
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon capture and storage (CCS): the way forward
Mai
Bui
ab,
Claire S.
Adjiman
bc,
André
Bardow
 d,
Edward J.
Anthony
e,
Andy
Boston
f,
Solomon
Brown
g,
Paul S.
Fennell
c,
Sabine
Fuss
h,
Amparo
Galindo
bc,
Leigh A.
Hackett
i,
Jason P.
Hallett
c,
Howard J.
Herzog
j,
George
Jackson
c,
Jasmin
Kemper
k,
Samuel
Krevor
lm,
Geoffrey C.
Maitland
cl,
Michael
Matuszewski
n,
Ian S.
Metcalfe
o,
Camille
Petit
c,
Graeme
Puxty
p,
Jeffrey
Reimer
q,
David M.
Reiner
r,
Edward S.
Rubin
s,
Stuart A.
Scott
t,
Nilay
Shah
bc,
Berend
Smit
qu,
J. P. Martin
Trusler
cl,
Paul
Webley
vw,
Jennifer
Wilcox
x and
Niall
Mac Dowell
*ab
d,
Edward J.
Anthony
e,
Andy
Boston
f,
Solomon
Brown
g,
Paul S.
Fennell
c,
Sabine
Fuss
h,
Amparo
Galindo
bc,
Leigh A.
Hackett
i,
Jason P.
Hallett
c,
Howard J.
Herzog
j,
George
Jackson
c,
Jasmin
Kemper
k,
Samuel
Krevor
lm,
Geoffrey C.
Maitland
cl,
Michael
Matuszewski
n,
Ian S.
Metcalfe
o,
Camille
Petit
c,
Graeme
Puxty
p,
Jeffrey
Reimer
q,
David M.
Reiner
r,
Edward S.
Rubin
s,
Stuart A.
Scott
t,
Nilay
Shah
bc,
Berend
Smit
qu,
J. P. Martin
Trusler
cl,
Paul
Webley
vw,
Jennifer
Wilcox
x and
Niall
Mac Dowell
*ab
aCentre for Environmental Policy, Imperial College London, South Kensington, London SW7 1NA, UK. E-mail: niall@imperial.ac.uk
bCentre for Process Systems Engineering, Imperial College London, South Kensington, London SW7 2AZ, UK
cDepartment of Chemical Engineering, Imperial College London, South Kensington, London, SW7 2AZ, UK
dChair of Technical Thermodynamics, RWTH Aachen University, 52056 Aachen, Germany
eCentre for Combustion, Carbon Capture & Storage, Cranfield University, Bedford, Bedfordshire MK43 0AL, UK
fRed Vector Ltd., Barrow Upon Soar, Loughborough, Leics LE12 8JY, UK
gDepartment of Chemical and Biological Engineering, The University of Sheffield, Sheffield S1 3JD, UK
hMercator Research Institute on Global Commons and Climate Change, 10829 Berlin, Germany
iIndustria Mundum AG, Zürich, Switzerland
jMassachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
kIEA Greenhouse Gas R&D Programme (IEAGHG), Pure Offices, Cheltenham Office Park, Hatherley Lane, Cheltenham, Gloucestershire GL51 6SH, UK
lQatar Carbonates and Carbon Storage Research Centre, Department of Chemical Engineering, Imperial College London, South Kensington Campus, London SW7 2AZ, UK
mDepartment of Earth Science & Engineering, Imperial College London, South Kensington, London, SW7 2AZ, UK
nUS Department of Energy, National Energy Technology Laboratory (NETL), Pittsburgh, PA, USA
oSchool of Chemical Engineering and Advanced Materials, Newcastle University, Newcastle-upon-Tyne NE1 7RU, UK
pCSIRO Energy, Mayfield West, New South Wales 2304, Australia
qDepartment of Chemical and Biomolecular Engineering, University of California Berkeley, Berkeley, CA 94720, USA
rEnergy Policy Research Group, Judge Business School, University of Cambridge, Cambridge, UK
sEngineering & Public Policy and Mechanical Engineering, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, USA
tUniversity of Cambridge, Department of Engineering, Cambridge CB2 1PZ, UK
uLaboratory of Molecular Simulation, Institut des Sciences et Ingénierie Chimiques, Valais Ecole Polytechnique Fédérale de Lausanne (EPFL), Rue de l'Industrie 17, CH-1951 Sion, Switzerland
vDepartment of Chemical Engineering, The University of Melbourne, Victoria 3010, Australia
wThe Peter Cook Centre for Carbon Capture and Storage, The University of Melbourne, Victoria 3010, Australia
xChemical and Biological Engineering Department, Colorado School of Mines, Golden, Colorado 80401, USA
First published on 12th March 2018
Abstract
Carbon capture and storage (CCS) is broadly recognised as having the potential to play a key role in meeting climate change targets, delivering low carbon heat and power, decarbonising industry and, more recently, its ability to facilitate the net removal of CO2 from the atmosphere. However, despite this broad consensus and its technical maturity, CCS has not yet been deployed on a scale commensurate with the ambitions articulated a decade ago. Thus, in this paper we review the current state-of-the-art of CO2 capture, transport, utilisation and storage from a multi-scale perspective, moving from the global to molecular scales. In light of the COP21 commitments to limit warming to less than 2 °C, we extend the remit of this study to include the key negative emissions technologies (NETs) of bioenergy with CCS (BECCS), and direct air capture (DAC). Cognisant of the non-technical barriers to deploying CCS, we reflect on recent experience from the UK's CCS commercialisation programme and consider the commercial and political barriers to the large-scale deployment of CCS. In all areas, we focus on identifying and clearly articulating the key research challenges that could usefully be addressed in the coming decade.
Broader contextCarbon capture and storage (CCS) is recognised as being vital to least cost pathways for climate change mitigation, and in particular the negative emissions technologies (NETs) that are key to limiting warming to “well below” 2C. However, it has not yet been deployed on the scale understood to be required, owing to a variety of technical, economic and commercial challenges. This paper provides a state-of-the-art update of each of these areas, and provides a perspective on how to the discipline forward, highlighting key research challenges that should be addressed over the course of the next decade. Importantly, this perspective balances scientific, policy and commercial priorities. |
1 Introduction
This paper is the third installment in a series of publications over several years in Energy & Environmental Science.1,2 The first (published in 2010) provided an introduction to CO2 capture technologies, with an overview of solvent-based chemisorption (amines and ionic liquids), carbonate looping, oxy-fuel combustion technologies, CO2 conversion and utilisation (CCU) and multi-scale process engineering of CCS.1 The second installment presented an update on developments in amine scrubbing, ionic liquids, oxy-combustion and calcium looping. New topics added in this second paper include chemical looping combustion, low temperature adsorbents, direct air capture technologies, flexible CCS operation, CO2 transport and storage, and a historical overview of the UK and EU CCS policy and legislation.2Distinct from the previous installments, this third paper sets out to comprehensively review the state-of-the-art developments in CCS, whilst also providing a holistic perspective on the role of CCS technologies in mitigating anthropogenic climate change. We first discuss the current status of CCS development and highlight key CCS technologies that are near commercialisation phase (Section 2). Then in Section 3 we contextualise CCS technology by considering its representation and utilisation in integrated assessment models (IAMs), challenging the view that it is a “bridging technology”, likely to be relevant for only a few decades. We then go on to quantify and qualify the role and value of CCS at a more granular level by evaluating the way in which CCS interacts with national scale electricity systems. This in turn helps us address the question of what service CCS provides to the electricity system, with whom is CCS competing and what technologies does CCS complement.
We then move on to consider the utility of CCS in decarbonising the industrial sector, with a focus on the key emitters – the production of iron and steel, cement and oil refining and petrochemicals. Throughout, we aim to challenge the perception that industrial CCS is uniquely costly, showing that, for example, the cost of decarbonising the refining sector is essentially “lost in the noise” of market fluctuations of the end use sectors.
Section 4 of the paper considers key post-combustion CCS technologies in detail. The purpose of this paper is not to enumerate the panoply of technologies that are available for capturing CO2. Rather, we focus on solid- and liquid-phase sorbents, and attempt to specify key research questions that need to be address in these areas. We then select three particularly promising alternative technologies for CCS in Section 5: chemical looping combustion, membranes and ionic liquids.
It is well known that the thermophysical and kinetic properties of the sorbents used for CO2 capture dictate both the capital and operating cost of the processes in which they are used. For this reason, there is a concerted effort to rationally design new sorbent materials, with the bulk of the effort in the development of liquid sorbents, where available theories are more readily applied. Thus, we present an assessment of SAFT-based approaches to model and design new materials in Section 6, with a focus on how efforts at the molecular and process scales might be linked.
Before CO2 can be safely and reliably sequestered, it must be transported from source to sink. Whilst the majority of studies assume pipeline transport, ship and rail transport are potential alternatives; these other transport options are discussed in Section 7. Similarly, despite the fact that CO2 transport by pipeline is exceptionally mature, the impact of capturing CO2 from a diverse set of power and industrial sources on the quality of CO2 being transported is sufficiently important to warrant careful consideration.
The typical fate of CO2 is to be sequestered, either in a saline aquifer or, potentially, used for enhanced oil recovery (EOR). The various challenges of operation, monitoring and verification of CO2 storage are discussed in Section 8, whereas Section 9 discusses CO2-EOR. A potential alternative to the storage of CO2 is its re-use – the valorisation of CO2 to produce marketable compounds. The argument is sometimes made that this can both contribute to climate change mitigation and provide an attractive revenue stream. Section 10 discusses the potential for CO2 conversion and utilisation (CCU), also its merits and challenges are presented and considered.
In light of the global commitment achieved in Paris in December, 2015,3 we have extended this paper to include key negative emissions technologies (Section 12); bioenergy with CCS (BECCS) and direct air capture of CO2 (DAC). These areas are of particular importance owing to their potential importance and their controversy.
Despite the fact that there are currently 37 CCS projects at various stages in the Americas, Europe, Middle East and Asia-Pacific,4 CCS continues to languish as an “orphan technology”.† With decades of technical experience across the entire value chain, it is clear that it is not a lack of technical expertise that is inhibiting the commercial deployment of CCS technology. Thus, we have devoted a section of this paper to consider “what needs to happen” from a commercial perspective (Section 13), drawing upon experience developed as part of the UK's most recent CCS commercialisation programme.5 Having provided this perspective from the private sector, we then complement this with an international analysis of the political economy of CCS (Section 14). Section 15 then concludes with a proposed approach to evaluate the utility of a “novel technology” and feasibility of particular targets by identifying limitations that might prove to be showstoppers.
2 Current status of CCS development
Carbon capture and storage is expected to play an important role in meeting the global warming targets set by the IPCC6 and at COP21.3 There is a suite of technologies being developed for the capture, transport, storage and utilisation of CO2. Typically, technology development will progress in a series of scale-up steps: (i) bench or laboratory scale, (ii) pilot-scale, (iii) demonstration scale, and lastly (iv) commercial scale.7Fig. 1 summarises the current development progress of different CCS technologies on the TRL scale.‡ As illustrated by Fig. 1, there is congestion of technologies at the TRL 3, TRL 6 and TRL 7 development phases. The progression of a technology beyond TRL 3 requires further research funding, whereas advancing technologies beyond TRL 5 and TRL 7 needs significant financial investment and/or commercial interest (e.g., in the case of polymeric membranes). Further detailed discussion on the technical development of the individual CCS technologies is presented in the following sections of this paper. Here in this section, we highlight the key CCS technologies that have reached (or close to reaching) the commercial phase of development.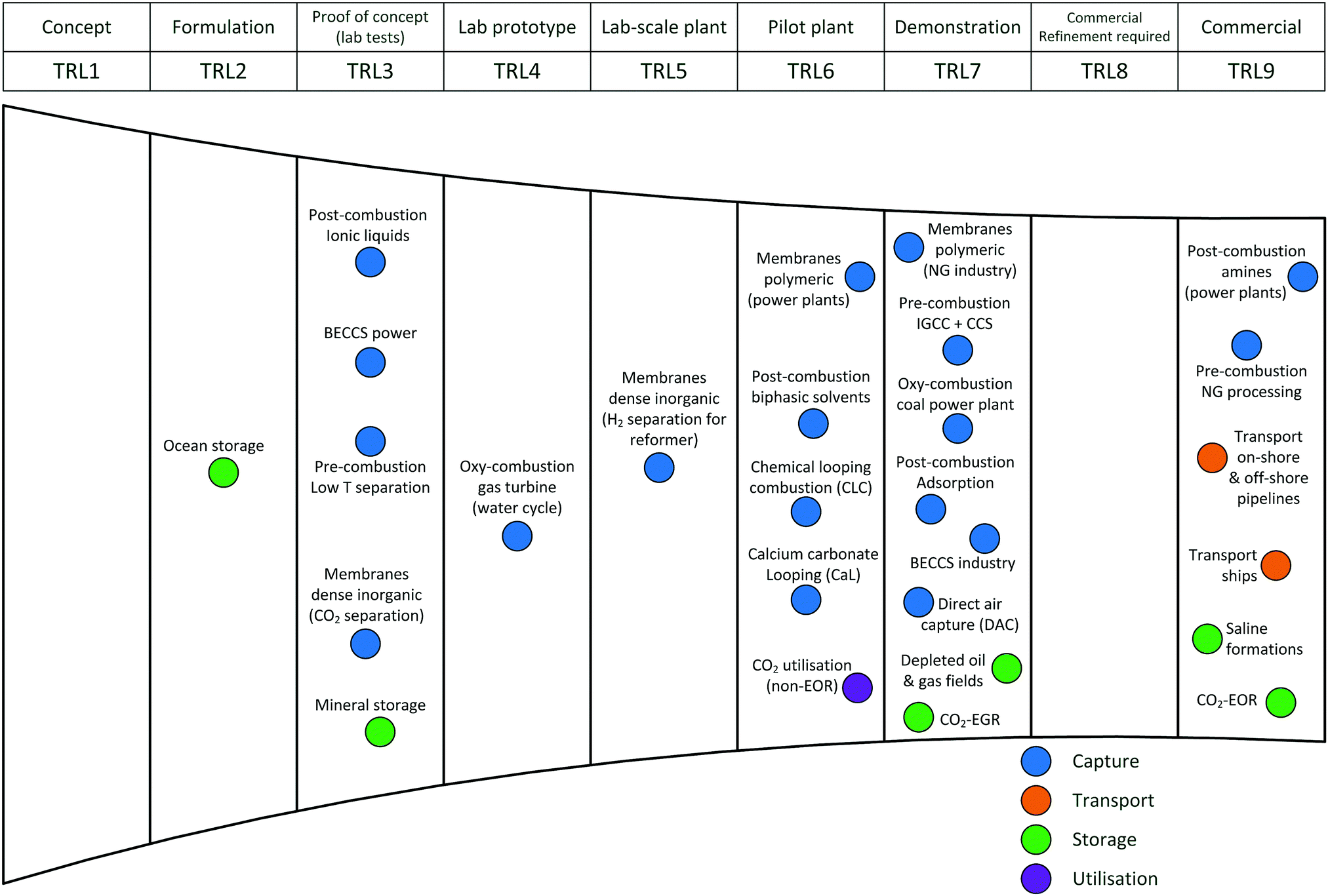 | ||
| Fig. 1 Current development progress of carbon capture, storage and utilisation technologies in terms of technology readiness level (TRL). BECCS = bioenergy with CCS, IGCC = integrated gasification combined cycle, EGR = enhanced gas recovery, EOR = enhanced oil recovery, NG = natural gas. Note: CO2 utilisation (non-EOR) reflects a wide range of technologies, most of which have been demonstrated conceptually at the lab scale. The list of technologies is not intended to be exhaustive. | ||
CO2 capture
Chemical absorption (e.g., using aqueous amine solutions) has been used to remove CO2 from natural gas for decades,11 thus, it is considered to have a TRL of 9. This technology has been utilised in two commercial-scale post-combustion capture facilities in coal-fired power plants, Boundary Dam12,13 and Petra Nova.14,15 Recent developments in polymeric membranes have enabled the technology to successfully achieve demonstration scale (TRL 7). The Polaris membrane is now available commercially and has been used for CO2 separation from syngas.16 Air Products are licensing a polymeric membrane developed at NTNU, which can be applied to coal-fired power plants and other combustion processes (still under development).17 Thus, The first “commercial-ready” direct air capture (DAC) plant recently opened in Hinwil, Switzerland on May 2017,18 with the support of cost contributions from the Swiss Federal Office of Energy. The plant supplies 900 tonnes of CO2 annually to a nearby greenhouse.19 Capture technologies that have also reached TRL 7 (demonstration) (e.g., oxy-combustion coal power plants, adsorption) could also potentially reached commercial status in the near future. In contrast to post-combustion capture, integrated gasification combined cycle (IGCC) with CCS has been less successful with the Kemper County IGCC Project being suspended recently.20 Southern Company's decision to halt the project came after encountering a series of problems, these include failure to meet the delivery deadline, severe technical issues and being majorly over budget.21,22CO2 transport
The technologies for CO2 transport are well established. There are >6500 km of CO2 pipelines worldwide (both on-shore and off-shore), most of which are associated with EOR operation in the United States.23 The technology for CO2 transport with ships is also relatively mature.24 As these transport technologies are currently being used in commercial applications, all have a TRL of 9.CO2 storage
As many commercial-scale CCS projects already use CO2-enhanced oil recovery (EOR), 13 of the 17 operating commercial-scale CCS projects, there is a significant amount of existing experience and knowledge, which has enabled CO2-EOR to reach TRL 9. Similarly, saline formations have been used for CO2 storage at commercial-scale project, including Sleipner CO2 Storage, Snøhvit CO2 Storage and Quest (on-shore and off-shore). In contrast, CO2 storage by enhanced gas recovery (EGR)25 and storage in depleted oil and gas fields have not reached operation at commercial-scale, thus, both are still at the demonstration phase (TRL 7). Ocean storage and mineral storage are still in the early phases of development.CO2 utilisation
There are a number of facilities that utilise CO2 for various applications. These commercial CO2 utilisation processes are TRL 9 as they are mature technologies. Most are in the food and beverage industry and some in chemical production (e.g., urea, methanol).26 Several projects utilise CO2 for mineral carbonation, for example, Searles Valley plant (US). In Saga City, Japan, CO2 capture from waste incineration is utilised for the cultivation of crops and algae.27 The CO2 for each project is mainly sourced from industrial processes (e.g., fertiliser production, ammonia production, ethylene glycol plants), but some projects capture the CO2 from power plant flue gas.26Commercial-scale CCS projects
Deployment of large scale CCS projects has been slow. Of the 37 major large scale CCS projects, 17 of these are in operation, 4 in construction and the remainder are in varying stages of development.4 As shown in Fig. 2 and 3, the majority of the commercial large-scale CCS projects are located in the United States. In terms of the project life cycle (i.e., identify, evaluate, define, execute and operate), the US also has the greatest proportion of projects in operation. For all but one of these projects, enhanced oil recovery is the primary storage for the captured CO2. Furthermore, the projects in the US have the largest CO2 capture capacity compared with projects in the rest of the world: Century Plant captures 8.4 MtCO2 per year, whereas Shute Creek Gas Processing Facility capture 7 MtCO2 per year.4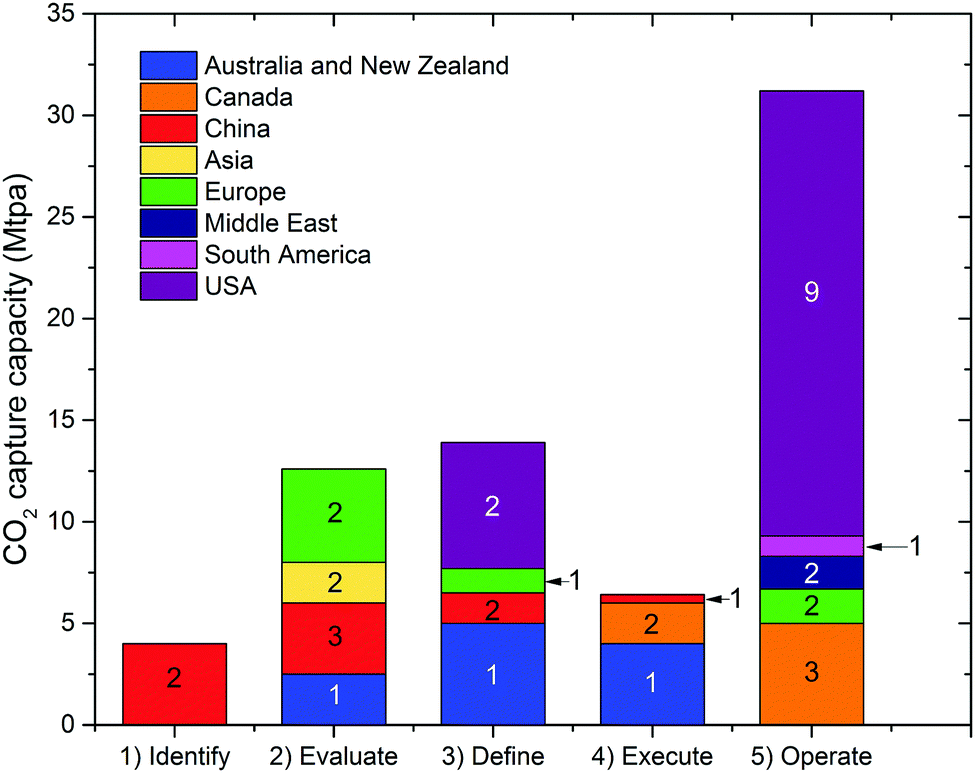 | ||
| Fig. 2 The CO2 capture capacity of commercial-scale CCS projects worldwide. The number labelled on each proportion of capture capacity corresponds to the number of projects. Data from the Global CCS Institute.4 | ||
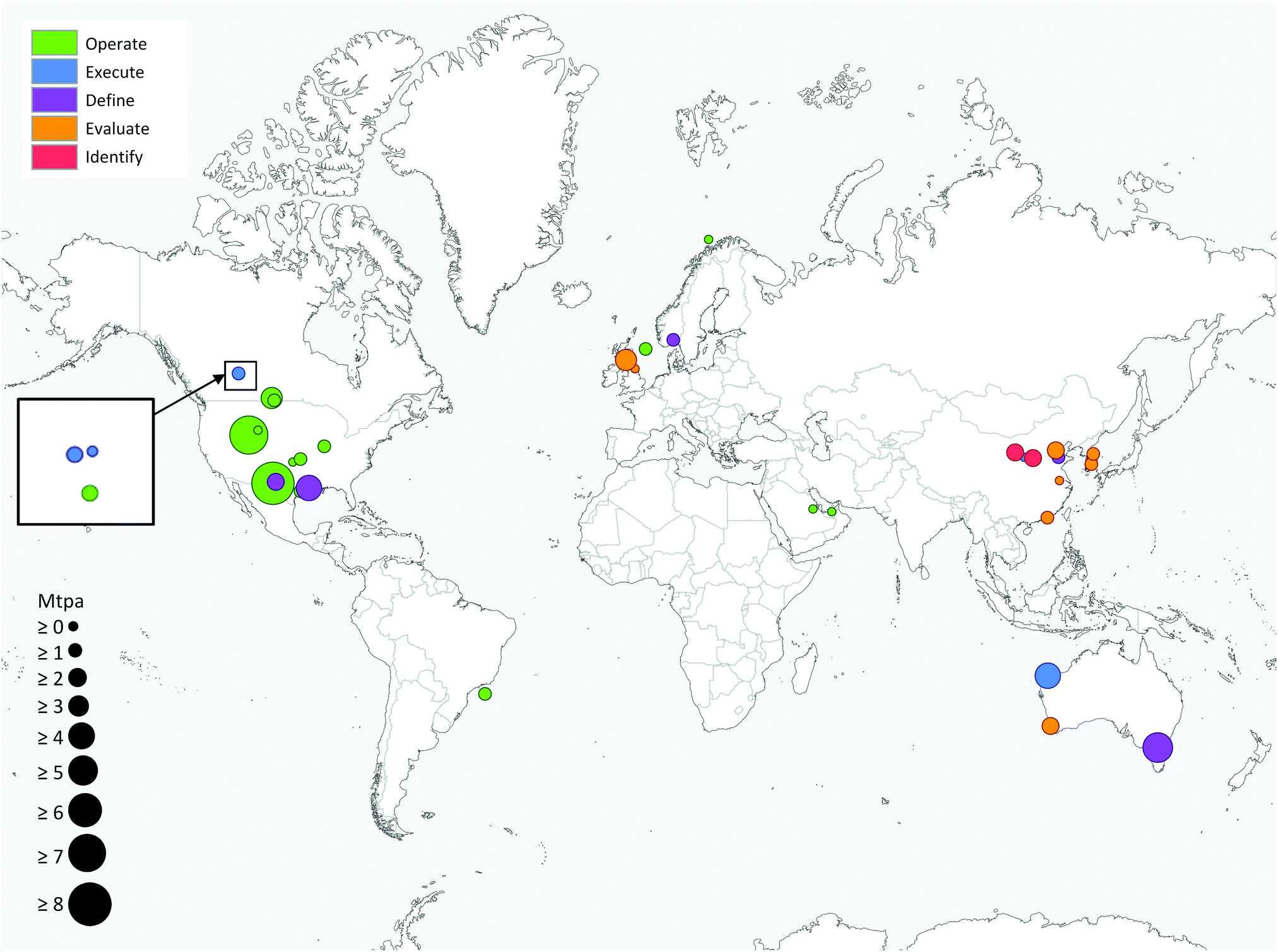 | ||
| Fig. 3 Commercial-scale integrated CCS projects around the world. Circle size is proportional to the CO2 capture capacity of the project and the colour indicates the lifecycle of the project. Data from the Global CCS Institute.4 | ||
Although China has the second highest number of projects, only one of these is in the execute phase (Yanchang Integrated CCS Demonstration), and most are in early stages of development (e.g., pre-feasibility, FEED studies). The CO2 capture capacity of the projects in China range between 0.4–2 MtCO2 per year. Europe has the third highest number of large-scale projects, with two operational projects in Norway: the Sleipner CO2 Storage Project captures 1 MtCO2 per year, and Snøhvit CO2 Storage Project 0.7 MtCO2 per year. Of the five projects in Canada, three are in operation: (i) Great Plains Synfuel Plant and Weyburn-Midale Project (3 MtCO2 per year), (ii) Boundary Dam CCS Project (1 MtCO2 per year), and (iii) Quest (∼1 MtCO2 per year). There are also operating CCS projects in Brazil, Saudi Arabia and United Arab Emirates with CO2 capture capacities ranging from 0.8–1 MtCO2 per year. A fundamental requirement for the success of CCS projects in all of these projects is the availability of safe geological storage for the capture CO2. Furthermore, other factors that can help bring CCS projects into operation phase include secure financial funding, as well as supportive policy and legislative frameworks.28
3 Role and value of CCS
3.1 Climate change mitigation
Integrated Assessment Models (IAMs) have been at the heart of the Intergovernmental Panel on Climate Change's (IPCC) assessment of pathways towards keeping average global warming to less than 2 °C within this century.6 They provide a means to explore the future role of particular technologies in meeting climate targets such as renewables or nuclear.CCS is one of the very attractive options in the IAMs mitigation portfolios, as it has a number of advantages. First, CCS can be integrated into existing energy systems without requiring large amendments to the system itself. Clearly, renewable technologies become more expensive at high penetration rates as a result of the need for the infrastructure to accommodate intermittency.29 Furthermore, CCS is a viable option for the decarbonisation of emission-intensive industries such as cement production (specific industrial CO2 capture costs are given in Section 3.3).30 And finally, CCS can be combined with low-carbon or carbon-neutral bioenergy (BECCS) to generate negative emissions,31i.e. while the cultivation of the feedstock biomass sequesters about as much CO2 as is generated during the process of producing energy (bio-electricity or biofuels), additionally capturing the latter leads to a withdrawal of CO2 from the atmosphere.32 BECCS has the double benefit of mitigating emissions and generating energy, making it attractive from the cost-optimisation perspective of an IAM.
Finally, concerning storage, while many models have a regional differentiation of storage capacity, only a few models allow for international trade in storage capacity. The maximum capacity ranges between 3500 GtCO2 cumulative, and unlimited storage. Transportation and storage cost (excluding capture cost) varied between 10–300 US$ per ton CO2, depending on model and storage type.33 All of the models considered transportation and storage costs at the lower end of this range. Models that also considered high transportation and storage cost include the POLES model (upper range value of $300 USD) and the GRAPE model (upper range cost of $262 USD).33 The higher values for storage cost are associated with options that were offshore, enhanced coal bed methane (ECBM) and at greater depths (e.g., 2000–3000m).34
The IAMs thus differ widely in their deployment of CCS, yet the model intercomparison, which is the basis for the numbers cited above, could not explain the divergence of results on the basis of model type, model assumptions or the way in which CCS has been modelled. So either these are not the drivers of the difference or their impact is confounded by other factors via system effects. Individual model studies find that CCS contributes 50% more to mitigation if technological learning is included (Riahi et al.35 cumulative storage of 150–250 GtCO2) and that the contribution of CCS is sensitive to its cost in 2050 but not in 2100.36
More specifically, a recent report on CCS by the IEA41 reviews the progress of the past 20 years and concludes that the current rate of progress is falling short of what is required to achieve climate goals. This is further underlined in the analysis of the INDCs by Spencer et al.:40 national and global scenarios based on the Paris pledges both show little deployment of CCS, with a share of CCS in electricity generation of only 3% in 2030 for the USA, China, Japan and the European Union. This is further exacerbated by the opposition against CCS, which is motivated by perceived uncertainties concerning its safety and the fear that it will serve to prolong the dependence on fossil fuels and be a barrier to greater utilisation of renewable power.42,43
The next section will present the current state-of-the-art knowledge on the role of CCS – and by extension BECCS – in IAMs. The review will first focus on an model intercomparison exercise of 18 IAMs33 (EMF27¶) and then widen towards the low stabilisation pathways in the IPCC's Fifth Assessment Report6 (AR5). Secondly, an investigation of the scenarios consistent with the more ambitious 1.5 °C climate goals adopted at COP21 in Paris (UNFCCC 2015), serves to underline the main insights on CCS and puts specifically BECCS into the spotlight.
State-of-the-art scenarios focusing on the 2 °C target. This subsection draws on the results of the model intercomparison presented in Koelbl et al.,33 as it is the most recent and most comprehensive assessment specifically targeted at the role of CCS in long-term climate change mitigation scenarios. The study itself draws on the output of the 27th Energy Model Forum (EMF), to which 18 IAMs contributed, thus providing an excellent opportunity for a systematic comparison of results with respect to the role of CCS.||
Koelbl et al.33 find that CCS plays an important role in all of the models’ mitigation portfolios that were investigated. While the range of CO2 captured varied widely between models (up to 3050 GtCO2 cumulatively until 2100 in some instances), none of them captured less than 600 GtCO2. Table 1 shows the ranges across scenarios with different stabilisation targets and renewables penetration by model type** based on Koelbl et al.33 While the authors cannot easily explain the large range across models by looking at individual model assumptions (see Section 3.1.1 and Table 1), the fact that models consistently capture a minimum of 600 GtCO2 cumulatively until 2100 – which would be more than half of the required emission reductions consistent with a 2 °C pathway†† – does give a sense for the magnitude and importance of the role of CCS in IAMs.
| Scenario | Model type | |||
|---|---|---|---|---|
| Hybrid | Macro-focus | Tech-focus | ||
| 1 | Cumulative storage 450 ppm | 730–2411 GtCO2 | — | 353–1629 GtCO2 |
| 2 | Cumulative storage 550 ppm | 655–2962 GtCO2 | 1262 GtCO2 | 846–1686 GtCO2 |
| 3 | Cumulative storage 450 ppm, limited renewables | 625–2447 GtCO2 | —— | 1232–1366 GtCO2 |
Furthermore, the authors do not find a decreasing role for CCS over time. On the contrary, the CCS share in primary energy is mostly higher in the second half of the century compared to the first. In particular, the ranges for capture rates in Koelbl et al.33 are 5–23 GtCO2 per year in 2050 and 8–50 GtCO2 per year in 2100. This undermines the reputation of CCS as a bridging technology and further underlines its importance in IAMs, which seek to achieve ambitious climate targets. The importance is further enhanced under pessimistic assumptions about technological development of renewable energy for a given climate target, indicating little flexibility for the cost-optimal deployment of alternatives.
Finally, the use of BECCS‡‡ in the models’ CCS fuel portfolio increases with the stringency of the target. This is mostly connected to substitution for coal and natural gas over time. In response to the concerns with respect to large-scale cultivation of biomass for BECCS and the reservations concerning CCS discussed above, the EMF models also produced a whole array of scenarios limiting the use of both biomass and CCS. Although these scenarios achieve the same target, they are consistently characterised by higher costs, which is consistent with earlier findings by e.g. Azar et al.49 and later confirmed by the results of the IPCC's AR5.6
In the absence of CCS, the total cost of climate change mitigation increased by 138%, whereas limited bioenergy availability increased cost by 64%.§§6 The integration of CCS into an energy system provides a significantly greater reduction in CO2 emissions compared to wind technology.50 With limited CCS and biomass availability, the deployment of nuclear, intermittent solar/wind, interconnection and gas-fired power needs to increase, consequently leading to higher total system cost.51 The increase in mitigation cost is associated with the delay in technology deployment6 (e.g., more time to establish infrastructure), use of more expensive technologies (nuclear), and maintaining grid stability (e.g., intermittency requires the addition of “back-up” capacity and part-load/flexible operation).50
In particular, the IPCC scenarios associated with a more than even chance of achieving the 2 °C target are characterised by average capture rates of 10 GtCO2 per year in 2050 and 25 GtCO2 per year in 2100 and cumulative storage of 800–3000 GtCO2 by the end of the century.29 With respect to finding more expensive mitigation strategies when CCS is not available, it is important to note that under these circumstances, there are actually a significant number of IAMs, which do not find a feasible solution at all: Riahi et al.52 conduct a model intercomparison, where a third of the IAMs do not find a feasible solution at 450 ppm without CCS under optimal circumstances. If there is further delay in mitigation, this share drops to a fifth. In other words, the target is not just more expensive to reach, but not reachable at all, given the current parameterisation of the models.
In addition, the AR5 scenarios have been under scrutiny for their deployment of CCS in conjunction with bioenergy. The 101 out of 116 scenarios leading to concentration levels of 430–480 ppm CO2-equivalent (CO2eq.) (considered to be consistent with a 66% probability of limiting warming to below 2 °C) require global net negative emissions between 2050 and 2100. About 50% of the scenarios feature BECCS exceeding 5% of primary energy supply.31
While these aspects of the IPCC scenarios have caused some people to doubt the feasibility of achieving the 2 °C target (e.g. Peters53), the role of CCS and particularly BECCS become even more important in light of the increased level of ambition following the 2015 Paris COP.54
Towards 1.5 °C. What is currently available in terms of 1.5 °C IAM scenarios is much less than what is presented above on 2 °C from the IPCC's AR5. This subsection draws on work from Rogelj et al.55 and Luderer et al.,56 which offer an assessment of what is currently available on 1.5 °C.¶¶
The most outstanding feature that systematically distinguishes the 1.5 °C from the 2 °C IAM scenarios examined in Rogelj et al.55 is that there is not a single pathway with a 50% probability of achieving the target without overshooting it until 2100. That is, the average global temperature increase will at some point exceed 1.5 °C, before returning to this level at the end of the century.
This implies that much of the CO2 emitted in the first half of the century will need to be removed from the atmosphere again. In other words, emissions have to be negative at some point. Indeed, the analysis in Rogelj et al.55 shows that there are no feasible 1.5 °C scenarios without negative emissions. In particular, the cumulative negative emissions are between 450 and 1000 GtCO2 until 2100. This is in stark contrast to some 2 °C scenarios, which do manage to reach their target without carbon removals. Luderer et al.56 point out that energy efficiency improvements can have this effect for 2 °C scenarios.
In the current IAMs, these negative emissions are primarily achieved by the deployment of BECCS.|||| This has triggered a discussion reflecting on large concerns not only about CCS (cf. discussion in Section 3.1), but also with respect to the implications of the large amounts of biomass that would be needed to achieve sufficient scales to reach the level of negative emissions needed for ambitious climate change mitigation. In an ex-post assessment of the amounts of negative emissions through BECCS in the IPCC's AR5, Smith et al.61 estimated the range of land area, costs, water and nutrients footprints and biophysical effects. They find that, indeed, the areas of land, which would be needed, are large (380–700 Mha by 2100).***
Relating the primary energy of the biomass (in EJ) used in BECCS to the amount of CO2 stored geologically is complex. This relationship strongly depends on the choices made in the cultivation, harvesting, transport and utilisation of the biomass throughout the BECCS supply chain. Assuming that all of the CO2 sequestered by the biomass is assumed to be released in the flue gas upon combustion, the amount of CO2 sequestered per MJ of biomass would then depend on the capture rate applied at the BECCS facility, the biomass carbon content, the biomass heating value and the biomass carbon footprint. Considering a capture rate between 60% and 90%, a biomass carbon content between 45%dry and 50%dry, an HHVdry between 18 and 20 MJ kg−1 (dry mass) and biomass carbon footprint between 0 and 36 gCO2 MJ−1, the amount of CO2 sequestered would be found to be between 14 and 92 gCO2 MJ−1. One EJ of biomass could thus capture between 14 and 92 MtCO2 per year, resulting in an annual requirement of between 130 and 860 EJ by 2100 to capture 12 GtCO2 per year,63 however, the total primary energy supply in 2100 is expected to grow to 1300–1800 EJ.64
Smith et al.61 also point to other negative emission technologies, which could complement BECCS to alleviate the pressure on land that is also needed to feed a growing population, host biodiversity and many other ecosystem services. The dominance of BECCS in the current scenarios may be due to the fact that other options (see Table 2) are not included in the models. Incorporating other negative emission technologies could potentially lead to a lower uptake of BECCS, assuming that these other technologies are cost-competitive in comparison to BECCS, especially in scenarios limiting CCS and/or biomass use.
| Technology | Description |
|---|---|
| Direct air capture (DAC) | Chemicals (e.g. amines or sodium hydroxide) are used to absorb CO2, which is then mineralised for solid storage, or is stored in geological formations. |
| Soil carbon sequestration (SCS) | Carbon soil sequestration is enhanced by increasing inputs or reducing losses (see Smith57). |
| Biochar | Through pyrolysis, biomass is made more resistant to decomposition and then added to the soil to store embedded carbon (see Smith57). |
| Enhanced weathering (EW) | Minerals like olivine that naturally absorb CO2 are ground and spread out to increase their surface area and make them absorb CO2 more rapidly. |
| Ocean fertilisation (OF) | Iron can be used to make ocean phytoplankton absorb more CO2 through photosynthesis, and then sink to the deep ocean and sequester carbon after their death. |
| Indirect ocean capture | Oceanic carbon uptake represents the largest sink for anthropogenic CO2, absorbing about 40% of CO2 emissions from the atmosphere since the start of the industrial era.58 The use of an efficient method for the extraction of CO2 (i.e. dissolved carbon) from seawater provides a method of CO2 removal from the atmosphere, for example, using a pH swing with bipolar membrane electrodialysis59 or electrolytic cation exchange units.60 |
The first is related to the above-mentioned concerns with respect to the high share of BECCS in low-stabilisation portfolios. More research along the lines of Smith et al.61 and Fajardy and Mac Dowell63 will help to shed light on the implications for other policy goals such as ensuring food security, as well as biodiversity and other ecosystem services. In addition, as can be seen in the adopted outline for the Special Report,65 climate change mitigation is closely embedded into a broader context of sustainable development, indicating that the new scenarios will also be designed to reflect a wider set of policy objectives.
The second development is the growing body of knowledge on other options for negative emissions and their interplay with what is currently included in the IAMs. There are already some IAMs that are experimenting with the integration of enhanced weathering66 and direct air capture (DAC) is also an important candidate for integration into the IAMs despite current uncertainty on technical performance and cost.67
The main CCS research priorities in IAMs include:
• More within-model studies to understand better the interactions between CCS characteristics and modelled deployment/cumulative storage, which are difficult to discern in model intercomparisons.33
• Update parameterisation with new insights from CCS research and demonstration.
• Within-model studies also to better understand system dynamics.
• Complement with geographically explicit techno-economic engineering approaches and geological suitability analysis to identify key areas for deployment and more realistic potentials.32,68
• Explore scenarios considering technology choice depending on institutional barriers and social acceptance.
• Include other negative emissions options (e.g. direct air capture, soil carbon sequestration, enhanced weathering) in addition to BECCS to decrease competition for storage capacity, and biomass (also other side effects, such as competition for land and water63).
Clearly, as ambitions become higher and action is further delayed, CCS will continue to play an important role in mitigation pathways. Broadening the portfolio of energy options to include CCS would improve the affordability of a near-zero emissions energy system.69 This is especially true in the case of combining it with bioenergy to generate negative emissions. Yet, especially with respect to negative emissions, many research gaps remain, which will need to be urgently addressed to keep this window of opportunity open.70
3.2 Integration of CCS into the electricity system
The following modelling assessment has been conducted in the context of the UK electricity system (i.e., uses data for the UK). There are a number of similar studies on the UK energy system which evaluate different scenarios.50,51,71–74 Also, energy systems in the context of other countries have been evaluated, for example, the US,75,76 Greece,77 Poland,78 or for Europe in general.79![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 82 to 100 g kWh−1.83–85 This will allow the partial decarbonisation of the heating and transport sectors via electrification. The UK has also adopted the 20–20–20 targets proposed by the European Commission.86 This requires approximately 30% of electricity to come from renewable sources by 2020 to achieve the UK's overall target of 15% of primary energy from renewable sources, e.g., solar, wind and biomass, however, excludes nuclear and CCS.86,87 This has led to a suite of policies that have subsidised the production of electricity from renewable sources. A significant proportion of this has come from intermittent sources such as wind and photovoltaic (PV). In financial year 2014/15, more than 50 TWh (representing 15% of the 340 TWh generated) was from intermittent renewable energy sources (IRES).88
82 to 100 g kWh−1.83–85 This will allow the partial decarbonisation of the heating and transport sectors via electrification. The UK has also adopted the 20–20–20 targets proposed by the European Commission.86 This requires approximately 30% of electricity to come from renewable sources by 2020 to achieve the UK's overall target of 15% of primary energy from renewable sources, e.g., solar, wind and biomass, however, excludes nuclear and CCS.86,87 This has led to a suite of policies that have subsidised the production of electricity from renewable sources. A significant proportion of this has come from intermittent sources such as wind and photovoltaic (PV). In financial year 2014/15, more than 50 TWh (representing 15% of the 340 TWh generated) was from intermittent renewable energy sources (IRES).88
It has been proposed that the UK could generate a very high proportion (if not all) its energy from IRES.89–91 However, there are a number of issues that are likely to arise that could be expensive to solve or could ultimately limit the penetration of IRES. The three main factors that may constrain IRES deployment:
(1) IRES technologies do not displace firm capacity on a one for one basis, nor do they typically provide ancillary services such as inertia, frequency response, or reserve capacity;50
(2) Their intermittent output and the relatively unpredictable element of their output demand more of these ancilliary services from the grid than conventional plants;50,51
(3) The highly correlated nature of the wind and sun across the UK means that at high penetration level, IRES output is weighted towards periods of surplus and away from times of system shortages. Consequently, the surplus causes wind output to be curtailed and become increasingly lower in value (market cannibalisation†††).92
Many studies have considered some of the costs that arise from integrating IRES.93 However, in addition to cost, it is vital to include the above issues and consider the need to balance energy, whilst also considering the margin of firm capacity over peak demand and the provision of response, reserve and inertial services. Using the BERIC model,94 we provide some new analysis of these issues here.
The carbon price was set at £70 per tCO2 for most runs described here, except for some sensitivities run at £100 per tCO2. Captured carbon had a total burial cost of £19 per tCO2 to cover all downstream costs as in the reports by PB.95,96 Other commodity costs were gas at 75 p per therm and biomass at £23.23 per MWh thermal HHV basis, making biomass-fuelled power plants slightly more expensive than a combined cycle gas turbine (CCGT) at full load.
Taking these inputs gave a full load cost of nuclear of £87 per MWh which compares well with Hinkley's nth of a kind strike price of £89.50 per MWh.§§§97,98 Onshore wind would need £81 per MWh which again compares well with payments under the Renewables Obligation (RO) which came out at an average of £84 per MWh in 2015/16.99 Note that the reported strike price for CCS varies from £90 per MWh (gas-CCS in 2030) to £100 per MWh (coal-CCS in 2030).100 However, if technology learning is taken into account, the cost of CCS may reduce to be £85 per MWh, enabling CCS to be competitive with other forms of clean energy.101
The availability profile for wind was based upon the generation reported to Elexon during 2012102 which, of the five years examined, had the most typical characteristics.¶¶¶ PV availability was simulated using a curve rising from zero at sunrise to maximum at noon back to zero at sunset. This was randomly scaled by a factor between zero and 1 to represent the daily variability of insolation, and scaled again to give the expected profile for monthly energies as predicted by the JRC online PV tool103 for Birmingham.
Demand data was based on 2012 outturns corrected for the small proportion of wind which is embedded and assumed to generate in line with the majority of the portfolio. This calculated consumer demand was then scaled to match the peak energy demand for 2030, derived from the National Grid's Slow Progression scenario,104 which gave an annual energy demand of 317 TWh.
Scenarios. The main modelling explored a matrix of scenarios covering all combinations of 9 levels of nuclear penetration (0–40 GW), 8 levels of wind (0–56 GW) and 7 levels of CCS (0–30 GW). For other technology, capacities were set at levels in National Grid's “Gone Green” scenario for 2030.104 Further sensitivity analysis looked at varying the capacity of each of the 17 technologies in the model one by one away from the central scenario. In all cases unabated gas-CCGT was treated as the “slack variable”, its capacity being adjusted to retain the same derated capacity margin over demand.
Methodology. BERIC is a linear program (LP), whose objective function is to minimise short run costs at each scheduling point in the scenario run.94 A sample set of 220 half hour “points” are scheduled independently from each other. The model is constrained to stay within the following bounds:
(1) Energy demand must be balanced exactly by generation. Demand is given by the 2012 shape scaled to meet peak energy demand of the 2030 Slow Progression scenario.
(2) There must be sufficient reserve to meet the requirement at all times. BERIC meets a reserve demand that represents the requirements for frequency response and faster reserve products covering timescales of seconds and minutes. Wind and PV generation creates a demand for reserve cover at a rate of 17% of output (similar to typical values used by National Grid).94
(3) There must be sufficient inertia to meet the requirement at all times. It is assumed that inertia levels will be allowed to drop from the current minimum level of 150 GW s down to 90 GW s following recent changes to the grid code that improved tolerance to a higher Rate Of Change Of Frequency (ROCOF).
Generation is scheduled in fleets according to type, so the fleet of CCGTs is scheduled as one, all wind turbines as another etc. However, the solver has freedom to assign any proportion of the fleet to one of four operating states (i) off, (ii) minimum stable generation, (iii) optimum level for providing spinning reserve, and (iv) full capacity. In effect, there are no quanta associated with individual units.
The effect of renewable energy. Fig. 4 shows the carbon intensity of the grid as a function of wind and nuclear capacity. Following the top blue line where no new nuclear (or CCS) is built, it can be seen that even with 56 GW of wind, CO2 emissions have only dropped to around 180 g kWh−1. The curvature of the line indicates that further wind build suffers from diminishing returns as emissions reduce – i.e., the available output of wind is increasingly ineffective at replacing fossil generation. This is due to curtailment when low carbon output exceeds demand. The ideal situation would be to have sufficient storage and/or demand side management to be able to make use of all curtailed output.51,74 This is represented by the dotted blue line that is tangential to the initial blue curve. Even without output curtailment, CO2 intensity is 133 g kWh−1 at 60 GW wind capacity, greater than the 50–100 g kWh−1 target by CCC,83–85 which is needed to enable decarbonisation of other sectors through electrification.
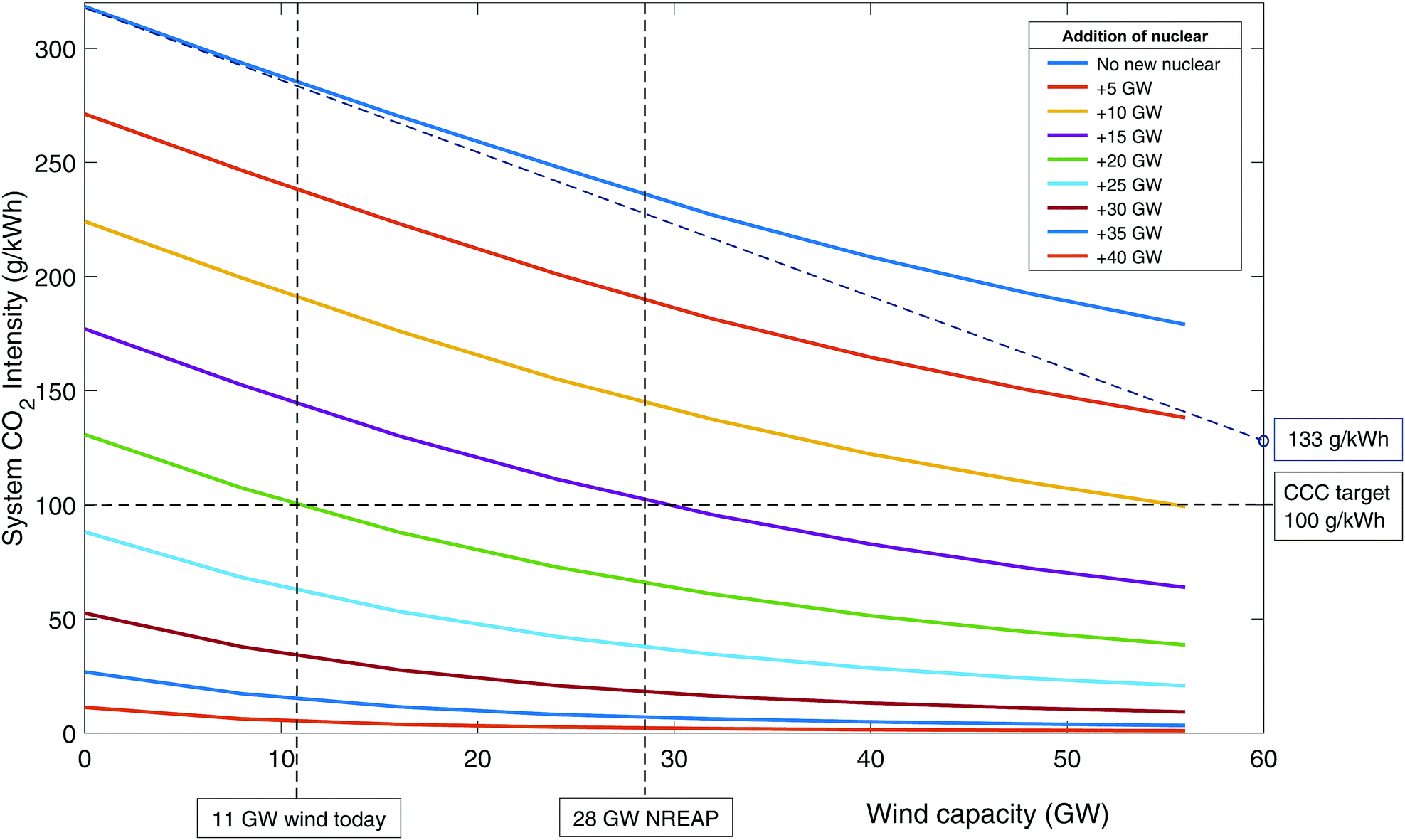 | ||
| Fig. 4 The CO2 emissions in 2030 as a function of wind and nuclear build when unabated CCGT is used as flexible back-up. | ||
The lower curves represent 5 GW increments of new nuclear build. It can be seen that with 20 GW of new nuclear then 100 g kWh−1 can be achieved with around 11 GW of wind. The National Renewable Energy Action Plan (NREAP) targets a wind build of 28 GW by 2020 (combined onshore and offshore capacity).105 It can be seen that if this is accompanied by about 15 GW of nuclear then 100 g kWh−1 is achievable. It should be noted that other firm low carbon plant (such as biomass and CCS) could achieve similar results, albeit with slightly higher capacities to account for their residual emissions.
Need for low carbon firm capacity. Fig. 5 shows the carbon intensity with different levels of nuclear, CCS and wind in the grid mix. The two surfaces represent the target of 50 g kWh−1 recommended by CCC for deep decarbonisation82 and the UK's Department of Business, Energy and Industrial Strategy (BEIS) central estimate of 100 g kWh−1.72 Meeting either of these targets would mean the solution would have to lie on the visible side of the surface. For example, point A is the pure nuclear solution meeting 50 g kWh−1, which corresponds to 31 GW of new nuclear. In the absence of gas-CCS at point C, 56 GW of wind is required, scaling back nuclear build to 18 GW. Adding gas-CCS is less effective at reducing emissions (it was modelled with 91% capture), so 30 GW will only displace 18 GW of nuclear build and achieve the same target grid intensity. This highlights the importance of considering the residual CO2 emissions. As decarbonisation targets become more stringent, there is the potential need for CO2 capture of 95% or more (Fig. 5), i.e. minimise/eliminate residual CO2 emissions.
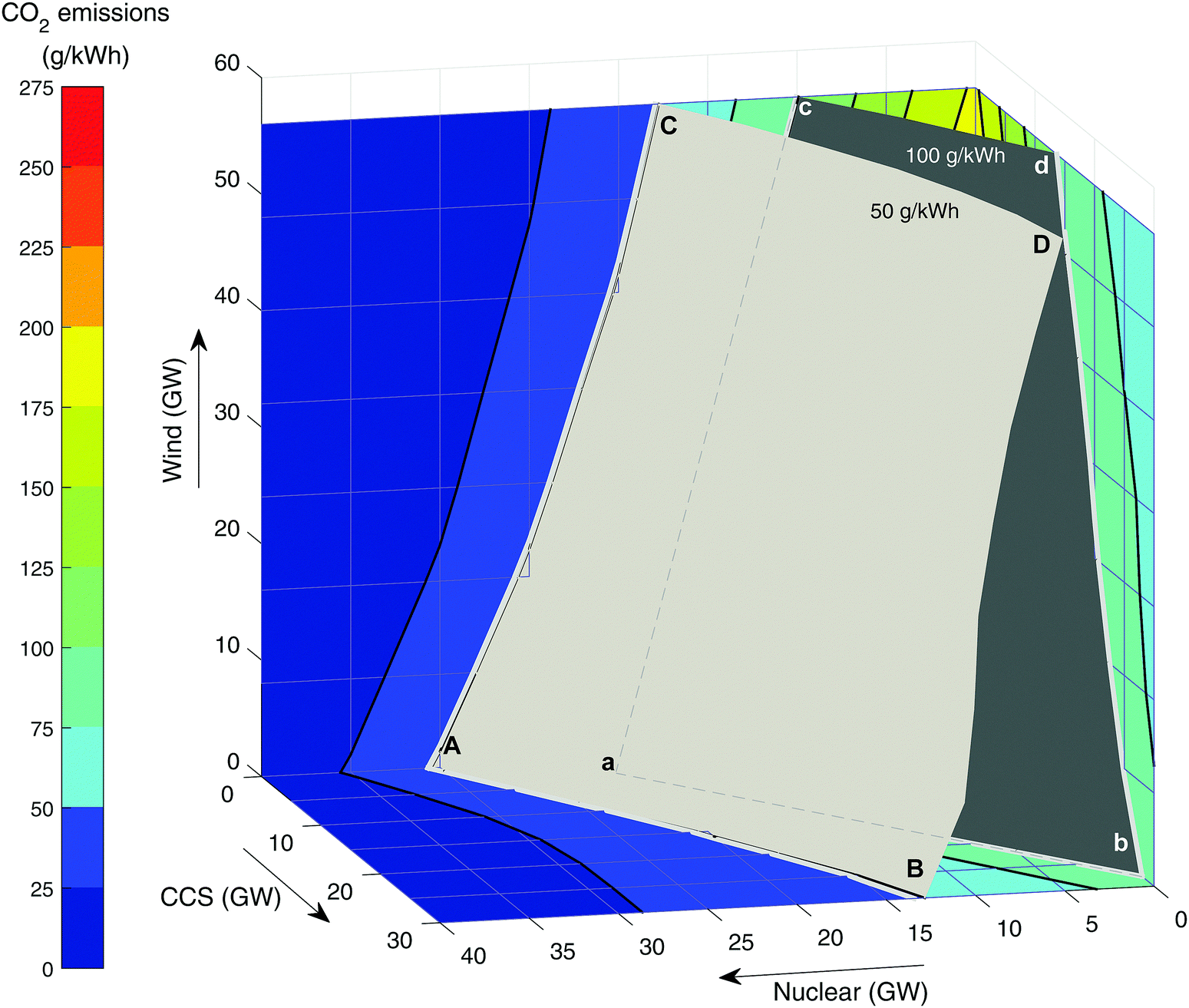 | ||
| Fig. 5 The CO2 emissions as a function of nuclear, CCS and wind build. The surfaces show the technology deployment requirements in order to meet the CCC targets for CO2 emissions of 100 g kWh−1 and 50 g kWh−1. | ||
Table 3 summarises the various technology adoption pathways that will meet the CCC targets for CO2 intensity (based on results in Fig. 5). Adopting the weaker 100 g kWh−1 target72,83–85 means a pure CCS (no wind, no new nuclear) solution is possible within the bounds modelled, at just 27 GW of gas-CCS. However, even with 56 GW of wind (“maximise wind” scenario), a significant amount of firm low carbon capacity is required, either 11 GW of new nuclear or 19 GW of CCS.
| Scenario | Build capacity | |
|---|---|---|
| 100 g kWh−1 target | 50 g kWh−1 target | |
| Maximise nuclear | 23 GW nuclear (a) | 31 GW nuclear (A) |
| Maximise CCS | 27 GW CCS (b) | 30 GW CCS & 13 GW nuclear (B) |
| Maximise wind | 56 GW wind & 11 GW nuclear (c) | 56 GW wind & 18 GW nuclear (C) |
| Maximise both wind and CCS | 56 GW wind & 19 GW CCS (d) | 56 GW wind, 30 GW CCS & 11 GW nuclear (D) |
Inherent storage and flexibility of the capture plant. Carbon capture plants of nearly all designs have some additional opportunities (over an unabated plant) to store energy by time shifting energy intensive processes.106 For post-combustion, the amine regeneration could be scheduled at times of excess power enabling output to be boosted when required,107–113 which could provide reserve, response or firm capacity services. Similarly for oxy-fuel or pre-combustion capture, an oxygen buffer would allow the air separation unit (ASU) to run independently of generation so energy was not sapped at times of high export value (e.g., operate ASU during off-peak electricity demand).114,115
In a system with a growing proportion of generation from low running cost options, such as IRES, the value of energy is likely to decline, and the supply of some grid services and firm capacity is likely to be limited whilst demand for them grows. Therefore, it is important to consider the balance between IRES and different services (firm capacity, reserve etc.) at the design stage so the full value of a CCS plant can be accessed.
Value of CCS, nuclear and wind. Many studies focus on the cost of technologies that can help decarbonise a system and often divide a discounted cash flow with a discounted energy output to give a levelised cost of electricity (LCOE) in £ per MWh. Although this approach has value for the comparison of a homogeneous set of thermal technologies providing a similar service (energy, firm capacity, reserve, response, inertia, etc.), it is no longer relevant when comparing technologies that deliver only a selection of these services, and nor does it account for the effects seen here where the effectiveness of a technology is strongly dependant on the existing grid mix.50,51,71,94,116
An alternative metric that works for all technologies is the value of technology addition (VOTA), also referred to as system value.51,116 VOTA is defined here as the reduction in annualised total system cost with the deployment of a technology and is in units of £ per MWh of capacity deployed in a given year. Fig. 6 illustrates VOTA of various energy technologies for a number of different scenarios in 2030. For a higher carbon price, the value is generally higher (Fig. 6b, d and f), as might be expected. It can also be seen that the VOTA profiles have a similar shape, following a path of continuous additions of a technology eventually leads to an accelerating decline in the VOTA. If the system already has a significant amount of another low carbon technology, the drop-off in the VOTA will start earlier and decline faster. This has also been observed in some recent work by Heuberger et al.51 The value of CCS is less affected by the addition of wind to the system than nuclear plant is (Fig. 6c–f). This is due to CCS having lower capital cost and greater flexibility compared to nuclear.
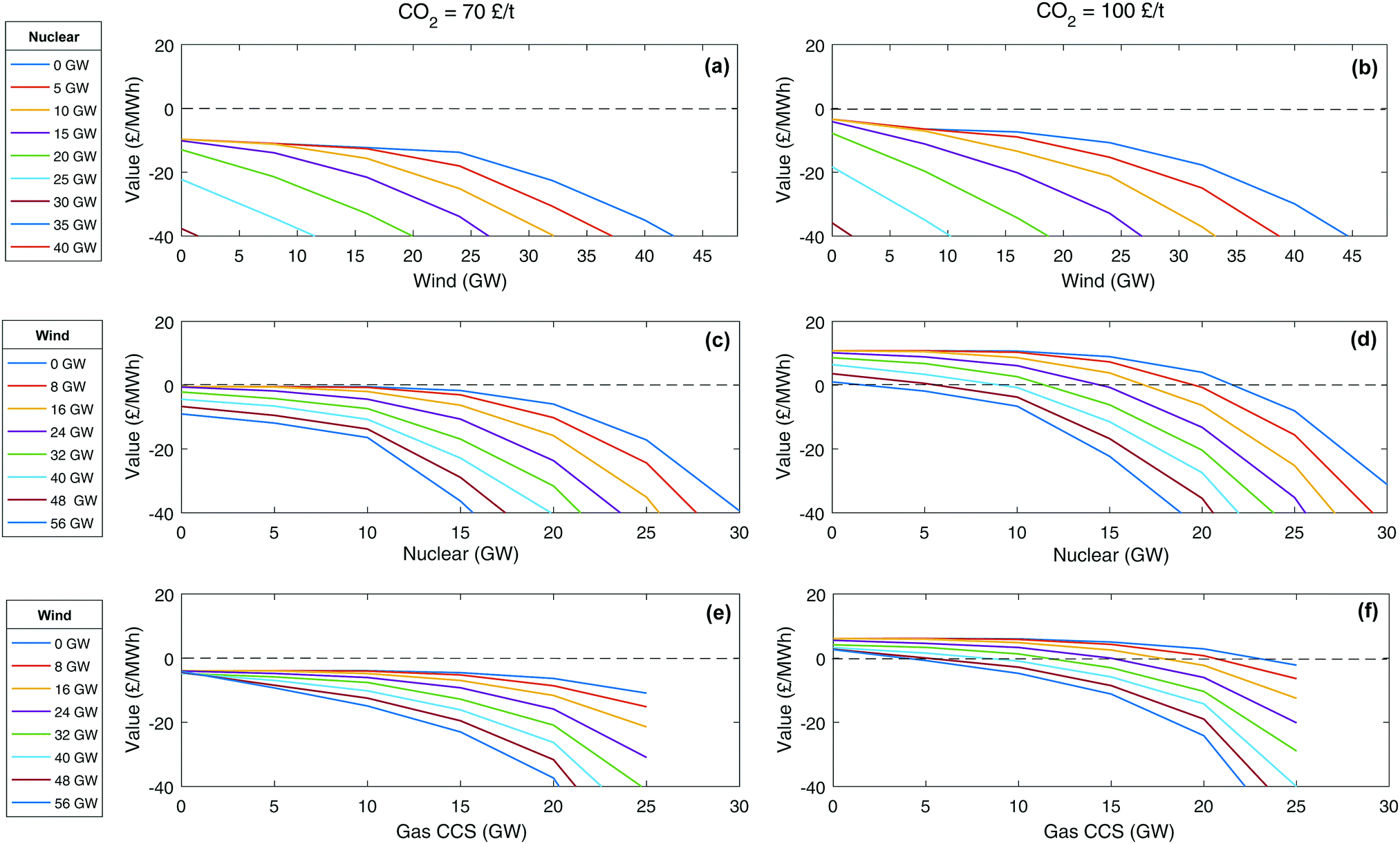 | ||
| Fig. 6 Value of technology addition (VOTA) or system value of building of wind, nuclear and gas CCS technology. | ||
Competitor technologies. Fig. 7 shows all the generation technologies that were modelled, starting with a 2030 system that meets 85 g kW−1 by incorporating 10 GW of new nuclear, 5 GW of gas-CCS, 28 GW of wind and 20 GW of PV. The trajectories for each technology represent the addition of more capacity. The left direction of Fig. 7 represents a reduction in emissions, whilst moving upwards corresponds to an increase in total system cost. Most technologies curve more steeply upwards as capacity is added (e.g. marine, PV, offshore wind), indicating the addition of cost whilst becoming increasingly less effective at reducing emissions. To achieve CO2 emissions of 50 g kWh−1, it would take an additional 28 GW of offshore wind, with an addition to total system cost of more than 25%. Adding nuclear would require 6 GW, whereas the addition of gas-CCS requires 10 GW, which results in an additional cost of 3–4%. Biomass moves directly left, indicating CO2 emissions reduce at no additional cost.
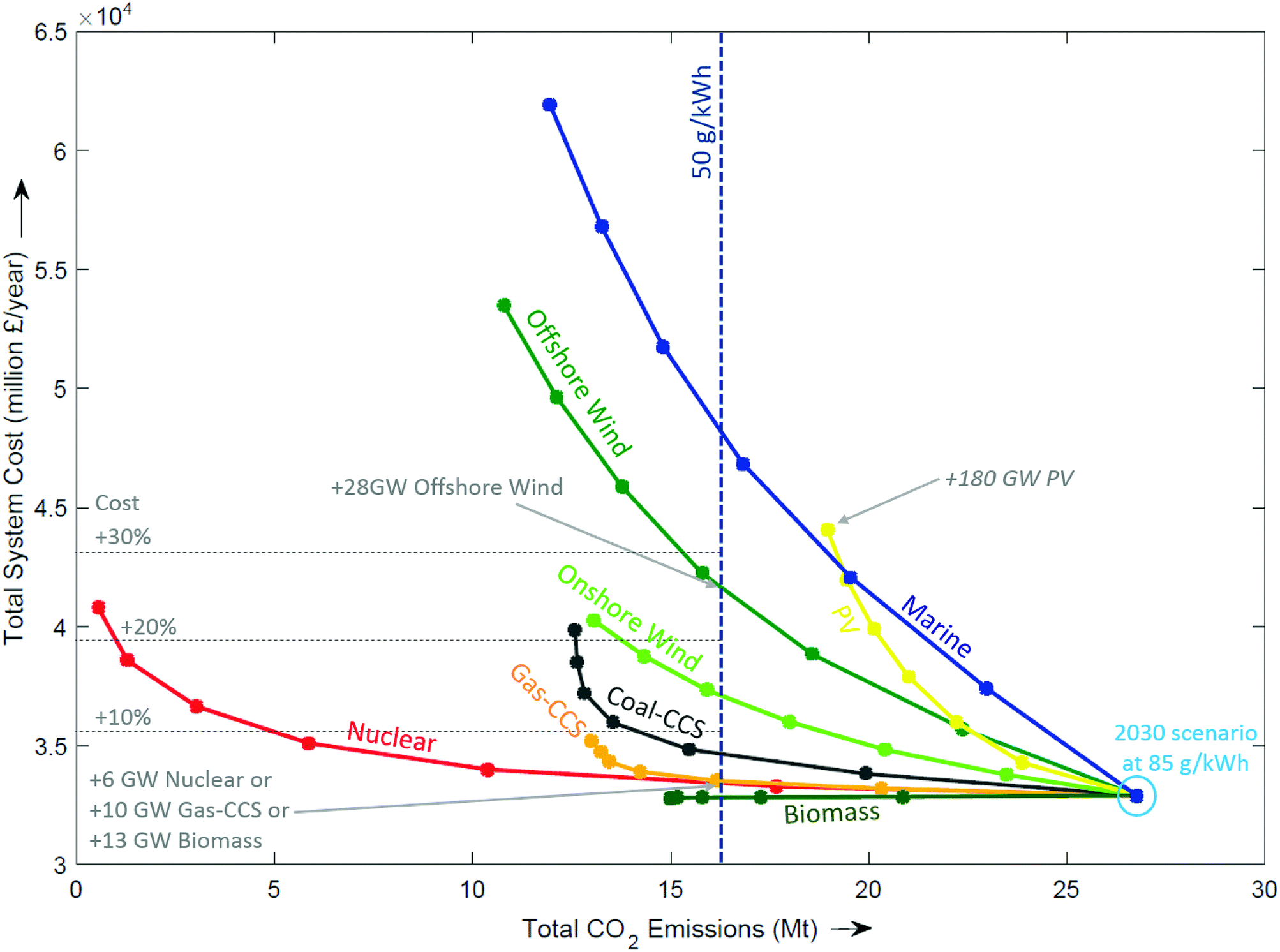 | ||
| Fig. 7 Effect of adding new technologies in 5 GW increments from a 2030 central scenario at the origin. | ||
The prediction of which technologies are cheapest is of course entirely dependent on the cost assumptions used. Although the absolute cost of a technology varies with the assumptions used, the curves have been independently shown to exhibit the same functional form. This demonstrates that there is a law of diminishing returns,94 and that this effect tends to be more pronounced for intermittent technologies than firm capacity as with increasing deployment the former delivers energy during increasingly congested periods.51,74,94 In summary, the three classes of technologies that can make significant reductions in emissions with only a small increase in total system costs are: (i) CCS, (ii) nuclear, and (iii) bioenergy.||||||
The value added to the system by a certain technology is dependent on the existing energy mix and the services that the technology can provide (e.g., inertia, reserve, firm capacity). Thus, a single number such as the LCOE cannot be used to characterise the performance of a technology. Similarly, value assessment of a single technology in isolation is inadequate. A whole systems approach to valuing technologies and their impact on total system cost would recognise the role of low-carbon technologies in balancing demand and cost. Such an approach would enable better characterisation (e.g. availability, controllability, economic benefit) of different generation technologies within a given energy mix.
3.3 Industrial CCS
A significant proportion of GHG emissions can be attributed to industrial processes,6 contributing 25% of the global CO2 emissions.119 Thus, decarbonisation of the industrial sector will be essential to meet the CO2 emissions targets by IPCC.6 Some key industrial sectors that have been the focus of CCS studies include cement, petroleum refining, iron and steel manufacturing, and pulp and paper,120,121 with iron and steel, cement and refining being especially “high-emitting”,119 together consuming 38% (43 EJ) of total industrial energy consumption.122 CCS is regarded as a cost effective option to reduce CO2 emissions from industrial processes.119,123 The physical properties, composition and gas volume flows are different for each industrial process.124 Thus, the suitability and selection of a CCS technology would depend on these stream properties, e.g., CO2 concentration, moisture content.125–127 The challenge for the industrial sector will be maintaining international competitiveness with the implementation of technologies that reduce CO2 emissions, but increase costs.128• Integrated steel mills, which uses the blast furnace-basic oxygen furnace (BF-BOF) process. Coke is used to reduce the iron ore in the blast furnace to form “pig iron”, which is then converted to liquid steel in the basic oxygen furnace (with an addition of ∼30% scrap steel).118
• Mini-mills using an electric arc furnace process and a feedstock consisting of scrap metal, direct reduced iron (DRI) and cast iron.118
The larger integrated steel mills are the main source of emissions and on average emit 3.5 Mt of CO2 annually, whereas the smaller mini-mill plants each emit <200 kt of CO2 annually.120,121 The average CO2 emissions from a typical steel mill is about 1.8 tCO2 per tonne of crude steel, where the major carbon sources are from coal and coke (1.7 tCO2) and limestone (0.1 tCO2).117Fig. 8 shows that there are multiple sources of the CO2 emissions within a steel mill process. Of these, the stream from the blast furnace contributes the greatest direct CO2 emissions (69%).117 However, this flue gas is not directed to a stack, but instead, the energy in the gas is recovered in the on-site power plant.118
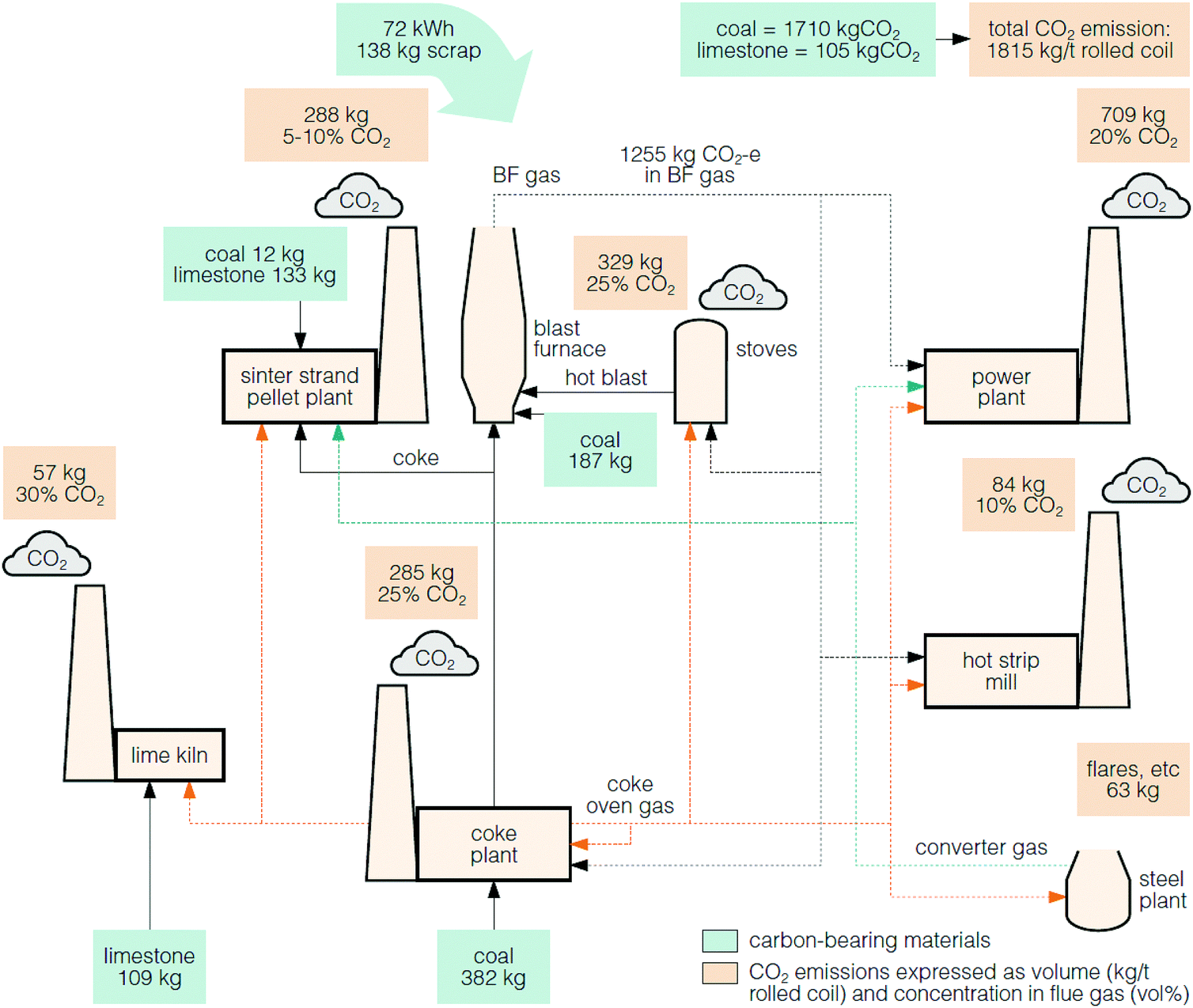 | ||
| Fig. 8 A typical steel mill and the CO2 emissions, which vary in concentration.117,118 | ||
The cost of CO2 capture in the iron and steel industry is dependant on the type of technology and the location within the process. Most of the research has focussed on applying CO2 capture in the blast furnace. Post-combustion capture from the BF has been estimated to cost between $65.1–119.2 per tonne of CO2 avoided, capturing 50–55% of emissions.129–131 A top-gas recycling blast furnace using post-combustion capture can capture 65% of emissions at $54–88 per tonne of CO2 avoided.132 The mean cost to capture 65% of total emissions from the blast furnace is $76.6 per tonne of CO2 avoided. Post-combustion capture from the coke oven will cost an average of $86.4 per tonne of CO2 avoided (27% of total emissions).121
In the short term, minimising energy consumption and improving energy efficiency is the most cost effective approach to reducing CO2 emissions.133–137 Some of the measures used to improve energy efficiency include heat loss reduction, heat recovery of waste energy, and efficient process design.133,137 Over the years, there have been efforts to reduce CO2 emissions from the overall production process, such approaches include increased recycling of steel scrap, use of biomass or renewable energy, utilisation of by-product fuels to reduce the use of coke and coal.118
The implementation of CCS technologies could further significantly reduce CO2 emissions. In integrated steel mills, it is possible to capture CO2 from the flue gas exiting the lime kiln, sinter plant, coke oven plant, stove, blast furnace and basic oxygen furnace. In the case of mini-mills, the main source of CO2 would be the electric arc furnace.130 Post-combustion capture technology can be applied to these gas streams without affecting the iron and steel making process. Alternatively, an “in-process” capture process could be employed, merging the iron/steel making and the CO2 capture processes.117,138 One such strategy is to use oxy-combustion conditions in the BF to produce flue gas of high CO2 concentration, which would enhance CO2 capture efficiency.117 Some commercial iron and steel facilities employ CO2 capture and removal as part of the production process, however, the CO2 is currently flared. For example, CO2 is captured as part of some DRI facilities,118 the Saldanha steel plant in South Africa,139 the Finex process (South Korea)140 and HIsarna process (Germany and Australia).141,142 To prevent this CO2 from being emitted to atmosphere, integration of CO2 storage would be necessary.
In November 2016, the world's first large-scale application of CCS in the iron and steel sector commenced operation as part of the Emirates Steel Industries (ESI) CCS Project (Phase 1 of the Abu Dhabi CCS Project).119,143 The system uses an amine-based absorption process with a capture capacity of 0.8 Mt of CO2 per year. The CO2 is subsequently transported through a 43 km onshore pipe to be injected for enhanced oil recovery (EOR) and stored.143
There are a number of different CCS technologies that are applicable to cement production; there are several variants of post-combustion CO2 capture, including solvent scrubbing or the use of solid sorbents, calcium looping, oxy-fuel and “direct capture”.145 In this context, the most obvious difference between cement production and power generation is that pre-combustion technologies are not applicable. This is because of the large quantity of process-related emissions from calcination of limestone that are not captured when pre-combustion is applied. The technologies are in general (with the exception of direct capture) conceptually similar to their counterparts in power generation, though it is notable that calcium looping utilises one of the feed stocks for cement production (CaO) as its main sorbent; this leads to significant synergies between the cement process and Ca looping. Direct capture has no obvious analogue; it utilises indirect radiative heating of the limestone-containing raw meal feed to the system to “directly” produce a pure stream of CO2. Both direct capture and oxy-fuelled systems have the potential for efficiency gains within the system, owing to either thermodynamic benefits (direct capture) or a reduction in the total amount of thermal “ballast” in the system by eliminating the nitrogen from air.
