
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct visible light activation of a surface cysteine-engineered [NiFe]-hydrogenase by silver nanoclusters†
Liyun
Zhang
 a,
Stephen E.
Beaton
a,
Stephen B.
Carr
bc and
Fraser A.
Armstrong
*a
a,
Stephen E.
Beaton
a,
Stephen B.
Carr
bc and
Fraser A.
Armstrong
*a
aInorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford OX1 3QR, Oxfordshire, UK. E-mail: fraser.armstrong@chem.ox.ac.uk
bResearch Complex at Harwell, Rutherford Appleton Laboratory, Harwell Oxford, Didcot, UK
cDepartment of Biochemistry, University of Oxford, Oxford OX1 3QU, Oxfordshire, UK
First published on 19th October 2018
Abstract
Genetically engineering a cysteine (thiolate) close to the distal [4Fe–4S] cluster of a [NiFe]-hydrogenase creates a highly specific target for attachment of Ag nanoclusters templated in polymethyl acrylate, the resulting ‘hard-wired’ enzyme catalysing rapid hydrogen evolution by visible light. The rate is further enhanced by binding to metal oxide nanoparticles – results of investigations focusing on P-25 TiO2 and including anatase TiO2, rutile TiO2, ZnO, SrTiO3 and ZrO2 leading to the proposal that these act as active or structural scaffolds to promote intra-assembly electron transfer.
Broader contextBiology long ago solved the problem of producing H2 efficiently under ambient conditions without using precious metals. Hydrogenases – enzymes having Fe (and Ni) at their active sites – have an interesting role in solar hydrogen research: (a) their high activities under minimal overpotentials directs attention to other limiting bottlenecks in integrated systems, such as light capture and inter-component electron transfers; (b) they can be genetically engineered to build in useful new features without compromising the structure and properties of the active site. Here, a surface thiol group (the amino acid, cysteine) has been engineered into a [NiFe]-hydrogenase close to the point at which electrons enter or leave the internal electron-transport chain, enabling the binding of a silver nanocluster (AgNC) that is excitable by visible light. Site selectivity is ensured by removing existing cysteines from the enzyme surface. The resulting enzyme–AgNC complex rapidly evolves H2 when illuminated, but equally significant is the observation that metal oxide nanoparticles, particularly TiO2 (P-25 or anatase), enhance the rate by another order of magnitude. Acting as a scaffold to bind the polymethyl acrylate-templated AgNCs, the nanoparticles smooth out intermittences and obstacles by electrically inter-connecting enzyme–AgNC complexes and greatly enlarging the capture area for sacrificial electron donors. |
Direct conversion of solar energy to hydrogen from water using artificial photosynthesis (‘Solar Fuels’) is considered an effective strategy to help meet future renewable energy requirements. One approach to motivate and inspire progress has been to use enzymes as the catalysts in model solar conversion devices and photocatalytic complexes.1–12 Enzymes are valuable because evolution has taken them close to perfection – their suitability being gauged in terms of their high substrate specificities, high turnover rates, low overpotential requirements and use of abundant elements (e.g. Fe, Mn, Ni and Cu) instead of precious metals.13 For instance, hydrogenases have catalytic activities comparable to Pt.14 In addition to the active site, the presence of internal electron relay systems comprising centres with low reorganization energies may enable catalytic electron transfer (ET) to compete more effectively with electron–hole recombination processes occurring at the semiconductor surface.8,11,15,16 Enzymes may therefore serve a unique role in the evaluation of integrated solar conversion systems, as they bring to the fore other steps of the process. Enzymes also offer structural definition that extends well beyond the immediate site at which reactants bind and they may be engineered in subtle ways. For example, using molecular biology procedures, it is possible to design and produce an enzyme for specific attachment of a suitable photosensitizer in such a position that allows rapid and direct electron injection into the enzyme's internal relay system. Examples of where enzymes have been used in model systems include their direct immobilization on semiconductors such as TiO2,11 CdX (X = S, Se, Te),1,8,9 and In2S3,17 electrodes (bulk or nanostructured carbon,18–20 TiO2,21,22 Au23) and molecular complexes with photoactive or conductive materials.1,7–10,12,24–26 In solar fuels research it is always necessary to close the cycle efficiently using an external electron donor: the latter is ideally water, but this requires a second special catalyst and knowledge has progressed mainly by focusing on one side of the cycle and using a ‘blunt tool’ in the form of an innocuous, non-selective agent such as an amine, present in high concentration.
To build a model system from the atomic level we sought to exploit the unique properties of silver nanoclusters (AgNCs), which bridge the gap between atoms and nanoparticles.27 First, AgNCs are powerful photoreductants, being able to reduce methyl viologen when illuminated with visible light;28 second, they should form stable complexes with soft ligands, particularly thiolates that can be positioned on the surface of enzymes, and allow good electronic coupling into nearby redox centres;29 third, their binding to a thiolate ligand can be detected by luminescence spectroscopy. To stabilize AgNCs, it is necessary to template them in a water-soluble matrix such as polymethacrylic acid (PMAA, average molecular mass 5 kDa, 60 monomer units) which can typically stabilize five AgNCs each containing <10 Ag atoms.30–33 The free carboxylates on PMAA that are not used to bind AgNCs also allow simultaneous attachment to the hard-acid surface of metal oxide supports.34,35 In a recent paper, we reported a much enhanced (100-fold) rate for CO formation by carbon monoxide dehydrogenase co-adsorbed on TiO2 (Degussa P-25) nanoparticles that had been modified with AgNCs–PMAA, in which Ag5 was the dominant species.33
In this Communication, we report a systematic approach which exploits the ability to re-engineer a [NiFe]-hydrogenase for selective surface attachment of AgNCs leading to a ‘hard-wired’ photoactive enzyme. The experiments are developed and extended to help understand the charge flow in a highly efficient system for photohydrogen production.
