responses of marine macroalgae to hydrogen-peroxide stress, J. Exp. Mar. Biol. Ecol., 2003, 289(1), 103–121 CrossRef CAS.
- K. Asada, K. Yoshikawa, M. Takahashi, Y. Maeda and K. Enmanji, Superoxide dismutases from a blue-green alga, Plectonema boryanum, J. Biol. Chem., 1975, 250(8), 2801–2807 CAS.
- C.-T. Shiu and T.-M. Lee, Ultraviolet-B-induced oxidative stress and responses of the ascorbate–glutathione cycle in a marine macroalga Ulva fasciata, J. Exp. Bot., 2005, 56(421), 2851–2865 CrossRef CAS PubMed.
- B. Halliwell and J. Gutteridge, Oxygen toxicity, oxygen radicals, transition metals and disease, Biochem. J., 1984, 219(1), 1 CrossRef CAS PubMed.
- R. Tocher and J. Craigie, Enzymes of marine algae: II. Isolation and identification of 3-hydroxytyramine as the phenolase substrate in Monostroma fuscum, Can. J. Bot., 1966, 44(5), 605–608 CrossRef CAS.
-
G. Noctor, J.-P. Reichheld and C. H. Foyer, ROS-related redox regulation and signaling in plants, in Semin. Cell Dev. Biol., Elsevier, 2017 Search PubMed.
- J. M. Diaz, C. M. Hansel, A. Apprill, C. Brighi, T. Zhang, L. Weber, S. McNally and L. Xun, Species-specific control of external superoxide levels by the coral holobiont during a natural bleaching event, Nat. Commun., 2016, 7, 1–10 CAS.
- M. Rubin, I. Berman-Frank and Y. Shaked, Dust-and mineral-iron utilization by the marine dinitrogen-fixer Trichodesmium, Nat. Geosci., 2011, 4(8), 529 CrossRef CAS.
- E. D. Melton, E. D. Swanner, S. Behrens, C. Schmidt and A. Kappler, The interplay of microbially mediated and abiotic reactions in the biogeochemical Fe cycle, Nat. Rev. Microbiol., 2014, 12(12), 797–808 CrossRef CAS PubMed.
- P. D. Tortell, M. T. Maldonado, J. Granger and N. M. Price, Marine bacteria and biogeochemical cycling of iron in the oceans, FEMS Microbiol. Ecol., 1999, 29(1), 1–11 CrossRef CAS.
- H. W. Rich and F. M. Morel, Availability of well-defined iron colloids to the marine diatom Thalassiosira weissflogii, Limnol. Oceanogr., 1990, 35(3), 652–662 CrossRef CAS.
- M. L. Wells, N. G. Zorkin and A. Lewis, The role of colloid chemistry in providing a source of iron to phytoplankton, J. Mar. Res., 1983, 41(4), 731–746 CrossRef CAS.
- M. L. Wells, Manipulating iron availability in nearshore waters, Limnol. Oceanogr., 1999, 44(4), 1002–1008 CrossRef CAS.
- D. A. Hutchins, A. E. Witter, A. Butler and G. W. Luther, Competition among marine phytoplankton for different chelated iron species, Nature, 1999, 400(6747), 858–861 CrossRef CAS.
- R. Nolting, L. Gerringa, M. Swagerman, K. Timmermans and H. De Baar, Fe(III) speciation in the high nutrient, low chlorophyll Pacific region of the Southern Ocean, Mar. Chem., 1998, 62(3), 335–352 CrossRef CAS.
- S. Soria-Dengg and U. Horstmann, Ferrioxamines B and E as iron sources for the marine diatom Phaeodactylum tricornutum, Mar. Ecol.: Prog. Ser., 1995, 127, 269–277 CrossRef CAS.
- M. T. Maldonado and N. M. Price, Utilization of iron bound to strong organic ligands by plankton communities in the subarctic Pacific Ocean, Deep Sea Res., Part II, 1999, 46(11), 2447–2473 CrossRef CAS.
- K. Deiss, K. Hantke and G. Winkelmann, Molecular recognition of siderophores: a study with cloned ferrioxamine receptors (FoxA) from Erwinia herbicola and Yersinia enterocolitica, BioMetals, 1998, 11(2), 131–137 CrossRef CAS PubMed.
- J. Martinez, M. Haygood and A. Butler, Identification of a natural desferrioxamine siderophore produced by a marine bacterium, Limnol. Oceanogr., 2001, 46(2), 420–424 CrossRef.
-
F. M. Morel and J. G. Hering, Principles and applications of aquatic chemistry, Wiley, New York, 1993 Search PubMed.
- R. J. Motekaitis and A. E. Martell, Stabilities of the iron(III) chelates of 1,2-dimethyl-3-hydroxy-4-pyridinone and related ligands, Inorg. Chim. Acta, 1991, 183(1), 71–80 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7em00497d |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*