
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Energetically efficient electrochemically tunable affinity separation using multicomponent polymeric nanostructures for water treatment†
Xianwen
Mao
,
Wenda
Tian
,
Yinying
Ren
,
Dexin
Chen
,
Sarah E.
Curtis
,
Marjorie T.
Buss
 ,
Gregory C.
Rutledge
and
T. Alan
Hatton
*
,
Gregory C.
Rutledge
and
T. Alan
Hatton
*
Department of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. E-mail: tahatton@mit.edu
First published on 25th July 2018
Abstract
We describe a water treatment strategy, electrochemically tunable affinity separation (ETAS), which, unlike other previously developed electrochemical processes, targets uncharged organic pollutants in water. Key to achieving ETAS resides in the development of multicomponent polymeric nanostructures that simultaneously exhibit the following characteristics: an oxidation-state dependent affinity towards neutral organics, high porosity for sufficient adsorption capacity, and high conductivity to permit electrical manipulation. A prototype ETAS adsorbent composed of nanostructured binary polymeric surfaces that can undergo an electrically-induced hydrophilic–hydrophobic transition can provide programmable control of capture and release of neutral organics in a cyclic fashion. A quantitative energetic analysis of ETAS offers insights into the tradeoff between energy cost and separation extent through manipulation of electrical swing conditions. We also introduce a generalizable materials design approach to improve the separation degree and energetic efficiency simultaneously, and identify the critical factors responsible for such enhancement via redox electrode simulations and theoretical calculations of electron transfer kinetics. The effect of operation mode and multistage configuration on ETAS performance is examined, highlighting the practicality of ETAS and providing useful guidelines for its operation at large scale. The ETAS approach is energetically efficient, environmentally friendly, broadly applicable to a wide range of organic contaminants of various molecular structures, hydrophobicity and functionality, and opens up new avenues for addressing the urgent, global challenge of water purification and wastewater management.
Broader contextSeparation processes are of paramount importance in the chemical and environmental industries, accounting for 10–25% of the world's energy consumption, and about a third of total capital and operation costs in industrial plants. The development of separation technologies for water treatment with high energy efficiency and low environmental impact has become a primary engineering challenge for the 21st century due to the worldwide occurrence of water contamination and its associated negative impacts on the environment and human health. Electrochemically controlled processes, such as capacitive deionization, have emerged as promising candidates for wastewater management and water desalination. However, since these previously developed electrochemical methods rely primarily on the electrostatic interaction between the electrode and the target pollutant, they only work for charged species (e.g., anions, cations), and are not applicable to uncharged organic pollutants, which constitute the majority of industrial and municipal water contaminants, including many dyes, pesticides, pharmaceuticals and carcinogenic aromatics. This study investigates a conceptually novel separation strategy that enables sensitive, programmable electrochemical control over the release and capture of uncharged organic pollutants in water resources. |
1 Introduction
The global prevalence of water resources contaminated by organic pollutants, such as pesticides, dyes, pharmaceuticals, and endocrine disrupting compounds, has raised concerns about potential deleterious effects on the environment (particularly aquatic ecosystems) as well as human health.1–6 Exposure to these pollutants has been found to be linked to increased predisposition toward diabetes, cancer, infertility, obesity, and other types of endocrine disorders.7–10 High separation efficiencies for the removal of organics from water have been achieved using conventional processes such as adsorption, stripping, distillation, and solvent extraction, as well as more recent technologies such as advanced oxidation treatment and membrane separation.11–13 However, the overall separation processes inherently associated with these methods usually involve energy-intensive steps (e.g. requirement for high temperature or pressure) and/or environmentally unfriendly processes (e.g., use of organic solvents and additives, generation of secondary pollutants). Therefore, it is of great interest to develop water treatment technologies that are more energy efficient and environmentally responsible. In a broader context, the development of novel separation strategies for chemical mixtures without the need for heating or the introduction of chemical reagents would significantly reduce energy consumption, emissions and pollution worldwide, and open up new ways for resource recovery and extraction.14Material systems that respond to external chemico-physical stimuli offer advanced control over a process with a high degree of tunability and flexibility, enabling a deeper understanding and thus optimization of that process.15–17 Recently, stimuli-responsive systems have been exploited in liquid-phase separation processes for water treatment and environmental clean-up. An azobenzene-modified membrane has been developed that exhibits “on/off” states for water penetration using alternating irradiation of the membranes with ultraviolet and visible light as the switch.18 In another example, suspended core–shell nanoparticles with adsorbed organic pollutants can lose colloidal stability and form macroscopic aggregates upon irradiation with ultraviolet light, allowing easy handling of bulkier materials as opposed to suspended nanomaterials.4 “Smart” systems with hydrophobicity tunable by external signals such as pH,19 chemical redox agent20 and CO2 concentration21 have also been developed for organics–water separation.
Here we introduce a conceptually new approach to water treatment using electrochemically tunable affinity separation (ETAS), which exploits electricity as the external stimulus for control over organics–water separation. ETAS relies on the use of a stimulus-responsive adsorbent system with a surface hydrophobicity that can be controlled by an electrical signal. Such an adsorbent displays an electrically programmable affinity toward neutral organic molecules, enabling the use of finely-tuned electrical swing to capture and release organics cyclically.
In terms of the working mechanism, and the nature of target species to be separated, ETAS is fundamentally distinct from previously developed electrochemical separation strategies for water treatment, such as capacitive deionization (CDI)22 and redox-mediated selective separation (RMSS)23 (Fig. 1). CDI and RMSS utilize electrostatic interactions as the primary driving force for separation, and the target species must possess electric charges, as insalt ions and charged organic species. In stark contrast, ETAS does not rely on electrostatic interactions at all, but rather exploits the hydrophobic interactions as the separation driving force. Hence, ETAS can be used to separate uncharged species, such as toxic pesticides/herbicides, industrial dyes, and endocrine disrupting compounds, which are primary pollutants found in water resources worldwide.1–10 ETAS represents a potentially significant addition to other electrochemical separation technologies for water treatment such as CDI22 and RMSS23 since it tackles a major category of water pollutants that cannot be addressed by these technologies. In fact, almost all known electrochemical separation technologies (e.g., not only CDI and RMSS, but others such as ion concentration polarization,24 and electrochemically switched ion-exchange25) must rely on electrostatic interactions. Therefore, as none of these conventional techniques is applicable to neutral species, ETAS is a conceptually new electrochemical separation strategy that represents the first attempt to incorporate fine electrical control into the separation of uncharged molecules.
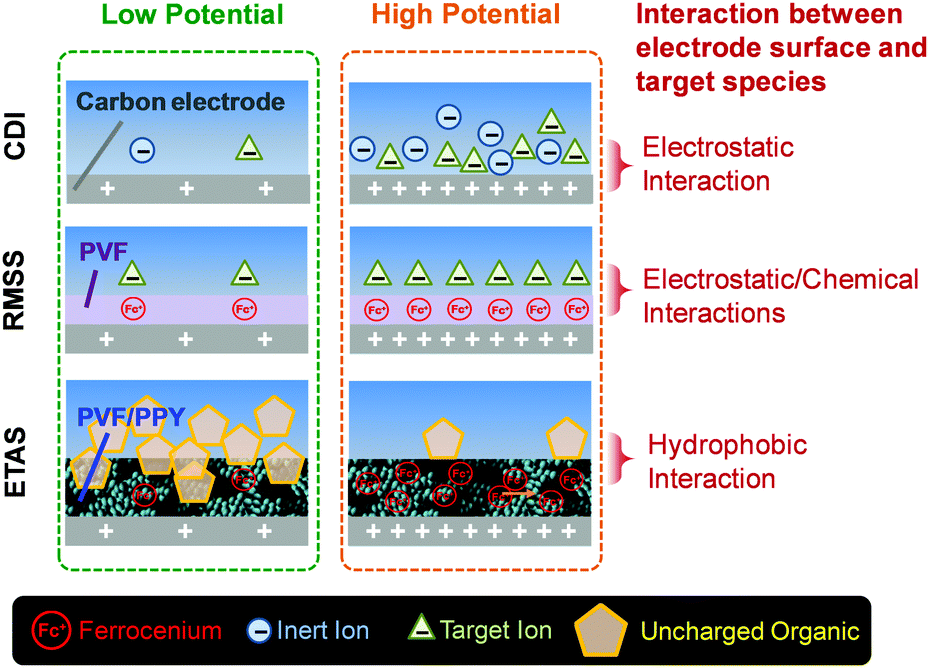 | ||
| Fig. 1 Schematic illustration of the ETAS concept, in comparison with capacitive deionization (CDI) and redox-mediated selective separation (RMSS). | ||
Notably, ETAS is a separation technology that is also fundamentally distinct from catalysis-based methods for treatment of organic species in water (e.g., electrocatalysis).26 Unlike catalysis-based water treatment methods, ETAS does not decompose organics chemically, and in fact can achieve nearly 100% recovery for many species tested here, which is often a key achievement goal for separation technologies. More importantly, ETAS possesses a number of advantageous characteristics not shared by electrocatalysis. ETAS does not result in any secondary pollution, a serious issue for electrocatalysis due to generation of multiple intermediates with long persistence, resulting from complicated and elusive degradation mechanisms. Moreover, the rationally engineered ETAS adsorbent is universally applicable to different types of organic species despite their vastly different redox potentials and chemical structures. However, for electrocatalysis, the catalyst properties and the operating potential must be customized individually for different organics, and one catalyst usually works for only one class of organics with very similar redox potentials and chemical structures. Also, many organics are extremely difficult to oxidize or reduce and thus a lengthy catalyst screening process is often needed to ensure efficient electrocatalytic degradation of these compounds.