The key issue for cement CCS is to ensure that the quality of the product remains the same after the CCS system has been applied. This is the main advantage for a scrubbing system based on post-combustion capture using alkanolamines (or other sorbent-based system). Unfortunately, cement plants are not in general endowed with sufficient low-grade heat to (without the addition of a CHP plant) allow capture of more than around 50% of the CO2 produced in a cement plant.145,149
One of the first references to a zero emission oxy-fuelled kiln was made in 2006.150 The European cement research association has identified oxy-firing of a kiln to be the most potentially beneficial process for the cement industry, and a long-term project has been working towards commercialisation of the process, starting in 2007 with basic literature review, followed by techno-economic feasibility studies. Initially, it was thought that the change in the kiln environment (from air/CO2 to O2/CO2) might change the volatilities of minor species within the process. Basic laboratory tests were conducted by the European Cement Research Academy (ECRA),151 no significant variations were found in cements produced under oxy-fuelled conditions as opposed to standard cements. Such findings were confirmed recently as part of a G8 project investigating the application of oxy-firing on cement.152
Similar to the conclusions above, minimal changes to cement quality have been observed when simulating at a laboratory scale the application of calcium looping to cement production.147,153,154 No changes to cement quality would be expected using solvent-based post-combustion capture. Potential changes to cement chemistry from direct capture have not been tested yet; it is a stated aim of the current EU LEILAC project to conduct such tests.155
The cost of CO2 capture on cement plants has been studied by a number of researchers, though (as with all other industrial processes) nowhere near as comprehensively as costs for the application of CCS on power generation.****120,121 There is a broad consensus in the literature that amine scrubbing is likely to be more expensive than the two other most studied technologies, oxy-firing the kiln and calcium looping. Some recent work by Leeson et al.120,121 found that, when adjusted to a consistent year and currency basis, costs for calcium looping were between $20 and $75 per tonne of CO2 avoided (central estimate ∼$40), oxy-fuel was around $60 (only one estimate was found in the academic literature) and amine scrubbing was significantly higher, between $65 and $165 with a central estimate of ∼$106. The single study referenced in Leeson et al.120 was conducted by Mott-MacDonald (with input from Whitehopleman and the British Cement Association) and was a comparison between post-combustion and oxy-fuel capture.157 Importantly, this study only considered “partial” oxy-firing, of only the pre-calciner, the sealing of the large rotating kiln, heat transfer changes within the kiln, etc., being considered to be sufficiently challenging that deployment in the “near future with moderate risk” was unlikely. However, it was stated that no “showstoppers” were found in the potential future deployment of a fully oxy-fired system, though this was an area for basic R&D. Post-combustion capture using amine scrubbing was shown to be nearly three times more expensive. This is because of two main issues – the requirement to protect the amines used from NOX and SO2, the clean-up of which necessitates a selective catalytic reduction and wet scrubbing system, respectively; and the heavily increased fuel-burn, owing to the paucity of low-grade heat in the system. One point of note is that because the kiln would not be converted to oxy-firing in the above study, the amine scrubbing system captured significantly more CO2 (74% vs. 61%). It was also noted that integration of a cement plant with a nearby power plant would significantly reduce the costs for post-combustion capture (allowing the transfer of steam from the power station to effect the regeneration of the amines), though presumably similar benefits would accrue if an oxy-fuelled plant were located next to an oxy-fuelled power station with an oversized ASU. A recent paper158 examining the application of calcium looping to cement manufacture found that calcium looping had a high avoided CO2 (94%) in comparison to an partially oxy-fuelled plant modelled (76%); the same group has modelled the integration of a calcium looping system with a power plant and export of the spent CaO to a cement works;159 this yields some of the lowest potential costs seen for CO2 avoidance in cement manufacture, though the fuel-based emissions in the cement plant are not fully addressed. The minimum cost of CO2 avoided in the combined system was approximately 27 £ per tonne. Of course, such integration would require the co-location of the two plants, which may be geographically challenging.
It was noted in Barker et al.157 that building a plant in a far-eastern location would be significantly cheaper (more than 50% cheaper) than building it in a European location. Another study160 investigating the application of post-combustion solvent scrubbing to cement production in a similar location (China), but for a retrofit, found a cost of $70 per tonne of CO2 avoided (in this case, a CHP plant was used to make up for the lack of low-grade heat to regenerate the amines). This paper was one of the first to make the case for “carbon capture readiness” for the cement industry. This subject was also explored by Hills et al.,145 who also considered the technology readiness levels (TRLs) for different CCS technologies applied to the cement industry. The TRLs for amine scrubbing, calcium looping and partial oxy-fuel were assessed to be at or near to 6, with full oxy-fuel being a little lower (4) and direct capture somewhere in between. This work also made it clear that because cement-plant renovation and capture plant construction are likely to take similar lengths of time, it would likely save time and money to synchronise these. Discussions with cement manufacturers161 similarly have underlined the long lifespans of cement plants and the razor-sharp margins in this industry; it has also long been known that some form of tariff is required to maintain a level playing field between regions with regulated CO2 emissions and those not subject to such controls.123,162
There is a renewed drive to apply CCS to industrial processes. Norcem (in collaboration with its parent company, Heidelberg Cement and the ECRA) has led the way163 in terms of testing amines on real flue gases (Aker Solutions) and are also testing solid sorbents (RTI International), and a membrane (DNV and others). As a part of the same test programme, Ca-looping (Alstom) will be tested at IFK, Stuttgart. A 1.9 MWth Ca-looping pilot plant is integrated with a cement works at ITRI (Industrial Technology Research Institute), Taiwan,164 and has demonstrated capture >85% in a 7 hour long test; the stated aim is to progress to a 30 MWth pilot plant. The LEILAC project, headed by Calix Europe155 is a 21 million euro, 5 year project aiming to demonstrate the direct capture process. Regarding oxy-fuel combustion, ECRA has a current stated aim165 to develop a 500–1000 tpd pilot plant to be operational by 2019, with a cost between 40 and 60 million euros. The key extra cost component was stated to be the oxygen production facility.
The sector is responsible for approximately 6% of total global CO2 emissions167 and these are distributed across the value chain from exploration and production, refining and downstream petrochemical production. Although in this analysis we are excluding the downstream use of the products, it is worth noting that the use of the industry's products in power generation, heating and transport is responsible for approximately 50% of global emissions.
The relevant experience of this sector is broad, with a comprehensive understanding of relevant issues to CCS: geology, licensing, site operation, safety, high pressure operation/transport, and offshore engineering. Particularly, the oil and gas industry has considerable experience in upstream processing, which involves gas sweetening and produced CO2, as well as CO2-enhanced oil recovery (CO2-EOR) with the associated CO2 transport and injection infrastructure.
An interesting macroeconomic feature of this industry sector is the large swings in crude oil price (e.g. between approximately $40 to $140 per bbl168) and that the price differentials that the industry must manage are considerably lower than the marginal cost of CCS applied to different elements of the value chain discussed below. For the sake of argument, assuming that approximately 17.2% of the feedstock is used as internal energy in the process for production, conversion and logistics (assuming an energy return on investment of 11:1169 and a refinery efficiency of 91%170,171) and a very conservative decarbonisation cost of $200 per tCO2, the additional cost per barrel of crude oil would be $13, a figure easily contained within the recent price fluctuations, which in turn have been well borne by the end-use sectors.
The downstream sector of the oil and gas industry was particularly challenging to decarbonise, while contributing around 4% of global CO2 emissions.172 Oil refineries offer a particular challenge here owing to their large, integrated nature, the heterogeneity of these facilities in general and finally the potentially large number of point sources in any given installation (e.g. heaters, furnaces, boilers, crackers and utilities), which themselves have the potential to be diverse in terms of flow rate and composition and which may need innovative concepts in retrofit and exploring trade-offs between aggregating flue gas sources for centralised capture and aggregating CO2 streams.
There are some point sources of CO2 at a refinery that are relatively easy to mitigate, such as catalytic crackers – decarbonisation of these units should be a high priority. It was further noted that decarbonising a complex refinery might require the use of more than one capture technology. A report produced for the UK Government in 2015 estimated that a new, efficient refinery exploiting CCS where most economic could have CO2 emissions which are 36% of a 2012 baseline.173 Refineries also benefit from locations favourable to CCS such as being near coasts and/or industrial hubs and therefore should have ready access to CCS infrastructure should it arise. Examples of such locations in Europe include Grangemouth and Rotterdam.
A variety of technologies for carbon capture have been considered for refining, including classical post-combustion capture (e.g. Andersson et al.,174 who explore how excess waste heat can be exploited), oxy-combustion (e.g. Escudero et al.,175 who consider utility boilers and find that it is an economically viable technology under certain scenario assumptions) and chemical looping combustion (CLC). The latter is interesting because the refinery light gases are suitable CLC fuels176 and from an engineering point of view, refinery designers and operators have experience of engineering and controlling hot solids looping processes in terms of fluid catalytic cracking.177 One obvious quick win is the hydrogen production plant178 (mainly used for fuel upgrading) which by its nature produces a relatively pure stream of CO2 which would require basic post-processing prior to compression and transport.
An important factor to consider is whether the end-use emissions would grow with an increase in hydrocarbon production (considering both conventional and unconventional oil and gas and in particular the scope for increased gas production), and how these emissions can be mitigated at source. This can for example be based on-site hydrocarbon reforming to produce hydrogen with simultaneous storage of the associated CO2 which may have the potential to be a more cost-effective option than the production of hydrocarbon and the subsequent capture and storage of CO2. Australia and Japan are planning such a supply chain, using Australian coal as the primary resource (initially without CCS) and shipping liquid hydrogen to Japan.179 More generally, while it is relatively commonplace for countries to produce and distribute CH4, in the future gas exporting countries might reform the CH4 as a matter of course, exporting the resulting H2 and using the CO2 for enhanced oil or gas recovery. This would have the effect of removing concern about CO2 – enhanced hydrocarbon recovery; if the carbon is being immediately returned to the subsurface, then there can be no subsequent CO2 emission when the hydrogen is being used for heat, power or transport.
4 Post-combustion capture technology
This section discusses capture technologies that have been demonstrated at pilot plant scale or higher (i.e., at TRL of 6 or greater).4.1 Liquid-phase chemisorption technology
The classic chemical absorbent for CO2 separation applications is 20–30 wt% aqueous monoethanolamine (MEA). It was proposed in the original patent from 1930 for an amine process to separate acid gases11 and has found widespread use in industry.180 MEA is particularly suited to low CO2 partial pressure applications and as a consequence has become the benchmark amine for CO2 capture from electricity generation. In a standard CO2 separation process applied to flue gas (10–15 kPa CO2) at 40 °C, and using 30 wt% MEA and 90% CO2 removal, typical minimum stripper reboiler duties are ∼3.6–4.0 GJ per tonne CO2 captured. This value has been validated at small to medium pilot scale in a number of studies.181–184 The reboiler energy requirement is not the only metric that defines the performance of an absorbent, but reducing this value is the primary goal of much chemical absorbent research, and new absorbents are typically benchmarked against the value for 30 wt% MEA. Rates of mass transfer, stability in the presence of oxygen and elevated temperature, volatility, solids formation, toxicity and biodegradability and price are also important in real world flue gas applications. New absorbents are also benchmarked against MEA in terms of these characteristics. MEA has good rates of CO2 mass transfer, is low cost and readily biodegradable but suffers from moderate rates of oxidative and thermal degradation and moderate levels of toxicity.180 It is also corrosive when used at higher concentrations.Research and development of new absorbents for flue gas applications has been ongoing for a number of decades. These new absorbents perform better than MEA in some or all of these characteristics. This suggests it may be time to move on from MEA and choose one of the new generation of absorbents for benchmarking purposes. As an example the formulation of aqueous piperazine (PZ) and 2-amino-2-methyl-1-propanol (AMP) has been extensively studied. This blend has achieved ∼3 GJ per tonne CO2 captured at pilot scale and also has favourable mass transfer and stability properties.185,186 Commercial solvent technologies are also potential benchmark solvents, e.g. Econamine FG+, KS-1, and Cansolv. These advanced solvents contain proprietary blends of amines187 and have been selected for use in commercial-scale projects.14,15,188
In the following sections state of the art amine-based and multi-phase chemical absorbents will be discussed. These will be limited to absorbents that have been characterised in detail and progressed from the lab scale at least to small pilot scale testing. The reason being that this is the critical step where absorbents that have performed well in the lab can suffer from unforeseen issues that limit their utility. When numbers for reboiler duty are quoted for amines these will be based on a standard absorber/stripper process configuration without absorbent specific or other process improvements (complete list provided in Table 4). By their very nature multi-phase absorbents require process modifications which makes the comparison less clear-cut.
| Solvent | Reboiler duty (GJ per tCO2) | Ref. |
|---|---|---|
| 30 wt% MEA | 3.6–4.0 | Cousins et al.,181 Kwak et al.,182 Mangalapally and Hasse,183 Stec et al.184 |
| 40 wt% MEA | 3.1–3.3 | Lemaire et al.205 |
| 40 wt% (8 molal) piperazine (PZ) | 2.9 | Cousins et al.196 |
| Cansolv | 2.3 | Singh and Stéphenne13 |
| 32 wt% EDA | 3.2–3.8 | Mangalapally and Hasse,185 Rabensteiner et al.204 |
| 28 wt% AMP + 17 wt% PZ | 3.0–3.2 | Mangalapally and Hasse,185 Rabensteiner et al.186 |
| MEA + MDEA (variable mix ratio) | 2.0–3.7 | Idem et al.,227 Sakwattanapong et al.231 |
| Aqueous ammonia (NH3) | 2.0–2.9* | Darde et al.,232 Dave et al.,233 Yang et al.234 |
| Aqueous potassium carbonate (K2CO3) | 2.0–2.5 | Anderson et al.,235,236 Smith et al.237 |
| Amino acids | 2.4–3.4* | Sanchez-Fernandez et al.238,239 |
| DEEA + MAPA | 2.1–2.4 | Raynal et al.,240 Liebenthal et al.241 |
| DMCA + MCA + AMP | 2.5 (not including extraction) | Zhang242 |
| CO2 + 2R1R2NH ↔ R1R2NCOO− + R1R2NH2+ | (1) |
| CO2 + R1R2NR3 ↔ HCO3− + R1R2NHR3+ (where R3 = –H or –CRaRbRc) | (2) |
Piperazine (PZ) is a cyclic diamine that has been used as a low concentration (<10 wt%) additive to increase the rates of absorption in aqueous solutions of the tertiary amine methyldiethanolamine (MDEA) by BASF since the 70's (BASF's activated MDEA).180 However, Xu et al.190 was one of the first investigations to tease out the specific impact of PZ on mass transfer in activated MDEA. Since then, it has been investigated extensively and concentrated PZ has been proposed as an absorbent in its own right at up to ∼40 wt% by Freeman et al.191 and Rochelle et al.192 Piperazine is of limited solubility unless some CO2 is present. It reacts very rapidly with CO2 resulting in fast mass transfer,193 and being a diamine its capacity for CO2 absorption is large.194 It has also been found to resist oxidative and thermal degradation,195 which allows for higher temperature stripper operation. In pilot scale testing it was able to achieve a 15% reduction in reboiler duty compared to 30 wt% MEA,196 giving a similar value to PZ/AMP formulations. The main challenges are the potential for precipitate formation191 and nitrosamine formation.197,198
Aqueous ethylenediamine (EDA) has also been extensively evaluated as an absorbent. It is analogous in structure to MEA but with the hydroxide group replaced by a second amino group. Similarly to PZ, it is a diamine, however both of the amine groups are primary, rather than secondary. The kinetics of the reaction between EDA and CO2 are slightly faster than MEA, but not as fast as PZ.199,200 As would be expected for a diamine its absorption capacity per molecule is larger than MEA, and it has a large enthalpy of CO2 absorption resulting in elevated CO2 pressures at stripping conditions.201,202 Its thermal and oxidative stability have been shown to be similar to MEA limiting stripper temperature to 120 °C.202 A drawback of EDA is that the replacement of the hydroxide group by an amino group results in larger volatility203 which will increase the demands on the washing sections during process operation and results in elevated absorbent losses. Pilot plant trials saw reductions in reboiler energy requirement of ∼8–11.5% relative to 30 wt% MEA.185,204
Lastly, MEA has itself made a resurgence. Advanced MEA processes are also being developed where MEA is used at concentrations greater than 30 wt%, and additives are used to control degradation and corrosion. For example, if MEA can be used at 40 wt% and other issues controlled via additives the reboiler energy demand can be reduced to 3.1–3.3 GJ per tCO2.205
In terms of new amines that are progressing through lab scale bench studies amines containing heterocyclic functionality (that is the amino group incorporated into a ring structure) are of particular interest. PZ is the first heterocyclic amine that has been investigated extensively, however other heterocycles (in particular piperidines) are also being investigated due to the inherent stability of cyclic structures, and the combination of steric hindrance furnishing large CO2 absorption capacities while retaining fast reaction with CO2.206–212
The blend of PZ and AMP is probably the best known and well characterised of new absorbent formulations. AMP is a very similar molecule to MEA, but with two additional methyl groups located at the α-carbon position. AMP is a sterically hindered amine highlighted in the work of Sartori and Savage.213 Being sterically hindered, it has greater absorption capacity than sterically unhindered primary and secondary amines, but it suffers from low rates of CO2 mass transfer at low CO2 loadings. Steric hindrance affects the absorption capacity of an amine by reducing the stability of the carbamate species formed either by crowding of the reactive site and/or electronic effects, resulting in increased bicarbonate formation. However in the case of crowding (which is the case for AMP), it may also affect mass transfer by reducing the intrinsic rate of reaction between CO2 and the amino group. Seo and Hong214 proposed the addition of PZ and so developed a PZ/AMP formulation. They demonstrated that the addition of PZ increased the rates of mass transfer significantly. Yang et al.215 then went on to show that the blend of PZ and AMP retains a large absorption capacity for CO2. In terms of stripper reboiler duties, this blend achieves values around ∼3.0 GJ per tCO2185,186 or ∼20% lower than 30 wt% MEA. Being a blend, there is some variability in these results depending upon the actual composition used. Typically the larger the PZ/AMP ratio, the faster mass transfer is, while the smaller the ratio the lower the reboiler duty.216 To avoid precipitation the total amine concentration for this blend is limited to about 40 wt% and the contribution of PZ to 10 wt%.217 Both PZ and AMP have low rates of oxidative and thermal degradation relative to MEA195,218 again with some variability depending upon the proportion of each amine.219
As mentioned previously PZ has been used as an additive to aqueous MDEA to increase CO2 mass transfer for decades. More recently, but in a similar approach, MEA has been assessed as an alternative rate promoting MDEA additive. MEA and MDEA are both well characterised independently. As a blend the overall CO2 absorption capacity is reduced relative to MDEA alone, but is improved at partial pressures relevant for flue gas capture compared to either MEA or MDEA.220,221 This blend can also match the rates of CO2 mass transfer determined for aqueous MEA alone.222–225 It does not suffer from the potential precipitation issues of PZ/AMP but does suffer from greater rates of oxidative degradation than either MEA or MDEA in isolation.226 In pilot scale testing using a synthetic flue gas, a reduction in reboiler duty of ∼6–12% was seen relative to 30 wt% MEA.227 However, in the same work when using real power station flue gas this benefit was lost. This was attributed to the accelerated degradation of the MEA/MDEA mixture in the harsher flue gas environment and highlights the importance of good chemical stability.
No other blends to-date have had the detailed results of trials at pilot scale published in the public domain. However a range of new blends are progressing through bench scale testing and assessment including ternary blends.228 These blends are generally constituted by amines that have first been investigated on an individual basis, as this is necessary to identify potential candidates for a blend.
Liquid–solid separation systems. Aqueous ammonia (NH3) is the most advanced of the multi-phase absorbent processes. At room temperature NH3 is a gas and aqueous NH3 solutions are solutions of a dissolved gas. In aqueous solutions NH3 reacts with CO2 to primarily form ammonium and bicarbonate243 ions and has a number of favourable properties:234 it does not thermally degrade or oxidise; it is low cost and readily available; it is non-corrosive; it has a good CO2 absorption capacity; and a low reboiler energy demand for stripping (2–3 GJ per tCO2). The challenge is the high NH3 vapour pressure and how losses and emissions to the environment can be controlled, and the formation of precipitates. Commercial technology vendors have been investigating chilled NH3 processes where absorption is carried out at 0–10 °C.244,245 The purpose of the low temperature is to increase the aqueous solubility of NH3 and concomitantly CO2, however the low temperatures reduces rates of mass transfer and results in solids formation (ammonium carbamate/bicarbonate) and a multi-phase process. In addition, the low reboiler duty must be considered alongside the additional complexity of liquid–solid processes (changes in design to gas–liquid contactors and heat exchangers and the introduction of liquid–solid separation equipment) and the cooling demand of chilled absorption.
Aqueous potassium carbonate (K2CO3) solutions have the advantage of being less volatile, non-toxic, less corrosive, lower cost and more resistant to oxidative degradation compared to amines.246 Other important advantages of K2CO3 is that absorption can occur at high temperatures, also it has a low heat of absorption (CO2 absorption in K2CO3 is 600 kJ kg−1, whereas MEA is 1900 kJ kg−1),246 reducing the thermal energy requirements of the regeneration process.247 However, the major challenge is the low reaction rate of K2CO3,246 resulting in poor CO2 mass transfer. Pilot plant tests using an unpromoted 30 wt% K2CO3 solution could only absorb between 20–25% of the CO2 from the flue gas.248 To improve CO2 mass transfer, K2CO3 requires the addition of a promoter or catalyst.247 The Cooperative Research Centre for Greenhouse Gas Technologies (CO2CRC) have developed and optimised the K2CO3 processes for post-combustion and also pre-combustion capture of CO2.237,248 The optimised K2CO3 technology, named UNO MK 3,235,249 can achieve a low regeneration energy of 2–2.5 GJ per tCO2 (much lower than conventional MEA).235–237 These promising results have led to the scale-up and commercialisation of this process by UNO Technology Pty Ltd.250
Amino acids, which are amines that also contain carboxylic acid functionality, have also been investigated as CO2 absorbents. They have favourable characteristics in terms of vapour pressure as they are ionic in their neutralised form and they are resistant to oxidative degradation.251 Amino acids undergo the same chemistry with CO2 as amines, with the amino group being the reactive centre. A challenge to their use is they are often of limited solubility and may form precipitates. To-date pilot plant trials of single phase amino acid based systems have not produced favourable results.252,253 As a consequence, amino acid based liquid–solid processes are also being developed.254 In this case the precipitate formed can be either the neutral amino acid or CO2 containing products depending upon the amino acid used. Similarly to aqueous ammonia reboiler duties are estimated to be in the 2–3 GJ per tCO2 range,238,239 but again the process complexity is increased and there is an additional heat duty to redissolve the precipitates for CO2 stripping. Commercial technology vendors are also pursuing amino acid based processes.246
Liquid–liquid phase separation systems. Though they are yet to reach pilot scale, liquid absorbents that undergo a phase separation upon reaction with CO2 are being investigated. Three types of dual-liquid systems exist: (i) low critical solution temperature (LCST), (ii) mutual solubility type, and (iii) extraction type.230 In LCST systems, the absorbent solution separates into two phases at a certain temperature range, providing opportunities to reduce energy consumption.255–257 However, years of technology research and development reveal that the lower phase absorbs most of the CO2 and has higher CO2 loading, however, the total amount of CO2 absorbed is very low. Thus, the performance of LCST systems is not as promising as anticipated.230
Mutual solubility systems consists of at least two amines, where the reaction products of one amine has a solubility limitation with CO2 in the other. As the reaction progresses, the CO2 loading and concentration of reaction products simultaneously increase, which drives the formation of two phases.230 A large range of mixtures exhibit this behaviour,258 for example the mixture of 2-(dimethylamino)-ethanol (DEEA) and 3-(methylamino)ethanol (MAPA) forms a single phase when CO2 free but two phases when CO2 is absorbed.259 The CO2 product is concentrated in the lower phase and the upper phase remains mostly DEEA. This formation of a phase concentrated in CO2 means only this phase need be sent to the stripper reducing the flow rate and sensible heat requirements.240 It is estimated this could yield reboiler duties of ∼2.1–2.4 GJ per tCO2.240,241 The third dual-liquid system is the extraction type, originally proposed by Zhang.257 Extraction dual-liquid systems can use either one amine or a mixture of amines. The separation into two phases occurs during regeneration, when the CO2-rich liquid has been heated to a specific temperature. The two phases formed are an upper organic phase and a lower aqueous phase. The organic phase acts as a solvent, extracting regenerated amine and driving the equilibrium towards desorption, which reduces the regeneration temperature to ∼80 °C.230 Solvent regeneration for extraction type systems only consumes ∼2.5 GJ per tCO2 (MCA + DMCA + AMP).†††† However, the additional extraction step, which involves further heating to recover solvent, consequently leads to a total energy requirement of 3.5 GJ per tCO2 for the whole process.242
Unlike the chilled ammonia and amino acid phase separation processes, which have additional cooling and heating duties respectively, along with other process modifications to deal with solids, these phase separation processes only require the introduction of a phase separation unit to the CO2 capture process.
In terms of developing the perfect amine for CO2 capture, this is a challenging task. The most common approach to-date has been to assess the performance of existing amine molecules. As a consequence of the knowledge gained doing this, there is now considerable understanding of the relationship between chemical structure and absorption performance and stability. The next generation of amines will be less a product of discovery and more a product of targeted task specific molecular design and synthesis, with multiple amino groups having complementary properties contained in single molecules. A few studies have started down this path260–263 with initial results looking promising. There is growing interest in water-lean solvents (e.g., ionic liquids, non-aqueous organic amine blends), which exhibit lower reboiler duty and higher mass transfer properties compared to aqueous formulations. However, water-lean solvent tend to be more expensive and testing has been limited to lab-scale (∼3 L).187 As expertise in the relevant synthetic chemistry increases, and these task specific molecules become available in larger quantities and at lower cost, they are likely to outperform and outlast the current suit of amines.
4.2 Adsorption processes for CCS
Adsorption processes were first considered in the early 1990's as an alternative to solvent processes for carbon capture.264–266 Since those initial studies, there has been a growing and sustained effort to develop adsorption technology for CO2 capture. By far, the greatest research efforts have been directed at developing improved adsorbents with higher working capacity for CO2, better selectivity, and better tolerance to impurities. New CO2 capture adsorbents are reported almost daily. The classical adsorbents (carbons, aluminas, silicas, zeolites) and modifications thereof have all been evaluated for their potential in CO2 capture applications and new adsorbents (metal organic frameworks, hydrotalcites, amine supported adsorbents, polymers, high temperature metal oxides) have all been explored for their application in a range of areas.267–270Leaving aside developments in adsorbents, important as they may be, there have also been important developments and progress in adsorption processes for CO2 capture over the last three decades. A large variety of cyclic processes have been developed, in which regeneration is accomplished by temperature, pressure, vacuum, steam or moisture, or combinations thereof. These processes have been comprehensively reviewed.269,271 Novel adsorbent structures and gas-adsorbent device geometries have been proposed and evaluated, such as hollow fibres, monoliths, radial beds, fluidised and moving beds. Hybrid adsorbent technologies have been investigated in which adsorption is coupled with other separation or reaction technologies either as a distinct unit operation or an integrated unit. The application areas for adsorption process have expanded from post and pre-combustion flue gas to process streams (e.g., food and beverage, cement, steel, petrochemical, pulp and paper, and natural gas industries) and direct capture of CO2 from air.
Considering the aforementioned strengths, pilot-scale CO2 adsorption projects have been proposed and implemented, enabling the community to acquire the knowledge, skills and expertise needed to improve the technological maturity of CO2 adsorption. Among them, there is the CO2CRC H3 capture project based at the International Power plant in Australia which operated from 2009 to 2011.277 Besides adsorption, the project also investigated the potential of absorption and membrane processes for CO2 capture from a coal-fired power plant. Publicly available information on the adsorbents used suggests they were provided by Monash University. The “CO2 Ultimate Reduction in Steelmaking Process by Innovative Technology for Cool Earth 50” (or COURSE 50) project was launched in Japan in 2008 at an industrial CCS to capture CO2 from the blast furnace stream at JFE Steel's West Japan Works.278,279 The capture system employs PSA and captures 3 ton per day of CO2 while also evaluating a number of zeolites and activated carbons as adsorbents.
The complexity of the screening challenge arising from the multitude of adsorbents is magnified by the plethora of performance criteria to be considered when designing an adsorbent. These criteria have been reviewed recently and are summarised in Table 5.282 In addition to the criteria detailed in Table 5, we add the energy required for regeneration (linked to OPEX) and chemical and thermal robustness. Although many adsorbents have been tested for CO2 capture, the focus has only been on their CO2 uptake, selectivity and recyclability; these criteria are insufficient for the technical confidence needed to move to higher technology readiness levels. Fortunately, researchers have started to combine experimental data with molecular simulation and process-scale modelling. Thus, incorporating multi-objective optimisation enables quantitative comparison of adsorbents against a number of process performance criteria (e.g. purity, recovery, energy consumption, productivity).290–296 For these studies, one needs to specify the process options, i.e. TSA, PSA or others, and rely on molecular simulation to provide sorption isotherms validated by experiments. It is interesting to note that while the challenge related to the numerous potential sorbents and performance criteria also exists when designing CO2 capture solvents (absorption), the complexity in the case of solid sorbents is increased because data on mass-transfer resistance and diffusion limitations are scarce, yet are required to model a full adsorption process.
| Criterion | Unit | Symbol, equation |
|---|---|---|
| Uptake under adsorption conditions | mol kg−1 | N ads1 |
| Working capacity | mol kg−1 | ΔN1 = Nads1 − Ndes1 |
| Regenerability | % |
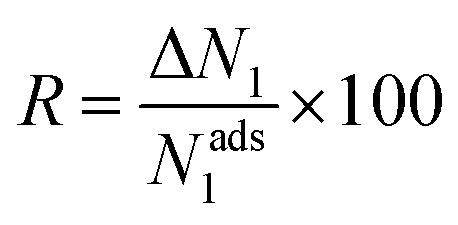
|
| Selectivity under adsorption conditions | — |
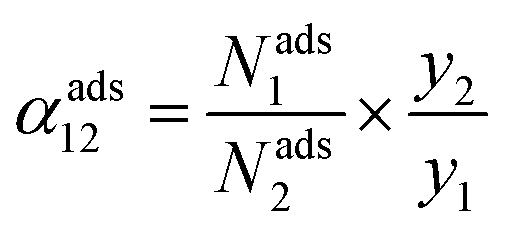
|
| Sorbent selection parameter | — |
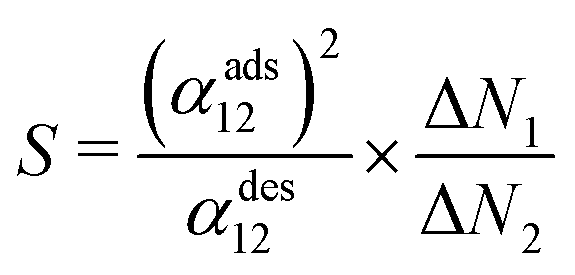
|
Adsorbents also face a number of process-related challenges. For instance, a pre-treatment step might be required as flue gas impurities, including water, can impair the performance of some adsorbents. In fact, this could be performed by a multi-layer adsorbent bed, as proposed in the CO2CRC H3 project.277 Heat effects in the adsorption beds poses an additional complication in adsorption process schemes because the inherent exothermic nature of adsorption implies potentially high bed temperature rises (especially in the case of chemisorption). These effects must be quantified, managed and possibly exploited in clever process schemes.297,298 Studies towards better heat integration are therefore of paramount importance.297–299 Another challenge pertains to the manufacturing of new adsorbents and the ability to develop new manufacturing processes that enables to strike a good balance between reducing particle size, to enhance good intra-particle diffusion kinetics, and increasing the particle size to limit pressure drop.
Pressure vacuum swing adsorption. Cyclic adsorption in fixed beds is now a relatively mature technology and all of the various modes of adsorbent regeneration have been applied to carbon capture. For post combustion flue gas at atmospheric pressure, vacuum swing adsorption (VSA) (also sometimes referred to as Pressure Vacuum Swing Adsorption (PVSA) since the feed stream might be slightly pressurised) is the logical choice amongst the pressure regeneration modes since the feed stream is mostly nitrogen. A large number of studies of VSA have been carried out, most typically using activated carbon300–302 and 13× zeolite adsorbents303–306 and a large number of adsorption cycles have been proposed and tested.307,308 The consensus is that if only a single VSA stage is used, significantly deep vacuum levels are required (at least 10 kPa) to achieve specification CO2 product purity (>95%) necessary for acceptable CO2 recovery using the popular 13× adsorbent. More exotic adsorbents, such as metal organic frameworks (Mg-MOF 74,309 UTSA-16292), may be able to improve this situation in the future but these MOFs are unlikely to be available at large scale in the near term. This has led researchers to examine two stage PVSA systems,300,310,311 in which different adsorbents can be used in each stage and there is scope for considerable integration. Of course, the penalty here is a substantial increase in cost. It is also worth pointing out that most PVSA studies have assumed a dry flue gas since the 13× adsorbent suffers severe CO2 capacity deterioration in the presence of water although Li et al.312 have shown that multilayer beds with 13× can be used (although at a significant energy penalty) to handle wet gas streams.
The energy demand of the VSA process is frequently cited as being considerably less than conventional amine processes. This is not correct. A range of specific energies from 100 kWh per tCO2 to more than 1000 kWh per tCO2 have been reported – the exact number depending greatly on specifics of the process. Most of these quoted numbers rely on unrealistic equations for vacuum pump energy consumption. In one of the few studies in which an experimental energy consumption of 340–580 kWh per tCO2 was reported,305 it was found that theoretical calculations significantly underestimated the true energy required. Reliable reported energies seem to fall between 1.5 to 3 GJ per tCO2 (electrical). This converts to about 4.5 to 9 GJ per tCO2 thermal, substantially more than conventional amine solvent processes (reboiler duty of 2–4 GJ per tCO2, shown in Table 4). Overall, VSA and PSA appears to be more suitable for smaller scale operation and it is difficult to see these fixed bed processes being extended beyond the 50 MW scale for post-combustion capture unless multiple trains are installed, losing economy of scale. Applications for VSA and PVSA are more likely to be in IGCC,313 petrochemical, and steel and cement processes where the gases have higher CO2 partial pressures and lower volumetric flow rates.
Temperature swing adsorption. A more attractive and scalable regeneration option for post combustion capture is temperature swing adsorption (TSA). Generally, both PSA and TSA are considered mature technologies and have been employed for a number of applications in industrial gas separation.266,301,314–316 Temperature swing adsorption has been used for removing trace amounts of CO2 and water from air in Air Separation Units and natural gas dehydration prior to liquefaction. Its application to bulk CO2 removal is however, in its infancy. Numerous studies have been reported from the groups of Mazzotti,317,318 Webley,319 and others (Korea,320,321 and RTI,322,323), which have highlighted the performance and obstacles of this approach. Joss et al.318 developed improved cycles for TSA operation attaining comparable regeneration energies to solvent based processes. In their study, additional purge and recycle steps as well as intermediate heating was used to produce very high purity CO2 (>99%) at high recovery (>95%). As their adsorbent was 13× zeolite, which strongly adsorbs water (thereby reducing its CO2 capacity), there was a need to dry the flue gas stream prior to their TSA. This adds 2–3 GJ per tCO2 to the energy requirement (already comparable to amine-based processes). Pre-drying the flue gas stream is therefore not a feasible option for large scale adsorption based CO2 capture, which suggests water tolerant adsorbents (in fact, water non-adsorbing adsorbents) may be needed. The voluminous work on amine-based sorbents fills this requirement.324
Conventional TSA run in packed beds incurs the significant penalty of long cycle times due to long heating and cooling requirements. To overcome these limitations, fluidised bed configurations are popular for TSA.325,326 The strong mixing enables rapid heat transfer but intense mixing and co-current gas-solid flow leads to lower average CO2 loadings (the adsorbent is at equilibrium with the gas leaving the fluidised bed). Pirngruber et al.327 has estimated a lowest heat of 2.1 GJ per tCO2 (thermal) for ideal adsorbents under isothermal conditions. More realistically, an energy of 3.2 GJ per tCO2 is likely. Sjostrom et al.328 recently developed and tested a supported amine sorbent in a circulating fluidised bed with adsorption using entrained flow, regeneration via a temperature swing with an option for a sweep gas, gas/solids separation, and cooling. A CO2 removal of 90% was achieved.
One important variant of the fluidised bed process is the SARC (swing adsorption reactor cluster) process,329 in which multiple fluidised bed adsorbers (each one consisting of several counter-current beds, similar to amine absorption) are cycled through adsorption, evacuation, regeneration (by heating) and cooling. The evacuation step removes nitrogen from the beds (vented to atmosphere) prior to the regeneration step in which the carbon dioxide is recovered. Heat pumps recover heat generated by adsorption and return it to the beds for regeneration. A process integration study of the proposed SARC process in a large-scale pulverised coal (PC) ultra-supercritical (USC) power plant was performed and showed an energy penalty of 9.6%-points for the base case with ammonia as the heat pump working fluid.329
In a process designed to mimic solvent systems (e.g, counter-current flow), moving beds have been investigated in temperature swing modes.297,330 The major challenge associated with large scale moving bed processes is that the gas velocity must be kept low enough to prevent the solids from becoming fluidised. This translates to beds with impractically large diameters, not to mention the mechanical difficulties in handling solids in large scale unit operations. Novel structured adsorbents (described in Section 4.2.4) may provide some solutions to these problems.
Steam, electrical and moisture regeneration. Many of the TSA systems described above rely on indirect heating or heating with hot gas for regeneration. If low-pressure steam is available, this is the preferred option for TSA implementation. Clearly, water tolerant adsorbents are required and functionalised adsorbents (e.g. amine-based sorbents) are ideal for this application.331 These adsorbents suffer from degradation at very high temperature and poor kinetics at low temperature. Therefore, the preferred operating window is between 60 and 100 °C. Fujiki et al.332 from RITE have overcome the diffusion limitation associated with low temperature operation of amine-based sorbents. This allowed them to use low temperature steam purge (which in prior studies degraded the sorbent and prevented high temperature operation) to assist a vacuum swing cycle. Through this means they were able to obtain high purity (>98% CO2) and high recovery (>93%) – difficult to achieve with VSA alone. The reported energy consumption of 1.47 GJ per tCO2 accounts only for the steam usage and apparently does not include vacuum power. A relatively modest vacuum level of 15 kPa was used. This is a promising approach and is worthy of continued effort. Avoiding TSA allowed these workers to retain the use of fixed beds.
Steam regeneration is at the heart of the VeloxoTherm™ Process of Inventys.333 This process is currently under testing to capture CO2 from the slipstream flue gas exiting a 10 MWe coal-fired unit. This technology utilises the rotary adsorption process with a structured adsorbent (Section 4.2.4) and is based on the design of an existing regenerative air preheater.
Electrical swing adsorption (ESA) has been stated to present an attractive option for rapid thermal (Joule) heating of an adsorbent.334 In this sense, it is a derivative of temperature swing adsorption (TSA). The effectiveness of this option depends greatly on the details of the adsorbent/electrical system. Since the adsorbent must be electrically conductive, both the material and its configuration must offer a continuous electrical path. Packed beds of adsorbent therefore are eliminated from consideration and carbon monoliths are the most common configuration. Since zeolites offer more benefits for temperature swing adsorption given their strong adsorption of CO2, they are more attractive for ESA applications. Efforts have therefore been made to integrate the zeolite into the carbon monolith walls or pack the zeolite within the carbon channels with some success.335 Grande et al.336 evaluated ESA for CO2 capture from a Natural Gas Combined Cycle power stations where the CO2 concentration of the flue gas is 3.5%. Using an adsorbent comprising of 70% zeolite and 30% of a binder conducting material to treat this flue gas, it was possible to obtain a concentrated stream with 80% CO2 with an energy consumption of 2.04 GJ per tCO2 (electrical). This is equivalent to ∼6 GJ per tCO2 thermal and is significantly higher than conventional amine processes (Table 4). Importantly, the cooling step could be eliminated. It is likely that ESA is more suited to small scale operation where a small process footprint is important.
Moisture Swing Adsorption (MSA) is an intriguing concept evaluated by Wang et al.337 In this scheme (ideally suited for direct capture of CO2 from air), an amine-based anion exchange resin dispersed in a flat sheet of polypropylene is prepared in alkaline form to enable CO2 capture from air when dry and releases it when wet. This is a moisture induced cycle, and is a new approach to regenerating CO2 sorbents – evaporation of water effectively provides the free energy that drives the cycle.
Hybrid systems – sorption enhanced capture and adsorbent/cryogenic systems. Adsorption processes have been integrated with reactors and membranes to exploit synergies between these technologies. Coupling adsorption with reaction is usually undertaken with the goal of shifting the equilibrium conversion by adsorption of CO2 (one of the products from the reaction). Thus, blending adsorbents with steam reformers,338,339 or water-gas shift reactors is popular.340,341
The latter process (denoted sorption enhanced water gas shift or SEWGS) uses a high temperature adsorbent (e.g., promoted hydrotalcite or CaO) to remove CO2, driving the reaction to the right hand size and maximising hydrogen production. The sorbent is then regenerated with steam.342 It is particularly suited to coal-based IGCC plants but is also applicable to natural gas plants. In the FP7 project, CAESAR, Air Products, BP, ECN, SINTEF and Politecnico di Milano worked together to develop the SEWGS process, improving the specific energy to between 0.8 to 1.0 GJ per tCO2 (electric). A new adsorbent named ALKASORB+ was developed with a high capacity resulting in cost of CO2 avoided for the IGCC application of approximately €23 per tonne of CO2 avoided. This is almost 40% lower cost than the conventional Selexol process.
The purity of CO2 produced from adsorbent processes is strongly dependent on operating parameters of the process. In contrast to chemical solvent systems in which 100% CO2 is produced in the stripper, the CO2 purity of the blow down gas is strongly dependent on blow down pressure, or, in the case of TSA, regeneration temperature. Therefore, there are considerable savings to be achieved by operating the adsorption system at lower CO2 purity. This makes adsorption technology suitable for use as a front end “rough” separator, upstream of a “polishing” separator. Conveniently, cryogenic CO2 capture systems become more cost effective as the CO2 concentration increases (above ∼70%). These systems are extremely attractive for producing liquid CO2, which may be pumped to high pressure at much lower energy than compression of CO2 gas. A hybrid process consisting of an adsorption process followed by a cryogenic process is therefore an ideal solution. Li Yuen Fong et al.343 explored this concept, using a vacuum swing adsorption (VSA) process as the initial CO2 recovery stage. A multi-objective optimisation (MOO) technique in combination with heat integration was used to optimise the total shaft work and the overall CO2 recovery rate of the capture process (including compression to 100 bar pressure). A minimum energy optimum was determined for the total specific shaft work required at an overall recovery rate of 88.9%, which consumes 1.40 GJ per tonne of CO2 captured.343 This is considerably lower than conventional processes. A simple CO2 liquefier was used in this study and there is considerable scope to employ a more sophisticated cryogenic process integrated with the adsorption process.
Zeolites. Zeolites are microporous aluminosilicate minerals that exhibit a crystalline structure with pore sizes typically between 4 and 15 Å and surface areas around 200–500 m2 g−1. Both natural and synthetic zeolites exist for carbon capture. The main physico-chemical mechanism for CO2 adsorption in non-modified zeolites derives from the large quadrupole moment of CO2, which enables the molecules to interact with the electric field created by the cations in zeolites. Because cations are introduced into zeolites by charge compensation of substituents, CO2 adsorption is governed by the zeolites framework structure and composition (i.e. Si/Al ratio), as well as the composition and location of extra-framework cations.344,345 For instance, the zeolite channel diameter, hence the topology, directly influences the dispersion interactions between CO2 and the zeolites walls. In addition, dual cation sites, i.e. sites where CO2 can interact simultaneously with two cations, are known to favour adsorption.344 An interesting phenomenon described recently is the so-called “selective trapdoor effect” or “cation gating effect”, whereby molecules able to interact with the cations located at the entrance of a channel, e.g. CO2, can permeate through the material and be adsorbed while other molecules that do not interact as strongly cannot.346 Adsorption can also be enhanced via modification with large and electropositive, polyvalent cations.344
There is considerable industrial knowledge of zeolite manufacturing and its applications in gas separations. From a more fundamental view point, zeolites are crystalline materials and hence can be relatively easily modelled, which can eventually reduce the time needed to evaluate their performance as outlined in Section 4.2.2. The CO2 uptake of zeolites is quite high and in fact, the synthetic zeolite 13× is often taken as the benchmark of CO2 (low T) adsorbents (capacity of about 3 mmol g−1 at 0.15 bar of CO2 and 313 K).347 Large scale screening of all known and over 100000 predicted zeolite structures has been achieved and has identified the best materials for CO2 separations.348 The key weakness of zeolites remains their sensitivity to moisture as water adsorbs strongly on zeolites, thereby reducing the CO2 uptake.
Metal–organic frameworks. Another family of porous crystalline adsorbents are metal–organic-frameworks (MOFs). MOFs are obtained via the self-assembly of metal ions and organic ligands (Fig. 9). They exhibit extraordinary surface areas and pore volumes. Typical ligands used to synthesise MOFs include carboxylate and imidazolate compounds, and the metal “nodes” span a considerable range of the periodic table. The size and shape of pores in MOFs can influence its adsorptive properties via a molecular sieving effect.349 In addition, chemisorption occurs either via interactions between, (i) CO2 molecules and open metal sites in the MOFs, i.e. uncoordinated metal sites, and/or (ii) interactions between CO2 molecules and functional groups located on the MOF ligands.282,285 For examples, studies have shown that one could vary the strength of CO2 adsorption by changing the type of uncoordinated metal sites.285 CO2 uptake can also be enhanced by using amine-containing ligands.285 In addition, CO2 adsorption in MOFs can be driven by the so-called breathing and gate-opening effects, though at present this behaviour is only observed at high pressures.350 In these materials, the pore of the flexible MOFs contracts or opens upon adsorption. The addition of extra framework cations is another factor enhancing CO2 capture in MOFs.282
 | ||
| Fig. 9 Example of a MOF. Here the structure of Mg-MOF-74 is shown. This MOF exhibits the highest reported CO2 uptake for unfunctionalised adsorbents. | ||
The inherent tunability of the MOFs chemistry and structure represent one of the key strengths of these materials, potentially allowing one to tune the CO2 uptake, selectivity and heat of adsorption. Like zeolites, their crystalline structure makes them ideal candidates for simulation studies. In 2013, MOFs held the record for CO2 adsorption capacity with MOF-74(Mg) exhibiting the highest reported uptake (5.5 mmol g−1 at 0.15 bar, 313 K) (Fig. 9).347 MOFs are often criticised for their chemical instability as they can react with flue gas components like water, NOX and SOX. In recent years though, a number of robust MOF structures have emerged, such as UiO-66 and SIFSIX-6_Zn.349,351,352 Unlike zeolites, most MOFs are not yet manufactured at a large scale and for those which are, they are most often supplied as powder rather than a structured adsorbent.
Carbonaceous materials. Carbon-based materials have also been investigated thoroughly for CO2 capture.286 This generic term represents a number of distinct materials whose structure is mostly composed of C atoms. For instance, we distinguish between low-cost pyrogenic carbon materials (e.g. charcoal, biochar), activated carbons, carbon molecular sieves, aerogels and carbon nanomaterials (e.g. graphene and carbon nanotubes). CO2 adsorption in these materials relies mostly on physisorption and hence porosity is the predominant characteristic, with a high volume of pores, and particularly micropores, increasing the uptake. It is worth noting that heteroatoms (i.e. O-containing groups) may be present in the materials as a result of the synthesis approach and these groups naturally influence the adsorption mechanisms by introducing desirable chemisorption interactions.
As with zeolites, many carbon-based materials (e.g. activated carbon) benefit from industrial maturity. With the exception of carbon nanomaterials such as nanotubes and graphene, carbonaceous materials are typically cheap and can be manufactured in large-scale. Owing to their hydrophobic nature, carbon-based materials are not strongly affected by moisture, though a decrease in capacity is often observed compared to the performance under dry conditions.353 The uptake and selectivity of non-functionalised carbon-based materials, however, are typically lower than those of zeolites. In fact, these performance metrics are limited at low pressure and non-functionalised carbon-based materials are thus preferred for high-pressure separations. Carbon-based materials are good candidates in ESA process protocols.271
We note that carbonaceous sorbents could include polymeric compounds, which have also been tested for CO2 capture. An example of such a polymeric adsorbent would be the so-called “polymers of intrinsic microporosity”.354,355 However, these materials are most often investigated in the context of membrane separation rather than adsorption.
Functionalised adsorbents. Chemisorption can be tailored to dominate carbon capture at low pressure, and thus a number of researchers have looked at ways to add functional groups and/or reactive species to the adsorbents described above. This is particularly true in the case of carbon-based adsorbents.356 N-Containing functionalities have been incorporated either into the carbonaceous structure or into the adsorbent pore space via immobilisation of amine-containing compounds. Amine functionalisation has also been reported for mesoporous silica.353,357 The modification of silica typically follows one of two routes: (i) porous silica is physically impregnated with amine-containing molecules,357 or (ii) amine-containing compounds are covalently grafted on the surface of porous silica.353 In the former case, polyethylenimine (PEI) is often selected to modify porous silica owing to its high density of amine groups. In the latter case, the surface of silica is pre-functionalised with a derivatised silane, which can be reacted with amines to form covalent bonds. MOFs have also been impregnated with amine-containing compounds.358–362 A famous example is the functionalisation of a triazolate-based MOF with ethylenediamine molecules.
The mechanisms of CO2 adsorption on amine-functionalised adsorbents are not as straightforward as one might expect and strongly depend on the type of amines as well as the type of support/adsorbent. These mechanisms include: (i) nucleophilic reaction with formation of a zwitterion or carbamate, (ii) base-catalysed hydration of CO2 with formation of bicarbonate and (iii) cooperative adsorption process between adjacent amine molecules.353,362 Functionalisation typically enhances CO2 uptake and selectivity but can limit gas diffusion when large organic molecules are used to modify the adsorbents. Chemical leaching is another typical issues that occur when N-containing compounds are only physically impregnated on the adsorbent. In many cases amine-modified adsorbents have either not been tested under process cycling schemes or have limited lifetimes under such testing conditions.
While the four classes of adsorbents described above typically form the core of materials used for CO2 capture, researchers have also tried to combine them and form composites to create synergistic effects and address one or more of the weaknesses of a given compound. Several of these composites are made of a carbon-based nanomaterial (i.e. graphene-based materials or CNTs) and a MOF.363,364
Developments in adsorbent structures. Conventional adsorbents are beaded or extruded of size 0.5 to 2 mm. While convenient for fixed beds, these are poor conductors of heat and prone to gas fluidisation or high pressure drop at high throughputs. For this reason, adsorbent structures have been investigated for CO2 capture applications.365,366 These structures include monoliths,367 laminated structures or hollow fibres.368–371 The latter have been employed effectively for very rapid temperature swing cycles using either hot water or steam as regeneration agent. The VeloxoTherm™ Process (discussed above) uses a rotary structured honeycomb adsorber for adsorption and desorption of large volumes of process gas. The temperature swing adsorption cycle is established by the rotation of the structured adsorbent, which completes a full revolution in about 60 seconds.
Thakkar et al.372 recently demonstrated how 3D printing techniques could be used to produce zeolitic adsorbent structures. 3D-printed monoliths with zeolite loadings as high as 90 wt% exhibited adsorption uptake that is comparable to that of powder sorbents. These are modest early steps but there is great promise for advanced manufacturing to allow creation of cheap, integrated adsorbent/flow devices to achieve unprecedented advances in system performance.
One key area that needs addressing is that of materials testing and screening. As the research community understands better the performance of adsorbents under equilibrium conditions using a ‘simple’ CO2/N2 mixture, there is now a need to ‘challenge’ the materials by running dynamic tests and use simulated flue gas streams. The former aspect will enable to derive the kinetic properties of the various adsorbents and hence provide a more realistic picture of their performance. It is recognised that some kinetic studies have already been reported but they are not yet performed systematically.353 A number of impurities contained in the flue gas could potentially impair the performance of adsorbents and therefore testing involving multicomponent streams are particularly informative. We highlight here recent work on the development of a high-throughput analyser enabling multicomponent equilibrium experiments.373 Using this set-up, it was found that of the adsorbents studied, those containing alkylamines performed well for CO2 capture in the presence of N2 and H2O. Further work around adsorber design is also crucial to allow faster cycles since it will directly influence the overall process. While the commonly proposed bed contactors include fixed bed, fluidised bed and moving bed,287 we note here the recent development of a rotary wheel adsorber to allow fast TSA cycles.374 Such rotary systems are incorporated in the Inventys© CO2 capture technology.
Industrial scale formulation of some of the novel advanced adsorbents (discussed in Section 4.2.4) will become increasingly important. To be integrated in a process, the materials must be manufactured as structured adsorbents, e.g. pellets, beads, monoliths, fibres. This is particularly true in the case of MOFs, which are still largely synthesised in a powder form. There are indications this is changing, as demonstrated by, for example, BASF and small companies such as the MOF company, NuMat, and Mosaic Materials. In the near future, we can expect the emergence of a number of spin-outs and start-ups producing MOF in different formats, e.g., Immaterial. Research studies have also started investigating the incorporation of MOFs into structured supports such as fibres and monoliths.375–381
As we have amassed more knowledge concerning CO2 adsorption, CO2 adsorbents and their performance metrics from different experimental and computational viewpoints, there is now a need to consolidate that knowledge and propose a combined multi-scale approach to the development of CO2 adsorbents and adsorption technologies. The examples of recent studies highlighted above are beginning to move towards that direction.
For mid-size CO2 capture applications, recent advances in adsorption technology are providing low cost and low energy options, thus potentially offering an attractive alternative to liquid scrubbing systems. Some promising developments include adsorbent structures, hybrid amine sorbents, low quality steam regeneration and rapid cycling. However, for large scale processes, it is unlikely that adsorbent technology will be competitive against established liquid scrubbing systems due to the complexity of large scale solids handling. Hybrid sorption enhanced reactive systems such as SEWGS have a strong role to play, particularly as hydrogen is promoted as an energy carrier in some economies. In a relatively short time, adsorption processes have developed rapidly and the future looks bright for further development and deployment in a range of CO2 capture applications.
4.3 Calcium looping technology
Calcium looping (CaL) technology is a relatively new alternative for post-combustion CO2 capture, and is based on the following reversible reaction:| CaCO3 ↔ CaO + CO2 ΔH = −178 kJ mol−1 | (3) |
Although the use of lime as a means for removing CO2 from hot gases is over 100 years old, the idea of using it in a reversible scheme to strip CO2 from flue gases is relatively new382 and can be represented schematically by Fig. 10. Implicit in such a cycle is the requirement that the lime product be used in multiple cycles in order to minimise the costs, and increase the overall efficiency of the process and this demands the use of a carbonator and a regenerator, normally envisaged as being a small oxy-fuel power plant to regenerate the spent sorbent and produce a pure stream of CO2 for storage, or possibly use (see Fig. 11).
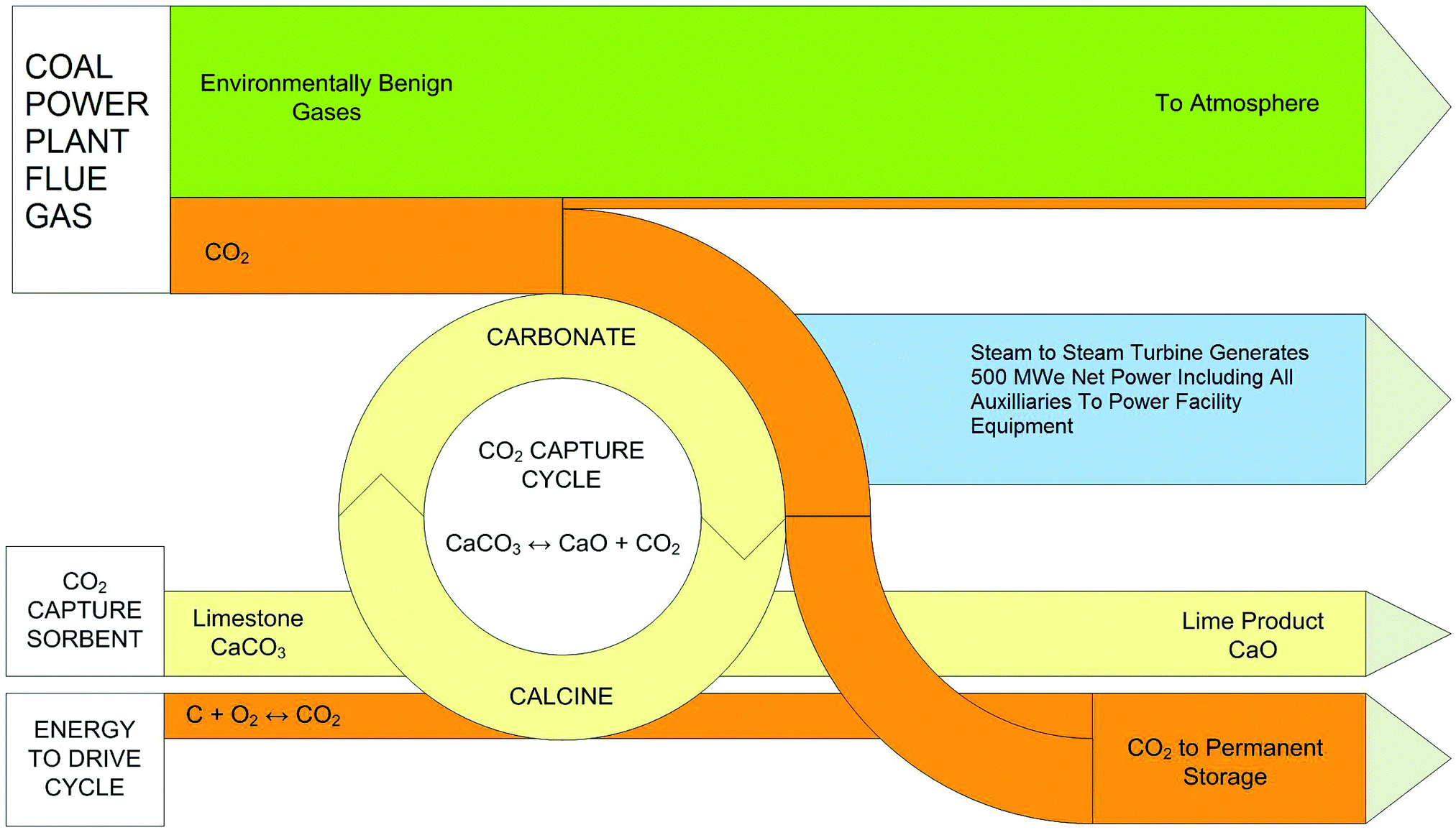 | ||
| Fig. 10 Schematic of the calcium looping (CaL) cycle. | ||
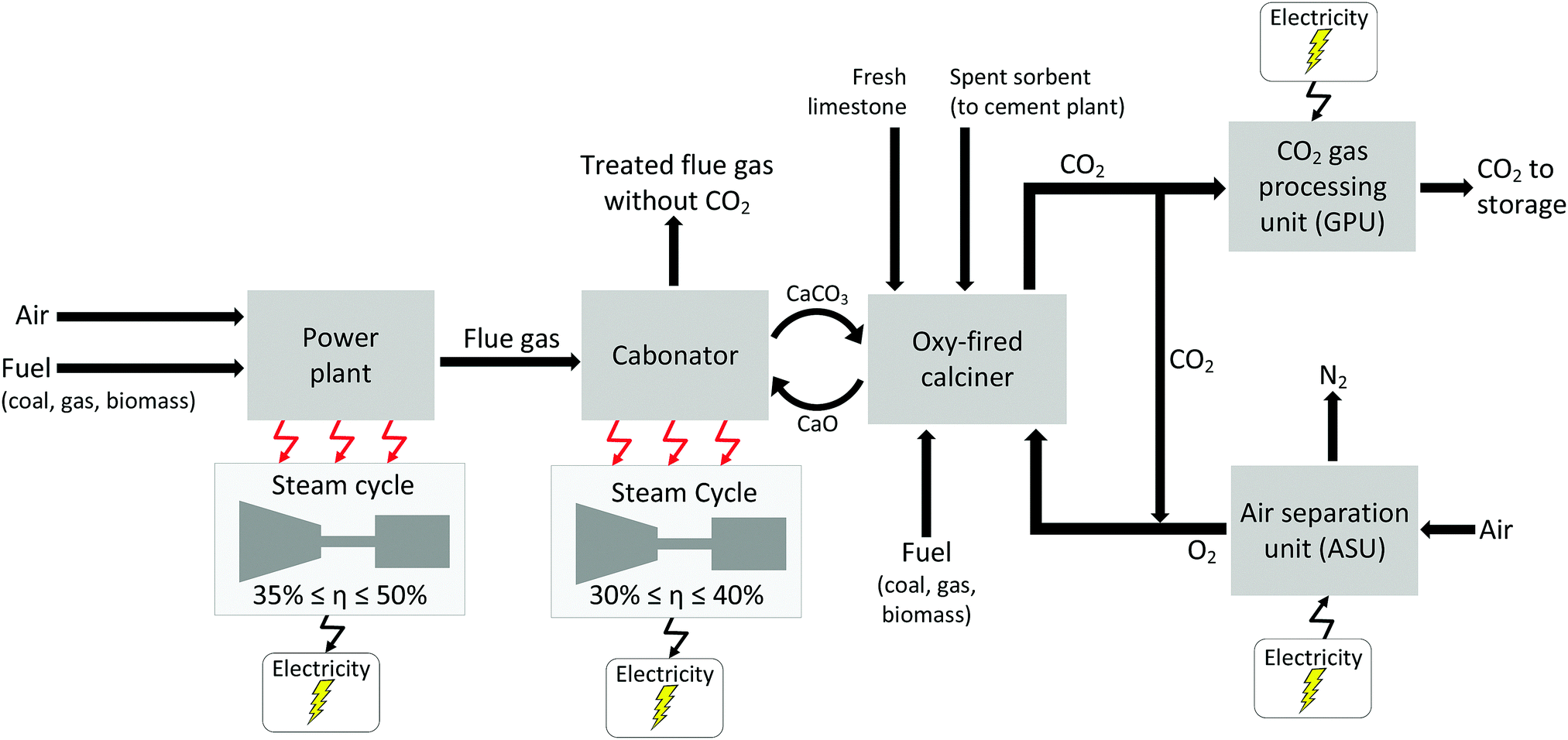 | ||
| Fig. 11 Calcium looping within a post-combustion capture process. Note that some units of operation may generate power (e.g., the carbonator), whereas the GPU and ASU requires a power supply. | ||
Three key factors distinguish CaL from the other CCS technologies. First, because the carbonator/calciner can serve as a heat source for a steam cycle to produce additional power, the energy penalty associated with the technology can be several percentage points lower than that of conventional amine scrubbing.‡‡‡‡383,384 Second, the sorbent used, namely limestone, is available in industrial quantities, and is also a non-hazardous chemical whose price is of the order of £10–20 per tonne (∼US$13–26 per tonne). In contrast, the cost of amine solvent MEA is much greater at US$1.8–2.9 kg−1.385,386 The third benefit of CaL is that there is a possibility of using the spent sorbent in industrial processes such as cement making, which, since lime manufacture represents 50% or more of the CO2 output in cement production, offers an approach to partially decarbonise the cement industry153 or even to achieve near-zero emissions by incorporating the technology into the cement manufacturing process.145,387–389 Finally, there exists substantial capacity worldwide to take most of the spent sorbent from CaL should it become a dominant technology.390 For instance, spent CaL sorbent can be used in the production of cement clinker,153,154 ocean liming, steel manufacturing (to make slag or capture CO2), or for flue gas desulphurisation.390
CaL technology has also been progressed to pilot scale. There are two major demonstration projects, one at the University of Darmstadt, in Germany391,392 and one in La Pereda, Spain,393 which have been used to extensively test circulating fluidised bed-based technology, and a 1.9 MWth pilot plant, which combines a bubbling fluidised bed carbonator and a rotary kiln calciner, in Taiwan that has been reported to have run for over 1 year.164 Based on its work, Industrial Technology Research Institute (ITRI) estimated that the integrated CaL process would offer a carbon capture cost of less than $30 per tCO2.394 These demonstration projects mean that the technology has achieved a technical readiness level of 6.395 Moreover, there is now an extensive number of small pilot plant facilities worldwide396 being used to address various aspects of the technology, from looking at aspects of CaL, such as sorbent attrition, and the behaviour of modified and synthetic sorbents to improve their overall performance, to the development of novel configurations for CaL applications.
It was gradually recognised that the typical TGA environment used to assess sorbent performance was associated with major flaws.397 In particular, the chemical environment was unrealistic as it missed both the positive effects of water addition on capture398–400 and the negative effects of SO2. More importantly, calcination in N2 or environments with low levels of CO2 at temperatures of 850 °C or below is unrealistic and tends to overestimate sorbent performance. In real systems, it appears that the water content typical of a combustion environment (10–20%) partially compensated for the presence of SO2,401 but higher temperatures necessary to drive calcination in the presence of nearly 100% CO2 always led to a significant deterioration in sorbent performance regardless of the modification process involved.402,403 It is also interesting to note that He et al.404 have reported a beneficial effect of steam on the ability of carbide slag to capture CO2. Finally, it is worth noting that steam has been shown to produce clear beneficial effects on the carbonation process; it also appears that it can produce significant benefits in improving sorbent performance when it is added to the calciner of a 100 kWth pilot plant.405 These benefits do not appear to be related to a lowering of the calcination temperature due to the reduction in partial pressure of CO2,406 but instead it has been suggested that they are associated with reduction in sintering produced by the lower CO2 levels in the calciner.
Despite ample evidence to the contrary, numerous studies on sorbent performance are compromised by the use of low calcination temperatures and unrealistic chemical environments. This has been pointed out again recently by Clough et al.,407 who have suggested a novel TGA protocol to ensure more realistic results are obtained on modified sorbent performance.
Sorbent attrition is another area which has received attention over an extended period. Although there are limestones that perform extremely poorly,408 the fact that there are both fully operational demonstration units and a large number of pilot plants396 is a clear indication that natural sorbents can perform adequately in CaL processes. Nonetheless, numerous attempts have been made to improve sorbent performance by various kinds of treatment, most notably pelletisation with a support material, often with mixed results (and a critical issue in such evaluations is again that tests be performed under realistic fluidised bed conditions).409 Another critical question for all such attempts is that the cost of such approaches may easily outweigh any benefit in terms of potentially superior performance and/or mechanical resistance.410 An interesting result from the work of Erans et al.411,412 is that some additives may actually weaken the resulting sorbent, and this is only apparent when tests are performed under fluidised bed conditions and that this phenomenon counteracts any reactivity benefit associated with the additive, in this case flour incorporated into the pelletised matrix, to serve as a representative form of biomass addition.
The importance of the final physical matrix in terms of ultimate performance of a Ca sorbent has been further demonstrated by mixing low levels of biomass into a pelletised matrix, and observing a significant improvement in sorbent performance,416 which as noted above does not necessarily lead to superior performance in a real fluidised bed system as the resulting material becomes more susceptible to fragmentation and attrition. It should also be noted that while this discussion is focused on limestone, there are many natural Ca-based materials, some of which may well have superior performance, and there is a significant body of literature on the potential of various such materials, some of which appear to have superior capture performance (e.g., waste marble powders417); however, the key issue here will be the overall amounts of such material available for significant removal of CO2 from industrial processes and power production, which is the primary reason for restricting this discussion to limestone and sorbents derived from it, rather than looking at other materials potentially available.
Hydration to form Ca(OH)2 at temperatures below 500 °C is beneficial due to the formation of cracks in the CaO particles creating paths to the interior of the particles and, therefore, improving CO2 capture.418–421 Another positive effect of hydration is the formation of larger pores; unfortunately, this is also associated with weakening the sorbent matrix, and so any potential benefit can easily be outweighed by sorbent loss due to attrition and elutriation. The hydration reaction:
| CaO + H2O ↔ Ca(OH)2 ΔH = −109 kJ mol−1 | (4) |
An alternative to steam reactivation is to recarbonate the spent sorbent. This was first suggested by Salvador et al.,422 who proposed that recarbonation in high concentrations of CO2 might be a reactivation strategy. Sun et al.423 subsequently showed that marginally increasing the carbonation times had a positive outcome on the capture capacity over several cycles. Chen et al.424 stated that extending the carbonation time substantially helped to recover some capture ability of the sorbents and although this recovery decreases with increasing number of cycles, the samples that experienced extended carbonation time showed better reactivity than those that did not. Further work demonstrated that carbonation time has a robust effect on carrying capacity. If the carbonation time increased, then the residual conversion also increased.425 More recently, the benefits of a recarbonation strategy were explored with the 1.7 MWth La Pereda plant in Spain, and it was reported that an improvement in sorbent performance of 10% in CO2 carrying capacity of the sorbent was achievable, if the solids were allowed sufficient residence time in the loop seal, which acted as a recarbonator in this work.426
In experiments with combined CaO/CuO/calcium aluminate cements, Rahman et al.432 have obtained results which suggest this is possible, albeit that they reported a decline in the oxidation potential of such pellets in a gasification environment. Duhoux et al.433 have carried out simulations for a process combining CaL and CLC for CO2 capture and report that a combined CaL–CLC process could show a 10% process efficiency gain, and significantly increased power output.
Another interesting possibility is combining CaL with thermal storage. At the simplest level, producing CaO from CaCO3 offers the possibility of thermal storage by itself.434 In principle, this can be combined with other thermal energy storage options and Hanak et al.434 suggest that this option with cryogenic O2 storage has the potential to increase the profitability of an integrated system over that of a reference coal-fired power plant without CO2 capture.
There has also been increasing interest in combining CaL technology with solar power.389,435–437 In this case the goal would be operate the calciner using solar energy, and if this is successfully achieved the calciner could serve as part of a conventional CaL cycle and/or a source of thermal energy storage.
5 Next generation CO2 capture processes
This section considers next generation CO2 capture technologies. These have been studied extensively, however, compared to conventional capture technologies (liquid-phase or solid-phase sorbents), they are in the earlier stages of development. These “new generation” technologies show particular promise in high temperature applications, with potential opportunities for use in process intensification.5.1 Chemical-looping, progress and prospective
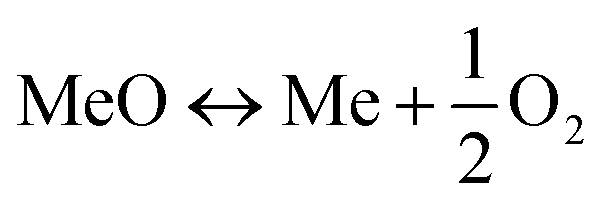 | (5) |
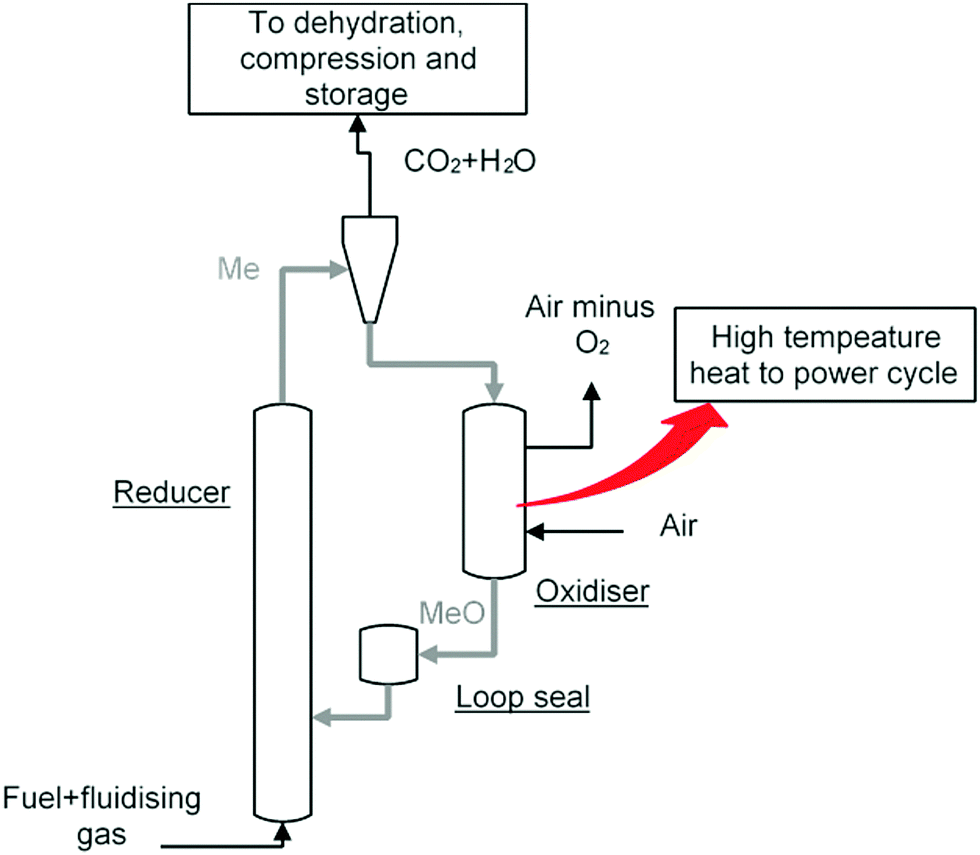 | ||
| Fig. 12 Typical chemical looping combustion (CLC) process. The metal oxide oxygen carrier (MeO) is circulated between two fluidised reactors. In the reducer, the MeO gives up oxygen to the fuel to produce CO2 and water. The reduced oxygen carrier (Me) is then carried over into a cyclone where the solid is separated from the gas phase and sent to an oxidiser where Me is regenerated, taking in oxygen from the air. The regenerated oxide MeO is then returned to the reducer for another cycle. | ||
One of the first uses of chemical looping was the Brin process used to manufacture gas phase oxygen (reaction (6)). The reaction has a sufficiently high oxygen partial pressure at ∼700 °C that gas phase oxygen can be produced in the forward reaction (at 0.05 atm). The reaction is reversed by lowering the temperature and increasing the partial pressure of oxygen.443 The Brin process fell out of favour with the introduction of the Linde process based on cryogenic air separation. More recently this approach has been investigated under various names, including ceramic auto-thermal reforming,444 and chemical looping air separation (CLAS);445 the former using perovskite material, and the latter using materials such as copper oxide.
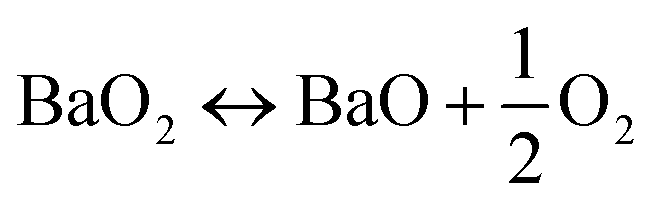 | (6) |
A similar argument can be made for chemical looping combustion, in which the fuel is brought into direct contact with the metal oxide. Here, the exergy loss associated with the combustion reaction itself is partially avoided. Chemical looping combustion was originally proposed as a way to increase the efficiency of fossil fuel power station because it avoided the exergy loss associated with combustion.447 Materials for chemical looping combustion need to operate with an equilibrium pO2 > O (10−7); this ensures that the partial pressure of CO (or other un-combusted fuel) is low at the exit of a well-mixed reactor. Iron oxide for example has several oxidation states which can be used to transfer oxygen, the Fe2O3 to Fe3O4 transition has a pO2 of ∼3.6 × 10−7 bar at 900 °C which means that, at equilibrium, the ratio of CO to CO2 would be ∼1 × 10−5, i.e. near complete combustion.
| C + H2O → CO + H2 | (7) |
| CO + H2 + 2MeO → H2O + CO2 + 2Me | (8) |
This can mean that the rate of solid fuel conversion is actually limited by the rate of gasification of the solid fuel.453,456 Research into the kinetics of the gasification reaction has a long history, beyond the scope of this discussion, however, two important features of relevance to chemical looping are: (i) the big differences in reactivity between different kinds of chars, and (ii) the effect of product inhibition. CO and H2 retard the rate of gasification, so when gasification is carried out in the presence of a solid oxygen carrier, the gasification rate is accelerated.456–458 This acceleration rate, although significant, does not lead to an order of magnitude change in the rate of gasification. This means that for coals which produce un-reactive chars, the build-up of char in the system is problematic. The problem is compounded by the use of interlinked reactors if the char is recirculated to the air reactor, where it can burn releasing CO2 back into the environment.
Two strategies for dealing with the low reactivity char are to: (i) physically separate the char from the oxygen carrier using carbon strippers,459 or (ii) increase the rate of char conversion using CLOU combustion.460 In the former, the difference in density is used to separate the char from the oxygen carrier and return it to the fuel reactor, allowing a larger inventory and increased carbon conversion rate. In the latter, oxygen carriers which release gas phase oxygen can be used to increase the carbon conversion rate. Copper(II) oxide461,462 and manganese based materials (e.g., mixed oxides of iron and manganese463 or perovskites based on calcium manganate) all have an equilibrium pO2 which is significant at fuel reactor temperatures and also allow the material to be re-oxidised by air containing 21% oxygen at air reactor temperatures.
Economic assessments must consider the cost and lifetime of the oxygen carrier material, and also the availability of the plant. Paper studies will make sensible assumptions about the availability of the plant, but will not consider the case where the technology fails (i.e., a very low plant availability). This latter point is perhaps what holds back chemical looping technology, in that a chemical looping combustion power plant will be much more complicated than a standard power station, and will require a massive upfront investment. For an investor the risk versus benefit argument becomes one of confidence in the technology at scale. Research effort has therefore been directed at precisely these issues, both in the (i) development of materials and understanding the costs, and in (ii) developing confidence at scale.
The contribution of the cost of the oxygen carrier to the overall operating cost is proportional to the supply cost and inversely proportional to the material lifetime; cheap easily degraded materials or expensive long lasting materials could perform equally as well. The conceptual design of a 1000 MW coal-fired system is given by,465 assuming a low cost ore (ilmenite or manganese ore) as the oxygen carrier. They assumed a lifetime of only 200 hours and concluded that the contribution to the cost of carbon capture of the oxygen carrier would be €1.3–4 per tCO2captured, less than cost of the final oxy-polishing step. For coal-based systems it is hard to see how a very expensive material could be used, since it will quickly become contaminated with the components of the coal ash. Natural gas systems are cleaner and therefore perhaps easier in this regard. Porrazzo et al.466 modelled the performance of a natural gas-fired CLC system, operating at 10 bar with an oxygen carrier consisting of NiO on alumina ($15.3 per kg). Given the difficulties of presenting consistent economic data, they explored the sensitivity to material lifetime. The levelised cost of electricity (LCOE) fell to a plateau (at around 500–1000 h of lifetime), at which point material cost was no longer significant. To break even with a NGCC system fitted with an amine scrubber, the particles would have to last around 500 to 700 hours. These lifetimes do not seem unreasonably difficult targets to achieve, and it is likely that materials for chemical looping can be made cost effective.
On building confidence, progress has been made on moving from laboratory tests, through to pilot scale, in order to answer the questions of (i) reliability, and (ii) durability of materials over long-term trials. Recent demonstrations have focussed on larger scale (e.g., 1 MWth CLC using ilmenite,448 3 MWth Alstom calcium sulphate process467 or longer trials (e.g., 99 hours of operation in a 10 kW CLC system using calcium manganate,468 200 hours in 25 kWth CDCL system469). All the indications from these trials suggest that chemical looping combustion and chemical looping hydrogen production have promise.
5.2 Membrane-based technology for CCS
Membrane processes for CO2 capture can, in a manner similar to other technologies, be classified as pre-combustion, oxy-combustion and post-combustion processes. Here, we focus our discussion on the opportunities for process intensification through the use of membranes in CO2 capture. Inorganic membranes are capable of high temperature operation. Intensification is achieved through the integration of these membrane processes with reforming, shift and oxidation reactions. Organic membranes are unsuitable for high temperature applications, and most are also unsuitable for low temperature shift (LTS) processes (around 180–250 °C).470 We confine ourselves to the consideration of dense inorganic membranes, as porous inorganic membranes do not currently have sufficient selectivity for application to the processes of interest here. For details on other membranes that may be applied to CO2 capture processes (e.g., organic membranes and porous inorganic membranes), the reader is referred to a number of reviews published previously.471–480 In the case of application to CCS processes we thus seek to exploit high temperature membranes that are selective for hydrogen, oxygen and CO2 permeation.Membrane permeability is considered an important property of membrane material and not associated with geometry. If the permeability of the membrane is known, along with its dimensions and the driving force across the membrane, then flux can be determined and process design calculations performed. However, this is assuming that transport within the membrane is the rate-determining step. This in general need not be the case; the rate may be determined by surface exchange processes or indeed mass transfer processes.
In Section 5.2.1 we describe the properties of a membrane that are important in conferring such CCS-relevant selectivity. Furthermore, we summarise the kinetic behaviour of such membranes through the use of permeabilities. Section 5.2.1 also discusses the classes of membrane that have the potential for use in process intensification of CCS. Section 5.2.2 describes how such membranes may fit into CCS processes, and the work that has been conducted to date. We conclude with the future outlook for membrane-based CCS processes.
Metallic membranes for hydrogen permeation. Hydrogen-selective membranes have potential applications in pre-combustion CO2 capture.475 Hydrogen selectivity is a result of the greater propensity for hydrogen, over other permanent gases, to dissolve and diffuse in metals.481 In addition to Pd, hydrogen will also diffuse through other transition metals such as Ti, V, Nb, Zr, Mn, Fe, Co and Ni.475 However, Pd and its alloys are most commonly used in membranes due to the ability of Pd to rapidly dissociate hydrogen while possessing the ability to incorporate a large amount of hydrogen and maintaining structural integrity.481 Pd–Ag is the most common alloy to use.
