
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Role of cobalt–iron (oxy)hydroxide (CoFeOx) as oxygen evolution catalyst on hematite photoanodes†‡
Jifang
Zhang
 a,
Rodrigo
García-Rodríguez
b,
Petra
Cameron
b and
Salvador
Eslava
*a
a,
Rodrigo
García-Rodríguez
b,
Petra
Cameron
b and
Salvador
Eslava
*a
aDepartment of Chemical Engineering, University of Bath, Bath, BA2 7AY, UK. E-mail: S.Eslava@bath.ac.uk
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK
First published on 18th July 2018
Abstract
Photoelectrochemical solar water splitting into hydrogen and oxygen offers an elegant and potentially efficient way to store solar energy in the chemical bonds of hydrogen, but the oxygen evolution rate is quite limited. The deposition of an oxygen evolution catalyst on the photoanode can enhance oxygen evolution, although the precise interplay between the semiconductor and the catalyst remains poorly understood and unoptimized. In this work, we use a combination of electrochemical approaches, including photoelectrochemical impedance spectroscopy and intensity modulated photocurrent spectroscopy, to unravel the nature of the interactions between different loadings of an electrocatalyst (CoFeOx) and a hematite (α-Fe2O3) semiconductor. A thin layer of CoFeOx mainly reduces surface charge recombination, while an extremely thin layer enhances charge transfer kinetics. Moreover, an interlayer of GaOx modifies the surface state distribution and increases the charge transfer rate even further. These findings point to new opportunities for understanding and manipulating complex photoanodes for oxygen evolution.
Broader contextThe increase in world population and its ever-increasing energy demands have made the use of fossil fuels a prominent threat to the global environment. Hydrogen fuel offers a clean and sustainable alternative, but current methods of production by steam reforming of natural gas creates a large carbon footprint. Photoelectrolysis of water for the production of hydrogen (and oxygen) shows great promise and utilizes energy from sunlight. Unfortunately, the rate of water photoelectrolysis is considerably limited by the oxygen evolution reaction, which is a four electron charge transfer process that takes seconds. This is in stark contrast to far more rapid charge recombination processes taking place in the bulk (μs) or at the surface (ms) of the semiconductor. To alleviate bulk recombination, nanostructuring has proved to be effective. To reduce surface recombination, electrocatalysts are used to accelerate the oxygen evolution reaction. Although the outcomes of using electrocatalysts often appear encouraging, the underlying cause of improvements in surface kinetics still remains poorly understood. This paper aims to deepen this understanding by studying the photoelectrochemical response of hematite photoanodes coated with cobalt–iron (oxy)hydroxide layers of various thicknesses as well as the role of surface states. |
Introduction
Photoelectrochemical (PEC) solar water splitting is a promising way to sustainably produce hydrogen.1,2 The key to optimizing a PEC cell to achieve efficient water splitting for hydrogen and oxygen lies in the choice of materials and the design of photoelectrodes. In particular, developing efficient photoanodes for the water oxidation side has been a more challenging task due to the slow kinetics of the four-electron process (2H2O → 4H+ + O2 + 4e−). Several approaches have been followed to enhance the intrinsic properties of the semiconductor (SC) light-absorbing layers in photoanodes, for example, doping and surface treatment.3–5 In addition, the construction of heterojunctions to enhance electron–hole pair separation has been achieved using different semiconductors to form a cascade of band energy levels,6–8 or adding other materials to make use of specific electronic phenomena such as piezoelectric or ferroelectric polarization.9,10 Notably, passivation of the semiconductor absorbing layers by water oxidation electrocatalysts, also known as oxygen evolution catalysts (OECs) that are conventionally used for water electrolyzers, has shown to be a particularly effective method to improve the photocurrents in photoanodes.11Mixed metal (oxy)hydroxides are promising candidates to replace noble metal oxides (e.g. IrO2 and RuO2) operating in alkaline solutions.12,13 First-row transition metal (e.g. Mn, Fe, Co, Ni) oxides or hydroxides attract widespread attention due to their elemental abundance and simple preparation techniques, including hydrothermal growth, photodeposition and electrodeposition.14–21 Simulations have pointed out that binary or ternary (oxy)hydroxides composed of Fe, Co and Ni have the highest activities which relate to their optimized M–OH bond strengths.13,22,23 This is supported by measured high turnover frequencies (TOFs) and low overpotentials.21,24
The deposition conditions for OEC on a semiconductor absorbing layer to obtain a better performing photoanode requires additional consideration compared to depositing on a highly conductive substrate (e.g. Au). For example, some electrodeposition methods using a strong negative potential at pH < 6 conditions on a hematite (α-Fe2O3) layer can lead to deterioration or dissolution of the hematite.16–18,25 Moreover, the loading level must be relatively low to prevent parasitic light absorption.19 Some progress has been achieved using anodic electrodeposition,20,21 but the understanding of the interaction between the OEC and semiconductor is still rather limited. Nellist et al. has modelled and observed experimentally that the permeability of electrolyte ions in an OEC plays an important role in the resulting photocurrent of a photoelectrode.26,27 For example, in semiconductors with a high density of surface states, as in hematite, these surface states and OEC can be simultaneously charged during operation, which can increase surface recombination if the OEC is not very efficient.27
The idea of integrating OECs with hematite semiconductor absorbing layers for enhanced photocurrent attracts great attention.20,28–33 The results often demonstrate a considerable improvement in photocurrent, which then leads to the conclusion that OECs accelerate the sluggish kinetics of the water oxidation reaction. The charge transfer efficiency when OECs are applied is commonly calculated relative to the assumed unity charge transfer efficiency where a hole scavenger (Na2SO3 or H2O2) is added in the electrolyte solution.30,33,34 This method is a reasonable representation of the effectiveness of the OEC used. However, further insight can only be attained through more advanced techniques such as (photo)electrochemical impedance spectroscopy (PEIS), intensity modulated photocurrent spectroscopy (IMPS) and transient absorption spectroscopy (TAS).32–34 These techniques show that surface electron–hole recombination is the dominating factor that accounts for a limited photocurrent, instead of a limited charge transfer rate. However, much more work is required for a clear insight of the interplay between OECs and semiconductor layers in photoanodes.
In this work, we study the role of cobalt–iron (oxy)hydroxide (CoFeOx, or cfox) as an OEC on mesoporous hematite (h) photoanodes. We use multiple electrochemical techniques to investigate h/CoFeOx composite photoanodes at varied applied voltages and different OEC loadings. A thin OEC layer leads to a cathodic shift in the onset potential due to inhibition of surface recombination and OEC charging but not due to a higher charge transfer rate to the electrolyte. However, we reveal that an extremely thin OEC layer achieves a higher hole transfer rate. We also show that the charge transfer process is further accelerated at low potentials with the assistance of an interlayer of GaOx that modifies the distribution of surface states.