Hydrogenase-2 from E. coli is a modest H2 producer, but one that is a valuable subject for research: notably, the ability to genetically engineer and produce this otherwise low-yielding enzyme has recently been greatly improved.36 In conventional experiments, the recombinant enzyme evolves H2 at a rate of approximately 6 s−1 (per active site) when driven by reduced methyl viologen at pH 6.36 The crystal structure (PDB: 6EN9) shows Hyd-2 to be a dimer of heterodimers (αβ)2 in which the [NiFe]-active site is located in each of the two large (α) subunits and an electron relay of three FeS clusters is housed in each small (β) subunit. On each α subunit there are three surface-exposed cysteine residues that could act as tether points to link Hyd-2 to PMAA-templated AgNCs. As shown in Fig. S1 (ESI†), the three cysteines (C197, C432 and C433) are all too far (>17 Å) from the [NiFe] active site or the nearest FeS cluster (the proximal [4Fe–4S] cluster) to allow fast, direct ET into the enzyme (the distances are summarized in Table S1, ESI†).37,38 However, the distal [4Fe–4S] cluster lies close to the protein surface, near to a tyrosine (Y′222; the apostrophe signifying a residue in the small subunit) which if replaced by cysteine should allow an AgNC to make a direct connection into the ET relay. The (αβ)2 structure places the two equivalent distal clusters on each half of the enzyme less than 14 Å apart, so two interaction sites should be available.
To investigate the physical and electronic connections between Hyd-2, Ag nanoclusters and TiO2 nanoparticles, we made a detailed comparison of the photoelectrocatalytic and luminescence properties of the native enzyme (Hyd-2) and variants produced by systematic site-directed mutagenesis. These variants are as follows: 3S-Hyd-2 (the triple variant C433S, C432S, C197S) in which all three naturally occurring surface cysteines are replaced by serine; Y′222C-Hyd-2 in which tyrosine-222 of the small subunit, which is located <10 Å from the distal [4Fe–4S] cluster, has been replaced by a cysteine; 3S1C-Hyd-2 (the quadruple variant C433S, C432S, C197S, Y′222C) in which all naturally-occurring surface cysteines are replaced by serine, and tyrosine 222 is replaced by cysteine. Details of the experimental methods are given in ESI† (Tables S2–S5 and Fig. S2).
The structure of the highly altered 3S1C-Hyd-2 variant was determined by X-ray crystallography. As shown in Fig. 1 and Fig. S2–S4 (ESI†), electron density maps for the 3S1C-Hyd-2 variant were of very high quality and clearly showed the positions of each variant amino acid. Each cysteine-to-serine mutation caused the side chain to adopt a different rotamer from the native protein with the more polar hydroxyl group pointing towards the solvent (Fig. 1). Replacing tyrosine-222 by cysteine had no effect on the conformation of the surrounding polypeptide backbone which could be superposed on that of native Hyd-2 with an r.m.s.d. of 0.16 Å. The side chain rotamer is also unaltered, but there was evidence of slight disorder in the position of the sulfhydryl group (Fig. S3 and S4, ESI†). Importantly, the distance between the S-atom of C222 and the closest S-atom of the distal cluster is less than 5 Å, making this a very promising binding site to attach a AgNC for photoactivation.
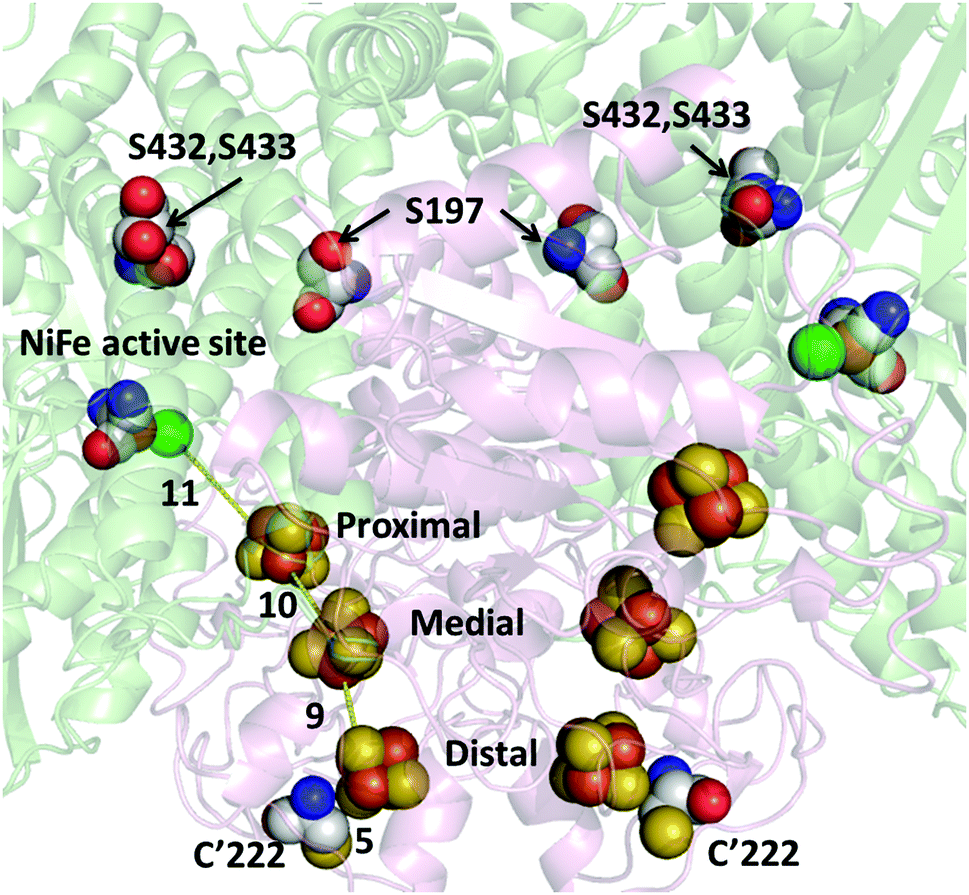 | ||
| Fig. 1 Structure of the active site and electron transfer relay in 3S1C-Hyd-2. The distance (Å) between the side chain sulfur atom of C222 and the distal cluster, and the distances between the nearest atoms of each FeS cluster are shown. The three Fe–S clusters, active site ligands and mutational sites (S197, S432, S433 and C′222) are shown as spheres with Ni atoms in green, Fe atoms in bronze and S atoms in yellow. C atoms are in grey, O atoms in red and N atoms in blue. | ||
Fig. 2 shows the results of experiments carried out in which various Hyd-2 enzymes had been incubated with AgNCs–PMAA before illumination (see ESI† for details). The AgNCs–PMAA was prepared as described previously and characterized by MALDI-TOF, absorption and emission spectroscopy.31,33 Briefly, 5 mL solutions contained in a sealed, thermostatted pressure vessel (22.5 mL volume) were irradiated with visible light using a 300 W Arc Lamp, (Newport 67005) fitted with a 420 nm filter and held 5 cm away. A high concentration (0.1 M) of triethanolamine (TEOA) was used as buffer and sacrificial electron donor. Production of H2 was monitored regularly by removing small volumes (20 μL) from the headspace for gas chromatography (GC) analysis (Fig. S5, ESI†). The two variants 3S1C-Hyd-2 and Y′222C-Hyd-2 in which tyrosine 222 has been exchanged for cysteine showed strong H2 production upon illumination, whereas the activity of Hyd-2 and 3S-Hyd-2 was very low (H2 production rates are summarised in Table 1).