ETAS is expected to show higher energy efficiencies and incur lower environmental costs than established methods for separation of neutral organics from water such as distillation, stripping, extraction, adsorption and filtration.11–13 It operates at ambient temperature and pressure, requires no need for organic solvents and additives for extraction and/or adsorbent regeneration, and produces no secondary pollutants over the course of the separation. Hence ETAS shows promise in terms of downstream waste sustainability and water economy.
Compared to other stimuli-responsive systems recently developed for water treatment, ETAS also exhibits a number of unique, advantageous features. First, in contrast to the previously developed pH-,19 redox agent-,20 light-,4,18 and CO221-responsive systems that exhibit only two levels of hydrophobicity (i.e., merely “on/off” bimodal control), ETAS can achieve multiple levels of hydrophobicity and thus affinity towards organics since the electrical signal (i.e., potential) can be tuned with high precision, permitting a systematic adjustment of the ratio between the hydrophobic and hydrophilic moieties. Such flexible modulation of affinity for target pollutants is key to achieving a balance between the separation degree and the energetic efficiency, as discussed later. Second, because the electrical potential of an ETAS adsorbent can be changed locally in real-time, the hydrophobicity of such an adsorbent can be modulated with high spatio-temporal resolution. Such precise spatial and temporal control over the hydrophobicity of a system is almost unattainable using responsive systems based on chemical (e.g., CO2, pH, reductant, oxidant) and thermal stimuli due to the presence of mass diffusion and/or heat dissipation processes that hamper the precise delivery of the stimulus over location and time. Moreover, ETAS-integrated devices enjoy common advantages with electrochemical systems, such as modularity, portability and low-cost. These features would make ETAS an economically viable platform technology for developing countries where water scarcity and environmental pollution pose an imminent threat to human health.
2 Results & discussion
2.1 ETAS design
The design and fabrication of an ETAS adsorbent is nontrivial. It should exhibit all the following characteristics: (i) an oxidation-state dependent affinity towards neutral organics, (ii) high porosity for sufficient adsorption capacity, and (iii) high conductivity to permit programmable electrical manipulation instead of merely redox control by chemical species. The first proof-of-concept ETAS adsorbent system we developed (Fig. 2a) consisted of a carbon cloth (CC) with a conformal coating of a polyvinylferrocene/polypyrrole (PVF/PPY) hybrid, fabricated via simultaneous electro-polymerization of pyrrole and electro-deposition of PVF.27 In the binary polymer film, the ferrocene moieties in PVF rendered redox-tunable hydrophobicity (i.e., the oxidized ferrocene is significantly more hydrophilic than its reduced form),28 while the conjugated PPY chains established electron transport pathways to permit electrical control. The CC, composed of bendable carbon microfibers, served as a flexible and robust conductive substrate. PVF, a localized state conductor with discrete redox centers, had a low conductivity (∼10−7 cm s−1),29 while our PVF/PPY hybrid exhibited a significantly higher conductivity (24 ± 7 cm s−1) measured by the four-probe method. Scanning electron microscopic (SEM) (Fig. 2b) and transmission electron microscopic (TEM) (Fig. 2c) imaging revealed that the PVF/PPY polymer film was comprised of nano-spheres with diameters of around 20 to 50 nm. Energy dispersive X-ray spectroscopic (EDS) elemental mapping of N and Fe (Fig. 2d and e) indicated the presence, with uniform spatial distribution, of both PPY and PVF in the hybrid polymer film.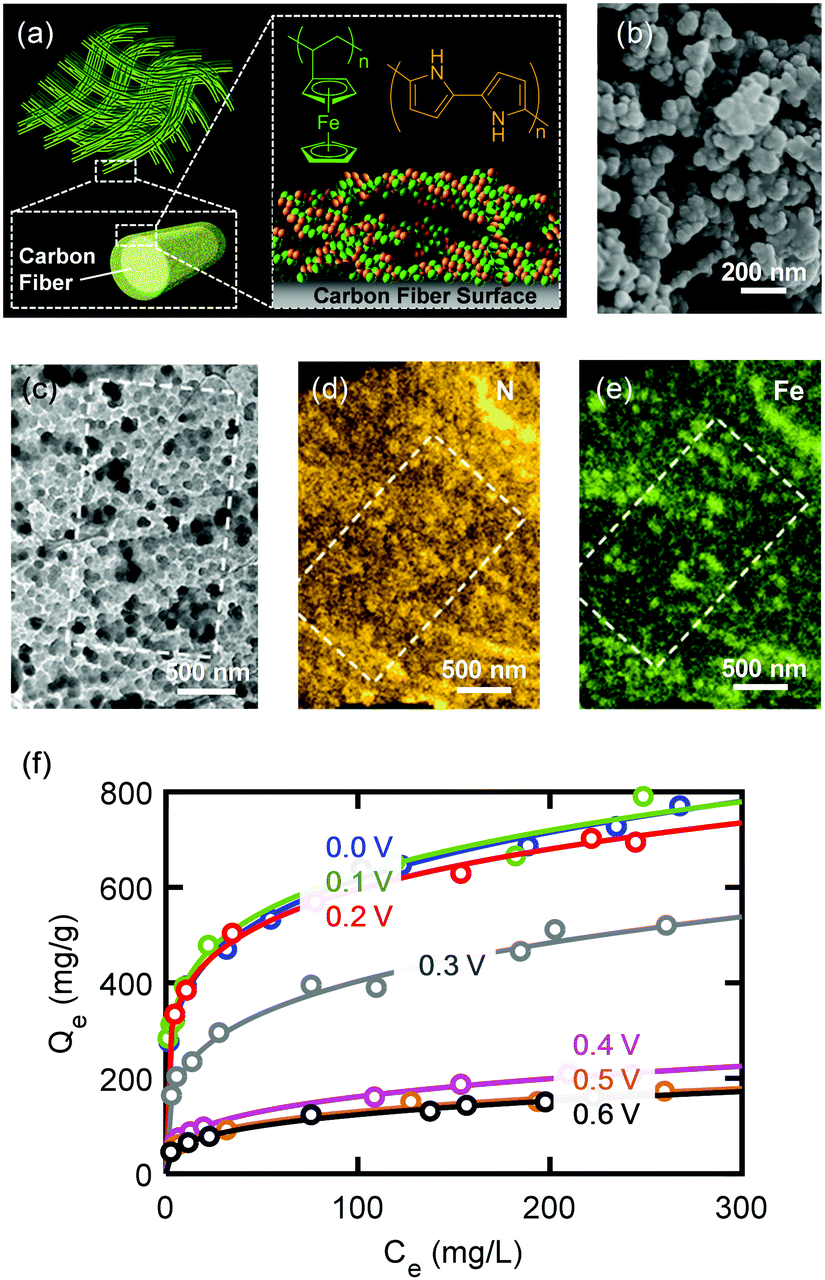 | ||
| Fig. 2 (a) Schematic illustration of the ETAS adsorbent composed of PVF/PPY coated on a flexible CC substrate. (b–e) SEM image (b), TEM image (c), EDS N map (d), and EDS Fe map (e) of PVF/PPY. (f) Adsorption isotherms of PVF/PPY at different potentials. Open symbol: experimental data. Solid lines: Freundlich fits. | ||
This ETAS adsorbent allows electrochemical control over adsorption of neutral organics: when the applied potential (E) is lower than the formal potential of ferrocene (E0 = 0.32 V, all potentials are referenced to Ag/AgCl), most ferrocene moieties are reduced, the ETAS adsorbent is hydrophobic, and organics can be captured from water. For E ≫ E0, most ferrocene moieties are oxidized, and the ETAS adsorbent becomes relatively hydrophilic, releasing neutral organics into a water stripping phase. The ratio (r) between the hydrophilic and hydrophobic moieties (i.e., oxidized and reduced ferrocene) can be tuned precisely by the applied potential according to the Nernst equation 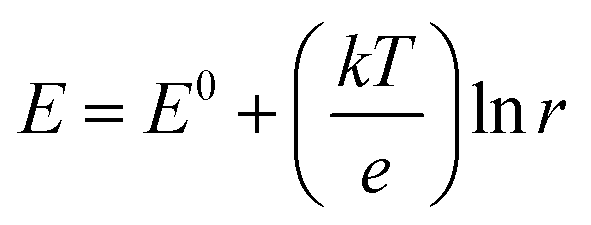 , where k is the Boltzmann constant, T the temperature, and e the elementary charge, and thus enables programmable adjustment of the adsorption affinity for organics.