The permeability of the membrane simply depends upon the product of the solubility of hydrogen in the membrane, SH, and an appropriate diffusion coefficient, DH:
| Permeabilityi = SHDH | (9) |
Pd and Pd-alloy membranes usually have permeabilities of around 10−8 mol m−1 s−1 Pa−0.5 (e.g., at 350 °C). Clearly, such membranes can be expensive. The cost of the materials is dependent upon the thickness of the active membrane layer, subsequently, most of the research tends to focus on fabricating membranes that are as thin as practical. For more information on hydrogen permeation in metals, the reader is referred to the review by Al-Mufachi et al.481 Also, the prospects for commercialisation of Pd membranes has been reviewed by Gallucci et al.482
Ion-transport membranes for oxygen and CO2 permeation. Materials exploiting solid-state ion conduction can also be used to fabricate highly selective membranes for use at high temperature. Selectivity is conferred by the fact that, e.g., an oxygen ion vacancy within an ionic oxide¶¶¶¶ is unlikely to be occupied by any species other than an oxygen ion due to the very specific chemical environment of that vacancy. However, it may be possible for similarly sized ions such as hydroxyl ions, or fluorine ions, to occupy the vacancy. Oxygen-ion transport may then occur by oxygen ions “hopping” from one vacancy to another. Appropriate oxides to consider for use as membranes include fluorites, pyrochlores, brownmillerites and perovskites. In many cases, such materials can be doped to create intrinsic oxygen defects (i.e., oxygen-ion vacancies in the case of our discussion), or employed under conditions where oxygen defects will be created as the result of gas-solid reactions with the prevailing atmosphere.
A membrane that shows pure oxygen vacancy diffusion cannot be employed as a gas separation membrane. A steady flux of ions across such a membrane cannot be achieved due to the development of a potential difference across the membrane. To achieve a steady flux, an equal and opposite flux of another charge carrying species must be permitted, either: (i) through the use of an external circuit (thus becoming a solid state electrochemical cell and not covered here), or (ii) by the introduction of a charge transfer pathway internal to the membrane. In the case that this additional charge carrier is an electron, the material is known as a mixed ion and electron conductor (MIEC). A second example of an ion-transport membrane includes the dual-phase ion-conducting gas separation membranes. These membranes will be discussed in more detail below.
Mixed ionic and electronic conducting (MIEC) membranes thus have both ionic charge carriers and electronic charge carriers. In the case of an oxygen-ion and electron conductor, if there is equilibrium on both sides of the membrane between gas-phase oxygen, oxygen ionic charge carriers and electrons, it can be shown that the flux of oxygen, jO, is:489
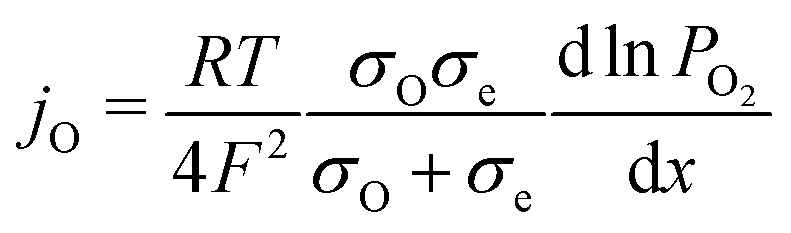 | (10) |
500 C mol−1), T is temperature, and PO2 is the partial pressure of O2. Application of the Nernst–Einstein equation relating conductivity to diffusion coefficient allows one to show that, once more, permeability depends upon a diffusion coefficient and a solubility. During permeation experiments, the permeate side of the membrane is often fed with an inert sweep gas, which leaves the logarithm of PO2 poorly controlled (and dependent upon an oxygen material balance on the permeate side). This is not an ideal way to perform a permeation experiment. Oxygen partial pressures should be controlled on both sides of the membrane (ideally, with the oxygen partial pressure difference being small). The flux is then determined by measuring what would be small changes in oxygen partial pressures over the membrane module. Such a technique has not been adopted by the research community possibly due to the difficulties of gas analysis but it would be a significant improvement in current experimental design.
MIEC oxide membranes tend to have oxygen fluxes of about 1 ml (STP) cm−2 min−1 or about 10−6 mol cm−2 s−1 at temperatures of around 800 °C. For a typical driving force of 104 Pa, the permeance can be estimated to be about 10−6 mol m−2 s−1 Pa−1. Assuming a membrane thickness of 1 mm would yield a permeability of 10−9 mol m−1 s−1 Pa−1. MIEC oxide membranes can be unstable in the presence of CO2 if the cations employed within the membrane have a propensity for carbonate formation. Thus materials selection must be considered carefully, e.g., in the case of perovskites, ABO3, La on the ‘A’ site is preferred over Ba in the presence of CO2.
There are a number of reviews covering the use of MIEC membranes for oxygen permeation alone,490–492 and for chemical production.489 MIEC membranes have been employed for oxygen permeation with good results over times scales of 1000 hours.493
Another application of MIEC oxide membranes is in hydrogen permeation. An oxygen vacancy in an oxide membrane has the possibility to react with water, forming a hydroxyl-like species and releasing a proton, which combines with a lattice oxygen species to produce a second hydroxyl species. Protons may then hop from one lattice oxygen site to another lattice oxygen site, leading to hydrogen permeation. The level of proton-conduction versus oxygen-ion conduction in such a material is a strong function of its degree of hydration. Hydration of the oxide lattice itself is exothermic, and thus, if high proton to oxygen-ion conductivity ratios are to be achieved, lower operating temperatures are necessary. However, lower temperature operation will limit flux. Hydrogen permeation using this class of membranes has not been exploited seriously in CCS applications to date. Interested readers are referred to a review by Phair and Badwal494 for further information.
The dual-phase ion-conducting class of membranes uses two phases to conduct charge carriers instead of one, i.e., one ion conducting phase and an electronic conducting phase. If both phases are solid, the membrane can suffer from thermal expansivity mismatch problems, leading to failure. There is a body of work investigating dual-phase systems with a solid oxygen-ion conductor and a solid electronic conducting membrane for oxygen permeation.490
One promising approach recently adopted uses a molten salt as one of the phases. This avoids problems with thermal expansivity mismatch and allows for the opportunity of tailoring membrane properties. Perhaps most interesting for the field of CO2 capture is the use of molten carbonate systems, where the carbonate is housed within a porous solid, providing a route for carbonate ion transport. The carbonate can be: (i) supported in an oxygen-ion conducting oxide, leading to pure CO2 permeation;495,496 or (ii) supported in an electron-conducting host, resulting in the co-permeation of CO2 and oxygen.497,498
Carbonate ions supported in oxygen-ion-conducting oxide are incorporated into the carbonate melt via the reaction of CO2 with oxygen ions within the ion conducting support:
| CO2 + O2− (oxygen-ion conducting support) → CO32− (carbonate melt) | (11) |
In contrast, carbonate ions in an electron-conducting support are incorporated via a reaction between CO2 and oxygen, with electrons from the electron-conducting support:
 | (12) |
:1 should be observed when employing this class of membrane, if this counter diffusion was the dominant mechanism. However, this is often not the case,499 indicated that the mechanism in such membranes is likely to be more complex.
Supported molten carbonate membranes have CO2 fluxes of about 10−8 mol cm−2 s−1 at temperatures of ∼650 °C. For a typical driving force of 5 × 104 Pa, the estimated permeance is approximately 2 × 10−9 mol m−2 s−1 Pa−1. For a membrane thickness of 1 mm, the permeability is around 2 × 10−12 mol m−1 s−1 Pa−1. However, Zhang et al.500 have achieved much higher permeances of approximately 10−10 mol m−1 s−1 Pa−1 at 650 °C through the application of highly interconnected three-dimensional channels, demonstrating that there is room for improvement.
There has been too little research on this class of membrane to clearly identify the major problems and modes of degradation under operating conditions. However, we might anticipate that possible problems with this class of membrane may include carbonate conversion to oxide in low CO2 partial pressure atmospheres. Although the oxide will initially remain dissolved in the carbonate, it will eventually solidify at common operating temperatures once the mole fraction reaches saturation. This will impact membrane function and will also eventually lead to gas leakage across the membrane. Furthermore, other gases are known to dissolve in molten carbonates and may also undergo reaction with the carbonate or dissolved oxide in the carbonate. The effects of any such processes will need to be studied and accounted for in the design of the membrane and process. It must be emphasised that this is a relatively new research area, and as a consequence, further work on the mechanism is required to develop good understanding of membrane behaviour.
| CCS process | Potential membrane processes | Application and conditions | Reducing or fuel side membrane | Oxidising or permeate side reaction | Nature of membrane | Overall membrane Process | Suitability for process intensification |
|---|---|---|---|---|---|---|---|
| Pre-combustion | Post-shift CO2 or hydrogen separation | Low temperatures, may require pressure difference driving force | Within a post shift mix: CO2 → CO2(M) | CO2(M) → CO2 | CO2 or hydrogen permeable membrane. Possibly organic membrane as post shift temperatures are low. | Simple separation | Limited. To date, work has not been performed on coupling these processes to, e.g., another chemical reaction such as hydrogenation. |
| H2 → 2H(M) | 2H(M) → H2 | ||||||
| Membrane integration into the shift process with or without reforming in the membrane unit prior to shift | Remove products as the shift reaction occurs, range of temperatures possible, may require trans-membrane pressure difference. | During shift reaction: CO2 → CO2(M) | With or without sweep gas: CO2(M) → CO2 2H(M) → H2 | Direct integration of a supported molten carbonate CO2 permeable membrane not demonstrated. Significant work on Pd hydrogen permeable membranes to higher TRL. | Simple separation but can be used to overcome equilibrium limitations of secondary reaction. | Yes. Separation is performed while the shift reaction is occurring, which removes equilibrium limitations to conversion of the shift reaction. | |
| H2 → 2H(M) | |||||||
| Unmixed reforming | High temperatures, water may be required with feed to limit carbon deposition. | Oxygen supply to a reforming reaction: CH4 + O(M) → CO + 2H2 | Oxygen consumption from water feed: H2O → H2 + O(M) | Oxygen permeable MIEC membrane | CH4 + H2O → CO + 2H2 + H2 | Yes. Separate syngas and hydrogen streams are produced. | |
| Unmixed shift with hydrogen separation | High temperature shift with no equilibrium limitation | Reformate is oxidised: CO + O(M) → CO2 | H2O → H2 + O(M) | Oxygen permeable MIEC membrane | CO + H2O → CO2 + H2 and hydrogen separation | Yes. Separate CO2 and hydrogen streams are produced. | |
| H2 + O(M) → H2O | |||||||
| Oxy-combustion | Air separation only | High temperature oxygen permeation |
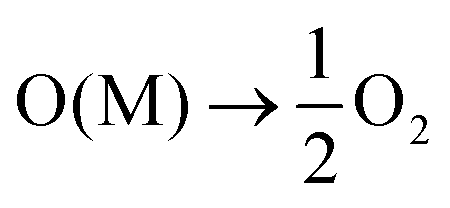
|
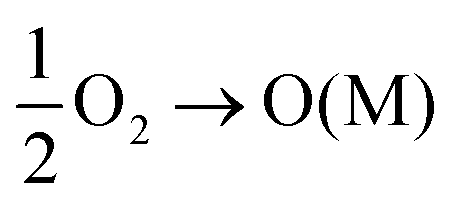
|
Oxygen permeable MIEC membrane | Simple separation | Limited |
| Air separation integrated with membrane oxy-combustion | Increased driving force for oxygen permeation, high temperature. | CH4 + 4O(M) → CO2 + 2H2O | 2O2 → 4O(M) | Oxygen permeable MIEC membrane | CH4 + 2O2 → CO2 + 2H2O | Yes. Nitrogen is not mixed in with combustion exhaust while chemical driving force of combustion drives permeation. | |
| Post-combustion | Capture of CO2 alone or co-permeation of CO2 and oxygen | Separation may occur at either high or low temperature, requires trans-membrane pressure difference. |
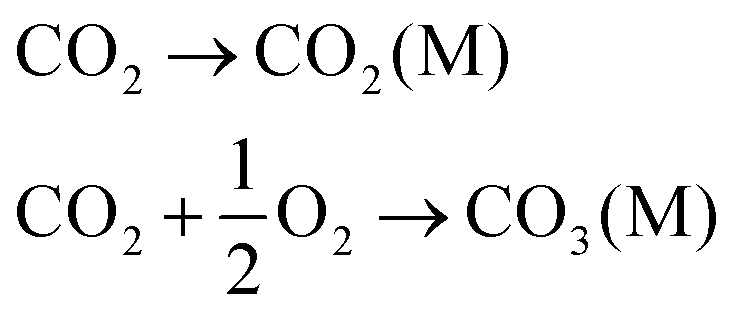
|
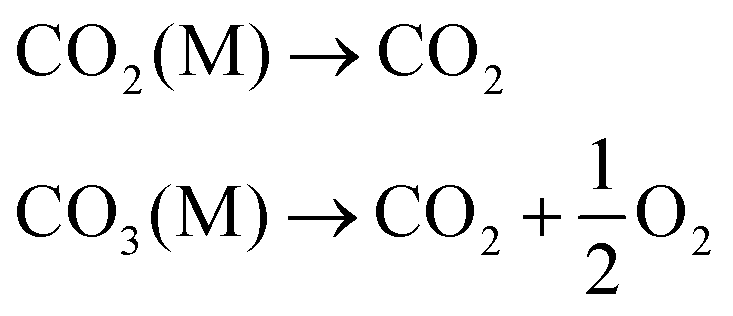
|
Supported molten carbonate CO2 permeable membrane and molten carbonate CO2 co-permeation membrane. | Separation only but co-permeation of CO2 and oxygen could be exploited for thermodynamic benefit. | May be possibilities but not explored yet in any detail. |
Membrane integration into the shift process. Membrane integration into the shift process requires CO2 or hydrogen permeable membranes, which would be used to remove these products whilst the reaction is occurring.475,501 This approach using membranes can overcome the equilibrium limitations associated with the water-gas shift (WGS) reaction, which enables higher equilibrium conversion at higher temperatures.|||||||| It is desirable to operate at higher temperatures in order to access favourable kinetics and reduce equipment footprint. Therefore, WGS processes would require CO2 or hydrogen permeable membranes that are stable within different temperature ranges. A trans-membrane pressure difference is required if high mole fractions on the permeate side are to be achieved,502 or if the system is to be operated in the absence of a permeate-side carrier gas (i.e., sweep gas). The sweep gas can be chosen to facilitate separation of the permeating gas from the sweep gas itself; water vapour is a common choice as it is condensable. Another possibility is to perform a reaction on the permeate side to consume the permeating gas, which would provide a chemical driving force for permeation, avoiding the need for a pressure difference across the membrane. Regardless of such considerations, there is a significant opportunity for intensification through removing the WGS equilibrium constraint. Ultimately, it is important to evaluate all process modifications via a whole-systems analysis with an aim to understand the impact of that modification on the cost per unit of decarbonised product, e.g., MWh, as discussed in Cabral and Mac Dowell.503
Early work on hydrogen permeation for WGS process intensification demonstrated the effectiveness of using a Pd membrane to overcome the WGS equilibrium. Since then, a large number of studies have investigated the importance of Pd membrane thickness, temperature of operation, nature of the WGS catalyst etc. From this large body of work, we present a small representative sample of the relevant work that has been performed. Uemiya et al.504 investigated a 20 micron thick Pd membrane supported on a porous glass cylinder operating at 400 °C with an argon sweep gas. A commercial iron-chromia catalyst was used in this system, which achieved carbon monoxide (CO) conversions in excess of those predicted from equilibrium calculations. More active catalysts (such as Pt-based systems) and thinner Pd–Ag membranes have been employed, e.g., Bi et al.505
Work has also been performed at the more-demanding lower temperatures of 200 to 300 °C,506 where higher hydrogen removal rates are required as the WGS equilibrium favours hydrogen production at lower temperature. Considered test conditions included both inert sweep gas and vacuum with no sweep gas at the permeate side of a Pd–Ag membrane. Lower temperatures and higher CO mole fraction can result in more significant co-adsorption of CO on the membrane surface, which can inhibit hydrogen adsorption and transport.507,508 However, under certain operating conditions, very high overall hydrogen recovery can be achieved, which is also associated with high CO2 mole fractions on the feed side of the membrane.506,508 Hydrogen production has also been demonstrated at larger scales in the laboratory,509 with membrane areas of 0.02 m2, thickness of 10 microns, and hydrogen production rates in the order of 0.25 N m3 h−1 at >99% purity. Operation temperature was in the range of 420 to 440 °C, at pressures up to 20 bar, and a ferrochrome catalyst was employed.
An alternative integration strategy is to use a combined reformer and membrane unit. Reforming occurs within the membrane unit to produce CO and H2, which is then followed by the shift step and hydrogen removal through the membrane. Tokyo Gas have successfully demonstrated an integrated reformer-shift-membrane unit rated for 40 Nm3 h−1 of hydrogen production and incorporating a Pd with rare-earth metal alloy film of less than 20 micron thickness supported on a stainless steel support.510 The unit has operated over 3000 hours with better than 99.99% hydrogen purity. The operating temperatures were in the range of 495 to 540 °C with a natural gas feed at 9.5 bar. A nickel–alumina catalyst in pellet form was used in the primary reformed catalyst bed and a further nickel–alumina catalyst in monolithic form was used in the vicinity of the membrane itself. Even at the highest natural gas supply rates, the CO2 mole fraction in the off-gas was above 60%.510
At the time of writing, there does not appear to have been any significant experimental work performed with organic membranes for hydrogen permeation coupled with the WGS reaction. Organic membranes are more suited for low temperatures operation, e.g., have the potential to be used for low temperature shift (around 180–250 °C470). Scholes et al.475 reviewed the opportunities for membrane integration with WGS processes, noting that there is the need for a WGS catalyst to be integrated with the membrane module. Furthermore, if the catalyst and membrane are fully integrated, then much of the membrane at the reactant inlet is relatively inactive due to the low hydrogen and CO2 mole fractions in this zone. As the work by Tokyo Gas demonstrates, this is easily avoided by the use of a primary reforming or shift process.510
The removal of CO2 from the WGS reaction mixture is possible with the use of CO2-permeable membranes. Supported molten carbonate membranes could be operated at temperatures in excess of those associated with high-temperature shift (HTS) processes, i.e., 350–450 °C.*****470 Molten carbonate membrane permeabilities are currently quite low, which is due to the necessity of solid state oxygen-ion diffusion to occur and temperatures in excess of 700 °C. Low-temperature shift could be integrated with a polymeric membrane, given that an appropriate catalyst is used, such opportunities have demonstrated through modelling work.511 The two approaches to combine CO2 permeation with WGS, i.e., organic and dual phase molten carbonate membranes, or low- and high-temperature shift, remain in the early stages of development and have not been demonstrated at scale.
Unmixed reforming and unmixed shift. As an alternative to catalytic steam reforming, the unmixed reforming process involves separate air and fuel/steam feeds to create a cyclic process (i.e., the air and fuel/steam feeds do not mix).512 Membranes can be used to facilitate unmixed reforming. The oxygen permeable membrane would operate at high temperature with feeds of methane and water to opposite sides of the membrane.††††† Synthesis gas would be produced on the methane side, and hydrogen on the water side; it is this hydrogen stream that can be used for a CO2 free combustion process. As this membrane process is endothermic, combustion of the synthesis gas may be required to provide energy. The combustion would only produce CO2 and water, provided oxy-combustion conditions are used, which would make CO2 separation relatively straight forward. High conversions can be achieved for steam-methane reforming (SMR) by using high temperatures, thereby avoiding the need for a trans-membrane pressure difference to increase hydrogen mole fraction. This process does have the potential for intensification through significant process simplification, e.g., combine reforming processes, HTS and LTS into one membrane reactor.
One approach for unmixed reforming is to direct the reformate or synthesis gas product to a membrane reactor that houses an oxygen permeable membrane with water being fed to the other side of the membrane. Alternatively, the two processes of unmixed reforming and subsequent unmixed shift could occur in series and operated in one membrane unit. The synthesis gas would provide the reducing gas to further drive water splitting on the water side of the membrane (overall this is a combined unmixed WGS and hydrogen purification). The WGS reaction is slightly exothermic, therefore no heat input is required. However, if the syngas produced from the unmixed reforming is not combusted, the overall process becomes endothermic. It is important to consider the energy requirement for such a process and avoid unacceptable CO2 emissions (e.g., due to syngas combustion). Similar to the previous unmixed reforming scenario, no trans-membrane pressure difference would be required to get high mole fractions of CO2 and water (on the reformate side) and hydrogen (on the water side). Also, process intensification is possible by combining the HTS and LTS in one membrane reactor.
The membrane-based processes for unmixed reforming and unmixed shift both result in hydrogen production using a reducing gas to provide, via an oxygen permeable membrane, the driving force to split water on the other side of the membrane. Some studies have begun to investigate membrane-based reforming and shift processes,513–515 however, further research is necessary. Jiang et al.513 tested a BCFZ (BaCoxFeyZr1−x−yO3) oxygen-permeable MIEC membrane at temperatures between 800 and 950 °C. There was a methane feed to the reducing side of the membrane, and a water feed to the oxidising side. A nickel-based catalyst was packed around the hollow fibre membrane in the reducing side chamber. The process produced syngas on the methane-feed side and hydrogen on the water-feed side.513 Using a BCF (Ba0.98Ce0.05Fe0.95O3) oxygen-permeable MIEC membrane, Li et al.515 tested slightly different conditions. Similarly, there was a methane feed to the reducing side, but in this case, a mixture of water and air was fed to the oxidising side. The membrane was operated between 800 and 925 °C with ruthenium-based catalysts present on both sides of the membrane. The oxygen flux was sufficient to remove all of the oxygen from the oxidising side as well as split the water to form hydrogen. Thus, the oxidising side produced a stream containing hydrogen and nitrogen suitable for ammonia synthesis. On the methane side, syngas was formed with an appropriate composition for methanol synthesis.515
Air separation integrated with membrane oxy-combustion. An oxygen-permeable membrane can be directly integrated into a fuel combustion chamber for oxy-combustion. Air is fed to one side of the membrane and fuel to the other. Oxygen permeation leads to combustion with a nitrogen-free exhaust gas, facilitating CO2 capture, produced on the fuel side of the membrane. Such a process is often referred to as the advanced zero emission power plant, AZEP.516,517 A number of papers have dealt with the optimisation of the AZEP concept using modelling approaches,518–521 and a number of review papers consider the nature and operation of such processes.518,522,523 Although there are many papers on oxygen permeation in MIEC and dual-phase membranes, the literature covering the application of such membranes in a membrane-based oxy-combustion is much more limited.
Any membrane that it is to be operated in AZEP must be stable in CO2-containing atmospheres. Subsequently, there is a body of work that evaluates the stability of oxygen permeation membranes in such atmospheres. However, these papers usually fall short of actually investigating membrane-based combustion. Carbon dioxide-tolerant single-phase MIEC membranes that have been investigated include (Nd0.9La0.1)2Ni0.74Cu0.21Ga0.05O4,524 and (Pr0.9La0.1)2(Ni0.74Cu0.21Ga0.05)O4.525–527 A number of studies have focussed on the use of CO2-tolerant dual phase membranes.528–530 Studies in which oxygen permeable membranes are actually subjected to combustion conditions are much more limited, these include experimental work of direct relevance,527,531 as well as investigations into how combustion chemistry couples with the permeation process.532,533
Post-combustion capture of CO2 alone or co-permeation of CO2 and oxygen. Although post-combustion capture can be considered to be a simple separation process with little opportunity for intensification, there may be some interesting unforeseen possibilities for improved processes. In the case of a membrane that exhibits co-permeation of CO2 and oxygen, we can reverse the direction of CO2 permeation such that it may proceed against its own chemical potential difference. This would require using an oxygen chemical potential difference of opposite sign that is more than double (based on the reaction stoichiometry of carbonate formation) the CO2 chemical potential difference. Although permeation results in an increase in CO2 chemical potential, it leads to a greater decrease in oxygen chemical potential. Papaioannou et al.498 have demonstrated such ‘uphill’ CO2 permeation. However, the use of oxygen co-permeation to ‘drive’ a post-combustion capture process has not been the subject of any other studies.
5.3 Ionic liquids for CO2 capture
Ionic liquids (ILs) are substances completely composed of ions and are arbitrarily liquid below 100 °C.534 Those ILs with melting points below room temperature are referred to as room temperature ionic liquids (RTILs).534 ILs are now widely used in various areas of chemistry (and are emerging in areas of chemical engineering) including as solvents for organic synthesis,534–538 as solvents for and/or as agents of catalysis,534,535,539–543 in separations,544–547 for the synthesis of nanomaterials,548–552 in energy applications,553–557 and for biofuel production.558–561 The reason that ILs have garnered such interest is that they possess unusual (and often extreme) physical properties which provide some advantages in handling and storage.534 The most often cited property is a vanishingly low vapour pressure,562 which is important for CO2 capture applications, but other common IL properties include high thermal and chemical stability (also of import for CCS),563,564 non-flammability,565 and high viscosity.566 Their main strength lies in their designation as “designer solvents” because it is possible to synthetically alter the cation and anion independently, allowing for customisation of many solvent properties, including polarity, acid/base character, density, viscosity and thermal stability.534 The high thermal stability and low volatility allow for use of ILs for CO2 capture in either a pressure-swing configuration567 where CO2 desorption is not accompanied by evaporative solvent losses, or temperature-swing desorption where the high thermal stability of ILs (typically over 300 °C)538 also negates degradative solvent losses. Combined, these properties provide an opportunity to regenerate the solvent at a very wide range of temperatures and pressures, providing an excellent opportunity for process optimisation that is not available using traditional aqueous liquid capture media.1 However, the viscosities of ionic liquids are high compared to conventional solvents (66 to 1110 cP at 293 to 298 K),2,568 which may cause processing issues as discussed later.ILs have been proposed for use in carbon capture for many years.569 This is mainly due to a perceived high capacity for CO2 dissolution, though as Carvalho et al.570 recently observed, this is only strictly true for certain subclasses of ILs.570 Most of the CO2 solubility work in ILs has been carried out at high pressures and with pure CO2 gas streams (in order to overcome low CO2 solubility during physisorption), conditions useful for scientific study, but unlikely to merit consideration for CCS.2,283,567,570–575 There has also been a marked emphasis on physical absorption of CO2,1,2,283,567,576–578 though many chemisorbing ILs have come to prominence recently.579–585 For the physisorption studies, the major driver of IL selection has been high capacity, with this parameter influenced mainly by the anion selection (and fluorine content) and the length of the cation alkyl chain,585 though the majority of studies have focussed on dialkylimidazolium cations.2 Despite several generation of ILs for CCS, the CO2 capacity at post-combustion CO2 partial pressures remains limited,585 unless referenced on a molar (mol L−1) rather than molal (mol kg−1) basis,570 suggesting a practical limitation that will not be easily overcome.
Due to the reduced capacity at flue gas type pressures and CO2 concentrations (<0.05 mole fraction at 0.15 bar CO2 partial pressure),575 it is no longer expected that conventional, physisorbant ILs will be feasible for large scale CCS applications.585 In order to increase capacity, a range of “task-specific” ILs585–592 have been designed with functionalities (such as amine groups587 or azolates592) capable of chemisorption of CO2, thereby significantly driving up capacity at ambient pressure (0.5–2 mol CO2 per mole of IL). This has led to a diversity of structures and numerous publications on the design of anions or cations for CO2 chemisorption with ILs.593 The ability to tune the physical and chemical properties of ILs through ion design is well established.534 However, the difficulty facing chemisorbing ILs is very slow mass transport due to the extremely high viscosity of most “task-specific” ILs.594 This led to a partial divergence of academic research efforts away from solving issues with IL CCS, and toward the design and synthesis of more novel (and inevitably ever more complex) anions and especially cations for CO2 sorption. More recently, several studies have looked at physicochemical properties of IL–CO2 systems, including thermodynamic modelling,595 transport,596,597 kinetics of CO2 uptake,582,598 and the mechanisms of CO2 capture.599,600 However, in order to take full advantage of the tunable synthesis of IL structures, chemical functionalisation remains the preferred route to increased capacity.
Amine-functionalised cations. Bates et al.586 reported the first synthesis of an IL specifically designed to chemically bind CO2 with the IL 1-propylamide-3-butylimidazolium tetrafluoroborate, which is essentially a cation-bound amino group. They reported up to 0.5 moles of CO2 could be captured per ion pair at atmospheric CO2 pressure. The mechanism they reported was similar to aqueous amines, though only one carbamate salt can form per two ion pairs.586 This highlights one of the drawbacks listed above for IL-based CCS: amine functionalised cations cannot bind as much CO2 per amine on a molar basis, much less compete on a mass or volume basis where the molecular weight of the IL outdistances that of a traditional amine several fold at similar densities. Further, as discussed below, the functionalisation of the cation inherently lowers electron density at the amine site, leading to reduced interactions with CO2. A similar IL, 1-(3-aminopropyl)-3-methylimidazolium tetrafluoroborate, demonstrated relatively slow kinetics for CO2 uptake compared to aqueous amines, due to the high viscosity of amine-functionalised ILs. However, although the viscosity increases dramatically upon complexation with CO2,601,602 the absorption of CO2 was significantly higher than for the analogous physisorbing IL 1-butyl-3-methylimidazolium tetrafluoroborate. Further studies confirmed the 1
:2 carbamate formation motif,603,604 suggesting a potential upper limit for CO2 capacity in amine-functionalised ILs. Indeed, given the much higher molar mass of the IL cation and anion (essentially “dead weight” for this application), the CO2 uptake on a mole per unit mass basis is obviously poor for any IL functionalised in this manner.570 Much cheaper and simpler anions (sulfonate) and cation units (ammonium) can be used to mitigate this effect, though only to a small extent.605 A final limitation on this approach lies in the chemical similarity to traditional amines: tertiary amine-based ILs can only perform physisorption, while primary amines can engage in chemisorption. While not surprising, this does confirm the chemical limitations of amino-functionalised ILs are analogous to those of the corresponding amines.
In order to overcome the stoichiometric limitations associated with amine-functionalised ILs, Wang et al.606 synthesised a series of tunable alkanolamine-functionalsied ILs coordinated to alkali metal ions in a quasi-aza-crown ether. This afforded an extra degree of freedom depending on the nature of the inorganic ions utilised. These led to molar ratios slightly above 1:2, but with much improved kinetics, indicating the carbamate mechanism still prevailed. Later, Yang and He607 used PEG-functionalised ILs chelated to Li ions to achieve molar ratios well above 1:2 (nearly up to 1:1). However, the molar mass of these ILs is very high, suggesting less improvement on a mole per unit mass basis. There are also general concerns surrounding IL cost – in order to mitigate cost concerns, Vijayraghavan et al.608 created a series of less expensive protic ionic liquids based on diamines to achieve CO2 loadings of 13% w/v, thus using lower molar mass to improve performance at the same carbamate limit. This aspect of IL design (cost) has only recently gained attention for potential large-scale application of ILs.609
Amine-functionalised anions. Since the anion of the IL is the more electron rich centre, it is a natural source for CO2 interactions. This has commonly involved the use of (deprotonated) amino acids with tethered amine groups (such as lysine) as the anionic component of the IL. While amino acids are naturally abundant, there are several misconceptions about their nature. For example, they are relatively high in cost (owing to difficult synthesis or isolation), and fairly toxic.610 However, they are unquestionably biodegradable, and their use is not regulated. Their incorporation into ILs is fairly difficult, however, due to the need to use a hydroxide intermediate,611 which is normally obtained through ion exchange chromatography. Also, amino acid ILs are highly viscous, highly basic and (as with amino acids themselves) have low thermal stability. In order to overcome the relatively poor mass transport in these ILs, Zhang et al.612 synthesised silica-supported amino acid ILs with an extra amine group and created 1
:2 complexes with CO2. The viscosity of these ILs was later reduced by using small teraalkylammonium cations in place of Zhang's tetraalkylphosphonium centres,613,614 with similar CO2 capacities. While a range of amino acids can be used, the necessity of a free amine group to increase capacity is obvious. This necessitates the use of the deprotonated, highly basic free amine form of the amino acid, with negative consequences for potential thermal or chemical stability of the IL. As the most common mechanism of IL degradation is nucleophilic attack of the cation by the anion (followed by dealkylation of the cation),615 the basic (and nucleophilic) nature of these amino anions is detrimental. More stable cations, such as tetraalkylphosphonium salts, can alleviate this effect to some extent,576,616 creating ILs which compete with aqueous amines on a (molar) capacity basis. Glycine and sarcosine ILs with phosphonium cations617,618 and tetraalkylammonium methionine619 have each shown essentially equimolar CO2 uptake, though not all amino acid ILs showed this level. Additionally, according to Luo et al.,620 functionalised methylbenzolate-based ILs and nicotinate-based ILs with an amino group at the para or ortho position exhibit both higher capacity and lower enthalpy than related structures. It was later pointed out that the absorption was significantly affected by the nature of anions, due to different entropic driving forces for the reaction with CO2.585
While the ILs mentioned thus far relied on long chain (trihexyltetradecyl phosphonium or ammonium) cations, traditional amino acid based ILs pair a much smaller cation. This can have disadvantages for transport properties, but more importantly bring the molal (mol kg−1) absorption values up due to smaller molecular mass. Tetrabutylphosphonium cations paired with a variety of amino acid anions have shown equimolar CO2 uptake (often at several bar of pressure),621 though alkylation of the amino acid removes chemisorption possibilities.622 While transport and stability are issues with the shorter cation systems, supporting the IL on silica can help.623
Aprotic heterocyclic anions (AHAs). Since amine-functionalised ILs chemisorb CO2 similarly to liquid amines (by making the carbamate half of a liquid amine complex), the resultant solutions always suffer from extremely high viscosities post-capture, limiting their potential as usable solvent systems.579,582,585,612 There are many alternative structures that can bind CO2 without forming carbamates – and after all, the synthetic flexibility of ILs is what attracts researchers to the field. Therefore other anions have been employed to bind CO2 without forming high-viscosity carbamate disalt structures.
Azolate ILs (aprotic heterocyclic anion ILs)624,625 capture CO2 in equimolar quantities, and the anions are fairly straightforward to obtain through neutralisation of an azole superbase. Examples such as tetraalkylphosphonium pyrazolate can capture equimolar CO2 at atmospheric pressure, while tetrazolate, triazolate, and even pyrrolidonate anions have been shown to be moderately effective, and the use of imidazolate as an anion provided a nice symmetry to the field of ILs by incorporating the most common cation structure as an anion.580 These very basic anions even sometimes show a slight decrease in viscosity upon CO2 absorption,580,626 though viscosities still typically range close to 1000 cP. Refining the structure of these salts led to the development of the aprotic heterocyclic anion (AHA) based ILs or azolide ILs.584,594,598,627–633 These anions absorb nearly equimolar amounts of CO2 at much lower viscosities than azolate ILs. Molecular dynamics simulations have shown that the CO2 uptake is enhanced through chemical interaction with the anion.624 Both kinetics625 and thermodynamics584,632 for CO2 uptake by tetraalkylphosphonium AHAs have been investigated. It is clear that substituted imidazolate anions can be used to vary reactivity, enthalpy of binding and CO2 capacity. This can clearly be attributed to the relative electron density (and therefore reactivity) of the active site on the anion. By varying the alkyl chain lengths on the cation (from 38 total carbons down to 14), the viscosity could be reduced to under 100 cP without impacting molar uptake.634 This was later attributed to differences in reaction entropy and ionicity,635 though it is additionally important to note that the uptake in moles per unit mass would now be significantly higher, resulting in an intensified process.570
What is clear from the AHAs is that the anion basicity has a controlling influence on the CO2 uptake. This is similar to the amine-functionalised cation dominance, and is unsurprising – the more electron-rich the reactive site of the ion, the more CO2 will be chemisorbed. However, traditional ILs with very basic anions become both highly viscous and unstable, and the AHAs appear to at least be able to avoid the viscosity issues. While tuning anion basicity does enable greater control over CO2 uptake, the highly complex nature of these ion structures does not lend much promise to industrial application for cost reasons. Additionally, higher capacity through electron density increase is normally associated with higher enthalpies of binding and therefore more energy on regeneration. One proposed method to break this co-dependence is to use alkali metal salts in conjunction with IL–PEG mixtures.629,636 This enables the use of less basic azolate anions, though PEG is also a highly viscous solvent. However, equimolar absorption can be achieved with lower desorption energies.636 This can be exploited to ensure easier reversibility,637 as the balance between physisorption and chemisorption can be manipulated, changing the Gibbs free energies of the capture between somewhat negative and slightly positive.585
:1 CO2 complex with the cation, rather than the anion, and is a reversible adduct. However, neither the NHC intermediate, nor the ILs themselves are especially stable,581 and the presence of impurities such as water has been noted as likely to prevent this mechanism from occurring.2 The absorption was shown to proceed through an NHC–CO2 complex based on NMR results,640 and it was later demonstrated that the C2 could be deprotonated by very basic anions alone.641 Unfortunately, the theoretical mole fraction of maximum capture relative to IL is only 1:3 for this mechanism,585 thus leading to only moderate sorption, but these salts are more readily available for other applications,642 and the complex itself is reversible.643,644 Interestingly, tetraalkylphosphonium acetates645 also demonstrate high CO2 sorption, despite the lack of any possible NHC adduct. This can be attributed to CO2–anion interactions, though the mechanism of this remains unclear.646 As this effect is restricted to highly basic anions, and these have already been mentioned as having low stability, it is not clear how this limitation can be effectively prevented. Wang et al.647 attempted this by mixing imidazolium ILs with superbases, forming a dicationic–dianionic complex (zwitterionic imidazolium carboxylate) wherein the imidazolium cation was deprotonated by the superbase during CO2 sorption. Alkylation of the C2 position was confirmed to prevent this effect entirely, thus establishing the mechanism of equimolar capture, though the stability of the salts was not analysed.
An interesting combination of the imidazolium-anion proton transfer concept involves the use of imidazolium azolates.648,649 Here the azolate anion is used to effect deprotonation of the imidazolium cation, resulting in CO2 carboxylation. Seo et al.649 proposed that the carbene intermediate was responsible through two distinct pathways (anion–CO2 binding and NHC–CO2 complexation). However, the complex resulted in 1:2 complexation of CO2 and was only reversible if the resulting carbamate salt was broken. The ability to mediate CO2 chemisorption through transfer of CO2 from the anion to the cation is an interesting concept, though it is again unlikely that these carboxylated cationic complexes will form in the presence of water from flue gas; the presence of water vapour is likely to prevent the NHC intermediate from forming or from being re-protonated, resulting in diminished sorption capacity. While the AHAs can also be used to create ILs where CO2 can react directly with a protonated cation,650,651 this provides a system that has a very high regeneration energy.585
A prior (similar) example of this approach was the series of “switchable” ILs demonstrated by Jessop et al.652 where a superbase (1,8-diazabicyclo-[5.4.0]undec-7-ene or tetramethylguanidine) can deprotonate the alkylcarbbonic acid formed from an alcohol reacting with CO2, forming a transiently stable salt. These CO2-binding organic liquids (CO2BOLs) have near equimolar capacity652–655 though regeneration is not trivial. Similar effects have been demonstrated for amino acid (OH) deprotonation by Wang et al.656 where a hydroxyl-functionalised IL cation and superbase combine to react with CO2. While equimolar capture is again possible, release is difficult under CO2 atmospheres, and heating can cause decomposition or volatilisation of the components. Some of the normal limitations, such as transport issues, can be obviated by using the alcohol as a diluent and reactant, as with β-amino acid anion-based ILs diluted in simple alcohols.657 This is a similar approach to MEA in water, and the methylcarbonate salt (formed in methanol) is key to the equimolar capture process (relative to the IL). A variety of cationic species were demonstrated as effective, including some relatively simple cations such as tetramethylammonium – here the small molecular weight of the cation could be advantageous for process intensification. However, there is a generic issue with superbase-IL CO2 capture – the need for releasing the CO2 under CO2-free atmospheres limits application.
An alternative approach is to directly capture the CO2-based protons through protonation of a sufficiently active amine base.658 In the presence of water, this becomes similar to aqueous amine capture processes, where a carbamate or carbonate-based equilibrium protonation will dominate, as the carbamate salt is hydrolysed into a bicarbonate salt, even with tertiary amines.659 This yields somewhat higher capacities for tertiary amines (1:1) vs. primary or secondary amines (1:2 to 1:1) though the absorption is very slow.1
Other alcohol-based cations include phenolate anions, which Wang reviewed recently.585 These can be prepared from the corresponding hydroxide salt by neutralisation with any substituted phenol.662 This is a general limitation of strongly basic ILs – the weakly acidic conjugate species must be deprotonated by a strong base such as hydroxide, and hydroxide salts are difficult to prepare at scale. However, this did demonstrate the utility of a series of substituted phenolate ILs for CO2 uptake and the surprising variation in CO2 sorption that could be achieved by varying the electron density in the anion, as was discussed above for azolate salts. The recurring theme of tuning CO2 capture through anion electronegativity or reactivity is a core part of the appeal of ILs for use in this field, though the synthetic complexity required to achieve this may render scale-up of these salts difficult.
Since anion functionisation is easier (and the anions come with built-in CO2-philicity due to the negative charge), multifunctional anions have been proposed where multiple capture sites are designed to be co-operative. This is analogous to the stabilising effects present in alkanolamines, though the methods differ. For example, high CO2 capacity and reversibility can be achieved by using a second interacting site on the AHA anions (phenolate, imidazolate) to stabilise the CO2 adduct.665,666 This includes hydroxypyridine (capacities up to 1.65 equivalents) where simultaneous carbamate/carbonate formation introduces added capacity, as demonstrated spectroscopically.666 This multi-functional cooperation could provide a blueprint for future anion design.
The use of IL solutions, where amines or water reduce the solution viscosity, are viable alternatives, though the loss of advantages in regeneration energy are noteworthy. The use of other blending agents (such as low molecular weight polymers) has shown less promise.624,685,686 It is clear that water remains the preferred dilution medium for reducing IL viscosity687 in CCS applications585 as the viscosity of both the pre- and post-capture solutions decreases with increasing water content,585,618,670 with minimal impact on absorption capacity,584 likely due to complex stabilisation through increased hydrogen bonding.624 An alternative to this approach is to directly tether an amine (such as MEA) onto the IL, with an (unsurprising) improved performance when tethering to the anion rather than the cation.665 Possibly the most forward-looking approach recently proposed is to attempt to tune the basicity (and therefore CO2 interaction strength) of functionalised ILs.585 This takes advantage of the inherent synthetic flexibility of ILs, one of their greatest strengths. The interaction strength can be tuned by weakening the cation–anion ion pairing (as has been done in chemical systems),534 and this has also been employed in gas sweetening.688,689 This idea shows great potential if cost considerations can be balanced successfully.
6 SAFT-based approaches for solvent design for CO2 capture; from molecules to processes
6.1 Chemical approaches to modelling the thermodynamics of aqueous amine solutions
The capability of the amine solvent to absorb CO2 is quantified by assessing both the specific chemistry of the complexation and the overall phase equilibria. The overall stoichiometry of the various reactions involved in CO2 absorption in aqueous MEA (forms a monoethanolammonium carbamate pair) can therefore be summarised as:| CO2 + 2(HO–CH2CH2–NH2) ⇌ [HO–CH2CH2–NH–CO2− + NH3+–CH2CH2–OH] | (13) |
:1 amine to CO2 stoichiometry forms a monoethanolammonium bicarbonate pair (shown between square brackets) and is characterised by the following:| CO2 + HO–CH2CH2–NH2 + H2O ⇌ [HCO3− + NH3+–CH2CH2–OH] | (14) |
Traditionally, the thermodynamics and phase equilibrium of reactive systems of this type are described with a so-called “chemical” approach first introduced by Dolezalek690 in the early twentieth century. In this case one has to identify the number of species that are present after assuming particular equilibrium schemes and then specify the state dependence of each of the equilibrium constants; the interactions between the various species are often treated at the level of an ideal mixture to simplify the description. As alluded to by van Laar691,692 the arbitrary manner in which the various species are assigned a priori and the number of thermodynamic variables required to describe the chemical and phase equilibria imply that chemical theories have limited predictive value. For example, in order to fully describe the thermodynamics and fluid-phase behaviour of the reactive mixture of CO2 in aqueous ammonia one would require 72 temperature-dependent interaction parameters;693 it is clearly challenging to estimate the necessary parameters reliably without extensive experimental data for the specific system under consideration, limiting the predictive capability of the method. This having been said, very successful methodologies have been developed to couple a description of the chemical equilibria with an activity coefficient model for the solution phase and an equation of state for the gas phase to incorporate “physical” effects arising from the non-ideality of the intermolecular interactions.694–696
In the particular case of the reactive equilibria of CO2 in aqueous amine solvents, the system is often treated at the level of a weak electrolyte solution, essentially involving the empirical determination of the chemical equilibrium constants for the various reactions (or equivalently the activity coefficients of all of the species). One of the most popular approaches employed in process modelling of carbon capture in alkanolamine solvents was presented by Austgen et al.,697,698 who combined the electrolyte non-random two liquid (eNRTL),699,700 description of the activity coefficients for the equilibrium ionic species in solution with the Soave–Redlich–Kwong (SRK)701 cubic equation of state for the fugacities of the species in the vapour phase; the vapour–liquid is not obtained by solving for phase equilibria but is described instead by using Henry's law constants. It is worth noting that the original eNRTL approach has been shown to lead to an inconsistent description of mixtures of mixed ions,702 and improved versions have now been developed and used703–705 to describe the complex speciation of CO2 in alkanolamine solvents. Another excellent example of this type of approach is the work of Faramarzi et al.706 who instead couple the extended universal quasi-chemical (UNIQUAC) approach for electrolyte solutions707 with the SRK equation of state for the vapour phase; a good description of the fluid-phase behaviour is obtained for the ternary mixture of CO2 in aqueous monoethanolamine (MEA) and other alkanolamines using a consistent determination of the vapour–liquid coexistence by imposing the conditions of phase equilibrium. Within chemical approaches, the first attempt to use a single equation of state to describe the properties of the vapour and the liquid phase is due to Kuranov et al.,708 who proposed a new method to treat the vapour–liquid equilibria of alkanolamine solutions with CO2 and H2S based on a hole quasichemical hole model (partially filled lattice model to deal with compressible fluid phases) modified to incorporate chemical reactions and electrostatic interactions in the liquid phase.
Methodologies which are based on a unified statistical mechanical treatment of both the liquid and vapour phases offer more promise as a predictive platform for the prediction of molecular thermodynamics and fluid-phase equilibria of complex fluid systems. The reversible reactions involved in carbon-capture in amine solvents can be represented as strong association equilibrium processes, where the properties of the mixture are attributed to large differences in the associative intermolecular interactions between the species which lead to aggregation. This type of “physical” perspective originally promoted by van Laar691,692 and his followers may at first seem diametrically opposed to the “chemical” view of the reacting system, leading to “harsh polemic” between the opposing camps from the very beginning.709 The two standpoints are but extreme representations of the real system and an unambiguous distinction between the role played by the chemical and physical interactions is often arbitrary and in many cases only a matter of taste or convenience.710
In the case of the readily reversible reactions involved in acid–gas scrubbing, it is reasonable to assume that the interactions characterising the associated species are not too dissimilar from those of the parent compounds and that a physical treatment is appropriate; when the products are significantly different from the reactants, a chemical perspective becomes essential. The statistical mechanics description of lattice models within the quasi-chemical approximation of Guggenheim711 is well suited to represent the properties of mixtures of strongly associating molecules (as exemplified by the early work of Barker and Fock712), gaining much popularity in engineering applications following the pioneering work of Abrams and Prausnitz,713 most notably with versatile group-contribution methods such as UNIFAC (universal quasi-chemical functional group activity coefficients)714 and its variants.715,716 As mentioned earlier, the related UNIQUAC approach has been used successfully to describe the thermodynamic properties and phase equilibria of CO2 absorption in amine solvents.706 It is, however, more appropriate to use equations of state which are firmly grounded in the fluid state rather than approaches based on lattice models because the latter are generally incompressible and as a consequence require a separate treatment of the vapour and liquid phases.
The statistical associating fluid theory (SAFT)717,718 is cast from the physical perspective of the interactions between the particles characterised by strong intermolecular forces responsible for hydrogen bonding or complexation. The description of the reactive phase equilibria of CO2 in aqueous alkanolamines with equations of state of the SAFT family is the central theme of our current review.
6.2 SAFT physical approach of chemisorption of CO2 in aqueous amine solvents
The SAFT equation of state is based on the Wertheim first-order perturbation theory (TPT1),719–724 which provides a compact platform to describe the thermodynamic properties of mixtures of associating species. The association interactions are mediated by off-centre bonding sites placed on the molecules, the number and nature of which control rich equilibrium association schemes including the formation of complex chain-like and network aggregates as well as simple dimerisation equilibria. The multiple associative equilibria are treated implicitly within the theory without having to specify the detailed equilibrium reactions, in contrast to a chemical treatment. The equivalence of a SAFT description of the associative equilibria and a chemical or quasi-chemical description has been demonstrated by Economou and Donohue,725 though care has to be taken with the stoichiometry of the reactions in the detailed comparison. The SAFT equation of state is finding ever increasing use in the accurate description of the thermodynamic properties of fluid mixtures for industrial applications. Examples of the more-popular versions of SAFT in current use include SAFT-VR for variable-range potentials,726–729 soft-SAFT,730,731 based on the Lennard-Jones potential, and the perturbed-chain PC-SAFT;732 the cubic plus association (CPA)733 equation of state, which couples the Wertheim TPT1 treatment of associating fluids with the SRK equation of state, is also worth a particular mention.An important advantage of employing a physically-based SAFT treatment is the significant reduction in number of parameters required to describe the associating or reacting system. Group-contribution approaches based on the chemical functionality of the molecules in the mixture (the interactions for which are estimated from the thermodynamic properties of systems comprising simpler target components) offer additional predictive capabilities; the SAFT-γ approach734–737 represents a recent reformulation of SAFT-VR equation of state within a group-contribution framework.
In order to exemplify the application of the physical approach inherent in the SAFT (and Wertheim) treatment, the molecular models employed to represent the reactions associated with the chemisorption of CO2 in aqueous monoethanolamine (MEA) are depicted in Fig. 13. Four association sites (two H to represent the hydrogens, and two e for the oxygen lone pair of electrons) are employed to mediate the hydrogen-bonding interactions between the electron lone pairs on the oxygen atom with the hydrogen atoms on different water molecules.738 Six association sites (one e and two H on the amine NH2 group, and two e and one H on the hydroxyl OH group) mediate the multiple hydrogen-bonding interactions between the MEA molecules.739,740
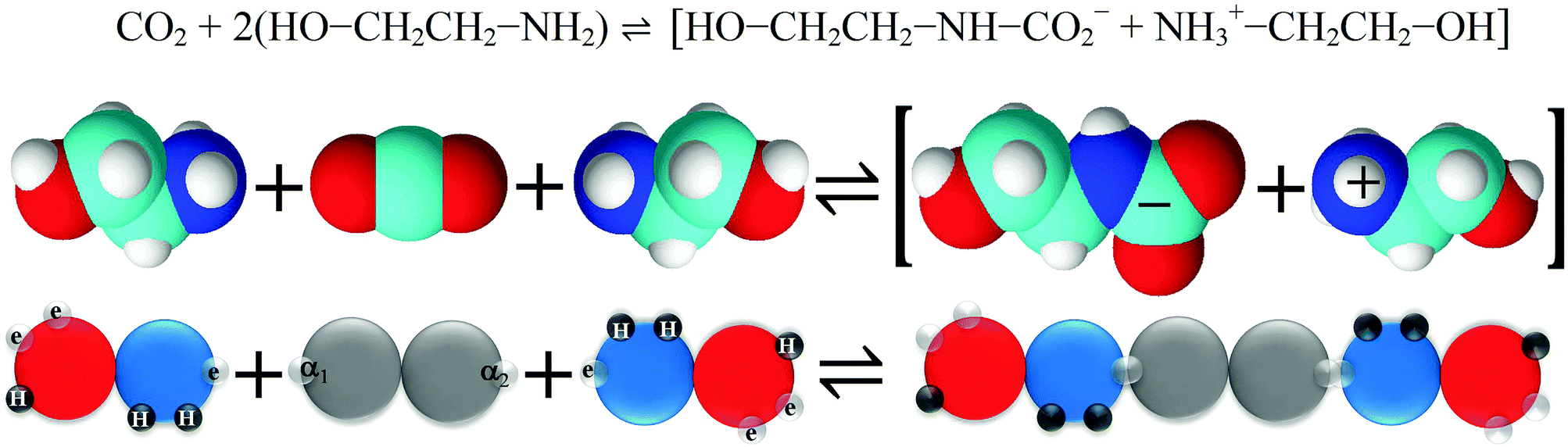 | ||
| Fig. 13 SAFT physical model of CO2 chemisorption in aqueous MEA solution: the amine groups are depicted in dark blue, the hydroxyl groups in red, and CO2 in grey; the sites labelled “e” are the hydrogen-bond acceptor electron-lone pairs of the electronegative atoms, the sites labelled “H” are the donor hydrogen sites, and the sites labelled “a” are the sites mediating the reactions with CO2. | ||
Details of the development of the SAFT models used to represent the asymmetric interactions between the MEA and CO2 (and H2O) molecules are provided in ref. 739 and 740. Just two physical “reaction” sites α1 and α2 on the CO2 molecules are employed to describe the formation of the monoethanolammonium carbamate complex (reaction (13)) by association with the hydrogen sites on the NH2 of MEA allowing for a 2:1 stoichiometry between MEA and CO2; association to the single α1 site can be used to quantify the formation of the monoethanolammonium bicarbonate pair (reaction (14)). A testament of the adequacy of this type of SAFT description of the complex chemical reactions between CO2 and MEA in aqueous solution can be seen in Fig. 14, where the experimental data,741,742 for the separate concentrations of the carbamate and bicarbonate products as a function of CO2 loading are compared with the theoretical predictions743 (note that in this example the calculations are carried out within the SAFT-γ SW group-contribution formalism).
 | ||
| Fig. 14 The carbamate and bicarbonate mole fractions as a function of the loading of CO2 (defined as the number of moles of CO2 absorbed in solution per mole of amine) in a 30 wt% MEA aqueous solution predicted with the SAFT-γ SW approach.743 The symbols correspond to the experimental data741,742 at 313.15 K (red circles) and 333.15 K (blue squares). The curves correspond to the SAFT-γ SW predictions: continuous curves for 313.15 K and dot-dashed curves for 333.15 K. | ||
The SAFT physical treatment has been used extensively to describe the thermodynamic properties and fluid-phase equilibria of the reactive systems associated with CO2 chemisorption in amine-based solvents including MEA,739,740,744,745 ammonia,746 2-amino-2-methyl-1-propanol (AMP),740,746 linear alkylamines,747 diethanolamine (DEA),740 and methyldiethanolamine (MDEA).740 We should also point out that that the eNRTL description of the activity coefficients of the ionic species in solution has been coupled with the PC-SAFT732 equation of state for the fugacities of the vapour phase to model the chemisorption of CO2 in aqueous MEA704 and MDEA.705 A group-contribution version of the theory (SAFT-γ SW)734,735 has now been deployed to assess the suitability of a broad family of multifunctional alkanolamine solvents including representative examples such as MEA, AMP, DEA, MDEA, methylmethanolamine (MMEA), ethylmethanolamine (EMEA), 3-amino-1-propanol (monopropanolamine, MPA), 5-amino-1-pentanol, and 6-amino-1-hexanol.743,748 An example of the quality of the SAFT-γ description of the degree of absorption of CO2 in aqueous MEA as a function of pressure and temperature can be seen in Fig. 15 in comparison with experimental data741,742,749,750 and with the correlation of Gabrielsen et al.751 It may now be apparent that a key advantage of the SAFT-γ group-contribution methodology provides a high degree of predictive capability allowing one to describe the thermodynamic properties of a large number of compounds and mixtures with a relatively small number of parameters (see, for example, Fig. 14 of ref. 743).
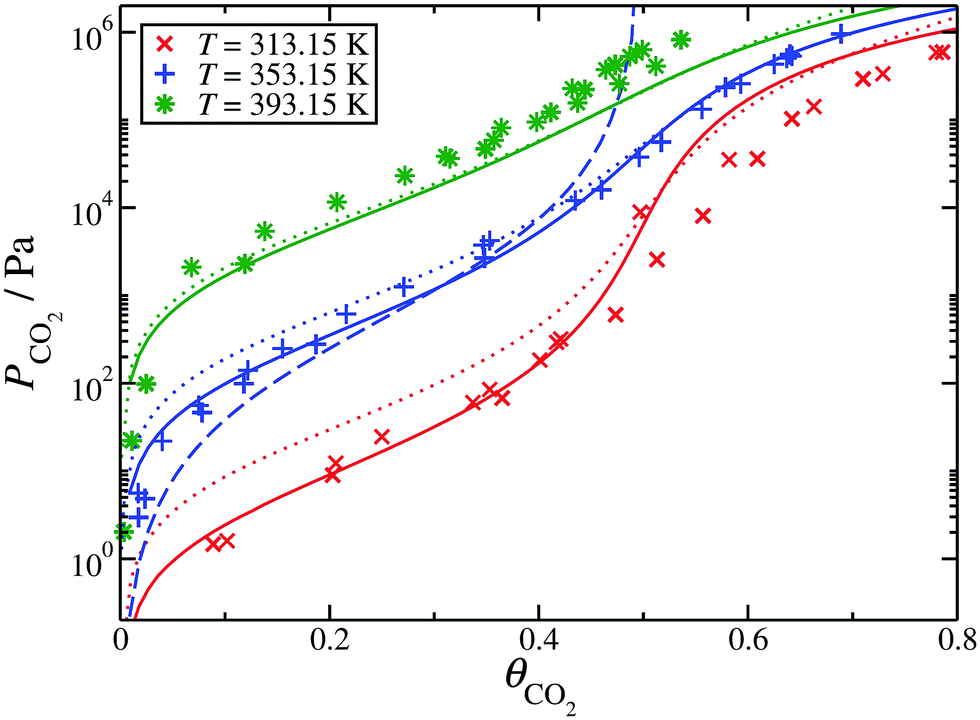 | ||
| Fig. 15 The solubility of CO2 (expressed as the number of moles of CO2 absorbed in solution per mole of amine) as a function of the partial pressure for a 30 wt% MEA aqueous solution predicted with the SAFT-γ approach.743 The symbols correspond to the experimental data741,742,749,750 at 313.15 K (red circles) and 333.15 K (blue squares), and 393.15 K (green asterisks). The continuous curves correspond to the SAFT-γ SW group-contribution calculations,743 the dotted curves to SAFT-VR SW calculations,740 and the dashed curve corresponds to the correlation of Gabrielsen et al.751 fitted to the data at 353.15 K. | ||
6.3 Explicit SAFT treatment of the electrolyte species
In the models described in the previous section, a SAFT treatment of the reactions associated with CO2 capture in amine solvents does not explicitly take into account the speciation products nor the electrolytic nature of the system. Though it is apparent that a good representation of the thermodynamics and fluid-phase equilibria can be obtained for the chemisorption of CO2 in a variety of amine solvents using SAFT without an explicit treatment of the polar and electrostatic interactions, a physical approach is expected to be increasingly inadequate when the reaction products are highly charged and chemically distinct from the parent reactants.As was mentioned in the introductory section, the eNRTL approach is often employed to represent the thermodynamic properties (activity) of the species formed in solutions of CO2 in aqueous amines697,698,703,704 and a different model (an equation of state) is used to treat the gas phase. Kuranov et al.708 treated these systems by extending the quasichemical hole model with electrostatics following the Pitzer752 modification of the Debye–Huckel approximation in a framework where, for the first time, the same equation of state was used for the liquid and gas phases. They obtain good correlative results in comparison to experiments with the use of a number of adjustable parameters. It is clearly advantageous to treat the liquid and gas phases within the same thermodynamic framework and Fürst and co-workers753,754 followed in this vein, using a different equation of state for electrolytes755 (which extends the capability of the SRK equation using the mean-spherical approximation, MSA,756 for the contribution to the free energy of the charged species) to represent the fluid-phase equilibria of in aqueous alkanolamine solutions. Unfortunately, the use of the SRK equation of state as the reference for the uncharged aqueous systems comes at the cost of a poor description of the properties of water which are dominated by the strong hydrogen-bonding association interactions. A natural step is the use of extensions of the SAFT approach to electrolytes to describe the chemical and phase equilibria of CO2 in aqueous alkanolamines.
The SAFT framework was first extended to treat electrolyte solutions by incorporation of free energy contributions to take into account the presence of charged species assuming fully dissociated charged species. A Coulombic term, usually written following the expressions of the primitive models of Debye and Hückel757 (DH) or the mean spherical approximation (MSA),756 is typically added to the classic SAFT Helmholtz free energy expression. The original SAFT equation was first combined with terms taking into account ion–dipole interactions, treated following a non-primitive perturbation approach758 as well as ion–ion interactions with the MSA of Blum and Høeye759 by Liu et al.760 In the same year, the SAFT-VR equation combined with an MSA term for electrolytes was presented761 and has been used in a number of works to study and model762 real systems incorporating different potentials.763–766 The free energy expression of the PC-SAFT equation has been combined with the DH theory and used to study solutions with charged species in several studies767–769 and models that incorporate explicitly ion–dipole interactions at different levels of approximation within SAFT frameworks have also been presented.770–772 In terms of a preference for the use of DH or MSA terms in combination with fluid equations of state to treat real electrolyte solutions, it has been shown that both approaches lead to a similar representation of the electrostatic effect when combined with an equation of state treatment.761,773
In all of the aforementioned SAFT models, one considers relatively simple so-called strong electrolytes and assume fully dissociated charged species as corresponds to the models underlying the DH and MSA models. In comparison to experimental data, they are found to be accurate in representing and predicting the phase equilibrium properties of the solutions, including the description of the vapour pressure, activity coefficients of the ionic species and osmotic coefficients and densities of the solutions. It is only relatively recently that approaches treating the chemical equilibria between the neutral and charged species have also been presented in SAFT frameworks, mostly with the ePC-SAFT equation,767 see for example ref. 769, 774 and 775. In particular, although there have been works modelling aqueous ethanol–amine solutions taking into account the phase and reaction equilibria,776,777 they treat the liquid phase as an ideal solution with respect to the activity coefficients of the charged species (i.e., making them equal to 1). Only recently, Uyan et al.778 have presented a model in which the activity coefficients of the ionic species are calculated, although in their approach the dielectric constant of the solution is not treated as dependent on the concentration of CO2.
In future, it will be of interest to continue developing approaches that combine the physical framework of Wertheim from which SAFT approaches stem and the speciation frameworks to deliver models better suited to study the reaction kinetics of amine solvent solutions with CO2 with increasing accuracy and predictive capability as these attributes are key for their use in process and solvent design and optimisation.
6.4 The role of thermodynamic models in the design of novel solvents for CO2 capture
Most thermodynamic models of CO2 absorption in aqueous amine solutions have been developed with the aim to enable the design and operation of chemical absorption and desorption processes, whether in the context of gas sweetening or in that of flue gas treatment. Detailed models based on chemical theory, such as those briefly discussed in Section 6.1 (e.g., Zhang et al.704), are very well-suited for this purpose: they take into account every chemical species in the postulated reaction mechanism and therefore allow the calculation of quantities that can have an impact on process performance when operating away from chemical and phase equilibrium. This is the case for instance of the ionic strength, which affects mass transfer, and of the concentrations of all species, which have an effect on reaction kinetics as well as mass transfer. Indeed, such detailed models have been used extensively and quite successfully in modelling pilot plant data without the need for fitting any model parameters to plant data;779,780 they have an important role to play in developing better processes.With the imperative to develop processes that can capture vast quantities of CO2 from post-combustion flue gas at low cost and with minimal environmental impact, however, there has been an increasing focus on effecting step-change improvements in the performance of absorption processes,781 through novel equipment (rotating packed beds)782 but also through novel solvents.783 In the latter case, extensive research has taken place to gain a better understanding of solvent mixtures or additives that have been found to provide some advantage, such as reduced degradation or increased reaction kinetics, as afforded for instance by the combination of methyldiethanolamine (MDEA) and PZ.784
The search for novel solvents has mainly been based on extensive experimental programs. For instance, within the CESAR project funded under the FP7 programme of the European Commission,785 experiments from laboratory to pilot plant scale were undertaken for monoethanolamine and several alternative mixtures. This painstaking work allowed the identification of PZ/AMP as a promising candidate. As discussed in Papadopoulos et al.,783 the thorough experimental investigation of a single solvent requires the deployment of significant resources over a number of years, hindering the search for improved amine-based carbon capture processes.
To support the search for novel solvents, there is thus a pressing need for predictive thermodynamic models of CO2 absorption that can be applied to solvents that have not been used for CO2 capture previously, or even to molecules that have never been synthesised. The prediction of the thermodynamic behaviour of novel solvents is an important enabling capability in overcoming the challenges to the implementation of solvent-based post-combustion capture. Even where new process topologies are sought, the benefits that can potentially be derived from flowsheet innovation are intimately connected to the nature of the solvent. This is the case for instance with phase-change solvents, which have been gaining increasing attention as a means to reduce the energetic cost of solvent regeneration, as discussed in Section 4.1.3. The performance of a process based on such a solvent depends greatly on the thermodynamic phase behaviour of solvent + CO2 mixtures as a function of composition, temperature and pressure. As seen in Section 6.2, the development of such models is a challenging task, but given that they are to be used to guide the search for novel solvents at an early stage, they need not be subject to the same stringent conditions on accuracy and fidelity as those models that are used for detailed process design.
In order to accelerate further the search for novel solvents and process configurations, systematic approaches to identifying candidate solvents have been proposed. These include rule-based approaches786,787 that can be used to identify promising classes of solvents. To enable a search among a wider set of compounds, computer-aided molecular design (CAMD) approaches have recently been proposed, based on the prediction of a range of properties.783,788–792 In all but two cases,783,791 the extent of CO2 absorption in the designed solvent and, by extension, the energetic requirements of desorption, were either not considered explicitly as a design target or were predicted on the basis of detailed thermodynamic models that were parameterised for other solvents, and thus likely to be of limited accuracy for novel solvents. The usefulness of CAMD approaches as a tool to identify the most promising solvents for experimentation is limited when such key solvent performance metrics are neglected during the generation of candidate molecules and mixtures.
The emergence of the SAFT-γ SW group-contribution models of CO2 absorption mixtures described in Section 6.2 has the potential to make the in silico identification of better solvents for CO2 chemisorption much more effective. To date, such models have been deployed as a final computational step in the solvent design methodology of Papadopoulos et al.783 A ranked list of solvents is first generated by considering a wide range of pure component properties, as well as reactivity and sustainability. The extent of CO2 absorption in the resulting solvents is then predicted with the SAFT-γ SW equation of state in order to refine the priority list of solvents to be investigated further. The explicit use of predicted CO2 absorption as a means of selecting solvents paves the way for a much more effective computational exploration of the molecular design space and more realistic design criterion.
Looking ahead, one can envisage the development of much more holistic approaches to the identification of promising solvents and solvent mixtures among the extremely large space of possibilities. Approaches for computer-aided molecular and process design (CAMPD),793,794 where molecular structure and process design are optimised simultaneously, have already been deployed successfully in the context of physical absorption processes for CO2 capture using the perturbed-chain polar-statistical-associating-fluid theory (PCP-SAFT) equation,795–798 the SAFT-VR SW equation799,800 and the SAFT-γ Mie equation.801,802
The various SAFT equations of state that have been used in these CAMPD approaches provide a reliable description of the phase equilibria of the mixtures involved and, in the case of the SAFT-γ Mie equation,737 of the caloric properties. Chemical absorption processes are, however, significantly more challenging than the physical absorption processes investigated so far with CAMPD due to the presence of chemical reactions that had until recently prevented the development of predictive models linking molecular structure to phase and chemical equilibria. Even with the advent of predictive SAFT-γ SW models, one must note that the use of a physical representation of the chemical reactions (see the scheme in Fig. 13) implies that the product concentrations are not explicit in the model calculations, that the ionic nature of the products is not explicitly taken into account and that the reactions are necessarily at equilibrium. In contrast, in many chemisorption processes, chemical equilibrium may not be reached in the liquid phase and, even if it is reached, the concentrations of product species and their charge impact on mass transfer rate and hence on overall process performance.
Nevertheless, it has been shown that it is possible to use such an implicit representation of product species within a process model of a CO2 absorption and to achieve good agreement with pilot plant data using monoethanolamine.745,746,803–807 These encouraging results suggest that SAFT-based physical models of aqueous amine and CO2 mixtures can be integrated within an iterative discovery and design process, as illustrated in Fig. 16. Design targets ranging from physical properties to economic performance and sustainability, as well as a design space consisting of atom groups (e.g., –NH2, >NH, –OH) are defined in step 1. One can then use SAFT-γ physical models of phase and chemical equilibria, in combination with other models, to link molecular structure and solvent composition to the design targets and apply CAMD or CAMPD. The outcome of this step (step 2 in Fig. 16) is a prioritised list of solvent candidates. Top candidates are then experimentally tested iteratively in step 3 in terms of their key physicochemical properties. For those that are confirmed to be most promising, additional data are gathered to build detailed thermodynamic and kinetic models that include speciation explicitly (step 4). Such models are detailed enough to help plan pilot plant studies and proceed with process design (step 5). If issues are identified at that stage, one can return to step 3 to test additional candidates. Otherwise, one can proceed with implementation (step 6). In this overall scheme to develop CO2 capture processes, the physical models of Fig. 13 and the chemical models are used in tandem to evolve from early stage molecular design to detailed process design.
 | ||
| Fig. 16 An overview of an approach to the design of solvents and processes for CO2 capture, highlighting the role of different modelling and experimental activities. | ||
7 CO2 transportation
Whilst at small scale other options are available, the significant volumes of CO2 requiring transport as a result of large scale carbon capture means that only two methods are practical, networks of pressurised pipelines and ship transport. The efficacy of either of these two depends to a great extent on the quantity of CO2 and the distance from its point of storage or utilisation; except over large distances (>1500 km)809,810 where it's expected that ship transport would be preferable, it is generally expected that the vast majority of transportation will occur via pipeline.2In both cases consideration must begin with the compression and/or liquefaction of the fluid, effectively the interface between capture and transport.811,812 During this process stage, the stream is transformed into a supercritical or dense-phase, i.e. above the critical pressure but below the critical temperature, to take advantage of the greater density in these phases.813 Of these, for CO2 transported by pipeline, dense-phase is likely to be preferred as keeping the temperature above critical temperature can in practice be problematic due to cooling through heat transfer effects.814,815 The actual amount of compression is chosen in concert with the expected flow rate and the pressure drop along the pipeline, which itself is dependent on the hydrodynamic and thermophysical properties of the fluid fed into the pipe. These properties are a function of the stream's composition, which therefore plays an important role in the analysis and design of transportation systems. Hence, for the cost-optimal design of a pipeline system, and to a lesser extent shipping, an understanding of the interaction between all of these factors is essential.
7.1 Composition of the CO2 stream
The particular components in the mixture as well as the amount of impurities remaining in the stream can significantly affect the thermophysical properties and phase equilibria of the fluid.816–818 The recent study of Porter et al.819 sought to define ranges of impurities expected from a number of carbon capture technologies. For the purposes of pipeline transport, particular impurities of concern include: water, which is important with regards to potential corrosion of the pipe steels, and non-condensable gases (i.e. N2, O2 or Ar) which can significantly alter the mixture's vapour–liquid equilibrium.Of practical importance for both shipping and pipeline transport is the impact that the composition has on the phase envelope. As Fig. 17 shows, as the amount of non-condensable gases in the mixture increases an appreciable expansion of the phase envelope is observed. This has both a considerable impact on the energy requirements for compression or liquefaction of the fluid as well as, for pipeline transport, the higher cricondenbar‡‡‡‡‡ necessitating higher pipeline operating pressures. While this is sufficient for steady state operation, the additional uncertainty in the behaviour of the fluid during transient operations has led to a great deal of focus on assessing the impact of the variations in the composition of the CO2 mixture on the dynamic flow behaviour.818,820
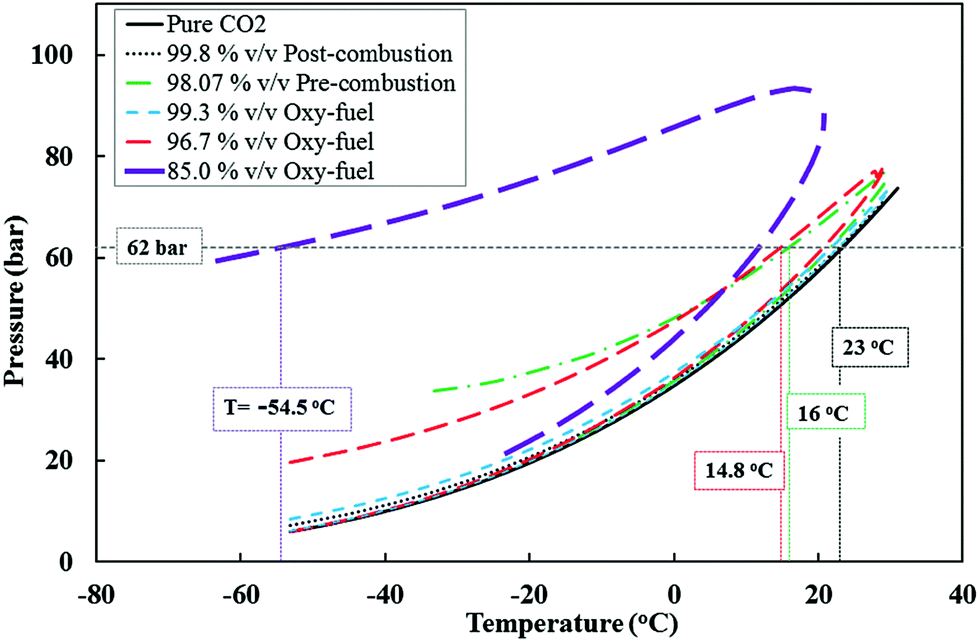 | ||
| Fig. 17 Boundaries of VLE region in pressure–temperature phase diagram for pure CO2, pre-combustion, post-combustion and oxy-fuel streams (85 and 96.7% v/v CO2) calculated using PR EoS.808 | ||
7.2 Compression
As the first stage in the transportation, the compression or liquefaction stage represents a significant use of energy, suggested to be as high as 12%821,822 of the loss of efficiency of a power plant; as such, the selection of the most efficient compression strategy823,824 is of significant importance to the overall performance of the CCS system. A number of studies have therefore sought to optimise this process, for example Witkowski et al.825 investigated 13 compression strategies ranging from various multistage compression with intercooling, compression coupled with liquefaction and pumping as well as more novel technologies such as supersonic shockwave compression. It was found, for example, that using integrally geared compressors could result in energy savings of more than 20% compared to conventional strategies. Other work has focussed on improving the efficiency by reusing the heat recovered as part of the intercooling system.822,826Recent work has sought to quantify the impact of composition on the energy and process requirements for the compression. The analysis of Martynov et al.808 found, using the compositions from various processes suggested by Porter et al.,819 that little difference is observed when dealing with relatively clean CO2 streams (>95% v/v purity), but large penalties (increases in power requirements of between 12–30%) are incurred for less concentrated streams. Similar findings were reported by Skaugen et al.815 where conditioning costs as a whole for an impure stream were increased by 13% or 2.3 € per tCO2. Given the substantial energy demand represented by the compression stage of transportation further work is required to continue to find efficiencies; additionally, given the likelihood of the dynamic operation of CCS plants the impact of this on the design of the compression train should be evaluated.
7.3 Pipeline transportation
Whilst CO2 pipelines must be designed and constructed as to ensure that they are reliable and safe to operate they must also be designed in as cost efficient fashion as possible. For the purposes of normal operation, the design requirements for a pipeline are primarily a function of the flowrates and of the hydrodynamic properties of the CO2 stream which it must transport,2 for example the density, compressibility and viscosity. One important constraint is that the phase transition should be avoided as, should it occur, it can result in operational problems such as liquid slugs. To avoid this, feed pressures and temperatures must be chosen so that the fluid remains in the single-phase region along the length of the pipe under normal operating conditions.Given that the stream fed into the network is also unlikely to be pure CO2, the composition of the CO2 mixture will impact the design by altering not only the fluids thermophysical properties but the vapour/liquid equilibrium, potentially altering the permissible operating envelope. Furthermore, the level and type of impurities can influence the material requirements of the steel used for the pipeline's construction; this includes both the steel strength needed to prevent fracture and the possibility of corrosion.
Network designs have generally assumed the use of a trunk pipeline to which various emitters can link.833–835 This has the benefit of providing a shared infrastructure that future sources can join as CCS is rolled out. It has been suggested that the trunk pipeline itself should be oversized to account for the growth of CCS, and therefore volume of CO2 requiring transportation.836
The ability of the networks designed to deal with intermittency of feed flowrate, such as predicted by Mac Dowell and Staffell,837 has received relatively little attention. Of the few available studies Liljemark et al.820 investigated the fluid transients during various load changes and found that for some cases phase transfer may occur, which, as in the case of steady state flow, is considered to be operationally problematic. Chaczykowski and Osiadacz838 showed that the composition of the CO2 stream may have a considerable effect on the dynamic behaviour of the fluid under such scenarios. However, these studies consider only single point to point pipelines, more work is required to understand these dynamics in networks containing multiple sources and evaluate operational risks. Finally, Mechleri et al.839 show that considering the evolution of the electricity system in the design of CO2 transport infrastructure can lead to non-negligible cost reductions without compromising safety or operability.
Furthermore, there are >6500 km of CO2 pipelines worldwide, most of which are associated with EOR operation in the United States.23 Whilst experience of CO2 pipelines exists,2,816 these tend to be away from densely populated regions. The deployment of CO2 pipelines closer to population centres has led to a body of work seeking to develop high fidelity tools in order to perform detailed quantitative risk assessments.840–845 Such analysis has the additional benefit of reducing unnecessary conservativeness in design by removing unwarranted safety concerns.
Fracture can occur in two modes: ductile and brittle,846 the majority of work has focused on the risk posed by the first of these. The process of ductile fracture involves significant deformation of the pipeline material and is driven primarily by the residual pressure of the fluid within the pipe, which is known to be a strong function of the mixture composition,847 exceeding the toughness of the steel.848 These are normally assessed using semi-empirical methods such as the Battelle Two Curve method,849 and an adequate toughness selected. However, the application of such a method has been shown to be insufficient for the purposes of obtaining conservative designs for linepipe by full scale fracture experiments performed in the UK.850 In order to avoid the burden of the costs associated with over-specification of the pipes structure, a number of works have sought to directly simulate the crack propagation/fluid decompression itself by coupling both fluid and structural models.851–853 While such an approach is computationally demanding, it offers the most convenient route of analysis given the lack of a simpler assessment method and the expense of full scale testing.
7.4 Transportation by ship
As compared to transportation by pipeline the use of shipping has received far less attention in the literature, summaries can be found in the recent reviews by Brownsort24 and Munkejord et al.818 While as described previously, shipping is considered to be the lower cost option over very large distance, this is also the case for small distributed sources,832,854,855 for example in a recent study, Kjärstad et al.856 have shown that for small sources around the Baltic, ships may provide the cost optimal solution. Discussion of relative costs is typically performed in terms of the point at which shipping becomes cheaper than pipeline for a given capacity, in both offshore and onshore storage scenarios.24,832,854 Interestingly, Roussanaly et al.832,854 found that this distance was a function of, amongst other things, the project duration; this results from the larger proportion of the total economic cost of shipping being operational expenditure.Contrary to the case of pipeline transport, where the capital cost is the main driver, the operating costs make up the bulk of total cost for shipping;857 the majority of which results from the liquefaction process. The reduction of this cost therefore represents a key technical challenge, along with the design and operation of the injection system.858 Only a limited comparative Life Cycle Assessment (LCA) for pipeline and shipping is available in the literature,858 the results of which are seemingly dependent on the precise design of pipeline system used as a reference. Given the importance of the greenhouse gas footprint of CCS systems, thorough analysis of this aspect is required.
A further issue raised in the literature is that, along with any technical difficulties, there are potential legislative difficulties in the use of ships for CCS859 in Europe, given that under the EU emissions trading system (ETS) this CO2 would be accounted for as released rather than stored.