Experimental
Preparation of photoanodes
Hematite films were prepared by a facile solution-based method. First, 2.16 g Pluronic 123 (P123, average Mn ∼ 5800) was dissolved in 6 g tetrahydrofuran (THF, Fisher Chemicals, 99.99%). In a separate vial, 6.06 g Fe(NO3)3·9H2O (Alfa Aesar, 98%) was dissolved in 6 g absolute ethanol (BDH Prolabo). The two solutions were mixed and stirred overnight. This precursor was then spin-coated onto fluorine-doped tin oxide (FTO) coated aluminoborosilicate glass (Solaronix, CH). The glass slides were previously cleaned by sonication in 2 vol% Hellmanex solution, 2-propanol and acetone, for 10 min each, sequentially. The spin coating was carried out at 1000 rpm for 5 s before ramping up to 6000 rpm and kept at this velocity for 30 s. The films were then calcined in air at 800 °C for 20 min in a preheated tube furnace. The spin coating and calcination were carried out twice to obtain sufficient thickness. The hematite films were then masked with black electric tape leaving a square area of 0.25 cm2 for PEC measurements.The loading method of CoFeOx was adapted from a previous study by Morales-Guio, using electrodeposition in a three-electrode system.21 In the present study, a Pt wire was used as a counter electrode coupled with an Hg/HgO/1 M NaOH reference electrode. The electrodeposition electrolyte was composed of 10 mM FeCl3·6H2O (Sigma Aldrich, 99+%), 16 mM CoCl2 (Alfa Aesar, anhydrous, 97%) and 0.1 M NaOAc (Sigma, 99%), dissolved in deionized water, without adjusting its pH. CoFeOx was coated with this electrolyte by positively sweeping voltage from 1.35 to 1.65 VRHE. The unidirectional linear sweeps were repeated for a controlled thickness. The sweeps were carried out three times for an extremely thin coating and up to thirty times for a standard thin coating. The bare hematite and 3–30 times CoFeOx-coated hematite photoanodes are denoted as h/cfox0, h/cfox3, h/cfox9, h/cfox18, and h/cfox30, respectively. An Ivium Compactstat potentiostat was used for all electrodepositions. GaOx layer between hematite and CoFeOx layers was fabricated by following Hisatomi's procedure.38 Briefly, hematite films were partly submerged into an aqueous solution containing 0.042 g Ga(NO3)3·nH2O, where 0.6 g urea was slowly added and subsequently stirred at 75 °C for 15 min. The films were then rinsed with DI water and calcined at 500 °C for 2 h, before CoFeOx deposition.
Physical characterization
X-ray diffraction (XRD) patterns were collected using a BRUKER AXS D8 advance diffractometer with a Vantec-1 detector and Cu Kα radiation (1.5418 Å). Film morphologies were examined by field emission scanning electron microscopy (FESEM, JEOL JSM-6301F) with an acceleration voltage of 5 keV. X-ray photoelectron spectroscopy (XPS) was performed on a Thermo Fisher Scientific K-alpha+ spectrometer. Samples were analyzed using a micro-focused monochromatic Al X-ray source (72 W) over an area of approximately 400 microns. Data was recorded at pass energies of 150 eV for survey scans and 40 eV for high resolution scans with 1 and 0.1 eV step sizes respectively. Charge neutralization of the sample was achieved using a combination of both low energy electrons and argon ions. No sputtering was carried out. Data analysis was performed in CasaXPS (2.3.19) using a Shirley type background and Scofield cross sections, with an energy dependence of −0.6. High-resolution transmission electron microscopy (HR-TEM) was used to examine the nanoparticles scraped from photoanode samples at 200 keV of electron beam energy (JEOL 2100 Plus).(Photo)electrochemical characterization
Photocurrent density measurements were carried out in the same setup used for electrodeposition of CoFeOx, replacing the electrolyte solution with 1 M NaOH (pH 13.4). Photocurrent densities were measured under chopped or continuous illumination of 100 mW cm−2 simulated sunlight (AM 1.5G) from the back side (glass side) using linear sweep voltammetry (LSV) at various scanning rates (5, 20 and 50 mV s−1) or using chronoamperometry. Incident photon-to-current efficiency (IPCE) measurements were performed from 300 to 700 nm with the same light source passing a monochromator (MSH-300F LOT QuantumDesign) without the AM 1.5G filter. The intensity of monochromatic light was calibrated by a SEL033/U photodetector (International Light Technologies). Transient photocurrent spectroscopy (TP) was carried out in the same PEC setup with a data acquisition interval of 1 ms and chopped simulated sunlight. PEIS was carried out in a frequency range from 105 to 0.1 Hz, with an AC voltage amplitude of 10 mV. Impedance spectra were obtained in the range from 0.6 to 1.2 VRHE, with 0.05 V steps, in 1 M NaOH, and under 1 sun irradiation unless otherwise specified. IMPS was conducted with a ModuLab XM PhotoEchem system (Solartron Analytical) under 470 nm LED (Thorlab M470L3) illumination (37.5 mW cm−2) at varying potentials from 0.6 to 1.3 VRHE at a step of 0.05 V. A modulation of 10% in light intensity was applied, over a frequency range from 103 to 0.1 Hz at each potential step. PEIS and IMPS spectra were fitted using Zview software (Scribner).Results and discussion
The facile photoanode preparation method used here produced high quality hematite films. The hematite phase was identified by XRD (Fig. S1, ESI‡). These photoanodes have a mesoporous worm-like morphology (Fig. S2, ESI‡), with feature sizes of 90 ± 19 nm (analyzed using ImageJ software). Thickness was approx. 1 μm.CoFeOx was deposited onto the hematite layer using from three up to thirty unidirectional LSV sweeps, for controlled thickness. As shown in Fig. 1, the photocurrent densities at high potential reach the highest values after three sweeps (h/cfox3). The photocurrent density then gradually decreases for heavier loading. Low potential photocurrent densities apparently improve with higher deposition repetitions. To understand the differences in PEC performance, we examined the properties of three representative photoanodes with none, three and thirty coating sweeps (h/cfox0, h/cfox3 and h/cfox30).