, where k is the Boltzmann constant, T the temperature, and e the elementary charge, and thus enables programmable adjustment of the adsorption affinity for organics.
We investigated the ETAS separation efficacy using a model water pollutant, Sudan Orange G (SOG), an uncharged organic dye molecule commonly used in the textile industry, which is the primary source of water contamination worldwide.30 We compared the adsorptive performance of PVF/PPY, PPY alone, and PVF alone for SOG based on the distribution coefficient, defined as Kd (mL g−1) = Qe/Ce, where Qe (mg g−1) and Ce (mg L−1) are the mass of adsorbed SOG per unit mass of polymer and the SOG concentration in the liquid phase at equilibrium, respectively. Significantly better adsorptive performance was found with PVF/PPY (Kd = 1.4 × 105 mL g−1, obtained at Ce = 2.5 mg L−1) than with PVF (Kd = 1.3 × 104 mL g−1) or PPY (Kd = 2.3 × 104 mL g−1), due to the more porous structure of the hybrid (Fig. S12, ESI†). The Brunauer–Emmett–Teller (BET) surface areas of PVF, PPY, and PVF/PPY were 18, 36, 168 m2 g−1, respectively.27Kd values around 105 mL g−1 indicate superb adsorbent performance.31
Fig. 2f shows that the adsorption isotherm (i.e., Qeversus Ce) of PVF/PPY for SOG followed the Freundlich adsorption model, and, more importantly, could be adjusted systematically through application of different electrochemical potentials. When the applied potential increased from 0 to 0.6 V, the adsorption capacity of PVF/PPY decreased monotonically. The adsorbent became increasingly hydrophilic at higher potentials at which more ferrocene moieties were oxidized. Such electrochemical tunability of the adsorption behavior is important for regeneration of the adsorbent without the use of solvent stripping, and permits cyclic operation of the ETAS-based separation process. Cyclic voltammetric (CV) scans at 0.1 V s−1 between 0.0 and 0.8 V showed that PVF/PPY could be oxidized and reduced for 300 cycles with negligible loss of the current signal (<1% decrease in the integrated CV area), indicating good electrochemical stability of the hybrid during the cyclic operation. For further enhancement of the performance stability of ETAS adsorbents, several previously developed methods such hydrothermal treatment27 or organic vapor deposition32 could be employed. Also, our control experiments show that application of an oxidative potential (0.6 V) did not result in degradation of SOG (Fig. S11, ESI†).
2.2 Multi-stage cyclic operation
We implemented ETAS in a multi-stage cyclic batch process (Fig. 3a), in which SOG was removed gradually from a 100 mL feed solution with an initial SOG concentration of 5 mg L−1 and transferred to a 5 mL receiving solution (i.e., water with no SOG initially). The mass of PVF/PPY on each electrode was 0.12 mg. For details, see Experimental. Briefly, two PVF/PPY adsorbents were subject to alternating oxidation–reduction cycles: at stage 1, adsorbent I was reduced and adsorbent II was oxidized, and at stage 2, the polarity of the two adsorbents was reversed. Such a cyclic operation was repeated until stage 24, permitting gradual transfer of SOG from the feed solution to the receiving solution. The feed solution was successively contacted with the reduced adsorbent (i.e., adsorbent I at stage 1, 3, 5…23; adsorbent II at stage 2, 4, 6…24), whereas the receiving solution was successively contacted with the oxidized adsorbent (i.e., adsorbent II at stage 1, 3, 5… 23; adsorbent I at stage 2, 4, 6…24). Fig. 3b and c show the SOG concentrations in the feed and receiving solutions (Cf and Cr), respectively, versus the number of stages, under three different potential pairs – a reductive potential to increase the hydrophobicity of PVF/PPY for capturing SOG and an oxidative potential to decrease the hydrophobicity of PVF/PPY for releasing SOG. The directly measured experimental data (open symbols) agreed with the calculated results (solid lines) obtained by numerical solution of the coupled Freundlich isotherms obtained at the paired potentials. This agreement indicates that the ability of ETAS to transfer