
Monolithic heterojunction quasi-solid-state battery electrolytes based on thermodynamically immiscible dual phases†

Abstract
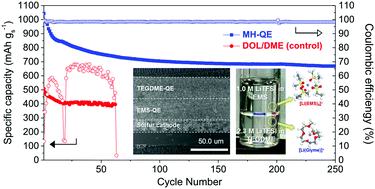
Traditional single-phase electrolytes, which are widely used in current state-of-the-art rechargeable batteries, have difficulties simultaneously fulfilling different chemical/electrochemical requirements of anodes and cathodes. Here, we demonstrate a new class of monolithic heterojunction quasi-solid-state electrolytes (MH-QEs) based on thermodynamically immiscible dual phases. As a proof-of-concept of the MH-QEs, their application to lithium–sulfur batteries is explored. Driven by combined effects of structural uniqueness and thermodynamic immiscibility, the electrode-customized MH-QEs provide exceptional electrochemical performance that lies far beyond those accessible with conventional battery electrolytes.
 Please wait while we load your content...
Please wait while we load your content...