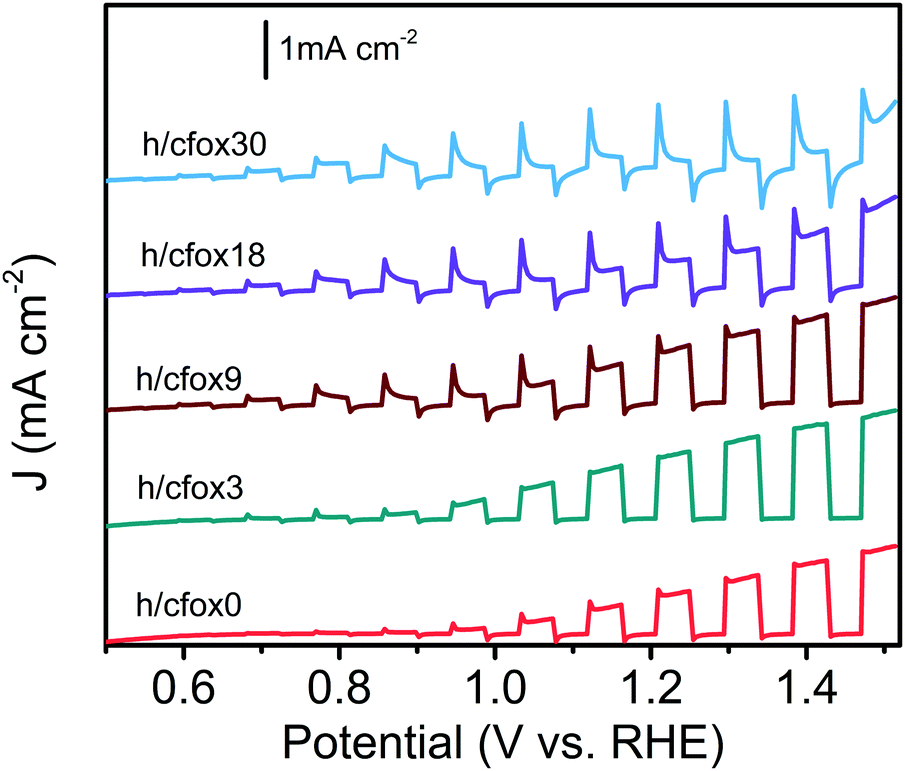 | ||
| Fig. 1 Linear sweep voltammograms of hematite photoanodes with 0–30 times CoFeOx loadings: h/cfox0 (red), h/cfox3 (green), and h/cfox9 (brown), h/cfox18 (purple) and h/cfox30 (blue). Measured under chopped AM 1.5G (100 mW cm−2) illumination at a scan rate of 20 mV s−1. | ||
The OEC on both h/cfox3 and h/cfox30 is very thin and parasitic light absorption is not observable. No apparent morphological changes in the hematite layer can be seen using FESEM even after thirty coating sweeps (Fig. S2, ESI‡). Successful electrodeposition of CoFeOx is observed however in HR-TEM (Fig. 2). The sample h/cfox0 shows hematite crystals with well-defined crystalline edges (Fig. 2a and b). The sample h/cfox3 shows hematite crystals with an extremely thin amorphous layer of ca. 0.7 nm, not ubiquitously covering all the hematite crystals (Fig. 2c and d). The sample h/cfox30 shows a highly uniform amorphous layer of 1.6 nm covering all the crystals (Fig. 2e and f). According to the current density maxima for each sweep during electrodeposition, the loading on h/cfox30 nearly approaches saturation, which corresponds to nearly 20 μg cm−2 (Fig. S3, ESI‡).21 This is comparable to or thinner than most OEC coatings in literature.16,39–41
 | ||
| Fig. 2 HR-TEM images of photoanodes h/cfox0 (a and b), h/cfox3 (c and d) and h/cfox30 (e and f). Scale bars represent 10 nm for left column and 5 nm for right column. | ||
CoFeOx deposition with thirty coating sweeps is also confirmed by characterizing the top surface of h/cfox30 with XPS and observing Co 2p peaks (Fig. 3a). A broad peak present between 775 and 795 eV in all photoanodes’ XPS spectra is ascribed to Fe LMM Auger lines.42 CoFeOx deposition with three coating sweeps (h/cfox3) is not confirmed by XPS on the top surface of h/cfox3, but confirmed following a direct deposition on solid FTO (Fig. 3b). Therefore, CoFeOx deposition on porous hematite layers must start closer to the FTO substrate, due to the gradient of potential across the porous hematite layer that requires multiple coating sweeps to cover the top surface with CoFeOx. CoFeOx loading in FTO/cfox3 is approximately 20% of that in FTO/cfox30, based on the peak areas of Co 2p (Fig. 2b). Similar CoFeOx ratio between h/cfox3 and h/cfox30 is expected.
 | ||
| Fig. 3 (a) Co 2p XPS spectra of h/cfox0 (red), h/cfox3 (green), and h/cfox30 (blue). The broad peak present at 785 eV is ascribed to Fe LMM Auger lines.42 (b) Co 2p XPS spectra (background subtracted) of CoFeOx coated three (green) or thirty times (blue) on FTO coated glass (FTO/cfox3 and FTO/cfox30, respectively). | ||
The photocurrent densities of these photoelectrodes were initially measured by LSV at 20 mV s−1 (Fig. 4a). The uncoated hematite sample (h/cfox0) shows a photocurrent density of 0.88 mA cm−2 at 1.23 VRHE with an onset potential of ca. 0.8 VRHE. When three layers of CoFeOx are coated (h/cfox3), the photocurrent density increases at all potentials, for example from 0.88 to 1.2 mA cm−2 at 1.23 VRHE. However, the onset potential has little shift. When thirty layers of CoFeOx are coated (h/cfox30), the photocurrent only increases at low potentials and there is a cathodic shift of the onset potential to approximately 0.6 VRHE. It also displays a high dark current above 1.1 VRHE and a strong peak centered at 1.18 VRHE. The IPCE of h/cfox0, h/cfox3 and h/cfox30 are shown in Fig. S4 (ESI‡) with similar projected photocurrent densities to the measured by LSV.
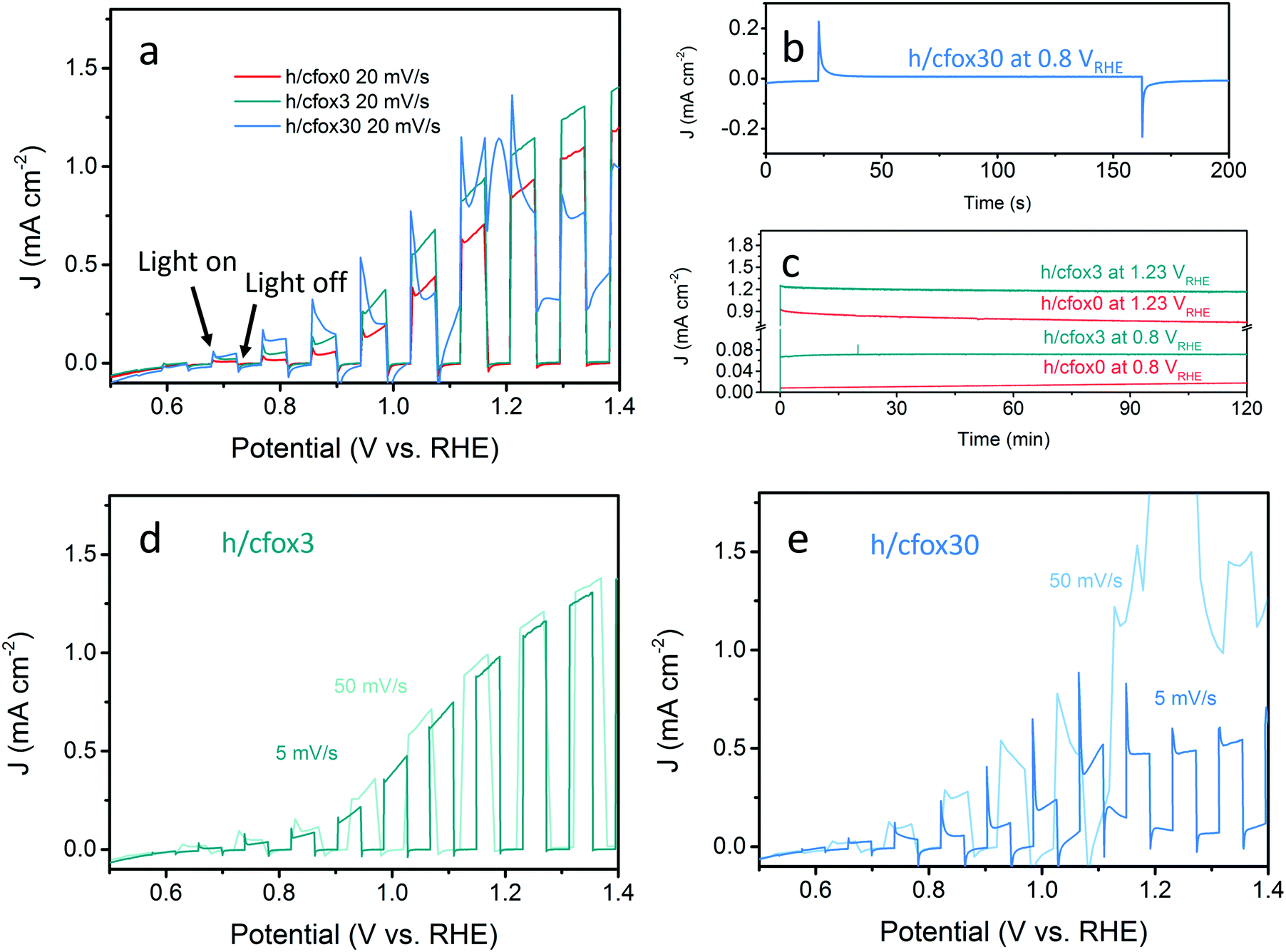 | ||
| Fig. 4 (a) Linear sweep voltammograms of photoanodes h/cfox0 (red), h/cfox3 (green), and h/cfox30 (blue) under chopped AM 1.5G (100 mW cm−2) illumination at a scan rate of 20 mV s−1. (b) Chronoamperometry of sample h/cfox30 showing its short-term stability. (c) 2 h stability tests for samples h/cfox0 (red line) and h/cfox3 (green line) at constant potentials of 0.8 and 1.23 VRHE. Linear sweep voltammograms of (d) h/cfox3 and (e) h/cfox30 under chopped light scanned at 5 mV s−1 (darker line) and 50 mV s−1 (lighter line). | ||