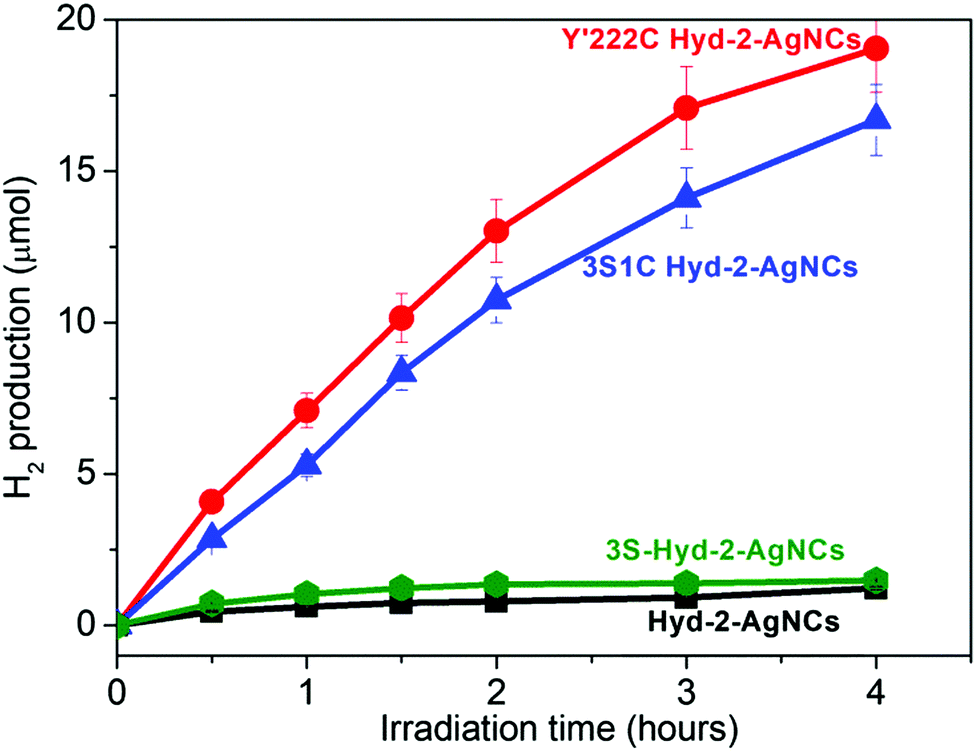 | ||
| Fig. 2 Photocatalysis of H2 generation by hydrogenase/AgNCs–PMAA complexes under visible light irradiation at pH 7.0, 25 °C. See ESI† for details. | ||
| Samplesa | H2b (μmol) h−1 | TOFc (s−1) | Average quantum yield (%) |
|---|---|---|---|
| a Unless otherwise stated, a standard condition was employed as follows: hydrogenase (0.25 nmol) was incubated with AgNCs (50 μL of 5 mg mL−1 solution) for 30 min, then the free AgNCs–PMAA was removed using a centrifugal concentrator with a 50 kDa molecular weight cut-off. Finally, the enzyme–AgNCs complex was diluted in 5 mL 0.1 M triethanolamine (TEOA) buffer containing 0.1 M NaCl at pH 7, 25 °C; or hydrogenase (0.25 nmol) was added to a 5 mL dispersion of AgNCs–PMAA/TiO2 (1 mg mL−1) in 0.1 M TEOA buffer containing 0.1 M NaCl at pH 7, 25 °C. Irradiation was carried out with a Newport 67005 arc lamp (300 W) fitted with a 420 nm filter, located 5 cm from the solution. b H2 production rate (μmol H2 h−1) based on the first h of irradiation. c Initial turnover frequency (TOF) expressed as molecules of H2 produced per second per active site. | |||
| Y′222C Hyd-2–AgNCs | 7.2 ± 0.5 | 9.1 ± 1.5 | 1.0 |
| 3S1C Hyd-2–AgNCs | 5.3 ± 0.3 | 6.8 ± 1.3 | 0.7 |
| 3S Hyd-2–AgNCs | 0.6 ± 0.1 | 0.6 ± 0.1 | <0.1 |
| Hyd-2–AgNCs | 0.4 ± 0.2 | 0.4 ± 0.1 | <0.1 |
| Y′222C Hyd-2/AgNCs–PMAA/TiO2 | 134.4 ± 8.5 | 193.3 ± 5.1 | 20.6 |
| 3S1C Hyd-2/AgNCs–PMAA/TiO2 | 112.2 ± 6.4 | 145.5 ± 3.5 | 15.5 |
| Hyd-2/AgNCs–PMAA/TiO2 | 100.2 ± 5.3 | 125.5 ± 5.1 | 13.4 |
| 3S Hyd-2/AgNCs–PMAA/TiO2 | 29.3 ± 2.5 | 30.0 ± 2.4 | 3.2 |
| Y′222C Hyd-2/TiO2 | 0.2 ± 0.1 | 0.2 ± 0.1 | <0.1 |
| 3S1C Hyd-2/TiO2 | 0.3 ± 0.1 | 0.4 ± 0.1 | <0.1 |
| Hyd-2/TiO2 | 0.2 ± 0.1 | 0.2 ± 0.1 | <0.1 |
| 3S Hyd-2/TiO2 | 0.2 ± 0.1 | 0.2 ± 0.1 | <0.1 |
| AgNCs–PMAA/TiO2 | 0.3 ± 0.1 | 0 | <0.1 |
Photoluminescence titrations were performed to define the interactions between AgNCs and hydrogenase variants. The dominant species was Ag5 along with smaller amounts of Ag9, Ag7 and Ag3.32 Details are given in ESI† (Fig. S6 and S7). Fig. 3 compares the results obtained for different Hyd-2 enzymes referenced against a control and glutathione. The quenching order is: control > 3S ≫ 3S1C > Hyd-2 > Y222C, all values being higher than glutathione. Notably, the greatly increased luminescence quenching observed for 3S1C-Hyd-2 compared to 3S-Hyd-2 shows that AgNCs must bind specifically at the new cysteine site,29,32 the Ag–S interactions being dynamic and transient to account for the virtual totality.
