
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient visible light-driven water oxidation and proton reduction by an ordered covalent triazine-based framework†
Jijia
Xie
 a,
Stephen A.
Shevlin
b,
Qiushi
Ruan
a,
Savio J. A.
Moniz
a,
Yangrong
Liu
ab,
Xu
Liu
a,
Yaomin
Li
ab,
Chi Ching
Lau
a,
Zheng Xiao
Guo
*b and
Junwang
Tang
*a
a,
Stephen A.
Shevlin
b,
Qiushi
Ruan
a,
Savio J. A.
Moniz
a,
Yangrong
Liu
ab,
Xu
Liu
a,
Yaomin
Li
ab,
Chi Ching
Lau
a,
Zheng Xiao
Guo
*b and
Junwang
Tang
*a
aDepartment of Chemical Engineering, University College London, Torrington Place, London, WC1E7JE, UK. E-mail: junwang.tang@ucl.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H0AJ, UK. E-mail: z.x.guo@ucl.ac.uk
First published on 12th April 2018
Abstract
Water oxidation is a rate-determining step in solar driven H2 fuel synthesis and is technically challenging to promote. Despite decades of effort, only a few inorganic catalysts are effective and even fewer are effective under visible light. Recently, attention has been paid to synthetic semiconducting polymers, mainly on graphitic C3N4, with encouraging hydrogen evolution performance but lower activity for water oxidation. Here, a highly ordered covalent triazine-based framework, CTF-1 (C8N2H4), is synthesised by a very mild microwave-assisted polymerisation approach. It demonstrates extremely high activity for oxygen evolution under visible light irradiation, leading to an apparent quantum efficiency (AQE) of nearly 4% at 420 nm. Furthermore, the polymer can also efficiently evolve H2 from water. A high AQE of 6% at 420 nm for H2 production has also been achieved. The polymer holds great potential for overall water splitting. This exceptional performance is attributed to its well-defined and ordered structure, low carbonisation, and superior band positions.
Broader contextSplitting water by sunlight is an attractive renewable approach to generate clean hydrogen for producing chemicals or powering vehicles. This ultra-pure hydrogen also avoids the dreaded catalyst poisoning, which occurs even with very low levels of CO residuals from fossil-fuel generated hydrogen. The key challenge to sustain continued hydrogen generation from this artificial photosynthesis process is to speed up the water oxidation reaction, which is hard to proceed due to multiple electron transfer steps. Oxidation catalysts based on polymeric semiconductors are particularly promising for this purpose because of their abundance leading to low cost, tunable band structure to match the solar spectrum and variable degree of conjugation to impact on the π→π* excitation for efficient electron transfer. With a moderate microwave assisted strategy, we are able to control the degree of conjugation and minimise the undesirable structural carbonisation of a new type of polymeric photocatalyst– covalent triazine-based framework. The optimised catalyst shows advantageous band positions, to capture a wide spectrum of visible light and enhance the charge separation efficiency, leading to very high water oxidation and hydrogen evolution capabilities. The overall discovery paves the way for the development of efficient and continuous clean hydrogen production from the renewable visible-light water splitting process. |
Introduction
Solar driven catalysis for clean chemical fuels, such as hydrogen, is highly promising to mitigate environmental issues caused by the combustion of fossil fuels and to meet increasing worldwide demands for energy.1,2 Since the discovery of water splitting by Fujishima and Honda on an n-type TiO2 electrode in 1972,3 water splitting by inorganic materials has gained the vast majority of attention. However, only a few inorganic semiconductors, such as GaN:ZnO4 and InNiTaO4,5 possess suitable band positions to drive both hydrogen and oxygen evolution from pure water under visible light irradiation.Due to the diverse molecular structures and tunable band positions, polymer photocatalysts are highly advantageous for solar H2 production from water.6 Furthermore, polymers are usually composed of abundant carbon, nitrogen and oxygen elements, which are economically more attractive than the reported metal-oxide photocatalysts. Recently, Zhang et al. reported overall water splitting achieved by modified g-C3N4 although with a relatively moderate efficiency due to the large bandgap of g-C3N4.7,8 Compared with the above single photocatalyst for pure water splitting, a Z-scheme composed of two photocatalysts responsible for two half reactions promises 10% higher solar-to-fuel conversion efficiency.9–13 Therefore, substantial efforts have recently been channeled into searching for efficient polymer photocatalysts for either proton reduction or water oxidation in an attempt to construct an efficient Z-scheme system. Therein, water oxidation has been widely accepted as the rate-determining step in water splitting, and thus it is more challenging and important than proton production.14,15 Metal-oxide photocatalysts have been widely studied in water oxidation, such as BiVO4, which shows very promising activity, though there is a concern about the heavy metal Bi used and difficulty in changing its band positions.6,8,16,17 For polymer based photocatalysts, up to now, there are many reports about proton reduction by either g-C3N4 or g-C3N4 doped by oxygen, boron, phosphorus, etc.6,9,18–21 For example, Wang et al. reported that graphitic carbon nitride presents great potential for photocatalytic proton reduction in 2009 and platinum decorated g-C3N4 shows ca. 10 μmol h−1 hydrogen evolution rate under visible light irradiation.6 Martin et al. further enhanced the degree of polymerisation of g-C3N4 and gained a much higher quantum efficiency of 26.5% at 400 nm for proton reduction.18 Later, the light absorbance of this polymer was extended to 800 nm by controlling the linker of carbon nitride and it generated H2 by light irradiation from the ultraviolet to near infrared regions.9 This progress in polymer photocatalysts for proton reduction has played a crucial role to stimulate research interest in polymer photocatalysts. However, the most challenging step, water oxidation, in water splitting has met with little success in polymer photocatalysis. Wang et al.6 first achieved polymer-based photocatalytic water oxidation by RuO2 loaded g-C3N4 and gained an evolution rate of ca. 1 μmol h−1 under visible light irradiation. After that, the same group further enhanced the activity by modifying g-C3N4, such as sulfur doping,22 molecular cobalt decoration23 and layered Co(OH)2 deposition,24 and gained an O2 evolution rate of 3 μmol h−1, 11 μmol h−1 and 26 μmol h−1 under visible light irradiation, respectively. Based on our knowledge, only Chu et al. have reported an apparent quantum yield (AQY) for polymer-based water oxidation of 0.2% (at 420 nm), which was achieved by a heptazine-based polymer, polyimide (PI).25 As by only addressing both water oxidation and proton reduction by polymer-based photocatalysts, one can construct a low cost and an efficient Z-scheme for entire water splitting, herein, this study concentrates on water oxidation following the previous success on proton reduction.