A short chronoamperometry test on h/cfox30 indicates that the enhancement at low potential (0.8 VRHE) is totally lost within 10 s, after which it stabilizes at 8 μA cm−2, a value almost identical to that on h/cfox0 at the same potential (Fig. 4b). In contrast, both h/cfox0 and h/cfox3 show remarkable stability over 2 h, and the significant improvement in h/cfox3 photocurrent over h/cfox0 is well maintained (Fig. 4c). We also performed LSV measurements at slower (5 mV s−1) and faster (50 mV s−1) scan rates. For h/cfox3, the J–V curves are consistent and independent of scanning rates (Fig. 4d). However, for h/cfox30, the current densities are highly dependent on the scanning rate (Fig. 4e). As scan rate increases, the intensity of the peak located near 1.18 VRHE increases roughly linearly (Table 1), which implies a surface immobilized redox reaction. Moreover, there is a anodic shift of the peak with increasing scan rate, indicating a potential driven process. Another feature to notice is that the position of the first photocurrent spikes are also dependent on the scan rate. The first relevant photocurrent spike is at 0.75, 0.85, and 0.92 VRHE for scan rates 5, 20 and 50 mV s−1, respectively. All these features are ascribed to the likely oxidation of CoFeOx from its hydroxide form to oxyhydroxide form [Co(OH)2 + OH− → CoOOH + H2O + e−].43 This oxidation appears to start at low potentials driven by photo-generated holes (during irradiation), and continues at higher potentials driven by both the applied potential and more photo-generated holes.
| Scan rate (mV s−1) | Peak intensity (mA cm−2) | Peak position (VRHE) |
|---|---|---|
| 5 | 0.2 | 1.12 |
| 20 | 1.2 | 1.18 |
| 50 | 3.0 | 1.23 |
We start our investigation by PEIS. A representative Nyquist plot (Fig. 5a) for a bare hematite photoanode (h/cfox0) contains two semicircles that can be fitted using a two-RC-unit equivalent circuit, as proposed by Klahr et al., where the oxygen evolution reaction (OER) is assumed to be driven by surface states.44 In this equivalent circuit, three resistances are used: a series resistance attributed to the electrolyte and conductive substrate layer, Rs; a trapping resistance at surface states where electron–hole pairs recombine, Rtrap; and a charge transfer resistance at the semiconductor–liquid junction, Rct (Fig. 5a inset). There are two capacitors used: a bulk capacitor mainly attributed to the space charge region, Cbulk, and a surface states capacitor, Css.
 | ||
| Fig. 5 (a) Nyquist plot for a typical PEIS measurement of h/cfox0 at 1.0 VRHE under 1 sun irradiation (AM 1.5G). Inset image shows the equivalent circuit used. HF and LF indicate high frequency and low frequency semicircles, respectively. (b) Fitting results of sample h/cfox0 as a function of applied potential. Black circles represent Rct; red squares represent Css; and green curve represents J–V curve (under the same irradiation condition). | ||
The PEIS spectra of h/cfox0 and h/cfox3 are similar (Fig. S5, ESI‡), so they are modelled using the same equivalent circuit depicted in the inset of Fig. 3a. However, the PEIS spectra of h/cfox30 shows different features (Fig. S6, ESI‡). Below 1.0 VRHE, the h/cfox30 PEIS spectra are akin to those observed for h/cfox0 and h/cfox3, so they are modelled using the same equivalent circuit, while the term Css is replaced with Ccat because charge transfer in h/cfox30 must take place mainly through the OEC, as suggested by Boettcher and Bisquert.40,45 Above 1.0 VRHE, there is an additional peak in the phase angle at low frequencies in the Bode plots, i.e. a third semicircle in Nyquist plots (Fig. S6, ESI‡) which requires another RC unit in its equivalent circuit (Fig. S7, ESI‡). These extra features observed above 1.0 VRHE are also observed in dark EIS measurements above 1.0 VRHE, coinciding with the peak onset in J–V curves (Fig. S8, ESI‡), which can be assigned to the oxidation of CoFeOx.39 Consequently, for this extra RC unit, the capacitance (Ccfox) represents the main pseudocapacitance from the redox reaction and the resistance (Rcfox) the ion diffusion during electrolyte permeation.46 Fitted parameters for h/cfox30 and the rest of the photoanodes studied are listed in Tables S1–S6 (ESI‡).
The PEIS spectra of our three representative electrodes (h/cfox0, h/cfox3, and h/cfox30) have good fit to the equivalent circuits and no constant phase elements are necessary. To confirm the validity of PEIS measurements and fittings, the total resistance (Rtot) for each sample is plotted and compared against differential resistance (dV/dJ) obtained from LSV curves; the Rtot of all three samples match the curves reasonably well (Fig. S9, ESI‡).
Fig. 5b shows Rct and Css fitted from PEIS and photocurrent measured at different potentials for h/cfox0. The presence of surface states, reported in literature for hematite,44,47,48 is confirmed with Css showing a Gaussian distribution centered at 0.95 V. This Gaussian distribution results from activation of OER intermediate species and appears close to the onset potential.49Rct also shows a local maximum at 0.7 VRHE. We assign the early Rct bending to accumulation of electrons at the electrode surface near the flat band potential (Efb),50 which is estimated to be ∼0.6 VRHE using Mott–Schottky equation (Note S1, ESI‡). To avoid this accumulation effect, further impedance results are analyzed from 0.7 VRHE.