the pollutant from the feed to the receiving solution was indeed the result of the potential-modulated affinity tuning. The net effect of the ETAS cyclic operation was the transfer of SOG from a dilute solution to a concentrated solution. Such a process is not thermodynamically spontaneous, and requires additional electrochemical work, which is related to the choice of the potential pair. At any given stage, a larger difference in the potential pair (0.0 V–0.6 V or 0.2 V–0.4 V) yielded lower Cf and higher Cr than did a smaller difference (0.2 V–0.3 V). This indicates that a larger potential difference was more effective in removing SOG from the feed solution and concentrating it in the receiving solution. However, the use of a larger potential difference was also associated with a higher energetic penalty, as discussed below.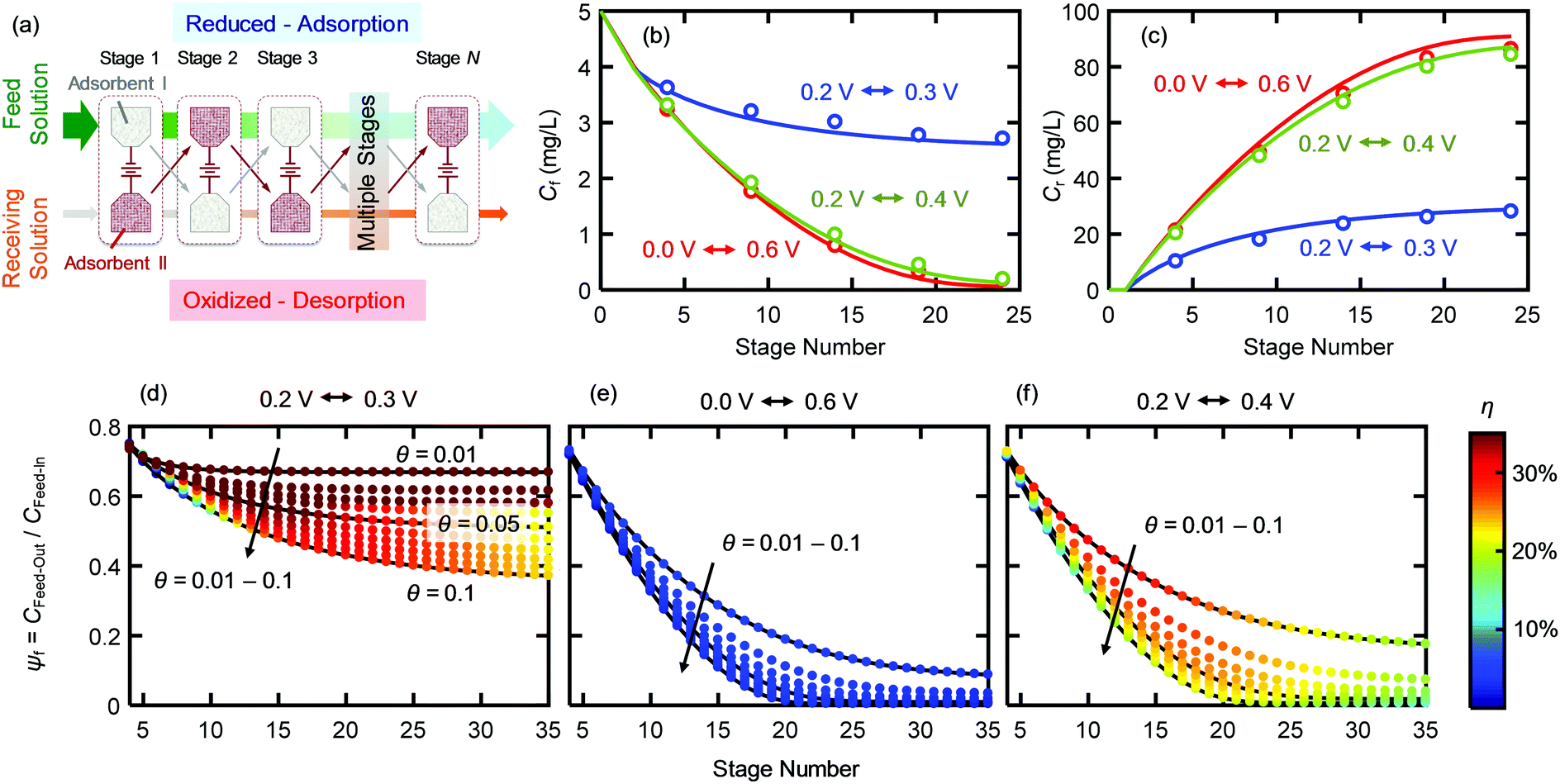 | ||
| Fig. 3 (a) ETAS multi-stage batch process. At stage 1, adsorbent I is reduced and contacts the feed solution for adsorption, whereas adsorbent II is oxidized and contacts the receiving solution for desorption. At stage 2, adsorbent II is reduced and contacts the feed solution for adsorption, whereas adsorbent I is oxidized and contacts the receiving solution for desorption. (b and c) Cf (b) and Cr (c) versus the stage number. (d–f) ψf–θ–η charts of PVF/PPY for selected potential pairs: 0.2 V–0.3 V (d), 0.0 V–0.6 V (e), and 0.2 V–0.4 V (f). | ||
2.3 Energetic analysis
A quantitative study was made on the effects of potential pairing and operating conditions on the energetic costs of ETAS. This overall separation process has two in-streams (feed-in: rich in SOG; receiving-in: pure water) and two out-streams (feed-out: lean in SOG; receiving-out: rich in SOG). The energetic efficiency (η) for this ETAS separation process is defined as the minimum work (i.e., thermodynamically reversible work, Wrev) needed to achieve a given change between the out-streams and the in-streams, divided by the actual energy consumed in the ETAS process (i.e., electrochemical work, Wec):33| η = Wrev/Wec × 100, | (1) |
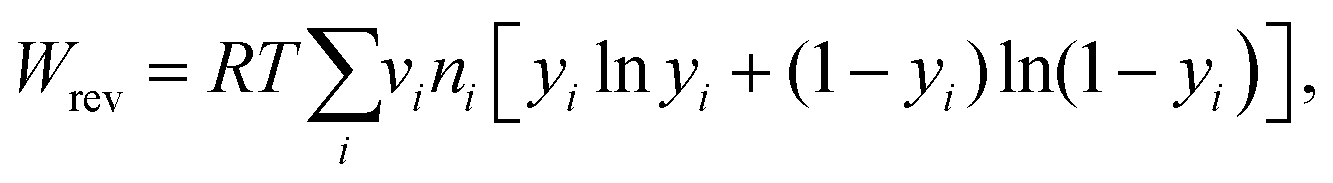 | (2) |
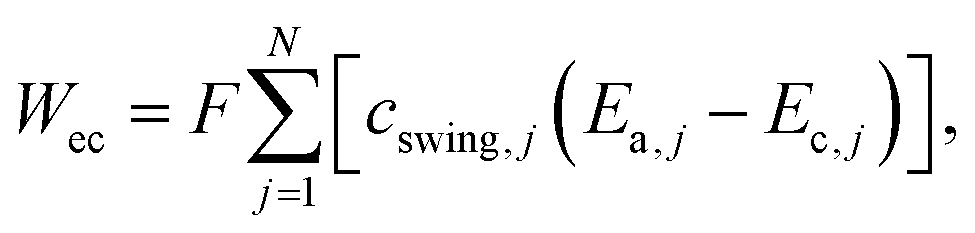 | (3) |
The energetic efficiencies should be compared at the same degree of separation and under identical operating conditions. The degree of separation is described by the extent of pollutant removal ψf = Cf,out/Cf,in, where Cf,out is the SOG concentration of the feed solution after a certain number of stages, and Cf,in is the initial SOG concentration of the feed solution. The ratio of the receiving and feed solution volumes, θ, is an important parameter determining the overall effectiveness of the separation process; in practice this ratio can be easily tuned and usually needs to be specified to achieve a desired degree of pollutant enrichment in the receiving solution. We introduce the ψf–θ–η chart (Fig. 3d–f) as the key metric for quantitative assessment of ETAS performance in terms of the coupled energetic efficiency and separation degree. To compare ETAS performance using different potential pairs, for each chart, we employ the same range of θ values (from 0.01 to 0.1) and the same number of stages (up to 35). For a given range of stage numbers and θ, it is desired to have a small ψf and a large η simultaneously. On comparison of Fig. 3d–f, it is evident that a smaller difference in the paired potentials yielded a higher energetic efficiency. However, the 0.2 V–0.3 V pair had a lower degree of separation than did the 0.2 V–0.4 V and 0.0 V–0.6 V pairs, while the 0.2 V–0.4 V pair yielded the best performance among the three cases in terms of the optimal combination of energetic efficiency and separation degree; even though the 0.0 V–0.6 V pair had essentially the same separation performance as the 0.2 V–0.4 V pair, the larger potential swing led to a significantly higher energetic penalty. Based on the ψf–θ–η chart, it is also apparent that with a lower feed to receiving ratio (i.e., a smaller θ), a higher energy efficiency is obtained, but more stages are required to reach a given separation extent.