The water oxidation potential of a polymer photocatalyst is determined by its valence band position, which is dominated by the N 2p orbitals for triazine-based units, e.g. g-C3N4.26–29 However, g-C3N4 shows a rather low activity for water oxidation due to its high valence band position.6,9,18 Controlling the ratio of N to C in such a polymer photocatalyst would induce both a band gap change and band position shift. Only a few reports have considered the N to C ratio by introducing nitrogen vacancies.30–32 However, most of those cannot lower the valence band of g-C3N4, because the N replacements are mostly on the linker, –NHx groups, mainly contributing to the n–π* transitions, which are far less active than the π–π* transitions of N in the heptazine units.9,33,34 A covalent triazine-based framework (CTF-1) is composed of alternating triazine units and phenyl groups in a 12-membered conjugated ring system, as shown in Fig. 1a. Thus, it is totally different from the recently studied g-C3N4, which is composed of heptazine units. CTF-1 also possesses an N to C ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, very different from the 4:3 ratio of g-C3N4. Jiang et al. have predicted that the valence band position of CTF-1 is located at ca. 2.0 eV (vs. the Neutral Hydrogen Electrode (NHE), at pH = 0) based on first principles calculations.35 Moreover, from a structural point of view, most of the reported carbon nitrides hold additional –NHx compared to the ideal graphitic carbon nitride and were suggested to be reasonably named as poly(aminoimino) heptazine or polymeric carbon nitride (PCN) in a critical review.36 These additional functional groups often cause defects and distortions to generate charge recombination centres.34,37 Herein, based on the different polymerisation strategies for the triazine-based polymer synthesis, the degree of conjugation has been investigated on CTF-1. The ordered in-plane structure has been controlled by a rapid while moderate microwave method to demonstrate the useful correlation between the degree of conjugation, carbonisation and photocatalytic activity for water splitting, which has not been investigated before. We have further identified that the novel polymer photocatalyst is far more active than g-C3N4 for both proton reduction and, in particular, water oxidation under visible light.
:4, very different from the 4:3 ratio of g-C3N4. Jiang et al. have predicted that the valence band position of CTF-1 is located at ca. 2.0 eV (vs. the Neutral Hydrogen Electrode (NHE), at pH = 0) based on first principles calculations.35 Moreover, from a structural point of view, most of the reported carbon nitrides hold additional –NHx compared to the ideal graphitic carbon nitride and were suggested to be reasonably named as poly(aminoimino) heptazine or polymeric carbon nitride (PCN) in a critical review.36 These additional functional groups often cause defects and distortions to generate charge recombination centres.34,37 Herein, based on the different polymerisation strategies for the triazine-based polymer synthesis, the degree of conjugation has been investigated on CTF-1. The ordered in-plane structure has been controlled by a rapid while moderate microwave method to demonstrate the useful correlation between the degree of conjugation, carbonisation and photocatalytic activity for water splitting, which has not been investigated before. We have further identified that the novel polymer photocatalyst is far more active than g-C3N4 for both proton reduction and, in particular, water oxidation under visible light.
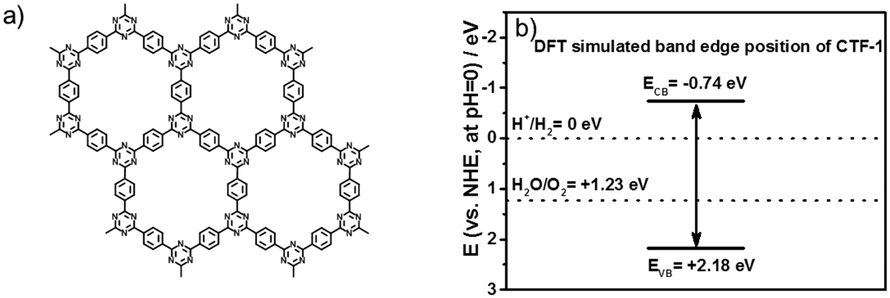 | ||
| Fig. 1 Structure of CTF-1 (a); the positions of the conduction band edge and valence band edge of CTF-1 (vs. NHE at pH = 0) predicted by density functional theory (DFT) calculations (b). | ||
Experimental
Material preparation
Covalent triazine-based frameworks (CTF-1) were fabricated by modified microwave chemistry.38 Briefly, in each typical experiment, 2 ml trifluoromethanesulfonic acid (Sigma-Aldrich, ≥99%) was added drop-wise to 0.4 g terephthalonitrile (Sigma-Aldrich, 98%) in a 10 ml glass reaction vessel (CEM, Discover and Explorer SP Vessels). A PTFE coated stirrer was added and sealed with a pressure control cap (CEM, ActiVent Pressure Control Technology). The mixture was pre-stirred at room temperature until terephthalonitrile was dissolved and a viscous red solution was obtained. Then the solution was subjected to a single-mode microwave irradiation at 20, 50, 100 or 200 W for 30 seconds, respectively. The safety temperature was set at 200 °C and the safety pressure was set at 200 psi (the time-on-line temperate and pressure profile are shown in Fig. S1 and S2, ESI,† respectively), which resulted in the formation of a dark yellow solid, named as CTF-1-20W, CTF-1-50W, CTF-1-100W and CTF-1-200W, respectively. The solid was ground carefully into powder and washed with 1 M ammonia solution, water, ethanol, acetone and THF, each of 50 ml, respectively, and dried in a vacuum oven at 180 °C to remove residual solvent and excess trifluoromethanesulfonic acid. The yield of this reaction was over 95%. In order to enhance the charge separation during water oxidation reactions, RuOx were loaded onto CTF-1s by a highly reproducible impregnation method. In each typical experiment, 6 mg RuCl3 (Sigma-Aldrich, Ru content 50%) was dissolved in 2 ml methanol. Then, the solvent was added drop-wisely into 100 mg CTF-1s to make a slurry. The mixture was evaporated at 50 °C with magnetic stirring (600 rpm) for 3 hours, then ground to a fine powder and calcined in a muffle furnace at 350 °C (ramping rate 5 °C min−1). Finally, the powder was washed by deionised water 3 times and then used for photocatalytic activity tests and physical characterisation.Material characterisation
Powder X-ray diffraction (PXRD) measurements were obtained using a SAXSLAB Ganesha 300XL small angle X-ray scattering (SAXS) system in wide angle X-ray scattering mode with a range from 2θ = 2°–40° (wavelength 0.154 nm, Cu-Kα radiation). ATR-FTIR spectroscopy was performed using a Perkin-Elmer 1605 FT-IR spectrometer in ATR mode with a range from 400–4000 cm−1. 13C cross-polarisation magic angle spinning (CPMAS) solid-state nuclear magnetic resonance (ssNMR) spectra were collected at ambient temperature on a BRUKER Advance 300 WB spectrometer (Bruker UK Ltd) with a 4 mm magic-angle spinning probe. X-ray photoelectron spectroscopy (XPS) was performed on a Thermo Scientific XPS K-alpha machine using monochromatic Al-Kα radiation. Survey scans were collected in the range of 0–1100 eV (binding energy) at a pass energy of 160 eV. Higher resolution scans were recorded for the main core lines at a pass energy of 20 eV. The analysis was performed on Casa XPS software. Specific surface areas were measured using the BET method with N2 absorption and the data were collected using a Micromeritics TriStar 3000 gas adsorption analyser. UV-Vis absorption spectra were obtained on a Shimadzu UV-Vis 2550 spectrophotometer fitted with an integrating sphere. Reflectance measurements were performed on powdered samples, using a standard barium sulphate powder as a reference. The reflection measurements were converted to absorption spectra using the Kubelka–Mulk transformation. Raman spectra were measured on a Renishaw InVia Raman Microscope, using a 325 nm excitation laser, between 100 and 3500 cm−1. The Mott–Schottky curves were measured at a certain DC potential range with an AC amplitude of 5 mV and a frequency of 500, 1000 and 2000 Hz under dark conditions in a conventional three-electrode cell using an electrochemical analyser (IVIUM Technologies).Photocatalytic analysis
The oxygen evolution half reaction was conducted in a 500 ml custom batch reactor with a top quartz window for light irradiation. Typically, a sample of 50 mg was dispersed in 200 ml of a 0.2 M aqueous AgNO3 solution (as the sacrificial electron scavenger). After ultra-sonication for 30 minutes, the reactor was sealed and purged with argon for 30 minutes and then irradiated under a 300 W Xeon lamp with a 420 nm long pass filter (Comar Optics 420 GY 50). Gas measurements were taken at regular intervals from the reactor headspace. The gas concentration was measured by GC (Varian 430-GC, TCD, argon carrier gas; BOC 99.999%) equipped with a molecular 5A column.The hydrogen evolution half reaction was carried out in a 500 ml custom-made batch reactor cell with a top quartz window for light irradiation. Typically, 50 mg of CTF-1 was first suspended and subsequently sonicated in a water/scavenger/dispersant mixture (230 ml total volume; consisting of 200 ml DI water, 23 ml triethanolamine (TEOA) and 7 ml methanol). In order to introduce Pt onto CTF-1, 0.56 g chloroplatinic acid hydrate (H2PtCl6·xH2O) (Sigma-Aldrich, 37% Pt) was added to the mixture, which would generate about 3% Pt on the polymer. After ultra-sonication for 30 minutes, the reactor was sealed and purged with argon (BOC 99.999%) for 30 minutes and irradiated for one hour under full arc irradiation using a 300 W Xenon lamp with stirring. Gas concentration analysis was performed by a GC to monitor the photo-deposition progress of Pt. The reactor was then purged a second time to conduct H2 evolution runs. For visible light irradiation, a 420 nm long-pass filter was used (Comar Optics 420 GY 50).