7.5 Outlook for CO2 transportation
The transportation system acts as the gateway between the CO2 emitters and the storage sites and imposes requirements on the design and operation of both. There remain uncertainties around material selection and operation which necessarily introduce conservativeness in design. The key challenge, however, is to understand the constraints for each transport technology to reduce the over-design and associated costs, as well as where restrictions placed on the feed streams, for example purity can be relaxed to allow a reduced-cost whole-system design, as observed by Kolster et al.860 Further, this must account for the evolution of the CCS network in order to avoid over-capacity that will not be utilised, as well as the dynamic use of the infrastructure to reduce risk.8 CO2 storage
8.1 Research challenges in subsurface CO2 storage
The state of knowledge around CO2 storage has seen a remarkable increase over the past 10 years.861 The injection and sequestration of CO2 at rates over 1 MtCO2 per year at individual sites is technically viable, demonstrated by 14 currently operating industrial scale projects, including three injecting into saline aquifer systems.862 The leading edge of research has thus moved beyond the viability of the technology which is now clearly demonstrated. Throughout all aspects of the injection of CO2 underground – thermophysics, geoscience, reservoir engineering, monitoring, and evaluation of large scale capacities and technology potential – research is addressing challenging questions where answers would lead to better characterisation, prediction of plume migration, lowering of uncertainty, managing the risks of leakage, and evaluation of the global role of CO2 storage in energy systems.8.2 Fluid properties and geoscience
Thermophysical properties of CO2 and its mixtures with reservoir fluids play an important role in storage-site selection, the design of injection strategies, and in predicting the long-term fate of injected CO2. Phase behaviour and diffusivity control the extent and rate of CO2 dissolution into the connate fluids. Interfacial tension (IFT) and fluid–fluid–mineral contact angles are key parameters influencing CO2 mobility at the pore scale. Finally, properties of the solutions formed when CO2 dissolves in the reservoir fluids play are role in determining field-scale convective flows. A great deal of effort has been expended on understanding the thermophysical properties of CO2–brine systems. The miscibility behaviour has been extensively studied at reservoir conditions and beyond, and reliable correlations are available.863,864 Diffusion coefficients have also been reported for CO2 in both water and brines865,866 and the effects of CO2-dissolution on both the density and viscosity are reasonably-well understood.867–870 The IFT of the baseline CO2–water system has been the subject of numerous studies. While there is a lack of agreement between some sources, recent high-quality measurements appear to be consistent.871–875 The addition of salts is found to result in a small increase of the IFT as demonstrated by Li et al.876,877 In summary, it can be said that the thermophysical properties of CO2–brine systems are well understood over relevant ranges of temperature and pressure and that these properties can be confidently predicted from knowledge of the temperature, pressure and brine chemistry. Current research is focused mainly on the effects on these properties of the impurities that are inevitably present in the CO2 stream.Dissolution of CO2 in the reservoir brines of course creates an acidic solution that can dissolve minerals (e.g. carbonates) at significant rates and transport them to regions of higher pH where they may then precipitate again. The dissolution rates of calcite and several other carbonate minerals have been measured experimentally at reservoir conditions by Peng et al.878,879 and used to help interpret pore-scale imaging of dynamic dissolution of limestones during injection of CO2-saturated brine.880
The presence of hydrocarbons, especially oil, complicates the problem substantially, mainly due to their diverse chemical compositions. Given the typically low miscibility of hydrocarbon and aqueous phases under reservoir conditions, the phases formed when CO2 dissolves in the reservoir fluids are primarily CO2–brine and CO2–hydrocarbon mixtures. To predict the thermophysical properties of the latter, bottom-hole oil samples are required; traditional techniques of reservoir fluid analysis and property modelling can then be applied.881
One of the most challenging properties to predict is the contact angle formed between partially miscible fluid phases and the mineral surfaces. A recent review of contact angle measurements in CO2–brine–silica systems shows the data scattered over a range of almost 90°.882 This variation is attributed to the variations in surface morphology and impurities. A perhaps more promising approach is direct imaging, by means of X-ray computed tomography (CT), of the contact angles formed in the pore space of a representative reservoir rock during controlled fluid injection.883,884
Much of the uncertainty around the petrophysics of CO2 storage, and particularly the relative permeability, has been resolved over the past 5 years.885,886 It has been established that drainage and imbibition processes are typical of water-wet systems in sandstones887–891 and carbonate rocks.892 One of the key field scale implications of this has been the confirmation that capillary trapping will be a significant mechanism for immobilisation of the CO2 plume in saline aquifer systems. This has been frequently estimated through modelling studies, e.g., Juanes et al.893 Another important implication is that the flow properties – relative permeability and residual trapping – are insensitive to reservoir conditions of pressure, temperature, and brine salinity.
Less well understood are the flow properties in systems altered by the presence of hydrocarbons. Research focused on petroleum systems has demonstrated that hydrocarbon-altered rock units trap less of the non-aqueous fluid phase.894,895 For carbon dioxide systems, Al-Menhali and Krevor892 and Al-Menhali et al.896 have shown that indeed residual trapping is significantly reduced in carbonate rocks with the “mixed-wet” state characteristic of many oil fields. There are no observations reported in the literature of the impact on relative permeability and capillary pressure functions, or dependencies on reservoir conditions. Injection of CO2 into oil reservoirs dominates the current suite of industrial scale projects, making up over 90% of the approximately 30 Mt per year of anthropogenic CO2 captured and sequestered and 11 of the 14 currently operating projects.862 It has been estimated that there is sufficient capacity for oil reservoirs to take up to 350 Gt of CO2, sufficient for the majority of the first generation of industrial scale CO2 storage.897 It is thus important that the petrophysics of these systems be more thoroughly investigated.
Incorporating the impact of natural reservoir rock heterogeneity on flow is a rapidly developing area of both laboratory petrophysics and larger scale flow modelling. Much of the uncertainty around the understanding of relative permeability in CO2–brine systems was due to the impact of rock heterogeneity on laboratory scale core flood experiments.890,898,899 It is increasingly understood that these small scale features can manifest in the reservoir at larger scales with significant impacts on flow900,901 and trapping.902–904 This may be an important source of discrepancies between predicted flow dynamics and observations of plume migration in field and industrial CO2 storage projects including the Sleipner project in the Norwegian North Sea,905,906 the Frio Brine experimental injection in Arizona, United States,907 and the Cranfield injection site in Mississippi, USA.908
8.3 Site development and reservoir engineering
Site characterisation involves the evaluation of a field with the aim of assessing its suitability for the injection of CO2. Most of the techniques were established in the petroleum industry.909 They include sampling and geophysical observational activities that allow for the construction of a geological model of the injection zone and overburden. This model is then used for dynamic reservoir simulations that assess possible outcomes of injection into the field, and to construct site development plans.The development practices for gas injection enhanced oil recovery projects are well established, whereas there is little experience with the development of dedicated CO2 storage projects injecting into saline aquifers. Industry field development plans for injection into a saline aquifer at the Gorgon storage injection project in Australia have been reported in Flett et al.910,911 and for the now cancelled Peterhead project in the UK.912 Detailed development plans, including simulations, have also been proposed in a screening study of prospective early storage development sites in the UK by Pale Blue Dot and Axis Well Technology.913 The presence of wells and previous hydrocarbon production activity is viewed as a significant factor in speeding up the development. Of the proposed development plans for five UK reservoirs in the report by Pale Blue Dot and Axis Well Technology,913 only two were deemed to require appraisal wells for initial site characterisation. A typical timeline inclusive of the use of appraisal wells, the final investment decision, and preparation for industrial scale injection will be 7–10 years.
Reservoir engineering for CO2 storage also largely follows practices established in petroleum engineering. The guiding principles are to meet injection targets while minimising the risk of leakage through overpressure and fracturing of the cap rock, or plume migration to zones with potential leakage pathways. Plume migration has in practice not been a concern due to the size of the geologic units relative to the injection volumes of the first-mover projects. Pressure increases that limited injection have occurred at the Snøhvit site in the Norwegian Barents Sea as well as at the In Salah site in Algeria.914 Both of these involve injection into saline aquifer systems, without the pressure alleviation provided by past or ongoing hydrocarbon production. In the case of the In Salah project, the overpressure resulted in a fracturing of the lower part of the sealing cap rock unit and a cessation of injection activity.915 At Snøhvit, the pressure buildup was not entirely unexpected given the uncertainty about compartmentalisation of the initial target reservoir unit. Contingency plans were in place and injection was successfully switched to an overlying formation from the original target.916 Other important industry scale projects injecting into saline formations, i.e. without hydrocarbon production, include the Sleipner project917 and the Quest project in Alberta, Canada.918 These have operated without incident or the need for brine production.
Pressure management through brine production is a key area of ongoing research. In many locations injection targets can be met without any pressure relief from the reservoir system.917 This is particularly the case for injection coupled with enhanced oil recovery – where pressure relief is provided by hydrocarbon production – and in depleted gas fields where prior production has lowered the pressure below hydrostatic.914 Brine production has not yet been implemented in an operating storage project but it is included in the development plans for the Gorgon project in Australia.910 It is also widely considered to be necessary to take full advantage of regional storage systems over decadal timescales.919 Engineering strategies for brine production focus on maximising pressure relief while minimising the risk to leakage through CO2 plume migration to the brine production wells, the analogue to gas or water breakthrough in oil production projects.919–922 Costs due to well construction and brine handling must also be minimised. A number of brine handling strategies have also been proposed, including those that enhance the rate of residual or dissolution trapping of CO2 through brine reinjection into the reservoir923,924 or dissolution of CO2 into brine at the surface prior to injection.925 These strategies remain at the research stage.
8.4 Monitoring, leak detection, and remediation
Monitoring of CO2 storage relies on a suite of technologies developed for petroleum production applications. Instrumentation in the well bores – down hole data includes pressure, temperature logging, fluid geochemical sampling, the use of tracers, near well geophysical saturation monitoring, and potentially crosswell seismic reservoir characterisation. Over large spatial scales, the use of seismic surveys has been demonstrated to be useful in monitoring the growth and migration of CO2 plumes. A major development was the successful demonstration of the use of InSAR technology at the In Salah field site to monitor movements in surface elevation over the injection site. This provided detailed constraints on the pressurisation of the subsurface. Jenkins et al.926 provides a comprehensive review of the state of the art.The main challenge in the further development of these technologies concern their use in ways that allow for quantitative estimation the amount of CO2 stored and the extent of the plume at the outer reaches of migration. This quantification is needed to verify the efficacy of storage. With seismic imaging, for example, there are significant limitations on quantifying fluid plume thickness and fluid saturation in the pore space.927,928 Additionally, CO2 plumes will frequently have long thin tongues migrating as a gravity current in response to buoyancy, difficult to detect with seismic monitoring. Current research efforts are focused on extending the interpretation of seismic surveys so that thin layer features may be identified and saturation quantified, e.g. Ghaderi and Landrø,929 Trani et al.930 Similarly, saturation quantitation is also being extended to down well geophysical measurements such as crosswell seismic931 and near well observations, e.g., pulsed-neutron logging.932
Another significant challenge for monitoring and verification lies in the development of techniques for quantifying the extent to which CO2 has partitioned into the aqueous phase or has become residually trapped. Geochemical techniques show the most promise, with the use of both artificial and naturally occurring partitioning tracers allowing for estimates of residual trapping and the extent of CO2 dissolution.933–936
Leak detection and remediation is a significant challenge for CO2 storage with little analogue in the petroleum industry. Pressure monitoring in injection and observation wells is a key tool due to its precision and responsiveness. At the In Salah site, pressure monitoring combined with InSar data was effective in diagnosing leakage of CO2 into the cap rock, with fractures in the cap rock induced by over pressuring of the system during injection.914 Pressure monitoring in overburden layers has been proposed and may also provide sensitive indicators of CO2 leakage away from the target formation.937 Environmental monitoring for CO2 flux at the surface over storage sites, either on land, or at the sea floor, has also been deployed.938,939 Techniques can be used to monitor CO2 concentrations or isotopic signatures in soil gas or the atmosphere over the site, or changes in other chemical signatures like pH. The challenges here reside in detection. The signature of CO2 leakage at the surface over a site is not well understood. It may be diffuse or focused through faults or abandoned wells.940 Similarly the isotopic composition of the fluid may have evolved as it percolated through the leakage pathways over the storage site. Once CO2 is released into the atmosphere, it is quickly diluted. Natural background variation in CO2 is also usually substantial, with daily and seasonal variations even in areas with little interference from industrial activities.
The mitigation of leakage risk and the remediation of leaks or their effects has recently emerged as an area of research interest.941 Applicable techniques developed in the hydrocarbon industry focus on the management of problematic wells and are directly applicable to CO2 storage. For leakage away from wells, management of the local hydrogeology through the injection of brine has been proposed,937,942 adapted from techniques used in groundwater protection. The use of chemical seals emplaced in the reservoir have also been proposed by Vialle et al.943 The major challenges in developing in-reservoir leak mitigation technology comes from the major uncertainties of the subsurface rock heterogeneity that will control the effectiveness of these strategies. Due to the significant costs of, e.g., drilling a single well, and time required to evaluate the response, the uncertainty around the success of these strategy must be significantly reduced before these can be implemented in an industry setting.
8.5 Storage capacity and the role of CO2 storage in energy systems
Storage capacity refers to the potential for a specific location or region to permanently store CO2 in the subsurface. Evaluating capacity for specific storage sites is seen as a routine aspect of the characterisation and reservoir management stages, e.g., Pale Blue Dot and Axis Well Technology.913 As injection progresses at a given location, models may be updated for more accurate pictures of the local storage capacity.The support for the importance of CO2 storage, however, is predicated on the large scale potential for managing CO2 emissions that would arise from the combined impact of sequestration across thousands of individual storage projects.6,944 This in turn is based on the assumption that there is far more pore space available to inject CO2 into the subsurface than would be required for substantial CO2 mitigation.897 Global scale assessments of CO2 storage capacity support that view. Recent compilations of the global distribution of storage capacity include those of Benson et al.,945 Dooley,946 and Cook and Zakkour.947 Most of the modelling underpinning the IPCC assessment reports are based on earlier estimates from Hendriks et al.34 or do not consider capacity limiting at all.897 The estimated capacities, however, are such that updates to more recent compilations would not substantially impact the models used for the IPCC.
These capacity estimates, however, are known as “static” capacity estimates. They are estimated assuming that some fraction of the total pore space of a geologic unit, usually between 1–10% and known as the storage efficiency, can ultimately be made available for CO2 storage.948 A more rigorous analysis of storage capacity would incorporate dynamic modelling of the pressure and plume migration during injection. A number of studies have shown that these are more likely to be limiting factors over decades to a century than the pore volume available in a geologic unit.949 Moreover, there is little correlation between static estimates of storage capacity and those making use of dynamic pressure and plume evolution (Fig. 18).
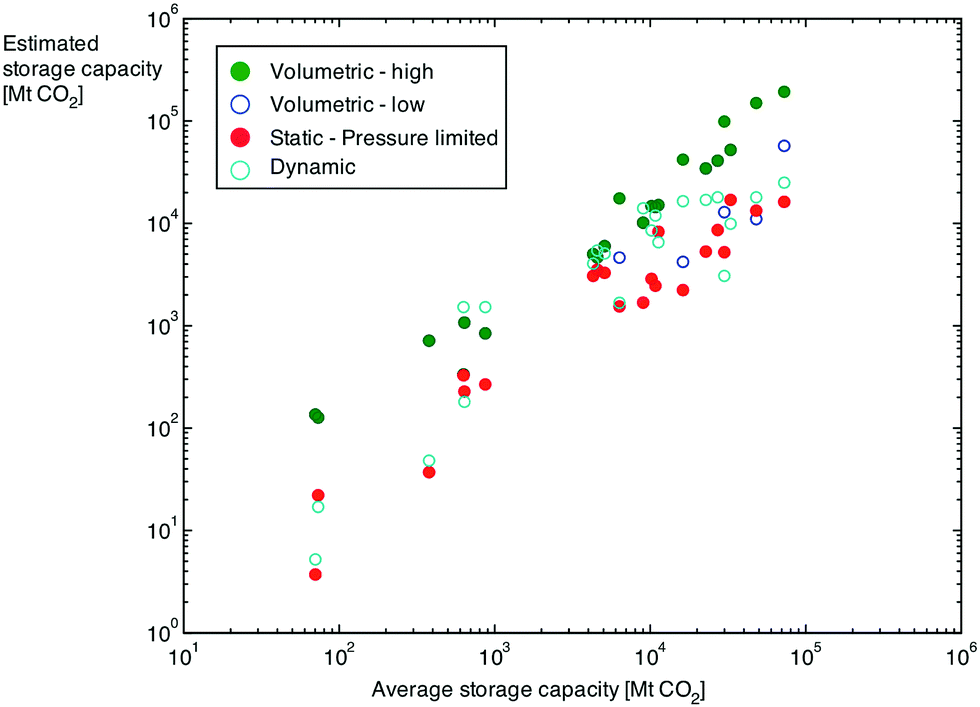 | ||
| Fig. 18 Storage capacity for fields and regions from studies where estimates have been made using three separate techniques – volumetric, pressure limited static, and dynamic. For a given region, estimates range across 1–2 orders of magnitude with little systematic correlation between estimate approaches. Data from Winkler et al.,950 Goodman et al.,951 Thibeau and Mucha,952 Bader et al.953 and Gorecki et al.954 | ||
Estimating the time-varying capacity of storage resources by modelling the dynamics of injection is known as a dynamic approach to capacity estimation. The most comprehensive regional evaluation of dynamic capacity has been performed for the Nordic region under the NORDICCS project.955 Capacity estimation was largely based on the use of reservoir simulation tools developed for the petroleum industry, e.g., ECLIPSE. Simplified analytic or semi-analytic models have also been proposed which capture the dynamics of plume migration, pressure evolution, average regional pressure buildup, and impacts on injectivity around a well.956–959 There is significant potential for the use of these simplified models for dynamic regional evaluations of CO2 storage, e.g., Szulczewski et al.,960 but there are also significant complications in the use of these models to represent regions with multiple injection sites.961 Assessments that evaluate the impact of regional development characteristics (e.g., number of injection sites, distance between injection sites, CO2 injection rate, frequency of rate variation) on CO2 storage962,963 provide valuable insight into improving models of CO2 storage. If these models could be successfully incorporated into systems level analysis of energy systems, e.g., IPCC,6 it would significantly deepen our understanding of the potential role for CCS in future scenarios of low carbon energy production.
8.6 Outlook for CO2 storage
CO2 storage research has progressed significantly over the last decade. The technical feasibility of CO2 storage has been demonstrated through a number of industrial scale projects, with most being EOR projects. Subsequently, much of the existing CO2 storage experience is based in the oil industry and knowledge about CO2 storage in saline aquifers is limited. Similarly, the technology for CO2 storage monitoring was originally developed for the petroleum industry. Further development of monitoring instruments is required to enable quantitative predictions of the amount of CO2 stored, the extent of plume migration, geophysical saturation, and the extent of CO2 trapping and dissolution. Although leak detection has not been a focus for the petroleum sector, advances in leak detection technology is required to ensure that the storage of CO2 is permanent. Sensitive CO2 leakage indicators include pressure monitoring and measuring CO2 flux at the surface over storage sites (either as CO2 concentration in the soil/atmosphere or pH at the sea floor). To prevent CO2 leakage, safety measures that have been developed are chemical seals and pressure management of the site (e.g., brine production to prevent over-pressure).The thermophysical and flow properties of CO2 are generally well understood. Furthermore, there are well established procedures for site characterisation and determining the suitability of an injection site (e.g., cap rock or rock heterogeneity that control/prevent leakage). However, to further de-risk CO2 storage, research is required on studying the effect of: (i) impurities introduced via the CO2 stream on the thermophysical properties of the CO2–brine system, (ii) hydrocarbons already present in the geological system, and (iii) heterogeneity of reservoir rock on flow properties. Ultimately, the capacity of permanent CO2 storage needs to be quantified on a global scale to ensure that all of the CO2 captured can be adequately stored. However, this has typically been evaluated using “static” capacity models, which does not account for the dynamics of injection. Recent advances in models now provide time-varying capacity estimates, which consider the influence of system dynamics, e.g., plume migration, injectivity, pressure build-up. Future work on integrating these dynamic storage capacity models into energy systems models could provide valuable insight into the impact of storage capacity on meeting demand for low carbon energy.
9 CO2 enhanced oil recovery (CO2-EOR)
Carbon dioxide enhanced oil recovery, CO2-EOR, has been practised for many decades as a means to enhance the recovery of oil from depleted reservoirs.965 The process (illustrated in Fig. 19) is most effective by operating in ‘miscible’ mode, whereby the injected CO2, usually in the liquid or supercritical state, is fully miscible with the oil phase in the reservoir, reduces the viscosity of the oil phase which is then displaced from the rock pores in a single-phase drainage process. This requires the temperature–pressure conditions in the reservoir to be above the minimum miscibility pressure (MMP) for the CO2–hydrocarbon mixture. This is typically about 75 bar for light crudes at reservoir temperatures of about 70 °C and rises with temperature. Whilst immiscible displacement using CO2 can also lead to moderate enhancement of recovery, this miscibility condition does mean that there is a limited window of opportunity to exploit the much more efficient miscible CO2-EOR process in depleting reservoirs before the pressure has declined below the MMP. Because of the lower viscosity and density of the injected CO2 relative to the in situ oil, there is usually significant fingering into the oil and breakthrough of the CO2 leading to about 50% recycle. Alternating injection of water and gas (WAG) is a widely practised technique to improve the displacement and recovery of oil.966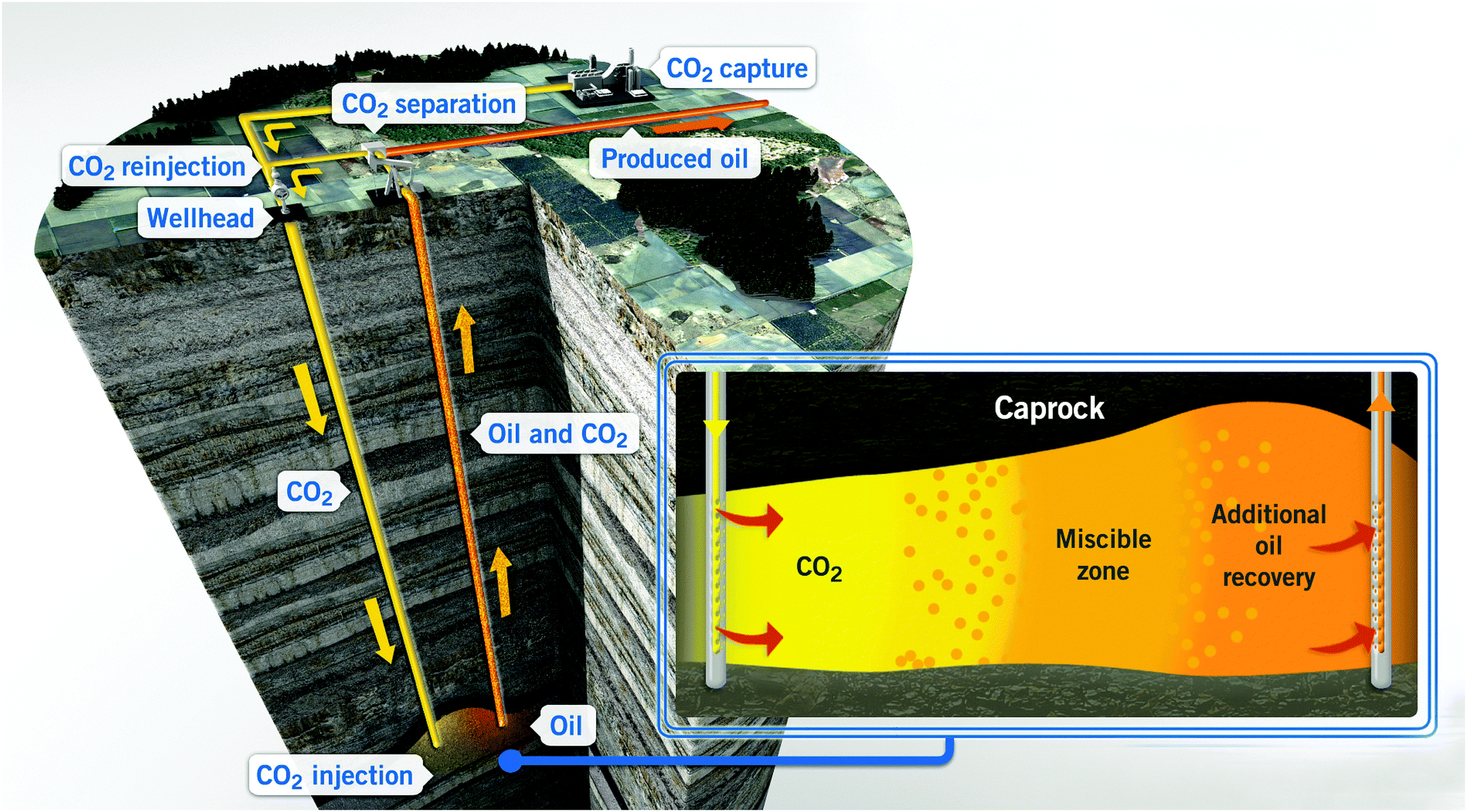 | ||
| Fig. 19 Schematic diagram of CO2-EOR process. Source: Global CCS Institute.964 | ||
Carbon capture and storage enhanced oil recovery, CCS-EOR, is a similar process but with the dual objective of recovering additional quantities of oil from reservoirs whose oil production has fallen below critical levels whilst at the same time storing some of the injected CO2 permanently in the depleted reservoir rather than pumping it back to surface. The driver here is to generate as much income as possible from incremental oil to offset the high costs of the CCS process – of order $70 per tonne of CO2 stored. Whereas in CO2-EOR, the objective is to produce as much incremental oil as possible using as little CO2 as possible, without any concern for CO2 retention in the reservoir (or in optimum scenarios to recover as much CO2 as possible so that it can be recycled), with CCS-EOR there clearly needs to be a balance between recovered hydrocarbon (to generate income) and the amount of CO2 stored, which for the process to be economically viable needs to be significantly greater than the optimal amount required for efficient miscible displacement. This balance turns out to be critical in determining the viability of CCS-EOR processes.
CCS-EOR is a form of CO2 utilisation967 and is currently the only way to add value to CO2 at the mega-tonne per annum scale. Typical incremental oil values for optimised CO2-EOR processes are 5–15% of the original oil in place (OOIP), and the typical utilisation is about 3 tonnes of CO2 purchased and injected leads to about 1 bbl of incremental oil.
9.1 Current CCS-EOR activity
Globally there are currently more than 140 CO2-EOR projects, producing about 300000 bbl of incremental oil per day, equivalent to about 0.35% of global oil production. Most of this production is in the United States Mid-West. The objective is mainly EOR, not CO2 storage, aiming to minimise net CO2 injection and maximise oil recovery. For the process to be a viable route for high volume CCS requires a paradigm shift where the business target is to maximise both oil recovery and CO2 storage. Since 2000 about six projects have been commissioned which are truly CCS-EOR, of which the largest are both in Saskatchewan, Canada: at Weyburn-Midale and at Estevan (Boundary Dam).
The Weyburn-Midale project,969,970 run by Cenovus and Apache, was fully monitored between 2000 to 2012 and verified as a CCS project. The incremental oil was approximately 220 Mbbl over the project lifetime, producing 3 bbl per tCO2 purchased. More than 20 MtCO2 have been stored to date and the expectation is that 40 MtCO2 will be stored over the project lifetime, 30 MtCO2 in the Weyburn field and 10 MtCO2 in Midale. The daily injection of CO2 consists of 6500 tCO2 of fresh feed accompanied by 6500 tCO2 of recycle; the CO2 feed comes from the North Dakota Beulah synfuels gas plant via a 320 km pipeline. The project has extended the lifetime of the fields by over 25 years.
By contrast the SaskPower Boundary Dam Integrated CCS Project is very recent.971 Opened in October 2014 as the world's first fully commercial CCS plant, it cost about $1.3bn, of which $300m was provided by government. It involves post-combustion capture (90% efficiency) of CO2 from a 110 MW coal-fired power station. Its target is to store 1 Mt of CO2 pa in steady state operation; it reached a total storage of 1 MtCO2 in August 2016 and is now operating close to the design storage rate. This is equivalent to removing 250000 cars from the road. The CO2 is pumped via a 66 km pipeline to the Weyburn depleted oil reservoirs where it is used for EOR before storage, hence adding significant value to the captured CO2. The remainder is stored in the 3.4 km deep Deadwood saline aquifer, which is only 2 km away, within the Aquistore project administered by the Petroleum Technology Research Centre (PTRC).
Other significant CO2-CCS-EOR (i.e., CO2-EOR combined with CCS) projects are summarised in Table 7. In 2004, IEAGHG identified 420 CO2-EOR ‘early opportunity’ candidates972 but relatively few of these have been exploited.
| Project | Description |
|---|---|
| Weyburn-Midale; IEAGHG, Cenovus, Apache (2000) | • CO2 from North Dakota Beulah synfuels gas plant via 320 km pipeline |
| • >20 MtCO2 injected to date; 40 MtCO2 target | |
| • Anticipated 220 Mbbl incremental oil from Weyburn-Midale fields | |
| Boundary Dam Unit 3, SaskPower, October 2014 (Saskatchewan, Canada) | • Continuous mode 110 MW coal (lignite)-fired power station |
| • Captures 95% of CO2 emissions (and 100% of the SO2) | |
| • 1 MtCO2 pa transported in 65 km pipeline to Weyburn field for EOR | |
| • Some CO2 stored in close (2 km) Deadwood saline aquifer (Aquistore Project) | |
| NRG Petra Nova, Texas (2015) | • 240 MW coal plant with gas post-combustion capture (90%) |
| • 1.4 MtCO2 pa injected into West Ranch oil field | |
| Petrobras, BG Brasil, Petrogal Brasil (2013) | • 0.7 MtCO2 pa from natural gas production |
| • Injected for EOR 5–7 km sub-sea, 300 miles offshore Rio | |
| • Deepest CO2 injection in the world | |
| Saudi Aramco Uthmaniyah Project, Saudi Arabia (2015) | • 0.8 MtCO2 from gas processing used for EOR from Uthmaniyah field |
9.2 Combining CO2-EOR with CCS)
In order to add CCS capability to a CO2-EOR project (CO2-CCS-EOR), several further issues need to be considered. Additional site characterisation and risk assessment is needed to assure storage integrity, together with additional measurements of gas venting and fugitive emissions associated with inadvertent gas releases from surface equipment. Long term integrity needs to be assured through additional monitoring of the subsurface and enhanced field surveillance using surface monitoring. Changes in abandonment processes to ensure long-term well seal integrity also need to be implemented. All this adds costs to conventional CO2-EOR.One of the most detailed analyses of the viability of combining CO2-EOR with CCS was carried out recently by the IEA.968 In their 2015 report they call this process ‘CO2-EOR+’ and identify three possible scenarios:
• CO2-EOR+ Conventional (or ‘Light’), where conventional CO2-EOR is supplemented by a full CCS risk assessment, monitoring and verification but little attempt is made to increase the amount of CO2 injected. Representative values given for this scenario are a net utilisation of 0.3 tCO2 per bbl oil produced with an incremental oil recovery of 6.5% of OOIP.
• CO2-EOR+ Advanced (or ‘Balanced’), where the aim is to increase both the amount of CO2 stored and also the incremental oil recovered – a win-win scenario. Here, typical values could be a net utilisation of 0.6 tCO2 per bbl oil produced for an incremental oil recovery of 13% OOIP.
• CO2-EOR+ Maximum Recovery (or ‘Heavy’) where there is a strong focus on CO2 storage, with representative values of injecting 0.9 tCO2 per bbl oil produced for the same incremental oil recovery of 13% OOIP as in the Balanced scenario. Here, there would be no produced water reinjection or CO2 recycle.
9.3 Is there enough storage volume for CO2-EOR+?
The brief answer to this question is ‘Yes, but maybe not in the right places’. Current CO2 emissions are about 36 Gt pa, equivalent to about 100 Mbbl CO2 per day, compared with oil production levels of about 90 Mbbl per day. We need to capture 15–20% of total CO2 emissions to meet global carbon mitigation targets (the remainder being accounted for by energy efficiency/savings measures [up to 50%] or through increased deployment of renewables and nuclear [about 30%]). CCS targets to meet the COP21 target of limiting mean global temperature rise to 1.5–2 °C through capping atmospheric CO2 levels at about 450 ppm require capacity to store 120–160 GtCO2 at a rate of about 10 GtCO2 pa by 2050, equivalent to ∼3000 major facilities. The storage requirement by 2100 may be as high as 1200–3300 GtCO2.The global storage potential for CO2-EOR+ Light has been estimated at 70–140 GtCO2, in principle, resulting in about 470bn bbl of incremental oil (see Table 8).973 However, this may be a highly optimistic estimate of the total deployable CO2-EOR capacity. As illustrated in Table 8 and Fig. 20, the majority of this capacity exists in the Middle East/North Africa and in the USA at 50% and 13% respectively, whereas the estimated CO2-EOR in South Asia is essentially zero with the Asia Pacific region accounting for only about 3%.
| Region name | Recovery (MMBO) | Basin count | CO2–oil ratio (tonnes per bbl) | CO2 stored (GtCO2) |
|---|---|---|---|---|
| Asia Pacific | 18376 |
6 | 0.27 | 2.7–5.0 |
| Central and South America | 31697 |
6 | 0.32 | 4.7–10.1 |
| Europe | 16312 |
2 | 0.29 | 2.5–4.7 |
| Former Soviet Union | 78715 |
6 | 0.27 | 11.8–21.6 |
| Middle East and North Africa | 230640 |
11 | 0.3 | 34.6–70.1 |
| North America/Non-US | 18080 |
3 | 0.33 | 2.7–5.9 |
| United States | 60204 |
14 | 0.29 | 9.0–17.2 |
| South Asia | — | 0 | N/A | — |
| Sub-Saharan Africa and Antarctica | 14505 |
2 | 0.3 | 2.2–4.4 |
| Total | 468529 |
50 | 0.296 | 70–139 |
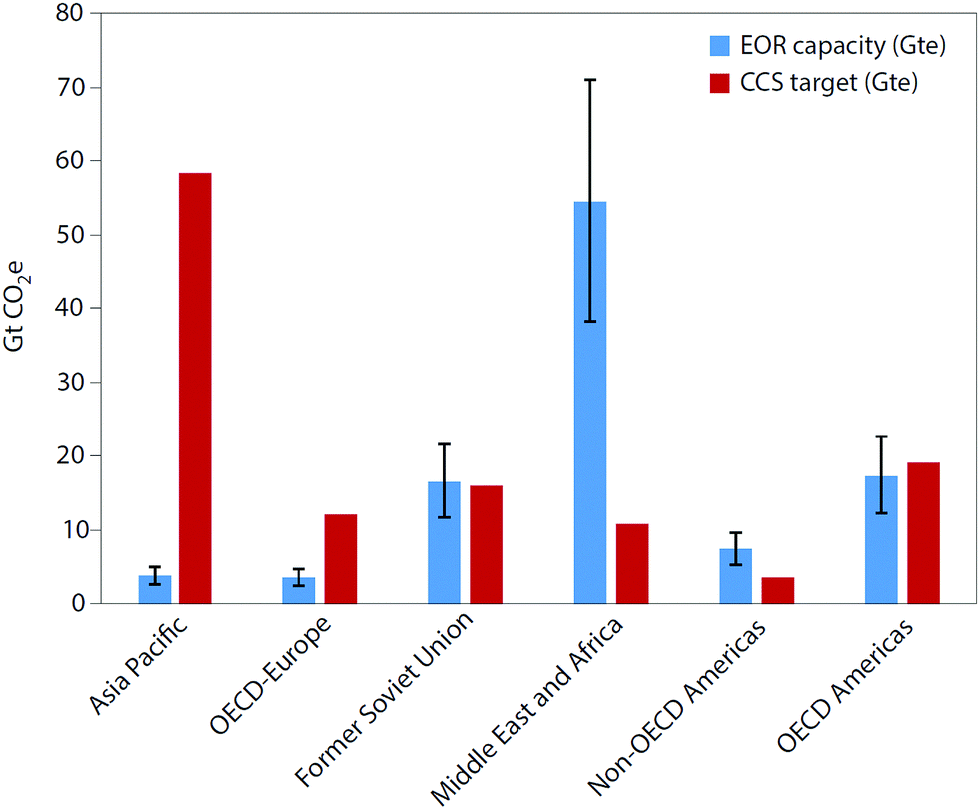 | ||
| Fig. 20 Global CO2-EOR capacity compared with CO2 mitigation targets. Figure reproduced from Mac Dowell et al.967 Data from Dooley et al.,974 IEA,975 and CIA.976 The error bars indicate an average calculated variance of 30%. The actual reported variance is in the range of 25–35%. | ||
In other words, as illustrated in Fig. 20, there appears to be an unfortunate disconnect between regions of substantial CO2-EOR potential and those regions with the largest anticipated population growth, dependence on fossil fuels and requirement to sequester CO2 over the course of the next century. In fact, the only region where it appears certain that there is sufficient CO2-EOR capacity to meet the CO2 storage requirements to 2050 is the Middle East and Africa – although the requirements are close in North America and the Former Soviet Union. Given the size and rate of growth of the CO2-EOR industry in the USA, it is likely that this region will be a leader. If we recognise that in some cases CO2 injection will be restricted by availability rather than by CO2-EOR capacity, a more realistic estimate is likely to be on the order of 40 GtCO2 injected and stored via CO2-EOR. This is in line with the recent IEA estimate. If we adjust this for the net emissions from the additional incremental oil (see later), the total capacity for CO2-EOR+ Light is about 35 GtCO2, which is ∼30% of the 2050 target and 4–5% of the total CO2 mitigation target.
The IEA analysis used the industry U-Cube capacity database977 on a field-by-field basis. It estimates that the global capacity for CO2-EOR+ ‘Balanced’ is ∼240 Gt and for CO2-EOR ‘Heavy’ is about 360 Gt, two and three times respectively the IEA's estimate of CO2 storage. Outside North America, the main capacity is focused on the Middle East (>100 Gt), Russia (>70 Gt), North Africa (∼35 Gt) and Central Asia (∼20 Gt). The potential for incremental oil for conventional CO2-EOR up to 2050 is 190bn bbl, whilst the ‘Balanced’ and ‘Heavy’ scenarios could deliver an additional 375bn bbls during that period, over ten times the current annual oil consumption. Therefore the technical global capacity of CO2-EOR+ to both store CO2 and to deliver significant incremental oil to deliver revenue through CCS is therefore considerable, capable of meeting current targets to meet the 2 °C scenario. The main issue is whether the economic drivers will be in place to make the process financially viable.
9.4 What about the economics?
Conventional CO2-EOR is profitable at ∼$65 bbl oil with CO2 costs at $30 per tCO2.968 CO2 prices are even lower than this for naturally occurring CO2 sources which explains why several of the existing CO2-EOR projects are located close to such sources. The major future drivers for CO2-EOR+ will be:• A regulatory requirement or fiscal incentive (carbon price through a tax or trading system) to store CO2.
• High oil prices – a major factor as this controls the value of the incremental oil produced per tonne of CO2 stored.
• Lower CO2 supply prices, which will be strongly dependent on the level of a carbon price. Currently the EOR operator pays the CO2 generator for acquisition of the CO2, whereas under a 2 °C regime supported by a realistic carbon price, by 2050 the generator could be paying the EOR operator a large fraction of the carbon price to store the CO2 generated in power production or industrial manufacture e.g. up to $125 per tonne CO2.
• Reductions in core EOR process costs, especially the additional cost requirements of CCS mentioned earlier.
The recent IEA study968 calculated the NPV of typical CO2-EOR+ projects for a range of ETP future scenarios where, as the mean global temperatures rise from 2 °C to 6 °C, both the oil price and the CO2 supply cost increase. Fig. 21 shows the sensitivity of the project profitability to the oil and CO2 prices.
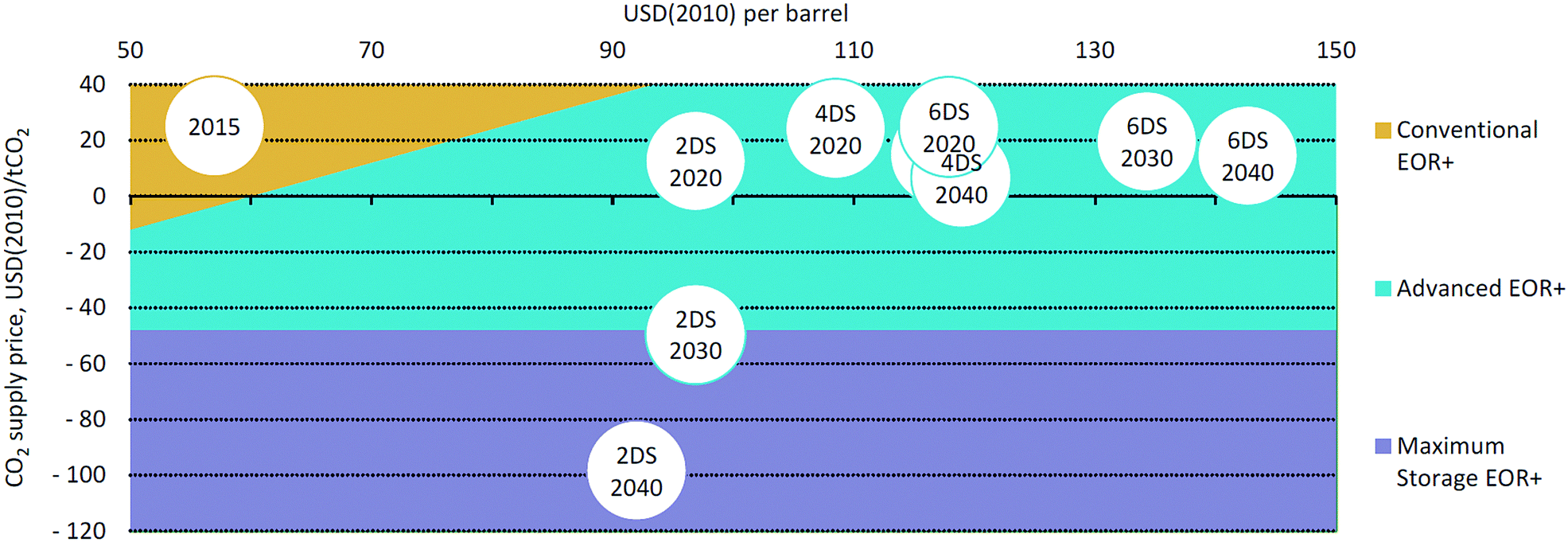 | ||
| Fig. 21 Sensitivity of economic viability and choice of CO2-CCS-EOR approach to oil and CO2 prices, reproduced from IEA.968 The circles indicate different ETP future scenarios: e.g. 2DS 2030 indicates where a 2 °C capped mean global temperature rise world may be positioned in 2030. | ||
The conventional CO2-EOR+ ‘Light’ process remains the best option for low oil and high CO2 prices (costs), down to the point where carbon pricing forces CO2 prices to be negative. The ‘Balanced’ scenario is the favoured option once CO2 prices become negative (CO2 producer pays) or oil rises above about $90 per bbl, which is where oil prices were 2008 to 2014, driven by income from both incremental oil and CO2 disposal payments. Once the latter rise further, with further rises in the carbon price, then the ‘Heavy’ CO2-EOR+ option becomes increasingly viable with increased CO2 disposal revenues. Hence the economic viability of CO2-EOR is closely linked to both the global oil-gas economy and to regional and global geopolitics governing the existence and level of a carbon price. In the absence of either high oil prices or high carbon prices, it is unlikely that CO2-CCS-EOR will move from its current position of opportunistic localised projects with minimal impact on reaching the required global CCS levels.978
9.5 Does CO2-CCS-EOR really reduce emissions?
A final critical issue for CO2-CCS-EOR, in the light of the production of incremental oil (and sometimes gas), which will be subsequently burned for fuel or power, is whether CO2 emissions are actually reduced by storing CO2 in the sub-surface alongside increasing oil production to partially offset the costs of CCS. Here it is crucial to carry out a life cycle analysis from the production of the original hydrocarbon, through its use, then its capture, transport and storage via CO2-EOR, including CO2 recycle and separation of produced natural gas liquids, to the eventual use of the incremental oil.Considering just the storage process itself, project level emissions are significantly carbon negative when disposing of anthropogenic CO2, ranging from −0.76 tonnes CO2-equivalent per tonne of CO2 delivered for CO2-EOR+ ‘Light’ to −0.86 tonne CO2-equivalent for the ‘Heavy’ process.968 When emissions from the use of the incremental oil are included, the ‘Balanced’ CO2-EOR+ process is essentially carbon neutral whereas the ‘Heavy’ scenario results in a net storage of ∼33% of the injected CO2. However, the additional oil produced by CO2-EOR can displace oil which would otherwise have been produced elsewhere with additional cost and additional CO2 footprint. Once these displaced emissions are taken into account, then the net CO2 emissions vary from about 0.7 to 0.8 tonne CO2-equivalent per tonne CO2 stored.968 This clearly depends on the type of oil recovered by EOR and displaced. In an era where new oil may consist of an increasing proportion of heavier crudes or unconventional tar sands or oil shales, the net emissions benefit of using CO2-EOR+ on conventional light oil reservoirs can rise to as much as 1.5 tonne CO2-equivalent per tonne CO2 stored.968
9.6 CO2-EOR: future challenges and opportunities
Overall, the emissions reductions resulting from the use of CO2-EOR+ will vary with the precise conditions, but they will always be negative and typically above 0.7 tonne CO2-equivalent per tonne CO2 stored. Yet the processes of CO2-EOR and CCS are currently far from optimal so better understanding and optimisation of miscible CO2 displacement processes in porous reservoirs for both enhanced recovery and storage979 should result in improved efficiencies and lower costs. There is an element of risk reduction and public reassurance that such processes are safe and secure, particularly on land. To enhance public reassurance and reduce risk, R&D and field trials focus on key assurance issues such as decommissioning, fugitive emissions monitoring and sub-surface monitoring of fluids migration and real-time process control. Although progress has been made, there is a need for further reconciliation of the legal frameworks for carbon storage and EOR, and ensuring CO2-CCS with EOR can remain part of the Clean Development Mechanism.There are significant CO2-CCS-EOR opportunities in unconventional gas recovery: enhanced coal-bed methane production,980 shale gas,981 even combined gas hydrate production and exchange/storage.982 These should be explored as potential routes to lower net costs of CO2 storage in coal seams, shale reservoirs and gas hydrate sediments, as well as accessing additional non-conventional gas reserves cost-effectively. In the absence of a strong carbon pricing driver, the linkage of CO2-CCS-EOR to relatively pure, low cost sources of CO2, such as that from the Leilac cement capture process983 or co-produced CO2 from gas wells, would be cost-effective routes to ramp up the approach.
10 CO2 conversion and utilisation (CCU)
10.1 The role of CCU in climate change mitigation
Carbon capture does not only enable CO2 storage but also utilisation and chemical conversion of the captured CO2. The resulting concept of carbon dioxide re-use (CDR) or CO2 conversion and utilisation (CCU) has been gaining significant attention in recent years.984–987 For a long time, CO2 has been used industrially for a variety of applications ranging from carbonated drinks to urea production.988 The recent interest in CCU is motivated by climate change mitigation as a societal issue989 but also by recent breakthroughs in catalysis for CO2 activation as technological driver.990,991 Several plants demonstrating novel pathways for carbon dioxide re-use are already in operation.992Carbon dioxide may be re-used directly as technological fluid, e.g., as solvent, by conversion to chemicals and fuels, and by mineralisation to solid inorganic carbonates984 (Fig. 22).
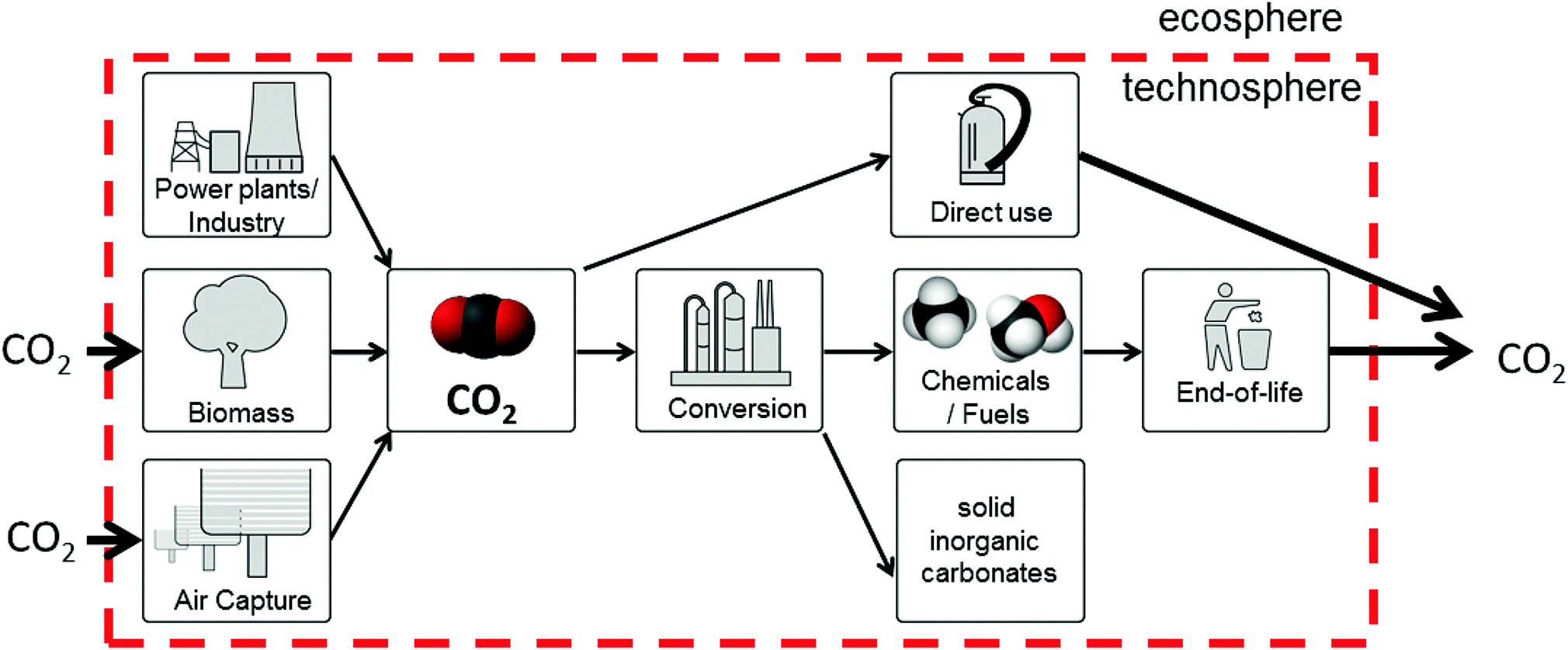 | ||
| Fig. 22 Life-cycle of carbon dioxide re-use with main classes of CO2 sources and utilisation pathways. The dashed box separates the technosphere from the ecosphere. | ||
The simplified life-cycle for CCU (shown in Fig. 22) identifies potential roles of CCU in climate change mitigation from a carbon-accounting perspective:
• Carbon-negative products: CCU can be carbon-negative, if and only if (1) atmospheric CO2 is used (either directly from direct air capture or via biomass)993 and (2) a solid inorganic carbonate is produced which is thermodynamically more stable than CO2 and thus provides long-term storage.994 If CO2 is captured from fossil sources such as power-plants or industry,995 CCU cannot be carbon-negative over its life cycle.
• Carbon-neutral products: CCU allows carbon-neutral pathways, if and only if atmospheric CO2 is used or CO2 is captured during the end-of-life treatment of a CO2-based product. Alternatively, converting fossil-based CO2 by mineralisation would also be conceptually carbon-neutral.
Beyond the simple carbon-accounting perspective, CCU could contribute to climate change mitigation via further mechanisms:
• Carbon-reducing products: even when a CCU pathway is not overall carbon-negative, it can be carbon-reducing by replacing an existing product with less greenhouse gas (GHG) emission intense alternative. A recent example is the production of polyethercarbonate polyols from CO2 (which can be processed further to polyurethane (PU) foams).996 The novel polyols contain about 20 mass% CO2 and could reduce GHG emissions by 11–19% compared to conventional polyether polyols.997 Often, the carbon reduction depends on the availability of hydrogen with low or even very low global warming impacts.998,999
• Temporary carbon storage: chemicals or fuels produced from CO2 offer temporary storage of CO2. While the storage duration is usually short for fuels, it would be longer for chemicals, e.g., polymers used for housing insulation. Still, permanent storage is usually only assumed for storage longer than 100000 years.1000 The impact of temporary CO2 storage on climate change is not sufficiently understood yet. Life-cycle assessment (LCA) does usually not account for emissions timing.1001 Several methods have been proposed for this purpose.1002–1004 In the absence of an accepted method, standards currently recommend to separately report the amount stored and the duration for temporary storage.1000
The life-cycle of CCU (Fig. 22) is also helpful to clarify the relation between carbon dioxide re-use and carbon capture and storage. Often, CCU has been contrasted to CCS and proposed as an alternative. This viewpoint may be tempting due the joint capture step and the similar acronyms CCS and CCU used for CO2 re-use in the literature. However, it is also misleading. Even though both CCU and CCS aim at mitigating climate change, the approaches are rather complementary than in conflict. While CCS addresses the end-of-life problem, CCU addresses a beginning-of-pipe problem, i.e., a raw materials problem, by providing a sustainable carbon source. Thus, the CCU life-cycle integrates naturally into the CCS life-cycle. A storage step could be added to all CCU routes where CO2 is released at the end-of-life. The debate about CCU should therefore be separated from CCS.1005
The preceding discussion of CCU focuses on its potential in climate change mitigation, which is also at the centre of the current debate in public and science. However, it has to be emphasised that one major environmental driver for CO2 utilisation is the provision of a non-fossil carbon feedstock for the chemical industry which helps to reduce depletion of resources.984 Avoiding fossil-based feedstock then often induces savings of GHG emissions as a secondary effect. An example is the replacement of fossil-based epoxides by CO2 in the production of polyols.997 In addition, CCU products may also lead to other environmental benefits. The direct synthesis of the potential fuel additive dimethoxymethane from CO2 has been recently demonstrated.1006 Dimethoxymethane is the first member of the homologous series of poly(oxymethylene) dimethyl ethers (OME) which have been shown to reduce soot formation during combustion and might thus lead to cleaner fuels.1007,1008
An additional driver can be economics as CCU leads to a valuable product and may thus generate a revenue in contrast to carbon storage.1009 It has therefore been argued that CCU might incentivise CO2 capture technologies. This argument, however, neglects the different scales of potential CCS installations and most CCU products.2 For most potential CCU products, even producing the global demand completely from CO2 would not be able to take up the CO2 generated by a single state-of-the-art coal-fired power plant. The current total global anthropogenic CO2 emissions from fuel use is 32.3 GtCO2 per year.1010 The eight most CO2-emitting thermal power plants in the European Union1011 alone generate sufficient CO2 to cover the current global demand for CO2 of about 200 Mt per year.1012 The upper limit of current CO2 demand for the manufacture of chemicals is estimated to be around 300 MtCO2 per year,988 with the potential to increase up to 500 MtCO2 per year.1012 Although CO2 demand is expected to increase, re-using CO2 for chemicals will always be limited since the total mass output of the chemical industry is 14–20 times smaller than the current output from the energy industry.1013 The production of fuels could increase the CO2 demand by up to 2050 Mt per year.1014 CO2-based fuels, however, usually require hydrogen that (a) comes from low-carbon sources to achieve environmental benefits,1015 and (b) is available at low cost to be economically viable.1016 One important point, the end-of-life for CO2-based fuels and chemicals will be the eventual release of CO2 (Fig. 22). Thus, CO2-based fuels/chemicals are carbon-neutral in the best case (as discussed earlier).
The discussion in the previous paragraph focused on the amount of CO2 utilised which is often used as a proxy for the amount of CO2 avoided (e.g., Otto et al.987). However, the amount of CO2 avoided might differ significantly from the amount of CO2 utilised as the discussion of climate-change-mitigation mechanisms above has shown. In fact, CCU might even increase CO2 emissions, e.g., if hydrogen from fossil sources is used to produce fuels.999 Carbon dioxide re-use can also reduce CO2 emissions beyond the amount of CO2 used. Aresta et al.988 estimate based on stoichiometric analysis that the production of ethane carbonate could avoid up to 17.9 tons of CO2 emissions for every ton of CO2 used; using data from an industrial pilot-plant, it was shown that using 1 ton CO2 captured from a lignite-fired power plant to produce polyols reduces CO2 emissions by up to 2.98 tons.997 Thus, the benefits have to be evaluated on a case-by-case basis for each CCU technology, along with the required setting to achieve them.
10.2 Future perspective and key research needs
Carbon dioxide re-use will have a role in climate-change mitigation – but the size of this role is still unclear. A number of key issues need to be addressed to make best use of the potential benefits from implementing CCU. These issues are discussed in the following.The scale of CCU will mainly depend on the large-scale implementation of CO2-based fuels since the potential for CO2 re-use in fuels is about 12–14 times higher than for chemicals.988 CO2-Based fuels have been shown to be able to reduce GHG emissions over the life cycle.998,999,1015 Several pilot-plants are already in operation for CO2-based methane and methanol.992 The George Olah Renewable Methanol Plant in Iceland is producing 4000 tonne per year.1017 However, the large-scale implementation of CO2-based fuels still requires to overcome several challenges.
The production of potential fuels such as methane, methanol by Fischer–Tropsch synthesis still requires major progress in catalysis991 and the design of efficient processes.1018 CO2-Based methanation is expected to increase the cost of natural gas by a factor of 2.4 based on the most optimistic estimate for the year 2050 while current worst estimates predict a cost increase as large as a factor of 30.1019 For methanol, CO2-based production are estimated to increase the selling price by a factor of 1.8.1020 Thus, cost efficiency has to be addressed in all aspects; most importantly for the hydrogen source but also for the feedstock CO2 and for investment cost. It has been argued that CO2-based fuels could provide an important link to the energy sector by providing chemical energy storage as well as flexibility to the electrical grid.1021 Thus, dynamic operation of CO2-based fuel production may provide opportunities but will require the development of novel technologies. In this case, hydrogen storage might become a cost and design factor. For methanation, the strong exothermic reaction provides opportunities for heat integration.1019 For methanol production, highly selective and long-term stable catalysts are required as well as more efficient separation technologies for the methanol–water mixture. Novel process concepts are tailored for CO2-based fuels.1022,1023 Integrating CO2 capture with conversion could provide efficiency gains. Developing suitable reaction systems is therefore important, which allow the conversion of CO2 directly in aqueous amine solutions used for CO2 capture.1024 The integrated capture and conversion can increase efficiency. However, an increased efficiency is not guaranteed because the solvent needs to be separation from the final product instead of CO2 which might be more difficult.1025 At the research level, electrocatalytic processes are gaining more interest since they allow the targeted integration of renewable energy into the conversion steps.1026 However, even with much improved future electrocatalytic processes, the cost of renewable energy is expected to remain the main obstacle for implementation.1027
The production of CO2-based fuels usually requires the reduction of the oxidation state of carbon to 2+ or lower. For this purpose, high energy exchanges are needed by strong reducing agents such as hydrogen.1026 Even assuming a conversion process that achieves ideal performance at the thermodynamic minimum, the availability of hydrogen at low-carbon impacts and low cost often remains the crucial element for CO2-based fuels. Thus, efficient production of hydrogen with low environmental impacts will be the key enabler for large-scale CCU. Machhammer et al.1028 compared the cost and carbon footprint related to the operation of hydrogen production technologies. The identified Pareto-efficient technologies for hydrogen production are: water electrolysis using wind power (zero carbon footprint for operation; high cost), methane pyrolysis (medium carbon footprint; medium cost) and conventional methane steam reforming (high carbon footprint; low cost). Taking a full life-cycle assessment into account suggests that methane pyrolysis could even become more CO2 efficient than water electrolysis driven by wind power if the produced carbon is stored.1029 In this case, however, the demand for fossil resources is more than doubled. Hydrogen production by water electrolysis has only low emissions if renewable surplus electricity is available. Renewable energy, however, will still be limited in most places for the foreseeable future. Thus, overall process efficiency remains a key performance indicator since power-to-fuel technologies would be competing with other pathways to utilise renewable energy. Most competing Power-to-X technologies currently offer a higher CO2 abatement per kWh electricity used than CO2-based fuels.1015 CO2-Based fuels would thus have to generate additional value, e.g., from the functionality as fuel or by avoiding expansion of the electricity grid. Systems level analysis of whole value chains is required to identify promising scenarios for CCU.1030 Here, regionalised concepts could help to identify promising locations combining sources for CO2 and H2 with suitable sites for fuel production. Hereby, the amount of fuels needed in the future is itself an open question due to electrification of the transportation sector and of heating. In such an analysis, CO2-based fuels need to be benchmarked to other low-carbon fuels such as biofuels,1031 or nitrogen-based fuels.1032 In this context, the integrated design of biorefineries with CO2 utilisation could provide a promising avenue for efficient carbon use.1033
Carbon dioxide can also be re-used while keeping the 4+ oxidation state of carbon. These routes target the production of urea, polymers, and inorganic carbonates.988 The production of solid inorganic carbonates by CO2 mineralisation seems a particularly promising target for market-entry of CO2-based products. Mineral carbonation can generate construction materials by conversion of suitable silicates. These routes are favoured by thermodynamics and lead to stable products. Mineralisation even offers opportunities to convert wastes, e.g., steel slags, with CO2 to valuable construction materials.1034 Thereby, the start-up company Carbon8 is able to charge its suppliers $190 per ton of waste, e.g., from fly ash, since the suppliers would pay more for landfill.992 The challenges for mineral carbonisation to be addressed are energy use, slow reaction rates and material handling.994 CO2 storage in solid carbonates is expected to enhance public acceptance since “this method of storage is highly verifiable and unquestionably permanent”.809
CCU has the potential to play a role as renewable carbon feedstock for the chemical industry. While the chemical industry contributes only 4% to the global GHG emissions from fossil fuel combustion, the emissions are large compared to other industries.1035 Thus, after the energy sector, the chemical industry is certainly one of the next targets to reduce GHG emissions. For the chemical industry, CCU could avoid emissions of several million tons of CO2, while at the same time decreasing the dependence on fossil fuels as carbon source for chemical production. The replacement of a fossil-based product with CO2-based production has no impact on the overall GHG emissions unless global fossil fuel demand is actually reduced. System-wide consequential life-cycle assessment1036,1037 is required to capture these interdependencies and to identify future tipping points away from fossil-based production of chemicals. For this purpose, a large variety of potential chemical products are currently explored to be produced from CO2.984–987 We expect the first implementations for processes where CCU improves the production of established products. The CO2-based polyols are such an example: 5000 tonne per year are currently produced commercially in a first demonstration plant.992 A further opportunity results from the production of formic acid1038 which can be synthesised directly from CO2 and H2991,1039 while several process steps are required in the conventional production. Several companies are therefore working on the production of CO2-based formic acid.1040 Cost are currently still estimated to be 2.5 times higher than fossil-based production due to hydrogen production and current catalysts.1041 For the production of the antifungal agent butenafine, CCU allows the switch to cheaper reactants, avoids potentially hazardous reagents and wastes, and reduces the number of process steps to one.1042 The formylation of amines with CO2 leads to formamide products which are versatile chemicals and key building blocks.1043 Organic carbonates provide a wide range of potential products ranging from low molecular weight products such as dimethylcarbonate1044,1045 to cycle carbonates1046 and finally polymeric compounds.1047,1048 Beyond the more efficient production of current products, advanced methods for CO2 conversion could generate novel products enabling environmental benefits such as the novel OME-fuels mentioned above.1006–1008
The systematic identification of such opportunities would be desirable. Systematic design methodologies could help to identify promising targets.1018 In order to support research at early design stages, predictive model approaches need to be developed which would allow to enable the in silico assessment of the potential of novel pathways and products.1030 By employing the quantum-chemistry-based thermodynamic model COSMO-RS, Jens et al.1049 were able to screen more than 100000 combinations of flowsheet layouts, solvents and chemical storage molecules for the conversion of CO2 to CO. Incorporating such model-based knowledge into the chemical design process would accelerate the development of novel CCU technologies.
Life-cycle assessment (LCA) of prospective products could help to guide research needs and to provide performance targets.1045,1050 Importantly, LCA should not only be limited to impacts on climate change. Instead, a wide range of environmental impacts should be considered to avoid problem shifting to other impact categories such as resource depletion. These trade-offs need to be analysed even though some CO2-based products reduce all environmental impacts as shown, e.g., for the novel polyethercarbonate polyols.997 Still, these products are not carbon negative in general and even often not close to carbon neutral due to the need of highly energetic reagents such as hydrogen or epoxides in the case of polyols. Identifying sustainable pathways for the co-reagents is thus a key challenge.1051
In order for life-cycle assessment to take on a guiding role, a methodological consensus has to be reached for the application to CO2 re-use technologies.1001 Currently, a wide variety of methodological choices are applied making it difficult to compare results from different studies.989 The LCA community can learn from the case of bio-based materials how (difficult it is) to apply LCA to renewable carbon feedstock.1052
To provide a better database for environmental assessment but in particular also for knowledge gaps regarding accurate costing and data on process technologies, large scale projects are helpful demonstrating the industrial application of novel CCU pathways.
Such large-scale projects would also be beneficial to learn about public acceptance of CCU technologies.1053,1054 Integrating insight from public acceptance studies into the research and development process could become crucial for the future implementation of CCU.
A major role in the transition to low-carbon fuels will be played by politics. Since many pathways are not yet economically viable, incentivising low-carbon fuels would be required. Carbon tax benefits or CO2 certificates could be related to the re-use of CO2 as means of climate change mitigation. For this purpose, carbon accounting methods would need to be adapted to provide a benefit from re-using carbon dioxide from the atmosphere or from flue gases. For a production technique for precipitated calcium carbonate, the Court of Justice of the European Union has recently ruled that the CO2 is chemically bound in a stable product, thus the CO2 source does not have to account for the CO2 emissions under the emissions trading system (ETS).1055
In order to contribute its potential share to climate change mitigation, CO2 re-use has to survive the hype cycle where it might currently be approaching the peak of inflated expectations. Sound and unbiased assessment of the benefits and disadvantages of CCU technologies should help to identify the plateau of productivity.
11 Technology learning and associated cost reduction
11.1 The theoretical basis for learning curves
Computer models used for energy-related planning and policy analysis typically employ one of two methods to represent technological change: either the future cost and performance of technologies are exogenously specified by the modeller, or a mathematical model is used to relate the future cost and performance of energy-related technologies to other model parameters. The latter method includes the use of “learning curves” (or experience curves) to project the future cost of technologies.In 1936, Wright1056 observed that the average time required to manufacture a given model of a Boeing aircraft decreased systematically with each unit produced. Wright1056 captured this phenomenon with an equation representing what he called a “progress curve” given by:
| Y = axb | (15) |
Wright's work remained relatively obscure until a decade later, when it was picked up by a group of economists at the then recently founded RAND Corporation, who applied his findings to the production of war materials and described the phenomenon as “learning-by-doing”. Subsequent work by the Boston Consulting Group (1968)1057 applied Wright's equation to the relationship between the average unit price and cumulative production of two dozen selected industrial products. When applied in this fashion to a class of product (rather than to a specific manufacturing process), the “learning curve” equation became referred to as an “experience curve”.
More recently, this formulation (eqn (15)) has been adopted in empirical studies to characterise learning phenomena in a broad range of sectors, including manufacturing, ship production, consumer products, energy supply technologies, fuel technology, energy demand technologies, and environmental control technologies.1058 In these applications, the dependent variable Y is typically the unit price or cost of a technology and x is its cumulative production or installed capacity. Eqn (15) also can be re-written as:
| logY = c + blogx | (16) |
Today, this log-linear form of the learning curve remains the most popular equation used to represent the expected cost improvements of a technology. A characteristic parameter is the “learning rate”, defined as the fractional reduction in cost for each doubling of cumulative production or capacity, and is given by:
| LR = 1 − 2b | (17) |
“Component-based learning curves” extend the one-factor learning model to represent the total cost of a technology as the sum of individual component or sub-system costs. This formulation seeks to account for the fact that different components of a complex technology (like a power plant) may have different levels of maturity and different rates of learning. Thus:
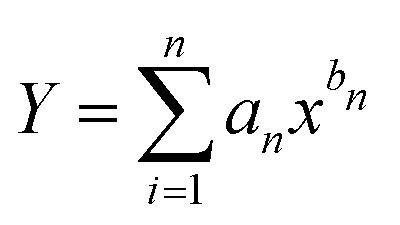 | (18) |
Research over the past few decades also has sought deeper insights into the underlying factors that contribute to cost reductions and technological change. One result is a variety of multi-factor learning models that have been developed to explicitly account for such factors such as R&D spending, knowledge spill-overs, increased capital investments, economies-of-scale, changes in input prices, labour costs, and other factors.1058 While such models provide more detailed descriptions of factors that affect a particular technology cost, they are not as prevalent as the one-factor model shown earlier, in large part because of data requirements and limitations.
For energy technologies, the most prevalent multi-factor model is a “two-factor learning curve” where the key drivers of cost reduction are assumed to be the cumulative expenditure for R&D on the technology, in addition to its cumulative installed capacity or production. In this formulation, eqn (17) is expanded to explicitly include the effect of cumulative R&D expenditures:
| logY = α + bLBD(logx) +bLBR(logR) | (19) |
An important caveat on learning curves is that the mathematical models outlined above may not correctly represent technology cost trends in all cases. Historical data show that for a variety of reasons the cost of a particular technology may increase with experience, especially in the early stages of deployment and adoption. Nor do cost reduction trajectories always follow a log-linear relationship.1058 Any use of learning curves for technology cost forecasting must take such uncertainties into account.
11.2 Learning rates for fossil fuel power plants
A recent literature review summarised the empirical learning rates reported for different types of electric power generation technologies.1060Table 9 summarises those results for combustion-based power plants fuelled by coal and natural gas. There is also considerable interest in the future cost of such plants equipped with CCS, as well as coal-based integrated gasification combined cycle (IGCC) plants with and without CCS. Since there is no significant historical basis from which to derive learning rates for these technologies, several studies have used the “bottom-up” component modelling approach outlined earlier to estimate the learning rates of future IGCC, PC and NGCC power plants with CCS based on analogous technologies. Table 9 shows the range of results from such studies.| Technology and energy source | No. of studies with one factor | No. of studies with two factors | One-factor modelsa | Years covered by studies | |
|---|---|---|---|---|---|
| Range of learning rates | Mean learning rate | ||||
| a Values in italics reflect model estimates, not empirical data. b No historical data for this technology. Learning rate values are estimated based on analogies. | |||||
| Coal | |||||
| PC | 4 | 0 | 5.6% to 12% | 8.3% | 1902–2006 |
| PC + CCSb | 2 | 0 | 1.1% to 9.9% | Projections | |
| IGCCb | 2 | 0 | 2.5% to 16% | Projections | |
| IGCC + CCSb | 2 | 0 | 2.5% to 20% | Projections | |
| Natural gas | |||||
| NGCC | 5 | 1 | −11% to 34% | 14% | 1980–1998 |
| Gas turbine | 11 | 0 | 10% to 22% | 15% | 1958–1990 |
| NGCC + CCSb | 1 | 0 | 2% to 7% | Projections | |
For CCS technologies, current commercial systems for post-combustion CO2 capture are often assumed to be technically analogous to post-combustion flue gas desulfurisation systems for SO2 capture, which had average historical learning rates of 12% for capital costs and 22% for O&M (operation and maintenance) costs, according to previous studies. Using a component-based learning curve (eqn (18)), Rubin et al.1059 derived composite (plant-level) learning rates from 1% to 4% for capital cost and from 2% to 5% for cost of electricity based on 100 GW of new plant capacity with CCS. Using a similar approach, Li et al.1061 projected learning rates of 5.7% to 9.9% for PC plants with CCS in China. For natural gas applications, van den Broek et al.1062 also used a component-based modelling approach to estimate future learning rates for NGCC plants with CCS. The resulting rates ranged from 2% to 7%, with a nominal value of 5%.
11.3 Implications for future CCS cost
The research on learning rates cited above suggests that the cost of CCS for power plant applications is expected to fall as such installations are more widely deployed. This is consistent with pronouncements from the Sask Power company in Canada and the NRG company in Texas, which operate the first two large-scale CCS projects at coal-fired power plants. Both companies project a roughly 20 percent cost reduction for a subsequent CCS installation based on the experience to date at the Boundary Dam and Petra Nova power plants, respectively.1063,1064Based on modelling studies that employ learning rates from Table 9, the magnitude of future cost reductions for power plants equipped with CCS will depend strongly on the nature and timing of policy drivers to achieve deep reductions in CO2 and other greenhouse gas emissions. One recent study projected reductions in 2050 of roughly 1% to 40% in the cost of electricity generation for power plants with CCS, and higher percentage reductions in the cost per ton of CO2 avoided.1062 However, such scenarios assumed increasing levels of a worldwide carbon price (tax) to incent markets for CCS technology. It remains to be seen whether strong policy drivers of this type will emerge to help drive significant reductions in future CCS cost.
12 Negative emissions technologies
12.1 Bioenergy with carbon capture and storage (BECCS)
Bioenergy with carbon capture and storage (BECCS) is a CO2 mitigation technology which combines bioenergy applications with carbon capture and storage. This concept is not consistently defined and can include a variety of industrial and energy technologies with different amounts of CO2 emissions, such as biomass combustion (dedicated or co-firing) for power production, biomass conversion to liquid and gaseous fuels, bio-refineries, pulp and paper production. Fig. 23 is a graphical representation of the BECCS concept (throughout the literature, the term Bio-CCS is also used as an alternative). In BECCS, CO2 originating from biomass, which has undergone a conversion process, is capture and stored in geological formations. CO2 utilisation concepts exist for BECCS as well (BECCUS or Bio-CCUS), where the CO2 is temporarily fixed in products, such as fuels, construction materials, chemicals, plastics etc. The concept of BECCS depends on the assumption that biomass binds CO2 from the atmosphere as it grows and, if captured and stored after conversion, results in a net removal of CO2 from the atmosphere. Technologies allowing for this net removal are referred to as negative emissions technologies (NETs), and include ocean fertilisation, mineral carbonation, afforestation and direct air capture (DAC) – discussed in Section 12.2. | ||
| Fig. 23 Concept of bioenergy with carbon capture and storage (BECCS), courtesy of Nature.1065 | ||
Sustainability, i.e., carbon neutrality, of the biomass feedstock is a decisive factor in terms of the extent of negative emissions a technology or other mitigation pathway can achieve.63 There are several factors that can make true carbon negativity difficult, e.g., emissions from land use change (LUC), production, pre-treatment and transport of biomass, conversion process and CCS process but also the issue of carbon debts, i.e. the amount of time required for carbon offsets to kick in. In comparison, CCS on fossil fuels (Fossil-CCS, i.e., Coal-CCS and Gas-CCS) is quite different, as it can at best lead to zero emissions. Fossil-CCS takes carbon from the geosphere and returns it there, while BECCS takes carbon from the atmosphere, puts it temporarily into the biosphere, and then permanently into the geosphere (assumed there is no major leakage from the storage reservoir). Without a CCS component, processes take carbon from either geosphere or biosphere and transfer it to the atmosphere, so can be at best carbon neutral (biomass resource) or will be net positive (fossil resources). Thus, BECCS can allow offsetting of emissions from sectors where CO2 reductions are hard to achieve due to technical, economic or political constraints (e.g., aviation, shipping, iron and steel). Many Fossil-CCS plants have the potential to become BECCS plants by switching their fuel feedstock, for example, a coal-fired power plant with CCS converted to co-fire biomass.
There are currently five operating BECCS projects worldwide that capture a total amount of 0.85 MtCO2 per year, compared to 16 CCS projects with a capacity of about 31 MtCO2 per year.4,1066 The Illinois Industrial CCS (IL-ICCS) project, capturing CO2 from Archer Daniels Midland's (ADM) corn ethanol plant in Decatur (Illinois, USA) and storing it in a sandstone formation, adds an additional 1 MtCO2 per year, when operation commenced April 2017.1067 The predecessor of the IL-ICCS project successfully captured and stored 1 MtCO2 per year over three years.1068–1070 Thus, the IL-CCS will be the largest BECCS demonstration. Other planned and existing BECCS projects are at significantly smaller scales. The currently operating projects are located in North America, where the main CO2 source is from ethanol fermentation plants and CO2 enhanced oil recovery (CO2-EOR) is the sink.1066,1071 Although the number of existing and planned projects appears promising, hundreds to thousands are needed1072 if BECCS is to make a significant contribution to global greenhouse gas (GHG) reduction targets. Necessary steps include the build-up of operational experience and confidence in the technology, as well as verification of the negative emissions potential.
The estimated technical potential for BECCS pathways, including a wide variety of applications in different sectors, varies from 3–20 GtCO2 per year.49,1066,1073–1080 This is significant compared to the global CO2 emissions of currently 36 GtCO2 per year.1081 The economic potential is usually only a fraction of this, as it considers the cost of resources, their competing use and the reference to fossil fuels.1074,1075 It is further limited by the land required to produce the required biomass in a sustainable way (refer to the designated section on land availability). The technical maturity and costs of BECCS are comparable to conventional Fossil-CCS technologies. Economic assessments published in the literature so far, have arrived at a ballpark range of 60–250 US$ per tCO2 for BECCS.6,1066,1076 Large-sale BECCS in power plants tends to be in the upper part of this range, whereas smaller niche applications, like ethanol fermentation, biomethane production and black liquor gasification, are on the lower end. Costs of BECCS are currently estimated to be about half the cost of DAC.1082 The important role that NETs play in future climate change mitigation becomes clear when one looks at integrated assessment models (IAMs). IAM studies predict that carbon prices are likely to be up to three times higher if key NETs (i.e. BECCS and DAC) are not available.1083 Due to the high uncertainties associated with future technology pathways and their cost development, it is currently not clear where cost effective BECCS deployment will take place.
To provide perspective on BECCS’ potential for atmospheric CO2 reduction, removing 0.5–1 ppm CO2 per year would require drawdown of 8–16 GtCO2 per year.49 Inefficiencies and losses along the supply chain play an important role as well. If considering all carbon losses along the chain for BECCS on a switchgrass gasification plant, the aim of storing 1 GtC, or 3.67 GtCO2, could require a fixation of up to 7.7 GtCO2.1084 There are several reasons for the uncertainty in BECCS’ potential and cost estimates, e.g., only considering CO2 but no other GHGs, omitting LUC emissions, insufficient carbon cycle models, lack of underlying data, differences in modelling assumptions, etc.1085
Biomass availability. Biomass broadly denotes material of biological origin that is derived from photosynthesis in a relatively short timeframe. Thus, it excludes material embedded in geological formations and material that is transformed into fossils or peat.1087 There are many different types of biomass feedstocks and they can be classified in many ways, e.g. terrestrial vs. marine, virgin vs. residues, agricultural vs. forest or dedicated vs. waste. One attempt of classification could look like the following,1088,1089 without claiming to be exhaustive:
(1) Dedicated energy crops
(a) Conventional annual crops
• Oil crops (palm, canola, sunflower, etc.)
• Sugar/starch crops (sugar cane, sugar beet, corn, all types of cereals, etc.)
(b) Perennial crops and energy grasses (Miscanthus, switchgrass, etc.)
(2) Forestry and forestry residues
(a) Short rotation forestry (alder, ash, Southern Beech, birch, eucalyptus, paper mulberry, Australian Blackwood, sycamore etc.)
(b) Short rotation coppice (willow, poplar, etc.)
(c) Forestry residues
• Primary (wood chips from branches/tips/poor quality stemwood etc.)
• Secondary (saw mill by-products: chips sawdust, bark etc.)
• Tertiary (material from municipal tree management, waste wood etc.)
(3) Other residues and wastes
(a) Agricultural crop residues (straw from cereals/oil seeds, bagasse etc.)
(b) Municipal organic waste (paper/cardboard, food, garden, textiles etc.)