2.4 Simultaneous enhancement of energetic efficiency and separation degree
The separation effectiveness and the energetic efficiency of an ETAS process were improved simultaneously through a generalizable materials design approach that relies on the development of a ternary heterogeneous nanostructure consisting of a metallic π-electron-rich CNT core and a binary polymer shell with redox-tunable hydrophobicity. Such a core–shell nanostructure allows for the use of a smaller potential difference to create a larger difference in the adsorption capacity between the reduced and oxidized states. This property of the ternary nanostructure is key to attaining enhanced ETAS performance. The model ternary nanostructure developed here (Fig. 4a) was composed of a metallic carbon nanotube (CNT) core with a conformal coating of PVF/PPY, formed by the non-covalent π stacking interaction between the three components.27,34 Hereafter the ternary nanostructure is denoted PVF/PPY/CNT. To construct ETAS adsorbents, we immobilized PVF/PPY/CNT onto the CC substrate by electrochemical deposition (Experimental). TEM imaging (Fig. 4b) shows that PVF/PPY/CNT exhibited a larger average tube diameter (∼50 nm) than that of the unmodified CNT (tube diameter: ∼10–15 nm, wall thickness: ∼2–4 nm; see Fig. S13 (ESI†) for a high-resolution TEM image). These TEM analyses suggested a conformal polymer coating existed around the nanotubes in the ternary hybrid. EDS elemental mapping of N and Fe (Fig. 4c) further confirmed that this polymer coating consisted of PPY and PVF. The adsorption isotherm of PVF/PPY/CNT for SOG shows a pronounced dependence on the applied potential (Fig. 4d), showing that this adsorbent should be highly effective in ETAS operation. Fig. 4e and f show the separation degree (= (1 − ψf) × 100) and energetic efficiency (η), respectively, of PVF/PPY/CNT for selected potential pairs at θ = 0.01 and 10 stages. The complete ψf–θ–η charts for PVF/PPY/CNT are shown in Fig. S14 (ESI†). It is evident that for PVF/PPY/CNT, among the different potential pairs, 0.2 V–0.35 V rendered the best performance, yielding both a high separation degree and a high energetic efficiency. More importantly, PVF/PPY/CNT showed markedly better performance than PVF/PPY. For instance, compared to the best-performing case with PVF/PPY operating under 0.2 V–0.4 V, PVF/PPY/CNT operating under 0.2 V–0.35 V, 0.2 V–0.4 V, and 0.25–0.4 V (marked with* in Fig. 4e and f) exhibited simultaneously enhanced separation degree and energetic efficiency.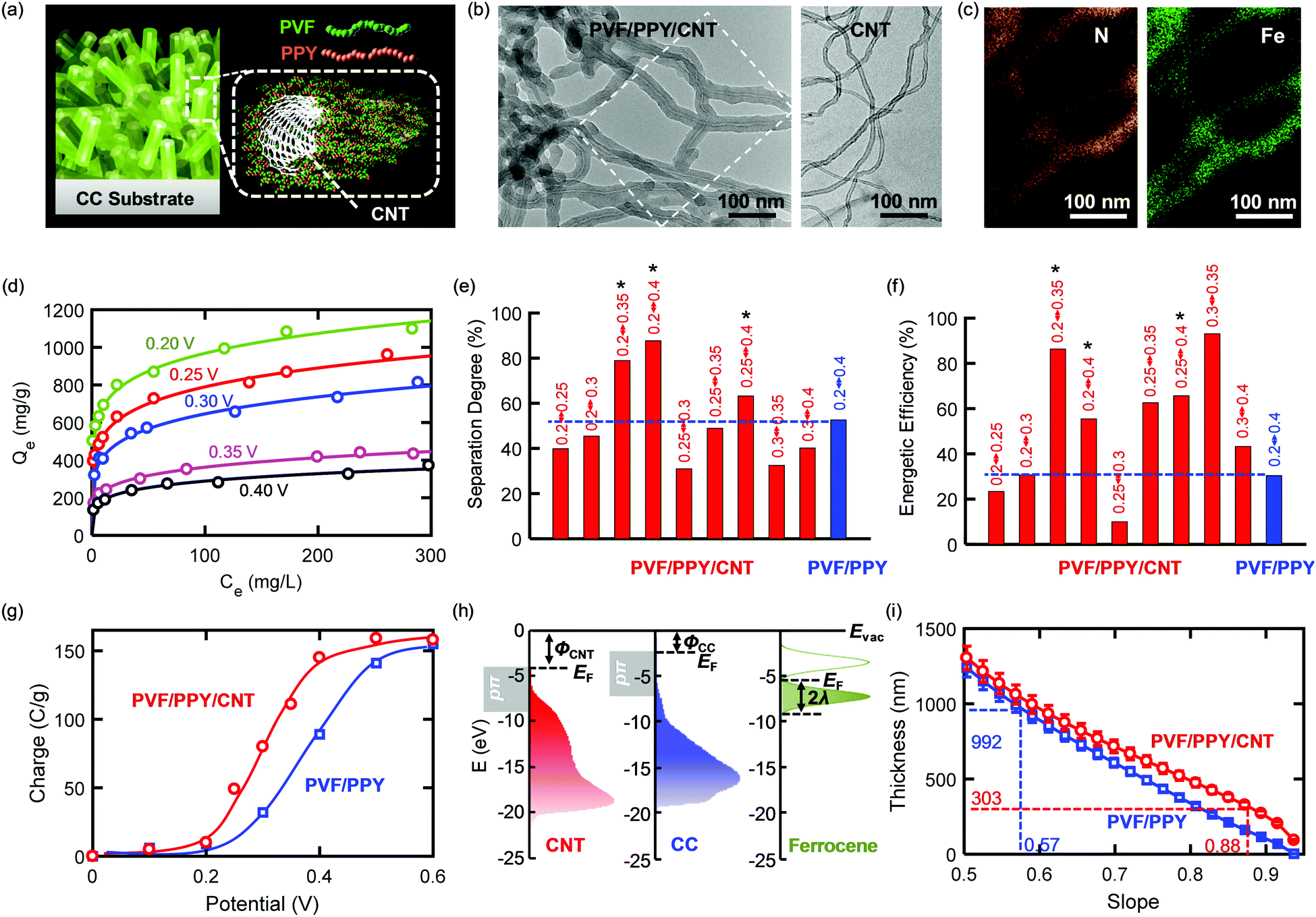 | ||
| Fig. 4 (a) Schematic illustration of the core-shell PVF/PPY/CNT ternary nanostructure deposited on a CC substrate. (b) TEM images of PVF/PPY/CNT and pristine CNT. (c) EDS elemental mapping of N and Fe for PVF/PPY/CNT for the area in (b) indicated by the dashed rectangle. (d) Adsorption isotherms of PVF/PPY/CNT obtained at different potentials from 0.2 to 0.4 V. (e and f) Comparison of separation degree (= (1 − ψf) × 100) (e) and energetic efficiency (η) (f) between PVF/PPY/CNT (red bar) and PVF/PPY (blue bar) for selected potential pairs (indicated by the numbers above each bar) at θ = 0.01 with the number of stages = 10. (g) Charge versus the applied potential for PVF/PPY and PVF/PPY/CNT. Solid lines are trend lines. (h) Energy diagram for CNT, CC and ferrocene. (i) The relationship between the polymer film thickness and the Siv value. | ||
The better ETAS performance with PVF/PPY/CNT than with PVF/PPY could be due to the following reasons. First, compared to PVF/PPY, PVF/PPY/CNT had higher adsorption capacities mainly due to its larger BET surface area (243 m2 g−1) than that of PVF/PPY (168 m2 g−1).27 The enhanced adsorption capabilities enabled a higher degree of separation during an ETAS operation. Second, incorporation of a conductive CNT core affected the electrochemical behavior of the key molecule with redox-tunable hydrophobicity (i.e., ferrocene), resulting in enhanced energetic efficiencies. The relationship between the charge passed upon oxidation from 0 V to the applied potential (Fig. 4g) suggests that ferrocene moieties were oxidized at lower potentials in PVF/PPY/CNT than in PVF/PPY. CV measurements (Fig. S15, ESI†) also confirmed a lower ensemble-averaged E0 of ferrocene in PVF/PPY/CNT (0.29 V) than in PVF/PPY (0.39 V). These results indicate that compared to PVF/PPY, PVF/PPY/CNT could be used with lower potentials to achieve the same extent of increase in hydrophilicity, yielding higher energetic efficiencies. It was found previously that ferrocene moieties embedded in thicker organic films or farther away from electrode surfaces were more difficult to oxidize, resulting in higher E0 values.35,36 In our case, with a uniform spatial distribution of ferrocene sites throughout the polymer film (as evidenced by EDS), a thicker film may have led to a larger population of ferrocene moieties that were either buried more deeply in an organic PPY environment or were located farther away from the conductive surfaces (either CC or CNT); hence a thicker polymer film should result in a higher ensemble-averaged E0 value. The thickness of a polymer film with uniformly distributed redox centers can be inferred from the slope (Siv) of a ln(CV peak current)–ln(scan rate) plot obtained with a redox polymer electrode (RPE) model to simulate current responses (Section S4,ESI†). PVF/PPY/CNT had a larger Siv value (0.88) than did PVF/PPY (0.57) (Fig. S16, ESI†), indicating that the former had a more surface-limiting charge transport behavior and thus a thinner polymer film. The RPE simulation requires a knowledge of the heterogeneous electron transfer kinetics at the polymer/electrode interface. As illustrated by the energy diagram (Fig. 4h), CNT had a larger valence band density of states (DOS) near the Fermi level (EF) compared to CC (pπ states, 0–5 eV downward from EF, measured by ultraviolet photoelectron spectroscopy). This larger DOS suggests that CNT could support faster electron transfer kinetics than CC. Gerischer-Marcus theoretical calculations (Section S5, ESI†) using the valence band DOS show that the use of CNT instead of CC as the electrode surface yielded a 5.4-fold increase in the standard electron transfer rate constant (k0ET). Incorporation of this difference in k0ET into the RPE simulation produced the Sivversus thickness relation (Fig. 4i), from which it can be estimated that PVF/PPY/CNT had a smaller average film thickness (303 ± 16 nm) than PVF/PPY (992 ± 48 nm). Additionally, electrochemical impedance spectroscopic measurements (Fig. S17, ESI†) show that compared to PVF/PPY, PVF/PPY/CNT has smaller charge-transfer resistance and solution resistance, indicating that the presence of conductive, nanosized CNTs may facilitate both electron transport and ion diffusion in the ternary hybrid system.