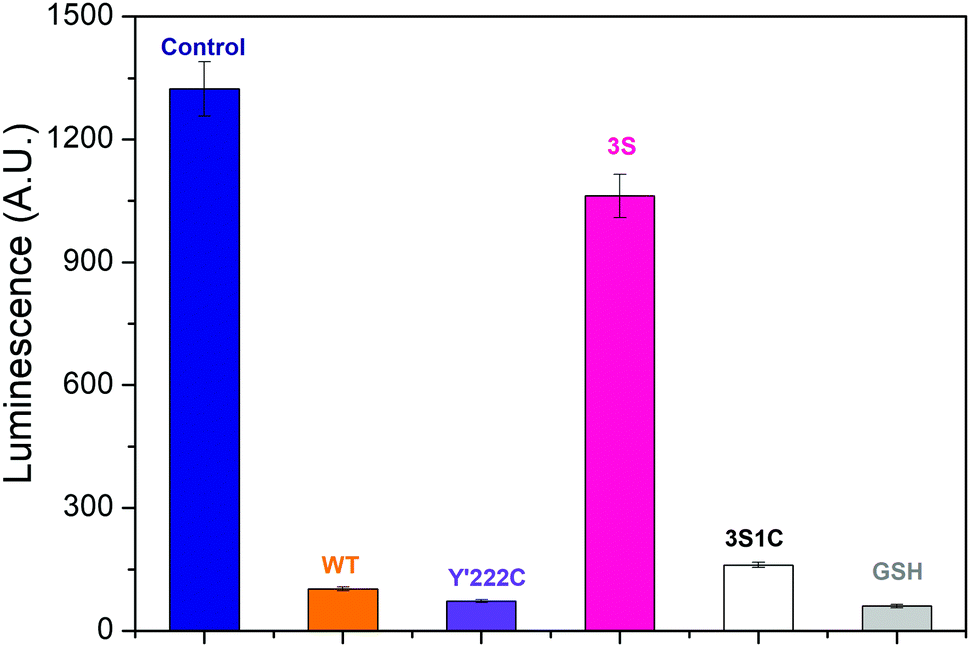 | ||
| Fig. 3 Interaction between Ag nanoclusters and Hyd-2 enzymes assayed by photoluminescence. Chart shows the photoluminescence intensity of AgNCs–PMAA/enzyme complexes (Ag atom 20 μM, enzyme or glutathione (GSH) 10 μM) at 550 nm in 0.2 M MES buffer. λex: 460 nm. The control experiment was carried out with a solution containing only AgNCs–PMAA. | ||
The special advantage that binding site specificity can confer in terms of delivering fast catalytic electron transfer was confirmed by protein film voltammetry experiments. A gold working electrode was available in the laboratory, allowing direct measurements to be made of the electrocatalytic activity obtained for each enzyme under a N2 atmosphere (see ESI†). Fig. 4 shows that both native Hyd-2 and 3S-Hyd-2 are inactive, whereas 3S1C Hyd-2 displays intense activity, the sharp peak obtained upon scanning in a positive direction being due to the oxidative depletion of H2 that has just been produced and yet to escape from the Au electrode surface at which the enzyme is bound (the electrode is stationary).39 The Y′222C variant which retains the three remote cysteines is less active: a plausible explanation for the low activity is that cysteines located far from the internal electron-transfer relay compete with C′222 for electrode surface binding, which results in the majority of enzyme molecules being poorly oriented with respect to the local electrode surface and unable to engage in catalytic electron transfer.