(c) Sewage sludge
(d) Animal manure
(e) Land fill gas
(4) Marine biomass (microalgae/phytoplankton and macroalgae/seaweed)
Although the number of potential feedstocks appears large, competition between different sectors for feedstock and competition with other ecosystem services, such as food production, could significantly limit their availability for BECCS. Fig. 24 shows a tree diagram of different biomass conversion technologies and the variety of end products for each conversion pathway. Currently, there is a high amount of food waste available, especially in developed countries, i.e., 1.3 Gt per year globally.1090 However, this amount could change over time in the long term, through improvements in agricultural production, storage, processing, distribution and consumer behaviour. Another issue with regards to using some of the above-mentioned feedstocks for large-scale BECCS is their seasonal availability due to harvesting schedules, which makes pre-treatment and storage necessary. Future availability will further depend on improvement in yields, cultivation methods, and growth in demand. Key drivers for biomass feedstock markets and supply chains are crude oil price, climate policy, energy policy, cost of primary energy production, infrastructure and development potential of rural areas.1091–1093 Finally, BECCS and other bioenergy applications might experience a feedstock limitation to so-called “additional biomass”. This term refers to biomass that can lead to a reduction in GHG emissions without displacing other ecosystem services, such as food or fibre production. Additional biomass includes: biomass grown in excess of what would have been grown anyway, biomass that would otherwise decompose, wastes/residues, and other biomass that does not interfere with important ecosystem services, especially food production.1094,1095 Except for wastes/residues and marine biomass, feedstock availability is highly dependent on land availability, which will be discussed in detail in the next section.
 | ||
| Fig. 24 Biomass feedstock conversion pathways and product tree.1086 | ||
Land availability. Land availability for biomass feedstock production is a key driver for large-scale BECCS implementation. Land demand for BECCS is relatively high and largely depends on the selected feedstock. Forest residues need 1.0–1.7 ha per tC,eq per year (0.27–0.46 ha per tCO2,eq per year), agricultural residues around 0.6 ha per tC,eq per year (0.16 ha per tCO2,eq per year), and dedicated energy crops 0.1–0.4 ha per ttC,eq per year (0.03–0.11 ha per tCO2,eq per year).61 For comparison, other NETs, like enhanced weathering of minerals (EW) and DAC, have significantly lower land demands: <0.01 ha per tC,eq per year (0.003 ha per tCO2,eq per year).61,1096,1097
To achieve removal of 3.3 GtC,eq per year (12 GtCO2,eq per year) through deployment of BECCS with dedicated energy crops, 380–700 Mha of land is required,61 or 500 Mha, which is in terms of a bioenergy deployment of 100 EJ per year.1080 According to other sources, for a range of 26–161 EJ per year, 133–990 Mha of land is necessary.1065 For comparison, DAC and EW need substantially lower land areas (below 10 Mha), and afforestation and reforestation (AR) needs a slightly higher amount of land (about 970 Mha).61,1084,1097,1098
The total land area for agriculture in 2014 was 4900 Mha, of which 1,585 Mha was used for arable land and permanent crops, and 3,315 Mha accounted for permanent pasture and meadows. A further 4002 Mha were designated as forest. Of the agricultural cropland, about 300–570 Mha are marginal lands.1099–1101 The total current amount of marginal land is relatively uncertain, as it depends on a definition that is rather inconsistent across literature, it ranges between 428–1035 Mha.1065,1102,1103 Any land or biomass supply limitation will very likely affect the costs of BECCS. Although current cost estimates for BECCS are lower than for DAC, these costs could rise steeply once land limitations are considered (in this case once removal rates reach 12 GtCO2 per year).1104,1105
To meet land requirements for BECCS (i.e., estimated to be 380–990 Mha), two important questions should be addressed: (i) how this land can be provided, and (ii) how much can be freed through other means. As discussed above, marginal lands can be used to partially meet land requirements. Another option that can free significant amounts of land is through dietary changes. The current average diet in the US contains a high amount of animal products (meat, dairy, eggs, fish) and has a land intensity of about 1.08 ha per year per person (of which, 0.74 ha per year is for pasture and 0.34 ha per year for cropland). In contrast, a vegetarian diet requires 0.14 ha per year per person (0.02 ha per year for pasture and 0.12 ha per year for cropland) and a fully plant-based diet needs 0.13 ha per year per person.1128 Assuming the current world population of 7.5 billion, a full transition to a vegetarian or plant-based diet could free around 605–685 Mha of cropland and 3165–3315 Mha of pasture. However, the likelihood of human society undergoing such a drastic change in behaviour appears unlikely. In addition, it is important to keep in mind that free allocations between cropland and pastures are usually not possible, i.e., only a certain proportion of pastures will be suitable as cropland. Other, less drastic scenarios estimate that a 40% cut in consumption of animal products by the 2.2 billion people currently on a US-type diet could free 140 Mha of cropland and 500 Mha of pasture.1129 Further options to free land are crop yield/livestock productivity improvements or reduction of food waste, as the land area associated with food waste totals ∼1400 Mha, for crop and animal commodities combined.1090,1111,1112,1130 In conclusion, we could make enough land available for large-scale BECCS deployment, or bioenergy deployment in general, but only with far-reaching changes to our diets and agricultural systems.
| Area | Key R&D needs |
|---|---|
| Biomass and land | • Identify and implement use of sustainable/additional biomass feedstocks, e.g. crops that need less fertiliser, grow in low quality soil, wastes/residues, 2nd generation bioenergy crops, winter cover crops. The main aim here should be to avoid competition with other ecosystem services, especially food production.6,1094,1095,1106 |
| • Identify BECCS pathways with a combined minimal water, carbon, energy and land footprint, e.g. through careful selection of crops, location, cultivation methods, pre-treatment and conversion technologies.1107,1108 | |
| • Improvement of pre-treatment processes to biomass (such as densification, dehydration and pelletisation) to remove geographical limitations for biomass supply, increase transport efficiency, reduce fossil fuel input and address supply chain emissions.64 | |
| • Develop innovations in farming methods to increase crop yields and decrease LUC emissions.64,1109 | |
| • Assess and implement ways for freeing land, e.g. through crop yield increases, food waste reduction and other demand side changes.1090,1110–1112 | |
| Technical | • Investigation of less mature BECCS technologies, like biomass gasification. |
| • Assess how to deal with the high moisture content and specific impurities of biomass during combustion/conversion, as they can lead to issues such as corrosion, fouling and slagging.1113,1114 | |
| • Evaluate high shares of biomass co-firing, i.e. in excess of 20%, regarding their implications for biomass pre-treatment and boiler modifications.1115 | |
| • Modify IAMs to adequately reflect the technical and economic potential of BECCS.1083 | |
| • Develop supply chains for sustainable biomass.1092,1093,1116,1117 | |
| Economic | • Design new financial mechanisms and incentives, apart from the Clean Development Mechanism (CDM), that acknowledge/reward negative emissions from BECCS.1106,1118,1119 |
| • Further investigate and validate a potential effect of overshoot scenarios on mitigation costs, i.e. in terms of a discount opportunity.1079,1120 | |
| • Quantify expected economies of scale for BECCS.809 | |
| • Identify the lowest cost BECCS pathways for every concerning sector.1121 | |
| • Clarify direction and timings of financial BECCS projects returns.1122 | |
| Policy | • Improve land management, forestry and monitoring systems, so they can properly account for LUC and related emissions.1118,1123 |
| • Task concerted efforts and research across all involved sectors to tackle biomass sustainability issues.1085,1124 | |
| Public perception | • Further research the public perception of BECCS in particular to understand how and to what extent perception of Fossil-CCS and stand-alone bioenergy applications influence this.42,1125,1126 |
| • Form a stronger collaboration between stakeholders of CCS, bioenergy and BECCS sectors.42,1125,1127 |
12.2 Direct air capture of CO2
Direct air capture (DAC) has gained a lot of interest mostly in popular media,1131–1134 because it appears to be an easy fix to our current climate crisis. The concept of placing DAC plants anywhere to remove CO2 from the air provides the mental picture of our atmosphere one day having a CO2 concentration as low as it was prior to the industrial revolution. However, this approach has many technical and economic caveats, primarily associated with the highly dilute nature of atmospheric CO2, 400 ppm, a factor of 100–300 times more dilute than the CO2 concentration in gas- and coal-fired power plants. In this section, we will summarise the technology, economics, and system considerations with an aim to objectively assess the state of DAC today.In order for DAC to result in a negative CO2 emissions scenario, it would have to be coupled to CO2 transport and sequestration infrastructure in order to ensure a positive climate impact. Several companies have emerged with small-scale applications,1135 but their impact on significant and permanent atmospheric CO2 reductions is minimal owing to their present scale.
There are a range of views regarding DAC as a realistic option for climate change mitigation. In particular, two reports have been published by the American Physical Society1097 and the National Academy of Sciences1136 that discuss the challenges associated with implementing DAC at a scale capable of impacting climate. To the authors’ knowledge, there have only been two studies that have proposed specific designs for DAC systems with estimated costs. In the work of Holmes and Keith,1137 an air–liquid contactor design based upon cooling tower technology was proposed, while in the work of Mazzotti et al.,1138 a more conventional contactor was proposed, which may be more suited for flue gas applications. Both designs are unique and the costs for CO2 capture range between $300 and $600 per tonne of CO2. House et al.67 demonstrated the relationship between CO2 concentration and the energy efficiency of a given separation process and determined that the more dilute a system is, the more unwanted material there is to be processed, leading directly to higher costs with an estimate on the order of $1000 per tCO2. Although there is general consensus in the community that DAC is significantly more expensive than conventional carbon capture from coal or natural gas-fired power plants, it is described by some as “insurance” against potential CO2 leakage from geologic storage sites or as a means to offset emissions from dispersed sources such as automobiles, ships, and airplanes.1139
| JCO2 = ciklE | (20) |
 | ||
| Fig. 25 Interfacial concentration of CO2, ci, based upon Henry's law for DAC (left) and the flue gas of natural gas and coal (right). | ||
Hence, to force the same amount of CO2 across the interface for DAC as one would have for the more concentrated cases, E may be up to two orders of magnitude higher to compensate since kl is a parameter solely influenced by the solvent and its relationship to the packing material. Increased enhancement can be achieved by choosing strong bases with fast kinetics, such as sodium hydroxide (NaOH)1141 or combinations of piperazine (PZ) with potassium carbonate (K2CO3).1142 However, the trade-off with having to choose a strong base is the increase in energy required for regeneration.
Due to the increased binding between CO2 and a strong base, a chemical shift process is required for regeneration rather than a simple thermal- or pressure-swing process. For instance, in the case of NaOH, sodium carbonate (NaCO3) is formed and in order to regenerate NaOH and to produce a near-pure stream of CO2, one can either react NaCO3 with lime (Ca(OH2))1143 or with TiO2, with the second process (titanate cycle) potentially being less energy-intensive.1144 In both reactions, NaOH is regenerated, but a final thermal decomposition step (calcination in the case of calcium carbonate) is also required for producing CO2.
As expected, similar to absorption separation processes, the low driving force inherent in DAC systems also affects CO2 separation using solid sorbents. Fig. 26 shows the relationship between pore size and CO2 concentration in the pore. Due to the low concentration of CO2 in air, no matter how small the pore is, CO2 will never saturate the pore. On the other hand, in the case of CO2 separation from the exhaust of a coal-fired power plant, which is 300 times more concentrated than air in CO2, saturation of CO2 takes place in the micropores and smaller mesopores. This state of saturation is an added driving force that can only take place for applications in which CO2 is sufficiently concentrated in the gas phase.
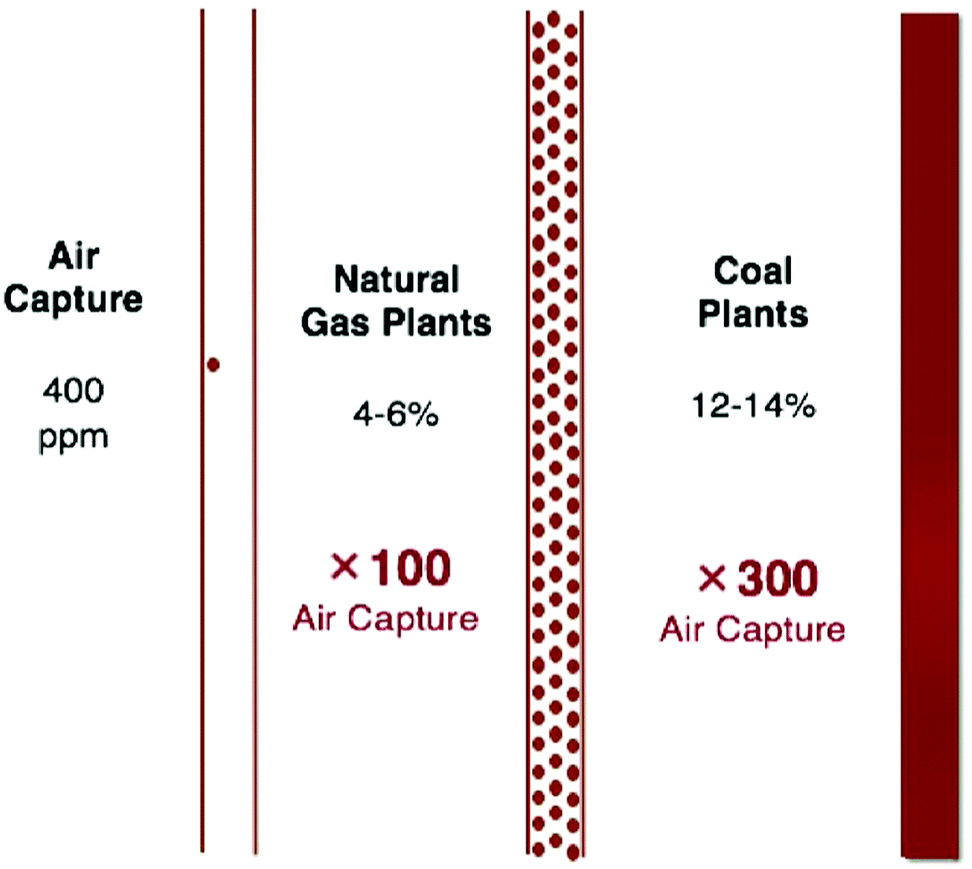 | ||
| Fig. 26 In the case of adsorption, the optimal pore size depends upon the dilution of CO2 in the gas mixture. | ||
An alternative way to show that energy increases with decreasing concentration is through examining the minimum work of a given separation process. The relationship between initial CO2 dilution and the energy required for purification can be shown by estimating the minimum work as a function of initial concentration, capture rate, and final purity. As shown in Fig. 27, the minimum work associated with separating CO2 from air is approximately 2, 3, and 5 times more energy-intensive than separating CO2 from the exhausts of natural gas combustion, coal combustion, and coal gasification, respectively.308 It is also important to note that a reduction in the capture rate combined with a reduction in CO2 purity will decrease the minimum work, but not significantly. Also, it is important to recognise that by decreasing the CO2 purity, there will be an additional expense associated with compression for transport.
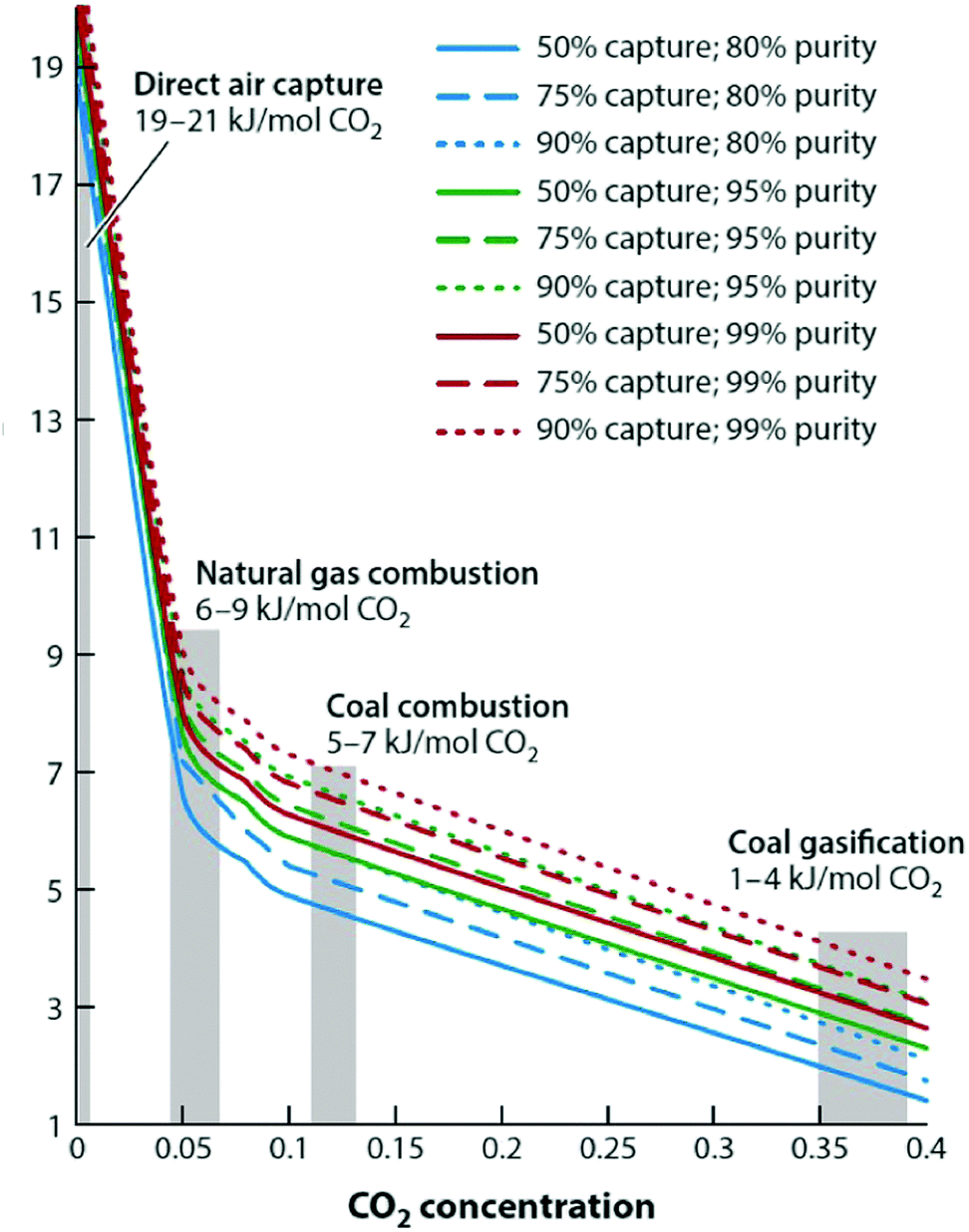 | ||
| Fig. 27 Minimum work required for CO2 capture based upon initial CO2 concentration, capture rate, and final CO2 purity.308 | ||
Another technical challenge associated with DAC is the amount of air that needs to be processed to capture a comparable amount of CO2 compared to a power plant. Assuming a capture rate of 50%, one is reducing a volume of gas from 400 ppm down to 200 ppm. In this case a tall contactor is not required, which is different than conventional carbon capture, where a common target is a capture rate of 90% from an exhaust stream with CO2 concentration ranging from approximately 6% to 12%. The degree of CO2 separation increases with column height, as does the pressure drop. The unique “short” design of a DAC plant is a consequence of the low degree of CO2 separation combined with minimising the energy required to overcome the pressure drop. However, there is also a lower limit to the pressure drop that should be avoided. Pressure drop allows for one to control how the flow of the solvent distributes across the system. Inadequate coating of the solvent across the packing material can impact the extent of mass transfer across the gas–liquid interface.
The contactor cross-sectional area can be estimated based upon the amount of air to be processed. For a given DAC plant to capture 1 MtCO2 per year at a 50% capture rate requires processing approximately 80000 m3 s−1 of air. Using a typical air velocity ranging between 2–3 m s−1 leads to surface areas on the order of 30000 m2. This is about 600 times the cross-sectional area of a large packed tower used for CO2 capture from a power plant flue gas. This large surface area requirement for DAC systems may well dominate the capital cost of the plant design. Overcoming the pressure drop across each of these units will require fan power. In conventional carbon capture systems for coal or natural gas exhaust streams, fan or blower power may only comprise up to 3% of the total energy of the separation process,1145 but may in fact dominate in the case of a DAC plant. An added expense is the need for an air filtration system. Due to the complexity of placing filtration units on each of the contactors, likely a central air handling unit would have to be in place prior to air distribution across the contactors.
There is strong evidence that the cost of CO2 capture rises with increasing initial dilution.121 The CO2 used for commercial markets is from high purity sources such as ammonia plants, ethanol plants and hydrogen production. The reason for this is that starting with high purity sources results in the lowest production costs. This relationship was quantified in an empirical correlation called the Sherwood Plot (see Fig. 28). Reasons for increased cost at lower dilution include smaller driving forces for mass transfer and greater amounts of material to process (see pressure drop discussion below).
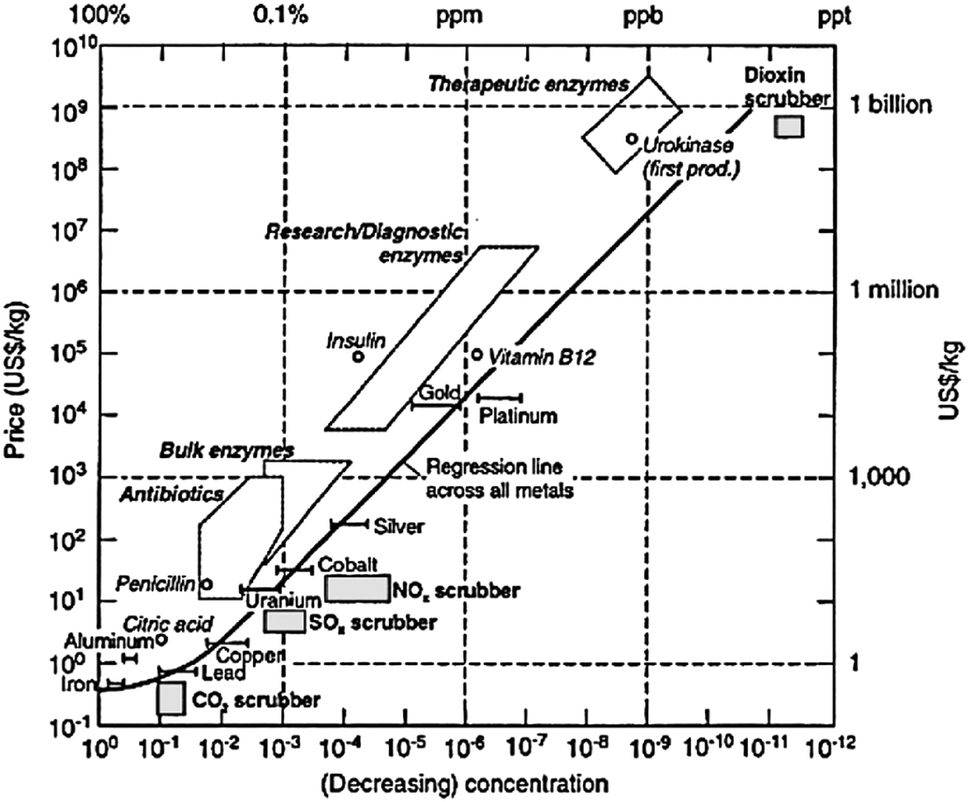 | ||
| Fig. 28 Sherwood plot exhibiting the relationship between the concentration of a target material in a feed stream versus the cost of its removal.67 | ||
Today, the cost of capture from a coal-fired power plant is on the order of $100 per tCO2 avoided.1146 If we knew the scaling factor, we could approximate the cost of DAC. The Sherwood Plot suggests a scaling factor on the order of 100, i.e., the ratio of the concentration of CO2 in the flue gas to the concentration in air. This results in a cost for DAC of $10000 per tCO2 avoided. Some proponents of DAC claim that the scaling factor should be based on minimum work, resulting in a cost of about $300 per tCO2 avoided. The truth probably lies somewhere between these numbers. In any case, the reported costs toward the lower end of the range in the literature (i.e., $20 per tCO2) just do not seem credible.
To illustrate the difficulty in estimating the cost of DAC systems, we calculate the amount of energy required to move air through the process. Assuming a concentration of CO2 in air of 400 ppm and a 50% recovery rate, we need to process 2.11 million m3 of air to capture 830 kg of CO2, approximately the amount of CO2 produced for every MWh generated at a supercritical coal-fired power plant. Based on air being an ideal gas and assuming no losses in the process, if we had a pressure drop of 0.016 bar (0.23 psi), we would need 1 MWh to move the air. In other words, at this pressure drop, just moving the air would require all the energy released in generating the CO2 in the first place. This means for a DAC process to be at all practical, pressure drops need to be limited to the order of 6.89 × 10−4 bar (0.01 psi) and/or that the energy source for DAC needs to be carbon-free. A pressure drop of 6.89 × 10−4 bar (0.01 psi) is extremely small and it is unclear whether it can be realised in a full-scale DAC system. A better use for carbon-free power today would be to replace fossil-fuel fired power and avoid putting the CO2 into the air in the first place.
The proponents of DAC make the case that there is a significant advantage to be able to theoretically site a DAC facility anywhere. Specifically, they suggest that it can be located near a storage site (reducing the pipeline cost), or away from populated areas. Siting is a multi-faceted, complex decision. While you may save some money building near a storage site, if there is no industrial infrastructure around, other costs will go up significantly. It is not at all clear that the fact that the feedstock for DAC (i.e., air) is found everywhere translates into any real economic advantage.
Proponents also suggest placement of a DAC plant nearby an EOR site may seem desirable. However, there are usually an abundance of CO2 sources that are more concentrated nearby EOR operations that can produce CO2 for much less cost than DAC.
Finally, for DAC to be at all practicable, the systems will need to operate at high capacity factors. Almost no work has been done on long-term operation of these systems. There are trace impurities in the air and since such a large amount of air is processed, they can have an adverse impact on DAC systems. Also, these systems must be able to stand up to the elements. Depending on where they are located, this includes water, wind, cold, and sandstorms. So far, the DAC literature is silent on these issues.
13 Commercialisation of CCS: what needs to happen?
This section is intended to provide insights into the challenges facing the development of a viable CCS industry highlighting new approaches and commercial models that could be deployed to realise the full potential of CCS in decarbonising future energy systems at lowest cost. Although based upon experiences from the recent UK CCS Commercialisation Programme and written mostly from a UK perspective with UK solutions in mind, the lessons learnt and proposed approaches can be applied globally.13.1 Current status
Since the late 1990s, a number of flagship government backed programmes have been set up around the world with the specific intent of demonstrating the commercial viability of carbon capture and storage (CCS) as an effective and affordable way to decarbonise power generation and other energy intensive industries (EII). Many of these programmes have featured financial support to off-set the costs of CCS as a means to encourage the private sector to invest in the development and deployment of CCS technology. Despite the ambition of these programmes and the scale of the support offered, progress has been minimal. To date, there are two “commercial scale” CCS projects in the power generation sector that are operational, the first is the Boundary Dam project in Canada at 110 MWe net output,188 second is the Petra Nova W.A. Parish CCS project designed to treat a 240 MW flue gas slipstream from a 610 MWnet coal-fired unit.14,15 The European Union's ambition for up to 12 CCS projects in operation by 20151148 supported firstly through the European Economic Programme for Recovery (EEPR) and latterly through the New Entrants Reserve (NER300) programme has failed to deliver a single CCS project. More success has been enjoyed in the United States through various programmes supported by the US Department of Energy. The Petra Nova W.A. Parish project commenced operation as planned in January 2017.14 However, Southern Company's Kemper County IGCC project (lignite power generation at 582 MWe net output) has encountered a number of problems with delivery delays, major technical issues and being significantly over-budget.21,22 Consequently, the clean coal component of the project has been suspended.20 In the United Kingdom two competitive CCS procurement programmes for power generation have been run by the UK Government since 2007 with both having being abandoned without success.The need for CCS as a key part of global strategies to reduce CO2 emissions may be great3 but so far this need has not been framed in a way that is attractive or rational for the private sector to respond to with investments in CCS projects. The physical and commercial risks associated with the development of large scale CCS projects and the associated CO2 transport and storage (T&S) infrastructure have so far outweighed the potential rewards on offer, as evidenced by the abandonment of many tens of promising CCS projects around the world.
With the failure of the various government-backed programmes to establish a viable CCS industry and in the absence of any private sector companies willing to expose their balance sheets to full chain CCS projects,1149 the question arises: what needs to happen to make CCS a commercial reality? The need for CCS is becoming ever more acute and new approaches to its commercial deployment are needed as a matter of urgency if we wish to meet our carbon targets in the most cost-effective manner.
One of the key attributes of CCS is that it can be applied to all main carbon emitting sectors and is therefore ideally suited to system-wide decarbonisation efforts. A key focus in the early stages of deployment will need to be on the development of CCS infrastructure to which multiple CO2 sources can connect so as to take advantage of economies of scale and to optimise the development pathway. In the UK regulatory and financial frameworks are already in place for low-carbon power which can be modified to fit CCS. This, together with the large volumes of CO2 available to support large scale CCS infrastructure development, makes the power generation sector, in an increasingly electrical future, the logical first mover sector for CCS.
13.2 The value of CCS
The value of CCS derives from the fact that it is the only technology that can simultaneously address carbon reduction objectives across all main carbon emitting sectors of the economy, without compromising their cost-effective provision of service. These sectors include power generation, industry, transport and heating.§§§§§For many industrial applications there is currently no alternative to CCS for reducing the CO2 emissions that are inherent to the manufacturing process. The decarbonisation of transport, including road transport, will inevitably involve increases in the numbers of electric vehicles. The resulting demand for electricity can be in part supplied from CCS enabled power stations. CCS in combination with hydrogen production could provide the low-cost route to the decarbonisation of heating as well as support the development of other aspects of the hydrogen economy including the use of fuel cells. CCS is also the only technology that can remove industrial quantities of CO2 from the atmosphere when combined with power generation from sustainable biomass combustion (so-called BECCS) creating room within carbon budgets for sectors more difficult to decarbonise, such as aviation. Indeed, in the UK, without CCS it is unlikely that the country's 4th and 5th carbon budgets can be met.85
The development of CCS, like all low-carbon technologies, will bring with it some additional costs. In a report prepared by the CCSA together with the TUC1150 however, it was estimated that the Gross Value Added¶¶¶¶¶ (GVA) benefits from CCS deployment in the UK would be in the region of £2bn–£4bn per year by 2030, with a cumulative market value of £15bn–£35bn (depending on whether 10 GW or 20 GW of CCS capacity is installed respectively). This is in addition to the creation of between 15000 and 30000 jobs.
If CCS is to form a key part of decarbonisation strategies it is important that the benefits of CCS across the economy at the total energy-system level are understood and that the long-term value-for-money case forms a central consideration in developing energy policy.
13.3 The cost of CCS
One of the most frequently expressed concerns regarding CCS is that it is too expensive. Indeed one of the primary reasons given for the discontinuation of the UK CCS competition was the view that the costs to consumers of the first CCS projects would be high and regressive1151 although it was acknowledged that the cost was likely to be higher for the first CCS projects as they provide transport and storage (T&S) infrastructure that could be used by subsequent projects. In the short term the cost of CCS for power generation will continue to be compared to alternative forms of low-carbon power generation even though those alternatives (and their intermittent output) will lead to higher system-wide costs in the long run.1152 In facing up to this challenge any new approaches to CCS commercialisation will need to deliver significant short-term reductions in the costs of first mover projects.The driving forces for cost reduction have been set out in the CCS cost reduction task force (CRTF) report1153 published in 2013 as part of the UK government's CCS roadmap,1154 including:
(1) investment in large CO2 storage hubs, supplying multiple CO2 sites connected through large, shared pipelines, with high load factors;
(2) investment in large power stations with progressive improvements in CO2 capture capability that should be available as from the early 2020s;
(3) a reduction in the cost of project capital through a set of measures to reduce risk and improve investor confidence in UK CCS projects; and
(4) exploiting potential synergies with CO2-based EOR in some Central North Sea oil fields.
All of these drivers are as relevant today as they were when the CRTF report was issued in 2013. Based upon technology progress in the intervening years and by applying the lessons learnt from the UK CCS Competition, significant reductions in the cost of CCS first mover projects are achievable. Success will depend on the development of large scale anchor projects that invest simultaneously in over-sized T&S infrastructure with third party access rights for follow on projects.
In addition, new commercial approaches will be required that balance multiple key risks (Fig. 29) and see a transfer of some of the CCS specific development and operational risk from the private sector to the public sector beyond that previously envisaged.1155
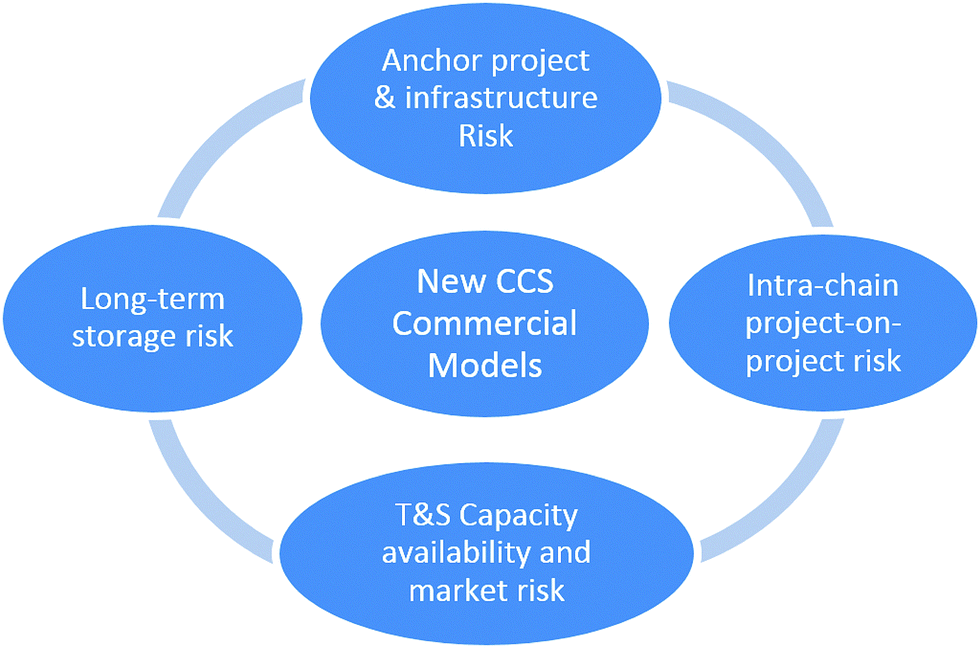 | ||
| Fig. 29 New commercial models need to balance multiple key risks. | ||
The CRTF predicted that the costs for CCS in the UK would be around £161 per MWh for the first mover projects and could approach £100 per MWh by the early 2020s, and achieve a cost significantly below £100 per MWh soon thereafter. The CRTF report was produced as part of the UK CCS roadmap and reflected the expected trajectory of cost reductions as experience and economies of scale grew against reducing capital and operating costs (discussed in Section 11). The UK CCS Commercialisation Programme, itself an integral part of the CCS roadmap, was aimed at attracting developers of first mover projects to invest in full chain CCS projects through a competitive process and offered a package of support in the form of capital grant funding, market price support through a contract for difference (CfD) and a share in the CCS specific risks.
The CRTF predictions for the first mover projects were largely borne out by the subsequent competition projects with the high prices largely a reflection of the adopted approach to risk allocation which crucially placed the full chain technical and commercial integration risk as well as significant CO2 storage risk with the private sector developers and operators. By adopting new commercialisation approaches that focus on the identified cost reduction drivers and include a modified risk approach that accommodates the lessons learnt from the competition projects, much of the cost reduction potential envisaged by the CRTF for subsequent projects could already be realised for the first mover projects albeit with a transfer of risk to the public sector. This would bring the cost of CCS to levels that are competitive with alternative forms of low-carbon power making CCS more affordable from the outset.
13.4 New approach to CCS commercialisation
Based on the UK lessons learnt1149 and the Key Knowledge Deliverables,1156 the CCS specific key risks that present the greatest challenges and could most benefit from additional public sector risk support to overcome barriers to CCS development and drive down costs through reduced risk premiums include:
(i) Cross chain default (also referred to as “project on project”) risk;
(ii) Post decommissioning CO2 storage risk;
(iii) Sub-surface CO2 storage performance risks impacting on storage rates and capacity;
(iv) Decommissioning cost sufficiency and financial securities related to the CO2 storage permit;
(v) Insurance market limitations for CO2 T&S operations.
Risk (i) applies to all individual chain link elements, whereas risks (ii) to (v) apply almost exclusively to the CO2 storage aspects. Risks (i) and (ii) would in all likelihood need to be absorbed by the public sector potentially for the lifetime of a specific CO2 T&S system, whereas risks (iii), (iv) and (v) may be time limited and transferrable back to the private sector as practical experience is gained and operating confidence increases. By introducing commercial models that entail a transfer of these risk categories to the public sector, not only can barriers be removed that have thus far prevented the private sector from investing in CCS, but also project financeability would increase and the risk premium added to the cost of capital funding would be significantly reduced.
With private sector confidence in the deliverability of CCS being at a low ebb presently, together with the current lack of appetite to invest in the development of storage capacity where all of the risks (i) to (v) apply, there is a strong argument for the public sector to take direct responsibility for the realisation of the T&S infrastructure. The creation of a publicly owned national transport and storage company (NT&SCo) to provide secure long term CO2 storage capacity, as recently recommended in a report by the parliamentary advisory group on CCS, chaired by Lord Oxburgh,101 would provide much needed certainty and boost confidence in the deliverability of CCS. Such a company would provide a strong counterparty and a significantly de-risked T&S infrastructure to potential private sector developers of generation and capture (G&C) assets. The use of public sector financing for the T&S assets would also bring benefits by lowering the overall cost of finance and as a consequence the cost of transporting and storing the CO2.
The Oxburgh report goes a step further and also considers public ownership of the G&C assets with a view to privatisation after a period of successful operation. Private sector investment at a later stage would still require sufficient financial shielding from shortfalls in the availability of the T&S infrastructure (cross chain default risk). This could be achieved through permitted unabated operation with assured revenue stream mechanisms for example through continuation of CfD payments or through switching to capacity market payments. Combining both CfD and capacity payment mechanisms for a single generator would however require amendments to current regulation. The private sector investor would also need to be shielded from liabilities associated with continued payment of T&S capacity reservation and use-of-system fees should the G&C assets suffer prolonged outages for example through contracting for capacity on a pay as you use basis with limitations of liability for non take-up.
Whether or not it is necessary for the public sector to take responsibility for the delivery of the G&C assets rather than the private sector will depend upon confidence in the deliverability of CCS in the UK and the degree to which CCS specific key risks are transferred to the public sector, whether that occurs at the outset or at a later stage following initial operations. Whichever route is followed it will be important to leverage the skills and competences of the private sector that has established a good track record in the delivery of power generation assets since privatisation of the electricity markets in 1990.
Though there are many ways to structure the commercial arrangements between the various stakeholders in a CCS network including direct public sector engagement, regulated asset based models, etc. Success will depend upon the appropriate balance of risk between the private sector and the public sector taking into account the listed CCS specific key risks. It will also be important that models form a robust template for the long term development of the CCS industry that is most likely to develop along the lines of clusters of users alongside CO2 T&S services providers with a clear transfer of liability for the CO2 to the T&S service provider, potentially a NT&SCo in the UK, at the factory boundary (Fig. 30).
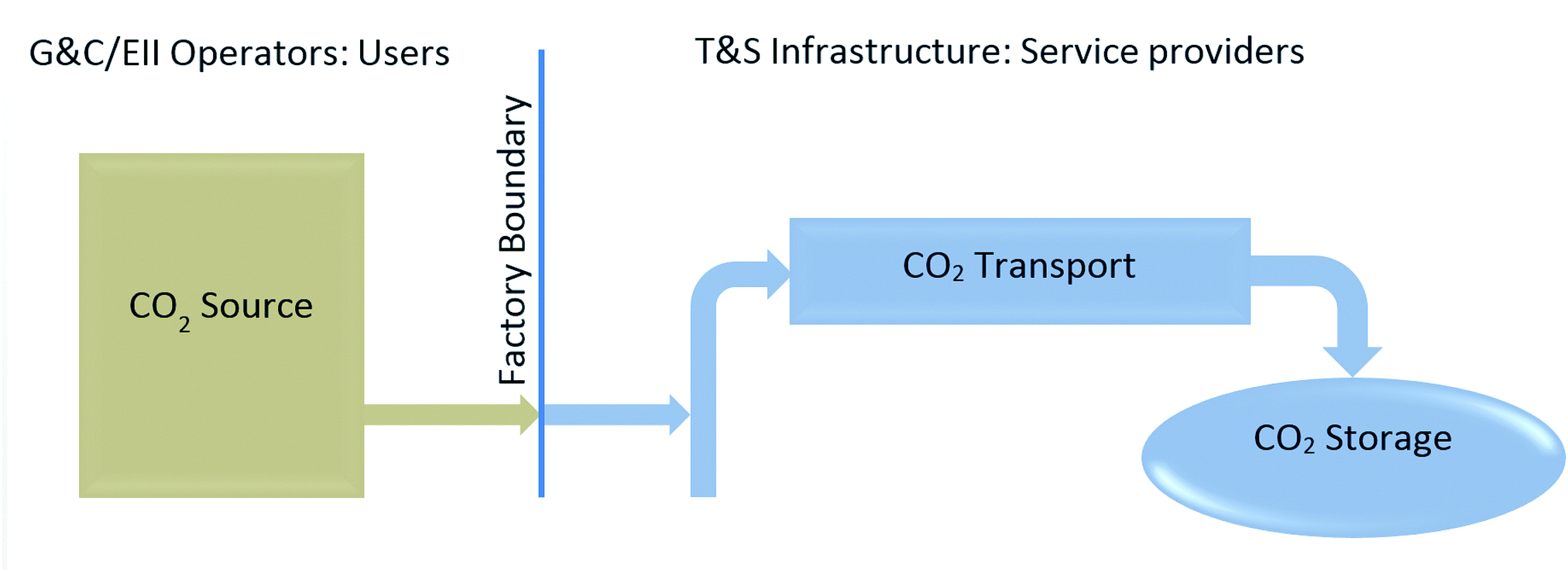 | ||
| Fig. 30 Industry market development, users and service providers. | ||
The use of Enhanced Oil Recovery (EOR) to boost production levels in the mature fields of the North Sea holds out the prospect that CO2 will, at some point in the future, command a material financial value potentially increasing the rewards available for the storage element of CCS. There is currently however no indication that these rewards would be sufficient for an EOR operator to underpin the associated development risks of the upstream elements of the CCS chain. It is more likely that EOR will develop in the North Sea once CCS is established in the power generation sector using sub-surface geological storage sinks, and hence only after reliable and predictable flows of CO2 become available off-shore.1157
To maximise the future benefit of the established T&S infrastructure, the anchor project should be sited in a CO2-intense industrial cluster. In the UK, there are several such clusters, located mostly along the east coast. This would also reduce transportation distances to the vast potential for CO2 storage sites in the Central and Southern North Sea. Keeping pipelines short and avoiding overland pipelines as far as possible will help to keep costs down and avoid protracted, complex and costly easement negotiations with a number of landowners.
13.5 Funding of CCS
The funding of CCS requires that a predictable and secure revenue stream is available to cover the costs of CCS and allow the developer to meet all of its financial needs. This will invariably require non-market derived sources of income and/or beneficial tax incentives for the generation of low-carbon power with CCS and the long-term storage of CO2.In the UK, power generation is currently the only sector for which existing regulation and financial frameworks are in place to support low-carbon technology through market price support mechanisms established through the electricity market reform (EMR) and as enshrined in the 2013 Energy Act.81 CCS is recognised as a low-carbon technology and as such qualifies for financial support through the CfD mechanism. Minimising the need for legislative adoption is an important factor in facilitating CCS rollout.
The development of a CCS project can take several years with costs running into several tens of millions of pounds. It is crucial therefore that the CfD allocation process provides developers with a high degree of certainty that a fully funded CfD will be available at the right strike price once they are ready to take a final investment decision on their projects. Even with such certainty however, a degree of public sector compensation of CCS project development costs is likely to be needed to mitigate to some degree the perceived political risk in such development programmes.
Much of the system-wide value of CCS derives from its ability to operate as flexible generation capacity alongside base load technologies like new nuclear and intermittent renewables. The CfD however as currently designed encourages base load operation as the marginal costs of production can always be covered. If the full value of CCS is to be realised mechanisms should be developed that reward flexibility.
The term of the CfD for CCS projects is set at 15 years in the generic CfD contract. By increasing the term to 20 years, significant reductions in the strike price can be achieved. Other design aspects that warrant further development include valuation and reward for negative emissions (BECCS) and application or alternative mechanism for industrial EII projects for which there is currently no CfD equivalent.
Alternative funding mechanisms across the full chain could be considered that would eliminate this bias. Currently, an unabated fossil fuel power generator can emit CO2 to atmosphere for a relatively low cost|||||||||| and, along with its customers, be forever freed from any future liability for the CO2 from the moment it leaves the stack. The CCS-enabled generator carries the cost of development of T&S infrastructure for its own and future users’ needs and has long-term liability for the safe and secure storage of the CO2 captured. Under the principle of the polluter pays consideration should be given to spreading the costs of the T&S infrastructure over all fossil fuelled power generators and potentially other CO2 emitters either through a hypothecation of carbon floor price levies, a carbon tax, or a form of CCS obligation certificate similar to the renewables obligation certificate first introduced in 2002 that was instrumental in supporting the early deployment of renewable technology in the UK.99
Such an alternative approach to funding of the T&S infrastructure would significantly reduce the strike price required by the CCS enabled generator to a level more competitive with alternative forms of low-carbon generation. It would also ensure that the value that CCS brings at the total energy system level in terms of decarbonising the economy is paid for more broadly across society and provide the economic drivers for further decarbonisation technology development using tax (or similar levies) as a behaviour modifier.
Grant funding. Grants provided by government as a means of promoting CCS projects bring many benefits. In addition to reducing the financial commitment from the private sector for CCS projects, it also demonstrates government CCS delivery commitment to developers, suppliers and financiers, etc. There remains however the question of how best to deploy grant funding, with most programmes providing grant funding to the developer of a single full chain project. Providing the grant in this way does not change the risk profile of the project, but serves only to reduce the developers’ financial exposure to full chain risks regardless of their nature including many business as usual risks. For future programmes it is worth considering targeting any grant funding to those risks in the full chain where there is a lack of market appetite particularly relating to the storage element. Deploying grant funding in this way for a multi-user store, without the requirement for a return on investment built in to the T&S capacity reservation and use-of-system fees, would provide several G&C and EII developers with low-cost CO2 T&S services representing a far better outcome for the public funding deployed.
Loan guarantees. Many private sector developers of a G&C asset including independent power producers (IPPs) are likely to look to limited or non-recourse debt finance structures (project finance) as the preferred approach to capital formation. The providers of project finance will in turn evaluate the credit worthiness of a CCS enabled power generation project on its stand-alone merits i.e. the ability of the project to meet its debt service obligations even when operating under certain adverse physical or economic conditions. The revenue certainty provided by the CfD mechanism, contracted through the low carbon contracts company (LCCC), is very attractive from a project finance perspective. However, to reach an investment grade rating in order to secure such finance, the investor group will need financial shielding from the risks associated with the transport and storage of CO2, as already discussed.
As additional support the availability of government backed loan guarantees for example through the UK Guarantees Scheme (UKGS) would help to increase the credit rating of a G&C project in turn reducing the cost of financing. The combination of the CfD, cross chain default risk support and loan guarantees could increase the credit rating of a G&C project sufficiently to open up the possibility of long-term funding from institutional investors and/or the debt capital markets further reducing the cost of capital.
13.6 Outlook for the commercialisation of CCS
To date, efforts around the world to develop a commercially viable CCS industry have largely failed despite the levels of government intervention and support that have been considered. If this trend is to be reversed the lessons of the past need to be learned and new approaches developed.The private sector is very unlikely to deliver fully integrated CCS infrastructure and projects without increased public sector support and clear government policy that supports CCS. It is imperative therefore for governments to take firm decisions on whether or not CCS technology will form a key part of their long-term low-carbon future energy strategy.
Where the case for CCS is made, a clear and stable CCS energy policy with a comprehensive roadmap for delivery will be required. This is necessary to build confidence in the deliverability of CCS and to attract the necessary private sector investment. In the UK a new strategy for CCS commercialisation is needed as a matter of urgency as each year of delay in deployment substantially increases the costs of decarbonisation of the UK economy in future years.
CCS can support carbon reduction efforts across all major carbon emitting sectors and represents an essential component of the low-cost pathway to energy-system-wide decarbonisation. Development of CCS will create some costs; however a vibrant CCS industry will bring significant GVA to the economy as well as generate substantial employment potential.
For CCS to take off as a commercially viable and financeable proposition, the public sector will need to accept more of the development and operational risks that have thus far proved to represent unsurmountable barriers for the private sector, most notably in terms of commercial integration of the full chain and the development and operation of storage sites in a multi-user environment.
By optimising the structure, scale, location, technology choices and introducing new commercial models with modified risk reward structures, on the basis of increased public sector allocation of certain CCS specific key risks, the cost of CCS can be reduced significantly. Strike prices that are competitive with alternative forms of low-carbon generation should be achievable including for the first mover anchor projects.
In the UK, the creation of a government backed national CO2 T&S company, with responsibility for the development of T&S infrastructure guaranteeing the long-term availability of CO2 storage capacity for G&C and EII users, would be necessary for the successful development of the CCS industry. The availability of a de-risked T&S infrastructure would provide a much firmer basis for the private sector to develop G&C and EII assets in the UK.
The financial viability of CCS in the power generation sector currently requires a source of funding out with that which can be derived solely from market trading to cover the extra capital and operating costs and provide investors with an adequate return for the risks involved. In the UK, the CfDs available to CCS-enabled power generators are a good example of how this can be achieved. As the market adjusts to further penetration of low carbon generation technologies, as CCS design and operating experience grows and capital and operation costs reduce, the additional funding required via the CfD will reduce accordingly. If CCS is to be successfully deployed by EII operators a comparable mechanism will need to be devised.
In order to reduce the costs of the first mover projects, large scale power generation anchor projects (ca. 1 GW) connected to multi-user T&S CCS infrastructure should be envisaged from the outset. CCS technology is ready for large scale deployment.
The benefits of CCS are economy wide however the costs have invariably been seen as the responsibility of the developer operator of a CCS project. Alternative funding mechanisms could be considered to spread the costs of CCS infrastructure across all major emitters. This would align with the principle of the polluter pays and also reduce the cost to the consumer of the low-carbon electricity generated.
UKGS financial guarantees should also be considered to support UK developers of G&C assets in securing the finance needed for their investment bringing increasing project credit ratings and reducing costs.
If the lessons of previous unsuccessful CCS development programmes are learnt and the remaining challenges to full commercialisation resolved though new commercial approaches, there is every chance that CCS will be able to play its envisaged key role in supporting the cost effective decarbonisation of energy use across the economy starting in the early 2020s.
14 Political economy of CCS: what needs to happen?
Compared to other leading alternatives for mitigating climate change such as nuclear energy, biofuels and renewable energy, carbon dioxide capture and storage (CCS) technologies is a relatively new and unfamiliar option for large-scale decarbonisation and, as such, the politics and economics are not yet settled. Debates over CCS have been embryonic and fairly tepid when compared to nuclear power for example, where entrenched views and social movements have led to vocal political opposition since the 1960s;1159 or biofuels, where non-governmental organisations (NGOs) have provoked heated disputes over potential competition with food and impacts on biodiversity and sustainability.1160 Even renewables, which are generally viewed more favourably on a national level, have also seen significant opposition, usually local, with opponents sometimes criticised for being driven by not-in-my-backyard (NIMBY) concerns.1161 In all cases, there have been longstanding government support mechanisms in the form of large scale R&D, subsidies and other support mechanisms, reflecting interests coalescing in support (or opposition) to specific options over the course of years and decades.1162By contrast, CCS has largely been far removed from attention of not only the public, but also of politicians and other key stakeholders.43,1163 Despite this wider neglect, the Intergovernmental Panel on Climate Change (IPCC) has led efforts to consolidate and disseminate knowledge on the subject and have highlighted the benefits (and to a lesser-extent the challenges) of large-scale deployment. Other leading analytical organisations such as the International Energy Agency (IEA)1164 or the UK's Committee on Climate Change (CCC), have found CCS to be critical to efforts to meet aggressive decarbonisation targets at least cost. For example, the CCC finds that without CCS, the cost of meeting the UK's 2050 targets would be twice as high as if CCS were to be included.89 As discussed in the introduction to this paper, the IPCC Fifth Assessment Report describes how leaving out CCS would result in far higher costs for an aggressive decarbonisation strategy than would be the case if similar limitations were imposed on other low-carbon technologies (e.g., costs would be on average 138% higher under a 450 ppm scenario if CCS were unavailable, compared to 7% higher if omitting nuclear power, 8% for limited penetration of solar/wind and 64% for bioenergy).45 Even more tellingly, when asked to solve such a stabilisation scenario, in the electricity sector only 5 models can even solve without recourse to CCS, compared to 36 models with CCS. In the industrial sector, only 3 models could solve (compared with 22 with CCS).
Indeed, there were many reasons to expect that deploying CCS technologies would be relatively straightforward in political economy terms. CCS is the rare option that could address many important policy goals simultaneously including: addressing concerns over security of supply by providing both base-load and flexible low-carbon power; appealing to major influential stakeholders in industry; and allowing for the possibility of decarbonising existing and planned infrastructure in major developing countries. Moreover, CCS is fairly unique in providing viable options for hard-to-reach sectors including process industries such as chemicals, cement, steel as well as offering a potential pathway for negative emissions technologies (NETs) with biomass energy plus CCS (BECCS). Still, in spite of the advent of a few individual projects, driven by local context and incentives, CCS has had, at best, a chequered track record over the past decade.39
Research into CCS dates back to the 1990s although the constituent parts have been tested over the course of many decades. Yet, CCS did not emerge as a potential energy option for low-carbon development until the 2000s. The IPCC Third Assessment Report (TAR) of 2001 did not devote more than one paragraph out of its 58 page Technical Summary to CCS. The first serious effort by the international scientific community to investigate the technologies was when the IPCC issued a 443 page Special Report on CCS in 2005 at the same time as the issue was receiving attention from many national governments and international institutions.
The optimism of 2005 was followed by a series of announcements and cancellations by both government and industry, but more recently, over the course of just the past two years, a number of operational large-scale projects have emerged. The full-chain (capture, transport and storage) exemplars that store roughly 1 million tons or more of CO2 per year have come on line include the Boundary Dam project in south-eastern Saskatchewan and the Shell Quest project in northern Alberta in Canada, the Petra Nova project in Texas, Emirates Steel in Abu Dhabi.1165 Other new projects slated to launch in 2017 include the Illinois Industrial CCS project (1 Mtpa), which claims to the first biomass energy with CCS (BECCS) project, the Gorgon LNG facility in Australia (capable of up to 4 Mtpa) and other CCS projects in Canada associated with the Alberta Trunk Line project. Prior to Boundary Dam beginning operations in 2014, however, the only large-scale efforts had been storage projects such as Sleipner and Snøhvit in Norway and In Salah in Algeria, which all used CO2 from gas processing facilities.
Looking forward however, the pipeline slows and little new CO2 capture capacity is expected between 2018 and 2022. Moreover, according to the IEA, very few national commitments on the advent of the 2015 Paris Climate Conference (i.e., intended nationally determined contributions or IDNC), even mention the possibility of using CCS.1166 In spite of its scant coverage in the IPCC's 2001 TAR, the reduction potential by 2020 was estimated at 150–750 MtCO2 primarily in the power sector, ‘split equally between coal and gas, and between developed and developing countries’. According to the Global CCS Institute, in 2020, large scale capture will amount to only 40 MtCO2 per year, virtually all of which will be in developed countries and the vast majority will be from gas processing and for use in enhanced oil recovery operations. The question therefore is how to explain this uneven and slower-than-expected rollout of CCS, first, the slow progress, followed by a spurt of new projects and then a drying up of projects before lessons can be learned from the first projects?
14.1 Stakeholder views
CCS has largely remained a technological and future-oriented solution and as a result, most firms and NGOs have kept a ‘watching brief’ on the issue but have not been involved deeply in advocacy (either in favour or against), with a few exceptions. In the early 2000s, under the leadership of John Browne, BP was the first major industry advocate for using CCS and sought to build a series of ‘decarbonised fossil’ (DF) plants, including DF-1 at Peterhead in Scotland, DF-2 at a petroleum coke plant in Carson, California, DF-3 at Kwinana in Australia and DF-4 at Hydrogen Power Abu Dhabi.1167 All of these projects failed for a variety of reasons including government reluctance to pick winners and local opposition. The one small success was the In Salah storage project at its facility in Algeria, which stored 1 Mtpa from 2004–2011. When Tony Hayward took over as CEO in 2007, however, BP largely abandoned its role as a strong advocate.In the meantime, other large energy firms became more deeply involved in supporting the technology, notably, Shell and Statoil. Other firms, which had taken a more active role in the expectation of growing demand for CCS include oil field services firm Schlumberger, which set up a carbon services division, power equipment manufacturers such as Alstom (now GE), and chemicals firms with air separation capabilities such as Air Products and BOC. Still, many other leading firms such as ExxonMobil or Halliburton have shied away from significant involvement. Finally, despite the initial focus being almost entirely on power sector decarbonisation, virtually all electric utilities (including those that had been early leaders and enthusiasts such as Vattenfall in Europe and AEP in the US) have given up on CCS due to a lack of political and financial support. Most recent projects have been led by oil and gas companies. For example, in the most recent UK Competition, the two finalists were not led by electric utilities, which were effectively junior partners. The one clear exception is the Boundary Dam project where SaskPower, as a Crown Corporation (owned by the provincial government) has a very different set of incentives and governance structure than any private sector power company.
The environmentalist view is probably best summarised by a 2006 position paper by the Climate Action Network Europe umbrella group, which argued that CCS ‘may have a role to play’ but ‘climate policy cannot wait for any one technology’ and ‘CCS must not divert public investments or political attention away from renewable energy and energy efficiency’.1168 There have been some NGOs that have taken a more positive stance. In 2011, a number of small to medium sized NGOs – Bellona and ZERO in Norway, Green Alliance, E3G and Sandbag in the UK, Pembina Institute in Canada, the Climate Institute in Australia and the Natural Resources Defence Council, Clean Air Task Force and World Resources Institute in the US – came together to form an ENGO network on CCS. The distribution of these NGOs also reflects the countries where CCS has received the greatest attention and support. The largest NGOs, such as WWF and Friends of the Earth, have taken a relatively positive if muted view, given the large diversity of their national branches.
Other NGOs, notably Greenpeace, have been more critical voices. For example, the only major example of open advocacy against CCS was their 2008 report False Hope.1169 Their concerns were that: (i) CCS ‘won’t deliver in time’ (i.e., before 2030); (ii) underground storage is risky and poses significant liability; (iii) CCS wastes energy, is expensive and undermines funding in sustainable solutions; and (iv) the world already has the solutions to the climate crisis in the form of renewables. Linked to these concerns is a view that CCS simply perpetuates fossil fuels, which is compounded by many of the first projects being part-financed by using the captured CO2 for enhanced oil recovery (EOR). Still, there is little evidence that NGO opposition has done much to shift support (for or against) CCS on specific projects or at a national level.
The one area where opposition has made a difference is when local concerns derailed the prospects of CCS, specifically in onshore projects in Germany and the Netherlands. For example, the effort by Shell in 2007–2010 to develop a pilot storage site at Barendrecht (outside of the Netherlands) in the face of significant public opposition ultimately led first to the project being abandoned and then to all onshore storage in the Netherlands being banned.1170,1171
14.2 The politics of CCS
Governments have been, if anything, less consistent in their support of CCS technologies than non-state actors. There have been a handful of countries or jurisdictions, all of which are reliant on fossil fuels, where CCS has moved up the political agenda to the point where it emerged onto the wider political stage. In Canada, resource-rich provinces of Saskatchewan and Alberta took the lead, and in the US, projects were pushed forward in Texas, Mississippi and Alabama, despite these regions being most sceptical of the need for action on climate change. In Europe, although domestic politics were more favourable towards climate action, the common denominator of the leaders, Norway, the Netherlands and the UK was that they were Europe's main natural gas producers. Australia, whose economy is almost completely dependent on resources, even tried to assert overall leadership by creating a Global CCS Institute funded at A$100 million per year.Nevertheless, here had been some indications that, even in leading jurisdictions, the politics of CCS would not be as easy as some had assumed and that turning expert consensus into action faced some serious political and economic obstacles. Although, as noted earlier, CCS is largely unknown to the public and many stakeholders, there have been cases where the subject has become politically salient, notably in these resource-rich economies and if CCS becomes an issue then there is a danger of being on the losing side.
Norway was the first nation to take CCS seriously as the government made the decision over whether CCS would be mandated on all fossil-fired generation, which at the time only involved a single gas-fired plant.1172 The technology continued to rise up the national agenda to the point where, in 2011, Jens Stoltenberg, the prime minister, declared CCS would be Norway's “Moon mission”.1173 The first major effort was focused on Statoil's Mongstad oil refinery, one of the largest point sources in the country, first on a test centre to be followed by full-scale capture. Unfortunately, the costs of Mongstad rose dramatically, leading to the larger ambitions for full-scale capture at Mongstad being first delayed in 2011 and then abandoned in 2013 (after an expenditure of over $1 billion) although the large Test Centre Mongstad continues.1174 This failure and criticism from Norway's Auditor General for cost overruns led the Norwegian government to completely revisit its approach to CCS before being relaunched in 2016, with a commitment to have a full-chain project operating by 2022.1175 There has been, therefore, fairly consistent support from one government to the next although with some division over specific details.
CCS became an issue in provincial elections in both Alberta and Saskatchewan with differing outcomes. In Alberta, the long-serving Conservative government had advocated for CCS. During the 2015 election campaign, the climate sceptic Wildrose Party opposed any further spending on CCS and promised to cancel the Quest project. Ultimately, the socialist New Democratic Party (NDP) won the election and although they had been sceptical of CCS and preferred renewables and carbon pricing, once in power the new left-wing government continued to support the project. In Saskatchewan, Brad Wall, the right-leaning premier who had championed CCS throughout his term, highlighted the Boundary Dam project in launching his 2016 re-election campaign. In response, the opposition NDP sought to highlight cost overruns and technical problems at Boundary Dam. Ultimately, Wall was easily re-elected with a 62% mandate and increased his majority to 51 of the 61 seats in the Legislative Assembly.
In some cases, the shift that followed an election was more dramatic. In Australia, the Tony Abbott Government cut the CCS budget by some 70% in its first budget (cutting A$460 million out of A$650 million), after having campaigned against Labour's climate-friendly agenda. In a stealthier manner, with a brief note to markets, the new majority Conservative government in the UK abandoned the 1 billion competition that had been initiated in 2011 when the Conservatives were in coalition with the Liberal Democrats.
In other cases, the barriers were more institutional than political. The MIT Future of Coal study in 2007, led by Ernie Moniz, the condemned the US Government's reliance on small-scale scale storage projects and called for 3–4 large-scale storage projects of greater than 1 million tons per year and significant investment in major demonstration projects.1176 Yet, as Secretary of Energy under President Obama, Moniz was unable to push through any major project, hemmed in by Congressional recalcitrance to take action on climate change and vested interests in the form of the existing Department of Energy (DOE) regional partnerships, each of which touted the benefits of their own small-scale storage experiments.
At a more technical level, governments can over-specify or poorly specify the rules and conditions and thereby reduce the viability of projects. The UK, which had been considered a leader in policy design pre-2015, has seen no less than three failed efforts to fund large-scale CCS demonstration projects. The first failure, BP's DF-1 project at Peterhead, failed in the early 2000s because the UK Government did not want to ‘pick winners’, which led to a first CCS competition. According to its own National Audit Office (NAO), the first Competition failed because of the government's insistence on mandating post-combustion coal thereby imposing unnecessary constraints on top of a poorly designed procurement process.1177 The second competition was cancelled in late 2015 and the government was criticised by the NAO for failing to properly quantify the costs of delaying large-scale deployment and take that account in their decision and pointed to inter-departmental battles with HM Treasury. The House of Commons Public Accounts Committee also issued a harsh report pointing to the additional cost of decarbonisation without CCS, the hole in the Government's long-term plans for decarbonisation and the damage to investor confidence from the hasty withdrawal of funding.1178 Despite the stern criticism from policy circles, the decision produced few political repercussions other than some criticism from the opposition (primarily from the Scottish National Party), but relatively minimal media coverage.1179
There have, of course, been cases where the problem was primarily technology choice and economics. The Kemper County project in Mississippi, which was intended to be a large 582 MW coal IGCC plant, was driven by interests in providing CO2 to nearby oil fields and taking advantage of the proximity of minemouth coal rather than climate ambition. Like earlier IGCC projects in the 1970s and 1980s which suffered from delays, cost overruns and reliability problems the Kemper project was delayed repeatedly. With costs projected to exceed $7 billion (some $5 billion more than the original estimate) and plagued by years of technical problems, low gas prices and a complicated supply chain, the plant owners, Southern Company, decided to halt the IGCC element and continue operating as a conventional natural gas power plant.20
Outside of the core leading countries or regions, the problem has been even more severe, in part because of the perception that CCS is at best of marginal interest and, at worst, would cannibalise support from preferred technologies. The Clean Development Mechanism (CDM) offers an example of the impact of CCS being perceived as being of relevance to only a select few.1180,1181 CDM was designed as a flexibility mechanism under the Kyoto Protocol in 1997 to allow public or private actors to get credit for abatement activities carried out in developing countries.1182 Many of the countries that benefited from CDM projects using existing approaches, for example, those receiving credit for afforestation projects in Latin America, were concerned that if CCS was included in the CDM, they would lose out. As a result, it took from 2005 to 2011 to officially accept even the possibility of using CCS as an option within the CDM and still not a single project has emerged.
Another instance of the marginalisation of CCS can be seen in the European debate, despite early ambitions. The European Union, as an institution, took some encouraging steps, such as issuing a CO2 Storage Directive (Directive 2009/31/EC) to encourage all member states to prepare appropriate regulations for storing CO2 supplemented by other support mechanisms (to which only a handful of member states responded). A zero emissions fossil fuel power plant (ZEP) technology platform was launched in 2005 with an aim to have up to twelve full-chain projects across Europe by 2020.
To that end, the EU created a new mechanism to provide financial support through its NER 300 programme, which reserved 300 million emissions permits from the New Entrants Reserve (NER) for auction.1183 The price of carbon in the ETS collapsed from over €20 to close to €5 and so much of the anticipated funding stream disappeared, but even more importantly, the scope of the NER300 was expanded to include innovative renewable technologies (IRTs). Unlike CCS, virtually every country had one or more small-scale IRT projects to advance. When NER300 projects were finally awarded under the first call, 15 diverse renewables projects in over a dozen member states had been selected, but not a single CCS project was funded.
Here too, part of the explanation was institutional since the priorities of the European Commission did not necessarily align with those of the member states that were expected to cover most of the bill. For example, the project rated highest of all by the Commission, the Hatfield/Don Valley project did not even make the shortlist of four projects that the UK Government was considering and so the potential of aligning sources of funding was missed.
14.3 Future challenges and opportunities
In the wake of the Paris agreement's reaffirmation of a 2 °C global target with an aim ‘to pursue efforts towards’ 1.5 °C, a rapid scale up of CCS (including BECCS and CCS for industry) should be crucial. Yet, the prospects for CCS technologies are problematic (and at a complete standstill in many countries), driven by the political economy challenge of decarbonising fossil fuels as much as by any technological or economic barriers. It is misleading though to describe the problems as primarily one of cost or economics. Some low-carbon technologies such as offshore wind receive generous subsidies of the scale that would be needed for CCS. Other technologies such as nuclear power have existed for over sixty years and yet at least some governments still willingly provide large subsidies.Undoubtedly, there are important technical, economic and commercial challenges, which help explain the slow rollout of CCS, but the political economy, which initially appeared promising, has proven to be more problematic than anticipated. As Lord Oxburgh has aptly described it, CCS is an ‘orphan technology’.1184 Unlike nuclear power (or onshore wind), there are no strong opponents, but equally there are few if any advocates willing to lobby strongly. If there were to be unambiguous, serious political commitment to meeting a 2 °C target, then all large energy firms would eagerly lobby for CCS, but for most (and many politicians), their preferred alternative is continued unabated fossil fuel use.
A few resource-rich countries such as Canada, the US, Australia and Norway have moved forward with CCS almost independent of (or despite) their level of commitment to climate change. The economic crisis of 2007–2008 and the stimulus spending that followed meant that CCS was carried along, which, in spite of numerous setbacks, allowed a half dozen large integrated facilities to emerge since 2014. Yet, the portfolio of new projects at even an advanced planning stage is diminishing. Given the long time-scales involved and significant possibility of governments or firms or both reneging on commitments, the cupboard is essentially bare.
The recent round of emergent projects offers an important opportunity for learning. Global R&D on CCS is increasing and in some countries, such as Norway and the US, R&D support has been particularly generous. There will therefore be technological progress; the question is whether the political economy dynamic will change. Ultimately, CCS provides a litmus test for how serious governments take the challenge of deep decarbonisation. If there is a genuine effort to meet ambitious climate targets then, if the many analyses are correct, the needed shifts in incentives and regulations will mean change the interests (and the economics) and large-scale deployment CCS will eventually follow.
15 R&D priorities for carbon emissions reduction in coal-based power generation
15.1 Benchmarking CO2 mitigation cost
For any process capturing CO2, costs are comprised of the capital to install carbon capture and storage (CCS) equipment, the fixed costs to operate, and the variable cost to operate which includes the electricity the facility would have otherwise generated had CCS not been implemented. The key challenge is to reduce, by the greatest extent possible, the increase in the cost of the decarbonised product, be this a tonne of low carbon steel or cement or a MWh or low carbon electricity. Whilst the perspectives and analyses presented in this section are general in nature, the remainder of this discussion will be constrained to the perspective of CO2 capture with subsequent storage in a saline aquifer implemented in the coal-fired power industry.Even in this context, there are a number of ways one can choose to calculate the CO2 capture cost that includes all three cost categories mentioned in some form. However, for the purposes of comparing the performance of one CCS technology versus another and for evaluating the most impactful CCS methods (i.e., more CO2 captured) this paper will formulate discussion around the following calculation:
 | (21) |
The effect of CCS on COE can be inferred through examination of:
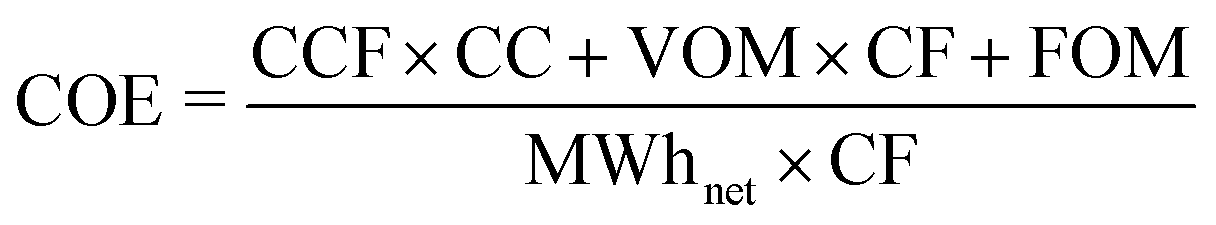 | (22) |
In general, without balance of plant improvements, and compared to a plant without CCS, CCS always increases COE. Practically, CCS adds to all cost terms in the numerator of eqn (22) (CC, VOM, FOM). Importantly, owing to the level of perceived risk associated with a “new technology”, the CCF is also likely to increase, at least for initial projects. Thermodynamically, CCS is proven via an entropy balance to always reduce the power generation term in the denominator (MWhnet).1185
15.2 Establishing a framework to evaluate CCS technology
In practice, there are numerous ways for a power plant to generate the power required to operate the CCS system. There are also numerous power generation sources of varying CO2 footprints (nuclear, solar, wind, natural gas, etc.) from which a plant can purchase electricity to operate a CCS system. In the interest of reducing confounding factors in the cost analysis, this discussion will not explicitly consider all options for electricity generation or its source. Instead, we imply in eqn (21) a formulation that the power required to operate the CCS equipment is generated by the base plant with CCS; i.e., when calculating COE of a plant with CCS, one does not use the MW of power generation of a plant without CCS in the denominator. Eqn (21) is then somewhat of an abstract comparison of plants with and without CCS, as it suggests a comparison of COE of one plant that is different in size than another. However, there are three main benefits of such a perspective.The first benefit is that each CCS technology can be evaluated independently of the method to provide the electrical load to operate it. The second is that each CCS technology is evaluated under consistent constraints to satisfy a given power demand and the resultant penalties in cost and performance. The third is that the balance of plant contribution to COE is calculated assuming the same equipment and costing methods, which more accurately isolates the true cost of implementing CCS (including larger equipment sizes and/or auxiliary equipment). While there a better metrics to assess the true financial burden on the entity installing and operating CCS on any specific plant, the metrics proposed here are ideal for objectively evaluating a range of CCS technologies.
With this in mind, the U.S. Department of Energy (DOE) has proposed near term goals for reducing the cost to capture CO2 and uses the above metrics in its assessment of the promise these technologies have for mitigating emissions from fossil-based power plants. In this framework, it is convenient to isolate two main factors that drive the cost to capture CO2: energy penalty to separate and compress the CO2, which result in a loss of power generation efficiency, and cost penalties, which are those costs required to build, install and operate the equipment. The energy penalty explicitly accounts for the electricity requirements to power the CCS equipment, as such embody the cost of lost power generation and associated lost revenue. Recall, the calculations assume the electricity required to run the CCS system is provided by the same plant fitted with the CCS system. The cost penalties required to operate the equipment do not include this lost revenue or power purchase costs, but instead includes the fixed and variable operating cost of supplies, personnel, maintenance, etc. If one acknowledges this distinction between the two cost types, the R&D efforts can be largely split between performance improvements and equipment/operating cost reductions. Decomposing costs in this manner is convenient from a systems perspective because the balance between performance improvements and their associated installation/operating cost increases can be explicitly analysed and optimised.
In the context of the equations presented above, the U.S. DOE has set a target for captured cost at $40 per tonne of CO2 captured and compressed to 152.7 bar (2214.7 psi) for sequestration in a saline aquifer. Cost and performance assumptions used to obtain this target are summarised in an extensive set of guidelines which can be found in Fisher et al.1186 Under these same assumptions and within the same calculational framework, today's cost to capture CO2 is $61 per tonne, assuming a given solvent based CO2 capture system detailed.1187 How to reduce the cost from $61 per tonne to $40 per tonne is a complex matter of extensive and multifaceted R&D efforts. However, one can somewhat simplify the landscape for determining a viable R&D direction by examining the relationship of CO2 Captured Cost to energy penalties and cost penalties.
Fig. 31 is adapted from Matuszewski et al.1188 and depicts the net electrical penalty of running CCS and the cost penalties of installing and operating CCS. The blue diagonal line represents all combinations that sum to a captured cost corresponding to the goal value of $40 per tonne set by the U.S. DOE. More details on its construction can be found in Matuszewski et al.1188 For reference, the cost to capture CO2 using today's baseline solvent technology for CCS is shown in Point A. Successful R&D will produce a trajectory from the reference value to any point on the line that corresponds to $40 per tonne capture; to Point B, for example. A continuum of cost combinations can achieve the DOE goal, however, these combinations are bound by thermodynamic laws and practical cost reductions. In fact, the key insight in this chart is the lower limit on thermodynamic costs associated with the unavoidable energetic investment of an ideal, reversible separation and compression process. This thermodynamically infeasible region was calculated by first determining the minimum energy required to overcome the entropy of ideal mixing that corresponds to 90% separation of CO2 out of a mixture that initially contains 14 mol% CO2.****** The power required for reversible compression from 1.014 bar to 152.7 bar was then added to the ideal separation energy requirement for a total minimum energy requirement to capture and compress CO2 from a typical flue gas. More details on this calculation can be found in McGlashan and Marquis.1185 The conclusion is that even if CCS equipment has zero installation and operation costs, there is a minimum energy penalty equating to ∼5–10% points of the base plant net power that manifests as an increase in COE, and thus a positive captured cost for a plant with even perfectly ideal CCS.
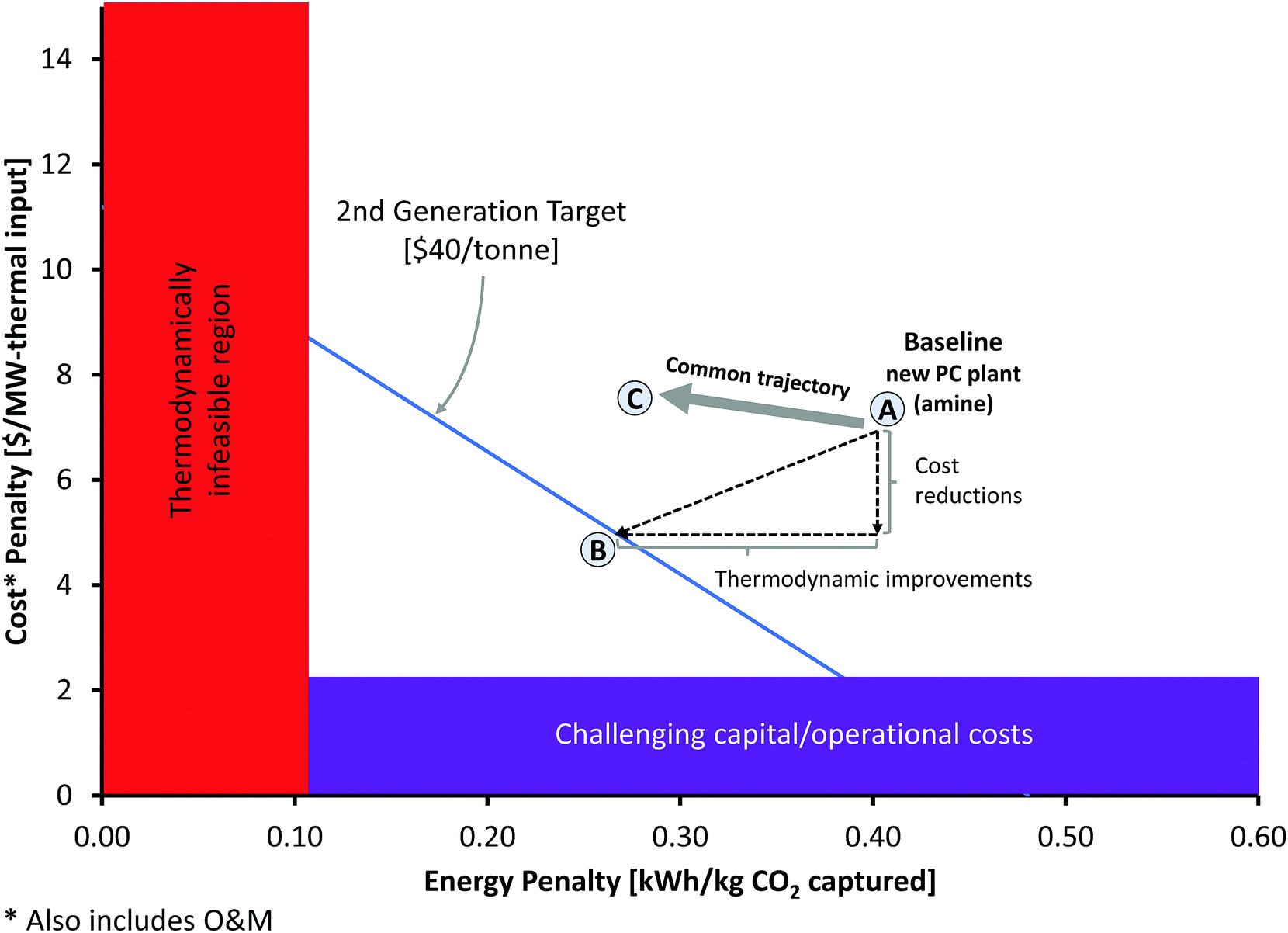 | ||
| Fig. 31 Cost and performance improvement trajectories to meet CCS goals. | ||
The scope of this analysis is on carbon capture and compression, therefore the thermodynamic limit in Fig. 31 is developed assuming the only processes added to the base plant are the minimum required for 90% CO2 separation from flue gas and compression to 152.7 bar. The scenarios depicted in Fig. 31 are specific to pulverised coal based power plants with post combustion carbon capture. While the ideal energy requirement to separate CO2 liberated by a process relying on complete combustion of coal cannot change, balance of plant improvements can reduce the net energy penalty as reflected in Fig. 31. For example, the minimum energy penalty of this separation and compression can be offset if implementing the CCS system provides a productive use of low grade heat not utilised in the base plant. Alternatively, changing the base power generation platform can also offset the net energy penalty by producing efficiency gains elsewhere in the process. It should be noted however that even power generation platforms projected to be among the most efficient options for power generation are only about one-third as effective at reducing CO2 emissions than CCS.†††††† So while thermodynamic penalties may be offset by adopting more efficient power generation, the need for implementing and improving CCS is certainly not removed.