2.5 Simulation of multi-unit stop-flow operation
The cyclic separation experiment described above (Fig. 3a) was in effect a single-unit batch operation in which a single pair of electrodes was cycled between the feed and receiving solutions. For practical applications at large scale, such separations are usually implemented in a continuous or semi-continuous mode with multiple separation units. To examine the potential utility of ETAS at scale, we performed simulations of ETAS performance under multi-unit stop-flow operation using multiple pairs of electrodes. Experimental demonstration of such an operation is beyond the scope of this work. Such simulations would allow an assessment to be made of the effects of parameters such as receiving-to-feed volume ratio (i.e., θ), electrode mass per volume of feed (ξ), number of units (Nunit), etc., to optimize the overall design of a separation process for a given task. Moreover, we can now evaluate the potential benefits of exploiting counter-current operations, which are generally more effective than co-current operations.The multi-unit stop-flow operation simulated here is illustrated in Fig. 5a for a counter-current process. The co-current process is similar, and is shown in Fig. S1 (ESI†). Details of the simulation are discussed in Section S6 (ESI†). The simulation results with Nunit = 3 are shown in Fig. 5b and c for PVF/PPY adsorbents switching between 0.2 and 0.4 V, expressed in terms of the final concentrations in the feed and receiving phases normalized by the initial feed concentration (left axis; ψct and ψco are for counter-current and co-current operations, respectively) with three separation units for different θ and ξ values. The right axis of each panel shows the relative difference in concentration between the counter-current and co-current operations, defined as (ψct − ψco)/ψco × 100. Simulations with other Nunit values yield similar trends (Fig. S3, ESI†). The removal efficiency improves with larger electrode mass loadings, ξ. The closer the receiving solution volume is to the feed solution volume, the better is the removal effectiveness; this is related to the capacity of the receiving solution for the solute – a smaller volume will result in an increased concentration in this phase at any given stage, and thus a larger residual adsorbed solute on the regenerated electrodes.
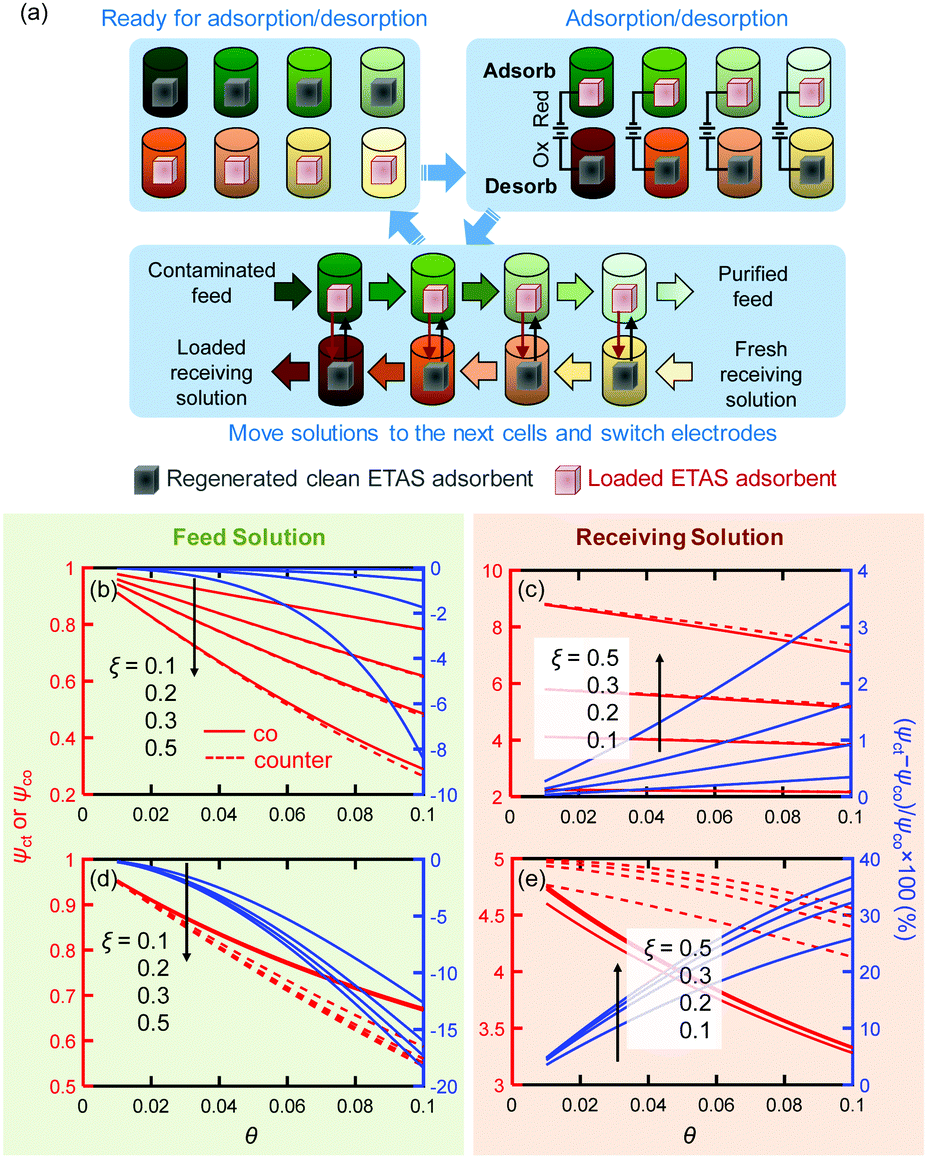 | ||
| Fig. 5 (a) Illustration of the multi-unit stop-flow operation in a counter-current fashion. A darker color of the solution in the cell indicates a higher pollutant concentration. (b–e) Simulated ψct or ψco values (left axis) with different θ and ξ for the nonlinear sorption isotherm case (b and c) and for the linear sorption isotherm case (d and e). Right axis: (ψct − ψco)/ψco × 100. | ||
An unexpected finding was that there appears to be little difference in performance under co- and counter-current conditions; usually counter-current operations are much more favorable for multi-unit processes. This is a reflection of the particular shapes of the isotherms, in which the difference in loadings (ΔQ) for the electrodes in the adsorption and desorption cells at any stage is almost independent of stage number (Fig. S4, ESI†). For other types of sorption isotherms, e.g., those following Henry's Law in which the adsorbed amount is directly proportional to the concentration in solution, ΔQ would be strongly concentration-dependent, and thus significant differences between the co-current and counter-current operations would be anticipated. To demonstrate this point, simulations were performed for an ETAS adsorbent having linear sorption isotherms Qe (mg g−1) = 100Ce (mg L−1) and Qr = 20Ce (mg L−1) for the reduced and oxidized states, respectively, with the same set of θ, Nunit and ξ values used for the nonlinear sorption isotherm case. It is evident that in the linear sorption isotherm case (Fig. 5d and e), the counter-current operation results in markedly improved separation effectiveness over the co-current case, with a lower final feed solution concentration and a higher final receiving solution concentration.