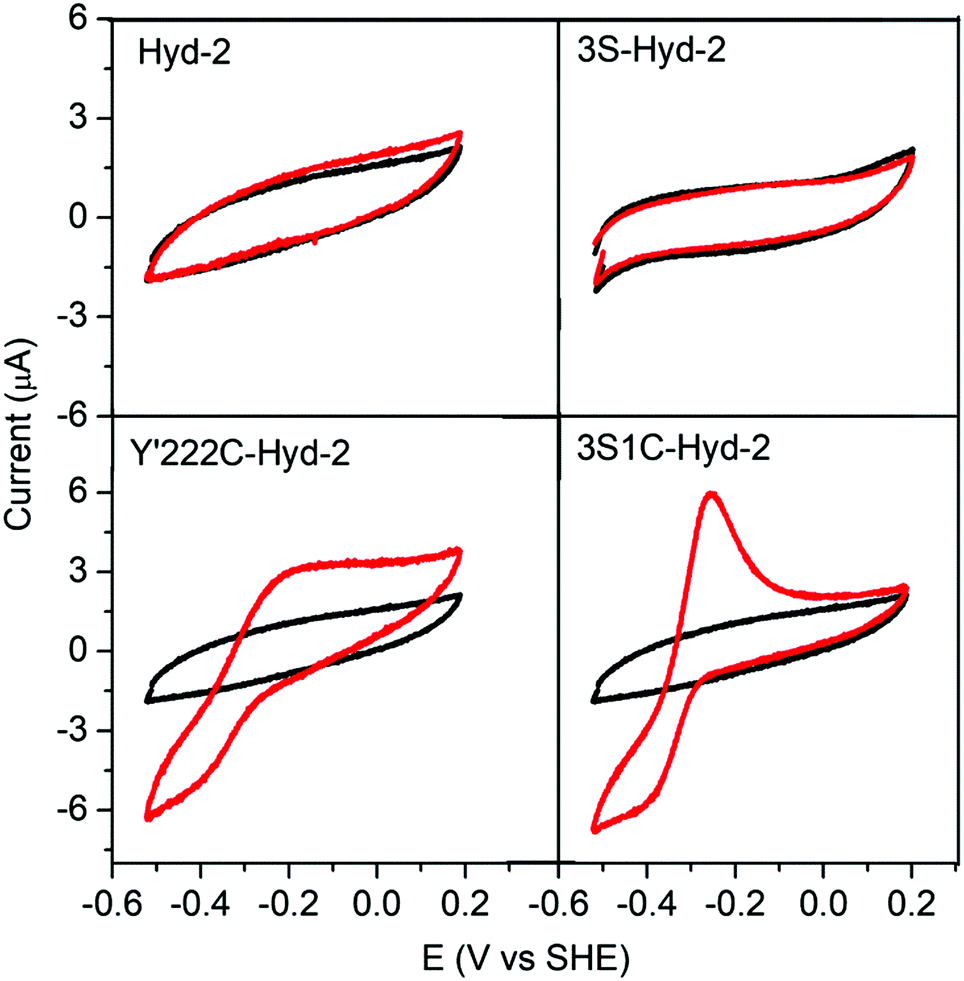 | ||
| Fig. 4 Cyclic voltammograms of Hyd-2 variants recorded at a stationary Au electrode under an atmosphere of 100% N2 (black traces correspond to bare Au electrode and red traces represent voltammograms obtained with enzyme adsorbed on the surface). All experiments were performed in 0.2 M MES (pH 5.0), 0.1 M NaCl, at 25 °C with a scan rate of 5 mV−1. | ||
Fig. 5A shows the results of experiments in which TiO2 nanoparticles (Degussa, P-25, characterized as described elsewhere40) were included to form an active scaffold for the AgNCs–PMAA/enzyme complexes.33 In all cases the catalytic activity is greatly increased compared to the complexes alone. Significantly, the activities of 3S1C-Hyd-2 and Y′222C-Hyd-2 are increased over 20-fold when TiO2 nanoparticles are added (the values of the initial turnover frequency are summarized in Table 1). The activity of Hyd-2 is much higher than that of 3S-Hyd-2 which contains no surface cysteines. In all cases the rate of H2 production decreased gradually over the course of 4 hours: this was partly due to product inhibition because the rate increased to a significant extent each time the headspace was refreshed (Fig. 5B). Even after 4 cycles of headspace flushing, the H2 evolution rate remained at a high level. An interesting and obvious outcome of the experiments shown in Fig. 5B is that after 8 hours, each molecule of Y′222C-Hyd-2 has undergone over 3 million turnovers. Estimated quantum yields for the 3S1C-Hyd-2 system (based on a wavelength of 500 nm) ranged from 0.7% to 15.5%, in the absence or presence of TiO2, respectively.
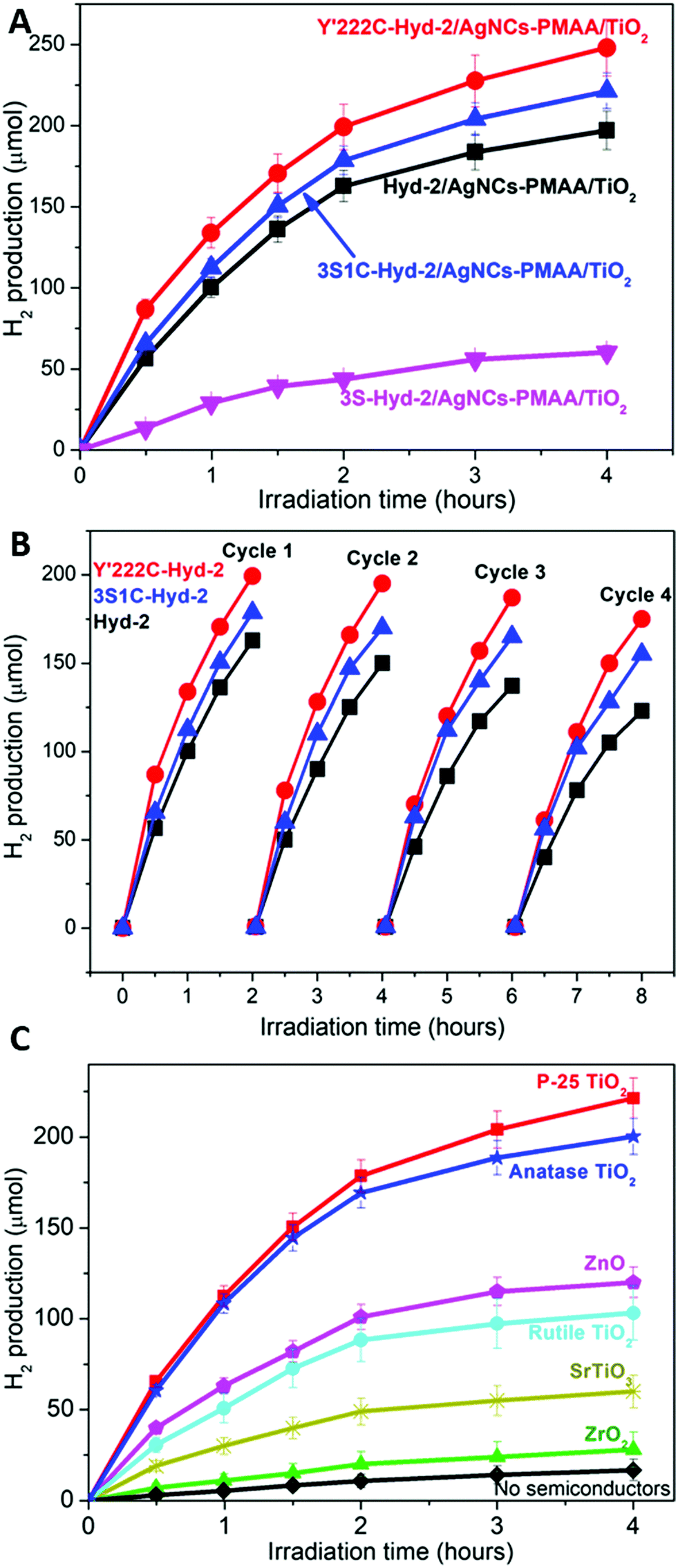 | ||
| Fig. 5 (A) Hydrogen generation by ternary enzyme/AgNCs–PMAA/TiO2 complexes under visible-light irradiation in 5 mL of 0.1 M TEOA buffer containing 5 mg of AgNCs–PMAA/TiO2 and 0.25 nmol enzyme at pH 7.0, 25 °C. (B) Production of H2 over the course of 8 hours, in which H2 was flushed out at 2 h intervals. (C) Hydrogen generation by various ternary 3S1C-Hyd-2/AgNCs–PMAA/metal oxide complexes under visible-light irradiation in 5 mL 0.1 M TEOA buffer containing 5 mg of AgNCs–PMAA/MO and 0.25 nmol 3S1C-Hyd-2 at pH 7.0, 25 °C. | ||
Such a large enhancement in rate, even for the 3S1C variant which is already optimized for specific AgNC binding, was puzzling. Further experiments were carried out to determine if other metal oxide semiconductor nanoparticles produced a similar effect. The photocatalytic H2 production results obtained with P-25 TiO2 for AgNCs–PMAA/3S1C were compared with analogous experiments carried out using similar quantities of other nanoparticles – pure anatase TiO2, pure rutile TiO2, ZnO, SrTiO3 and ZrO2. Fig. 5C shows that all metal oxide nanoparticles enhanced the rate to different levels above that of AgNC/PMAA–3S1C Hyd-2 alone. The order (to be considered in qualitative terms only) was P-25 TiO2 ∼ anatase TiO2 > ZnO > rutile TiO2 > SrTiO3 > ZrO2, the latter showing relatively little rate enhancement above the level of AgNCs–PMAA/3S1C. The decrease in rate with time was similar in all case, showing that the loss of activity (partly attributable to product inhibition, as mentioned above) does not depend on the identity of the metal oxide.
Native Hyd-2 is inactive for H2 evolution at TiO2 nanoparticles that have been photosensitized to visible light using Ru complexes,11 and shows no electrochemical activity at a P-25 TiO2 electrode (Fig. S8, ESI†). Voltammograms obtained for electrodes constructed by depositing P-25 TiO2 on indium tin oxide glass (see ESI† and Fig. S9–S12) showed that the binding of AgNCs–PMAA to TiO2 introduces a surface-confined reduction peak at a position coinciding with the onset of H2 evolution activity observed when each of the Hyd-2 enzymes is also adsorbed. Consequently, there exists a rapid electron pathway through the sequence [TiO2 → AgNCs–PMAA → enzyme]. We were unable to obtain analogous electrodes with ZnO, SrTiO3 or ZrO2 owing to instability.