Because zero cost equipment is unrealistic, one can also presume a maximum capital/operating cost reduction that will limit the ultimate success of R&D in this category, similar to the thermodynamic limit previously discussed. A maximum cost reduction is less rooted in fundamental theory and therefore is a more subjective limit that depends on technology, material, manufacturing improvement and even engineering intuition. While the exact value of maximum cost reduction will almost certainly be subject to significant debate, as an R&D trajectory brings costs closer to zero, additional gains will become more difficult to attain and should be considered when devising an R&D plan. Fig. 31 assumes an 80% reduction limit on the cost per gross thermal input to the system.
History has shown in many cases (in particular those cases that do not involve manufacturing or materials breakthroughs) that as thermodynamic improvements are made (via more integrated system configurations, better materials, or better devices) that equipment costs, installation costs, and/or operational costs generally increase. See the notional trajectory from Point A to Point C in Fig. 31. This phenomenon suggests some degree of tolerance has typically been required for cost penalties as performance is improved, or vice versa. However, Fig. 31 suggests a limit on tolerance for both types of penalties. Insight in this context can be gained by noting where the line that represents the DOE goal crosses into space that is thermodynamically-infeasible or cost-infeasible. The point at which these crossovers occur correlates to maximum acceptable penalty values on the vertical and horizontal axes. That is, if one penalty is too high, reducing the other such that the cost reduction goals are met requires an impossible value in infeasible space.
15.3 Framing future CCS R&D strategies
The location of the reference technology on Fig. 31 suggests that most likely a combination of both cost and performance improvements are required to reach the DOE goal; at this point additional thermodynamic penalty will ultimately result in an infeasible path to the DOE target, with a similar conclusion for the cost penalties, albeit there seems to be a bit more room for cost excursions when targeting the DOE goal. To date, most R&D for CCS has explored various ways to improve thermodynamics, independent of cost or manufacturing improvements. In fact, some early stage technologies are projecting a 50% reduction in net energy penalty compared to reference technology. This is remarkable, but unfortunately remains insufficient to reach the DOE target.Cost projections in most of the CCS screening studies done to date carry a fairly large uncertainty range (±30–50%). However even with the error acknowledged in most studies performed, the trends still indicate performance enhancements tend to result in slightly increased capital/operating costs. To date, nearly all of the cost penalties of CCS technology are due to capital and installation costs. This suggests a need for more focused attention on reducing capital cost while preserving thermodynamic improvements. If additional capital cost penalties cannot be prevented, thermodynamic laws may ultimately prevent the CCS industry from reaching the DOE goal. The maturity of readily available equipment and state of the art manufacturing processes has left little room for equipment cost improvements. However, recent progress in advanced manufacturing and process intensification may provide additional options for realising cost improvements. New manufacturing techniques will allow production of multi-functional unit operations which require complexities in geometry that until recently were infeasible to build. Combining simultaneous thermodynamic operations in such a way may allow improved efficiencies in smaller equipment, with less raw material costs. For example, 3D printing techniques have great freedom in building multiple independent, yet adjacent flow paths in one unit operation which may allow nearly arbitrarily close integration of heat exchange, reaction, and mass exchange operations.
Reaching an optimal combination of cost and performance improvements requires a detailed understanding and ability to accurately simulate the underlying thermodynamics and kinetic phenomena in a manner sufficient for holistic optimisation of the entire system. Evaluating a CO2 capture material in a system optimised for its performance is critical to properly assessing its promise and intelligently informing future R&D pathways. Little work of this extent has been done to date. However, recent efforts in the Carbon Capture Simulation Initiative (CCSI) funded by the U.S. DOE's Office of Fossil Energy have begun to address this issue rigorously.1189
Understanding the scale at which certain CCS technology platforms outperform others is also critical. The DOE goal of $40 per tonne of CO2 captured is formulated around a plant that is sized to produce 550 MW of net power generation, which is on the larger size of most point source generators of CO2. Indeed, over half of the CO2 from coal fired power generation in the U.S. is produced by power plants smaller than 550 MW, with normalised CCS capital costs that can triple compared to those required for the baseline 550 MW plant. The implication is that achieving the DOE goal of $40 per tonne will become exponentially more difficult at smaller plant sizes.
At large scales, the need for thermodynamic performance improvements will likely dominate, because economies of scale are healthy. Still, process intensification strategies will almost certainly be required to shrink equipment sizes or collapse some of the larger, single-purpose unit operations into more multi-purpose unit operations that require less material and space for similar performance. However, as point source sizes become one to two orders of magnitude smaller, economies of scale disappear and the cost to capture a unit mass of CO2 increases dramatically. At those smaller scales, adequately reducing capital costs will likely require a dramatic shift in paradigm to uncover cost effective solutions. Advanced, mass manufacturing strategies will likely be required to generate low capital cost solutions for small scale, modular CCS. Furthermore, the degree to which cost savings can be realised will almost certainly depend on technology platform. Regardless of platform, and in contrast to larger scales, cost savings at smaller sizes are likely to come at the expense of thermodynamic performance. Whether process intensification, mass manufacturing, advanced manufacturing or a combination of all three are employed to reduce cost, a firm understanding of which technology platform will be most cost effective at each scale after optimisation is first required. An important counterfactual in this context is that, for some hard to reach point sources, it may be preferable to simply off-set these emissions via the use of negative emission or greenhouse gas removal (GGR) technologies.
In tandem with the stated need to reduce cost and energy penalties of CCS, simultaneous performance improvements in power generation itself are also critical as they reduce the amount of CO2 liberated to satisfy a given power demand in the first place. However, the opportunity for improvements in the power generation efficiency of an existing facility is extremely limited without a major repowering effort. This requires a shift in perspective to greenfield designs. To reveal where the most opportunity for improvements in power generation improvements exist, one can extend the exergy analysis used to generate the thermodynamically infeasible region associated with CCS in Fig. 31 to the remainder of the subsystems in a Pulverised Coal (PC) plant with CCS.
The methodology for exergy analysis in McGlashan and Marquis1185 was used to calculate the work lost due to thermodynamic non-idealities in each subsystem of the power plant. In identifying the sources of these non-idealities, one also identifies areas with the most room for improvement. A typical analysis will reveal that for an average PC power plant equipped with amine based CCS, the ordered priority for improvements above the base PC plant is as follows:
(1) Fuel chemical energy transfer (50–60% loss of theoretically ideal work)
(2) Steam turbine (∼10% loss)
(3) CO2 capture and compression (5–10% loss)
Of particular interest is that operating the CCS system results in less of a power penalty than sources of thermodynamic inefficiency elsewhere in the plant. The transfer of coal combustion heat to the steam cycle by far exhibits the largest losses, due primarily to the large temperature difference in the associated heat exchange process. The steam turbine is the next largest source of lost work. The lost work associated with these two processes in the standard coal based power generation platform process suggests the best opportunity to decrease lost work of low carbon power generation (i.e., to increase power generation efficiency) is to first improve the base power generation technology platform, rather than to increase the efficiency of the CCS process.
Indeed, there is great opportunity in greenfield plant designs to integrate advanced power generation with CCS in manners that render CO2 separation more inherent (e.g., Chemical Looping, Fuel Cells) or that have higher driving forces for CO2 separation & compression (e.g., IGCC, Pressurised Oxycombustion). Nevertheless, in addition to offsetting CCS penalties with high efficiency power generation platforms, improving the CCS processes not completely inherent in power generation remains critical. Most work to date focuses on CCS processes that extract CO2 from a flue gas or syngas already generated by the power generation process, and are in that context largely independent of power generation platform. Transformational, low carbon fossil power generation will need to simultaneously generate power and concentrated CO2 more efficiently. As a result, the current sequential, or even semi-integrated, approaches to CCS are likely to become obsolete in the long term.
In the near term, where applications for CCS to be retrofit abound, developers should continue to preserve the currently projected thermodynamic improvements to CCS. However, kinetic improvements including heat and mass transfer as well as precisely controlled reaction/separation networks should garner more attention to decrease overall equipment size. Furthermore, as most state of the art CCS systems are sequential and/or only semi integrated, there is great untapped potential to apply concepts of process intensification to even near-term CCS designs. The intent here is to transcend the limitations of conventional, generalised equipment designs to produce geometries and configurations for new equipment that are highly specific to and efficient for CCS application. While an increase in equipment complexity has often resulted in an increase in cost, we again mention that the impending revolution in advanced manufacturing has the potential to cost effectively bring this designer equipment to market.
In the long term, as retirement of existing PC plant designs increases, the need to replace them with advanced baseline fossil power generation platforms like those mentioned above will increase. At this point, the power generation industry will need to implement a new paradigm of CCS; one where the line separating power generation from CO2 capture is significantly blurred.
16 Conclusion
16.1 IAMs and negative emissions
In the context of meeting the climate change commitments of limiting warming to less than 2 °C, most integrated assessment models (IAMs) cannot find a solution without carbon capture and storage (CCS). In other words, CCS is not just vital to the cost-optimal solution, it is vital to the solution, period. Intergovernmental Panel on Climate Change (IPCC) scenarios associated with a more than even chance of achieving the 2 °C target are characterised by average capture rates of 10 GtCO2 per year in 2050, 25 GtCO2 per year in 2100 and cumulative storage of 800–3000 GtCO2 by the end of the century.Further, IAMs do not find a decreasing role for CCS over time. On the contrary, the CCS share in primary energy is mostly higher in the second half of the century compared to the first. This undermines the reputation of CCS as a bridging technology and further underlines its importance in IAMs, which seek to achieve ambitious climate targets. In other words, CCS is likely to be important for the long term.
The more stringent our climate targets, the more important bioenergy with CCS (BECCS), and other negative emissions technologies (NETs), become. In 2100, it is more likely than not that BECCS will provide more than 5% of total global primary energy. For a 1.5 °C scenario, the cumulative negative emissions are between 450 and 1000 GtCO2 until 2100. This is in stark contrast to some 2 °C scenarios, which do manage to reach their target without carbon removal technologies. Thus, in the context of meeting a 1.5 °C target, all the evidence points to an overshoot in temperature, which will need to be brought back under control via NETs. There are no feasible scenarios which do not involve NETs. Importantly, in addition to removing CO2 from the atmosphere, NETS allow the offsetting of emissions from hard-to-reach areas, like shipping or aviation. Importantly, the dominance of BECCS may well be a function of the lack of other options within IAMs, and therefore incorporating other NETs in IAMs is a priority. Of the range of potential NETs, BECCS and direct air capture (DAC) are two that are of potential significance.
BECCS is currently industrially deployed, with five plants operating worldwide. The technical maturity and costs of BECCS are comparable to conventional Fossil-CCS technologies, with BECCS costs typically estimated to be on the order of half that of direct air capture.
The technical potential for BECCS pathways is estimated to be in the range of 3–20 GtCO2 per year. However, it is becoming increasingly clear that the resource requirement to deliver BECCS is case-specific. Between 380–700 Mha of land may be required by 2100, and depending on the choices made throughout the biomass supply chain, capturing 12 GtCO2 per year would require between 130–860 EJ. Land availability for biomass feedstock production is a key driver for large-scale BECCS implementation. Land demand for BECCS largely depends on the selected feedstock.
It is finally important to note that the large-scale deployment of BECCS is contingent on first having access to a mature CCS industry. Given the challenges associated with delivering CCS, the additional challenges of securing sufficient sustainably sourced biomass and the policy questions around incentivising and regulating negative emissions facilities would render attempting large-scale BECCS in the absence of a mature CCS industry exceptionally challenging.
The direct capture of CO2 from the air is possible, but technically and economically challenging, primarily as a result of the extremely dilute nature of atmospheric CO2. Owing to the low concentration of atmospheric CO2, costs of DAC are likely to be very substantial, and perhaps as much as two orders of magnitude greater than CO2 capture from power plant exhaust gases. However, at the time of writing, a DAC facility is operating on a commercial basis in Switzerland, and is selling the captured CO2 for utilisation.18 However, given that the CO2 is not geologically stored, this does not constitute a negative emissions project.
One key way in which the dilute nature of atmospheric CO2 affects process design is the required affinity of the sorbent for CO2 – this would need to be as much as two orders of magnitude greater than is the case for “standard” capture with chemical solvents such as amines. This in turn makes sorbent regeneration that much more challenging and necessitates a chemical shift process as opposed to a simple temperature or pressure swing. DAC costs are likely to be dominated by the requirement to treat vast volumes of air in order to capture a meaningful amount of CO2, with the capture of 1 MtCO2 per year necessitating the processing of 80000 m3 s−1 of air. The actual cost of DAC is likely to be in the range of $600–1000 per tCO2. In order to have a realistic assessment of the likely utility of DAC, transparent and realistic estimates of the cost of this technology are urgently required. Given the present level of uncertainty, rational decision making or inclusion in IAMs is nigh impossible.
16.2 Industrial CCS
In addition to decarbonising the power sector, CCS also plays an important role in decarbonising the industry sector. Decarbonising the industrial sector presents a unique set of challenges, in that there are no obvious alternatives to CCS (cf. renewable energy in the power sector) and the international nature of industry. Particularly important industrial sectors are iron and steel, cement, and oil refining, with large integrated mills, for example, emitting 3.5 MtCO2 per year, on the order of 1.8 tCO2 per tsteel. Cement production is also a carbon intense activity, with a carbon intensity in the range of 0.6–1.0 tCO2 per tcement, with approximately 60% of this CO2 associated with the calcination step, i.e., even if the energy required to operate the process was entirely zero carbon, this would only reduce the CO2 intensity by 40%.Decarbonisation of cement production via amine scrubbing has been observed to be ∼3 times more energy intense than oxy-firing and the demonstration of full oxy-fired cement production has been identified as a priority for future research, though Calcium looping is another important technology. In the context of industrial decarbonisation, industrial symbiosis, e.g., recovering waste heat from neighbouring facilities, has been observed to be key to cost reduction. Owing to their unique skill sets, the application of CCS to any industrial sector is likely to involve the petrochemical industry. Moreover, when it comes to decarbonising the refining sector, this cost is well within the large swings in oil price. Put another way, the end-use sectors can easily afford to pay for decarbonising this sector. However, relative to the power sector, there is a paucity of academic studies of industrial carbon capture, despite the fact that much of the real-world deployment is happening in the industrial sector. This is therefore an area that warrants further research.
16.3 Technology development
The rate at which new materials progress from the lab- or bench-scale to the pilot-scale is too slow. It is critical that this rate be increased. One potential impediment is that the current sorbent benchmark is still 30 wt% MEA, which was originally proposed in 1930. However, this option is now significantly outclassed by blends of solvents, such as the formulation of piperazine (PZ) and 2-amino-2-methyl-1-propanol (AMP). It is time to update this benchmark, preferably with reference to current industrial best practice. The fact that new materials continue to be compared with an obsolete benchmark is potentially limiting progress in this area. Efforts should also be made to ensure that laboratory-scale work investigates materials under conditions somewhat representative of the real world-high CO2 partial pressure for desorption, and eventually including the presence of steam and trace and minor species.Ionic liquids (ILs) have shown some promise as next-generation CO2 capture solvents, mainly through their highly desirable stability and low volatility, combined with reasonable absorption capacity. However, there are issues with poor gas uptake kinetics, stemming from high viscosity. Also, the high molar mass of most ILs dictates that, even with high molar CO2 capacities, the mass uptake rate remains inferior to aqueous alkanolamines, particularly at low pressures. It would seem prudent for future research to focus on reducing the molar mass and viscosity of functionalised ILs. By employing functionalised ions (azolate, phenolate, amino acid) there remains the possibility of increased capacity without necessitating higher cost. If future designs can take into consideration cost aspects in combination with reduced capture energy and higher capacity, there could be justification for the use of these novel solvents.
Metal organic frameworks (MOFs) are a promising class of sorbent materials. However, they are not typically manufactured at large scale, and for those that are, they are typically supplied as a powder rather than a structured adsorbent. Developing an understanding of the large-scale production of MOFs in a form suitable for practical application is key to moving these materials forward towards industrial deployment.
There are an immense number of possible materials which could be used for CCS – testing them all at pilot scale is not practicable. The development of high throughput modelling and simulation approaches which combine molecular- and process-scale information for material screening is therefore vital. Importantly, data describing mass transfer resistance and diffusion limitations are required to enable this, but are particularly scarce.
CO2 transport remains an over-designed element of the CCS chain, owing to uncertainties around material selection and process operation. A better understanding of the role of CO2 composition in fracture propagation is vital for derisking this element of this technology. Importantly, if purity constraints can be relaxed, this can lead to non-negligible reductions in whole-system cost.
When assessing an improvement in CCS technology, it is important to recall that whilst the CCS community want to reduce the cost of CO2 capture, the owner of the facility from which the CO2 must be captured prioritise minimising the cost of their low carbon product. This is especially true of the power sector where they are operating to meet a fixed, and largely inflexible, demand.
With existing technologies, the majority of “CCS costs” are, in fact, associated with increases in capital cost, as opposed to operating cost. This implies that whilst a continued focus on improving thermodynamic performance is helpful, priority must be given to research which promises reduced capital costs.
In this context, in addition to improving the CCS process, the value of improving the underlying process, i.e., the power plant is also vital. From a thermodynamic perspective, 50–60% of theoretical losses occur in the conversion of chemical energy to electrical energy. By comparison, CCS is only 5–10% (except for the cases of high temperature cycles, which integrate the capture efficiency loss into the lost work from the conversion of heat to work, and are therefore more efficient). Moreover, as the power plant increases in efficiency, less CO2 is produced per MWh thus reducing the cost of the starting point. Thus, where new facilities are built, the deployment of state-of-the-art facilities should be a given.
Thus, R&D initiatives aimed at “improving” CCS should take a whole systems approach and focus on reducing the cost per unit of decarbonised product (e.g., steel, cement, power), and how this decarbonised process will, itself, compete in the market, i.e., what will displace what. This may well be distinct from focusing exclusively on minimising the cost of capturing the CO2.
16.4 CO2 storage
In the past 5 years, great progress has been made in the area of CO2 storage. Outstanding challenges in the area of CO2 storage monitoring and verification include the development of technologies to allow for the quantification of the amount of CO2 stored and plume migration. Approaches for obtaining quantitative insight into the extent to which CO2 has partitioned into the aqueous phase, or has become residually trapped are also a key research priority. Leak detection and remediation remain key areas for research, hampered by the lack of analogue in the petroleum industry.A final key area for urgent research is the development of better understanding of regional CO2 storage capacity, how it changes with use and how this capacity might evolve over time. Indeed, the lack of such insight is a key hurdle to a better representation of CCS in IPCC-type assessments.
16.5 CO2 conversion and utilisation (CCU)
There is a common narrative that CCU can enable CCS. In the US, where the CO2 transport and storage infrastructure is available and largely written off, this may be true in the case of CO2 enhanced oil recovery (CO2-EOR). In this context, the potential of residual oil zones (ROZs) is significant owing to their likely high CO2 injected to oil recovered ratios. Thus, in the US, the primary objective is driving down the $ per tCO2 cost so that CO2-EOR, combined with the existing tax credit scheme, represents an attractive investment in an era of relatively low oil prices.In other parts of the world, like the UK or EU, the CO2 transport and storage infrastructure does not exist at the same scale, nor is there a sufficient investment incentive to induce its deployment (e.g., EOR may not be an option). Thus, in these regions, the key barriers are the lack of infrastructure, with the cost of capture a secondary barrier. Consequently, the UK/EU region would be better advised to focus on deploying transport and storage infrastructure and derisking that element of the investment. Innovations to reduce the $ per tCO2 cost of capture will continue to come from the global academic community, and can be imported on an as-needed basis. Infrastructure cannot, however CO2-EOR can potentially enable the deployment of CCS infrastructure. However, the extent to which CO2-EOR will actually store CO2 strongly depends on the way in which the EOR operation is managed, and also on the kind of oil which is recovered. It is important, therefore, to carry out thorough life cycle analyses, both attributional and consequential, in order to develop this kind of insight.
The magnitude of the role that CCU might play in climate change mitigation is likely to be very small, relative to that played by CCS. However, CCU might offer very cost efficient options for CO2 mitigation, even yielding a profit in some cases. One option which might be deployable at scale is the conversion of CO2 to a fuel product (e.g., via Fischer–Tropsch processes). However, this requires major progress in catalysis and process design, additionally this route does not store CO2 long-term but would offer carbon-neutral fuels in a best case future scenario. The primary source of cost in CO2 to fuels processes is that of hydrogen, with the cost of CO2 coming second. Thus, in order to move CCU forwards, a key area for research is the development of reduced cost approaches for producing renewable hydrogen. Another key constraint is that, in order to avoid partial decarbonisation scenarios, the CO2 used must (a) ideally not come from a fossil source, and (b) be recaptured after the, e.g., CO2-fuel is used. This will have the effect of enabling CCU to be a key element of a coherent circular economy narrative.
16.6 Policy considerations
Current and medium-term UK and EU decarbonisation targets are expressed in terms of a percentage of renewable energy. This is essentially confusing ends (sustainable, affordable and reliable energy) with means (deployment of specific technologies) and is distinct to a technology-agnostic aim of deploying low carbon electricity. As a consequence, the deployment of CCS is disadvantaged from a policy perspective. Better would be to define low carbon energy, e.g., 50 g kWh−1, and replace renewables targets with low carbon targets, e.g., x% of power to come from low carbon targets by a given date. This would allow individual states the flexibility to realise these goals in a locally optimal manner.It is vital to recognise that, as we move to a more diverse energy system, not all power generation technologies provide the same services to the system, and thus attempting to value them on a basis of levelised cost of electricity (LCOE) is, at best, misguided. Better to evaluate the value of each technology to the energy system on an individual basis, noting that this value varies with the composition of the system. In this context, CCS, nuclear and bioenergy are notable for their ability to significantly reduce CO2 emissions at a marginal increase in total system cost. Thus, these technologies must compete amongst themselves. In other words; CCS and intermittent renewable energy generators are not competing to provide the same set of services.
It is evident that, despite substantial public and private effort to commercialise and deploy CCS technology, progress is lagging behind what is commonly considered to be required to meet climate targets. This is despite ample evidence that CCS will both reduce whole-system energy costs, and thus the cost to the consumer and also create a significant number of jobs.
One key issue with the CCS commercialisation models that have been followed thus far is that the private sector should manage all of the technical and commercial integration risks across the full CCS chain (capture, transport and storage). Whilst the private sector can manage and competitively price many risks, there is a lack of proven models for commercialising CCS (distinct to the CO2-EOR industry, which is much more straightforward). This lack of a proven commercial model across the full chain, means that the market will either only accept at a premium, or not accept, whatever the price.
The key commercial risks that require public support are (i) cross chain default, (ii) post decommissioning CO2 storage risk, (iii) CO2 storage performance risks, (iv) decommissioning cost and financial securities related to the CO2 storage permit, and finally (v) insurance market limitations for CO2 T&S operations. A commercial model that entails a transfer of risks iii–v categories to the public sector will both remove the key barriers that have thus far prevented the private sector from investing in CCS and also improve financeability and consequently significantly reduce the risk premium added to the cost of capital funding. This model is in line with what was suggested by the 2016 Oxburgh Report.101
However, it is not necessary to have public ownership of the generation and capture assets, and with the transfer of the aforementioned risks iii-v to the public sector, the private sector can likely deliver CCS without any change to existing regulation. This latter element is particularly important; wherever possible initiatives should ideally fall within existing regulatory frameworks.
Whilst the technical elements of CCS are well-understood, and, as has been discussed in this paper, the financial models are becoming increasingly clear, public acceptability and the consequent impact on the political economy are as yet at an embryonic stage. This is despite substantial evidence of the economy-wide GDP and employment benefits associated with the deployment of CCS.
It is misleading to describe the problems with CCS commercialisation as primarily one of cost. Some low-carbon technologies such as offshore wind receive generous subsidies of the scale that would be needed for CCS. Other technologies such as nuclear power have existed for over sixty years and yet, some governments still willingly provide large subsidies. Uniquely for CCS, the political economy has proven to be more problematic than anticipated. Unlike nuclear power or onshore wind, there are no strong opponents, but neither are there advocates willing to lobby strongly. If there were to be unambiguous, serious political commitment to meeting a 2 °C target, then all large energy firms would eagerly lobby for CCS, but for most (and many politicians), their preferred alternative is continued unabated fossil fuel use.
Ultimately, CCS provides a litmus test for how serious governments take the challenge of deep decarbonisation. If there is a genuine effort to meet ambitious climate targets then, if the many analyses are correct, the needed shifts in incentives and regulations will mean change in the interests (and the economics) and large-scale deployment CCS will eventually follow.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors contributed individual sections to this paper. It is noted that co-authorship does not imply group consensus or endorsement of the entire paper. Views are those of the individual authors and do not necessarily reflect the views of their organisation and/or its members. We are grateful for the support of the UK Foreign and Commonwealth Office Prosperity Fund in contributing to the CCS Forum 2016 which led to this paper. The authors would also like to acknowledge funding from the RCUK under grants EP/M001369/1 (MESMERISE-CCS), EP/M015351/1 (Opening New Fuels for UK Generation), EP/N024567/1 (CCSInSupply), and NE/P019900/1 (GGR Opt). André Bardow acknowledges that part of this work was supported by the Cluster of Excellence “Tailor-Made Fuels from Biomass”, which is funded under contract EXC 236 by the Excellence Initiative by the German federal and state governments to promote science and research at German universities. Paul Fennell acknowledges that part of this work was conducted with funding from Project LEILAC, which has gratefully received €12m of funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 654465. Sabine Fuss would like to acknowledge that this synthesis is part of the efforts under the Global Carbon Project's MaGNET Intitiative. Leigh Hackett would like to express his appreciation to a number of people that providing knowledgeable, constructive and insightful feedback during the preparation of this paper including Richard Simon-Lewis, James Whitney, and Saurabh Kapoor. Responsibility for any errors or misinterpretations lies solely with the author. Sam Krevor acknowledges that part of this work was conducted under the Qatar Carbonates and Carbon Storage Research Centre, jointly funded by Shell, Qatar Petroleum, and the Qatar Science and Technology Foundation. Parts of the work were conducted under NERC Grant NE/N016173/1 Migration of CO2 through North Sea Geological Carbon Storage Sites and under EPSRC Grant EP/M001369/1 MESMERISE-CCS. Berend Smit acknowledges funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No 666983, MaGic). Requests for data may be made through http://www.imperial.ac.uk/people/niall.References
- N. Mac Dowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010, 3, 1645–1669 CAS.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 CAS.
- COP21 Paris Agreement, European Commission, http://ec.europa.eu/clima/policies/international/negotiations/paris/index_en.htm.
- GCCSI, Large-scale CCS projects, Global CCS Institute, http://www.globalccsinstitute.com/projects/large-scale-ccs-projects, accessed July 2017.
- BEIS, UK carbon capture and storage: Government funding and support, Department for Business, Energy & Industrial Strategy (BEIS), London, UK, http://https://www.gov.uk/guidance/uk-carbon-capture-and-storage-government-funding-and-support, accessed June 2017.
- IPCC, Climate Change 2014: Mitigation of Climate Change. Working Group III Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2014 Search PubMed.
- A. Cousins, L. Wardhaugh and A. Cottrell, Pilot plant operation for liquid absorption-based post-combustion CO2 capture, Absorption-based Post-combustion Capture of Carbon Dioxide, Woodhead Publishing, Cambridge, UK, 2016, pp. 649–684 Search PubMed.
- R. Sanchez, Technology Readiness Assessment Guide, U.S. Department of Energy, Washington, DC, 2011, http://https://www.directives.doe.gov/directives-documents/400-series/0413.3-EGuide-04-admchg1 Search PubMed.
- GCCSI, CO2 capture technologies: Technology options for CO2 capture, Global CCS Institute, Canberra, Australia, 2012, http://https://www.globalccsinstitute.com/publications/technology-options-co2-capture Search PubMed.
- NDA, Guide to Technology Readiness Levels for the NDA Estate and its Supply Chain, Nuclear Decommissioning Authority, Cumbria, UK, 2014, http://https://www.gov.uk/government/news/guidance-on-technology-readiness-levels Search PubMed.
- R. R. Bottoms, US Pat., application 1783901, 1930 Search PubMed.
- M. Campbell, Energy Procedia, 2014, 63, 801–807 CrossRef CAS.
- A. Singh and K. Stéphenne, Energy Procedia, 2014, 63, 1678–1685 CrossRef CAS.
- DOE, Petra Nova – W.A. Parish Project, Office of Fossil Energy, U.S. Department of Energy (DOE), http://energy.gov/fe/petra-nova-wa-parish-project, accessed May 2017.
- MIT, Petra Nova W.A. Parish Fact Sheet: Carbon Dioxide Capture and Storage Project, Carbon Capture and Sequestration Technologies program at MIT, 2016, http://https://sequestration.mit.edu/tools/projects/wa_parish.html.
- MTR, PolarisTM membrane: CO2 removal from syngas, Membrane Technology & Research, http://www.mtrinc.com/co2_removal_from_syngas.html, accessed June 2017.
- Chemical Processing, Air Products and NTNU Enter Licensing Agreement for Carbon Capture Technology, 2017.
- J. Owen-Jones, Grand opening of Climeworks commercial DAC plant, Gasworld, 2017, http://https://www.gasworld.com/grand-opening-worlds-first-dac-plant/2012895.article.
- Climeworks, Climeworks launches world's first commercial plant to capture CO2from air, Press Release, 2017, http://www.climeworks.com/wp-content/uploads/2017/05/01_PR-Climeworks-DAC-Plant-Opening.pdf.
- D. Wagman, The three factors that doomed Kemper County IGCC, IEEE Spectrum, 2017, http://spectrum.ieee.org/energywise/energy/fossil-fuels/the-three-factors-that-doomed-kemper-county-igcc, accessed July 2017.
- Mississippi Power, Mississippi Power issues statement regarding Kemper County energy facility progress and schedule, MississippiPower News Center, http://mississippipowernews.com/2017/02/22/mississippi-power-issues-statement-regarding-kemper-county-energy-facility-progress-and-schedule-2/, accessed May 2017.
- K. E. Swartz, Southern Co.'s clean coal plant hits a dead end, E&E News - Energywire, 2017, http://https://www.eenews.net/stories/1060056418, accessed July 2017.
- P. Noothout, F. Wiersma, O. Hurtado, P. Roelofsen and D. Macdonald, CO2 Pipeline Infrastructure, IEA Greenhouse Gas R&D Programme (IEAGHG), 2014, http://ieaghg.org/docs/General_Docs/Reports/2013-18.pdf.
- P. Brownsort, Ship transport of CO2 for Enhanced Oil Recovery-Literature survey, January, Scottish Carbon Capture & Storage (SCCS).
- Y. Gou, Z. Hou, H. Liu, L. Zhou and P. Were, Acta Geotechnica, 2014, 9, 49–58 CrossRef.
- GCCSI, Projects Database: CO2utilisation, Global CCS Institute, http://https://www.globalccsinstitute.com/projects/co2-utilisation-projects, accessed July 2017.
- GCCSI, Saga City Waste Incineration Plant, Global CCS Institute, 2016, http://www.globalccsinstitute.com/sites/www.globalccsinstitute.com/files/content/page/122975/files/Saga%20City%20Waste%20Incineration%20Plant_0.pdf.
- GCCSI, Strategic analysis of the global status of carbon capture and storage. Report 1: Status of carbon capture and storage projects globally, Global CCS Institute, 2009.
- D. van Vuuren, E. Kriegler, K. Riahi, M. Tavoni, B. S. Koelbl and M. van Sluisveld, The use of carbon capture and storage in mitigation scenarios—An integrated assessment modelling perspective. Our Common Future Under Climate Change, International Scientific Conference, Paris, France, 2015 Search PubMed.
- E. Benhelal, G. Zahedi, E. Shamsaei and A. Bahadori, J. Cleaner Prod., 2013, 51, 142–161 CrossRef.
- S. Fuss, J. G. Canadell, G. P. Peters, M. Tavoni, R. M. Andrew, P. Ciais, R. B. Jackson, C. D. Jones, F. Kraxner, N. Nakicenovic, C. Le Quere, M. R. Raupach, A. Sharifi, P. Smith and Y. Yamagata, Nat. Clim. Change, 2014, 4, 850–853 CrossRef CAS.
- F. Kraxner, S. Fuss, V. Krey, D. Best, S. Leduc, G. Kindermann, Y. Yamagata, D. Schepaschenko, A. Shvidenko, K. Aoki and J. Yan, The role of bioenergy with carbon capture and storage (BECCS) for climate policy, John Wiley & Sons, Ltd, UK, 2015, vol. 3, pp. 1465–1484 Search PubMed.
- B. S. Koelbl, M. A. van den Broek, A. P. C. Faaij and D. P. van Vuuren, Clim. Change, 2014, 123, 461–476 CrossRef CAS.
- C. Hendriks, W. Graus and F. van Bergen, Global carbon dioxide storage potential and costs, Ecofys and TNO, Utrecht, The Netherlands, 2004, http://www.ecofys.com/files/files/ecofys_2004_globalcarbondioxidestorage.pdf Search PubMed.
- K. Riahi, E. S. Rubin, M. R. Taylor, L. Schrattenholzer and D. Hounshell, Energy Econ., 2004, 26, 539–564 CrossRef.
- A. Kurosawa, Energy Econ., 2004, 26, 675–684 CrossRef.
- V. Scott, S. Gilfillan, N. Markusson, H. Chalmers and R. Stuart Haszeldine, Nat. Clim. Change, 2013, 3, 105–111 CrossRef CAS.
- R. van Noorden, Nature, 2013, 493, 141–142 CrossRef CAS PubMed.
- D. M. Reiner, Nat. Energy, 2016, 1, 15011 CrossRef.
- T. Spencer, R. Pierfederici, H. Waisman, M. Colombier, C. Bertram, E. Kriegler, G. Luderer, F. Humpenöder, A. Popp, O. Edenhofer, M. D. Elzen, D. van Vuuren, H. van Soest, L. Paroussos, P. Fragkos, M. Kainuma, T. Masui, K. Oshiro, K. Akimoto, B. S. Tehrani, F. Sano, J. Oda, L. Clarke, G. Iyer, J. Edmonds, T. Fei, F. Sha, J. Kejun, A. C. Köberle, A. Szklo, A. F. P. Lucena, J. Portugal-Pereira, P. Rochedo, R. Schaeffe, A. Awasthy, M. K. Shrivastava, R. Mathur, J. Rogelj, J. Jewell, K. Riah, A. Garg and I. M. P. Consortium, Beyond the numbers: Understanding the transformation induced by INDCs. Study Number 05/15, IDDRI – MILES Project Consortium, Paris, France, 2015 Search PubMed.
- IEA, 20 Years of carbon capture and storage: Accelerating future deployment, Organisation for Economic Co-operation and Development (OECD) and International Energy Agency (IEA), Paris, France, 2016 Search PubMed.
- P. Upham and T. Roberts, Int. J. Greenhouse Gas Control, 2011, 5, 1359–1367 CrossRef.
- D. M. Reiner, T. E. Curry, M. A. de Figueiredo, H. J. Herzog, S. D. Ansolabehere, K. Itaoka, F. Johnsson and M. Odenberger, Environ. Sci. Technol., 2006, 40, 2093–2098 CrossRef CAS PubMed.
- A. Löschel, Ecol. Econ., 2002, 43, 105–126 CrossRef.
- IPCC, in Climate Change 2014: Synthesis Report of the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, ed. Core Writing Team, R. K. Pachauri and L. Meyer, Intergovernmental Panel on Climate Change (IPCC), 2014 Search PubMed.
- G. P. Peters, R. M. Andrew, S. Solomon and P. Friedlingstein, Environ. Res. Lett., 2015, 10, 105004 CrossRef.
- GCP, Global Carbon Budget 2016, Global Carbon Project, 2016, http://www.globalcarbonproject.org/carbonbudget/16/files/GCP_CarbonBudget_2016.pdf.
- C. Le Quéré, R. M. Andrew, J. G. Canadell, S. Sitch, J. I. Korsbakken, G. P. Peters, A. C. Manning, T. A. Boden, P. P. Tans, R. A. Houghton, R. F. Keeling, S. Alin, O. D. Andrews, P. Anthoni, L. Barbero, L. Bopp, F. Chevallier, L. P. Chini, P. Ciais, K. Currie, C. Delire, S. C. Doney, P. Friedlingstein, T. Gkritzalis, I. Harris, J. Hauck, V. Haverd, M. Hoppema, K. Klein Goldewijk, A. K. Jain, E. Kato, A. Körtzinger, P. Landschützer, N. Lefèvre, A. Lenton, S. Lienert, D. Lombardozzi, J. R. Melton, N. Metzl, F. Millero, P. M. S. Monteiro, D. R. Munro, J. E. M. S. Nabel, S. I. Nakaoka, K. O'Brien, A. Olsen, A. M. Omar, T. Ono, D. Pierrot, B. Poulter, C. Rödenbeck, J. Salisbury, U. Schuster, J. Schwinger, R. Séférian, I. Skjelvan, B. D. Stocker, A. J. Sutton, T. Takahashi, H. Tian, B. Tilbrook, I. T. van der Laan-Luijkx, G. R. van der Werf, N. Viovy, A. P. Walker, A. J. Wiltshire and S. Zaehle, Earth Syst. Sci. Data, 2016, 8, 605–649 CrossRef.
- C. Azar, K. Lindgren, M. Obersteiner, K. Riahi, D. P. van Vuuren, K. M. G. J. den Elzen, K. Möllersten and E. D. Larson, Clim. Change, 2010, 100, 195–202 CrossRef CAS.
- C. F. Heuberger, I. Staffell, N. Shah and N. Mac Dowell, Energy Environ. Sci., 2016, 9, 2497–2510 CAS.
- C. F. Heuberger, I. Staffell, N. Shah and N. Mac Dowell, Comput. Chem. Eng., 2017, 107, 247–256 CrossRef CAS.
- K. Riahi, E. Kriegler, N. Johnson, C. Bertram, M. den Elzen, J. Eom, M. Schaeffer, J. Edmonds, M. Isaac, V. Krey, T. Longden, G. Luderer, A. Méjean, D. L. McCollum, S. Mima, H. Turton, D. P. van Vuuren, K. Wada, V. Bosetti, P. Capros, P. Criqui, M. Hamdi-Cherif, M. Kainuma and O. Edenhofer, Technol. Forecase. Soc., 2015, 90(Part A), 8–23 CrossRef.
- G. P. Peters, Nat. Clim. Change, 2016, 6, 646–649 CrossRef.
- UNFCCC, Adoption of the Paris Agreement, United Nations Framework Convention on Climate Change (UNFCCC), Paris, France, 2015, http://unfccc.int/resource/docs/2015/cop21/eng/l09r01.pdf, accessed December 2016.
- J. Rogelj, G. Luderer, R. C. Pietzcker, E. Kriegler, M. Schaeffer, V. Krey and K. Riahi, Nat. Clim. Change, 2015, 5, 519–527 CrossRef.
- G. Luderer, R. C. Pietzcker, C. Bertram, E. Kriegler, M. Meinshausen and O. Edenhofer, Environ. Res. Lett., 2013, 8, 34033 CrossRef.
- P. Smith, Global Change Biol., 2016, 22, 1315–1324 CrossRef PubMed.
- T. DeVries, M. Holzer and F. Primeau, Nature, 2017, 542, 215–218 CrossRef CAS PubMed.
- M. D. Eisaman, K. Parajuly, A. Tuganov, C. Eldershaw, N. Chang and K. A. Littau, Energy Environ. Sci., 2012, 5, 7346–7352 CAS.
- H. D. Willauer, F. DiMascio, D. R. Hardy and F. W. Williams, Ind. Eng. Chem. Res., 2014, 53, 12192–12200 CrossRef CAS.
- P. Smith, S. J. Davis, F. Creutzig, S. Fuss, J. Minx, B. Gabrielle, E. Kato, R. B. Jackson, A. Cowie, E. Kriegler, D. P. van Vuuren, J. Rogelj, P. Ciais, J. Milne, J. G. Canadell, D. McCollum, G. Peters, R. Andrew, V. Krey, G. Shrestha, P. Friedlingstein, T. Gasser, A. Grubler, W. K. Heidug, M. Jonas, C. D. Jones, F. Kraxner, E. Littleton, J. Lowe, J. R. Moreira, N. Nakicenovic, M. Obersteiner, A. Patwardhan, M. Rogner, E. Rubin, A. Sharifi, A. Torvanger, Y. Yamagata, J. Edmonds and C. Yongsung, Nat. Clim. Change, 2016, 6, 42–50 CrossRef CAS.
- FAO, Land under cereal production (hectares), Food and Agriculture Organization, The World Bank Group, http://data.worldbank.org/indicator/AG.LND.CREL.HA, accessed July 2017.
- M. Fajardy and N. Mac Dowell, Energy Environ. Sci., 2017, 10, 1389–1426 CAS.
- T. Bruckner, I. A. Bashmakov, Y. Mulugetta, H. Chum, A. de la Vega Navarro, J. Edmonds, A. Faaij, B. Fungtammasan, A. Garg, E. Hertwich, D. Honnery, D. Infield, M. Kainuma, S. Khennas, S. Kim, H. B. Nimir, K. Riahi, N. Strachan, R. Wiser and X. Zhang, Energy Systems, in Climate Change 2014: Mitigation of Climate Change. Contribution of Working Group III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, 2014 Search PubMed.
- IPCC, Proposed outline of the IPCC special report on the impacts of global warming of 1.5 °C, Forty-fourth session of the Intergovernmental Panel on Climate Change (IPCC), Bangkok, Thailand, 2016 Search PubMed.
- J. Strefler, N. Bauer, T. Amann, E. Kriegler and J. Hartmann, Enhanced weathering and BECCS-are carbon dioxide removal technologies complements or substitutes? International Energy Workshop, 2015.
- K. Z. House, A. C. Baclig, M. Ranjan, E. A. van Nierop, J. Wilcox and H. J. Herzog, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20428–20433 CrossRef CAS PubMed.
- F. Kraxner, K. Aoki, S. Leduc, G. Kindermann, S. Fuss, J. Yang, Y. Yamagata, K.-I. Tak and M. Obersteiner, Renewable Energy, 2014, 61, 102–108 CrossRef.
- C. T. M. Clack, S. A. Qvist, J. Apt, M. Bazilian, A. R. Brandt, K. Caldeira, S. J. Davis, V. Diakov, M. A. Handschy, P. D. H. Hines, P. Jaramillo, D. M. Kammen, J. C. S. Long, M. G. Morgan, A. Reed, V. Sivaram, J. Sweeney, G. R. Tynan, D. G. Victor, J. P. Weyant and J. F. Whitacre, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6722–6727 CrossRef CAS PubMed.
- S. Fuss, C. D. Jones, F. Kraxner, G. P. Peters, P. Smith, M. Tavoni, D. P. van Vuuren, J. G. Canadell, R. B. Jackson, J. Milne, J. R. Moreira, N. Nakicenovic, A. Sharifi and Y. Yamagata, Environ. Res. Lett., 2016, 11, 115007 CrossRef.
- C. F. Heuberger, I. Staffell, N. Shah and N. Mac Dowell, Levelised value of technology-A systemic approach to technology valuation, 26th European Symposium on Computer Aided Process Engineering (ESCAPE 26), Computer Aided Chemical Engineering 38, 2016, pp. 721–726 Search PubMed.
- G. Strbac, M. Aunedi, D. Pudjianto, P. Djapic, S. Gammons and R. Druce, Understanding the Balancing Challenge, report for the Department of Energy and Climate Change (DECC), Imperial College London and NERA Economic Consulting, London, UK, 2012 Search PubMed.
- G. Strbac, M. Aunedi, D. Pudjianto, P. Djapic, F. Teng, A. Sturt, D. Jackravut, R. Sansom, V. Yufit and N. Brandon, Strategic Assessment of the Role and Value of Energy Storage Systems in the UK Low Carbon Energy Future, Energy Futures Lab, Imperial College London, 2012, http://https://www.carbontrust.com/media/129310/energy-storage-systems-role-value-strategic-assessment.pdf.
- D. Pudjianto, M. Aunedi, P. Djapic and G. Strbac, IEEE Trans. Smart Grid, 2014, 5, 1098–1109 CrossRef.
- R. G. Richels and G. J. Blanford, Energy Econ., 2008, 30, 2930–2946 CrossRef.
- J. Nelson, J. Johnston, A. Mileva, M. Fripp, I. Hoffman, A. Petros-Good, C. Blanco and D. M. Kammen, Energy Policy, 2012, 43, 436–447 CrossRef.
- N. E. Koltsaklis, A. S. Dagoumas, G. M. Kopanos, E. N. Pistikopoulos and M. C. Georgiadis, Appl. Energy, 2014, 115, 456–482 CrossRef.
- M. Wierzbowski, W. Lyzwa and I. Musial, Appl. Energy, 2016, 169, 93–111 CrossRef.
- R. Green and I. Staffell, Oxford Review of Economic Policy, 2016, 32, 282–303 Search PubMed.
- Climate Change Act, Part 1: Carbon target and budgeting, Parliament of the United Kingdom, 2008, http://www.legislation.gov.uk/ukpga/2008/27/pdfs/ukpga_20080027_en.pdf.
- Energy Act 2013 – Chapter 32, Department of Energy & Climate Change, Parliament of the United Kingdom, 2013.
- CCC, Fourth carbon budget review – Part 2: The cost-effective path to the 2050 target, Committee on Climate Change (CCC), London, UK, 2013, http://https://www.theccc.org.uk/wp-content/uploads/2013/12/1785a-CCC_AdviceRep_Singles_1.pdf.
- CCC, The Fifth Carbon Budget: The next step towards a low-carbon economy, Committee on Climate Change (CCC), London, UK, 2015, http://https://www.theccc.org.uk/wp-content/uploads/2015/11/Committee-on-Climate-Change-Fifth-Carbon-Budget-Report.pdf.
- CCC, Sectoral scenarios for the Fifth Carbon Budget: Technical report, Committee on Climate Change (CCC), London, UK, 2015, http://https://www.theccc.org.uk/wp-content/uploads/2015/11/Sectoral-scenarios-for-the-fifth-carbon-budget-Committee-on-Climate-Change.pdf.
- CCC, Meeting Carbon Budgets-2016 Progress Report to Parliament, Committee on Climate Change, London, UK, 2016, http://https://www.theccc.org.uk/publication/meeting-carbon-budgets-2016-progress-report-to-parliament/.
- House of Lords, The EU's Target forRenewable Energy: 20% by 2020, 27th report of session 2007–08, European Union Committee, House of Lords, London, UK, 2008, http://https://www.publications.parliament.uk/pa/ld200708/ldselect/ldeucom/175/175.pdf.
- 2Co Energy, Making the business case for CCS, Global CCS Institute, 2Co Energy, European Union, 2012, http://www.globalccsinstitute.com/publications/making-business-case-ccs.
- BEIS, Digest of United Kingdom energy statistics 2016, National Statistics, Department for Business, Energy & Industrial Strategy, London, UK, 2016.
- CCC, Power sector scenarios for the fifth carbon budget, Committee on Climate Change (CCC), London, UK, 2015, http://https://www.theccc.org.uk/publication/power-sector-scenarios-for-the-fifth-carbon-budget/.
- T. Page, i-manager's Journal on Power Systems Engineering, 2016, 4, 1–17 Search PubMed.
- M. Z. Jacobson, M. A. Delucchi, M. A. Cameron and B. A. Frew, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15060–15065 CrossRef CAS PubMed.
- F. Genoese, E. Drabik and C. Egenhofer, The EU power sector needs long-term price signals, Special report number 135, Centre for European Policy Studies (CEPS) Energy Climate House, Brussels, Belgium, 2016 Search PubMed.
- R. Gross, P. Heptonstall, D. Anderson, T. Green, M. Leach and J. Skea, The costs and impacts of intermittency: An assessment of the evidence on the costs and impacts of intermittent generation on the British electricity network, UK Energy Research Centre, Imperial College London, UK, 2006, http://www.ukerc.ac.uk/programmes/technology-and-policy-assessment/the-intermittency-report.html.
- A. Boston and H. K. Thomas, Managing flexibility whilst decarbonising the GB electricity system, Energy Research Partnership, London, UK, 2015, http://erpuk.org/project/managing-flexibility-of-the-electricity-sytem/.
- K. Foy, Electricity Generation Cost Model – 2013 Update of Non-Renewable Technologies, Report number 3512649A, Parsons Brinckerhoff, Prepared for Department of Energy and Climate Change (DECC), UK, 2013 Search PubMed.
- J. Munro and H. Windebank, Electricity Generation Costs Model – 2013 Update of Renewable Technologies, Report number 3511633B, Parsons Brinckerhoff, Prepared for Department of Energy and Climate Change (DECC), UK, 2013 Search PubMed.
- DECC, Oral statement to parliament: Agreement reached on new nuclear power station at Hinkley, Department of Energy & Climate Change (DECC), UK, 2013, http://https://www.gov.uk/government/speeches/agreement-reached-on-new-nuclear-power-station-at-hinkley.
- DECC, Press release: Initial agreement reached on new nuclear power station at Hinkley, Department of Energy & Climate Change (DECC), UK, 2013, http://https://www.gov.uk/government/news/initial-agreement-reached-on-new-nuclear-power-station-at-hinkley.
- Ofgem, Renewables Obligation (RO), http://https://www.ofgem.gov.uk/environmental-programmes/ro, accessed October 2016.
- E. Durusut, S. Slater, S. Murray and P. Hare, CCS Sector Development Scenarios in the UK, Final report prepared for the Energy Technologies Institute, Element Energy and Pöyry, 2015, http://www.eti.co.uk/library/ccs-sector-development-scenarios-in-the-uk.
- R. Oxburgh, Lowest cost decarbonisation for the UK: The critical role of CCS, Report to the Secretary of State for Business, Energy and Industrial Strategy from the Parliamentary Advisory Group on Carbon Capture and Storage (CCS), 2016.
- Elexon, Electricity generation data for Great Britain from the National Grid, reported during 2012, 2014.
- JRC, Photovoltaic geographic information system, web tool for photovoltaic output, European Commission, Joint Research Centre (JRC), Brussels, Belgium, 2014, http://re.jrc.ec.europa.eu/pvgis/apps4/pvest.php.
- National Grid, UK Future Energy Scenarios – July 2013 edition, Warwick, UK, 2013, http://www2.nationalgrid.com/WorkArea/DownloadAsset.aspx?id=10451.
- DECC, National Renewable Energy Action Plan for the United Kingdom, Article 4 of the Renewable Energy Directive 2009/28/EC, Department of Energy & Climate Change, United Kingdom, 2009.
- M. R. Haines and J. E. Davison, Energy Procedia, 2009, 1, 1457–1464 CrossRef CAS.
- H. Chalmers and J. Gibbins, Fuel, 2007, 86, 2109–2123 CrossRef CAS.
- S. M. Cohen, G. T. Rochelle and M. E. Webber, Int. J. Greenhouse Gas Control, 2012, 8, 180–195 CrossRef.
- D. Patiño-Echeverri and D. C. Hoppock, Environ. Sci. Technol., 2012, 46, 1243–1252 CrossRef PubMed.
- P. C. van der Wijk, A. S. Brouwer, M. van den Broek, T. Slot, G. Stienstra, W. van der Veen and A. P. C. Faaij, Int. J. Greenhouse Gas Control, 2014, 28, 216–233 CrossRef CAS.
- T. Van Peteghem and E. Delarue, Int. J. Greenhouse Gas Control, 2014, 21, 203–213 CrossRef CAS.
- N. Mac Dowell and N. Shah, Comput. Chem. Eng., 2015, 74, 169–183 CrossRef CAS.
- E. Mechleri, P. S. Fennell and N. Mac Dowell, Int. J. Greenhouse Gas Control, 2017, 59, 24–39 CrossRef CAS.
- J.-P. Tranier, R. Dubettier, A. Darde and N. Perrin, Energy Procedia, 2011, 4, 966–971 CrossRef.
- Y. Hu, X. Li, H. Li and J. Yan, Appl. Energy, 2013, 112, 747–754 CrossRef CAS.
- C. F. Heuberger, I. Staffell, N. Shah and N. Mac Dowell, Valuing Flexibility in CCS Power Plant. Final report on the FlexEVAL project, International Energy Agency Greenhouse Gas R&D Programme (IEAGHG), 2017, http://www.ieaghg.org/exco_docs/2017-09.pdf.
- J. P. Birat, Steel sectoral report, contribution to the UNIDO roadmap on CCS (fifth draft). Prepared for the UNIDO Global Technology Roadmap for CCS in Industry-Sectoral Experts Meeting in Amsterdam, 24 September 2010, 2010.
- A. Carpenter, CO2 abatement in the iron and steel industry, report CCC/193, IEA Clean Coal Centre, London, UK, 2012.
- GCCSI, Global status of CCS. Special report: Introduction to industrial carbon capture and storage, Global CCS Institute, Melbourne, Australia, 2016.
- D. Leeson, P. Fennell, N. Shah, C. Petit and N. Mac Dowell, A techno-economic analysis and systematic review of carbon capture and storage (CCS) applied to the iron and steel, cement, oil refining and pulp and paper industries. 13th International Conference on Greenhouse Gas Control Technologies (GHGT-13), Lausanne, Switzerland. Energy Procedia, 2016.
- D. Leeson, N. Mac Dowell, N. Shah, C. Petit and P. S. Fennell, Int. J. Greenhouse Gas Control, 2017, 61, 71–84 CrossRef CAS.
- T. A. Napp, A. Gambhir, T. P. Hills, N. Florin and P. S. Fennell, Renewable Sustainable Energy Rev., 2014, 30, 616–640 CrossRef CAS.
- IEA and UNIDO, Technology roadmap: Carbon capture and storage in industrial applications, International Energy Agency and United Nations Industrial Development Organisation, 2011, http://www.iea.org/publications/freepublications/publication/ccs_industry.pdf, accessed February 2017.
- M. Bui, I. Gunawan, V. Verheyen and E. Meuleman, Dynamic operation of liquid absorbent-based postcombustion CO2 capture plants, Absorption-based Post-combustion Capture of Carbon Dioxide, Woodhead Publishing, Cambridge, 2016, pp. 589–621 Search PubMed.
- N. Mahasenan and D. R. Brown, Beyond the big picture: Characterizaton of CO2-laden streams and implications for capture technologies, 7th International Conference on Greenhouse Gas Control Technologies, Oxford, UK, 2005, pp. 1817–1820.
- M. M. F. Hasan, R. C. Baliban, J. A. Elia and C. A. Floudas, Ind. Eng. Chem. Res., 2012, 51, 15642–15664 CrossRef CAS.
- M. M. F. Hasan, R. C. Baliban, J. A. Elia and C. A. Floudas, Ind. Eng. Chem. Res., 2012, 51, 15665–15682 CrossRef CAS.
- B. Fais, N. Sabio and N. Strachan, Appl. Energy, 2016, 162, 699–712 CrossRef.
- A. Arasto, E. Tsupari, J. Kärki, E. Pisilä and L. Sorsamäki, Int. J. Greenhouse Gas Control, 2013, 16, 271–277 CrossRef CAS.
- D. E. Wiley, M. T. Ho and A. Bustamante, Energy Procedia, 2011, 4, 2654–2661 CrossRef.
- E. Tsupari, J. Kärki, A. Arasto and E. Pisilä, Int. J. Greenhouse Gas Control, 2013, 16, 278–286 CrossRef CAS.
- T. Kuramochi, A. Ramrez, W. Turkenburg and A. Faaij, Prog. Energy Combust. Sci., 2012, 38, 87–112 CrossRef CAS.
- N. Pardo and J. A. Moya, Energy, 2013, 54, 113–128 CrossRef CAS.
- G. F. Porzio, B. Fornai, A. Amato, N. Matarese, M. Vannucci, L. Chiappelli and V. Colla, Appl. Energy, 2013, 112, 818–833 CrossRef.
- J.-C. Brunke and M. Blesl, Energy Policy, 2014, 67, 431–446 CrossRef.
- N. Karali, T. Xu and J. Sathaye, Appl. Energy, 2014, 120, 133–146 CrossRef CAS.
- J. A. Moya and N. Pardo, J. Cleaner Prod., 2013, 52, 71–83 CrossRef CAS.
- J. P. Birat, Carbon dioxide (CO2) capture and storage technology in the iron and steel industry, in Developments and innovation in carbon dioxide (CO2) capture and storage technology, Carbon dioxide (CO2) capture, transport and industrial applications, Woodhead Publishing Ltd., Cambridge, UK, 2010, vol. 1 Search PubMed.
- T. Kuramochi, A. Ramírez, W. Turkenburg and A. Faaij, Energy Procedia, 2011, 4, 1981–1988 CrossRef.
- Posco and Primetals Technologies, The Finex process: Economic and environmentally safe ironmaking, Posco Ltd. (Incheon, South Korea) and Primetals Technologies Ltd. (Linz, Austria), 2015, http://primetals.com/en/technologies/ironmaking/finex%C2%AE/Lists/FurtherInformation/The%20Finex%20process.pdf, accessed March 2017.
- K. Meijer, C. Guenther and R. J. Dry, HIsarna Pilot Plant Project, METEC Conference, Germany, 2011, http://www.riotinto.com/documents/_Iron%20Ore/HIsarna_0711_METEC_Conference.pdf.
- J. van der Stel, K. Meijer, C. Teerhuis, C. Zeijlstra, G. Keilman and M. Ouwehand, Update to the Developments of HIsarna: An ULCOS alternative ironmaking process, IEAGHG/IETS Iron and steel industry CCUS and process integration workshop, IEA Greenhouse Gas R&D Programme, 2013.
- GCCSI, Abu Dhabi CCS Project (Phase 1 being Emirates Steel Industries (ESI) CCS Project), Global CCS Institute, 2016, http://https://www.globalccsinstitute.com/projects/abu-dhabi-ccs-project-phase-1-being-emirates-steel-industries-esi-ccs-project.
- Globalcement.com, CEMENT 101-An introduction to the World's most important building material, accessed February 2017.
- T. Hills, D. Leeson, N. Florin and P. Fennell, Environ. Sci. Technol., 2016, 50, 368–377 CrossRef CAS PubMed.
- C. C. Dean, J. Blamey, N. Florin, M. J. Al-Jeboori and P. S. Fennell, Chem. Eng. Res. Des., 2011, 89, 836–855 CrossRef CAS.
- C. C. Dean, D. Dugwell and P. S. Fennell, Energy Environ. Sci., 2011, 4, 2050–2053 CAS.
- IEA, Global action to advance carbon capture and storage: A focus on industrial applications-Annex to tracking clean energy progress, International Energy Agency, 2013, http://https://www.iea.org/publications/freepublications/publication/CCS_Annex.pdf.
- O. Graff, CCS in Aker Solutions with focus on cement industry, Norcem International CCS Conference, 2015.
- F. S. Zeman and K. S. Lackner, International Cement Review, 2006, 55–58 Search PubMed.
- ECRA, TR-ECRA-119/2012 Technical Report on Phase III of ECRA CCS Project, European Cement Research Academy, 2012, http://https://www.ecra-online.org/fileadmin/redaktion/files/pdf/ECRA_Technical_Report:CCS_Phase_III.pdf, accessed 15/02/17.
- L. Zheng, T. P. Hills and P. Fennell, Faraday Discuss., 2016, 192, 113–124 RSC.
- C. Dean, T. Hills, N. Florin, D. Dugwell and P. S. Fennell, Energy Procedia, 2013, 37, 7078–7090 CrossRef CAS.
- A. Telesca, D. Calabrese, M. Marroccoli, M. Tomasulo, G. L. Valenti, G. Duelli and F. Montagnaro, Fuel, 2014, 118, 202–205 CrossRef CAS.
- T. Hills, M. Sceats, D. Rennie and P. Fennell, LEILAC: Low cost CO2 capture for the cement and lime industries, 13th International Conference on Greenhouse Gas Control Technologies (GHGT-13), Lausanne, Switzerland, Energy Procedia, 2016.
- S. Evans, Around the world in 22 carbon capture projects, CarbonBrief Clear on Climate, 2014, http://https://www.carbonbrief.org/around-the-world-in-22-carbon-capture-projects.
- D. J. Barker, S. A. Turner, P. A. Napier-Moore, M. Clark and J. E. Davison, Energy Procedia, 2009, 1, 87–94 CrossRef CAS.
- M. C. Romano, M. Spinelli, S. Campanari, S. Consonni, M. Marchi, N. Pimpinelli and G. Cinti, Energy Procedia, 2014, 61, 500–503 CrossRef CAS.
- M. C. Romano, M. Spinelli, S. Campanari, S. Consonni, G. Cinti, M. Marchi and E. Borgarello, Energy Procedia, 2013, 37, 7091–7099 CrossRef CAS.
- X. Liang and J. Li, Energy Convers. Manage., 2012, 64, 454–465 CrossRef CAS.
- DECC, Industrial Decarbonisation & Energy Efficiency Roadmaps to 2050, prepared by Parsons Brinkerhoff and DNV GL for UK Departments of Energy and Climate Change and Business, Innovation and Skills, 2015.
- T. A. Napp, K. S. Sum, T. Hills and P. Fennell, Attitudes and barriers to deployment of CCS from industrial sources in the UK-Grantham Report 6, Grantham Institute for Climate Change, Imperial College London, 2014, http://https://www.imperial.ac.uk/media/imperial-college/grantham-institute/public/publications/institute-reports-and-analytical-notes/Attitudes-and-Barriers-to-CCS-GR6.pdf.
- L.-M. Bjerge and P. Brevik, Energy Procedia, 2014, 63, 6455–6463 CrossRef CAS.
- M.-H. Chang, W.-C. Chen, C.-M. Huang, W.-H. Liu, Y.-C. Chou, W.-C. Chang, W. Chen, J.-Y. Cheng, K.-E. Huang and H.-W. Hsu, Energy Procedia, 2014, 63, 2100–2108 CrossRef CAS.
- M. Schneider, ECRA's Oxyfuel project, Norcem International CCS Conference, Langesund, Norway, 2015.
- OGCI, Oil and Gas Climate Initiative (OGCI), accessed January 2017.
- S. Nyquist and J. Ruys, CO2abatement: Exploring options for oil and natural gas companies, McKinsey & Company: Oil & Gas, 2010, http://www.mckinsey.com/industries/oil-and-gas/our-insights/co2-abatement-exploring-options-for-oil-and-natural-gas-companies.
- Macrotrends, Crude Oil Prices-70 Year Historical Chart, accessed January 2017.
- M. C. Guilford, C. A. S. Hall, P. O'Connor and C. J. Cleveland, Sustainability, 2011, 3, 1866–1887 CrossRef.
- L. Bredeson, R. Quiceno-Gonzalez, X. Riera-Palou and A. Harrison, Int. J. Life Cycle Assess., 2010, 15, 817–826 CrossRef CAS.
- I. Palou-Rivera, J. Han and M. Wang, Updates to petroleum refining and upstream emissions, Center for Transportation Research Argonne National Laboratory, 2011, http://https://greet.es.anl.gov/files/petroleum, accessed January 2017.
- K. Harland, H. Pershad, S. Slater, G. Cook and J. Watt, Potential for the application of CCS to UK industry and natural gas power generation. Final report, Issue 3, prepared for Committee on Climate Change, Element Energy Limited, Cambridge UK, 2010, http://www.element-energy.co.uk/wordpress/wp-content/uploads/2012/05/CCS_on_gas_and_industry_2010.pdf.
- Parsons Brinckerhoff and DNV GL, Industrial decarbonisation & energy efficiency roadmaps to 2050-Oil refining, prepared for the Department of Energy and Climate Change and the Department for Business, Innovation and Skills, 2015, http://https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/416671/Oil_Refining_Report.pdf.
- V. Andersson, P. Franck and T. Berntsson, Int. J. Greenhouse Gas Control, 2016, 45, 130–138 CrossRef CAS.
- A. I. Escudero, S. Espatolero and L. M. Romeo, Int. J. Greenhouse Gas Control, 2016, 45, 118–129 CrossRef CAS.
- J. Adánez, C. Dueso, L. F. de Diego, F. García-Labiano, P. Gayán and A. Abad, Ind. Eng. Chem. Res., 2009, 48, 2509–2518 CrossRef.
- M. T. Shah, R. P. Utikar, V. K. Pareek, G. M. Evans and J. B. Joshi, Chem. Eng. Res. Des., 2016, 111, 403–448 CrossRef CAS.
- Q. Q. Song, Q. Z. Jiang, Z. Z. Song, B. Yuan and W. J. Song, Adv. Mater. Res., 2014, 864–867, 1725–1731 Search PubMed.
- F. Untalan, Australia signs contract with Japan to ship hydrogen, International Business Times, http://www.ibtimes.com.au/australia-signs-contract-japan-ship-hydrogen-1539612, January 2017.
- A. Kohl and R. Nielsen, Gas Purification, Gulf Publishing Company, Houston, Texas, 5th edn, 1997 Search PubMed.
- A. Cousins, A. Cottrell, A. Lawson, S. Huang and P. H. M. Feron, Greenhouse Gases: Sci. Technol., 2012, 2, 329–345 CrossRef CAS.
- N. S. Kwak, J. H. Lee, I. Y. Lee, K. R. Jang and J. G. Shim, Energy, 2012, 47, 41–46 CrossRef CAS.
- H. P. Mangalapally and H. Hasse, Chem. Eng. Res. Des., 2011, 89, 1216–1228 CrossRef CAS.
- M. Stec, A. Tatarczuk, L. Wieclaw-Solny, A. Krotki, T. Spietz, A. Wilk and D. Spiewak, Clean Technol. Environ. Policy, 2016, 18, 151–160 CrossRef CAS.
- H. P. Mangalapally and H. Hasse, Chem. Eng. Sci., 2011, 66, 5512–5522 CrossRef CAS.
- M. Rabensteiner, G. Kinger, M. Koller and C. Hochenauer, Int. J. Greenhouse Gas Control, 2016, 51, 106–117 CrossRef CAS.
- D. J. Heldebrant, P. K. Koech, V.-A. Glezakou, R. Rousseau, D. Malhotra and D. C. Cantu, Chem. Rev., 2017, 117, 9594–9624 CrossRef CAS PubMed.
- SaskPower, The world's first post-combustion coal-fired CCS facility, http://www.saskpowerccs.com, accessed October 2016.
- G. Puxty and M. Maeder, The fundamentals of post combustion capture, Absorption-based Post-combustion Capture of Carbon Dioxide, Woodhead Publishing, Cambridge, 2016, pp. 13–33 Search PubMed.
- G. Xu, C. Zhang, S. Qin and Y. Wang, Ind. Eng. Chem. Res., 1992, 31, 921–927 CrossRef CAS.
- S. A. Freeman, R. Dugas, D. H. Van Wagener, T. Nguyen and G. T. Rochelle, Int. J. Greenhouse Gas Control, 2010, 4, 119–124 CrossRef CAS.
- G. Rochelle, E. Chen, S. Freeman, D. Van Wagener, Q. Xu and A. Voice, Chem. Eng. J., 2011, 171, 725–733 CrossRef CAS.
- R. E. Dugas and G. T. Rochelle, J. Chem. Eng. Data, 2011, 56, 2187–2195 CrossRef CAS.
- S. K. Dash, A. Samanta, A. N. Samanta and S. S. Bandyopadhyay, Fluid Phase Equilib., 2011, 300, 145–154 CrossRef CAS.
- S. A. Freeman, J. Davis and G. T. Rochelle, Int. J. Greenhouse Gas Control, 2010, 4, 756–761 CrossRef CAS.
- A. Cousins, S. Huang, A. Cottrell, P. H. M. Feron, E. Chen and G. T. Rochelle, Greenhouse Gases: Sci. Technol., 2015, 5, 7–16 CrossRef CAS.
- A. Cousins, P. T. Nielsen, S. Huang, R. Rowland, B. Edwards, A. Cottrell, E. Chen, G. T. Rochelle and P. H. M. Feron, Int. J. Greenhouse Gas Control, 2015, 37, 256–263 CrossRef CAS.
- N. Dai, A. D. Shah, L. H. Hu, M. J. Plewa, B. McKague and W. A. Mitch, Environ. Sci. Technol., 2012, 46, 9793–9801 CrossRef CAS PubMed.
- H. Hikita, A. Asai, H. Ishikawa and M. Honda, Chem. Eng. J., 1977, 14, 27–30 CrossRef CAS.
- J. L. Li, A. Henni and P. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2007, 46, 4426–4434 CrossRef CAS.
- R. H. Weiland and O. Trass, Can. J. Chem. Eng., 1971, 49, 767–772 CrossRef CAS.
- S. Zhou, X. Chen, T. Nguyen, A. K. Voice and G. T. Rochelle, ChemSusChem, 2010, 3, 913–918 CrossRef CAS PubMed.
- T. Nguyen, M. Hilliard and G. T. Rochelle, Int. J. Greenhouse Gas Control, 2010, 4, 707–715 CrossRef CAS.
- M. Rabensteiner, G. Kinger, M. Koller, G. Gronald and C. Hochenauer, Int. J. Greenhouse Gas Control, 2014, 27, 1–14 CrossRef CAS.
- E. Lemaire, P. A. Bouillon and K. Lettat, Oil Gas Sci. Technol., 2014, 69, 1069–1080 CrossRef.
- K. Ballerat-Busserolles, M. R. Simond, Y. Coulier and J. Y. Coxam, Pure Appl. Chem., 2014, 86, 233–243 CrossRef CAS.
- W. Conway, Y. Beyad, M. Maeder, R. Burns, P. Feron and G. Puxty, Ind. Eng. Chem. Res., 2014, 53, 16715–16724 CrossRef CAS.
- Y. Coulier, A. Lowe, P. R. Tremaine, J. Y. Coxam and K. Ballerat-Busserolles, Int. J. Greenhouse Gas Control, 2016, 47, 322–329 CrossRef CAS.
- L. Dubois and D. Thomas, Chem. Eng. Technol., 2012, 35, 513–524 CrossRef CAS.
- H. L. Liu, G. Y. Chen and Z. W. Liang, Int. J. Greenhouse Gas Control, 2016, 50, 206–217 CrossRef CAS.
- B. J. Sherman, A. F. Ciftja and G. T. Rochelle, Chem. Eng. Sci., 2016, 153, 295–307 CrossRef CAS.
- P. D. Vaidya and S. G. Jadhav, Can. J. Chem. Eng., 2014, 92, 2218–2227 CrossRef CAS.
- G. Sartori and D. W. Savage, Ind. Eng. Chem. Fundam., 1983, 22, 239–249 CAS.
- D. J. Seo and W. H. Hong, Ind. Eng. Chem. Res., 2000, 39, 2062–2067 CrossRef CAS.
- Z. Y. Yang, A. N. Soriano, A. R. Caparanga and M. H. Li, J. Chem. Thermodyn., 2010, 42, 659–665 CrossRef CAS.
- A. A. Khan, G. N. Halder and A. K. Saha, Int. J. Greenhouse Gas Control, 2016, 44, 217–226 CrossRef CAS.
- P. Bruder, A. Grimstvedt, T. Mejdell and H. F. Svendsen, Chem. Eng. Sci., 2011, 66, 6193–6198 CrossRef CAS.
- F. Bougie and M. C. Iliuta, Int. J. Greenhouse Gas Control, 2014, 29, 16–21 CrossRef CAS.
- T. L. Wang and K. J. Jens, Int. J. Greenhouse Gas Control, 2014, 24, 98–105 CrossRef CAS.
- M. H. Li and K. P. Shen, Fluid Phase Equilib., 1993, 85, 129–140 CrossRef CAS.
- K. P. Shen and M. H. Li, J. Chem. Eng. Data, 1992, 37, 96–100 CrossRef CAS.
- M. Edali, A. Aboudheir and R. Idem, Int. J. Greenhouse Gas Control, 2009, 3, 550–560 CrossRef CAS.
- A. Naami, T. Sema, M. Edali, Z. W. Liang, R. Idem and P. Tontiwachwuthikul, Int. J. Greenhouse Gas Control, 2013, 19, 3–12 CrossRef CAS.
- N. Ramachandran, A. Aboudheir, R. Idem and P. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2006, 45, 2608–2616 CrossRef CAS.
- T. Sema, A. Naami, K. Y. Fu, M. Edali, H. L. Liu, H. C. Shi, Z. W. Liang, R. Idem and P. Tontiwachwuthikul, Chem. Eng. J., 2012, 209, 501–512 CrossRef CAS.
- O. Lawal, A. Bello and R. Idem, Ind. Eng. Chem. Res., 2005, 44, 1874–1896 CrossRef CAS.
- R. Idem, M. Wilson, P. Tontiwachwuthikul, A. Chakma, A. Veawab, A. Aroonwilas and D. Gelowitz, Ind. Eng. Chem. Res., 2006, 45, 2414–2420 CrossRef CAS.
- C. Nwaoha, C. Saiwan, P. Tontiwachwuthikul, T. Supap, W. Rongwong, R. Idem, M. J. AL-Marri and A. Benamor, J. Nat. Gas Sci. Eng., 2016, 33, 742–750 CrossRef CAS.
- L. V. van der Ham, E. L. V. Goetheer, E. S. Fernandez, M. R. M. Abu-Zahra and T. J. H. Vlugt, Precipitating amino acid solutions, Absorption-Based Post-combustion Capture of Carbon Dioxide, Woodhead Publishing, Cambridge, 2016, pp. 103–119 Search PubMed.
- S. Wang and Z. Xu, Dual-liquid phase systems, Absorption-Based Post-combustion Capture of Carbon Dioxide, Woodhead Publishing, Cambridge, 2016, pp. 201–223 Search PubMed.
- R. Sakwattanapong, A. Aroonwilas and A. Veawab, Ind. Eng. Chem. Res., 2005, 44, 4465–4473 CrossRef CAS.
- V. Darde, K. Thomsen, W. J. M. van Well and E. H. Stenby, Energy Procedia, 2009, 1, 1035–1042 CrossRef CAS.
- N. Dave, T. Do, G. Puxty, R. Rowland, P. H. M. Feron and M. I. Attalla, Energy Procedia, 2009, 1, 949–954 CrossRef CAS.
- N. Yang, H. Yu, L. C. Li, D. Y. Xu, W. F. Han and P. Feron, Ind. Eng. Chem. Fundam., 2014, 69, 931–945 Search PubMed.
- C. Anderson, T. Harkin, M. Ho, K. Mumford, A. Qader, G. Stevens and B. Hooper, Energy Procedia, 2013, 37, 225–232 CrossRef CAS.
- C. Anderson, B. Hooper, A. Qader, T. Harkin, K. Smith, K. Mumford, J. Pandit, M. Ho, A. Lee, N. Nicholas, Indrawan, J. Gouw, J. Xiao, N. Thanumurthy, N. Temple, G. Stevens and D. Wiley, Energy Procedia, 2014, 63, 1773–1780 CrossRef CAS.
- K. Smith, G. Xiao, K. Mumford, J. Gouw, I. Indrawan, N. Thanumurthy, D. Quyn, R. Cuthbertson, A. Rayer, N. Nicholas, A. Lee, G. da Silva, S. Kentish, T. Harkin, A. Qader, C. Anderson, B. Hooper and G. Stevens, Energy Fuels, 2014, 28, 299–306 CrossRef CAS.
- E. Sanchez-Fernandez, K. Heffernan, L. van der Ham, M. J. G. Linders, E. L. V. Goetheer and T. J. H. Vlugt, Energy Procedia, 2014, 63, 727–738 CrossRef CAS.
- E. Sanchez-Fernandez, K. Heffernan, L. van der Ham, M. J. G. Linders, D. W. F. Brilman, E. L. V. Goetheer and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2014, 53, 2348–2361 CrossRef CAS.
- L. Raynal, P. Alix, P. A. Bouillon, A. Gomez, M. L. de Nailly, M. Jacquin, J. Kittel, A. di Lella, P. Mougin and J. Trapy, Energy Procedia, 2011, 4, 779–786 CrossRef CAS.
- U. Liebenthal, D. D. D. Pinto, J. G. M. S. Monteiro, H. F. Svendsen and A. Kather, Energy Procedia, 2013, 37, 1844–1854 CrossRef CAS.
- J. Zhang, Study on CO2 capture using thermomorphic biphasic solvents with energy-efficient regeneration, PhD thesis, TU Dortmund University, 2013 Search PubMed.
- C. K. Ahn, H. W. Lee, Y. S. Chang, K. Han, J. Y. Kim, C. H. Rhee, H. D. Chun, M. W. Lee and J. M. Park, Int. J. Greenhouse Gas Control, 2011, 5, 1606–1613 CrossRef CAS.
- V. Darde, K. Thomsen, W. J. M. van Well and E. H. Stenby, Int. J. Greenhouse Gas Control, 2010, 4, 131–136 CrossRef CAS.
- P. M. Mathias, S. Reddy and J. P. O'Connell, Int. J. Greenhouse Gas Control, 2010, 4, 174–179 CrossRef CAS.
- K. A. Mumford, Y. Wu, K. H. Smith and G. W. Stevens, Front. Chem. Sci. Eng., 2015, 9, 125–141 CrossRef CAS.
- K. H. Smith, N. J. Nicholas and G. W. Stevens, Inorganic salt solutions for post-combustion capture, Absorption-based Post-combustion Capture of Carbon Dioxide, Woodhead Publishing, Cambridge, 2016, pp. 145–166 Search PubMed.
- K. A. Mumford, K. H. Smith, C. J. Anderson, S. Shen, W. Tao, Y. A. Suryaputradinata, A. Qader, B. Hooper, R. A. Innocenzi, S. E. Kentish and G. W. Stevens, Energy Fuels, 2012, 26, 138–146 CrossRef CAS.
- K. Endo, G. Stevens, B. Hooper, S. Kentish and C. Anderson, A process and plant for removing acid gases, EP20110771408, 2011 Search PubMed.
- UNO, UNO Technology Pty Ltd, http://unotech.com.au/, 2014.
- U. E. Aronu, H. F. Svendsen and K. A. Hoff, Int. J. Greenhouse Gas Control, 2010, 4, 771–775 CrossRef CAS.
- M. Rabensteiner, G. Kinger, M. Koller, G. Gronald, S. Unterberger and C. Hochenauer, Int. J. Greenhouse Gas Control, 2014, 29, 1–15 CrossRef CAS.
- M. Rabensteiner, G. Kinger, M. Koller and C. Hochenauer, Int. J. Greenhouse Gas Control, 2015, 42, 562–570 CrossRef CAS.
- E. S. Fernandez, K. Heffernan, L. V. van der Ham, M. J. G. Linders, E. Eggink, F. N. H. Schrama, D. W. F. Brilman, E. L. V. Goetheer and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2013, 52, 12223–12235 CrossRef.
- R. M. Stephenson, J. Chem. Eng. Data, 1993, 38, 634–637 CrossRef CAS.
- Y. H. Tan, Study of CO2-absorption into thermomorphic lipophilic amine solvents, PhD thesis, TU Dortmund University, 2010 Search PubMed.
- X. Zhang, Studies on multiphase CO2 capture systems, PhD thesis, TU Dortmund University, 2007 Search PubMed.
- Q. Ye, X. L. Wang and Y. Q. Lu, Int. J. Greenhouse Gas Control, 2015, 39, 205–214 CrossRef CAS.
- A. F. Ciftja, A. Hartono and H. F. Svendsen, Chem. Eng. Sci., 2013, 102, 378–386 CrossRef CAS.
- F. A. Chowdhury, H. Yamada, T. Higashii, Y. Matsuzaki and S. Kazama, Energy Procedia, 2013, 37, 265–272 CrossRef CAS.
- H. C. Shi, T. Sema, A. Naami, Z. W. Liang, R. Idem and P. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2012, 51, 8608–8615 CrossRef CAS.
- S. Singto, T. Supap, R. Idem, P. Tontiwachwuthikul, S. Tantayanon, M. J. Al-Marri and A. Benamor, Sep. Purif. Technol., 2016, 167, 97–107 CrossRef CAS.
- Q. Yang, G. Puxty, S. James, M. Bown, P. Feron and W. Conway, Energy Fuels, 2016, 30, 7503–7510 CrossRef CAS.
- E. S. Kikkinides, R. T. Yang and S. H. Cho, Ind. Eng. Chem. Res., 1993, 32, 2714–2720 CrossRef CAS.
- K. T. Chue, J. N. Kim, Y. J. Yoo, S. H. Cho and R. T. Yang, Ind. Eng. Chem. Res., 1995, 34, 591–598 CrossRef CAS.
- M. Ishibashi, H. Ota, N. Akutsu, S. Umeda, M. Tajika, J. Izumi, A. Yasutake, T. Kabata and Y. Kageyama, Energy Convers. Manage., 1996, 37, 929–933 CrossRef CAS.
- I. Ahmed and S. H. Jhung, Chem. Eng. J., 2017, 310, 197–215 CrossRef CAS.
- A. Sayari and Y. Belmabkhout, US Pat., US9314730 B1, 2016 Search PubMed.