The apparent quantum efficiency (AQE) was measured by inserting an appropriate band pass filter (400, 420, 500, 600 nm, λ ± 10 nm at 10% of peak height, Comar Optics) in front of a 150 W Xe lamp. For these tests, 50 mg of photocatalyst was used. The light intensity was measured at 5 different points to obtain an average intensity using a calibrated photodiode coupled with an optical power meter (Newport, Model 1908-R). The average intensities at each wavelength are 111 μW cm−1 (400 nm), 119 μW cm−1 (420 nm), 311 μW cm−1 (500 nm) and 276 μW cm−1 (600 nm). The apparent quantum efficiency was calculated using the following formula:16
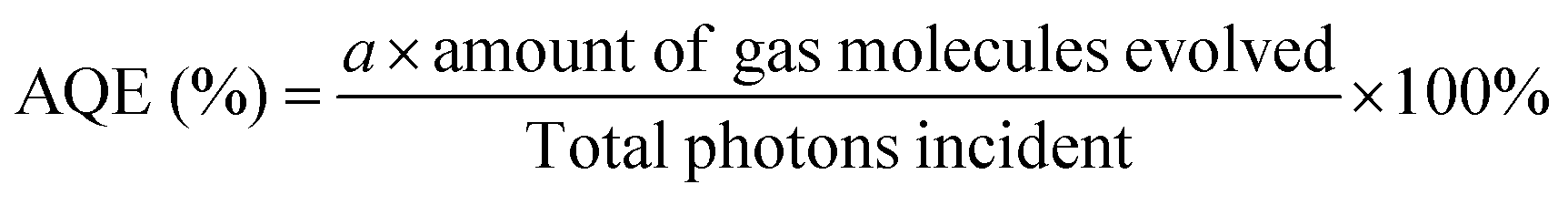
Computational simulations
The electronic and optical properties of the covalent triazine framework CTF-1 were investigated using periodic Density Functional Theory (DFT), using the VASPcode.39 A plane wave cutoff of 520 eV was used, with the projector augmented wave methods used to treat the core electrons.40 The GGA functional was used for structural optimisation and thermodynamic calculation. van der Waals interactions were included by using the DFT-D2 method of Grimme et al.41 All atoms were fully relaxed until the change in force upon ionic displacement was less than 0.01 eV Å−1, with the change in energies no greater than 10−5 eV. The crystal structure of CTF-1 was generated using an AA stacking motif with lattice parameters obtained using the same methodology as above. The lowest energy vibrationally stable structure was found to have space group Ci, with lattice parameters a = 3.689 Å, b = 13.672 Å, and c = 14.304 Å with α = 117.3°, β = 93.4° and γ = 96.6°. The triazine–benzene sheets are tilted in this cell with respect to the bc-plane, however, the intrasheet lattice vector is 14.566 Å, which is in excellent agreement with the experiment. A Monkhorst–Pack k-point mesh of (7 × 7 × 3) was used for the g-C3N4 bulk system, whereas for the CTF-1 bulk system a k-point mesh of (3 × 3 × 2) k-points was used. In order to accurately align the KS eigenvalues for both systems, we performed single sheet calculations of the ground state structure with a vacuum gap of a 20 Å. A (1 × 1 × 1) supercell was used for the CTF-1 sheet. As a large vacuum spacing is used the planar averaged electrostatic potential converges to a constant value far from the surface. These converged values may be taken as the reference level with which the Kohn–Sham (KS) eigenvalues are aligned.42 The NHE is taken to be at −4.44 eV. The photoelectrochemical efficiency is exquisitely dependent on the position of the band edges, therefore, we used a hybrid functional for the band alignment calculations, specifically a modified version of the HSE06 functional.43Results and discussion
Firstly, we modelled the band positions of the CTF-1 polymer to understand if it has appropriate potential for water oxidation and proton reduction. Fig. 1b presents the estimated band edge alignments of single sheet CTF-1, based on first principles calculations. The simulated conduction band of CTF-1 lies at −0.74 eV (vs. NHE, at pH = 0) and a valence band at +2.18 eV (vs. NHE, at pH = 0). Compared with the redox potential of proton reduction (at 0 eV vs. NHE at pH = 0) and water oxidation (at +1.23 eV vs. NHE at pH = 0), it thus has enough driving force for both water oxidation and proton reduction. More importantly, the simulated valence band position of CTF-1 is ca. 0.6 eV deeper than the widely researched polymer photocatalyst, g-C3N4.7,18 It is thus predicted to have a larger driving force for the water oxidation process than g-C3N4, indicating a more efficient photocatalytic activity for oxygen generation.The CTF-1 polymers were then prepared by the single mode microwave assisted approach at different powers (20–200 W). The molecular structures were first confirmed by X-ray photoelectron spectroscopy (XPS) and elemental analysis (EA). As shown in Fig. 2a, the XPS carbon 1s peaks are found at ca. 286 eV and the small peaks at 400 eV are assigned to nitrogen 1s.9 The XPS survey spectra show that there are almost no other impurity elements remaining from the precursors such as, fluorine, oxygen and sulphur. However, the carbon peaks in the XPS spectra are larger than the ideal carbon concentration due to the adventitious carbon used for calibration.44 Thus, EA measurements were used to further prove the elemental ingredients. As shown in Fig. 2b, CTF-1-20W, -50W and -100W have a carbon:nitrogen:hydrogen ratio of ca. 75:21:3.7 wt%, which is comparable to the ideal ratio (75.0:21.9:3.1 wt%). Hence, there is barely any additional “free” residual carbon in these three samples. Upon increasing the microwave power to 200 W, the C:N:H ratio changes to 78.5:18.3:3.2 wt%, which indicated the carbonisation process during high microwave power irradiation. Fourier transform infrared spectroscopy (FTIR) and solid state nuclear magnetic resonance (ssNMR) further confirm the chemical state of the carbon and nitrogen elements. As shown in Fig. 2c, all the