The bulk electronic properties of these metal oxides have been extensively studied, and conduction band energies, despite some variation among results from different researchers, offer some explanation for our results. Scanlon and coworkers recently concluded that the electron affinity (work function) of rutile is approximately 0.4 eV lower than that of anatase.41 Although previous consensus had favored the opposite order, the more recent experiments and calculations could account for why electrons flow spontaneously across a phase boundary from rutile to anatase. For SrTiO3, the most recent literature suggests an electron affinity 0.4 eV lower than anatase42 (although much lower relative values, 0.6 eV and 1.0 eV have been reported43,44). Greater qualitative certainty surrounds ZrO2 which has a much lower electron affinity due to its 4d0 configuration,43,44 while values for ZnO place its electron affinity close to, or 0.2 eV lower than anatase.42–45 On the basis of the most recent assessments, the resulting ordering of conduction band energies and, equivalently, the decreasing ease of electron transfer from AgNCs would be: anatase < ZnO < rutile < SrTiO3 ≪ ZrO2. The broad agreement with the results shown in Fig. 5C suggests that a viable pathway exists from AgNC to TiO2 and then to non-specifically bound hydrogenase. Whereas anatase, ZnO, and rutile could assist H2 production by using their conduction band to assist the transfer and distribution of electrons from excited AgNCs, this would be less likely with SrTiO3 and impossible with ZrO2.
It is unlikely that any of the oxides could engage the valence band in a way that could enhance TEOA oxidation. On the other hand, defects which form at the surfaces of metal oxides may be important for mediating charge transfer within and between bound AgNC-PMAA–enzyme units.46 Such restricted conductivity would not only be important for improving electron transfer between AgNCs and poorly connected enzyme molecules, but the greatly enlarged ‘collection’ area available on the nanoparticle could also be important for increasing the efficiency of electron donation from TEOA. Among the metal oxides studied, ZrO2 stands out as being extremely stable and such defects are unlikely. The observation that ZrO2 shows any enhancement effect at all suggests that it serves as a structural scaffold, binding AgNC/PMAA–enzyme units in close proximity to each other and improving efficiency by optimizing connections at a local level.
A general mechanism for photocatalytic H2 production by AgNC/PMAA–enzyme and the enhancement by metal oxide nanoparticles, most notably P-25 TiO2, can be proposed, which is outlined in Scheme 1. The primary mechanistic argument begins with 3S1C-Hyd-2 and Y′222C-Hyd-2, for which introduction of a cysteine so close to the distal [4Fe–4S] cluster provides a unique site at which AgNCs bind and inject electrons directly into the FeS relay. Since this site is occupied (and obscured) by a AgNC, the primary function of the supporting nanoparticles must be to mediate electron transfer from sacrificial electron donors which now have a much greater surface area at which to react (Scheme 1). The second mechanistic feature must apply to variants lacking C′222: here, the connections formed between AgNCs and the nanoparticle surface also allows the photoexcited electron to transfer indirectly into the enzyme by non-specific interactions that bring other AgNCs in the PMAA template to within tunneling distance of the distal cluster. Evidence that dynamic electron mediation depends on the presence of AgNC is obtained from the fact that no catalytic reduction current is observed when Hyd-2 is introduced to a PMAA–TiO2 electrode: consequently, PMAA alone cannot facilitate electron transfer or enzyme binding (Fig. S13, ESI†).