- P. A. Webley, Adsorption, 2014, 20, 225–231 CrossRef CAS.
- J. Duan, W. Jin and S. Kitagawa, Coord. Chem. Rev., 2017, 332, 48–74 CrossRef CAS.
- C. A. Grande and A. E. Rodrigues, Int. J. Greenhouse Gas Control, 2008, 2, 194–202 CAS.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- A. Kumar, D. G. Madden, M. Lusi, K.-J. Chen, E. A. Daniels, T. Curtin, J. J. Perry and M. J. Zaworotko, Angew. Chem., Int. Ed., 2015, 54, 14372–14377 CrossRef CAS PubMed.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 4228 CAS.
- J. D. Figueroa, T. Fout, S. Plasynski, H. McIlvried and R. D. Srivastava, Int. J. Greenhouse Gas Control, 2008, 2, 9–20 CrossRef CAS.
- M. Olivares-Marín and M. M. Maroto-Valer, Greenhouse Gases: Sci. Technol., 2012, 2, 20–35 CrossRef.
- CO2CRC, The CO2CRC H3 Capture Project, http://http://old.co2crc.com.au/research/demo_postcombustion.html, CO2CRC Limited, The University of Melbourne, Australia, accessed January 2017.
- S. Tonomura, Energy Procedia, 2013, 37, 7160–7167 CrossRef CAS.
- W. H. Saima, Y. Mogi and T. Haraoka, Energy Procedia, 2013, 37, 7152–7159 CrossRef CAS.
- S. D. Kenarsari, D. Yang, G. Jiang, S. Zhang, J. Wang, A. G. Russell, Q. Wei and M. Fan, RSC Adv., 2013, 3, 22739–22773 RSC.
- Z. Zhang, Z.-Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868–2899 CAS.
- Y.-S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS PubMed.
- X. Zhang, X. Zhang, H. Dong, Z. Zhao, S. Zhang and Y. Huang, Energy Environ. Sci., 2012, 5, 6668–6681 CAS.
- J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2011, 112, 724–781 CrossRef PubMed.
- Q. Wang, J. Luo, Z. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42–55 CAS.
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438–1463 CrossRef CAS.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- J. M. Huck, L.-C. Lin, A. H. Berger, M. N. Shahrak, R. L. Martin, A. S. Bhown, M. Haranczyk, K. Reuter and B. Smit, Energy Environ. Sci., 2014, 7, 4132–4146 CAS.
- M. Hefti, L. Joss, Z. Bjelobrk and M. Mazzotti, Faraday Discuss., 2016, 192, 153–179 RSC.
- R. Haghpanah, A. Majumder, R. Nilam, A. Rajendran, S. Farooq, I. A. Karimi and M. Amanullah, Ind. Eng. Chem. Res., 2013, 52, 4249–4265 CrossRef CAS.
- R. Haghpanah, R. Nilam, A. Rajendran, S. Farooq and I. A. Karimi, AIChE J., 2013, 59, 4735–4748 CrossRef CAS.
- B. J. Maring and P. A. Webley, Int. J. Greenhouse Gas Control, 2013, 15, 16–31 CrossRef CAS.
- G. N. Nikolaidis, E. S. Kikkinides and M. C. Georgiadis, Ind. Eng. Chem. Res., 2016, 55, 635–646 CrossRef CAS.
- E. L. First, M. M. F. Hasan and C. A. Floudas, AIChE J., 2014, 60, 1767–1785 CrossRef CAS.
- Y. G. Chung, D. A. Gómez-Gualdrón, P. Li, K. T. Leperi, P. Deria, H. Zhang, N. A. Vermeulen, J. F. Stoddart, F. You, J. T. Hupp, O. K. Farha and R. Q. Snurr, Sci. Adv., 2016, 2, e1600909 Search PubMed.
- K. Kim, Y. Son, W. B. Lee and K. S. Lee, Int. J. Greenhouse Gas Control, 2013, 17, 13–24 CrossRef CAS.
- R. P. Lively, R. R. Chance and W. J. Koros, Ind. Eng. Chem. Res., 2010, 49, 7550–7562 CrossRef CAS.
- P. Bollini, N. A. Brunelli, S. A. Didas and C. W. Jones, Ind. Eng. Chem. Res., 2012, 51, 15145–15152 CrossRef CAS.
- C. Shen, Z. Liu, P. Li and J. Yu, Ind. Eng. Chem. Res., 2012, 51, 5011–5021 CrossRef CAS.
- C. Z. Shen, J. G. Yu, P. Li, C. A. Grande and A. E. Rodrigues, Adsorption, 2011, 17, 179–188 CrossRef CAS.
- D. Xu, P. Xiao, J. Zhang, G. Li, G. Xiao, P. A. Webley and Y. Zhai, Chem. Eng. J., 2013, 230, 64–72 CrossRef CAS.
- D. Ko, Ind. Eng. Chem. Res., 2016, 55, 8967–8978 CrossRef CAS.
- M. C. Campo, A. M. Ribeiro, A. F. P. Ferreira, J. C. Santos, C. Lutz, J. M. Loureiro and A. E. Rodrigues, Fuel Process. Technol., 2016, 143, 185–194 CrossRef CAS.
- S. Krishnamurthy, V. R. Rao, S. Guntuka, P. Sharratt, R. Haghpanah, A. Rajendran, M. Amanullah, I. A. Karimi and S. Farooq, AIChE J., 2014, 60, 1830–1842 CrossRef CAS.
- J. Ling, A. Ntiamoah, P. Xiao, P. A. Webley and Y. Zhai, Chem. Eng. J., 2015, 265, 47–57 CrossRef CAS.
- A. D. Ebner and J. A. Ritter, Sep. Sci. Technol., 2009, 44, 1273–1421 CrossRef CAS.
- J. Wilcox, R. Haghpanah, E. C. Rupp, J. He and K. Lee, Annu. Rev. Chem. Biomol. Eng., 2014, 5, 479–505 CrossRef CAS PubMed.
- M. Khurana and S. Farooq, AIChE J., 2017, 1–9 Search PubMed.
- G. N. Nikolaidis, E. S. Kikkinides and M. C. Georgiadis, Ind. Eng. Chem. Res., 2017, 56, 974–988 CrossRef CAS.
- K. T. Leperi, R. Q. Snurr and F. You, Ind. Eng. Chem. Res., 2016, 55, 3338–3350 CrossRef CAS.
- G. Li, P. Xiao, D. Xu and P. A. Webley, Chem. Eng. Sci., 2011, 66, 1825–1834 CrossRef CAS.
- J. Schell, N. Casas and M. Mazzotti, Energy Procedia, 2009, 1, 655–660 CrossRef CAS.
- S. Garcia, M. V. Gil, J. J. Pis, F. Rubiera and C. Pevida, Int. J. Greenhouse Gas Control, 2013, 12, 35–43 CrossRef CAS.
- Z. Liu, C. A. Grande, P. Li, J. G. Yu and A. E. Rodrigues, Sep. Purif. Technol., 2011, 81, 307–317 CrossRef CAS.
- V. Mulgundmath and F. H. Tezel, Adsorption, 2010, 16, 587–598 CrossRef CAS.
- D. Marx, L. Joss, M. Hefti and M. Mazzotti, Ind. Eng. Chem. Res., 2016, 55, 1401–1412 CrossRef CAS.
- L. Joss, M. Gazzani and M. Mazzotti, Chem. Eng. Sci., 2017, 158, 381–394 CrossRef CAS.
- A. Ntiamoah, J. Ling, P. Xiao, P. A. Webley and Y. Zhai, Ind. Eng. Chem. Res., 2016, 55, 703–713 CrossRef CAS.
- B. C. Shin, H. Kwak and K. M. Lee, Korean Chem. Eng. Res., 2012, 50, 646–653 CrossRef CAS.
- K. Kim, S. Yang, J. B. Lee, T. H. Eom, C. K. Ryu, H.-J. Lee, T.-S. Bae, Y.-B. Lee and S.-J. Lee, Korean J. Chem. Eng., 2015, 32, 677–684 CrossRef CAS.
- T. O. Nelson, D. A. Green, P. Box, R. P. Gupta, G. Henningsen, B. S. Turk and R. T. I. International, Carbon dioxide capture from flue gas using dry regenerable sorbents, RTI International, Research Triangle Institute, USA, 2009 Search PubMed.
- P. Chaiwang, D. Gidaspow, B. Chalermsinsuwan and P. Piumsomboon, Chem. Eng. Sci., 2014, 105, 32–45 CrossRef CAS.
- L. A. Darunte, K. S. Walton, D. S. Sholl and C. W. Jones, Curr. Opin. Chem. Eng., 2016, 12, 82–90 CrossRef.
- W. Zhang, H. Liu, C. Sun, T. C. Drage and C. E. Snape, Chem. Eng. J., 2014, 251, 293–303 CrossRef CAS.
- G. Schöny, F. Dietrich, J. Fuchs, T. Pröll and H. Hofbauer, Powder Technol., 2017, 316, 519–527 CrossRef.
- G. D. Pirngruber, F. Guillou, A. Gomez and M. Clausse, Int. J. Greenhouse Gas Control, 2013, 14, 74–83 CrossRef CAS.
- S. Sjostrom, H. Krutka, T. Starns and T. Campbell, Energy Procedia, 2011, 4, 1584–1592 CrossRef.
- A. Zaabout, M. C. Romano, S. Cloete, A. Giuffrida, J. Morud, P. Chiesa and S. Amini, Int. J. Greenhouse Gas Control, 2017, 60, 74–92 CrossRef CAS.
- Y. Son, W. Won, T. Lee and K. S. Lee, Int. J. Greenhouse Gas Control, 2016, 49, 34–46 CrossRef CAS.
- S. Hammache, J. S. Hoffman, M. L. Gray, D. J. Fauth, B. H. Howard and H. W. Pennline, Energy Fuels, 2013, 27, 6899–6905 CrossRef CAS.
- J. Fujiki, F. A. Chowdhury, H. Yamada and K. Yogo, Chem. Eng. J., 2017, 307, 273–282 CrossRef CAS.
- Inventys, VeloxoTherm™ process, Burnaby, BC Canada, 2016, http://inventysinc.com/.
- N. Tlili, G. Grévillot, A. Latifi and C. Vallières, Ind. Eng. Chem. Res., 2012, 51, 15729–15737 CrossRef CAS.
- R. P. P. L. Ribeiro, C. A. Grande and A. E. Rodrigues, Chem. Eng. Sci., 2013, 104, 304–318 CrossRef CAS.
- C. A. Grande, R. P. P. L. Ribeiro and A. E. Rodrigues, Energy Fuels, 2009, 23, 2797–2803 CrossRef CAS.
- T. Wang, K. S. Lackner and A. Wright, Environ. Sci. Technol., 2011, 45, 6670–6675 CrossRef CAS PubMed.
- J. R. Fernandez, J. C. Abanades and R. Murillo, Chem. Eng. Sci., 2012, 84, 1–11 CrossRef CAS.
- Y. J. Wu, P. Li, J. G. Yu, A. F. Cunha and A. E. Rodrigues, Chem. Eng. Technol., 2013, 36, 567–574 CrossRef CAS.
- M. Gazzani, E. Macchi and G. Manzolini, Fuel, 2013, 105, 206–219 CrossRef CAS.
- G. Manzolini, E. Macchi and M. Gazzani, Fuel, 2013, 105, 220–227 CrossRef CAS.
- D. Jansen, E. van Selow, P. Cobden, G. Manzolini, E. Macchi, M. Gazzani, R. Blom, P. P. Henriksen, R. Beavis and A. Wright, Energy Procedia, 2013, 37, 2265–2273 CrossRef CAS.
- J. C. Li Yuen Fong, C. J. Anderson, G. Xiao, P. A. Webley and A. F. A. Hoadley, J. Cleaner Prod., 2016, 111(part A), 193–203 CrossRef CAS.
- L. Grajciar, J. Čejka, A. Zukal, C. Otero Areán, G. Turnes Palomino and P. Nachtigall, ChemSusChem, 2012, 5, 2011–2022 CrossRef CAS PubMed.
- J. Kim, L. C. Lin, J. A. Swisher, M. Haranczyk and B. Smit, J. Am. Chem. Soc., 2012, 134, 18940–18943 CrossRef CAS PubMed.
- M. M. Lozinska, J. P. S. Mowat, P. A. Wright, S. P. Thompson, J. L. Jorda, M. Palomino, S. Valencia and F. Rey, Chem. Mater., 2014, 26, 2052–2061 CrossRef CAS.
- T. H. Bae, M. R. Hudson, J. A. Mason, W. L. Queen, J. J. Dutton, K. Sumida, K. J. Micklash, S. S. Kaye, C. M. Brown and J. R. Long, Energy Environ. Sci., 2013, 6, 128–138 CAS.
- L.-C. Lin, A. H. Berger, R. L. Martin, J. Kim, J. A. Swisher, K. Jariwala, C. H. Rycroft, A. S. Bhown, M. W. Deem, M. Haranczyk and B. Smit, Nat. Mater., 2012, 11, 633–641 CrossRef CAS PubMed.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
- S. Couck, J. F. M. Denayer, G. V. Baron, T. Rémy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326–6327 CrossRef CAS PubMed.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- C. Wang, X. Liu, N. Keser Demir, J. P. Chen and K. Li, Chem. Soc. Rev., 2016, 45, 5107–5134 RSC.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- C. R. Mason, L. Maynard-Atem, N. M. Al-Harbi, P. M. Budd, P. Bernardo, F. Bazzarelli, G. Clarizia and J. C. Jansen, Macromolecules, 2011, 44, 6471–6479 CrossRef CAS.
- C. R. Mason, L. Maynard-Atem, K. W. J. Heard, B. Satilmis, P. M. Budd, K. Friess, M. Lanc, P. Bernardo, G. Clarizia and J. C. Jansen, Macromolecules, 2014, 47, 1021–1029 CrossRef CAS PubMed.
- M. Sevilla, P. Valle-Vigon and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781–2787 CrossRef CAS.
- X. C. Xu, C. S. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Energy Fuels, 2002, 16, 1463–1469 CrossRef CAS.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8786 CrossRef CAS PubMed.
- T. M. McDonald, D. M. D'Alessandro, R. Krishna and J. R. Long, Chem. Sci., 2011, 2, 2022–2028 RSC.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed.
- W. R. Lee, S. Y. Hwang, D. W. Ryu, K. S. Lim, S. S. Han, D. Moon, J. Choi and C. S. Hong, Energy Environ. Sci., 2014, 7, 744–751 CAS.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- M. Anbia and V. Hoseini, Chem. Eng. J., 2012, 191, 326–330 CrossRef CAS.
- Y. Zhao, H. Ding and Q. Zhong, Appl. Surf. Sci., 2013, 284, 138–144 CrossRef CAS.
- F. Rezaei and P. Webley, Chem. Eng. Sci., 2009, 64, 5182–5191 CrossRef CAS.
- F. Rezaei, A. Mosca, P. Webley, J. Hedlund and P. Xiao, Ind. Eng. Chem. Res., 2010, 49, 4832–4841 CrossRef CAS.
- D. P. Vargas, L. Giraldo, J. Silvestre-Albero and J. C. Moreno-Piraján, Adsorption, 2011, 17, 497–504 CrossRef CAS.
- R. P. Lively, D. P. Leta, B. A. DeRites, R. R. Chance and W. J. Koros, Chem. Eng. J., 2011, 171, 801–810 CrossRef CAS.
- R. P. Lively, N. Bessho, D. A. Bhandari, Y. Kawajiri and W. J. Koros, Int. J. Hydrogen Energy, 2012, 37, 15227–15240 CrossRef CAS.
- F. Rezaei, S. Subramanian, J. Kalyanaraman, R. P. Lively, Y. Kawajiri and M. J. Realff, Chem. Eng. Sci., 2014, 113, 62–76 CrossRef CAS.
- M. D. Determan, D. C. Hoysall, S. Garimella, R. Lenz and D. P. Leta, Ind. Eng. Chem. Res., 2016, 55, 2119–2127 CrossRef CAS.
- H. Thakkar, S. Eastman, A. Hajari, A. A. Rownaghi, J. C. Knox and F. Rezaei, ACS Appl. Mater. Interfaces, 2016, 8, 27753–27761 CAS.
- J. A. Mason, T. M. McDonald, T.-H. Bae, J. E. Bachman, K. Sumida, J. J. Dutton, S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2015, 137, 4787–4803 CrossRef CAS PubMed.
- J. A. A. Gibson, E. Mangano, E. Shiko, A. G. Greenaway, A. V. Gromov, M. M. Lozinska, D. Friedrich, E. E. B. Campbell, P. A. Wright and S. Brandani, Ind. Eng. Chem. Res., 2016, 55, 3840–3851 CrossRef CAS.
- E. V. Ramos-Fernandez, M. Garcia-Domingos, J. Juan-Alcañiz, J. Gascon and F. Kapteijn, Appl. Catal., A, 2011, 391, 261–267 CrossRef CAS.
- M. G. Schwab, I. Senkovska, M. Rose, M. Koch, J. Pahnke, G. Jonschker and S. Kaskel, Adv. Eng. Mater., 2008, 10, 1151–1155 CrossRef CAS.
- P. Küsgens, A. Zgaverdea, H.-G. Fritz, S. Siegle and S. Kaskel, J. Am. Ceram. Soc., 2010, 93, 2476–2479 CrossRef.
- C. L. Calvez, M. Zouboulaki, C. Petit, L. Peeva and N. Shirshova, RSC Adv., 2016, 6, 17314–17317 RSC.
- M. R. Armstrong, K. Y. Y. Arredondo, C. Y. Liu, J. E. Stevens, A. Mayhob, B. H. Shan, S. Senthilnathan, C. J. Balzer and B. Mu, Ind. Eng. Chem. Res., 2015, 54, 12386–12392 CrossRef CAS.
- D. Bradshaw, A. Garai and J. Huo, Chem. Soc. Rev., 2012, 41, 2344–2381 RSC.
- R. Ostermann, J. Cravillon, C. Weidmann, M. Wiebcke and B. M. Smarsly, Chem. Commun., 2011, 47, 442–444 RSC.
- T. Shimizu, T. Hirama, H. Hosoda, K. Kitano, M. Inagaki and K. Tejima, Chem. Eng. Res. Des., 1999, 77, 62–68 CrossRef CAS.
- D. P. Hanak, C. Biliyok, E. J. Anthony and V. Manovic, Int. J. Greenhouse Gas Control, 2015, 42, 226–236 CrossRef CAS.
- D. P. Hanak and V. Manovic, Energy, 2016, 102, 343–353 CrossRef CAS.
- M. Iijima, Mitsbubishi Heavy Industries Flue Gas CO2 Recovery Technology. Presentation, Global Climate & Energy Project Energy Workshop on Carbon Capture and Separation, Stanford University, 2004, http://https://gcep.stanford.edu/pdfs/energy_workshops_04_04/carbon_iijima.pdf.
- D. A. Jones, T. F. McVey and S. J. Friedmann, Technoeconomic evaluation of MEA versus mixed amines for CO2 removal at near-commercial scale at Duke Energy Gibson 3 plant. Report prepared for the U.S. Department of Energy, Lawrence Livermore National Laboratory, 2012, http://https://e-reports-ext.llnl.gov/pdf/700272.pdf.
- D. C. Ozcan, H. Ahn and S. Brandani, Int. J. Greenhouse Gas Control, 2013, 19, 530–540 CrossRef CAS.
- K. Atsonios, P. Grammelis, S. K. Antiohos, N. Nikolopoulos and E. Kakaras, Fuel, 2015, 153, 210–223 CrossRef CAS.
- A. Perejón, L. M. Romeo, Y. Lara, P. Lisbona, A. Martínez and J. M. Valverde, Appl. Energy, 2016, 162, 787–807 CrossRef.
- J. Blamey and B. Anthony, Chapter 8: End use of lime-based sorbents from calcium looping systems, Calcium and Chemical Looping Technology for Power Generation and Carbon Dioxide (CO2) Capture, Woodhead Publishing Ltd., UK, 2015, pp. 153–169 Search PubMed.
- J. Kremer, A. Galloy, J. Ströhle and B. Epple, Chem. Eng. Technol., 2013, 36, 1518–1524 CrossRef CAS.
- J. Ströhle, M. Junk, J. Kremer, A. Galloy and B. Epple, Fuel, 2014, 127, 13–22 CrossRef.
- B. Arias, M. E. Diego, J. C. Abanades, M. Lorenzo, L. Diaz, D. Martínez, J. Alvarez and A. Sánchez-Biezma, Int. J. Greenhouse Gas Control, 2013, 18, 237–245 CrossRef CAS.
- H.-W. Hsu, Calcium-looping CO2Capture Technology, Industrial Technology Research Institute (ITRI), http://https://www.itri.org.tw/eng/Content/MsgPic01/Contents.aspx?SiteID=1&MmmID=620170236661141772&MSid=620170263150637304, accessed December 2016.
- J. C. Abanades, B. Arias, A. Lyngfelt, T. Mattisson, D. E. Wiley, H. Li, M. T. Ho, E. Mangano and S. Brandani, Int. J. Greenhouse Gas Control, 2015, 40, 126–166 CrossRef CAS.
- D. P. Hanak, E. J. Anthony and V. Manovic, Energy Environ. Sci., 2015, 8, 2199–2249 CAS.
- J. Blamey, E. J. Anthony, J. Wang and P. S. Fennell, Prog. Energy Combust. Sci., 2010, 36, 260–279 CrossRef CAS.
- V. Manovic and E. J. Anthony, Ind. Eng. Chem. Res., 2010, 49, 9105–9110 CrossRef CAS.
- F. Donat, N. H. Florin, E. J. Anthony and P. S. Fennell, Environ. Sci. Technol., 2012, 46, 1262–1269 CrossRef CAS PubMed.
- G. Duelli, A. Charitos, M. E. Diego, E. Stavroulakis, H. Dieter and G. Scheffknecht, Int. J. Greenhouse Gas Control, 2015, 33, 103–112 CrossRef CAS.
- R. T. Symonds, D. Y. Lu, V. Manovic and E. J. Anthony, Ind. Eng. Chem. Res., 2012, 51, 7177–7184 CrossRef CAS.
- V. Manovic and E. J. Anthony, Ind. Eng. Chem. Res., 2010, 49, 6916–6922 CrossRef CAS.
- Y. Hu, W. Liu, H. Chen, Z. Zhou, W. Wang, J. Sun, X. Yang, X. Li and M. Xu, Fuel, 2016, 181, 199–206 CrossRef CAS.
- Z. He, Y. Li, X. Ma, W. Zhang, C. Chi and Z. Wang, Int. J. Hydrogen Energy, 2016, 41, 4296–4304 CrossRef CAS.
- S. Champagne, D. Y. Lu, R. T. Symonds, A. Macchi and E. J. Anthony, Powder Technol., 2016, 290, 114–123 CrossRef CAS.
- M. Kavosh, K. Patchigolla, E. J. Anthony and J. E. Oakey, Appl. Energy, 2014, 131, 499–507 CrossRef CAS.
- P. T. Clough, M. E. Boot-Handford, M. Zhao and P. S. Fennell, Fuel, 2016, 186, 708–713 CrossRef CAS.
- L. Jia, R. Hughes, D. Lu, E. J. Anthony and I. Lau, Ind. Eng. Chem. Res., 2007, 46, 5199–5209 CrossRef CAS.
- F. N. Ridha, D. Y. Lu, R. T. Symonds and S. Champagne, Powder Technol., 2016, 291, 60–65 CrossRef CAS.
- M. Erans, V. Manovic and E. J. Anthony, Appl. Energy, 2016, 180, 722–742 CrossRef CAS.
- M. Erans, T. Beisheim, V. Manovic, M. Jeremias, K. Patchigolla, H. Dieter, L. Duan and E. J. Anthony, Faraday Discuss., 2016, 192, 97–111 RSC.
- M. Erans, F. Cerciello, A. Coppola, O. Senneca, F. Scala, V. Manovic and E. J. Anthony, Fuel, 2017, 187, 388–397 CrossRef CAS.
- F. N. Ridha, V. Manovic, Y. Wu, A. Macchi and E. J. Anthony, Int. J. Greenhouse Gas Control, 2013, 17, 357–365 CrossRef CAS.
- P. S. Fennell, R. Pacciani, J. S. Dennis, J. F. Davidson and A. N. Hayhurst, Energy Fuels, 2007, 21, 2072–2081 CrossRef CAS.
- M. J. Al-Jeboori, M. Nguyen, C. Dean and P. S. Fennell, Ind. Eng. Chem. Res., 2013, 52, 1426–1433 CrossRef CAS.
- F. N. Ridha, Y. Wu, V. Manovic, A. Macchi and E. J. Anthony, Chem. Eng. J., 2015, 274, 69–75 CrossRef CAS.
- C. I. C. Pinheiro, A. Fernandes, C. Freitas, E. T. Santos and M. F. Ribeiro, Ind. Eng. Chem. Res., 2016, 55, 7860–7872 CrossRef CAS.
- P. S. Fennell, J. F. Davidson, J. S. Dennis and A. N. Hayhurst, J. Energy Inst., 2007, 80, 116–119 CrossRef CAS.
- Y. Wu, J. Blamey, E. J. Anthony and P. S. Fennell, Energy Fuels, 2010, 24, 2768–2776 CrossRef CAS.
- F.-C. Yu, N. Phalak, Z. Sun and L.-S. Fan, Ind. Eng. Chem. Res., 2012, 51, 2133–2142 CrossRef CAS.
- J. Blamey, V. Manovic, E. J. Anthony, D. R. Dugwell and P. S. Fennell, Fuel, 2015, 150, 269–277 CrossRef CAS.
- C. Salvador, D. Lu, E. J. Anthony and J. C. Abanades, Chem. Eng. J., 2003, 96, 187–195 CrossRef CAS.
- P. Sun, C. J. Lim and J. R. Grace, AIChE J., 2008, 54, 1668–1677 CrossRef CAS.
- Z. Chen, H. S. Song, M. Portillo, C. J. Lim, J. R. Grace and E. J. Anthony, Energy Fuels, 2009, 23, 1437–1444 CrossRef CAS.
- B. Arias, J. C. Abanades and G. S. Grasa, Chem. Eng. J., 2011, 167, 255–261 CrossRef CAS.
- M. E. Diego, B. Arias, A. Méndez, M. Lorenzo, L. Díaz, A. Sánchez-Biezma and J. C. Abanades, Int. J. Greenhouse Gas Control, 2016, 50, 14–22 CrossRef CAS.
- F. Yin, K. Shah, C. Zhou, P. Tremain, J. Yu, E. Doroodchi and B. Moghtaderi, Energy Fuels, 2016, 30, 1730–1740 CrossRef CAS.
- X. Wang, J. Gao and L. Jiang, Int. J. Hydrogen Energy, 2016, 41, 12000–12018 CrossRef CAS.
- W. Wu, S.-C. Chen, P.-C. Kuo and S.-A. Chen, J. Cleaner Prod., 2017, 140(part 3), 1049–1059 CrossRef CAS.
- V. Manovic, Y. Wu, I. He and E. J. Anthony, Ind. Eng. Chem. Res., 2011, 50, 12384–12391 CrossRef CAS.
- C. Qin, V. Manovic, J. Ran and B. Feng, Fuel, 2016, 181, 522–530 CrossRef CAS.
- R. A. Rahman, P. Mehrani, D. Y. Lu, E. J. Anthony and A. Macchi, Energy Fuels, 2015, 29, 3808–3819 CrossRef CAS.
- B. Duhoux, P. Mehrani, D. Y. Lu, R. T. Symonds, E. J. Anthony and A. Macchi, Energy Technol., 2016, 4, 1158–1170 CrossRef CAS.
- D. P. Hanak, C. Biliyok and V. Manovic, Energy Environ. Sci., 2016, 9, 971–983 CAS.
- C. Tregambi, F. Montagnaro, P. Salatino and R. Solimene, Sol. Energy, 2015, 120, 208–220 CrossRef CAS.
- R. Chacartegui, A. Alovisio, C. Ortiz, J. M. Valverde, V. Verda and J. A. Becerra, Appl. Energy, 2016, 173, 589–605 CrossRef CAS.
- R. Zhai, C. Li, J. Qi and Y. Yang, Energy Convers. Manage., 2016, 117, 251–263 CrossRef CAS.
- Y. Lara, A. Martínez, P. Lisbona and L. M. Romeo, Energy, 2016, 116(part 1), 956–962 CrossRef CAS.
- E. J. Anthony, Private communication from Cranfield University, December, 2016.
- J. Adanez, A. Abad, F. Garcia-Labiano, P. Gayan and L. F. de Diego, Prog. Energy Combust. Sci., 2012, 38, 215–282 CrossRef CAS.
- L.-S. Fan, L. Zeng, W. Wang and S. Luo, Energy Environ. Sci., 2012, 5, 7254 CAS.
- A. Murugan, A. Thursfield and I. S. Metcalfe, Energy Environ. Sci., 2011, 4, 4639–4649 CAS.
- W. B. Jensen, J. Chem. Educ., 2009, 86, 1266–1267 CrossRef CAS.
- M. Bülow, J. Böer, W. Burckhardt, H. U. Guth, H. Ullmann and V. V. Vashook, US Pat., US7338549B2, 2008 Search PubMed.
- C. Zhou, K. Shah, H. Song, J. Zanganeh, E. Doroodchi and B. Moghtaderi, Energy Fuels, 2016, 30, 1741–1755 CrossRef CAS.
- A. Thursfield, A. Murugan, R. Franca and I. S. Metcalfe, Energy Environ. Sci., 2012, 5, 7421–7459 CAS.
- M. Ishida, D. Zheng and T. Akehata, Energy, 1987, 12, 147–154 CrossRef CAS.
- J. Ströhle, M. Orth and B. Epple, Appl. Energy, 2015, 157, 288–294 CrossRef.
- H. Jin and M. Ishida, Ind. Eng. Chem. Res., 2002, 41, 4004–4007 CrossRef CAS.
- H. Jin and M. Ishida, Fuel, 2004, 83, 2411–2417 CrossRef CAS.
- T. Mattisson, F. García-Labiano, B. Kronberger, A. Lyngfelt, J. Adánez and H. Hofbauer, Int. J. Greenhouse Gas Control, 2007, 1, 158–169 CrossRef CAS.
- N. Berguerand and A. Lyngfelt, Energy Fuels, 2009, 23, 5257–5268 CrossRef CAS.
- H. Leion, T. Mattisson and A. Lyngfelt, Int. J. Greenhouse Gas Control, 2008, 2, 180–193 CrossRef CAS.
- J. S. Dennis, C. R. Müller and S. A. Scott, Fuel, 2010, 89, 2353–2364 CrossRef CAS.
- R. Siriwardane, H. Tian, D. Miller, G. Richards, T. Simonyi and J. Poston, Combust. Flame, 2010, 157, 2198–2208 CrossRef CAS.
- H. Leion, T. Mattisson and A. Lyngfelt, Fuel, 2007, 86, 1947–1958 CrossRef CAS.
- M. Saucedo, J. Dennis and S. Scott, Proc. Combust. Inst., 2014, 35, 2785–2792 CrossRef.
- S. A. Scott, J. S. Dennis, A. N. Hayhurst and T. Brown, AIChE J., 2006, 52, 3325–3328 CrossRef CAS.
- C. Linderholm, M. Schmitz, P. Knutsson, M. Källén and A. Lyngfelt, Energy Fuels, 2014, 28, 5942–5952 CrossRef CAS.
- T. Mattisson, A. Lyngfelt and H. Leion, Int. J. Greenhouse Gas Control, 2009, 3, 11–19 CrossRef CAS.
- M. Arjmand, M. Keller, H. Leion, T. Mattisson and A. Lyngfelt, Energy Fuels, 2012, 26, 6528–6539 CrossRef CAS.
- L. Xu, J. Wang, Z. Li and N. Cai, Energy Fuels, 2013, 27, 1522–1530 CrossRef CAS.
- G. Azimi, H. Leion, M. Rydén, T. Mattisson and A. Lyngfelt, Energy Fuels, 2013, 27, 367–377 CrossRef CAS.
- C. Ekström, F. Schwendig, O. Biede, F. Franco, G. Haupt, G. de Koeijer, C. Papapavlou and P. E. Røkke, Energy Procedia, 2009, 1, 4233–4240 CrossRef.
- A. Lyngfelt and B. Leckner, Appl. Energy, 2015, 157, 475–487 CrossRef.
- R. Porrazzo, G. White and R. Ocone, Faraday Discuss., 2016, 192, 437–457 RSC.
- I. Abdulally, C. Beal, H. Andrus, B. Epple, A. Lyngfelt and B. Lani, Alstom's Chemical Looping Prototypes, Program Update, 37th International Technical Conference on Clean Coal & Fuel Systems, Clearwater, FL, USA.
- P. Hallberg, M. Hanning, M. Rydén, T. Mattisson and A. Lyngfelt, Int. J. Greenhouse Gas Control, 2016, 53, 222–229 CrossRef CAS.
- S. C. Bayham, H. R. Kim, D. Wang, A. Tong, L. Zeng, O. McGiveron, M. V. Kathe, E. Chung, W. Wang, A. Wang, A. Majumder and L.-S. Fan, Energy Fuels, 2013, 27, 1347–1356 CrossRef CAS.
- D.-W. Jeong, W.-J. Jang, J.-O. Shim, W.-B. Han, H.-S. Roh, U. H. Jung and W. L. Yoon, Renewable Energy, 2014, 65, 102–107 CrossRef CAS.
- E. E. McLeary, J. C. Jansen and F. Kapteijn, Microporous Mesoporous Mater., 2006, 90, 198–220 CrossRef CAS.
- S. Sridhar, B. Smitha and T. M. Aminabhavi, Sep. Purif. Rev., 2007, 36, 113–174 CrossRef CAS.
- M. A. Aroon, A. F. Ismail, T. Matsuura and M. M. Montazer-Rahmati, Sep. Purif. Technol., 2010, 75, 229–242 CrossRef CAS.
- A. Brunetti, F. Scura, G. Barbieri and E. Drioli, J. Membr. Sci., 2010, 359, 115–125 CrossRef CAS.
- C. A. Scholes, K. H. Smith, S. E. Kentish and G. W. Stevens, Int. J. Greenhouse Gas Control, 2010, 4, 739–755 CrossRef CAS.
- E. Favre, Chem. Eng. J., 2011, 171, 782–793 CrossRef CAS.
- N. Du, H. B. Park, M. M. Dal-Cin and M. D. Guiver, Energy Environ. Sci., 2012, 5, 7306–7322 CAS.
- C. A. Scholes, G. W. Stevens and S. E. Kentish, Fuel, 2012, 96, 15–28 CrossRef CAS.
- B. Belaissaoui and E. Favre, Oil Gas Sci. Technol., 2014, 69, 1005–1020 CrossRef.
- A. Brunetti, E. Drioli, Y. M. Lee and G. Barbieri, J. Membr. Sci., 2014, 454, 305–315 CrossRef CAS.
- N. A. Al-Mufachi, N. V. Rees and R. Steinberger-Wilkens, Renewable Sustainable Energy Rev., 2015, 47, 540–551 CrossRef CAS.
- F. Gallucci, E. Fernandez, P. Corengia and M. V. Annaland, Chem. Eng. Sci., 2013, 92, 40–66 CrossRef CAS.
- C. Chi, X. Wang, Y. Peng, Y. Qian, Z. Hu, J. Dong and D. Zhao, Chem. Mater., 2016, 28, 2921–2927 CrossRef CAS.
- O. M. Ilinitch, A. A. Lapkin and K. I. Zamaraev, J. Membr. Sci., 1995, 98, 287–290 CrossRef CAS.
- S. Jiao and Z. Xu, ACS Appl. Mater. Interfaces, 2015, 7, 9052–9059 CAS.
- V. V. Kharton, A. A. Yaremchenko, A. V. Kovalevsky, A. P. Viskup, E. N. Naumovich and P. F. Kerko, J. Membr. Sci., 1999, 163, 307–317 CrossRef CAS.
- J. Sunarso, S. S. Hashim, N. Zhu and W. Zhou, Prog. Energy Combust. Sci., 2017, 61, 57–77 CrossRef.
- D. Yepes, L. M. Cornaglia, S. Irusta and E. A. Lombardo, J. Membr. Sci., 2006, 274, 92–101 CrossRef CAS.
- A. Thursfield and I. S. Metcalfe, J. Mater. Chem., 2004, 14, 2475–2485 RSC.
- J. Sunarso, S. Baumann, J. M. Serra, W. A. Meulenberg, S. Liu, Y. S. Lin and J. C. D. da Costa, J. Membr. Sci., 2008, 320, 13–41 CrossRef CAS.
- K. Zhang, J. Sunarso, Z. Shao, W. Zhou, C. Sun, S. Wang and S. Liu, RSC Adv., 2011, 1, 1661–1676 RSC.
- P. M. Geffroy, J. Fouletier, N. Richet and T. Chartier, Chem. Eng. Sci., 2013, 87, 408–433 CrossRef CAS.
- Z. Shao, W. Yang, Y. Cong, H. Dong, J. Tong and G. Xiong, J. Membr. Sci., 2000, 172, 177–188 CrossRef CAS.
- J. W. Phair and S. P. S. Badwal, Ionics, 2006, 12, 103–115 CrossRef CAS.
- Y. Li, Z. Rui, C. Xia, M. Anderson and Y. Lin, Catal. Today, 2009, 148, 303–309 CrossRef CAS.
- Z. Rui, M. Anderson, Y. Lin and Y. Li, J. Membr. Sci., 2009, 345, 110–118 CrossRef CAS.
- S. J. Chung, J. H. Park, D. Li, J. I. Ida, I. Kumakiri and J. Y. S. Lin, Ind. Eng. Chem. Res., 2005, 44, 7999–8006 CrossRef CAS.
- E. I. Papaioannou, H. Qi and I. S. Metcalfe, J. Membr. Sci., 2015, 485, 87–93 CrossRef CAS.
- J. Tong, X. Lei, J. Fang, M. Han and K. Huang, J. Mater. Chem. A, 2016, 4, 1828–1837 CAS.
- L. Zhang, N. Xu, X. Li, S. Wang, K. Huang, W. H. Harris and W. K. S. Chiu, Energy Environ. Sci., 2012, 5, 8310–8317 CAS.
- G. Barbieri, A. Brunetti, G. Tricoli and E. Drioli, J. Power Sources, 2008, 182, 160–167 CrossRef CAS.
- A. Brunetti, A. Caravella, G. Barbieri and E. Drioli, J. Membr. Sci., 2007, 306, 329–340 CrossRef CAS.
- R. P. Cabral and N. Mac Dowell, Appl. Energy, 2017, 205, 529–539 CrossRef CAS.
- S. Uemiya, N. Sato, H. Ando and E. Kikuchi, Ind. Eng. Chem. Res., 1991, 30, 585–589 CrossRef CAS.
- Y. Bi, H. Xu, W. Li and A. Goldbach, Int. J. Hydrogen Energy, 2009, 34, 2965–2971 CrossRef CAS.
- D. Mendes, V. Chibante, J. M. Zheng, S. Tosti, F. Borgognoni, A. Mendes and L. M. Madeira, Int. J. Hydrogen Energy, 2010, 35, 12596–12608 CrossRef CAS.
- J. Catalano, M. G. Baschetti and G. C. Sarti, J. Membr. Sci., 2010, 362, 221–233 CrossRef CAS.
- D. Mendes, S. Sa, S. Tosti, J. M. Sousa, L. M. Madeira and A. Mendes, Chem. Eng. Sci., 2011, 66, 2356–2367 CrossRef CAS.
- J. Catalano, F. Guazzone, I. P. Mardilovich, N. K. Kazantzis and Y. H. Ma, Ind. Eng. Chem. Res., 2013, 52, 1042–1055 CrossRef CAS.
- Y. Shirasaki, T. Tsuneki, Y. Ota, I. Yasuda, S. Tachibana, H. Nakajima and K. Kobayashi, Int. J. Hydrogen Energy, 2009, 34, 4482–4487 CrossRef CAS.
- J. Huang, L. El-Azzami and W. S. W. Ho, J. Membr. Sci., 2005, 261, 67–75 CrossRef CAS.
- A. Lima da Silva and I. L. Müller, J. Power Sources, 2011, 196, 8568–8582 CrossRef CAS.
- H. Jiang, H. Wang, S. Werth, T. Schiestel and J. Caro, Angew. Chem., Int. Ed., 2008, 47, 9341–9344 CrossRef CAS PubMed.
- S. B. Abdullah, Hydrogen production via simultaneous methane reforming and water splitting processes using membrane reactor, PhD thesis, Newcastle University, Newcastle upon Tyne, 2014 Search PubMed.
- W. Li, X. Zhu, S. Chen and W. Yang, Angew. Chem., Int. Ed., 2016, 128, 8708–8712 CrossRef.
- R. Bredesen, K. Jordal and A. Bolland, Chem. Eng. Process., 2004, 43, 1129–1158 CrossRef CAS.
- S. S. Hashim, A. R. Mohamed and S. Bhatia, Renewable Sustainable Energy Rev., 2011, 15, 1284–1293 CrossRef CAS.
- M. A. Habib, H. M. Badr, S. F. Ahmed, R. Ben-Mansour, K. Mezghani, S. Imashuku, G. J. la O', Y. Shao-Horn, N. D. Mancini, A. Mitsos, P. Kirchen and A. F. Ghoneim, Int. J. Energy Res., 2011, 35, 741–764 CrossRef CAS.
- N. D. Mancini and A. Mitsos, Phys. Chem. Chem. Phys., 2011, 13, 21351–21361 RSC.
- N. D. Mancini and A. Mitsos, Energy, 2011, 36, 4701–4720 CrossRef CAS.
- S. Gunasekaran, N. D. Mancini and A. Mitsos, Energy, 2014, 70, 338–354 CrossRef CAS.
- X. Dong and W. Jin, Curr. Opin. Chem. Eng., 2012, 1, 163–170 CrossRef CAS.
- Y. Wei, W. Yang, J. Caro and H. Wang, Chem. Eng. J., 2013, 220, 185–203 CrossRef CAS.
- Q. Zheng, J. Xue, Q. Liao, Y. Wei, Z. Li and H. Wang, Chem. Eng. Sci., 2013, 101, 240–247 CrossRef CAS.
- J. Tang, Y. Wei, L. Zhou, Z. Li and H. Wang, AIChE J., 2012, 58, 2473–2478 CrossRef CAS.
- Y. Wei, O. Ravkina, T. Klande, H. Wang and A. Feldhoff, J. Membr. Sci., 2013, 429, 147–154 CrossRef CAS.
- Y. Wei, Y. Wang, J. Tang, Z. Li and H. Wang, AIChE J., 2013, 59, 3856–3862 CrossRef CAS.
- J. Xue, Q. Liao, Y. Wei, Z. Li and H. Wang, J. Membr. Sci., 2013, 443, 124–130 CrossRef CAS.
- X. Zhu, Y. Liu, Y. Cong and W. Yang, Solid State Ionics, 2013, 253, 57–63 CrossRef CAS.
- F. Liang, H. Luo, K. Partovi, O. Ravkina, Z. Cao, Y. Liu and J. Caro, Chem. Commun., 2014, 50, 2451–2454 RSC.
- X. Tan, K. Li, A. Thursfield and I. S. Metcalfe, Catal. Today, 2008, 131, 292–304 CrossRef CAS.
- J. Hong, P. Kirchen and A. F. Ghoniem, J. Membr. Sci., 2013, 445, 96–106 CrossRef CAS.
- J. Hong, P. Kirchen and A. F. Ghoniem, J. Membr. Sci., 2015, 488, 1–12 CrossRef CAS.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- H. Olivier-Bourbigou and L. Magna, J. Mol. Catal. A: Chem., 2002, 182, 419–437 CrossRef.
- H. Zhao and S. V. Malhotra, Aldrichimica Acta, 2002, 35, 75–83 CrossRef CAS.
- C. Chiappe and D. Pieraccini, J. Phys. Org. Chem., 2005, 18, 275–297 CrossRef CAS.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, John Wiley & Sons, Germany, 2nd edn, 2008 Search PubMed.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772–3789 CrossRef CAS PubMed.
- C. M. Gordon, Appl. Catal., A, 2001, 222, 101–117 CrossRef CAS.
- R. Sheldon, Chem. Commun., 2001, 2399–2407 RSC.
- T. Welton, Coord. Chem. Rev., 2004, 248, 2459–2477 CrossRef CAS.
- Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13, 2619–2637 RSC.
- J. G. Huddleston, H. D. Willauer, R. P. Swatloski, A. E. Visser and R. D. Rogers, Chem. Commun., 1998, 1765–1766 RSC.
- X. Han and D. W. Armstrong, Acc. Chem. Res., 2007, 40, 1079–1086 CrossRef CAS PubMed.
- A. Berthod, M. J. Ruiz-Angel and S. Carda-Broch, J. Chromatogr. A, 2008, 1184, 6–18 CrossRef CAS PubMed.
- O. Kuzmina and J. Hallett, Application, Purification, and Recovery of Ionic Liquids, Elsevier, London, UK, 2016 Search PubMed.
- Y. Zhou, Curr. Nanosci., 2005, 1, 35–42 CrossRef CAS.
- S. Z. El Abedin, M. Pölleth, S. A. Meiss, J. Janek and F. Endres, Green Chem., 2007, 9, 549–553 RSC.
- M. Smiglak, A. Metlen and R. D. Rogers, Acc. Chem. Res., 2007, 40, 1182–1192 CrossRef CAS PubMed.
- Z. Li, Z. Jia, Y. Luan and T. Mu, Curr. Opin. Solid State Mater. Sci., 2008, 12, 1–8 CrossRef CAS.
- Z. Ma, J. Yu and S. Dai, Adv. Mater., 2010, 22, 261–285 CrossRef CAS PubMed.
- T. Sato, G. Masuda and K. Takagi, Electrochim. Acta, 2004, 49, 3603–3611 CrossRef CAS.
- J. F. Wishart, Energy Environ. Sci., 2009, 2, 956–961 CAS.
- L. Zhao, Y. Hu, H. Li, Z. Wang and L. Chen, Adv. Mater., 2011, 23, 1385–1388 CrossRef CAS PubMed.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 CAS.
- S. Zhang, J. Sun, X. Zhang, J. Xin, Q. Miao and J. Wang, Chem. Soc. Rev., 2014, 43, 7838–7869 RSC.
- M. Mora-Pale, L. Meli, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2011, 108, 1229–1245 CrossRef CAS PubMed.
- H. Tadesse and R. Luque, Energy Environ. Sci., 2011, 4, 3913–3929 CAS.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- A. M. da Costa Lopes, K. G. João, A. R. C. Morais, E. Bogel-uukasik and R. Bogel-uukasik, Sustainable Chem. Processes, 2013, 1, 3 CrossRef.
- M. J. Earle, J. M. S. S. Esperança, M. A. Gilea, J. N. C. Lopes, L. P. N. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegren, Nature, 2006, 439, 831 CrossRef CAS PubMed.
- H. L. Ngo, K. LeCompte, L. Hargens and A. B. McEwen, Thermochim. Acta, 2000, 357, 97–102 CrossRef.
- M. Kosmulski, J. Gustafsson and J. B. Rosenholm, Thermochim. Acta, 2004, 412, 47–53 CrossRef CAS.
- M. Smiglak, W. M. Reichert, J. D. Holbrey, J. S. Wilkes, L. Sun, J. S. Thrasher, K. Kirichenko, S. Singh, A. R. Katritzky and R. D. Rogers, Chem. Commun., 2006, 2554–2556 RSC.
- H. Niedermeyer, J. P. Hallett, I. J. Villar-Garcia, P. A. Hunt and T. Welton, Chem. Soc. Rev., 2012, 41, 7780–7802 RSC.
- J. E. Bara, T. K. Carlisle, C. J. Gabriel, D. Camper, A. Finotello, D. L. Gin and R. D. Noble, Ind. Eng. Chem. Res., 2009, 48, 2739–2751 CrossRef CAS.
- J. S. Kanel, Overview: Industrial application of ionic liquids for liquid extraction, Chemical Industry Vision 2020 Technology Partnership Workshop, New York, New York, 2003.
- L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Nature, 1999, 399, 28 CrossRef.
- P. J. Carvalho, K. A. Kurnia and J. A. P. Coutinho, Phys. Chem. Chem. Phys., 2016, 18, 14757–14771 RSC.
- L. A. Blanchard, Z. Gu and J. F. Brennecke, J. Phys. Chem. B, 2001, 105, 2437–2444 CrossRef CAS.
- R. E. Baltus, B. H. Culbertson, S. Dai, H. Luo and D. W. DePaoli, J. Phys. Chem. B, 2004, 108, 721–727 CrossRef CAS.
- J. L. Anthony, J. L. Anderson, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2005, 109, 6366–6374 CrossRef CAS PubMed.
- M. J. Muldoon, S. N. V. K. Aki, J. L. Anderson, J. K. Dixon and J. F. Brennecke, J. Phys. Chem. B, 2007, 111, 9001–9009 CrossRef CAS PubMed.
- M. Ramdin, T. W. de Loos and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2012, 51, 8149–8177 CrossRef CAS.
- J. F. Brennecke and B. E. Gurkan, J. Phys. Chem. Lett., 2010, 1, 3459–3464 CrossRef CAS.
- M. Hasib-ur Rahman, M. Siaj and F. Larachi, Chem. Eng. Process., 2010, 49, 313–322 CrossRef CAS.
- F. Karadas, M. Atilhan and S. Aparicio, Energy Fuels, 2010, 24, 5817–5828 CrossRef CAS.
- Y. Zhang, S. Zhang, X. Lu, Q. Zhou, W. Fan and X. Zhang, Chem. – Eur. J., 2009, 15, 3003–3011 CrossRef CAS PubMed.
- C. Wang, H. Luo, D. Jiang, H. Li and S. Dai, Angew. Chem., Int. Ed., 2010, 49, 5978–5981 CrossRef CAS PubMed.
- G. Gurau, H. Rodríguez, S. P. Kelley, P. Janiczek, R. S. Kalb and R. D. Rogers, Angew. Chem., Int. Ed., 2011, 50, 12024–12026 CrossRef CAS PubMed.
- C. Wang, X. Luo, H. Luo, D. Jiang, H. Li and S. Dai, Angew. Chem., Int. Ed., 2011, 50, 4918–4922 CrossRef CAS PubMed.
- C. Wang, X. Luo, X. Zhu, G. Cui, D.-e. Jiang, D. Deng, H. Li and S. Dai, RSC Adv., 2013, 3, 15518–15527 RSC.
- S. Seo, M. Quiroz-Guzman, M. A. DeSilva, T. B. Lee, Y. Huang, B. F. Goodrich, W. F. Schneider and J. F. Brennecke, J. Phys. Chem. B, 2014, 118, 5740–5751 CrossRef CAS PubMed.
- G. Cui, J. Wang and S. Zhang, Chem. Soc. Rev., 2016, 45, 4307–4339 RSC.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- J. H. J. Davis, Chem. Lett., 2004, 33, 1072–1077 CrossRef CAS.
- J. Huang and T. Rüther, Aust. J. Chem., 2009, 62, 298–308 CrossRef CAS.
- D. Wappel, G. Gronald, R. Kalb and J. Draxler, Int. J. Greenhouse Gas Control, 2010, 4, 486–494 CrossRef CAS.
- M. Petkovic, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Chem. Soc. Rev., 2011, 40, 1383–1403 RSC.
- Z.-Z. Yang, Y.-N. Zhao and L.-N. He, RSC Adv., 2011, 1, 545–567 RSC.
- M. S. Shannon and J. E. Bara, Sep. Sci. Technol., 2012, 47, 178–188 CrossRef CAS.
- R. Giernoth, Angew. Chem., Int. Ed., 2010, 49, 2834–2839 CrossRef CAS PubMed.
- B. Gurkan, B. F. Goodrich, E. M. Mindrup, L. E. Ficke, M. Massel, S. Seo, T. P. Senftle, H. Wu, M. F. Glaser and J. K. Shah, J. Phys. Chem. Lett., 2010, 1, 3494–3499 CrossRef CAS.
- X. Zhang, Z. Liu and W. Wang, AIChE J., 2008, 54, 2717–2728 CrossRef CAS.
- M. B. Shiflett and A. Yokozeki, Ind. Eng. Chem. Res., 2005, 44, 4453–4464 CrossRef CAS.
- C. Moya, J. Palomar, M. Gonzalez-Miquel, J. Bedia and F. Rodriguez, Ind. Eng. Chem. Res., 2014, 53, 13782–13789 CrossRef CAS.
- J. de Riva, J. Suarez-Reyes, D. Moreno, I. Díaz, V. Ferro and J. Palomar, Int. J. Greenhouse Gas Control, 2017, 61, 61–70 CrossRef CAS.
- P. J. Carvalho, V. H. Álvarez, B. Schröder, A. M. Gil, I. M. Marrucho, M. Aznar, L. M. Santos and J. A. P. Coutinho, J. Phys. Chem. B, 2009, 113, 6803–6812 CrossRef CAS PubMed.
- M. B. Shiflett, D. W. Drew, R. A. Cantini and A. Yokozeki, Energy Fuels, 2010, 24, 5781–5789 CrossRef CAS.
- G. Yu, S. Zhang, G. Zhou, X. Liu and X. Chen, AIChE J., 2007, 53, 3210–3221 CrossRef CAS.
- K. E. Gutowski and E. J. Maginn, J. Am. Chem. Soc., 2008, 130, 14690–14704 CrossRef CAS PubMed.
- P. Sharma, S. Do Park, K. T. Park, S. C. Nam, S. K. Jeong, Y. I. Yoon and I. H. Baek, Chem. Eng. J., 2012, 193, 267–275 CrossRef.
- Y. S. Sistla and A. Khanna, J. Ind. Eng. Chem., 2014, 20, 2497–2509 CrossRef CAS.
- M. D. Soutullo, C. I. Odom, B. F. Wicker, C. N. Henderson, A. C. Stenson and J. H. Davis, Chem. Mater., 2007, 19, 3581–3583 CrossRef CAS.
- C. Wang, Y. Guo, X. Zhu, G. Cui, H. Li and S. Dai, Chem. Commun., 2012, 48, 6526–6528 RSC.
- Z.-Z. Yang and L.-N. He, Beilstein J. Org. Chem., 2014, 10, 1959–1966 CrossRef CAS PubMed.
- R. Vijayraghavan, S. J. Pas, E. I. Izgorodina and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2013, 15, 19994–19999 RSC.
- L. Chen, M. Sharifzadeh, N. Mac Dowell, T. Welton, N. Shah and J. P. Hallett, Green Chem., 2014, 16, 3098–3106 RSC.
- K. S. Egorova, M. M. Seitkalieva, A. V. Posvyatenko and V. P. Ananikov, Toxicol. Res., 2015, 4, 152–159 RSC.
- H. Ohno and K. Fukumoto, Acc. Chem. Res., 2007, 40, 1122–1129 CrossRef CAS PubMed.
- J. Zhang, S. Zhang, K. Dong, Y. Zhang, Y. Shen and X. Lv, Chem. – Eur. J., 2006, 12, 4021–4026 CrossRef CAS PubMed.
- Y.-Y. Jiang, G.-N. Wang, Z. Zhou, Y.-T. Wu, J. Geng and Z.-B. Zhang, Chem. Commun., 2008, 505–507 RSC.
- H. Yu, Y.-T. Wu, Y.-Y. Jiang, Z. Zhou and Z.-B. Zhang, New J. Chem., 2009, 33, 2385–2390 RSC.
- M. T. Clough, K. Geyer, P. A. Hunt, J. Mertes and T. Welton, Phys. Chem. Chem. Phys., 2013, 15, 20480–20495 RSC.
- B. E. Gurkan, J. C. de la Fuente, E. M. Mindrup, L. E. Ficke, B. F. Goodrich, E. A. Price, W. F. Schneider and J. F. Brennecke, J. Am. Chem. Soc., 2010, 132, 2116–2117 CrossRef CAS PubMed.
- B. F. Goodrich, J. C. de la Fuente, B. E. Gurkan, D. J. Zadigian, E. A. Price, Y. Huang and J. F. Brennecke, Ind. Eng. Chem. Res., 2010, 50, 111–118 CrossRef.
- B. F. Goodrich, J. C. de la Fuente, B. E. Gurkan, Z. K. Lopez, E. A. Price, Y. Huang and J. F. Brennecke, J. Phys. Chem. B, 2011, 115, 9140–9150 CrossRef CAS PubMed.
- S. Saravanamurugan, A. J. Kunov-Kruse, R. Fehrmann and A. Riisager, ChemSusChem, 2014, 7, 897–902 CrossRef CAS PubMed.
- X. Y. Luo, F. Ding, W. J. Lin, Y. Q. Qi, H. R. Li and C. M. Wang, J. Phys. Chem. Lett., 2014, 5, 381–386 CrossRef CAS PubMed.
- K. Anderson, M. P. Atkins, J. Estager, Y. Kuah, S. Ng, A. A. Oliferenko, N. V. Plechkova, A. V. Puga, K. R. Seddon and D. F. Wassell, Green Chem., 2015, 17, 4340–4354 RSC.
- S. Kasahara, E. Kamio, A. Otani and H. Matsuyama, Ind. Eng. Chem. Res., 2014, 53, 2422–2431 CrossRef CAS.
- G. E. Romanos, P. S. Schulz, M. Bahlmann, P. Wasserscheid, A. Sapalidis, F. K. Katsaros, C. P. Athanasekou, K. Beltsios and N. K. Kanellopoulos, J. Phys. Chem. C, 2014, 118, 24437–24451 CAS.
- H. Wu, J. K. Shah, C. M. Tenney, T. W. Rosch and E. J. Maginn, Ind. Eng. Chem. Res., 2011, 50, 8983–8993 CrossRef CAS.
- B. E. Gurkan, T. R. Gohndrone, M. J. McCready and J. F. Brennecke, Phys. Chem. Chem. Phys., 2013, 15, 7796–7811 RSC.
- A. Li, Z. Tian, T. Yan, D.-e. Jiang and S. Dai, J. Phys. Chem. B, 2014, 118, 14880–14887 CrossRef CAS PubMed.
- M. Breugst, T. Tokuyasu and H. Mayr, J. Org. Chem., 2010, 75, 5250–5258 CrossRef CAS PubMed.
- C. Wu, T. P. Senftle and W. F. Schneider, Phys. Chem. Chem. Phys., 2012, 14, 13163–13170 RSC.
- J. Ren, L. Wu and B.-G. Li, Ind. Eng. Chem. Res., 2013, 52, 8565–8570 CrossRef CAS.
- H. Tang and C. Wu, ChemSusChem, 2013, 6, 1050–1056 CrossRef CAS PubMed.
- X. Lei, Y. Xu, L. Zhu and X. Wang, RSC Adv., 2014, 4, 7052–7057 RSC.
- S. Seo, L. D. Simoni, M. Ma, M. A. DeSilva, Y. Huang, M. A. Stadtherr and J. F. Brennecke, Energy Fuels, 2014, 28, 5968–5977 CrossRef CAS.
- E. Kamio, T. Matsuki, F. Moghadam and H. Matsuyama, Sep. Sci. Technol., 2017, 52, 197–208 CrossRef CAS.
- P. Brown, B. E. Gurkan and T. A. Hatton, AIChE J., 2015, 61, 2280–2285 CrossRef CAS.
- S. Seo, M. A. DeSilva, H. Xia and J. F. Brennecke, J. Phys. Chem. B, 2015, 119, 11807–11814 CrossRef CAS PubMed.
- S. Zhang, Y. Li, Y. Zhang, L. He, B. Yu, Q. Song and X. Lang, ChemSusChem, 2014, 7, 1484–1489 CrossRef CAS PubMed.
- D. S. Firaha, O. Hollóczki and B. Kirchner, Angew. Chem., Int. Ed., 2015, 54, 7805–7809 CrossRef CAS PubMed.
- M. I. Cabaço, M. Besnard, Y. Danten and J. A. P. Coutinho, J. Phys. Chem. A, 2012, 116, 1605–1620 CrossRef PubMed.
- C. J. Mathews, P. J. Smith and T. Welton, Chem. Commun., 2000, 1249–1250 RSC.
- E. J. Maginn, J. Phys.: Condens. Matter, 2009, 21, 373101 CrossRef CAS PubMed.
- H. Rodríguez, G. Gurau, J. D. Holbrey and R. D. Rogers, Chem. Commun., 2011, 47, 3222–3224 RSC.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- M. B. Shiflett, D. J. Kasprzak, C. P. Junk and A. Yokozeki, J. Chem. Thermodyn., 2008, 40, 25–31 CrossRef CAS.
- M. B. Shiflett, B. A. Elliott, S. R. Lustig, S. Sabesan, M. S. Kelkar and A. Yokozeki, ChemPhysChem, 2012, 13, 1806–1817 CrossRef CAS PubMed.
- W. Shi, R. L. Thompson, E. Albenze, J. A. Steckel, H. B. Nulwala and D. R. Luebke, J. Phys. Chem. B, 2014, 118, 7383–7394 CrossRef CAS PubMed.
- Y. Chen, J. Han, T. Wang and T. Mu, Energy Fuels, 2011, 25, 5810–5815 CrossRef CAS.
- C. Wang, H. Luo, X. Luo, H. Li and S. Dai, Green Chem., 2010, 12, 2019–2023 RSC.
- Y. Zhang, Z. Wu, S. Chen, P. Yu and Y. Luo, Ind. Eng. Chem. Res., 2013, 52, 6069–6075 CrossRef CAS.
- S. Seo, M. A. DeSilva and J. F. Brennecke, J. Phys. Chem. B, 2014, 118, 14870–14879 CrossRef CAS PubMed.
- T. R. Gohndrone, T. B. Lee, M. A. DeSilva, M. Quiroz-Guzman, W. F. Schneider and J. F. Brennecke, ChemSusChem, 2014, 7, 1970–1975 CrossRef CAS PubMed.
- T. B. Lee, S. Oh, T. R. Gohndrone, O. Morales-Collazo, S. Seo, J. F. Brennecke and W. F. Schneider, J. Phys. Chem. B, 2015, 120, 1509–1517 CrossRef PubMed.
- P. G. Jessop, D. J. Heldebrant, X. Li, C. A. Eckert and C. L. Liotta, Nature, 2005, 436, 1102 CrossRef CAS PubMed.
- D. J. Heldebrant, P. G. Jessop, C. A. Thomas, C. A. Eckert and C. L. Liotta, J. Org. Chem., 2005, 70, 5335–5338 CrossRef CAS PubMed.
- D. J. Heldebrant, C. R. Yonker, P. G. Jessop and L. Phan, Energy Environ. Sci., 2008, 1, 487–493 CAS.
- L. Phan, D. Chiu, D. J. Heldebrant, H. Huttenhower, E. John, X. Li, P. Pollet, R. Wang, C. A. Eckert and C. L. Liotta, Ind. Eng. Chem. Res., 2008, 47, 539–545 CrossRef CAS.
- C. Wang, S. M. Mahurin, H. Luo, G. A. Baker, H. Li and S. Dai, Green Chem., 2010, 12, 870–874 RSC.
- S. Y. Hong, Y. Cheon, S. H. Shin, H. Lee, M. Cheong and H. S. Kim, ChemSusChem, 2013, 6, 890–897 CrossRef CAS PubMed.
- J. Benitez-Garcia, G. Ruiz-Ibanez, H. A. Al-Ghawas and O. C. Sandall, Chem. Eng. Sci., 1991, 46, 2927–2931 CrossRef CAS.
- P. D. Vaidya and E. Y. Kenig, Chem. Eng. Technol., 2007, 30, 1467–1474 CrossRef CAS.
- R. Safdar, A. A. Omar, L. Ismail and B. Lal, Appl. Mech. Mater., 2014, 625, 549–552 CrossRef.
- Y. Zhao, B. Yu, Z. Yang, H. Zhang, L. Hao, X. Gao and Z. Liu, Angew. Chem., 2014, 126, 6032–6035 CrossRef.
- C. Wang, H. Luo, H. Li, X. Zhu, B. Yu and S. Dai, Chem. – Eur. J., 2012, 18, 2153–2160 CrossRef CAS PubMed.
- J. Zhang, C. Jia, H. Dong, J. Wang, X. Zhang and S. Zhang, Ind. Eng. Chem. Res., 2013, 52, 5835–5841 CrossRef CAS.
- P. Hu, R. Zhang, Z. Liu, H. Liu, C. Xu, X. Meng, M. Liang and S. Liang, Energy Fuels, 2015, 29, 6019–6024 CrossRef CAS.
- F. Ding, X. He, X. Luo, W. Lin, K. Chen, H. Li and C. Wang, Chem. Commun., 2014, 50, 15041–15044 RSC.
- X. Luo, Y. Guo, F. Ding, H. Zhao, G. Cui, H. Li and C. Wang, Angew. Chem., Int. Ed., 2014, 53, 7053–7057 CrossRef CAS PubMed.
- Z. Feng, F. Cheng-Gang, W. You-Ting, W. Yuan-Tao, L. Ai-Min and Z. Zhi-Bing, Chem. Eng. J., 2010, 160, 691–697 CrossRef.
- J.-w. Ma, Z. Zhou, F. Zhang, C.-g. Fang, Y.-t. Wu, Z.-b. Zhang and A.-m. Li, Environ. Sci. Technol., 2011, 45, 10627–10633 CrossRef CAS PubMed.
- Y. Zhang, P. Yu and Y. Luo, Chem. Eng. J., 2013, 214, 355–363 CrossRef CAS.
- J. L. McDonald, R. E. Sykora, P. Hixon, A. Mirjafari and J. H. Davis, Environ. Chem. Lett., 2014, 12, 201–208 CrossRef CAS.
- H. Guo, Z. Zhou and G. Jing, Int. J. Greenhouse Gas Control, 2013, 16, 197–205 CrossRef CAS.
- B.-S. Guo, G.-H. Jing and Z.-M. Zhou, Int. J. Greenhouse Gas Control, 2015, 34, 31–38 CrossRef CAS.
- G. Wang, W. Hou, F. Xiao, J. Geng, Y. Wu and Z. Zhang, J. Chem. Eng. Data, 2011, 56, 1125–1133 CrossRef CAS.
- S. Stevanovic, A. Podgorsek, L. Moura, C. C. Santini, A. A. H. Padua and M. F. C. Gomes, Int. J. Greenhouse Gas Control, 2013, 17, 78–88 CrossRef CAS.
- D. Camper, J. E. Bara, D. L. Gin and R. D. Noble, Ind. Eng. Chem. Res., 2008, 47, 8496–8498 CrossRef CAS.
- M. M. Taib and T. Murugesan, Chem. Eng. J., 2012, 181, 56–62 CrossRef.
- Z. Feng, M. Jing-Wen, Z. Zheng, W. You-Ting and Z. Zhi-Bing, Chem. Eng. J., 2012, 181, 222–228 CrossRef.
- Z. Feng, G. Yuan, W. Xian-Kun, M. Jing-Wen, W. You-Ting and Z. Zhi-Bing, Chem. Eng. J., 2013, 223, 371–378 CrossRef.
- Y. Gao, F. Zhang, K. Huang, J.-W. Ma, Y.-T. Wu and Z.-B. Zhang, Int. J. Greenhouse Gas Control, 2013, 19, 379–386 CrossRef CAS.
- Z. Zhou, G. Jing and L. Zhou, Chem. Eng. J., 2012, 204–206, 235–243 CrossRef CAS.
- B. Lv, Y. Shi, C. Sun, N. Liu, W. Li and S. Li, Chem. Eng. J., 2015, 270, 372–377 CrossRef CAS.
- B. Lv, C. Sun, N. Liu, W. Li and S. Li, Chem. Eng. J., 2015, 280, 695–702 CrossRef CAS.
- G. Yu, D. Zhao, L. Wen, S. Yang and X. Chen, AIChE J., 2012, 58, 2885–2899 CrossRef CAS.
- M. Atilhan, J. Jacquemin, D. Rooney, M. Khraisheh and S. Aparicio, Ind. Eng. Chem. Res., 2013, 52, 16774–16785 CrossRef CAS.
- Z.-Z. Yang, Q.-W. Song and L.-N. He, Capture and Utilization of Carbon Dioxide with Polyethylene Glycol, Springer Science & Business Media, Verlag Berlin Heidelberg, 2012 Search PubMed.
- X. Li and D. Deng, J. Chem. Thermodyn., 2014, 79, 230–234 CrossRef CAS.
- K. R. Seddon, A. Stark and M.-J. Torres, Pure Appl. Chem., 2000, 72, 2275–2287 CrossRef CAS.
- G. Cui, W. Lin, F. Ding, X. Luo, X. He, H. Li and C. Wang, Green Chem., 2014, 16, 1211–1216 RSC.
- G. Cui, F. Zhang, X. Zhou, H. Li, J. Wang and C. Wang, Chem. – Eur. J., 2015, 21, 5632–5639 CrossRef CAS PubMed.
- F. Dolezalek, Z. Phys. Chem., 1908, 64, 727–747 Search PubMed.
- J. J. van Laar, Z. Phys. Chem., 1910, 72, 723–751 CAS.
- J. J. van Laar, Z. Phys. Chem., 1913, 83, 599–608 Search PubMed.
- G. Maurer, Fluid Phase Equilib., 1986, 30, 337–352 CrossRef CAS.
- R. A. Heidemann and J. M. Prausnitz, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 1773–1776 CrossRef CAS.
- J. M. Prausnitz, R. N. Lichtenthaler and E. G. de Azevedo, Molecular Thermodynamics of Fluid Phase Equilibria, Prentice-Hall, New Jersey, US, 3rd edn, 1999 Search PubMed.
- B. E. Poling, J. M. Prausnitz and J. P. O'Connell, The Properties of Gases and Liquids, McGraw-Hill, New York, US, 5th edn, 2004 Search PubMed.
- D. M. Austgen, G. T. Rochelle, X. Peng and C. C. Chen, Ind. Eng. Chem. Res., 1989, 28, 1060–1073 CrossRef CAS.
- D. M. Austgen, G. T. Rochelle and C. C. Chen, Ind. Eng. Chem. Res., 1991, 30, 543–555 CrossRef CAS.
- C.-C. Chen, H. I. Britt, J. F. Boston and L. B. Evans, AIChE J., 1982, 28, 588–596 CrossRef CAS.
- C.-C. Chen and L. B. Evans, AIChE J., 1986, 32, 444–454 CrossRef CAS.
- G. Soave, Chem. Eng. Sci., 1972, 27, 1197–1203 CrossRef CAS.
- G. M. Bollas, C. C. Chen and P. I. Barton, AIChE J., 2008, 54, 1608–1624 CrossRef CAS.
- E. T. Hessen, T. Haug-Warberg and H. F. Svendsen, Chem. Eng. Sci., 2010, 65, 3638–3648 CrossRef CAS.
- Y. Zhang, H. Que and C.-C. Chen, Fluid Phase Equilib., 2011, 311, 67–75 CrossRef CAS.
- Y. Zhang and C.-C. Chen, Ind. Eng. Chem. Res., 2011, 50, 163–175 CrossRef CAS.
- L. Faramarzi, G. M. Kontogeorgis, K. Thomsen and E. H. Stenby, Fluid Phase Equilib., 2009, 282, 121–132 CrossRef CAS.
- K. Thomsen and P. Rasmussen, Chem. Eng. Sci., 1999, 54, 1787–1802 CrossRef CAS.
- G. Kuranov, B. Rumpf, G. Maurer and N. Smirnova, Fluid Phase Equilib., 1997, 136, 147–162 CrossRef CAS.
- E. D. Eastman and J. H. Hildebrand, J. Am. Chem. Soc., 1914, 36, 2020–2030 CrossRef CAS.
- H. G. Harris and J. M. Prausnitz, Ind. Eng. Chem. Fundam., 1969, 8, 180–188 CAS.
- E. A. Guggenheim, Mixtures: The Theory of the Equilibrium Properties of Some Simple Classes of Mixtures, Solutions and Alloys, Clarendon Press, Oxford, UK, 1952 Search PubMed.
- J. A. Barker and W. Fock, Discuss. Faraday Soc., 1953, 15, 188–195 RSC.
- D. S. Abrams and J. M. Prausnitz, AIChE J., 1975, 21, 116–128 CrossRef CAS.
- A. Fredenslund, R. L. Jones and J. M. Prausnitz, AIChE J., 1975, 21, 1086–1099 CrossRef CAS.
- A. Fredenslund and J. M. Sørensen, Group contribution estimation methods, Models for Thermodynamic and Phase Equilibria Calculations, Marcel Dekker, New York, 1994 Search PubMed.
- V. Papaioannou, C. S. Adjiman, G. Jackson and A. Galindo, Group contribution methodologies for the prediction of thermodynamic properties and phase behavior in mixtures, Process Systems Engineering, Molecular Systems Engineering, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, vol. 6, pp. 135–172 Search PubMed.
- W. G. Chapman, K. E. Gubbins, G. Jackson and M. Radosz, Fluid Phase Equilib., 1989, 52, 31–38 CrossRef CAS.
- W. G. Chapman, K. E. Gubbins, G. Jackson and M. Radosz, Ind. Eng. Chem. Res., 1990, 29, 1709–1721 CrossRef CAS.
- M. S. Wertheim, J. Stat. Phys., 1984, 35, 19–34 CrossRef.
- M. S. Wertheim, J. Stat. Phys., 1984, 35, 35–47 CrossRef.
- M. S. Wertheim, J. Stat. Phys., 1986, 42, 477–492 CrossRef.
- M. S. Wertheim, J. Stat. Phys., 1986, 42, 459–476 CrossRef.
- G. Jackson, W. G. Chapman and K. E. Gubbins, Mol. Phys., 1988, 65, 1–31 CrossRef CAS.
- W. G. Chapman, G. Jackson and K. E. Gubbins, Mol. Phys., 1988, 65, 1057–1079 CrossRef CAS.
- I. G. Economou and M. D. Donohue, AIChE J., 1991, 37, 1875–1894 CrossRef CAS.
- A. Gil-Villegas, A. Galindo, P. J. Whitehead, S. J. Mills, G. Jackson and A. N. Burgess, J. Chem. Phys., 1997, 106, 4168–4186 CrossRef CAS.
- A. Galindo, L. A. Davies, A. Gil-Villegas and G. Jackson, Mol. Phys., 1998, 93, 241–252 CrossRef CAS.
- T. Lafitte, A. Apostolakou, C. Avendaño, A. Galindo, C. S. Adjiman, E. A. Müller and G. Jackson, J. Chem. Phys., 2013, 139, 154504 CrossRef PubMed.
- S. Dufal, T. Lafitte, A. J. Haslam, A. Galindo, G. N. I. Clark, C. Vega and G. Jackson, Mol. Phys., 2015, 113, 948–984 CrossRef CAS.
- F. J. Blas and L. F. Vega, Mol. Phys., 1997, 92, 135–150 CrossRef CAS.
- F. J. Blas and L. F. Vega, Ind. Eng. Chem. Res., 1998, 37, 660–674 CrossRef CAS.
- J. Gross and G. Sadowski, Ind. Eng. Chem. Res., 2001, 40, 1244–1260 CrossRef CAS.
- G. M. Kontogeorgis, E. C. Voutsas, I. V. Yakoumis and D. P. Tassios, Ind. Eng. Chem. Res., 1996, 35, 4310–4318 CrossRef CAS.
- A. Lymperiadis, C. S. Adjiman, A. Galindo and G. Jackson, J. Chem. Phys., 2007, 127, 234903 CrossRef PubMed.
- A. Lymperiadis, C. S. Adjiman, G. Jackson and A. Galindo, Fluid Phase Equilib., 2008, 274, 85–104 CrossRef CAS.
- V. Papaioannou, T. Lafitte, C. Avendaño, C. S. Adjiman, G. Jackson, E. A. Müller and A. Galindo, J. Chem. Phys., 2014, 140, 54107 CrossRef PubMed.
- S. Dufal, V. Papaioannou, M. Sadeqzadeh, T. Pogiatzis, A. Chremos, C. S. Adjiman, G. Jackson and A. Galindo, J. Chem. Eng. Data, 2014, 59, 3272–3288 CrossRef CAS.
- G. N. I. Clark, A. J. Haslam, A. Galindo and G. Jackson, Mol. Phys., 2006, 104, 3561–3581 CrossRef CAS.
- N. Mac Dowell, F. Llovell, C. S. Adjiman, G. Jackson and A. Galindo, Ind. Eng. Chem. Res., 2010, 49, 1883–1899 CrossRef CAS.
- J. Rodriguez, N. Mac Dowell, F. Llovell, C. S. Adjiman, G. Jackson and A. Galindo, Mol. Phys., 2012, 110, 1325–1348 CrossRef CAS.
- F.-Y. Jou, A. E. Mather and F. D. Otto, Can. J. Chem. Eng., 1995, 73, 140–147 CrossRef CAS.
- W. Böttinger, M. Maiwald and H. Hasse, Fluid Phase Equilib., 2008, 263, 131–143 CrossRef.
- A. Chremos, E. Forte, V. Papaioannou, A. Galindo, G. Jackson and C. S. Adjiman, Fluid Phase Equilib., 2016, 407, 280–297 CrossRef CAS.
- J. K. Button and K. E. Gubbins, Fluid Phase Equilib., 1999, 158–160, 175–181 CrossRef CAS.
- N. Mac Dowell, N. J. Samsatli and N. Shah, Int. J. Greenhouse Gas Control, 2013, 12, 247–258 CrossRef CAS.
- N. Mac Dowell, A. Galindo, G. Jackson and C. S. Adjiman, Comput.-Aided Chem. Eng., 2010, 28, 1231–1236 CAS.
- N. Mac Dowell, F. E. Pereira, F. Llovell, F. J. Blas, C. S. Adjiman, G. Jackson and A. Galindo, J. Phys. Chem. B, 2011, 115, 8155–8168 CrossRef CAS PubMed.
- A. Chremos, E. Forte, V. Papaioannou, A. Galindo, G. Jackson and C. S. Adjiman, Chem. Eng. Trans., 2013, 35, 427–432 Search PubMed.
- U. E. Aronu, S. Gondal, E. T. Hessen, T. Haug-Warberg, A. Hartono, K. A. Hoff and H. F. Svendsen, Chem. Eng. Sci., 2011, 66, 6393–6406 CrossRef CAS.
- M. W. Arshad, H. F. Svendsen, P. L. Fosbøl, N. von Solms and K. Thomsen, J. Chem. Eng. Data, 2014, 59, 764–774 CrossRef CAS.
- J. Gabrielsen, M. L. Michelsen, E. H. Stenby and G. M. Kontogeorgis, Ind. Eng. Chem. Res., 2005, 44, 3348–3354 CrossRef CAS.
- K. S. Pitzer, J. Phys. Chem., 1973, 77, 268–277 CrossRef CAS.
- G. Vallée, P. Mougin, S. Jullian and W. Fürst, Ind. Eng. Chem. Res., 1999, 38, 3473–3480 CrossRef.
- L. Chunxi and W. Fürst, Chem. Eng. Sci., 2000, 55, 2975–2988 CrossRef CAS.
- W. Fürst and H. Renon, AIChE J., 1993, 39, 335–343 CrossRef.
- L. Blum, Mol. Phys., 1975, 30, 1529–1535 CrossRef CAS.
- P. Debye and E. Hückel, Phys. Z., 1923, 24, 185–206 CAS.
- D. Henderson, L. Blum and A. Tani, Equation of state of ionic fluids., Equations of State, Theories and Applications, ACS Symposium Series, American Chemical Society, Washington, DC, 1986, ch. 13, vol. 300, pp. 281–296 Search PubMed.
- L. Blum and J. S. Høeye, J. Phys. Chem., 1977, 81, 1311–1316 CrossRef CAS.
- W.-B. Liu, Y.-G. Li and J.-F. Lu, Fluid Phase Equilib., 1999, 158-160, 595–606 CrossRef CAS.
- A. Galindo, A. Gil-Villegas, G. Jackson and A. N. Burgess, J. Phys. Chem. B, 1999, 103, 10272–10281 CrossRef CAS.
- A. Gil-Villegas, A. Galindo and G. Jackson, Mol. Phys., 2001, 99, 531–546 CrossRef CAS.
- B. H. Patel, P. Paricaud, A. Galindo and G. C. Maitland, Ind. Eng. Chem. Res., 2003, 42, 3809–3823 CrossRef CAS.
- B. Behzadi, B. H. Patel, A. Galindo and C. Ghotbi, Fluid Phase Equilib., 2005, 236, 241–255 CrossRef CAS.
- J. M. A. Schreckenberg, S. Dufal, A. J. Haslam, C. S. Adjiman, G. Jackson and A. Galindo, Mol. Phys., 2014, 112, 2339–2364 CrossRef CAS.