synthesised CTFs shows similar FTIR spectra. Two strong absorption bands at 1501 cm−1 and 1345 cm−1 are associated with the triazine ring.45,46 The peaks at 1500–1480 cm−1 and 1600–1590 cm−1 can be assigned to the phenyl groups, whilst the stretching modes of the terminal C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group result in small peaks at 2226 cm−1.45,46 Increasing the microwave power from 20 W to 200 W significantly decreases the terminal CN groups, which is associated with the higher degree of polymerisation.45 Fig. S3 (ESI†) presents the full spectra of FT-IR; two more peaks at 810 and 1011 cm−1 are associated with the aromatic C–H out-of plane bending vibration and the aromatic C–H bending vibration, respectively.45 Solid-state 13C NMR spectra (Fig. 2d) also confirmed the successful synthesis of the conjugated triazine rings. The carbon atoms in the triazine units correspond to the signal at 170 ppm.45 The peaks at 139 ppm are assigned to the carbon atom directly connected to the triazine ring.45 The strongest peaks at 129 ppm represent the aromatic ring.45 The weakest peaks at 116 ppm are characteristic of the two types of carbon atom in the terminating groups,45 which shows that the concentration of terminals decreases on increasing the microwave power. Therefore, both FTIR and NMR represent that all the CTF-1 samples are composed of triazine and benzene units while higher microwave power results in a higher degree of polymerisation. Even for the lowest power sample (CTF-1-20W), it only has a tiny response of terminal nitrile groups, indicating a good polymerisation.45
N group result in small peaks at 2226 cm−1.45,46 Increasing the microwave power from 20 W to 200 W significantly decreases the terminal CN groups, which is associated with the higher degree of polymerisation.45 Fig. S3 (ESI†) presents the full spectra of FT-IR; two more peaks at 810 and 1011 cm−1 are associated with the aromatic C–H out-of plane bending vibration and the aromatic C–H bending vibration, respectively.45 Solid-state 13C NMR spectra (Fig. 2d) also confirmed the successful synthesis of the conjugated triazine rings. The carbon atoms in the triazine units correspond to the signal at 170 ppm.45 The peaks at 139 ppm are assigned to the carbon atom directly connected to the triazine ring.45 The strongest peaks at 129 ppm represent the aromatic ring.45 The weakest peaks at 116 ppm are characteristic of the two types of carbon atom in the terminating groups,45 which shows that the concentration of terminals decreases on increasing the microwave power. Therefore, both FTIR and NMR represent that all the CTF-1 samples are composed of triazine and benzene units while higher microwave power results in a higher degree of polymerisation. Even for the lowest power sample (CTF-1-20W), it only has a tiny response of terminal nitrile groups, indicating a good polymerisation.45
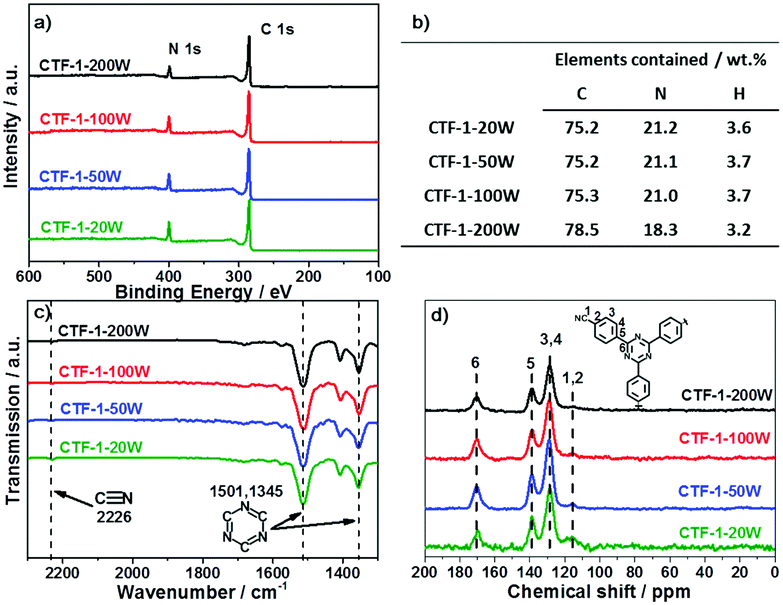 | ||
| Fig. 2 XPS survey spectra (a); elemental analysis (b); selected zone FT-IR spectra (c) and full spectra in Fig. S3 (ESI†); and 13CCP MAS ssNMR spectra (d) of the series of CTF-1. | ||
The degree of conjugation of CTF-1 has been investigated by Raman, XRD and HRTEM. Fig. 3a shows the Raman spectra of the series of CTF-1. The strongest G+ peak at 1613 cm−1 is an indication of a well-ordered sp2 planar structure. The G− peak and D peak at 1519 cm−1 and 1416 cm−1 relate to the distortion and carbon defects in each layer, respectively.47 The G/D ratio is enhanced by higher microwave power from 20 W to 100 W. Thus, higher microwave power gives fewer defects. Meanwhile, CTF-1-100W and CTF-1-200W show the same high G/D ratio of 12:1. The smallest peaks at 980 cm−1 relate to the Raman effects of the terminate nitrile groups.48 The powder X-ray diffraction (PXRD) patterns are shown in Fig. 3b. The peaks assigned as [100] at ca. 7° are associated with the hexagonal unit cell (a = b = 13.524 Å).38 The [100] peak intensity increases when the microwave power enhances from 20 W to 100 W. Thus, a higher microwave power induces a higher degree of crystallisation and a better ordered structure. However, CTF-1-200W presents a relatively lower intensity of the [100] peaks compared with CTF-1-100W. Hence, further higher microwave power can destroy the hexagonal units. The other obvious peak assigned as [001] in the PXRD patterns at ca. 26° represents the layer distance of the series of CTF-1. According to the Bragg's law, CTF-1-20W, CTF-1-50W and CTF-1-100W show a similar layer distance of ca. 3.4 Å. But, CTF-1-200W presents a larger layer distance of ca. 3.7 Å. Therefore, the highest microwave power can distort the crystallised structure by both breaking the hexagonal unit and extending the layer distance. The porosity of the materials has been studied by nitrogen adsorption and desorption experiments at 77K, as shown in Fig. 3c. CTF-1-200W exhibits a steep rise at a high relative pressure (P/P0 ∼ 1), indicating macrospores formed between highly aggregated particles,49 consistent with the low surface area.50,51 Among all samples, CTF-1-100W has the highest ordered structure both in plane and interlayer. Therefore, the morphology of CTF-1-100W was observed by TEM as shown in Fig. 3d. It shows a clear multilayer structure with a lattice space of ca. 0.34 nm calculated from the TEM contrast intensity profile and the Fast Fourier Transforms (FFT), which is consistent with the [001] peak shown in the PXRD patterns.52