Fig. 6 shows Rct and Css or Ccat at different potentials for the three representative hematite photoanodes (h/cfox0, h/cfox3, and h/cfox30). For h/cfox0 and h/cfox3, Rct–V curves show the same behavior from 0.7 to 1.2 VRHE, but h/cfox3 has generally lower values than h/cfox0 below 1.05 VRHE, suggesting easier charge transfer. The Gaussian distribution of Css for h/cfox3 is depressed due to partial replacement of sluggish surface states with active CoFeOx sites.
 | ||
| Fig. 6 (a) Rct and (b) Css, Ccat obtained from EIS fitting as a function of applied potential for h/cfox0 (red squares), h/cfox3 (green circles) and h/cfox30 (blue diamonds). | ||
The Ccat–V curve of h/cfox30 is fundamentally different to the one of h/cfox3 (Fig. 6b). The curve shape observed for h/cfox30 is similar to that obtained for a NiFeOx/Fe2O3 photoanode.27 The high and flat region of Ccat for h/cfox30 at lower potential indicates that almost all photogenerated holes are transferred to the CoFeOx layer as opposed to h/cfox3 where the loading is so low that only a small fraction of photogenerated holes is sufficient to oxidize the OEC. Rct of h/cfox30 decreases dramatically with potential. At voltages below 1.0 VRHE, Rct decreases as photogenerated holes transform hydroxides into oxyhydroxides, which is known to be a more effective OEC.51 Above 1.0 VRHE, this process is further accelerated with the assistance of applied voltage. The significant decrease in Rct of h/cfox30 above 1.0 VRHE is, nevertheless, not accompanied by an improvement in photocurrent. Therefore, EIS data must be analyzed from a kinetic perspective.
In a simplified model, a hematite photoanode surface has two competing processes that determine the rate of water oxidation, namely charge transfer and surface recombination.47 Its Nyquist plot typically exhibits two semicircles at different frequency domains [high frequency (HF) and low frequency (LF)] as shown in Fig. 5a. The rate constants of these two processes (kct for charge transfer and krec for surface recombination) can be calculated using a phenomenological model developed by Peter and co-workers.47,52 The formal equivalence of this kinetic model and the EIS elements used previously is demonstrated in Note S2 of ESI.‡
Assuming the space charge capacitance of the semiconductor is much smaller than the capacitance across the Helmholtz layer at electrode surface (CSC ≪ CH), kct is inversely related to the time constant of the low frequency semicircle:53
 | (1) |
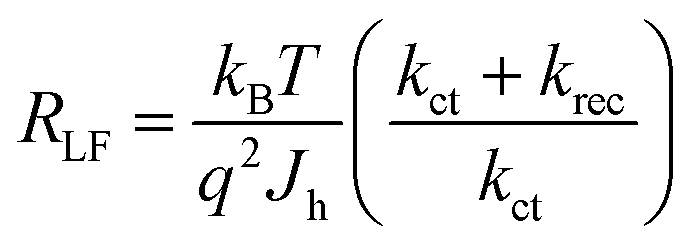 | (2) |
 | (3) |
The resistance of the high frequency semicircle (RHF) is Rtrap and can be calculated as:
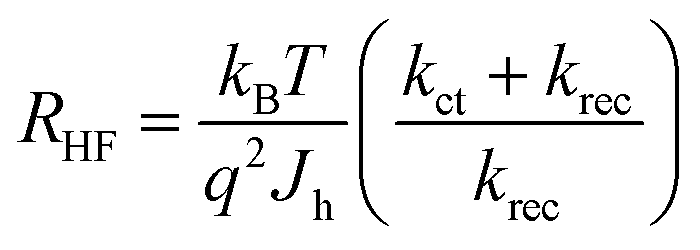 | (4) |
 | (5) |
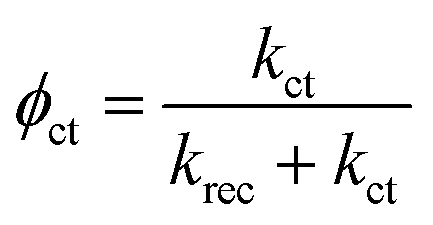 | (6) |
| Jest. = Jhϕct | (7) |
The kinetic results estimated from eqn (1)–(7) are plotted in Fig. 7. All measurements, where possible, are carried out on a single substrate for better comparability. According to Peter's model, at a high concentration of positively charged surface states, kct first increases then saturates as limited by light intensity. In our case, the saturation point is not reached possibly because of strong light intensity (100 mW cm−2).52 In contrast to the reported model that predicts a plummet of krec, our photoanodes show nearly constant values. Such unusual behavior is, again, an indication of strong light intensity that induces complete Fermi level pinning, where the band edge is unpinned and the degree of band bending is constant.54 We also measured the impedance response under a weaker light intensity of 10 mW cm−2 and kct was indeed constant while krec decreased with potential (Fig. S10, ESI‡), thereby proving that total Fermi level pinning happens under strong illumination which creates a high density of surface states. For our hematite photoanode, the extraordinary trend of increased krec (Fig. S11, ESI‡) above 1.0 VRHE suggests a fundamental change to the semiconductor. One explanation is the formation of a deep depletion region where the semiconductor surface behaves like an insulator, based on the observation that Css starts to decrease at 0.95 VRHE, and intensified band bending.55 However, this explanation seems unlikely considering the strong Fermi level pinning effect. Another possible cause is a reversible modification of the surface states under strong illumination and high potential.56 Under this circumstance, water oxidation mechanism is different and the kinetic model loses its continuity. As such, our kinetic analysis only considers potentials up to 1.0 VRHE for simplicity.
 | ||
| Fig. 7 Calculated (a) surface recombination and (b) charge transfer rates for photoanodes h/cfox0 (red squares), h/cfox3 (green circles) and h/cfox30 (blue diamonds) from PEIS. The estimated photocurrent densities are compared with measured J–V curves in (c). | ||
To justify the applicability of this model for our photoanodes at relatively low potentials, the measured photocurrent densities were compared to estimated ones calculated using charge transfer efficiencies and hole fluxes (Fig. 7c and Fig. S12, ESI‡). Despite lower values, the estimated photocurrent densities follow the same trends of real J–V curves measured at 5 mV s−1, showing that this model is at least useful to compare the trends of rate constants.
Fig. 7a and b show that both krec and kct of h/cfox3 increase with respect to h/cfox0 at all potentials, especially kct. Thus, the estimated photocurrent (Jest.) increases (Fig. 7c). The marginally increased krec could be a result of interphase charge trapping or fitting error.57 It should be noted that the moderate increase in krec below 0.8 VRHE is unexpected but is a result of potential before photocurrent onset and therefore can be ignored. In contrast, both krec and kct of h/cfox30 have lower values. The higher estimated photocurrent density for h/cfox30 is mainly ascribed to the significantly reduced krec and subsequently improved charge transfer efficiency, ϕct (Fig. S12a, ESI‡). However, kct increases much less with potential than lightly or even uncoated photoanodes especially above 0.85 VRHE. The photocurrent thus falls behind the other two despite the subdued recombination. Therefore, the effect of CoFeOx is highly dependent on its thickness. These data, combined with chronoamperometry results shown previously, indicate that the characteristics of lowered onset potential and depressed photocurrent at high potential on h/cfox30 are associated with the relatively high thickness of CoFeOx. In this situation, the OEC undergoes oxidation, stores charges and influences the photocurrent measured.