2.6 Generality of ETAS
Our ETAS strategy is applicable to a range of neutral or near-neutral organic water pollutants of varying molecular structure, hydrophobicity, and functionality, including pesticides, pharmaceuticals and carcinogenic aromatics. 2,4-Dichlorophenol (DCP), 2-naphthol (NT), and 1-naphthylamine (NA) are known carcinogenic pollutants, and intermediates for herbicide manufacturing and synthesis of industrial dyes.3,37 Bisphenol A (BA) and bisphenol S (BS) are anthropogenic pollutants that appear to be endocrine disruptors with long-term environmental persistence.38 Metolachlor (MC) is considered to be one of the leading pesticides responsible for groundwater contamination.39 Ethinyl estradiol (EE) and propranolol hydrochloride (PH) are widely used pharmaceuticals, with the former being a common contraceptive that has led to the collapse of fish populations,40 and the latter being a β-blocker for treatment of hypertension.41 Methyl orange (MO) and Rhodamine B (RB) are common industrial dyes identified as municipal water contaminants, particularly in developing countries.42,43Fig. 6a shows the Kd values of these organic pollutants measured at three different potentials (0.0, 0.3, 0.5 V) using PVF/PPY adsorbents. Pronounced Kd dependences on the applied potential for DCP, NT, NA, BA, BS, MC, EE and PH were observed, indicating that ETAS is suitable for the mitigation of these organic pollutants also.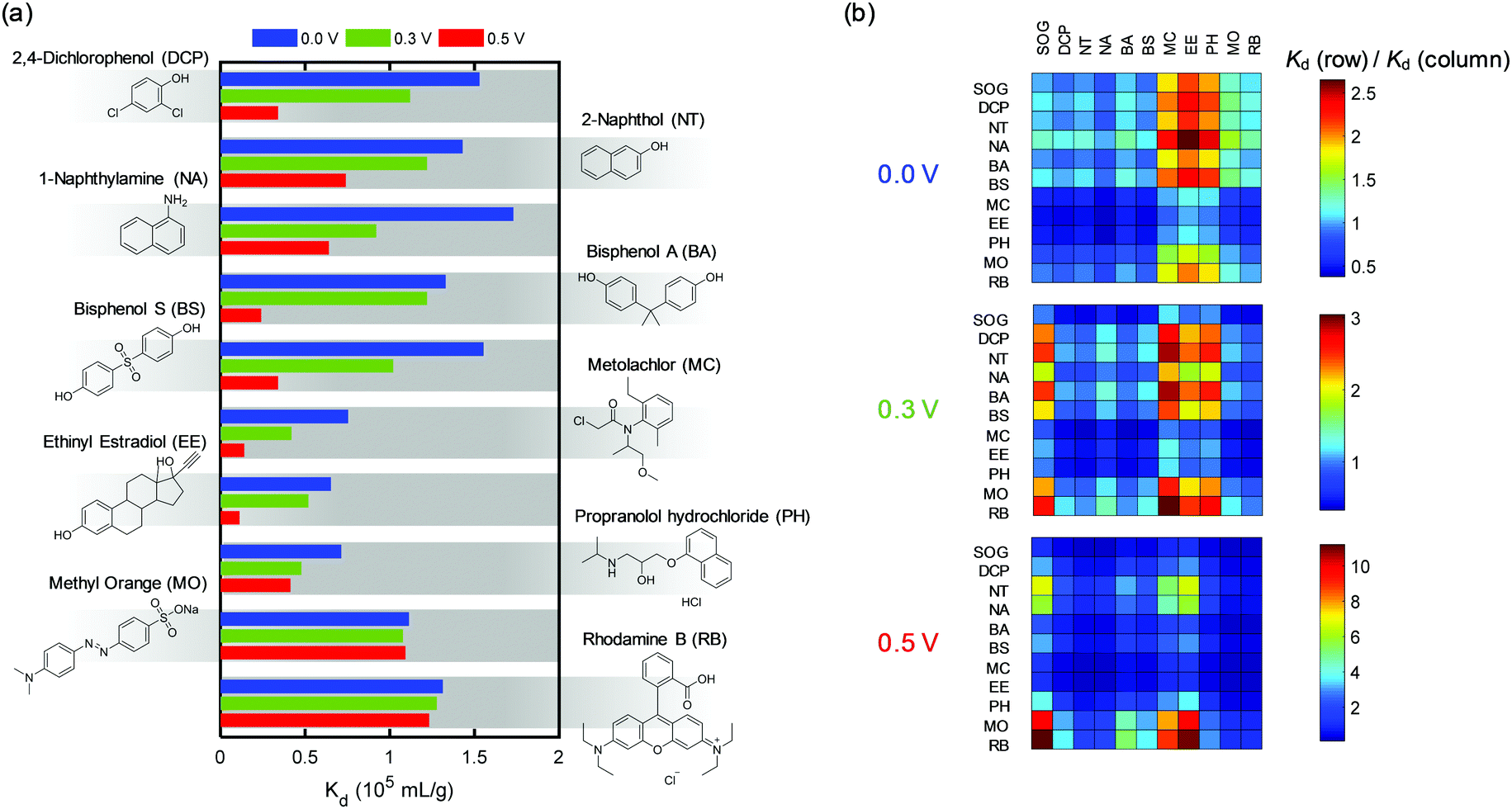 | ||
| Fig. 6 (a) Kd values for a range of uncharged organic pollutants obtained at 0.0, 0.3 and 0.5 V. (b) Heat maps of the ratio of Kd values between two different organics for 0.0, 0.3 and 0.5 V. | ||
The difference in Kd between different organics reflects the selectivity of the adsorbent towards them. Fig. 6b shows the heat map of the ratio of Kd for any two organics at three potentials. A ratio significantly different from unity indicates a high selectivity. Moreover, interestingly, the ratio and the spatial pattern of the heat map change with the potential. This indicates that the affinity of the ETAS adsorbent towards two competing organics can be tuned by electrical means, suggesting a novel strategy for selectivity control. Furthermore, since the potential of an electrode can be changed locally and independently, the overall selectivity of an ETAS adsorptive system composed of multiple electrodes can be modulated by applying different potentials to different electrodes (Fig. S18, ESI†).
3 Conclusions
Our study introduced a new separation concept, ETAS, that allows the use of finely-tuned, programmable electrical swings to remove neutral organics from water. We present a detailed quantitative analysis of ETAS that elucidates the relation between the energetic efficiency and the separation degree, providing valuable insights into the critical factors dictating ETAS performance. A materials design approach was demonstrated to improve the separation extent and energetic efficiency simultaneously during ETAS operation through the creation of a ternary heterogeneous nanostructure that consists of a π-electron-rich metallic CNT core and a binary polymer shell with redox-tunable hydrophobicity. We further examined the effects of operating mode and device configuration on the performance of ETAS, and demonstrated its effectiveness for a range of uncharged organic pollutants with varying chemical and structural characteristics, highlighting the practicality and broad applicability of ETAS. Additionally, we estimate that the energy consumption and cost per unit mass of organics removed by ETAS may be significantly less than those associated with conventional thermal swing adsorption/desorption technologies (Section S7, ESI†), indicating that ETAS could potentially be economically viable at large scale.Our work represents a potentially important step toward the development of controllable, energetically efficient, and environmentally friendly separation processes that target uncharged organics, which constitute the majority of industrial and municipal water contaminants. ETAS may offer a possible solution to the challenges associated with the water-energy nexus.44 The ETAS concept could be extended to a range of other judiciously devised redox-responsive adsorbent materials with varying chemical, structural and mechanical properties, allowing further interrogation of the fundamental physico-chemical behaviour of ETAS adsorbents. For instance, on-going work from our group develops an electrochemically responsive adsorbent based on surfactant-doped conducting polymers, in which the change in the surfactant orientation relative to the polymer backbone upon electrical stimulation is studied in detail with combined experimental and molecular modelling approaches.45 More broadly, the method proposed here for quantifying the energetic cost and the effect of operating conditions offers a useful, general framework for evaluating the overall efficacy of electrically modulated separation processes beyond ETAS, such as ion concentration polarization and capacitive deionization. Furthermore, the electrically responsive polymeric interfaces developed here could be used for molecular systems wherein the controllability of surface hydrophobicity is important, such as lubricants, biosensors, actuators, and drug delivery systems, with broad implications for diverse areas of inquiry, ranging from devising high-performance electrochemical systems to rationalizing fundamental electrostatic interactions in biologically relevant environments.