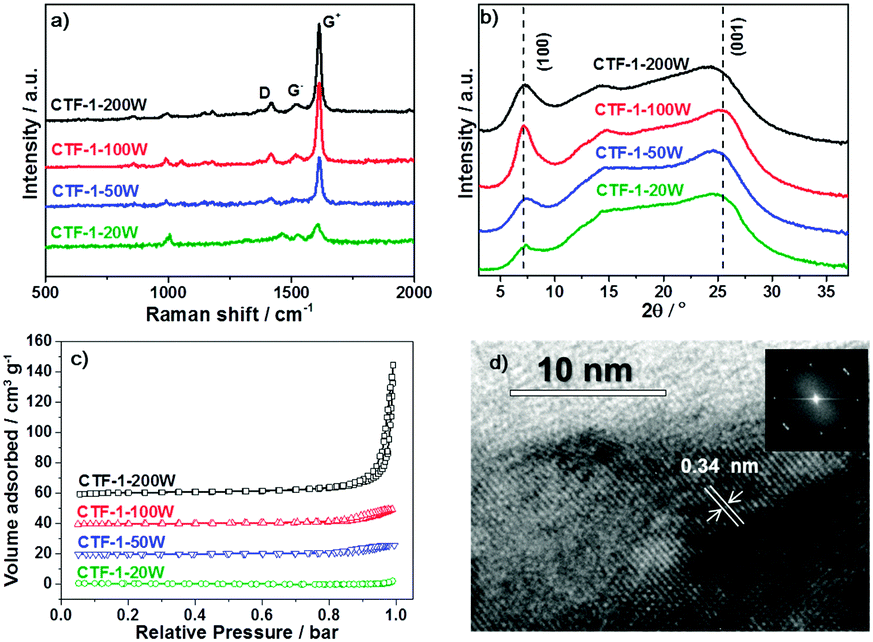 | ||
| Fig. 3 Raman spectra (a); powder X-ray diffraction (PXRD) patterns (b); nitrogen adsorption and desorption isotherms measured at 77 K (c) of the series of CTF-1. Transmission electron microscopy (TEM) image with Fast Fourier Transforms (FFT) of CTF-1-100W(d). | ||
The ability of the CTF-1 samples to evolve oxygen from water, as the focus of the study, was investigated using silver ions (from AgNO3) as a sacrificial electron acceptor, which is widely used in this half reaction to fairly assess catalysts’ activity. Fig. 4a shows the water oxidation ability of CTF-1-20W, -50W, -100W and -200W under visible light irradiation and the reference materials g-C3N4. RuOx as a widely used cocatalyst for water oxidation, was also utilised in this study. 50 mg 3 wt%RuOx/CTF-1-100W shows the highest oxygen evolution rate of ca.140 μmol g−1 h−1 under visible light irradiation (λ ≥ 420 nm). Furthermore, such bare CTF-1-100W without any cocatalyst shows even 3 times higher activity than RuOx decorated g-C3N4 (Fig. S4, ESI†) and 20 times better when a RuOx cocatalyst was loaded on our new catalyst. This strongly indicates the promising potential of the new polymer for photocatalytic water oxidation under visible irradiation, which is owing to both the deeper valence band and narrower bandgap of CTF-1 than g-C3N4 clearly, the degree of the polymers’ crystallisation impacts their photocatalytic activity and a good correlation between the photocatalytic water oxidation activity and the degree of crystallisation has been observed. The stability of CTF-1-100W for prolonged O2 evolution under visible light was investigated for 18 hours (composed of three 6 hour runs) and it was found that there was no obvious decrease in activity, which suggests that the material is quite stable (Fig. 4b). The ruthenium species on the best sample RuOx/CTF-1-100W has been further investigated by XPS (as shown in Fig. S5, ESI†). The ruthenium to oxygen ratio is ca. 1:1.85 and the ruthenium to nitrogen ratio is ca. 5.1%. The Ru 3d XPS spectrum (as shown in Fig. S6, ESI†) indicates that the ruthenium species are RuO2.53 Thus, 2.6 wt%RuO2 nanoparticles have been loaded onto CTF-1-100W. According to the TEM image (as shown in Fig. S7, ESI†), the RuO2 particles are of 1 to 6 nm. The total amount of oxygen elements contained in the 2.6 wt%RuO2/CTF-1-100W was calculated by XPS as well. For 50 mg samples, the O elementary amount is lower than 40 μmol, thus even the first run produces two times more oxygen than that contained in the 2.6 wt%RuO2/CTF-1-100W photocatalyst and a total of three runs give six times more oxygen than that in the photocatalyst, proving that oxygen gas can only be generated from water. In order to further exclude that the O2 gas was due to air leaking, N2 was used as an inert standard. As shown in Fig. S8 (ESI†), gas chromatography shows that N2 remains almost stable and O2 increases almost linearly.
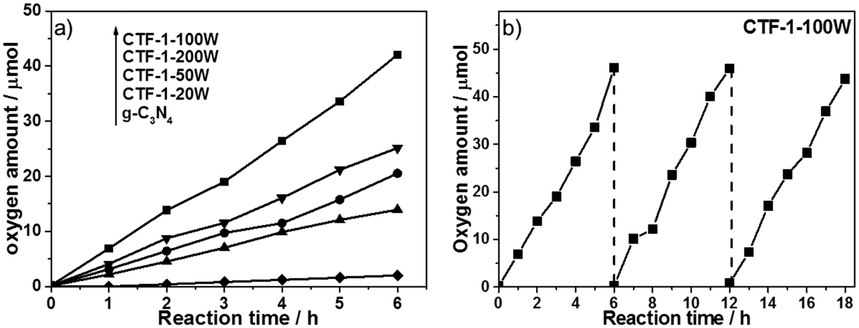 | ||
| Fig. 4 Oxygen production from water using 50 mg 3 wt% RuOx on CTF-1-20W, -50W, -100W, -200W and g-C3N4 containing 0.05 M AgNO3 as an electron scavenger under visible irradiation (λ ≥ 420 nm) for 6 h (a) and the best sample CTF-1-100W under same conditions for 3 runs (b). | ||
The photocatalytic activity for hydrogen evolution on the best O2-evolution sample, CTF-1-100W was also tested to demonstrate the potential of the material for proton reduction. Pt has been utilised as the co-catalyst. According to the XPS spectrum (as shown in Fig. S9 and S10, ESI†), the platinum to nitrogen ratio is 0.68%, thus 2.01 wt%Pt has been loaded onto the CTF-1-100W. Most of the Pt species are Pt metal and have ca. 18% partial reduced Pt(II) and ca. 5% unreacted Pt(IV).54 The TEM image (as shown in Fig. S11, ESI†) of Pt/CTF-1-100W shows that the Pt particles are 1–5 nm and highly dispersed on CTF-1. The hydrogen evolution rate for 2.01 wt% Pt/CTF-1-100W under visible irradiation (λ ≥ 420 nm) is 5500 μmol g−1 h−1, which is 50 times higher than the widely used reference polymer photocatalyst 3 wt%Pt/g-C3N4, as shown in Fig. 5a.6,18 It also presents a near 3 times higher hydrogen evolution rate than the bench mark reported very recently under identical conditions.55 More importantly, even without loading the widely used platinum co-catalyst onto CTF-1-100W, it can catalyse proton reduction, exhibiting a stable hydrogen evolution rate of 102 μmol g−1 h−1 under visible irradiation (λ ≥ 420 nm), as shown in Fig. 5b. Furthermore, the polymer has been found to be stable after a 15 hour test.