- D. K. Eriksen, G. Lazarou, A. Galindo, G. Jackson, C. S. Adjiman and A. J. Haslam, Mol. Phys., 2016, 114, 2724–2749 CrossRef CAS.
- L. F. Cameretti, G. Sadowski and J. M. Mollerup, Ind. Eng. Chem. Res., 2005, 44, 3355–3362 CrossRef CAS.
- C. Held, L. F. Cameretti and G. Sadowski, Fluid Phase Equilib., 2008, 270, 87–96 CrossRef CAS.
- C. Held and G. Sadowski, Fluid Phase Equilib., 2009, 279, 141–148 CrossRef CAS.
- H. Zhao, M. C. dos Ramos and C. McCabe, J. Chem. Phys., 2007, 126, 244503 CrossRef PubMed.
- S. Herzog, J. Gross and W. Arlt, Fluid Phase Equilib., 2010, 297, 23–33 CrossRef CAS.
- J. Rozmus, J.-C. de Hemptinne, A. Galindo, S. Dufal and P. Mougin, Ind. Eng. Chem. Res., 2013, 52, 9979–9994 CrossRef CAS.
- B. Maribo-Mogensen, G. M. Kontogeorgis and K. Thomsen, Ind. Eng. Chem. Res., 2012, 51, 5353–5363 CrossRef CAS.
- T. Reschke, S. Naeem and G. Sadowski, J. Phys. Chem. B, 2012, 116, 7479–7491 CrossRef CAS PubMed.
- C. Held, T. Reschke, S. Mohammad, A. Luza and G. Sadowski, Chem. Eng. Res. Des., 2014, 92, 2884–2897 CrossRef CAS.
- K. Nasrifar and A. H. Tafazzol, Ind. Eng. Chem. Res., 2010, 49, 7620–7630 CrossRef CAS.
- H. Pahlavanzadeh and S. Fakouri Baygi, J. Chem. Thermodyn., 2013, 59, 214–221 CrossRef CAS.
- M. Uyan, G. Sieder, T. Ingram and C. Held, Fluid Phase Equilib., 2015, 393, 91–100 CrossRef CAS.
- L. Kucka, I. Müller, E. Y. Kenig and A. Górak, Chem. Eng. Sci., 2003, 58, 3571–3578 CrossRef CAS.
- C. Kale, A. Górak and H. Schoenmakers, Int. J. Greenhouse Gas Control, 2013, 17, 294–308 CrossRef CAS.
- C.-H. Yu, C.-H. Huang and C.-S. Tan, Aerosol Air Qual. Res., 2012, 12, 745–769 CAS.
- M. Wang, A. S. Joel, C. Ramshaw, D. Eimer and N. M. Musa, Appl. Energy, 2015, 158, 275–291 CrossRef CAS.
- A. I. Papadopoulos, S. Badr, A. Chremos, E. Forte, T. Zarogiannis, P. Seferlis, S. Papadokonstantakis, A. Galindo, G. Jackson and C. S. Adjiman, Mol. Syst. Des. Eng., 2016, 1, 313–334 CAS.
- F. Closmann, T. Nguyen and G. T. Rochelle, Energy Procedia, 2009, 1, 1351–1357 CrossRef CAS.
- European Commission, Final Report – CESAR (CO2 Enhanced Separation and Recovery), Community Research and Development Information Service (CORDIS), http://cordis.europa.eu/publication/rcn/13962_en.html, accessed July 2017.
- P. Singh and G. F. Versteeg, Process Saf. Environ. Prot., 2008, 86, 347–359 CrossRef CAS.
- G. Puxty, R. Rowland, A. Allport, Q. Yang, M. Bown, R. Burns, M. Maeder and M. Attalla, Environ. Sci. Technol., 2009, 43, 6427–6433 CrossRef CAS PubMed.
- S. Bommareddy, N. G. Chemmangattuvalappil, C. C. Solvason and M. R. Eden, Comput. Chem. Eng., 2010, 34, 1481–1486 CrossRef CAS.
- N. G. Chemmangattuvalappil and M. R. Eden, Ind. Eng. Chem. Res., 2013, 52, 7090–7103 CrossRef CAS.
- J. Salazar, U. Diwekar, K. Joback, A. H. Berger and A. S. Bhown, Energy Procedia, 2013, 37, 257–264 CrossRef.
- A. I. Papadopoulos, S. Badr, A. Chremos, E. Forte, T. Zarogiannis, P. Seferlis, S. Papadokonstantakis, C. S. Adjiman, A. Galindo and G. Jackson, Chem. Eng. Trans., 2014, 39, 211–216 Search PubMed.
- P. Limleamthong, M. Gonzalez-Miquel, S. Papadokonstantakis, A. I. Papadopoulos, P. Seferlis and G. Guillen-Gosalbez, Green Chem., 2016, 18, 6468–6481 RSC.
- C. S. Adjiman, A. Galindo and G. Jackson, Comput.-Aided Chem. Eng., 2014, 34, 55–64 CAS.
- J. Schilling, D. Tillmanns, M. Lampe, M. Hopp, J. Gross and A. Bardow, Mol. Syst. Des. Eng., 2017, 2, 301–320 CAS.
- A. Bardow, K. Steur and J. Gross, Ind. Eng. Chem. Res., 2010, 49, 2834–2840 CrossRef CAS.
- B. Oyarzún, A. Bardow and J. Gross, Energy Procedia, 2011, 4, 282–290 CrossRef.
- M. Stavrou, M. Lampe, A. Bardow and J. Gross, Ind. Eng. Chem. Res., 2014, 53, 18029–18041 CrossRef CAS.
- M. Lampe, M. Stavrou, J. Schilling, E. Sauer, J. Gross and A. Bardow, Comput. Chem. Eng., 2015, 81, 278–287 CrossRef CAS.
- F. E. Pereira, E. Keskes, A. Galindo, G. Jackson and C. S. Adjiman, Integrated design of CO2 capture processes from natural gas, Process Systems Engineering, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, pp. 231–248 Search PubMed.
- F. E. Pereira, E. Keskes, A. Galindo, G. Jackson and C. S. Adjiman, Comput. Chem. Eng., 2011, 35, 474–491 CrossRef CAS.
- J. Burger, V. Papaioannou, S. Gopinath, G. Jackson, A. Galindo and C. S. Adjiman, AIChE J., 2015, 61, 3249–3269 CrossRef CAS.
- S. Gopinath, G. Jackson, A. Galindo and C. S. Adjiman, AIChE J., 2016, 62, 3484–3504 CrossRef CAS.
- C. V. Brand, J. Rodrguez, A. Galindo, G. Jackson and C. S. Adjiman, Comput.-Aided Chem. Eng., 2012, 31, 930–934 CAS.
- A. Arce, N. Mac Dowell, N. Shah and L. F. Vega, Int. J. Greenhouse Gas Control, 2012, 11, 236–250 CrossRef.
- N. Mac Dowell and N. Shah, Int. J. Greenhouse Gas Control, 2013, 13, 44–58 CrossRef CAS.
- A. Alhajaj, N. Mac Dowell and N. Shah, Int. J. Greenhouse Gas Control, 2016, 44, 26–41 CrossRef CAS.
- C. V. Brand, E. Graham, J. Rodriguez, A. Galindo, G. Jackson and C. S. Adjiman, Faraday Discuss., 2016, 192, 337–390 RSC.
- S. B. Martynov, N. K. Daud, H. Mahgerefteh, S. Brown and R. T. J. Porter, Int. J. Greenhouse Gas Control, 2016, 54, 652–661 CrossRef CAS.
- IPCC, IPCC Special Report on Carbon Dioxide Capture and Storage, Prepared by Working Group III of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, UK and New York, NY, USA, 2005, p. 442 Search PubMed.
- ZEP, The costs of CO2transport: Post-demonstration CCS in the EU, Zero emissions platform (ZEP), 2011.
- A. Aspelund and K. Jordal, Int. J. Greenhouse Gas Control, 2007, 1, 343–354 CrossRef CAS.
- A. Aspelund, M. Mølnvik and G. De Koeijer, Chem. Eng. Res. Des., 2006, 84, 847–855 CrossRef CAS.
- S. McCoy and E. Rubin, Int. J. Greenhouse Gas Control, 2008, 2, 219–229 CrossRef CAS.
- S. Brown, H. Mahgerefteh, S. Martynov, V. Sundara and N. Mac Dowell, Int. J. Greenhouse Gas Control, 2015, 43, 108–114 CrossRef CAS.
- G. Skaugen, S. Roussanaly, J. Jakobsen and A. Brunsvold, Int. J. Greenhouse Gas Control, 2016, 54, 627–639 CrossRef CAS.
- A. Oosterkamp and J. Ramsen, State-of-the-art overview of CO2pipeline transport with relevance to offshore pipelines, January, Polytec, 2008.
- A. Chapoy, M. Nazeri, M. Kapateh, R. Burgass, C. Coquelet and B. Tohidi, Int. J. Greenhouse Gas Control, 2013, 19, 92–100 CrossRef CAS.
- S. T. Munkejord, M. Hammer and S. W. Løvseth, Appl. Energy, 2016, 169, 499–523 CrossRef CAS.
- R. T. Porter, M. Fairweather, M. Pourkashanian and R. M. Woolley, Int. J. Greenhouse Gas Control, 2015, 36, 161–174 CrossRef CAS.
- S. Liljemark, K. Arvidsson, M. T. Mc Cann, H. Tummescheit and S. Velut, Energy Procedia, 2011, 4, 3040–3047 CrossRef.
- J. J. Moore, A. Lerche, H. Delgado, T. Allison and J. Pacheco, Proceedings of the Fortieth Turbomachinery Symposium, 2011, pp. 107–120 Search PubMed.
- P. Pei, K. Barse, A. J. Gil and J. Nasah, Int. J. Greenhouse Gas Control, 2014, 30, 86–96 CrossRef CAS.
- P. A. Calado, Modeling and design synthesis of a CCS compression train system via MINLP optimization, Tecnico Lisboa, 2012, pp. 1–122 Search PubMed.
- A. Witkowski and M. Majkut, Arch. Mech. Eng., 2012, 59, 343–360 Search PubMed.
- A. Witkowski, A. Rusin, M. Majkut, S. Rulik and K. Stolecka, Energy Convers. Manage., 2013, 76, 665–673 CrossRef CAS.
- L. M. Romeo, I. Bolea, Y. Lara and J. M. Escosa, Appl. Therm. Eng., 2009, 29, 1744–1751 CrossRef CAS.
- R. S. Middleton and J. M. Bielicki, Energy Policy, 2009, 37, 1052–1060 CrossRef.
- A. Alhajaj, N. Mac Dowell and N. Shah, Energy Procedia, 2013, 37, 2552–2561 CrossRef CAS.
- T. Lazic, E. Oko and M. Wang, Proc. Inst. Mech. Eng., Part E, 2013, 228, 210–225 CrossRef.
- G. Fimbres Weihs and D. Wiley, Int. J. Greenhouse Gas Control, 2012, 8, 150–168 CrossRef CAS.
- R. S. Middleton, M. J. Kuby and J. M. Bielicki, Comput., Environ. Urban Syst., 2012, 36, 18–29 CrossRef.
- S. Roussanaly, J. P. Jakobsen, E. H. Hognes and A. L. Brunsvold, Int. J. Greenhouse Gas Control, 2013, 19, 584–594 CrossRef.
- B. Wetenhall, J. Race and M. Downie, Int. J. Greenhouse Gas Control, 2014, 30, 197–211 CrossRef CAS.
- M. K. Chandel, L. F. Pratson and E. Williams, Energy Convers. Manage., 2010, 51, 2825–2834 CrossRef CAS.
- M. Knoope, W. Guijt, A. Ramírez and A. Faaij, Int. J. Greenhouse Gas Control, 2014, 22, 25–46 CrossRef.
- Z. Wang, G. A. Fimbres Weihs, G. I. Cardenas and D. E. Wiley, Int. J. Greenhouse Gas Control, 2014, 31, 165–174 CrossRef CAS.
- N. Mac Dowell and I. Staffell, Int. J. Greenhouse Gas Control, 2015, 48, 327–344 CrossRef.
- M. Chaczykowski and A. J. Osiadacz, Int. J. Greenhouse Gas Control, 2012, 9, 446–456 CrossRef CAS.
- E. Mechleri, S. Brown, P. S. Fennell and N. Mac Dowell, Chem. Eng. Res. Des., 2017, 119, 130–139 CrossRef CAS.
- R. Cooper and J. Barnett, Energy Procedia, 2014, 63, 2412–2431 CrossRef CAS.
- J. Gale and J. Davison, Energy, 2004, 29, 1319–1328 CrossRef CAS.
- D. Shuter, M. Bilio, J. Wilday, L. Murray and R. Whitbread, Energy Procedia, 2011, 4, 2261–2268 CrossRef.
- S. Connolly and L. Cusco, IChemE Symposium Series, 2007, pp. 1–5.
- R. Woolley, M. Fairweather, C. Wareing, C. Proust, J. Hebrard, D. Jamois, V. Narasimhamurthy, I. Storvik, T. Skjold, S. Falle, S. Brown, H. Mahgerefteh, S. Martynov, S. Gant, D. Tsangaris, I. Economou, G. Boulougouris and N. Diamantonis, Int. J. Greenhouse Gas Control, 2014, 27, 221–238 CrossRef CAS.
- H. W. M. Witlox, J. Stene, M. Harper and S. H. Nilsen, Energy Procedia, 2011, 4, 2253–2260 CrossRef CAS.
- M. Bilio, S. Brown, M. Fairweather and H. Mahgerefteh, CO2 pipelines material and safety considerations, IChemE Symposium Series: Hazards XXI Process Safety and Environmental Protection, Manchester, 2009, pp. 423–429.
- H. Mahgerefteh, S. Brown and G. Denton, Chem. Eng. Sci., 2012, 74, 200–210 CrossRef CAS.
- A. Cosham and R. Eiber, Journal of Pipeline Engineering, 2008, 7, 115–124 Search PubMed.
- W. A. Maxey, Fracture initiation, propagation and arrest, Proceedings of the 5th Symposium in Line Pressure Research, Houston, 1974.
- A. Cosham, D. G. Jones, K. Armstrong, D. Allason and J. Barnett, Analysis of two dense phase carbon dioxide full-scale fracture propagation tests, 10th International Pipeline Conference, American Society of Mechanical Engineers, 2014, pp. 1–15.
- H. Mahgerefteh, S. Brown and P. Zhang, Journal of Pipeline Engineering, 2010, 9, 265–276 Search PubMed.
- H. Nordhagen, S. Kragset, T. Berstad, A. Morin, C. Dørum and S. Munkejord, Comput. Struct., 2012, 94-95, 13–21 CrossRef.
- E. Aursand, S. Dumoulin, M. Hammer, H. I. Lange, A. Morin, S. T. Munkejord and H. O. Nordhagen, Eng. Struct., 2016, 123, 192–212 CrossRef.
- S. Roussanaly, A. L. Brunsvold and E. S. Hognes, Int. J. Greenhouse Gas Control, 2014, 28, 283–299 CrossRef CAS.
- M. Knoope, A. Ramírez and A. Faaij, Int. J. Greenhouse Gas Control, 2015, 41, 174–193 CrossRef.
- J. Kjärstad, R. Skagestad, N. H. Eldrup and F. Johnsson, Int. J. Greenhouse Gas Control, 2016, 54, 168–184 CrossRef.
- R. Skagestad, N. Eldrup, H. R. Hansen, S. Belfroid, A. Mathisen, A. Lach and H. A. Haugen, Ship transport of CO2, 3918, Tel-Tek, 2014.
- T. N. Vermeulen, Knowledge Sharing Report – CO2Liquid Logistics Shipping Concept (LLSC): Overall Supply Chain Optimization, Global CCS Institute (GCCSI), 2011.
- N. Rydberg and D. Langlet, CCS in the Baltic Sea region – Bastor 2, Elforsk, 2014.
- C. Kolster, E. Mechleri, S. Krevor and N. Mac Dowell, Int. J. Greenhouse Gas Control, 2017, 58, 127–141 CrossRef CAS.
- J. Gale, J. C. Abanades, S. Bachu and C. Jenkins, Int. J. Greenhouse Gas Control, 2015, 40, 1–5 CrossRef.
- GCCSI, The global status of CCS: 2015, Global CCS Institute, Melbourne, Australia, 2015.
- Z. Duan, R. Sun, C. Zhu and I. M. Chou, Mar. Chem., 2006, 98, 131–139 CrossRef CAS.
- N. Spycher and K. Pruess, Transp. Porous Media, 2010, 82, 173–196 CrossRef CAS.
- S. P. Cadogan, G. C. Maitland and J. P. M. Trusler, J. Chem. Eng. Data, 2014, 59, 519–525 CrossRef CAS.
- S. P. Cadogan, J. P. Hallett, G. C. Maitland and J. P. M. Trusler, J. Chem. Eng. Data, 2015, 60, 181–184 CrossRef CAS.
- S. Bando, F. Takemura, M. Nishio, E. Hihara and M. Akai, J. Chem. Eng. Data, 2004, 49, 1328–1332 CrossRef CAS.
- M. Fleury and H. Deschamps, J. Chem. Eng. Data, 2008, 53, 2505–2509 CrossRef CAS.
- M. McBride-Wright, G. C. Maitland and J. P. M. Trusler, J. Chem. Eng. Data, 2015, 60, 171–180 CrossRef CAS.
- C. Calabrese, M. McBride-Wright, G. C. Maitland and J. P. M. Trusler, Viscosity and density of NaCl(aq) and CaCl2(aq) with dissolved CO2 at temperatures from (275 to 449) K and at pressures up to 100 MPa, J. Chem. Eng. Data, 2017 Search PubMed , submitted.
- A. Hebach, A. Oberhof, N. Dahmen, A. Kogel, H. Ederer and E. Dinjus, J. Chem. Eng. Data, 2002, 47, 1540–1546 CrossRef CAS.
- P. Chiquet, J. L. Daridon, D. Broseta and S. Thibeau, Energy Convers. Manage., 2007, 48, 736–744 CrossRef CAS.
- C. Chalbaud, M. Robin, J. M. Lombard, F. Martin, P. Egermann and H. Bertin, Adv. Water Resour., 2009, 32, 98–109 CrossRef CAS.
- A. Georgiadis, G. Maitland, J. P. M. Trusler and A. Bismarck, J. Chem. Eng. Data, 2010, 55, 4168–4175 CrossRef CAS.
- Y. T. F. Chow, G. C. Maitland and J. P. M. Trusler, J. Chem. Thermodyn., 2016, 93, 392–403 CrossRef CAS.
- X. Li, E. Boek, G. C. Maitland and J. P. M. Trusler, J. Chem. Eng. Data, 2012, 57, 1078–1088 CrossRef CAS.
- X. Li, E. S. Boek, G. C. Maitland and J. P. M. Trusler, J. Chem. Eng. Data, 2012, 57, 1369–1375 CrossRef CAS.
- C. Peng, J. P. Crawshaw, G. C. Maitland and J. P. M. Trusler, Chem. Geol., 2015, 403, 74–85 CrossRef CAS.
- C. Peng, B. U. Anabaraonye, J. P. Crawshaw, G. C. Maitland and J. P. M. Trusler, Faraday Discuss., 2016, 192, 545–560 RSC.
- H. P. Menke, B. Bijeljic, M. G. Andrew and M. J. Blunt, Environ. Sci. Technol., 2015, 49, 4407–4414 CrossRef CAS PubMed.
- K. S. Pedersen, P. L. Christensen and S. J. Azeem, Phase Behavior of Petroleum Reservoir Fluids, CRC Press, Boca Raton, FL, USA, Second edn, 2015, p. 450 Search PubMed.
- S. Iglauer, A. Salamah, M. Sarmadivaleh, K. Liu and C. Phan, Int. J. Greenhouse Gas Control, 2014, 22, 325–328 CrossRef CAS.
- M. Andrew, B. Bijeljic and M. J. Blunt, Adv. Water Resour., 2014, 68, 24–31 CrossRef CAS.
- K. Singh, B. Bijeljic and M. J. Blunt, Water Resour. Res., 2016, 52, 1716–1728 CrossRef.
- S. Benson, R. Pini, C. Reynolds and S. Krevor, Relative permeability analyses to describe multi-phase flow in CO2storage reservoirs, Global CCS Institute, 2013.
- S. M. Benson, F. Hingerl, L. Zuo, R. Pini, S. Krevor, C. Reynolds, B. Niu, R. Calvo and A. Niemi, Relative permeability for multi-phase flow in CO2storage reservoirs. Part II: resolving fundamental issues and filling data gaps, Global CCS Institute, 2015.
- P. Egermann, C. A. Chalbaud, J. Duquerroix and Y. Le Gallo, An integrated approach to parameterize reservoir models for CO2 injection in aquifers, SPE Annual Technical Conference and Exhibition, Society of Petroleum Engineers, Paper SPE-102308-MS, San Antonio, Texas, USA, 2006.
- J. C. Manceau, J. Ma, R. Li, P. Audigane, P. X. Jiang, R. N. Xu, J. Tremosa and C. Lerouge, Water Resour. Res., 2015, 51, 2885–2900 CrossRef.
- B. Niu, A. Al-Menhali and S. C. Krevor, Water Resour. Res., 2015, 51, 2009–2029 CrossRef CAS.
- C. A. Reynolds and S. Krevor, Water Resour. Res., 2015, 51, 9464–9489 CrossRef.
- S. Krevor, M. J. Blunt, S. M. Benson, C. H. Pentland, C. Reynolds, A. Al-Menhali and B. Niu, Int. J. Greenhouse Gas Control, 2015, 40, 221–237 CrossRef CAS.
- A. S. Al-Menhali and S. Krevor, Environ. Sci. Technol., 2016, 50, 2727–2734 CrossRef CAS PubMed.
- R. Juanes, E. J. Spiteri, F. M. Orr and M. J. Blunt, Water Resour. Res., 2006, 42, W12418 CrossRef.
- R. A. Salathiel, J. Pet. Technol., 1973, 25, 1216–1224 CrossRef.
- E. J. Spiteri, R. Juanes, M. J. Blunt and F. M. Orr, Soc. Pet. Eng. J., 2008, 13, 277–288 Search PubMed.
- A. S. Al-Menhali, H. P. Menke, M. J. Blunt and S. C. Krevor, Environ. Sci. Technol., 2016, 50, 10282–10290 CrossRef CAS PubMed.
- B. S. Koelbl, M. A. van den Broek, B. J. van Ruijven, A. P. C. Faaij and D. P. van Vuuren, Int. J. Greenhouse Gas Control, 2014, 27, 81–102 CrossRef CAS.
- J.-C. Perrin and S. Benson, Transp. Porous Media, 2010, 82, 93–109 CrossRef CAS.
- M. Krause, S. Krevor and S. M. Benson, Transp. Porous Media, 2013, 98, 565–588 CrossRef.
- B. Li and S. M. Benson, Adv. Water Resour., 2015, 83, 389–404 CrossRef CAS.
- A. Rabinovich, K. Itthisawatpan and L. J. Durlofsky, J. Pet. Sci. Eng., 2015, 134, 60–75 CrossRef CAS.
- T. A. Meckel, S. L. Bryant and P. Ravi Ganesh, Int. J. Greenhouse Gas Control, 2015, 34, 85–96 CrossRef CAS.
- S. C. M. Krevor, R. Pini, B. Li and S. M. Benson, Geophys. Res. Lett., 2011, 38, L15401 CrossRef.
- E. Saadatpoor, S. L. Bryant and K. Sepehrnoori, Transp. Porous Media, 2010, 82, 3–17 CrossRef CAS.
- R. A. Chadwick and D. J. Noy, Geological Society, London, Petroleum Geology Conference series, 2010, 7, pp. 1171–1182.
- A. J. Cavanagh and R. S. Haszeldine, Int. J. Greenhouse Gas Control, 2014, 21, 101–112 CrossRef CAS.
- S. D. Hovorka, S. M. Benson, C. Doughty, B. M. Freifeld, S. Sakurai, T. M. Daley, Y. K. Kharaka, M. H. Holtz, R. C. Trautz, H. S. Nance, L. R. Myer and K. G. Knauss, Environ. Geosci., 2006, 13, 105–121 CrossRef.
- J. Lu, P. J. Cook, S. A. Hosseini, C. Yang, K. D. Romanak, T. Zhang, B. M. Freifeld, R. C. Smyth, H. Zeng and S. D. Hovorka, J. Geophys. Res.: Solid Earth, 2012, 117, B03208 Search PubMed.
- A. C. Gringarten, Evolution of reservoir management techniques: From independent methods to an integrated methodology. Impact on petroleum engineering curriculum, graduate teaching and competitive advantage of oil companies, SPE Asia Pacific Conference on Integrated Modelling for Asset Management, Society of Petroleum Engineers, Paper SPE-39713-MS, Kuala Lumpur, Malaysia, 1998.
- M. A. Flett, G. J. Beacher, J. Brantjes, A. J. Burt, C. Dauth, F. M. Koelmeyer, R. Lawrence, S. Leigh, J. McKenna, R. Gurton, W. F. Robinson and T. Tankersley, Gorgon Project: Subsurface evaluation of carbon dioxide disposal under Barrow Island. SPE Asia Pacific Oil and Gas Conference and Exhibition, Society of Petroleum Engineers, Paper SPE-116372-MS, Perth, Australia, 2008.
- M. Flett, J. Brantjes, R. Gurton, J. McKenna, T. Tankersley and M. Trupp, Energy Procedia, 2009, 1, 3031–3038 CrossRef CAS.
- Shell, Peterhead CCS Project Storage Development Plan, Document number PCCS-00-PT-AA-5726-00001, Shell UK Limited, 2015.
- ETI, Progressing Development of the UK's Strategic Carbon Dioxide Storage Resource, Energy Technologies Institute (ETI), Pale Blue Dot Energy & Axis Well Technology, 2016.
- J. P. Verdon, J.-M. Kendall, A. L. Stork, R. A. Chadwick, D. J. White and R. C. Bissell, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E2762–E2771 CrossRef CAS PubMed.
- J. A. White, L. Chiaramonte, S. Ezzedine, W. Foxall, Y. Hao, A. Ramirez and W. McNab, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8747–8752 CrossRef CAS PubMed.
- S. Grude, M. Landrø and J. Dvorkin, Int. J. Greenhouse Gas Control, 2014, 27, 178–187 CrossRef CAS.
- R. A. Chadwick, D. J. Noy and S. Holloway, Pet. Geosci., 2009, 15, 59–73 CrossRef CAS.
- Shell, Quest carbon capture and storage project reaches significant one-year milestone, Shell Canada News Press Release, http://www.shell.ca/en_ca/media/news-and-media-releases/news-releases-2016/shell_s-quest-carbon-capture-and-storage-project-reaches-signifi.html, accessed October 2016.
- P. E. Bergmo, D. Wessel-Berg and A. A. Grimstad, Energy Procedia, 2014, 63, 5114–5122 CrossRef CAS.
- J. T. Birkholzer, A. Cihan and Q. Zhou, Int. J. Greenhouse Gas Control, 2012, 7, 168–180 CrossRef CAS.
- T. A. Buscheck, Y. Sun, M. Chen, Y. Hao, T. J. Wolery, W. L. Bourcier, B. Court, M. A. Celia, S. J. Friedmann and R. D. Aines, Int. J. Greenhouse Gas Control, 2012, 6, 230–245 CrossRef CAS.
- A. Cihan, J. T. Birkholzer and M. Bianchi, Int. J. Greenhouse Gas Control, 2015, 42, 175–187 CrossRef CAS.
- R. Qi, T. C. LaForce and M. J. Blunt, Int. J. Greenhouse Gas Control, 2009, 3, 195–205 CrossRef CAS.
- Y. Leonenko and D. W. Keith, Environ. Sci. Technol., 2008, 42, 2742–2747 CrossRef CAS PubMed.
- M. Burton and S. L. Bryant, SPE Reservoir Eval. Eng., 2009, 12, 399–407 CrossRef CAS.
- C. Jenkins, A. Chadwick and S. D. Hovorka, Int. J. Greenhouse Gas Control, 2015, 40, 312–349 CrossRef CAS.
- R. A. Chadwick, R. Arts and O. Eiken, Geological Society, London, Petroleum Geology Conference series, 2005, 6, pp. 1385–1399.
- R. Pevzner, E. Caspari, B. Gurevich, T. Dance and Y. Cinar, Geophysics, 2015, 80, B105–B114 CrossRef.
- A. Ghaderi and M. Landrø, Geophysics, 2009, 74, O17–O28 CrossRef.
- M. Trani, R. Arts, O. Leeuwenburgh and J. Brouwer, Geophysics, 2011, 76, C1–C17 CrossRef.
- J. B. Ajo-Franklin, J. Peterson, J. Doetsch and T. M. Daley, Int. J. Greenhouse Gas Control, 2013, 18, 497–509 CrossRef CAS.
- T. Dance and L. Paterson, Int. J. Greenhouse Gas Control, 2016, 47, 210–220 CrossRef CAS.
- S. M. V. Gilfillan, B. S. Lollar, G. Holland, D. Blagburn, S. Stevens, M. Schoell, M. Cassidy, Z. Ding, Z. Zhou, G. Lacrampe-Couloume and C. J. Ballentine, Nature, 2009, 458, 614–618 CrossRef CAS PubMed.
- S. M. V. Gilfillan, M. Wilkinson, R. S. Haszeldine, Z. K. Shipton, S. T. Nelson and R. J. Poreda, Int. J. Greenhouse Gas Control, 2011, 5, 1507–1516 CrossRef CAS.
- M. Myers, L. Stalker, B. Pejcic and A. Ross, Appl. Geochem., 2013, 30, 125–135 CrossRef CAS.
- T. LaForce, J. Ennis-King, C. Boreham and L. Paterson, Int. J. Greenhouse Gas Control, 2014, 26, 9–21 CrossRef CAS.
- D. A. Cameron, L. J. Durlofsky and S. M. Benson, Int. J. Greenhouse Gas Control, 2016, 52, 32–43 CrossRef.
- J. L. Lewicki, C. M. Oldenburg, L. Dobeck and L. Spangler, Geophys. Res. Lett., 2007, 34, L24402 CrossRef.
- K. Shitashima, Y. Maeda and A. Sakamoto, Int. J. Greenhouse Gas Control, 2015, 38, 135–142 CrossRef CAS.
- M. Bickle, N. Kampman and M. Wigley, Rev. Mineral. Geochem., 2013, 77, 15–71 CrossRef CAS.
- J. C. Manceau, D. G. Hatzignatiou, L. de Lary, N. B. Jensen and A. Réveillère, Int. J. Greenhouse Gas Control, 2014, 22, 272–290 CrossRef CAS.
- A. Esposito and S. M. Benson, Int. J. Greenhouse Gas Control, 2012, 7, 62–73 CrossRef CAS.
- S. Vialle, J. L. Druhan and K. Maher, Int. J. Greenhouse Gas Control, 2016, 44, 11–25 CrossRef CAS.
- S. Pacala and R. Socolow, Science, 2004, 305, 968–972 CrossRef CAS PubMed.
- S. M. Benson, K. Bennaceur, P. Cook, J. Davison, H. de Coninck, K. Farhat, A. Ramirez, D. Simbeck, T. Surles, P. Verma and I. Wright, Carbon Capture and Storage, Global Energy Assessment–Toward a Sustainable Future, 2012, ch. 13, pp. 993–1068 Search PubMed.
- J. J. Dooley, Energy Procedia, 2012, 37, 5141–5150 CrossRef.
- G. Cook and P. Zakkour, CCS deployment in the context of regional developments in meeting long-term climate change objectives, Report 2015/TR3, IEA Greenhouse Gas R&D Programme (IEAGHG), 2015.
- S. Bachu, Int. J. Greenhouse Gas Control, 2015, 40, 188–202 CrossRef CAS.
- J. T. Birkholzer, C. M. Oldenburg and Q. Zhou, Int. J. Greenhouse Gas Control, 2015, 40, 203–220 CrossRef CAS.
- M. Winkler, R. Abernathy, M. Nicolo, H. Huang, A. Wang, S. Zhang, A. Simon, C. Clark, S. Crouch, H. De Groot, R. El Mahdy, M. Smith, S. Malik, S. Bourne, R. Pierpont and V. Hugonet, The dynamic aspect of formation-storage use for CO2 sequestration, SPE International Conference on CO2 Capture, Storage, and Utilization, Society of Petroleum Engineers, Paper SPE-139730-MS, New Orleans, Louisiana, USA, 2010.
- A. Goodman, G. Bromhal, B. Strazisar, T. Rodosta, W. F. Guthrie, D. Allen and G. Guthrie, Int. J. Greenhouse Gas Control, 2013, 18, 329–342 CrossRef CAS.
- S. Thibeau and V. Mucha, Oil Gas Sci. Technol., 2011, 66, 81–92 CrossRef CAS.
- A. G. Bader, S. Thibeau, O. Vincké, F. Delprat Jannaud, S. Saysset, G. H. Joffre, F. M. Giger, M. David, M. Gimenez, A. Dieulin and D. Copin, Energy Procedia, 2014, 63, 2779–2788 CrossRef CAS.
- C. D. Gorecki, S. C. Ayash, G. Liu, J. R. Braunberger and N. W. Dotzenrod, Int. J. Greenhouse Gas Control, 2015, 42, 213–225 CrossRef CAS.
- A. Lothe, B. U. Emmel, P. Bergmo, G. M. Mortensen and P. Frykman, Updated estimate of storage capacity and evaluation of Seal for selected Aquifers (D26), NORDICCS Technical Report D 6.3.1401 (D26), Nordic CCS Competence Centre (NORDICCS), 2015.
- J. M. Nordbotten, M. A. Celia and S. Bachu, Transp. Porous Media, 2005, 58, 339–360 CrossRef CAS.
- Q. Zhou, J. T. Birkholzer, C.-F. Tsang and J. Rutqvist, Int. J. Greenhouse Gas Control, 2008, 2, 626–639 CrossRef CAS.
- S. A. Mathias, P. E. Hardisty, M. R. Trudell and R. W. Zimmerman, Transp. Porous Media, 2009, 79, 265–284 CrossRef CAS.
- M. J. Golding, J. A. Neufeld, M. A. Hesse and H. E. Huppert, J. Fluid Mech., 2011, 678, 248–270 CrossRef CAS.
- M. L. Szulczewski, C. W. MacMinn, H. J. Herzog and R. Juanes, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5185–5189 CrossRef CAS PubMed.
- X. Huang, K. W. Bandilla, M. A. Celia and S. Bachu, Int. J. Greenhouse Gas Control, 2014, 20, 73–86 CrossRef CAS.
- S. Agada, S. Jackson, C. Kolster, N. Mac Dowell, G. Williams, H. Vosper, J. Williams and S. Krevor, Int. J. Greenhouse Gas Control, 2017, 65, 128–136 CrossRef.
- C. Kolster, S. Agada, N. Mac Dowell and S. Krevor, Int. J. Greenhouse Gas Control, 2018, 68, 77–85 CrossRef CAS.
- Global CCS Institute, CCS images, Understanding CCS Resources, http://www.globalccsinstitute.com/understanding-ccs/information-resource, Melbourne, Australia, accessed January 2017.
- F. Gozalpour, S. R. Ren and B. Tohidi, Oil Gas Sci. Technol., 2005, 60, 537–546 CAS.
- S. Chen, H. Li, D. Yang and P. Tontiwachwuthikul, J. Can. Pet. Technol., 2010, 49, 75–82 CrossRef CAS.
- N. Mac Dowell, P. S. Fennell, N. Shah and G. C. Maitland, Nat. Clim. Change, 2017, 7, 243–249 CrossRef CAS.
- IEA, Storing CO2through enhanced oil recovery-Combining EOR with CO2storage (EOR+) for profit, International Energy Agency Insights Series, Paris, France, 2015.
- B. Hitchon, Best Practices for Validating CO2 Geological Storage: Observations and Guidance from the IEAGHG Weyburn-Midale CO2 Monitoring and Storage Project, Geoscience Publishing Ltd., Sherwood Park, Alberta, Canada, 2012 Search PubMed.
- MIT, Weyburn-Midale Fact Sheet: Carbon Dioxide Capture and Storage Project, http://https://sequestration.mit.edu/tools/projects/weyburn.html, Carbon Capture and Sequestration Technologies program at MIT, 2016.
- MIT, Boundary Dam Fact Sheet: Carbon Dioxide Capture and Storage Project, http://https://sequestration.mit.edu/tools/projects/boundary_dam.html, Carbon Capture and Sequestration Technologies program at MIT, 2016.
- F. van Bergen, J. Gale, K. J. Damen and A. F. B. Wildenborg, Energy, 2004, 29, 1611–1621 CrossRef CAS.
- M. L. Godec, Global technology roadmap for CCS in industry: Sectoral assessment CO2 enhanced oil recovery, Advanced Resources International, Inc. and United Nations Industrial Development Organization (UNIDO), 2011 Search PubMed.
- J. J. Dooley, R. T. Dahowski, C. L. Davidson, M. A. Wise, N. Gupta, S. H. Kim, E. L. Malone and B. M. Institute, Carbon dioxide capture and geologic storage: A core element of a global energy technology strategy to address climate change, The Global Energy Technology Strategy Program, Battelle Memorial Institute, USA, 2006 Search PubMed.
- IEA, Technology roadmap: Carbon capture and storage, International Energy Agency, Paris, France, 2013, 2013 edn Search PubMed.
- CIA, The World Factbook, http://https://www.cia.gov/library/publications/the-world-factbook/rankorder/2241rank.html, Central Intelligence Agency, United States, 2014.
- Rystad Energy, UCube Upstream Database, http://https://www.rystadenergy.com/Products/EnP-Solutions/UCube, Oslo, Norway, 2017.
- C. Kolster, M. S. Masnadi, S. Krevor, N. Mac Dowell and A. R. Brandt, Energy Environ. Sci., 2017, 10, 2594–2608, 10.1039/C7EE02102J.
- QCCSRC, Qatar Carbonates and Carbon Storage Research Centre, http://www.imperial.ac.uk/qatar-carbonates-and-carbon-storage, Imperial College London, UK, 2017.
- M. Mazzotti, R. Pini and G. Storti, J. Supercrit. Fluids, 2009, 47, 619–627 CrossRef CAS.
- X. Li and D. Elsworth, J. Nat. Gas Sci. Eng., 2015, 26, 1607–1619 CrossRef CAS.
- G. Ersland, J. Husebø, A. Graue and B. Kvamme, Energy Procedia, 2009, 1, 3477–3484 CrossRef CAS.
- LEILAC, Low Emissions Intensity Lime & Cement, A European Union Horizon 2020 Research & Innovation Project, http://www.project-leilac.eu/, Calix Europe Ltd, 2017.
- M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed.
- E. A. Quadrelli, G. Centi, J.-L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS PubMed.
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Mueller, Energy Environ. Sci., 2012, 5, 7281–7305 CAS.
- A. Otto, T. Grube, S. Schiebahn and D. Stolten, Energy Environ. Sci., 2015, 8, 3283–3297 CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- R. M. Cuellar-Franca and A. Azapagic, J. CO2 Util., 2015, 9, 82–102 CrossRef CAS.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 CAS.
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS PubMed.
- A. Scott, Chem. Eng. News, 2015, 93, 10–16 Search PubMed.
- A. Goeppert, M. Czaun, G. K. Surya Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833–7853 CAS.
- A. Sanna, M. Uibu, G. Caramanna, R. Kuusik and M. M. Maroto-Valer, Chem. Soc. Rev., 2014, 43, 8049–8080 RSC.
- N. von der Assen, L. J. Mueller, A. Steingrube, P. Voll and A. Bardow, Environ. Sci. Technol., 2016, 50, 1093–1101 CrossRef CAS PubMed.
- J. Langanke, A. Wolf, J. Hofmann, K. Boehm, M. A. Subhani, T. E. Mueller, W. Leitner and C. Guertler, Green Chem., 2014, 16, 1865–1870 RSC.
- N. von der Assen and A. Bardow, Green Chem., 2014, 16, 3272–3280 RSC.
- A. Sternberg and A. Bardow, ACS Sustainable Chem. Eng., 2016, 4, 4156–4165 CrossRef CAS.
- C. van der Giesen, R. Kleijn and G. J. Kramer, Environ. Sci. Technol., 2014, 48, 7111–7121 CrossRef CAS PubMed.
- European Commission Joint Research Centre and Institute for Environment and Sustainability, International Reference Life Cycle Data System (ILCD) Handbook-General guide for Life Cycle Assessment-Detailed guidance, Publications Office of the European Union, Luxembourg, 1st edn, 2010 Search PubMed.
- N. von der Assen, J. Jung and A. Bardow, Energy Environ. Sci., 2013, 6, 2721–2734 CAS.
- A. Levasseur, P. Lesage, M. Margni, L. Deschênes and R. Samson, Environ. Sci. Technol., 2010, 44, 3169–3174 CrossRef CAS PubMed.
- G. P. Peters, B. Aamaas, M. T. Lund, C. Solli and J. S. Fuglestvedt, Environ. Sci. Technol., 2011, 45, 8633–8641 CrossRef CAS PubMed.
- M. Brandão, A. Levasseur, M. U. F. Kirschbaum, B. P. Weidema, A. L. Cowie, S. V. Jørgensen, M. Z. Hauschild, D. W. Pennington and K. Chomkhamsri, Int. J. Life Cycle Assess., 2013, 18, 230–240 CrossRef.
- T. Bruhn, H. Naims and B. Olfe-Kraeutlein, Environ. Sci. Policy, 2016, 60, 38–43 CrossRef CAS.
- K. Thenert, K. Beydoun, J. Wiesenthal, W. Leitner and J. Klankermayer, Angew. Chem., Int. Ed., 2016, 55, 12266–12269 CrossRef CAS PubMed.
- B. Lumpp, D. Rothe, C. Pastötter, R. Lämmermann and E. Jacob, MTZ worldwide eMagazine, 2011, 72, 34–38 CrossRef.
- N. Schmitz, J. Burger, E. Stroefer and H. Hasse, Fuel, 2016, 185, 67–72 CrossRef CAS.
- F. D. Meylan, V. Moreau and S. Erkman, J. CO2 Util., 2015, 12, 101–108 CrossRef CAS.
- IEA, Key world energy statistics, International Energy Agency, http://www.iea.org/publications/freepublications/publication/KeyWorld2017.pdf, 2017.
- K. Gutmann, J. Huscher, D. Urbaniak, A. White, C. Schaible and M. Bricke, Europe's Dirty 30: How the EU's coal-fired power plants are undermining its climate efforts, Climate Action Network (CAN)Europe, Health and Environment Alliance (HEAL), WWF European Policy Office, European Environmental Bureau (EEB) and Climate Alliance Germany, Brussels, Belgium, 2014.
- M. Aresta, A. Dibenedetto and A. Angelini, J. CO2 Util., 2013, 3–4, 65–73 CrossRef CAS.
- S. F. Mitchell and D. F. Shantz, AIChE J., 2015, 61, 2374–2384 CrossRef CAS.
- H. Naims, Environ. Sci. Pollut. Res., 2016, 23, 22226–22241 CrossRef CAS PubMed.
- A. Sternberg and A. Bardow, Energy Environ. Sci., 2015, 8, 389–400 CAS.
- I. Dimitriou, P. Garcia-Gutierrez, R. H. Elder, R. M. Cuellar-Franca, A. Azapagic and R. W. K. Allen, Energy Environ. Sci., 2015, 8, 1775–1789 CAS.
- Carbon Recycling International, World's Largest CO2Methanol Plant, Kopavogur, Iceland, http://carbonrecycling.is/george-olah/2016/2/14/worlds-largest-co2-methanol-plant, accessed March 2017.
- K. Roh, J. H. Lee and R. Gani, Int. J. Greenhouse Gas Control, 2016, 47, 250–265 CrossRef CAS.
- M. Götz, J. Lefebvre, F. Mörs, A. M. Koch, F. Graf, S. Bajohr, R. Reimert and T. Kolb, Renewable Energy, 2016, 85, 1371–1390 CrossRef.
- M. Pérez-Fortes, J. C. Schöneberger, A. Boulamanti and E. Tzimas, Appl. Energy, 2016, 161, 718–732 CrossRef.
- J. Klankermayer and W. Leitner, Philos. Trans. R. Soc., A, 2016, 374, 1–8 CrossRef PubMed.
- A. A. Kiss, J. J. Pragt, H. J. Vos, G. Bargeman and M. T. de Groot, Chem. Eng. J., 2016, 284, 260–269 CrossRef CAS.
- S. Rönsch, J. Schneider, S. Matthischke, M. Schlüter, M. Götz, J. Lefebvre, P. Prabhakaran and S. Bajohr, Fuel, 2016, 166, 276–296 CrossRef.
- M. Scott, B. Blas Molinos, C. Westhues, G. Franciò and W. Leitner, ChemSusChem, 2017, 10, 1085–1093 CrossRef CAS PubMed.
- C. M. Jens, L. Müller, K. Leonhard and A. Bardow, To integrate or not to integrate – Techno-economic and life cycle assessment of CO2 capture and conversion to methyl formate using methanol, The Royal Society of Chemistry, submitted Search PubMed.
- A. J. Martin, G. O. Larrazabal and J. Perez-Ramirez, Green Chem., 2015, 17, 5114–5130 RSC.
- J. A. Herron and C. T. Maravelias, Energy Technol., 2016, 4, 1369–1391 CrossRef CAS.
- O. Machhammer, A. Bode and W. Hormuth, Chem. Eng. Technol., 2016, 39, 1185–1193 CrossRef CAS.
- S. Postels, A. Abánades, N. von der Assen, R. K. Rathnam, S. Stückrad and A. Bardow, Int. J. Hydrogen Energy, 2016, 41, 23204–23212 CrossRef CAS.
- Z. Yuan, M. R. Eden and R. Gani, Ind. Eng. Chem. Res., 2016, 55, 3383–3419 CrossRef CAS.
- W. Leitner, J. Klankermayer, S. Pischinger, H. Pitsch and K. Kohse-Höinghaus, Angew. Chem., Int. Ed., 2017, 56, 5412–5452 CrossRef CAS PubMed.
- A. Grinberg Dana, O. Elishav, A. Bardow, G. E. Shter and G. S. Grader, Angew. Chem., Int. Ed., 2016, 55, 8798–8805 CrossRef CAS PubMed.
- M. Sharifzadeh, L. Wang and N. Shah, Renewable Sustainable Energy Rev., 2015, 47, 151–161 CrossRef CAS.
- S.-Y. Pan, R. Adhikari, Y.-H. Chen, P. Li and P.-C. Chiang, J. Cleaner Prod., 2016, 137, 617–631 CrossRef CAS.
- OECD, OECD Environmental Outlook for the Chemicals Industry, Organisation for Economic Co-operation and Development, Paris, France, 2001 Search PubMed.
- J. M. Earles and A. Halog, Int. J. Life Cycle Assess., 2011, 16, 445–453 CrossRef.
- A. Kätelhön, A. Bardow and S. Suh, Environ. Sci. Technol., 2016, 50, 12575–12583 CrossRef PubMed.
- W. Leitner, Angew. Chem., Int. Ed., 1995, 34, 2207–2221 CrossRef CAS.
- S. Moret, P. J. Dyson and G. Laurenczy, Nat. Commun., 2014, 5, 4017 CAS.
- T. Schaub and R. A. Paciello, Angew. Chem., Int. Ed., 2011, 50, 7278–7282 CrossRef CAS PubMed.
- M. Pérez-Fortes, J. C. Schöneberger, A. Boulamanti, G. Harrison and E. Tzimas, Int. J. Hydrogen Energy, 2016, 41, 16444–16462 CrossRef.
- K. Beydoun, G. Ghattas, K. Thenert, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2014, 53, 11010–11014 CrossRef CAS PubMed.
- A. Tlili, E. Blondiaux, X. Frogneux and T. Cantat, Green Chem., 2015, 17, 157–168 RSC.
- E. Leino, P. Mäki-Arvela, V. Eta, D. Y. Murzin, T. Salmi and J.-P. Mikkola, Appl. Catal., A, 2010, 383, 1–13 CrossRef CAS.
- I. Garcia-Herrero, R. M. Cuéllar-Franca, V. M. Enríquez-Gutiérrez, M. Alvarez-Guerra, A. Irabien and A. Azapagic, ACS Sustainable Chem. Eng., 2016, 4, 2088–2097 CrossRef CAS.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- C. M. Jens, K. Nowakowski, J. Scheffczyk, K. Leonhard and A. Bardow, Green Chem., 2016, 18, 5621–5629 RSC.
- N. von der Assen, P. Voll, M. Peters and A. Bardow, Chem. Soc. Rev., 2014, 43, 7982–7994 RSC.
- N. von der Assen, A. Sternberg, A. Kaetelhoen and A. Bardow, Faraday Discuss., 2015, 183, 291–307 RSC.
- P. Pawelzik, M. Carus, J. Hotchkiss, R. Narayan, S. Selke, M. Wellisch, M. Weiss, B. Wicke and M. K. Patel, Resour., Conserv. Recycl., 2013, 73, 211–228 CrossRef.
- C. R. Jones, D. Kaklamanou, W. M. Stuttard, R. L. Radford and J. Burley, Faraday Discuss., 2015, 183, 327–347 RSC.
- J. van Heek, K. Arning and M. Ziefle, Energy Policy, 2017, 105, 53–66 CrossRef CAS.
- InfoCuria, Judgement of the Court (First Chamber) of 19 January 2017, Schaefer Kalk GmbH & Co. KG versus Bundesrepublik Deutschland. Document ECLI:EU:C:2017:29, Case-law of the Court of Justice, http://curia.europa.eu/juris/liste.jsf?language=en&num=C-460/15, 2017.
- T. P. Wright, J. Aeronaut. Sci., 1936, 3, 122–128 CrossRef.
- Boston Consulting Group, Perspectives on Experience, Boston Consulting Group Inc., Boston, MA, United States, 1972.
- S. Yeh and E. S. Rubin, Energy Econ., 2012, 34, 762–771 CrossRef.
- E. S. Rubin, S. Yeh, M. Antes, M. Berkenpas and J. Davison, Int. J. Greenhouse Gas Control, 2007, 1, 188–197 CrossRef CAS.
- E. S. Rubin, I. M. L. Azevedo, P. Jaramillo and S. Yeh, Energy Policy, 2015, 86, 198–218 CrossRef.
- S. Li, X. Zhang, L. Gao and H. Jin, Appl. Energy, 2012, 93, 348–356 CrossRef.
- M. van den Broek, R. Hoefnagels, E. Rubin, W. Turkenburg and A. Faaij, Prog. Energy Combust. Sci., 2009, 35, 457–480 CrossRef CAS.
- M. Monea, Plenary presentation, 12th International Conference on Greenhouse Gas Control Technologies (GHGT-12), Austin, Texas, US, 2014.
- J. Schwartz, High-Stakes Test for Carbon Capture, New York Times, 3 January, 2017.
- J. G. Canadell and E. D. Schulze, Nat. Commun., 2014, 5, 5282 CrossRef PubMed.
- J. Kemper, Int. J. Greenhouse Gas Control, 2015, 40, 401–430 CrossRef CAS.
- Archer Daniels Midland Company, ADM Begins Operations for Second Carbon Capture and Storage Project, http://https://www.adm.com/news/news-releases/adm-begins-operations-for-second-carbon-capture-and-storage-project-1, accessed June 2017.
- S. Gollakota and S. McDonald, Greenhouse Gases: Sci. Technol., 2012, 2, 346–351 CrossRef CAS.
- R. Finley, Greenhouse Gases: Sci. Technol., 2014, 4, 571–579 CrossRef.
- R. Jones and R. McKaskle, Greenhouse Gases: Sci. Technol., 2014, 4, 617–625 CrossRef.
- H. Karlsson and L. Byström, Global Status of BECCS Projects 2010, Global CCS Institute and Biorecro, http://https://www.globalccsinstitute.com/publications/global-status-beccs-projects-2010, 2011.
- K. Whiriskey, Carbon dioxide removal-Necessary but unloved. Insight to upcoming report on CO2removal. Presentation, 13th International Conference on Greenhouse Gas Control Technologies (GHGT-13), Lausanne, Switzerland, 2016.
- D. Woolf, J. Amonette, F. Street-Perrott, J. Lehmann and S. Joseph, Nat. Commun., 2010, 1, 56 Search PubMed.
- IEAGHG, Potential for Biomass and Carbon Dioxide Capture and Storage. Report 2011/06, IEA Greenhouse Gas R&D Programme, Cheltenham, UK, 2011.
- IEAGHG, Potential for Biomethane Production and Carbon Dioxide Capture and Storage. Report 2013/11, IEA Greenhouse Gas R&D Programme, Cheltenham, UK, 2013.
- D. McLaren, Process Saf. Environ. Prot., 2012, 90, 489–500 CrossRef CAS.
- D. van Vuuren, S. Deetman, J. van Vliet, M. Berg, B. Ruijven and B. Koelbl, Clim. Change, 2013, 118, 15–27 CrossRef.
- A. Arasto, K. Onarheim, E. Tsupari and J. Kärki, Energy Procedia, 2014, 63, 6756–6769 CrossRef CAS.
- B. Caldecott, G. Lomax and M. Workman, Stranded carbon assets and negative emissions technologies. Working paper, Smith School of Enterprise and the Environment, University of Oxford, 2015.
- NRC, Climate Intervention: Carbon Dioxide Removal and Reliable Sequestration, National Research Council and National Academy of Sciences, The National Academies Press, Washington, DC, United States, 2015 Search PubMed.
- Global Carbon Project, Global Carbon Atlas: CO2emissions, accessed March 2017.
- D. Keith, M. Ha-Duong and J. Stolaroff, Clim. Change, 2006, 74, 17–45 CrossRef CAS.
- S. Rose, R. Beach, K. Calvin, B. McCarl, J. Petrusa, B. Sohngen, R. Youngman, A. Diamant, F. de la Chesnaye, J. Edmonds, R. Rosenzweig and M. Wise, Estimating global greenhouse gas emissions offset supplies: Accounting for investment risks and other market realties, Electric Power Research Institute (EPRI), Palo Alto, CA, USA, 2013.
- L. Smith and M. Torn, Clim. Change, 2013, 118, 89–103 CrossRef CAS.
- L. Clarke, K. Jiang, K. Akimoto, M. Babiker, G. Blanford, K. Fisher-Vanden, J.-C. Hourcade, V. Krey, E. Kriegler, A. Löschel, D. McCollum, S. Paltsev, S. Rose, P. R. Shukla, M. Tavoni, B. van der Zwaan and D. van Vuuren, Assessing Transformation Pathways. In: Climate Change 2014: Mitigation of Climate Change. Contribution of Working Group III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, United Kingdom, and New York, NY, USA, 2014.
- S. Sukumara, A multidisciplinary techno-economic decision support tool for validating long-term economic viability of biorefining processes, PhD thesis, University of Kentucky, Lexington, KY, USA, 2014 Search PubMed.
- H. Chum, A. Faaij, J. Moreira, G. Berndes, P. Dharnija, H. Dong and B. Gabrielle, Bioenergy. In: IPCC Special Report on Renewable Energy Sources and Climate Change Mitigation. Prepared by Working Group III of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, UK, New York, NY, USA, 2011, pp. 209–332 Search PubMed.
- R. Slade, R. Saunders, R. Gross and A. Bauen, Energy from biomass: the size of the global resource. An assessment of the evidence that biomass can make a major contribution to future global energy supply, Imperial College Centre for Energy Policy and Technology and UK Energy Research Centre, London, United Kingdom, 2011.
- IEA and FAO, How2Guide for Bioenergy: Roadmap Development and Implementation, International Energy Agency and the Food and Agriculture Organisation of the United Nations, 2017.
- FAO, Food wastage footprint: Impacts on natural resources. Summary report, Food and Agriculture Organization (FAO) of the United Nations, http://www.fao.org/docrep/018/i3347e/i3347e.pdf, 2013.
- A. Welfle, P. Gilbert and P. Thornley, Biomass Bioenergy, 2014, 70, 249–266 CrossRef.
- J. Seay and F. You, 4-Biomass supply, demand, and markets, Biomass Supply Chains for Bioenergy and Biorefining, Woodhead Publishing, 2016, pp. 85–100 Search PubMed.
- D. Yue and F. You, 7-Biomass and biofuel supply chain modeling and optimization, Biomass Supply Chains for Bioenergy and Biorefining, Woodhead Publishing, 2016, pp. 149–166 Search PubMed.
- European Environment Agency (EEA), Opinion of the EEA Scientific Committee on Greenhouse Gas Accounting in Relation to Bioenergy, 2011.
- T. Searchinger and R. Heimlich, Avoiding bioenergy competition for food crops and land, Working paper, Installment 9 of Creating a Sustainable Food Future, World Resources Institute, Washington, DC, United States, 2015.
- J. Hartmann and S. Kempe, Naturwissenschaften, 2008, 95, 1159–1164 CrossRef CAS PubMed.
- R. Socolow, M. Desmond, R. Aines, J. Blackstock, O. Bolland, T. Kaarsberg, N. Lewis, M. Mazzotti, A. Pfeffer, K. Sawyer, J. Siirola, B. Smit and J. Wilcox, Direct Air Capture of CO2 with Chemicals-A Technology Assessment for the APS Panel on Public Affairs, American Physical Society (APS) Physics, 2011.
- R. D. Schuiling and P. Krijgsman, Clim. Change, 2006, 74, 349–354 CrossRef CAS.
- S. Wood, K. Sebastian and S. J. Scherr, Pilot analysis of global ecosystems: Agroecosystems, International Food Policy Research Institute and World Resources Institute, Washington, DC, United States, 2000.
- S. Kang, W. Post, J. Nichols, D. Wang, T. West, V. Bandaru and R. Izaurralde, J. Agric. Sci., 2013, 5, 129–139 Search PubMed.
- FAOSTAT, FAOSTAT land database, accessed February 2017.
- J. Campbell, D. Lobell, R. Genova and C. Field, Environ. Sci. Technol., 2008, 42, 5791–5794 CrossRef CAS PubMed.
- S. Fritz, L. See, M. van der Velde, R. A. Nalepa, C. Perger, C. Schill, I. McCallum, D. Schepaschenko, F. Kraxner, X. Cai, X. Zhang, S. Ortner, R. Hazarika, A. Cipriani, C. Di Bella, A. H. Rabia, A. Garcia, M. Vakolyuk, K. Singha, M. E. Beget, S. Erasmi, F. Albrecht, B. Shaw and M. Obersteiner, Environ. Sci. Technol., 2013, 47, 1688–1694 CAS.
- K. Lackner, Sci. Am., 2010, 302, 66–71 CrossRef CAS PubMed.
- E. Kriegler, O. Edenhofer, L. Reuster, G. Luderer and D. Klein, Clim. Change, 2013, 118, 45–57 CrossRef CAS.
- P. Smith, M. Bustamante, H. Ahammad, H. Clark, H. Dong, E. Elsiddig, H. Haberl, R. Harper, J. House, M. Jafari, O. Masera, C. Mbow, N. Ravindranath, C. Rice, C. Robledo Abad, A. Romanovskaya, F. Sperling and F. Tubiello, Agriculture, Forestry and Other Land Use (AFOLU). In: Climate Change 2014: Mitigation of Climate Change. Contribution of Working Group III to the FifthAssessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, United Kingdom, and New York, NY, USA, 2014.
- M. Fajardy, Investigating the water-energy-carbon and land nexus of bio-energy and CCS (BECCS). Presentation, 13th International Conference on Greenhouse Gas Control Technologies (GHGT-13), Lausanne, Switzerland, 2016.
- N. Mac Dowell and M. Fajardy, Faraday Discuss., 2016, 192, 241–250 RSC.
- M. Flugge, J. Lewandrowski, J. Rosenfeld, C. Boland, T. Hendrickson, K. Jaglo, S. Kolansky, K. Moffroid, M. Riley-Gilbert and D. Pape, A life-cycle analysis of the greenhouse gas emissions of corn-based ethanol. Report prepared by ICF under USDA Contract No. AG-3142-D-16-0243, US Department of Agriculture, Climate Change Program Office, Washington, DC, http://https://www.usda.gov/oce/climate_change/mitigation_technologies/USDAEthanolReport_20170107.pdf, 2017.
- A. M. Thomson, K. V. Calvin, L. P. Chini, G. Hurtt, J. A. Edmonds, B. Bond-Lamberty, S. Frolking, M. A. Wise and A. C. Janetos, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19633–19638 CrossRef CAS PubMed.
- J. Gustavsson, C. Cederberg, U. Sonesson, R. van Otterdijk and A. Meybeck, Global food losses and food waste-Extent, causes and prevention, Food and Agriculture Organization (FAO) of the United Nations, Rome, Italy, http://www.fao.org/docrep/014/mb060e/mb060e00.pdf, 2011.
- HLPE, Food losses and waste in the context of sustainable food systems, High Level Panel of Experts on Food Security and Nutrition of the Committee on World Food Security, Rome, Italy, 2014.
- K. Al-Qayim, W. Nimmo and M. Pourkashanian, Int. J. Greenhouse Gas Control, 2015, 43, 82–92 CrossRef CAS.
- M. Pourkashanian, J. Szuhanszki and K. Finney, BECCS-Technical challenges and opportunities. Presentation, UKCCSRC BECCS Specialist Meeting, London, 2016.
- C. Gough and P. Upham, Biomass energy with carbon capture and storage (BECCS): A review. Working Paper 147, The Tyndall Centre, University of Manchester, Manchester Institute of Innovation Research, 2010.
- P. Luckow, M. Wise, J. Dooley and S. Kim, Int. J. Greenhouse Gas Control, 2010, 4, 865–877 CrossRef CAS.
- C. Hamelinck, Fact checks for the biofuels sustainability debate, Ecofys Webinar, http://www.slideshare.net/Ecofys/factsheets-on-the-sustainability-of-biofuels, 2014.
- IEAGHG, Biomass and CCS-Guidance for accounting for negative emissions. Report 2014/05, IEA Greenhouse Gas R&D Programme, Cheltenham, UK, 2014.
- J. Dooley, Keynote II-3, Industrial CO2removal: CO2capture from ambient air and geological. In: Meeting Report of the Intergovernmental Panel on Climate Change Expert Meeting on Geoengineering, IPCC Working Group III Technical Support Unit, Potsdam Institute for Climate Impact Research, Potsdam, Germany, 2012, pp. 30–33.
- G. Lomax, M. Workman, T. Lenton and N. Shah, Energy Policy, 2015, 78, 125–136 CrossRef.
- J. Meerman, M. Knoope, A. Ramirez, W. Turkenburg and A. Faaij, Int. J. Greenhouse Gas Control, 2013, 16, 311–323 CrossRef CAS.
- S. Thomas, P. Dargusch, S. Harrison and J. Herbohn, Land Use Policy, 2010, 27, 880–887 CrossRef.
- R. Sands, S. Malcolm, S. Suttles and E. Marshall, Dedicated energy crops and competition for agricultural land, ERR-223, U.S. Department of Agriculture, Economic Research Service, 2017.
- M. Wise, K. Calvin, A. Thomson, L. Clarke, B. Bond-Lamberty, R. Sands, S. Smith, A. Janetos and J. Edmonds, Science, 2009, 324, 1183–1186 CrossRef CAS PubMed.
- P. Upham and T. Roberts, Public perceptions of CCS: the results of Near CO2European focus groups, Tyndall Centre, The University of Manchester, accessed March 2015, 2010.
- S. Mander, D. Polson, T. Roberts and A. Curtis, Energy Procedia, 2011, 4, 6360–6367 CrossRef.
- A.-M. Dowd, M. Rodriguez and T. Jeanneret, Energies, 2015, 8, 4024–4042 CrossRef.
- C. Peters, J. Picardy, A. Darrouzet-Nardi, J. Wilkins, T. Griffin and G. Fick, Elementa, 2016, 4, 000116 Search PubMed.
- J. Ranganathan, D. Vennard, R. Waite, B. Lipinski, T. Searchinger, P. Dumas, A. Forslund, H. Guyomard, S. Manceron, E. Marajo-Petitzon, C. Le Mouël, P. Havlik, M. Herrero, X. Zhang, S. Wirsenius, F. Ramos, X. Yan, M. Phillips and R. Mungkung, Shifting diets for a sustainable food future. Working paper, Installment 11 of Creating a Sustainable Food Future, World Resources Institute, Washington, DC, United States, 2016.
- S. Wirsenius, C. Azar and G. Berndes, Agr. Syst., 2010, 103, 621–638 CrossRef.
- C. Mooney, The suddenly urgent quest to remove carbon dioxide from the air, The Washington Post, 2016.
- D. Biello, 400 PPM: Can Artificial Trees Help Pull CO2from the Air?, Scientific American, 2013.
- E. Kolbert, Can carbon-dioxide removal save the world?, The New Yorker, 2017.
- Sucking up carbon, Greenhouse gases must be scrubbed from the air, The Economist, 2017.
- M. Gunther, Startups have figured out how to remove carbon from the air. Will anyone pay them to do it?, The Guardian, 2015.
- M. K. McNutt, W. Abdalati, K. Caldeira, S. C. Doney, P. G. Falkowski, S. Fetter, J. R. Fleming, S. P. Hamburg, G. Morgan, J. E. Penner, R. T. Pierrehumbert, P. J. Rasch, L. M. Russell, J. T. Snow, D. W. Titley and J. Wilcox, Climate Intervention: Carbon Dioxide Removal and Reliable Sequestration, The National Academies Press, Washington, D. C., USA, 2015 Search PubMed.
- G. Holmes and D. W. Keith, Philos. Trans. R. Soc., A, 2012, 370, 4380 CrossRef CAS PubMed.
- M. Mazzotti, R. Baciocchi, M. J. Desmond and R. H. Socolow, Clim. Change, 2013, 118, 119–135 CrossRef CAS.
- K. S. Lackner, S. Brennan, J. M. Matter, A.-H. A. Park, A. Wright and B. van der Zwaan, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13156–13162 CrossRef CAS PubMed.
- J. Wilcox, P. Rochana, A. Kirchofer, G. Glatz and J. He, Energy Environ. Sci., 2014, 7, 1769–1785 CAS.
- J. K. Stolaroff, D. W. Keith and G. V. Lowry, Environ. Sci. Technol., 2008, 42, 2728–2735 CrossRef CAS PubMed.
- J. T. Cullinane, Thermodynamics and Kinetics of Aqueous Piperazine with Potassium Carbonate for Carbon Dioxide Absorption, PhD thesis, The University of Texas at Austin, 2005 Search PubMed.
- F. Zeman, Environ. Sci. Technol., 2007, 41, 7558–7563 CrossRef CAS PubMed.
- M. Mahmoudkhani and D. W. Keith, Int. J. Greenhouse Gas Control, 2009, 3, 376–384 CrossRef CAS.
- E. S. Rubin, C. Chen and A. B. Rao, Energy Policy, 2007, 35, 4444–4454 CrossRef.
- E. S. Rubin, J. E. Davison and H. J. Herzog, Int. J. Greenhouse Gas Control, 2015, 40, 378–400 CrossRef CAS.
- J. Wilcox, P. C. Psarras and S. Liguori, Environ. Res. Lett., 2017, 12, 065001 CrossRef.
- EU, Brussels European Council 8/9 March 2007 Presidency Conclusions, Council of the European Union, 2007.
- P. Dixon and T. Mitchell, Lesson Learned: Lessons and Evidence Derived from UK CCS Programmes, 2008–2015, Carbon Capture and Storage Association, 2016.
- TUC, The economic benefits of carbon capture and storage in the UK, Carbon Capture & Storage Association (CCSA) & Trades Union Congress (TUC), 2014.
- NAO, Briefing for the House of Commons Environmental Audit Committee: Sustainability in the spending review, National Audit Office, 2016.
- D. Radov, A. Carmel, H. Fearnehough, C. Koenig, S. Forrest, G. Strbac, M. Aunedi and D. PudjiantoUK Renewable Subsidies and Whole System Costs: The Case for Allowing Biomass Conversion for a CfD, NERA Economic Consulting & Imperial College London, 2016 Search PubMed.
- DECC, CCS Cost Reduction Taskforce Final Report, UK Carbon Capture and Storage Cost Reduction Task Force, London, UK, 2013.
- DECC, CCS Roadmap: Supporting deployment of carbon capture and storage in the UK, Department of Energy & Climate Change, 2012.
- Capture Power Limited, White Rose: K.04 Full-chain FEED lessons learnt, White Rose Carbon Capture & Storage Project, Department of Energy and Climate Change, 2016.
- Capture Power Limited, White Rose: K.01 Full chain FEED summary report, White Rose Carbon Capture & Storage Project, Department of Energy and Climate Change, 2016.
- SCCS, CO2storage and Enhanced Oil Recovery in the North Sea: Securing a low-carbon future for the UK, Scottish Carbon Capture & Storage, 2015.
- E. Davey, Government agreement on energy policy sends clear, durable signal to investors, Department of Energy & Climate Change, http://https://www.gov.uk/government/news/government-agreement-on-energy-policy-sends-clear-durable-signal-to-investors, 2012.
- H. P. Kitschelt, Br. J. Polit. Sci., 1986, 16, 57–85 CrossRef.
- D. Kennedy, Science, 2007, 316, 515 CrossRef CAS PubMed.
- S. Krohn and S. Damborg, Renewable Energy, 1999, 16, 954–960 CrossRef.
- P. C. Stern, B. K. Sovacool and T. Dietz, Nature Climate Change, 2016, 6, 547–555 CrossRef.
- P. Ashworth, S. Wade, D. Reiner and X. Liang, Int. J. Greenhouse Gas Control, 2015, 40, 449–458 CrossRef.
- IEA, Carbon capture and storage: The solution for deep emissions reductions, Organisation for Economic Co-operation and Development (OECD) and International Energy Agency (IEA), Paris, France, 2015.
- GCCSI, The global status of CCS: 2016 summary report, Global CCS Institute, Melbourne, Australia, 2016.
- IEA, Energy and Climate Change: World Energy Outlook Special Briefing for COP21, Organisation for Economic Co-operation and Development (OECD) and International Energy Agency (IEA), Paris, France, 2015.
- GCCSI and UNIDO, Carbon capture and storage in industrial applications: Technology synthesis report, Global CCS Institute and United Nations Industrial Development Organization (UNIDO), Vienna, 2010.
- CAN, Climate Action Network Europe (CAN Europe) position paper on CO2capture and storage, http://www.caneurope.org/publications/can-europe-positions/90-carbon-capture-and-storage, 2006.
- E. Rochon, E. Bjureby, P. Johnston, R. Oakley, D. Santillo, N. Schulz and G. von Goerne, False Hope: Why carbon capture and storage won't save the climate, Greenpeace International, Amsterdam, The Netherlands, 2008 Search PubMed.
- B. W. Terwel, E. ter Mors and D. D. L. Daamen, Int. J. Greenhouse Gas Control, 2012, 9, 41–51 CrossRef.
- E. Cuppen, S. Brunsting, U. Pesch and Y. Feenstra, Environment and Planning A, 2015, 47, 1963–1978 CrossRef.
- A. Tjernshaugen, Environ. Polit., 2011, 20, 227–245 CrossRef.
- K. Buhr and A. Hansson, Global Environ. Change, 2011, 21, 336–345 CrossRef.
- A. Doyle, Norway drops carbon capture plan it had likened to “Moon landing”, Reuters, http://www.reuters.com/article/us-norway-carbon-idUSBRE98J0QB20130920, 2013.
- G. Fouche, Norway says could achieve full carbon capture and storage by 2022, Reuters, http://www.reuters.com/article/us-norway-ccs-idUSKCN0ZK1LW, 2016.
- S. Ansolobehere, J. Beer, J. Deutch, A. D. Ellerman, S. J. Friedman, H. Herzog, H. D. Jacoby, P. L. Joskow, G. McRae, R. Lester, E. J. Moniz and E. Steinfeld, The future of coal: Options for a carbon-constrained world, Massachusetts Institute of Technology (MIT), Cambridge, MA, US, 2007 Search PubMed.
- NAO, Carbon capture and storage: lessons from the competition for the first UK demonstration, National Audit Office (NAO), 2012.
- Committee of Public Accounts, Carbon Capture and Storage inquiry, Sixty-fourth Report of Session 2016–17, 28 April 2017, House of Commons, London, UK, http://https://publications.parliament.uk/pa/cm201617/cmselect/cmpubacc/1036/1036.pdf, accessed July 2017.
- R. Syal, Carbon capture scheme collapsed 'over government department disagreements', The Guardian, http://https://www.theguardian.com/environment/2017/jan/20/carbon-capture-scheme-collapsed-over-government-department-disagreements, 2017.
- T. Krüger, Energy Policy, 2017, 100, 58–67 CrossRef.
- H. de Coninck, Energy Policy, 2008, 36, 929–936 CrossRef.
- M. Wara, Nature, 2007, 445, 595–596 CrossRef CAS PubMed.
- M. Lupion and H. J. Herzog, Int. J. Greenhouse Gas Control, 2013, 19, 19–25 CrossRef.
- R. Oxburgh, Parliamentary Debate, House of Lords, 9 September 2015, column 1443, http://www.publications.parliament.uk/pa/ld201516/ldhansrd/text/150909-0001.htm, accessed December 2016.
- N. D. McGlashan and A. J. Marquis, J. Mech. Eng. Sci., 2007, 221, 1057–1065 CAS.
- J. Fisher, A. Zoelle, M. Turner and V. Chou, Quality Guidelines for Energy System Studies: Performing a Techno-economic Analysis for Power Generation Plants, National Energy Technology Laboratory (NETL), 2015.
- T. Fout, A. Zoelle, D. Keairns, L. Pinkerton, M. Turner, M. Woods, N. Kuehn, V. Shah and V. Chou, Cost and Performance Baseline for Fossil Energy Plants Volume 1a: Bituminous Coal (PC) and Natural Gas to Electricity Revision 3, National Energy Technology Laboratory, 2015.
- M. Matuszewski, J. Ciferno, J. Marano and S. Chen, Research and Development Goals for CO2 Capture Technology, National Energy Technology Laboratory, http://https://www.netl.doe.gov/research/energy-analysis/search-publications/vuedetails?id=817, 2011.
- D. C. Miller, D. A. Agarwal, D. Bhattacharyya, J. Boverhof, Y. W. Cheah, Y. Chen, J. Eslick, J. Leek, J. Ma, P. Mahapatra, B. Ng, N. Sahinidis, C. Tong and S. E. Zitney, Innovative computational tools and models for the design, optimization of control of carbon capture processes, 26th European Symposium on Computer Aided Process Engineering (ESCAPE 26), Computer Aided Chemical Engineering, 2016, pp. 2391–2396.
Footnotes |
| † Anecdotally attributed to Lord Ronald Oxburgh of the United Kingdom House of Lords. |
| ‡ The “technology readiness level” (TRL) system provides a means of tracking the status of technologies during their progression through different stages of research and development (R&D). It is a nine-point scaling system used to qualitatively evaluate the maturity level of a technology.8–10 |
| § Endogenous learning occurs through learning curves in these models, i.e. cumulative capacity determines the cost reductions, while other models assume cost reductions according to an exogenously given factor. |
| ¶ 27th round of the Energy Modelling Forum: http://https://emf.stanford.edu/projects/emf-27-global-model-comparison-exercise. |
| || It has to be noted, however, that in most cases, results were only available for the full time horizon and scenarios considered for 12 models out of the 18 ones that participated, thus the authors conclude that more research is needed to substantiate some of the more detailed findings, which this section will not go into. |
| ** The technology-focussed models are engineering-based models which consider a large number of energy technologies. They are typically used to calculate the least cost approach to meet a given demand (e.g., emission reduction target). In contrast, macro-econometric models consider production costs at an industry level, offering more economic detail but lack structural detail. A hybrid model combines both technology-based and macro-economic approaches.44 |
| †† To ensure global warming stays below 2 °C, the cumulative emissions from 1870 must remain less than 3650 GtCO2.45 Of this quota, the total remaining emissions from 2017 is estimated to be around 800 GtCO2.46–48 At current emission rates, global emissions is expected to exceed the 800 GtCO2 budget within 20 years.47 |
| ‡‡ The models currently only include BECCS and some of them afforestation. Please refer to section for a discussion of this and to Table 2 for an overview of alternative negative emission technologies. |
| §§ In contrast, limited nuclear and solar/wind availability only increased mitigation costs by 7% and 6%, respectively.6 |
| ¶¶ It has to be noted that these scenarios are characterised by different probabilities than the 2 °C scenarios reviewed above, which means that the focus here should be on the qualitative results and not a direct comparison of numbers. |
| |||| There are a few that also consider large-scale afforestation, i.e. CO2 is sequestered in additionally grown vegetation. |
| *** For comparison, the land area of 380–700 Mha is equivalent to 53–97% of the total land used for cereal production worldwide (∼720 Mha).62 The land intensity of BECCS is 0.1–0.6 ha per tC,eq per year (energy crops and agricultural residues), requiring more land than other NETs, e.g., enhanced weathering requires <0.01 ha per tC,eq per year, direct air capture needs <0.001 ha per tC,eq per year.61 |
| ††† Market cannibalisation refers to the effect of decreasing market price that occurs with increased production of intermittent renewable energy. The reduction in market price is due the following reasons: (i) highest production of wind and solar energy does not coincide with the peak electricity demand, and (ii) market value tends to reduce with increased market share. |
| ‡‡‡ In July 2016, the Department of Energy and Climate Change (DECC) merged with the Department for Business, Innovation and Skills (BIS) to form the Department for Business, Energy and Industrial Strategy (BEIS). |
| §§§ The agreed strike price of 89.50 per MWh has been fully indexed to the Consumer Price Index. It also includes a price reduction benefit, which is based on the assumption that EDF Energy will distribute the first of a kind costs of the reactors across the Hinkley Point C and Sizewell sites. However, if EDF decides not to invest in Sizewell C, the strike price for Hinkley Point C alone will be 92.50 per MWh.97,98 |
| ¶¶¶ Examination of the profiles for the last five years showed that the profiles for 2012/13/14 were very similar in shape. However, 2011 had significant fluctuations (more peaks) and 2010 had substantially lower load factors. Thus, 2012 was chosen to represent a typical availability profile. |
| |||||| Assuming that the embodied energy in the biomass supply chain are not themselves great. |
| **** This is in contrast to activity on commercial deployment of large-scale CCS, where the vast majority of operational projects are in the industrial sector,119,156 15 of the 17, and only two are in the power sector.4 |
| †††† Note the solvent blend in this study included an absorption activator is N-methylcyclohexylamine, MCA, regeneration promoter is N,N-dimethylcyclohexylamine, DMCA, and solubiliser is AMP. |
| ‡‡‡‡ This assessment was performed with 30 wt% MEA as the amine system. As was noted in Section 4, this technology was originally proposed in 1930, with much superior solvent systems currently available. |
| §§§§ Perovskites have a cubic structure and formula ABO3−δ where A and B are the ions at the vertices and centres respectively (e.g., in the mineral perovskite A = Ca2+, B = Ti4+). Their usefulness in chemical looping arises from the fact that they can show variable non-stoichiometry δ allowing a limited amount of oxygen to be transferred without reconstructive phase change. The variable stoichiometry also allows conduction of oxygen ions through the lattice. |
| ¶¶¶¶ Oxides are by far the most common class of material to have been studied, e.g., as gas separation membranes,483–488 but also for chemical looping and adsorption (discussed in earlier sections). |
| |||||||| The WGS reaction is mildly exothermic and has lower equilibrium conversions at higher temperature. |
| ***** Note that supported molten carbonate membranes are confined to the class of membranes that utilises an oxide support as there is no oxygen available for co-permeation. |
| ††††† It is likely that the methane would need to be fed with some water to avoid carbon deposition. |
| ‡‡‡‡‡ The highest pressure at which liquid–vapour mixtures can exist. |
| §§§§§ Transport and heating through increased electrification and/or hydrogen production with CCS. |
| ¶¶¶¶¶ A measure of the goods and services produced in any region, industry or economic sector of an economy. |
| |||||||||| September 2016, ETS (€4.5–€5 per tCO2), UK carbon floor price £23 per tCO2. |
| ****** This assumes a conventional CO2 capture process, where the capture process is additional to the power generation process. In the context of, for example, chemical looping combustion the separation of CO2 is intrinsic to the combustion process, therefore, no additional energy penalty is imposed for the separation of CO2. In all cases, compression of CO2 will impose a penalty. |
| †††††† Consider the CO2 reduction below a baseline PC plant (CO2 foot print of 816.5 kg MWhnet−1) provided by Integrated Gasification Fuel Cell (IGFC) technologies with a CO2 footprint of 612.3 kg MWhnet−1versus the same baseline PC plant with CCS and a CO2 footprint of ∼91 kg MWhnet−1. |
| This journal is © The Royal Society of Chemistry 2018 |