To confirm this hypothesis, transient photocurrents (TP) were investigated by converting it to a normalized parameter D (Fig. 8), which can be calculated as:58
| D = (Jt − Jst)/(Jin − Jst) | (8) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D = −1. We then approximate τ as:53
D = −1. We then approximate τ as:53| τ = (krec + kct)−1 | (9) |
 | ||
| Fig. 8 (a) Photocurrent transients of photoanodes h/cfox0 (red), h/cfox3 (green) and h/cfox30 (blue) measured at 0.8 VRHE. (b) ln(D) as a function of time for h/cfox0 (red), h/cfox3 (green) and h/cfox30 (blue). Phase 1 and 2 represent surface charge recombination and OEC charging, respectively. | ||
The ratio of Jst and Jin is given by
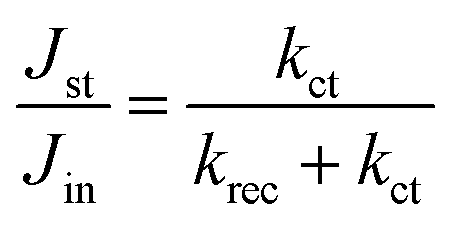 | (10) |
Thus, kct and krec can be estimated from photocurrent transients. We compare both rate constants using this simple method with those obtained from PEIS in Table 2. TP and PEIS methods produce good agreement overall except for kct of h/cfox30, where PEIS gives a value nearly one magnitude higher than TP. The cause of such difference can be found in lnD–t curves measured at 0.8 VRHE. The shapes of these curves resemble TAS results (Fig. 8b).37 For h/cfox30, two decay phases can be distinguished as opposed to only one for h/cfox0 and h/cfox3. The high frequency decay from 1 ms (recording limit) to about 50 ms is assigned to surface charge recombination (Phase 1 of Fig. 8b).37 It is clear that CoFeOx in h/cfox30 effectively slows down this decay rate. The second decay stage in h/cfox30 is associated with the retention of photocurrent because of charging of CoFeOx, which indicates this interfacial charge transfer from the semiconductor to the OEC is more rapid than the water oxidation (Phase 2 of Fig. 8b). In Table 2, charge transfer efficiencies calculated from rate constants by PEIS and TP (using eqn (6)) are compared with values obtained from the ratio of photocurrent densities measured in NaOH without and with a hole scavenger H2O2 (0.5 M). Notably, ϕ(H2O2) are much lower compared to TP and PEIS methods. The differences can be understood by a stronger degree of band bending when H2O2 is present, where recombination at space charge region is minimized, hence giving a more accurate estimation of maximum photocurrent density. This effect is more pronounced in h/cfox0 due to its slower kinetics. PEIS and TP give similar results for h/cfox0 and h/cfox3 whereas TP has a more accurate approximation for h/cfox30. Therefore, we believe that the overestimation of kct with PEIS is a result of the AC environment during measurements which takes charging current of CoFeOx in h/cfox30 for water oxidation current. This analysis confirms that the higher photocurrent density is at least partly a result of OEC charging.
| Method | h/cfox0 | h/cfox3 | h/cfox30 | |||
|---|---|---|---|---|---|---|
| TP | EIS | TP | EIS | TP | EIS | |
| k ct (s−1) | 1.95 | 1.62 | 2.62 | 3.82 | 0.26 | 2.40 |
| k rec (s−1) | 39.72 | 46.71 | 35.84 | 65.04 | 12.09 | 18.92 |
| ϕ(est.) | 4.7% | 3.4% | 6.8% | 5.5% | 2.1% | 11.3% |
| ϕ(H2O2) | 0.6% | 3.7% | 0.6% | |||
IMPS was also carried out to complement PEIS and TP outcomes. The theory behind IMPS is briefly introduced in Note S3 (ESI‡) and more thoroughly explained elsewhere.36,54 IMPS applies small perturbations of light intensity at a fixed potential and probes the photocurrent response from the PEC system. In contrast to PEIS, the redox reaction of CoFeOx by external voltage perturbation is avoided, which allows us to see if photo-generated holes are able to oxidize the OEC.
The complex IMPS plots of h/cfox0 and h/cfox3 show well defined low frequency semicircles at all potentials with no apparent flattening (Fig. 9a, with full dataset of IMPS complex available in Fig. S13, ESI‡). The smaller low frequency semicircle of h/cfox3 clearly shows better charge transfer efficiency at high potentials. On the other hand, h/cfox30 shows distinct characteristics at 0.85 VRHE. Two semicircles can be distinguished in the first quadrant, with the lower frequency part overlapping that of h/cfox0 and h/cfox3 and hence we attribute it to water oxidation. The appearance of another semicircle is indicative of an additional PEC process. Here, the only possible explanation is the oxidation of CoFeOx by holes. The high frequency intercept point of h/cfox30 is notably higher than the other two samples, meaning a higher hole flux to the surface. In h/cfox30, the band bending is more pronounced in the space charge region thanks to rapid charge transfer from hematite to CoFeOx. Consequently, less recombination at the space charge region occurs and a higher hole flux reaches the surface. At 1.2 VRHE, as most CoFeOx is oxidized by external bias, this is no longer an advantage, so high frequency intercepts become close again. Here only one semicircle is measured, meaning the absence of CoFeOx photo-oxidation. This semicircle is, however, the biggest among all, meaning a lower charge transfer efficiency. Its poor performance with respect to the others can be understood by calculating the rate constants from these complex plots (steps illustrated in Note S3, ESI‡).
 | ||
| Fig. 9 (a) IMPS complex plots for h/cfox0 (red squares) h/cfox3 (green circles) and h/cfox30 (blue diamonds) at 0.85 (solid symbols) and 1.2 VRHE (open symbols). (b) Rate constants for h/cfox0, h/cfox3 and h/cfox30 (same color scheme) calculated with IMPS plots at various potentials. Solid symbols represent kct and open symbols represent krec. | ||
The rate constants calculated with IMPS are displayed in Fig. 9b. Unlike PEIS at 100 mW cm−2 irradiation, IMPS gives decreasing krec for h/cfox0 and h/cfox3. As discussed before, this is a result of a weaker light intensity used (37.5 mW cm−2, cf. Fig. S10, ESI‡). The values of krec for h/cfox0 and h/cfox3 are similar, while kct is higher for h/cfox3 at all potentials, in excellent agreement with PEIS. Rate constants below 0.8 VRHE are not investigated since they are below the photocurrent onset potential. As applied potential increases, kct for both h/cfox0 and h/cfox3 increase continuously until surpassing krec at 1.05 VRHE, beyond which charge transfer is more favored, which lead to high ϕct. The fitting for h/cfox30 requires more attention since the low frequency parts are convoluted with two semicircles at low voltages. To obtain meaningful rate constants, the lowest frequency semicircles must be disregarded. As such, the fitted rate constants are representative of photo-oxidation of CoFeOx rather than water. Above 0.95 VRHE, water oxidation rate constants can be successfully fitted again. From the plots it can be seen that the kinetics of CoFeOx oxidation is faster than water oxidation as previously suggested. When CoFeOx is fully functional after being oxidized, charge transfer is slowed as well as surface recombination, which agrees remarkably with PEIS results. The charge transfer efficiencies at 1.25 VRHE calculated from IMPS rate constants are, relatively, in good agreement compared to ϕct obtained with the hole scavenging approach (Table 3). Higher values produced by IMPS result from differences in band bending as discussed before for PEIS.
| h/cfox0 (%) | h/cfox3 (%) | h/cfox30 (%) | |
|---|---|---|---|
| ϕ(IMPS) | 85.9 | 91.8 | 36.7 |
| ϕ(H2O2) | 70.7 | 85.0 | 12.0 |
All four electrochemical methods applied in this work lead to the finding that photogenerated holes are used for CoFeOx oxidation. The results show that enhancement of charge transfer rate and reduction of surface recombination for water oxidation cannot be harnessed simultaneously. Therefore, in an attempt to decrease krec without sacrificing the increase in kct, we passivated the hematite film by adding a layer of GaOx by chemical bath deposition (denoted as h/GaOx).38,59 Then, CoFeOx was electrodeposited with three coating sweeps as before (denoted as h/GaOx/cfox3). The J–V curves for h/cfox0, h/cfox3, h/GaOx and h/GaOx/cfox3 are displayed in Fig. 10a. The improvement at low potentials with the GaOx coating is similar with cfox3 coating. When the two treatments are combined, the photocurrent density is significantly enhanced. We then conducted PEIS measurements and kinetic analyses for GaOx treated samples, which are compared with h/cfox3 (Fig. 10b).