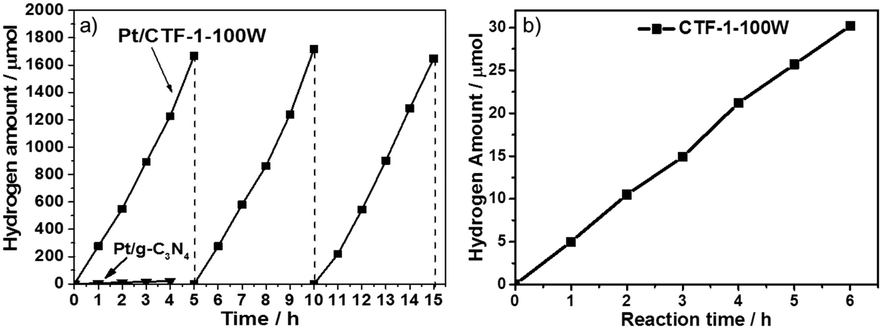 | ||
| Fig. 5 Hydrogen production from water using 50 mg 3 wt%Pt on both CTF-1-100W and g-C3N4 photocatalysts containing 3 vol% methanol as the dispersant and 10 vol% triethanolamine (TEOA) as a hole scavenger under visible light irradiation (λ ≥ 420 nm) (a) and hydrogen production from water using 50 mg CTF-1-100W containing 3 vol% methanol as the dispersant and 10 vol% TEOA as a hole scavenger but without a cocatalyst under visible light irradiation (λ ≥ 420 nm) (b). | ||
To further prove the efficiency of our polymer for water oxidation and proton reduction, the apparent quantum efficiencies (AQE) of CTF-1-100W for both half reactions were measured using band-pass filters as shown in Fig. 6a and b. The AQE is ca. 4% for oxygen evolution at 400 nm and 3.8% at 420 nm. In parallel, for hydrogen evolution, the AQE is 6.3% at 400 nm and 6% at 420 nm, respectively. These are consistent with the optical absorption of the polymer and furthermore, both O2 and H2 evolution are observed up to 600 nm incident illumination. The control experiment of pure CTF-1 in water without light irradiation was carried out, which resulted in no O2 or H2 evolution, indicating that this is a light driven process. As the photocatalyst shows excellent activity for both half reactions, its potential for overall water splitting was also studied by using 3 wt%Pt and 3 wt%RuOx as co-catalysts without any sacrificial agent in pure water. During 6 hours of visible light irradiation, in total ∼5 μmol of H2 and ∼7 μmol of O2 were generated from pure water, which is not an ideal ratio likely due to the inappropriate reduction co-catalyst Pt used, which resulted in a competitive reaction between Pt4+ reduction and proton reduction. However, the results proved that the new polymer has strong potential to be either an efficient proton reduction or water oxidation photocatalyst in a Z-scheme for water splitting, which is underway. To the best of our knowledge, the photocatalytic activity of CTF-1-100W shows much superior quantum efficiency for water oxidation to any reported carbon based or carbon-nitride based polymer photocatalyst, according to the recently published reviews.21,56 As shown in Table S1 (ESI†), the new polymer photocatalyst synthesised by our approach almost presents both the highest H2 and O2 evolution rates under visible light irradiation,22,45,57–62 which is attributed to the well-controlled crystallisation by tuning the microwave power and the avoidance of carbonisation.
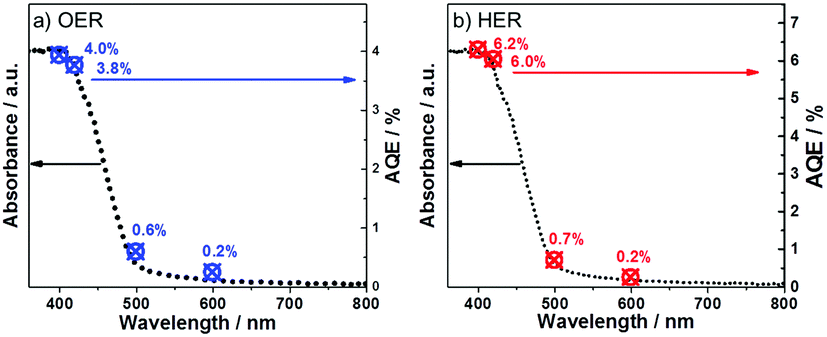 | ||
| Fig. 6 UV-Vis spectra and apparent quantum yield of CTF-1 for the oxygen evolution reaction (a) and for the hydrogen evolution reaction (b). | ||
The band structure of CTF-1 was experimentally confirmed by the UV-Vis spectra, valence band XPS and Mott–Schottky spectra as shown in Fig. 7. According to the UV-Vis spectra as shown in Fig. 7a, CTF-1 has a band gap at ca. 2.48 eV, which is 0.49 eV narrower than that of g-C3N4, thus, CTF-1 can absorb more photo-energy from the visible region. The experimental result of the band gap is smaller than that predicted by DFT, due to the quantum effect of the single sheet used in the DFT calculation. Fig. 7b shows the valence band XPS of both g-C3N4 and CTF-1. CTF-1 is more positive by 0.3 eV than g-C3N4. As the valence band of g-C3N4 is reported at ca. +1.5 eV (vs. NHE, at pH = 0),7,18 the top of the valence band of CTF-1 could be estimated at ca. +1.8 eV (vs. NHE, at pH = 0). The conduction band position of CTF-1 was further confirmed by the Mott–Schottky spectra of the electrode in neutral electrolyte. As shown in Fig. 7c, the conduction band bottom can be estimated to be very close to −0.7 eV (vs. NHE, at pH = 0) and the valence band position should be located at ca. +1.8 eV based on its band gap measured by UV-Vis spectra, which is consistent with the valence band measurement by XPS. Thus, the synthesised polymer has an appropriate band position to reduce protons (0 eV vs. NHE, at pH = 0). More importantly, as shown in Fig. 7d, CTF-1-100W shows ca. 0.3 eV deeper valence band position than bulk g-C3N4 and generates holes with stronger oxidisation potential than that in bulk g-C3N4.62 Thus, higher efficiency water oxidation activity can be achieved by CTF-1, which is consistent with our DFT calculations.