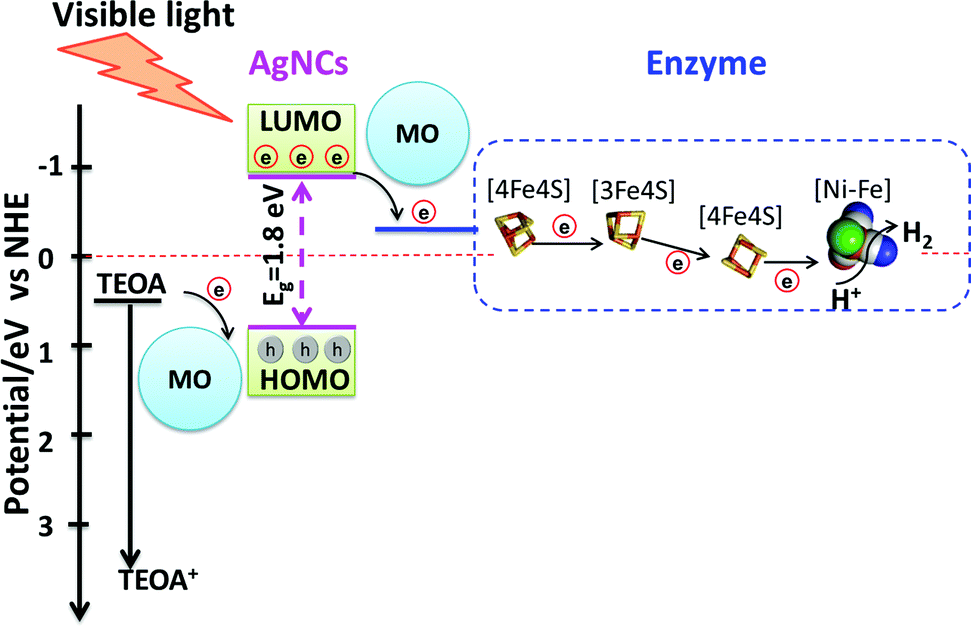 | ||
| Scheme 1 Schematic diagram showing the energies and electron-transfer pathways in AgNCs–PMAA and points of mediation by the metal oxide (MO) nanoparticles. The Eg of AgNCs was estimated from the absorption spectrum and the potential of TEOA, AgNCs LUMO and enzyme are referred to in the literature.47–49 | ||
An important outcome of this study has been to quantify the primary advantage achieved by site-specific ‘hard-wiring’ of an enzyme catalyst to its excitation apparatus. However, the clear difference compared to non-‘hard-wired’ enzyme is only seen when a metal oxide nanoparticle scaffold is absent. Addition of metal oxide nanoparticles must improve the rate of electron supply from electron donors in solution: apart from this, they also introduce an active non-specific pathway that results from their ability to connect AgNCs–PMAA and enzyme molecules together in such a way as to facilitate ET regardless of whether hard-wiring is achieved. The activities of the metal oxide nanoparticles appear to correlate with conduction band potential (accessible yet sufficiently reducing to produce H2) and ease of surface defect formation. The fact that the rates of photoelectrochemical H2 evolution displayed by the ternary complexes are so much higher than when measured in solution using methyl viologen also shows that the traditional approach to gauging hydrogenase activity may be misleading, as even relatively poor H2 producers may excel once their electron supply chain is improved. Attachment to any source that can provide and support continuous (non-interrupted) electron flow to a catalyst is seen to be highly advantageous, as remarked recently by Brown and co-workers in regard to their discovery of NH3 formation by nitrogenase attached to CdS nanocrystals.26
Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. Zhang thanks the Royal Society and Newton fund for a Newton International Fellowship. S. E. Beaton is grateful to the Dakota Foundation for the award of a Holaday Scholarship held at Exeter College, Oxford. Research on hydrogenases has been supported by grants from the UK Biotechnology and Biological Sciences Research Council, particularly BB/N006321/1. F. A. A. thanks the Royal Society for a Wolfson Research Merit Award (2013–2018).Notes and references
- B. Chica, C.-H. Wu, Y. Liu, M. W. W. Adams, T. Lian and R. B. Dyer, Energy Environ. Sci., 2017, 10, 2245–2255 RSC.
- D. Adam, L. Bosche, L. Castaneda-Losada, M. Winkler, P. Apfel and T. Happe, ChemSusChem, 2017, 10, 894–902 CrossRef CAS PubMed.
- K. F. Wu and T. Q. Lian, Chem. Soc. Rev., 2016, 45, 3781–3810 RSC.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z. X. Guo and J. W. Tang, Energy Environ. Sci., 2015, 8, 731–759 RSC.
- S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- T. Sakai, D. Mersch and E. Reisner, Angew. Chem., Int. Ed., 2013, 52, 12313–12316 CrossRef CAS PubMed.
- B. L. Greene, C. A. Joseph, M. J. Maroney and R. B. Dyer, J. Am. Chem. Soc., 2012, 134, 11108–11111 CrossRef CAS PubMed.
- K. A. Brown, M. B. Wilker, M. Boehm, G. Dukovic and P. W. King, J. Am. Chem. Soc., 2012, 134, 5627–5636 CrossRef CAS PubMed.
- C. E. Lubner, P. Knörzer, P. J. N. Silva, K. A. Vincent, T. Happe, D. A. Bryant and J. H. Golbeck, Biochemistry, 2010, 49, 10264–10266 CrossRef CAS PubMed.
- E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 18457–18466 CrossRef CAS PubMed.
- K. A. Brown, S. Dayal, X. Ai, G. Rumbles and P. W. King, J. Am. Chem. Soc., 2010, 132, 9672–9680 CrossRef CAS PubMed.