 | ||
| Fig. 10 (a) Linear sweep voltammograms under 1 sun chopped light of h/cfox0 (red), h/cfox3 (light green), h/GaOx (grey) and h/GaOx/cfox3 (purple). (b) Rate constants calculated from PEIS for h/cfox0 (red squares), h/cfox3 (light green circles) h/GaOx (grey triangles) and h/GaOx/cfox3 (purple stars). Empty symbols indicate krec and filled symbols indicate kct. | ||
Firstly, the effect of GaOx agrees with previously reported results obtained via IMPS, i.e. kct remains similar and krec drops.60 However, when three sweeps of electrodeposition of CoFeOx are carried out on h/GaOx, there is a marginal upshift in krec possibly due to interphase recombination. On the other hand, kct of h/GaOx/cfox3 is much larger than that of h/cfox3 at any potential (Fig. 10b). This rise in kct with a GaOx interlayer is a result of an alteration of the distribution of intermediate surface states (i-ss), as evidenced by a cathodic shift of Css for h/cfox0 compared with h/GaOx and h/GaOx/cfox3 (Fig. S14, ESI‡). A similar observation was reported by Wang et al. in the case of Al2O3 passivation of hematite photoanodes.49 The energetics of altered surface states, as a consequence, may be more in favor of OER by CoFeOx. This way, by adding a GaOx interlayer, the photocurrent density can be greatly improved at relatively low potentials (∼0.5 mA cm−2 at 1.0 VRHE) owing to the additional enhancement of kct.
Discussion
Our understanding of the role of CoFeOx on hematite photoanodes is summarized in Fig. 11. For a bare hematite photoanode, surface charge recombination takes place at a much faster time scale than charge transfer at semiconductor–liquid junction. Introducing CoFeOx is found to have different impact at different loading levels. | ||
| Fig. 11 Energetic schemes for (a) h/cfox0 showing surface recombination krec and charge transfer kct through i-ss; (b) h/cfox3 where kct increase relative to h/cfox0 and density of i-ss reduces; (c) h/cfox30 where both rate constants are decreased; (d) h/GaOx/cfox3 with significantly increased kct and increased density of i-ss. | ||
An extremely thin layer of CoFeOx does not strongly affect recombination but significantly accelerates the charge transfer. In this case, CoFeOx assists the intermediate surface states by acting as a more efficient shuttle for holes thereby improving the OER charge transfer kinetics. When thickness is relatively higher, both the charge transfer and surface recombination slow down but especially the surface recombination, which is in good agreement with other studies on OECs of similar thickness on semiconductors.35,36,60 Since CoFeOx fully covers the hematite electrode, as well as having a lower oxidation potential compared to i-ss, the photogenerated holes mainly charge the catalyst. The higher photocurrent density at low potential is partly attributed to a slower recombination but also to this pseudocapacitive OEC charging. In this situation, only the holes reaching the catalyst with energies between quasi-Fermi level (EF*) and E(H2O/O2) are capable of carrying out water oxidation, which only takes a small proportion. This detrimental effect is, on the other hand, alleviated by the drop of krec, giving a photocurrent density comparable to h/cfox0. Accordingly, if CoFeOx loading is even higher, it is possible that all photogenerated holes oxidize the catalyst and EF* moves higher than E(H2O/O2) (Fig. S15a, ESI‡). If that happens, photo-assisted water oxidation will become energetically impossible, which can be evidenced by the absence of net photocurrent after the catalyst is sufficiently oxidized. In this situation, photogenerated holes have no other pathways but recombination. Indeed, this expected J–V behavior is observed on an h/cfox film containing a very thick CoFeOx layer deposited at 1.8 VRHE for 25 min (Fig. S15b, ESI‡). It shows little net photocurrent and strong spikes at higher applied potentials. Although higher applied potential can oxidize CoFeOx thus avoiding photo-charging of it, the low charge transfer rate constants prevent rapid improvement of photocurrent density.
Additional improvement of photoactivity can be achieved by adding an interlayer of GaOx. The result is ascribed to remarkable enhancement of kct without the strong OEC charging effect due to a redistribution of i-ss. This configuration can also be seen as an “adaptive junction” on top of a “buried junction” as proposed by Nellist and co-workers, although GaOx is not catalytically active.27 Despite this promising improvement of photocurrent density, GaOx is unstable in strong alkaline solutions. Therefore, a more stable material to form a “buried junction” with the light absorbing layer is of research interest.
Our theory on the effect of different thicknesses has been tested to be applicable on some other Co or Ni containing OEC species (data not shown). Nevertheless, when CoPi is coated on our hematite photoanodes, the capacitive behavior is totally absent. We believe in this case, the Co2+/Co3+ reaction may be stabilized by phosphate ions and photo-charging is prevented. This topic deserves more attention.
Conclusion
We have investigated the effect of OEC coating thickness on hematite photoanodes using a promising OEC candidate, CoFeOx. The research outcomes suggest that to improve interfacial charge transfer properties, the loading of the OEC must be carefully controlled to an extremely thin level as the oxidation current of CoFeOx can easily introduce an “illusion” of increase in photocurrent density. A slow scan rate is therefore preferred for LSV measurements, while light chopping sometimes provides extra information. According to our kinetic analysis, some traditional OEC coatings are perhaps too thick to take advantage of the rapid water oxidation kinetics of the OEC. We have also revealed that an interlayer of GaOx between hematite and OEC can enhance hole transfer rates further compared to an OEC coating alone. Our work has found a new way of improving charge transfer kinetics at photoanode surfaces and helps understand the interplay between a semiconductor and an electrocatalyst. Future research will target at deeper understanding of semiconductor–catalyst junctions and creating more efficient complex photoanodes.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
XPS data collection was performed at the EPSRC National Facility for XPS (‘HarwellXPS’), operated by Cardiff University and UCL, under contract No. PR16195. JZ would like to thank Dr David Morgan's assistance on XPS data analysis. JZ also greatly acknowledges discussion with Prof. L. M. Peter and Prof. F. Marken at University of Bath. The authors thank the Microscopy and Analysis Suite (MAS) at University of Bath for assistance with HR-TEM imaging.References
- K. Sivula and R. Van De Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef.
- K. Sivula, J. Phys. Chem. Lett., 2015, 6, 975–976 CrossRef PubMed.
- G. Wang, Y. Ling, D. a. Wheeler, K. E. N. George, K. Horsley, C. Heske, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3503–3509 CrossRef PubMed.
- S. Shen, J. Zhou, C. L. Dong, Y. Hu, E. N. Tseng, P. Guo, L. Guo and S. S. Mao, Sci. Rep., 2014, 4, 1–9 Search PubMed.
- J. W. Jang, C. Du, Y. Ye, Y. Lin, X. Yao, J. Thorne, E. Liu, G. McMahon, J. Zhu, A. Javey, J. Guo and D. Wang, Nat. Commun., 2015, 6, 7447 CrossRef PubMed.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 RSC.
- K. Sivula, F. Le Formal and M. Gratzel, Chem. Mater., 2009, 21, 2862–2867 CrossRef.