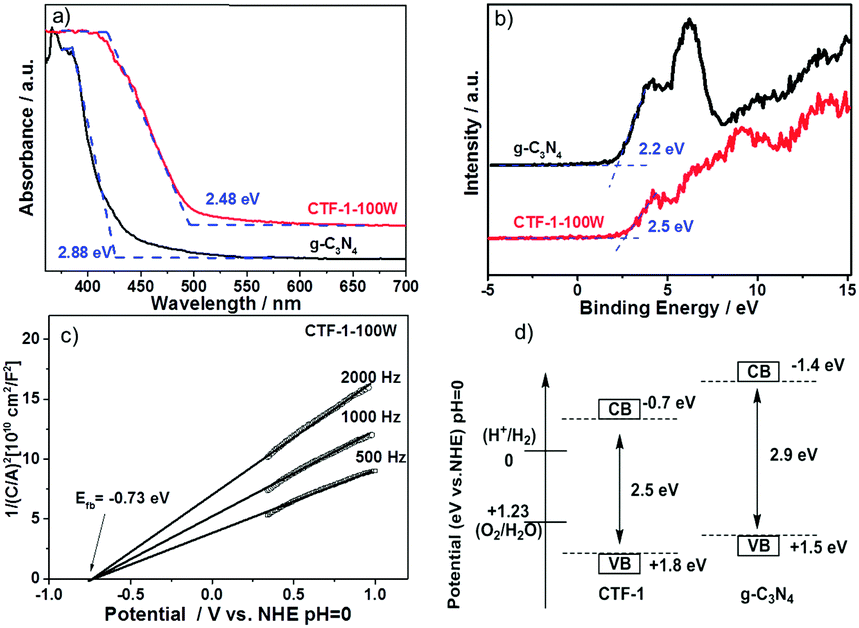 | ||
| Fig. 7 UV-Vis spectra (a); valence band XPS (b) of both g-C3N4 and CTF-1; Mott–Schottky plots of the CTF-1 electrode in 0.1 M Na2SO4, pH = 7 (c), and the band structure of CTF-1 (d). | ||
Photoluminescence (PL) spectroscopy was further undertaken by 325 nm laser excitation in order to study the charge recombination during light irradiation. As shown in Fig. 8a, CTF-1-20W showed the strongest charge recombination, followed by CTF-1-50W. However, the higher microwave power dramatically mitigates the charge recombination of CTF-1 photocatalysts. CTF-1-100W and CTF-1-200W show an order of magnitude smaller charge recombination than the lower power-produced samples, likely indicating a much better charge separation due to the better ordered structure as indicated in Fig. 3. Fig. 8b presents that CTF-1-100W shows much weaker charge recombination than the reference material g-C3N4, indicating a much better charge separation on CTF-1-100W than bulk g-C3N4 under irradiation.9,52,63 This is consistent with the highest photocatalytic activity in both water oxidation and proton reduction. Thus, a higher degree of polymer crystallisation shows less charge recombination, consistent with the higher photocatalytic activity.
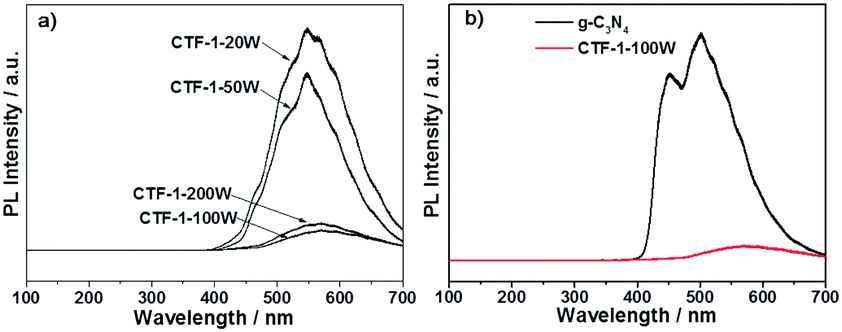 | ||
| Fig. 8 PL spectra of the series of CTFs (a) and the reference material g-C3N4 with the best sample (b). | ||
The stability of CTF-1-100W under light irradiation during water splitting was already assessed by testing the material for O2 and H2 evolution over three cycles in three days (Fig. 4b and 5a). The overall activity is identical during the three runs. According to the post-test FTIR, XPS and Raman spectra (see Fig. S2–S14, ESI†), there are no obvious changes to the material's structure after these reactions. Furthermore, the microwave conditions have been moderated to obtain the most ordered sample and low degree of carbonisation, both of which contribute to high activity of the best polymer.50,51 The BET specific surface area of the CTF-1-100W is around 1 m2 g−1, indicating that its activity can be further improved if the surface area could be improved.
Conclusions
In conclusion, we present an effective polymer, ordered CTF-1, to achieve proton reduction and more importantly, water oxidation. The CTF-1-100W sample, readily fabricated under very moderate conditions, shows a well-defined layered morphology. It exhibits an oxygen evolution rate of 140 μmol g−1 h−1, 20 times higher than the reference polymer g-C3N4 under visible light irradiation (λ ≥ 420 nm), even with a rather low surface area of 1 m2 g−1. It also exhibits a hydrogen evolution rate of 5500 μmol g−1 h−1, which is 50 times higher than that of g-C3N4. The photocatalytic activity enhancement is owing to the well-controlled degree of conjugation which gives better charge separation in an optimised more planar structure. More importantly, compared to the reference material, bulk g-C3N4, CTF-1 has a deeper valence band position and smaller bandgap, thus showing a stronger potential to oxidise water. We also confirm that the AQE is approximately 3.8% at 420 nm for oxygen production and 6% at 420 nm for hydrogen evolution. The structure of CTF-1 remained intact after 3-day runs of water splitting. Its potential for overall water splitting has also been indicated by a 6-hour test, although we targeted at a Z-scheme water splitting system. Our synthetic approach can be further applied to other polymer photocatalysts for efficient solar fuel synthesis by controlling crystallinity, carbonisation and ordering. In addition, this CTF-1 could also be used in other applications such as solar water treatment and organic solar cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All the authors acknowledge financial support from EPSRC (EP/N009533/1 – JX, QR, JT and EP/K021192/1 – SAS, ZXG). QR and JT thank the Royal Society-Newton Advanced Fellowship grant (NA170422) and JT acknowledges the Leverhulme Trust (RPG-2017-122). Computational work was carried out using the UCL Legion High Performance Computing Facility (Legion@UCL), with additional computer support from the ARCHER National Computing Facility by a Resource Allocation Project (Project e454). QR and YL acknowledge financial support from the China Scholarship Council (201406370157 –YL).References
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 CAS.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286–8287 CrossRef CAS PubMed.
- Z. Zou, J. Ye, S. Kazuhiro and A. Hironori, Nature, 2001, 414, 625–627 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- G. Zhang, Z.-A. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- Y. Wang, M. K. Bayazit, S. J. A. Moniz, Q. Ruan, C. C. Lau, N. Martsinovich and J. Tang, Energy Environ. Sci., 2017, 10, 1643–1651 CAS.
- J. R. Bolton, S. J. Strickler and J. S. Connolly, Nature, 1985, 316, 495–500 CrossRef CAS.
- S. Hu, C. Xiang, S. Haussener, A. D. Berger and N. S. Lewis, Energy Environ. Sci., 2013, 6, 2984–2993 CAS.