- T. W. Woolerton, S. Sheard, Y. S. Chaudhary and F. A. Armstrong, Energy Environ. Sci., 2012, 5, 7470–7490 RSC.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- M. W. Ratzloff, M. B. Wilker, D. W. Mulder, C. E. Lubner, H. Hamby, K. A. Brown, G. Dukovic and P. W. King, J. Am. Chem. Soc., 2017, 139, 12879–12882 CrossRef CAS PubMed.
- M. B. Wilker, K. E. Shinopoulos, K. A. Brown, D. W. Mulder, P. W. King and G. Dukovic, J. Am. Chem. Soc., 2014, 136, 4316–4324 CrossRef CAS PubMed.
- C. Tapia, S. Zacarias, I. A. C. Pereira, J. C. Conesa, M. Pita and A. L. De Lacey, ACS Catal., 2016, 6, 5691–5698 CrossRef CAS.
- D. Svedružić, J. L. Blackburn, R. C. Tenent, J.-D. R. Rocha, T. B. Vinzant, M. J. Heben and P. W. King, J. Am. Chem. Soc., 2011, 133, 4299–4306 CrossRef PubMed.
- M. Hambourger, M. Gervaldo, D. Svedruzic, P. W. King, D. Gust, M. Ghirardi, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2008, 130, 2015–2022 CrossRef CAS PubMed.
- M. A. Alonso-Lomillo, O. Rüdiger, A. Maroto-Valiente, M. Velez, I. Rodríguez-Ramos, F. J. Muñoz, V. M. Fernández and A. L. De Lacey, Nano Lett., 2007, 7, 1603–1608 CrossRef CAS PubMed.
- S. Morra, F. Valetti, S. J. Sadeghi, P. W. King, T. Meyer and G. Gilardi, Chem. Commun., 2011, 47, 10566–10568 RSC.
- E. Reisner, J. C. Fontecilla-Camps and F. A. Armstrong, Chem. Commun., 2009, 550–552 RSC.
- C. Gutiérrez-Sánchez, D. Olea, M. Marques, V. M. Fernández, I. A. C. Pereira, M. Vélez and A. L. De Lacey, Langmuir, 2011, 27, 6449–6457 CrossRef PubMed.
- L. E. Roth, J. C. Nguyen and F. A. Tezcan, J. Am. Chem. Soc., 2010, 132, 13672–13674 CrossRef CAS PubMed.
- C. A. Caputo, L. Wang, R. Beranek and E. Reisner, Chem. Sci., 2015, 6, 5690–5694 RSC.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters, L. C. Seefeldt and P. W. King, Science, 2016, 352, 448–450 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- W.-T. Chen, Y.-J. Hsu and P. V. Kamat, J. Phys. Chem. Lett., 2012, 3, 2493–2499 CrossRef CAS PubMed.
- L. Shang and S. Dong, Biosens. Bioelectron., 2009, 24, 1569–1573 CrossRef CAS PubMed.
- H. Xu and K. S. Suslick, ACS Nano, 2010, 4, 3209–3214 CrossRef CAS PubMed.
- I. Diez, M. Pusa, S. Kulmala, H. Jiang, A. Walther, A. S. Goldmann, A. H. E. Muller, O. Ikkala and R. H. A. Ras, Angew. Chem., Int. Ed., 2009, 48, 2122–2125 CrossRef CAS PubMed.
- L. Shang and S. Dong, Chem. Commun., 2008, 1088–1090 RSC.
- L. Zhang, M. Can, S. W. Ragsdale and F. A. Armstrong, ACS Catal., 2018, 8, 2789–2795 CrossRef CAS.
- D. G. Brown, P. A. Schauer, J. Borau-Garcia, B. R. Fancy and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 1692–1695 CrossRef CAS PubMed.
- B. J. Brennan, M. J. L. Portoles, P. A. Liddell, T. A. Moore, A. L. Moore and D. Gust, Phys. Chem. Chem. Phys., 2013, 15, 16605–16614 RSC.
- S. E. Beaton, R. M. Evans, A. J. Finney, C. M. Lamont, F. A. Armstrong, F. Sargent and S. B. Carr, Biochem. J., 2018, 475, 1353–1370 CrossRef CAS PubMed.
- B. L. Greene, G. E. Vansuch, C. H. Wu, M. W. W. Adams and R. B. Dyer, J. Am. Chem. Soc., 2016, 138, 13013–13021 CrossRef CAS PubMed.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS PubMed.
- F. A. Armstrong, R. M. Evans, S. V. Hexter, B. J. Murphy, M. M. Roessler and P. Wulff, Acc. Chem. Res., 2016, 49, 884–892 CrossRef CAS PubMed.
- T. Ohno, K. Sarukawa, K. Tokieda and M. Matsumura, J. Catal., 2001, 203, 82–86 CrossRef CAS.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed.
- J.-i. Fujisawa, T. Eda and M. Hanaya, Chem. Phys. Lett., 2017, 685, 23–26 CrossRef CAS.
- M. A. Butler and D. S. Ginley, J. Electrochem. Soc., 1978, 125, 228–232 CrossRef CAS.
- Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CrossRef CAS.
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS PubMed.
- S. Lany, A. Zakutayev, T. O. Mason, J. F. Wager, K. R. Poeppelmeier, J. D. Perkins, J. J. Berry, D. S. Ginley and A. Zunger, Phys. Rev. Lett., 2012, 108, 016802 CrossRef CAS PubMed.
- Y. Pellegrin and F. Odobel, C. R. Chim, 2017, 20, 283–295 CrossRef CAS.
- H. J. Chen, Q. Wang, M. Q. Lyu, Z. Zhang and L. Z. Wang, Chem. Commun., 2015, 51, 12072–12075 RSC.
- M. J. Lukey, A. Parkin, M. M. Roessler, B. J. Murphy, J. Harmer, T. Palmer, F. Sargent and F. A. Armstrong, J. Biol. Chem., 2010, 285, 3928–3938 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, methods and any associated references. See DOI: 10.1039/c8ee02361a |
| This journal is © The Royal Society of Chemistry 2018 |