- S. Kment, F. Riboni, S. Pausova, L. Wang, L. Wang, H. Han, Z. Hubicka, J. Krysa, P. Schmuki and R. Zboril, Chem. Soc. Rev., 2017, 46, 3716–3769 RSC.
- H. Li, Y. Yu, M. B. Starr, Z. Li and X. Wang, J. Phys. Chem. Lett., 2015, 6, 3410–3416 CrossRef PubMed.
- W. Yang, Y. Yu, M. B. Starr, X. Yin, Z. Li, A. Kvit, S. Wang, P. Zhao and X. Wang, Nano Lett., 2015, 15, 7574–7580 CrossRef PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 3 CrossRef.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- L. Cai, J. Zhao, H. Li, J. Park, I. S. Cho, H. S. Han and X. Zheng, ACS Energy Lett., 2016, 1, 624–632 CrossRef.
- J. A. Seabold and K. S. Choi, J. Am. Chem. Soc., 2012, 134, 2186–2192 CrossRef PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef PubMed.
- C. Du, X. Yang, M. T. Mayer, H. Hoyt, J. Xie, G. McMahon, G. Bischoping and D. Wang, Angew. Chem., Int. Ed., 2013, 52, 12692–12695 CrossRef PubMed.
- C. G. Morales-Guio, M. T. Mayer, A. Yella, S. D. Tilley, M. Grätzel and X. Hu, J. Am. Chem. Soc., 2015, 137, 9927–9936 CrossRef PubMed.
- C. G. Morales-Guio, L. Liardet and X. Hu, J. Am. Chem. Soc., 2016, 138, 8946–8957 CrossRef PubMed.
- M. Bajdich, M. García-Mota, A. Vojvodic, J. K. Nørskov and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 13521–13530 CrossRef PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef PubMed.
- M. S. Burke, S. Zou, L. J. Enman, J. E. Kellon, C. A. Gabor, E. Pledger and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 3737–3742 CrossRef PubMed.
- M. D. Merrill and R. C. Dougherty, J. Phys. Chem. C, 2008, 112, 3655–3666 CrossRef.
- N. Guijarro, M. S. Prévot and K. Sivula, Phys. Chem. Chem. Phys., 2015, 17, 15655–15674 RSC.
- M. R. Nellist, F. A. L. Laskowski, F. Lin, T. J. Mills and S. W. Boettcher, Acc. Chem. Res., 2016, 49, 733–740 CrossRef PubMed.
- M. G. Ahmed, T. a. Kandiel, A. Y. Ahmed, I. Kretschmer, F. Rashwan and D. Bahnemann, J. Phys. Chem. C, 2015, 119, 5864–5871 CrossRef.
- A. Y. Ahmed, M. G. Ahmed and T. a. Kandiel, J. Phys. Chem. C, 2016, 120, 23415–23420 CrossRef.
- A. J. Abel, A. M. Patel, S. Y. Smolin, B. Opasanont and J. B. Baxter, J. Mater. Chem. A, 2016, 4, 6495–6504 RSC.
- F. Malara, A. Minguzzi, M. Marelli, S. Morandi, R. Psaro, V. Dal Santo and A. Naldoni, ACS Catal., 2015, 5, 5292–5300 CrossRef.
- J. W. Moir, E. V. Sackville, U. Hintermair and G. A. Ozin, J. Phys. Chem. C, 2016, 120, 12999–13012 CrossRef.
- L. Wang, N. T. Nguyen and P. Schmuki, ChemSusChem, 2016, 9, 2048–2053 CrossRef PubMed.
- M. Chhetri, S. Dey and C. N. R. Rao, ACS Energy Lett., 2017, 2, 1062–1069 CrossRef.
- G. M. Carroll and D. R. Gamelin, J. Mater. Chem. A, 2016, 4, 2986–2994 RSC.
- C. Zachäus, F. F. Abdi, L. M. Peter and R. van de Krol, Chem. Sci., 2017, 8, 3712–3719 RSC.
- F. Le Formal, S. R. Pendlebury, M. Cornuz, S. D. Tilley, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2014, 136, 2564–2574 CrossRef PubMed.
- T. Hisatomi, F. Le Formal, M. Cornuz, J. Brillet, N. Tétreault, K. Sivula and M. Grätzel, Energy Environ. Sci., 2011, 4, 2512–2515 RSC.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef PubMed.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, J. Am. Chem. Soc., 2012, 134, 16693–16700 CrossRef PubMed.
- L. Badia-Bou, E. Mas-Marza, P. Rodenas, E. M. Barea, F. Fabregat-Santiago, S. Gimenez, E. Peris and J. Bisquert, J. Phys. Chem. C, 2013, 117, 3826–3833 CrossRef.
- Y. B. Hu, C. F. Dong, M. Sun, K. Xiao, P. Zhong and X. G. Li, Corros. Sci., 2011, 53, 4159–4165 CrossRef.
- R. Wang, X. Yan, J. Lang, Z. Zheng and P. Zhang, J. Mater. Chem. A, 2014, 2, 12724–12732 RSC.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, T. Hamann and J. Bisquert, J. Am. Chem. Soc., 2012, 134, 4294–4302 CrossRef PubMed.
- J. Qiu, H. Hajibabaei, M. R. Nellist, F. A. L. Laskowski, T. W. Hamann and S. W. Boettcher, ACS Cent. Sci., 2017, 3, 1015–1025 CrossRef PubMed.
- F. Lin, B. F. Bachman and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 2427–2433 CrossRef PubMed.
- K. G. Upul Wijayantha, S. Saremi-Yarahmadi and L. M. Peter, Phys. Chem. Chem. Phys., 2011, 13, 5264–5270 RSC.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, Energy Environ. Sci., 2012, 5, 7626–7636 RSC.
- Z. Wang, F. Fan, S. Wang, C. Ding, Y. Zhao and C. Li, RSC Adv., 2016, 6, 85582–85586 RSC.
- L. M. Peter, L. H. Wong and F. F. Abdi, J. Electroanal. Chem., 2018, 819, 447–458 CrossRef.
- M. Zhang, M. De Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 CrossRef PubMed.
- L. M. Peter, E. A. Ponomarev and D. J. Fermín, J. Electroanal. Chem., 1997, 427, 79–96 CrossRef.
- H.-J. Lewrenz and L. Peter, Photoelectrochemical Water Splitting: Materials, Processes and Arhitectures, RSC Publishing, 2013 Search PubMed.
- L. M. Peter, Chem. Rev., 1990, 90, 753–769 CrossRef.
- R. Van de Krol and M. Gratzel, Photoelectrochemical Hydrogen Production, Springer, 2012 Search PubMed.
- E. A. Ponomarev and L. M. Peter, J. Electroanal. Chem., 1995, 397, 45–52 CrossRef.
- L. M. Peter, J. Li and R. Peat, J. Electroanal. Chem., 1984, 165, 29–40 CrossRef.
- D. Tafalla, P. Salvador and R. M. Benito, J. Electrochem. Soc., 1990, 137, 1810–1815 CrossRef.
- L. Steier, I. Herraiz-Cardona, S. Gimenez, F. Fabregat-Santiago, J. Bisquert, S. D. Tilley and M. Grätzel, Adv. Funct. Mater., 2014, 24, 7681–7688 CrossRef.
- J. E. Thorne, J.-W. Jang, E. Y. Liu and D. Wang, Chem. Sci., 2016, 7, 3347–3354 RSC.
Footnotes |
| † All data created during this research are available upon reasonable request. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ee01346b |
| This journal is © The Royal Society of Chemistry 2018 |