- H. Döscher, J. F. Geisz, T. G. Deutsch and J. A. Turner, Energy Environ. Sci., 2014, 7, 2951–2956 Search PubMed.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martinc, M. Higashib, D. Kong, R. Abeb and J. Tang, Chem. Rev., 2018 DOI:10.1021/acs.chemrev.7b00286.
- J. Tang, J. R. Durrant and D. R. Klug, Water, 2008, 127, 13885–13891 Search PubMed.
- W. Luo, C. Jiang, Y. Li, S. A. Shevlin, X. Han, K. Qiu, Y. Cheng, Z. Guo, W. Huang and J. Tang, J. Mater. Chem. A, 2017, 5, 2021–2028 CAS.
- W. Luo, Z. Yang, Z. Li, J. Zhang, J. Liu, Z. Zhao, Z. Wang, S. Yan, T. Yu and Z. Zou, Energy Environ. Sci., 2011, 4, 4046 CAS.
- W. Luo, J. Wang, X. Zhao, Z. Zhao, Z. Li and Z. Zou, Phys. Chem. Chem. Phys., 2013, 15, 1006–1013 RSC.
- D. Martin, K. Qiu, S. Shevlin, A. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- V. W. Lau, D. Klose, H. Kasap, F. Podjaski, M.-C. Pignié, E. Reisner, G. Jeschke and B. V. Lotsch, Angew. Chem., Int. Ed., 2017, 56, 510–514 CrossRef CAS PubMed.
- Q. Ruan, W. Luo, J. Xie, Y. Wang, X. Liu, Z. Bai, C. J. Carmalt and J. Tang, Angew. Chem., Int. Ed., 2017, 56, 8221–8225 CrossRef CAS PubMed.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- L. Li, W. Fang, P. Zhang, J. Bi, Y. He, J. Wang and W. Su, J. Mater. Chem. A, 2016, 4, 12402–12406 CAS.
- G. Zhang, C. Huang and X. Wang, Small, 2015, 11, 1215–1221 CrossRef CAS PubMed.
- G. Zhang, S. Zang and X. Wang, ACS Catal., 2015, 5, 941–947 CrossRef CAS.
- S. Chu, Y. Wang, Y. Guo, J. Feng, C. Wang, W. Luo, X. Fan and Z. Zou, ACS Catal., 2013, 3, 912–919 CrossRef CAS.
- L. Sun, X. Zhao, C.-J. Jia, Y. Zhou, X. Cheng, P. Li, L. Liu and W. Fan, J. Mater. Chem., 2012, 22, 23428–23438 RSC.
- X. Chen, C. Zhou, W. Zhao, H. He, R. Li and W. Zou, Comput. Mater. Sci., 2017, 134, 84–92 CrossRef CAS.
- W.-J. Ong, L.-L. Tan, S.-P. Chai and S.-T. Yong, Chem. Commun., 2015, 51, 858–861 RSC.
- M. Li, Y. Wang, P. Tang, N. Xie, Y. Zhao, X. Liu, G. Hu, J. Xie, Y. Zhao, J. Tang, T. Zhang and D. Ma, Chem. Mater., 2017, 29, 2769–2776 CrossRef CAS.
- P. Niu, G. Liu and H. M. Cheng, J. Phys. Chem. C, 2012, 116, 11013–11018 CAS.
- Q. Tay, P. Kanhere, C. F. Ng, S. Chen, S. Chakraborty, A. C. H. Huan, T. C. Sum, R. Ahuja and Z. Chen, Chem. Mater., 2015, 27, 4930–4933 CrossRef CAS.
- X. Bai, L. Wang, R. Zong and Y. Zhu, J. Phys. Chem. C, 2013, 117, 9952–9961 CAS.
- Y. Chen, B. Wang, S. Lin, Y. Zhang and X. Wang, J. Phys. Chem. C, 2014, 118, 29981–29989 CAS.
- R. Godin, Y. Wang, M. A. Zwijnenburg, J. Tang and J. R. Durrant, J. Am. Chem. Soc., 2017, 139, 5216–5224 CrossRef CAS PubMed.
- X. Jiang, P. Wang and J. Zhao, J. Mater. Chem. A, 2015, 3, 7750–7758 CAS.
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 17030 CrossRef CAS.
- Y. Miyamoto, M. L. Cohen and S. G. Louie, Solid State Commun., 1997, 102, 605–608 CrossRef CAS.
- S. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Adv. Mater., 2012, 24, 2357–2361 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Walsh and C. R. A. Catlow, J. Mater. Chem., 2010, 20, 10438 RSC.
- A. V. Krukau, G. E. Scuseria, J. P. Perdew and A. Savin, J. Chem. Phys., 2008, 129, 124103 CrossRef PubMed.
- S. N. Talapaneni, T. H. Hwang, S. H. Je, O. Buyukcakir, J. W. Choi and A. Coskun, Angew. Chem., Int. Ed., 2016, 55, 3106–3111 CrossRef CAS PubMed.
- K. Schwinghammer, S. Hug, M. B. Mesch, J. Senker and B. V. Lotsch, Energy Environ. Sci., 2015, 8, 3345–3353 CAS.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- M. S. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, Phys. Rep., 2005, 409, 47–99 CrossRef.
- L. Liu, Y. Xia and J. Zhang, RSC Adv., 2014, 4, 59102–59105 RSC.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. S. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- M. Hu, J. Reboul, S. Furukawa, N. L. Torad, Q. Ji, P. Srinivasu, K. Ariga, S. Kitagawa and Y. Yamauchi, J. Am. Chem. Soc., 2012, 134, 2864–2867 CrossRef CAS PubMed.
- T. Ben, Y. Li, L. Zhu, D. Zhang, D. Cao, Z. Xiang, X. Yao and S. Qiu, Energy Environ. Sci., 2012, 5, 8370–8376 CAS.
- H. Ou, L. Lin, Y. Zheng, P. Yang, Y. Fang and X. Wang, Adv. Mater., 2017, 29, 1700008 CrossRef PubMed.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- M. A. Matin, E. Lee, H. Kim, W.-S. Yoon and Y.-U. Kwon, J. Mater. Chem. A, 2015, 3, 17154–17164 CAS.
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6, 3921–3931 CrossRef CAS.
- G. Zhang, Z.-A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- J. Bi, W. Fang, L. Li, J. Wang, S. Liang, Y. He, M. Liu and L. Wu, Macromol. Rapid Commun., 2015, 36, 1799–1805 CrossRef CAS PubMed.
- S. Kuecken, A. Acharjya, L. Zhi, M. Schwarze, R. Schomäcker and A. Thomas, Chem. Commun., 2017, 53, 5854–5857 RSC.
- K. Wang, L. M. Yang, X. Wang, L. Guo, G. Cheng, C. Zhang, S. Jin, B. Tan and A. Cooper, Angew. Chem., Int. Ed., 2017, 56, 14149–14153 CrossRef CAS PubMed.
- Z. A. Lan, Y. Fang, Y. Zhang and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 470–474 CrossRef CAS PubMed.
- H. Ou, P. Yang, L. Lin, M. Anpo and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 10905–10910 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ee02981k |
| This journal is © The Royal Society of Chemistry 2018 |