4 Experimental
Two types of ETAS adsorbents, carbon cloth (CC)-supported PVF/PPY and PVF/PPY/CNT, and two control samples, CC-supported PVF and PPY, were prepared by electrochemical deposition under different conditions (see Section S3, ESI†). Adsorption isotherms were obtained through adjustment of the concentration of organic pollutants in solution and measurement of the subsequent sorption over contact times of ∼12–15 h. The concentrations of organic pollutants were determined by UV-Vis spectroscopy or fluorescence spectroscopy. The masses of the polymer materials deposited on the carbon cloth substrates were determined by thermogravimetric analysis (TGA) with a final temperature of 900 °C with a heating rate of 5 °C min−1. Samples for TGA measurements were dried in a vacuum oven at 60 °C for 8 to 12 h. For the case of PVF/PPY/CNT ternary hybrid, the mass ratio between the polymer components and the CNT component were determined by TGA using free-standing PVF/PPY/CNT films peeled off stainless steel substrates. In the ETAS cyclic operation using a multi-stage batch process for the transfer of SOG from a feed solution to a receiving solution, the volumes of the feed solution and the receiving solution were 100 and 5 mL, respectively. At stage zero (i.e., before the ETAS cyclic operation), the SOG concentrations of the feed solution and the receiving solution were 5 and 0 mg L−1, respectively. During each stage, the PVF/PPY adsorbent with a nominal surface area of 1 cm2 and polymer mass of 0.12 mg at a selected potential was contacted with either the feed or the receiving solution for ∼5 h, which was sufficiently long to reach adsorption equilibrium for small quantities of adsorbents used in the multi-stage process.Conflicts of interest
There are no conflicts to declare.References
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Marinas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef PubMed.
- R. P. Schwarzenbach, B. I. Escher, K. Fenner, T. B. Hofstetter, C. A. Johnson, U. von Gunten and B. Wehrli, Science, 2006, 313, 1072–1077 CrossRef PubMed.
- A. Alsbaiee, B. J. Smith, L. L. Xiao, Y. H. Ling, D. E. Helbling and W. R. Dichtel, Nature, 2016, 529, 190–U146 CrossRef PubMed.
- F. Brandl, N. Bertrand, E. M. Lima and R. Langer, Nat. Commun., 2015, 6 Search PubMed.
- S. D. Richardson and S. Y. Kimura, Anal. Chem., 2016, 88, 546–582 CrossRef PubMed.
- K. E. Murray, S. M. Thomas and A. A. Bodour, Environ. Pollut., 2010, 158, 3462–3471 CrossRef PubMed.
- A. M. Soto and C. Sonnenschein, Nat. Rev. Endocrinol., 2010, 6, 364–371 Search PubMed.
- A. Nadal, Nat. Rev. Endocrinol., 2013, 9, 9–10 CrossRef PubMed.
- P. Alonso-Magdalena, I. Quesada and A. Nadal, Nat. Rev. Endocrinol., 2011, 7, 346–353 CrossRef PubMed.
- S. M. Rappaport and M. T. Smith, Science, 2010, 330, 460–461 CrossRef PubMed.
- R. W. Rousseau, Handbook of separation process technology, J. Wiley, New York, 1987 Search PubMed.
- J. R. Werber, C. O. Osuji and M. Elimelech, Nat. Rev. Mater., 2016, 1 Search PubMed.
- R. Y. Li, L. B. Zhang and P. Wang, Nanoscale, 2015, 7, 17167–17194 RSC.
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef PubMed.
- A. Doring, W. Birnbaum and D. Kuckling, Chem. Soc. Rev., 2013, 42, 7391–7420 RSC.
- M. Fujiwara and T. Imura, ACS Nano, 2015, 9, 5705–5712 CrossRef PubMed.
- Z. G. Xu, Y. Zhao, H. X. Wang, H. Zhou, C. X. Qin, X. G. Wang and T. Lin, ACS Appl. Mater. Interfaces, 2016, 8, 5661–5667 CrossRef PubMed.
- A. Akhoury, L. Bromberg and T. A. Hatton, ACS Appl. Mater. Interfaces, 2011, 3, 1167–1174 CrossRef PubMed.
- H. L. Che, M. Huo, L. Peng, T. Fang, N. Liu, L. Feng, Y. Wei and J. Y. Yuan, Angew. Chem., Int. Ed., 2015, 54, 8934–8938 CrossRef PubMed.
- M. E. Suss, S. Porada, X. Sun, P. M. Biesheuvel, J. Yoon and V. Presser, Energy Environ. Sci., 2015, 8, 2296–2319 RSC.
- X. Su, K. J. Tan, J. Elbert, C. Ruttiger, M. Gallei, T. F. Jamison and T. A. Hatton, Energy Environ. Sci., 2017, 10, 1272–1283 RSC.
- S. J. Kim, S. H. Ko, K. H. Kang and J. Han, Nat. Nanotechnol., 2010, 5, 297–301 CrossRef PubMed.
- Z. D. Wang, Y. T. Feng, X. G. Hao, W. Huang and X. S. Feng, J. Mater. Chem. A, 2014, 2, 10263–10272 RSC.
- S. De, J. G. Zhang, R. Luque and N. Yan, Energy Environ. Sci., 2016, 9, 3314–3347 RSC.
- W. D. Tian, X. W. Mao, P. Brown, G. C. Rutledge and T. A. Hatton, Adv. Funct. Mater., 2015, 25, 4803–4813 CrossRef.
- X. W. Mao, G. C. Rutledge and T. A. Hatton, Langmuir, 2013, 29, 9626–9634 CrossRef PubMed.
- X. W. Mao, E. H. Yan, G. C. Rutledge and T. A. Hatton, Chem. Mater., 2016, 28, 543–548 CrossRef.
- K. G. Zhou, D. McManus, E. Prestat, X. Zhong, Y. Y. Shin, H. L. Zhang, S. J. Haigh and C. Casiraghi, J. Mater. Chem. A, 2016, 4, 11666–11671 RSC.
- C. W. Abney, J. C. Gilhula, K. Lu and W. Lin, Adv. Mater., 2014, 26, 7993–7997 CrossRef PubMed.
- X. W. Mao, A. D. Liu, W. D. Tian, X. X. Wang, K. K. Gleason and T. A. Hatton, Adv. Funct. Mater., 2018, 28 Search PubMed.
- E. L. Cussler and B. K. Dutta, AIChE J., 2012, 58, 3825–3831 CrossRef.
- X. Mao, F. Simeon, D. S. Achilleos, G. C. Rutledge and T. A. Hatton, J. Mater. Chem. A, 2013, 1, 13120–13127 RSC.
- E. R. Dionne, T. Sultana, L. L. Norman, V. Toader and A. Badia, J. Am. Chem. Soc., 2013, 135, 17457–17468 CrossRef PubMed.
- G. K. Rowe and S. E. Creager, J. Phys. Chem., 1994, 98, 5500–5507 CrossRef.
- D. E. Latch, J. L. Packer, B. L. Stender, J. VanOverbeke, W. A. Arnold and K. McNeill, Environ. Toxicol. Chem., 2005, 24, 517–525 CrossRef PubMed.
- M. Ike, M. Y. Chen, E. Danzl, K. Sei and M. Fujita, Water Sci. Technol., 2006, 53, 153–159 CrossRef PubMed.
- J. Benner, D. E. Helbling, H. P. E. Kohler, J. Wittebol, E. Kaiser, C. Prasse, T. A. Ternes, C. N. Albers, J. Aamand, B. Horemans, D. Springael, E. Walravens and N. Boon, Water Res., 2013, 47, 5955–5976 CrossRef PubMed.
- K. A. Kidd, P. J. Blanchfield, K. H. Mills, V. P. Palace, R. E. Evans, J. M. Lazorchak and R. W. Flick, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8897–8901 CrossRef PubMed.
- M. S. Kostich, A. L. Batt and J. M. Lazorchak, Environ. Pollut., 2014, 184, 354–359 CrossRef PubMed.
- J. Guo, X. Chen, Y. Shi, Y. Q. Lan and C. Qin, PLoS One, 2015, 10 Search PubMed.
- R. G. Liu, X. Li, Y. Q. Li, P. F. Jin, W. Qin and J. Y. Qi, Biosens. Bioelectron., 2009, 25, 629–634 CrossRef PubMed.
- E. Karatairi and S. Darling, MRS Bull., 2018, 43, 404–405 CrossRef.
- Y. Ren, Z. Lin, X. Mao, W. Tian, T. Van Voorhis and T. A. Hatton, Adv. Funct. Mater., 2018 DOI:10.1002/adfm.201801466.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ee02000k |
| This journal is © The Royal Society of Chemistry 2018 |