
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemperature dependence of the spin state and geometry in tricobalt paddlewheel complexes with halide axial ligands†
Anandi
Srinivasan
ab,
Xiaoping
Wang
 c,
Rodolphe
Clérac
ab,
Mathieu
Rouzières
ab,
Larry R.
Falvello
d,
John E.
McGrady
e and
Elizabeth A.
Hillard
*ab
c,
Rodolphe
Clérac
ab,
Mathieu
Rouzières
ab,
Larry R.
Falvello
d,
John E.
McGrady
e and
Elizabeth A.
Hillard
*ab
aCNRS, CRPP, UMR 5031, 33600 Pessac, France. E-mail: hillard@crpp-bordeaux.cnrs.fr
bUniv. of Bordeaux, CRPP, UMR 5031, F-33600 Pessac, France
cNeutron Scattering Division, Neutron Sciences Directorate, Oak Ridge National Laboratory, Oak Ridge, TN, USA
dDept. of Inorganic Chemistry and ICMA, Univ. of Zaragoza, Zaragoza, Spain
eDepartment of Chemistry, University of Oxford, Oxford, UK
First published on 15th November 2018
Abstract
Trinuclear cobalt paddlewheel complexes, [Co3(dpa)4X2] (dpa = the anion of 2,2′-dipyridylamine, X = Cl−, Br−, –NCS−, –CN−, (NC)2N−), are known to demonstrate a thermally-induced spin-crossover (SCO). Despite a wealth of structural and magnetic information about such complexes, the role of the axial ligand on the characteristic SCO temperature (T1/2) remains ambiguous. The situation is complicated by the observation that the solid state geometry of the complexes, symmetric or unsymmetric, with respect to the central cobalt ion, also appears to influence the SCO behavior. In order to seek trends in the relationship between the nature of the axial ligand, geometry and magnetic properties, we have prepared the first examples of tricobalt paddlewheel complexes with axial fluorido and iodido ligands, as well as two new chlorido and bromido solvates. Their SCO properties are discussed in the context of an examination of previously reported chlorido and bromido adducts. The main conclusions are: (1) T1/2 values follow the trend I− < Br− ≈ Cl− < F−; (2) while the molecular geometry is predominantly guided by crystal packing for the Cl−, Br− and I− derivatives, the presence of an axial fluoride may favor a more symmetric core; (3) the magnetic characterization of a second example of an unsymmetric complex supports the observation that they display dramatically lower T1/2 values than their symmetric analogues; and (4) SCO in crystallographically symmetric compounds apparently occurs without loss of molecular or crystallographic symmetry, while a gradual geometric transformation linking the temperature dependence of quasi-symmetric to unsymmetric in crystallographically unconstrained compounds was found.
Introduction
Complexes consisting of three or more linearly-disposed metal ions, “extended metal atom chains” (EMACs) or “metal strings”, have been intensively studied with respect to their electronic, magnetic, and single-molecule conductive properties.1–3 The simplest examples are trinuclear complexes supported by four 2,2′-dipyridylamine anions (dpa), where the axial positions are most often occupied by halide or pseudohalide ligands. Chromium and cobalt derivatives have the particularity of presenting either symmetric metal cores, where ΔM–M, the difference between the intermetallic distances, is less than 0.05 Å,1 or dramatically unsymmetric cores, where ΔM–M has been found to reach up to 0.65 Å.4 In trinuclear chromium complexes, the preferred geometry appears to depend on the nature of the axial ligands; stronger σ-donors tend to stabilize the symmetric arrangement and weaker donors the unsymmetric form.5,6The effect of the axial ligand on the conformation of the tricobalt core is less well understood. Remarkably, [Co3(dpa)4Cl2] can present a symmetric (s-[Co3(dpa)4Cl2]) or unsymmetric core (u-[Co3(dpa)4Cl2]), depending on the crystalline environment (Chart 1).7,8 All evidence to date points to this variability being a solid state phenomenon; in solution, only s-[Co3(dpa)4Cl2] has been detected,9 while a Hirshfeld surface analysis demonstrated the importance of intermolecular interactions on the geometry of [Co3(dpa)4Cl2].10 Geometric plasticity is not restricted to the chlorido adducts; in the case of [Co3(dpa)4Br2], for which three solvates have been crystallographically characterized, two of the structures are rigorously symmetric at room temperature, while the third is markedly unsymmetric.11 Conversely, all of the reported complexes with stronger-field axial ligands (–CN−, –NCS−, N(CN)2−) are highly symmetric12 suggesting that, like for the trichromium analogues, the ligand field may play a role in the core geometry.
 | ||
| Chart 1 | ||
This question is not purely academic, as it directly influences the spin-crossover (SCO) behaviour found in many tricobalt complexes.7,8 For example, while the dichloromethane solvates of both s- and u-[Co3(dpa)4Cl2] show a thermally-induced SCO, there are significant differences in their solid state magnetic signatures. Symmetric [Co3(dpa)4Cl2]·CH2Cl2 was reported to display an S = 1/2 ground state with g ≈ 2.3, and the temperature where 50% of the complexes are in the high spin state, T1/2, equal to 329 K.7 On the other hand, the unsymmetric form, [Co3(dpa)4Cl2]·2CH2Cl2, presented a low-temperature plateau with χT ≈ 1 cm3 K mol−1, indicating an S = 1/2 ground state with g ≈ 3.2. This material displays a SCO at relatively low temperature, with an asymptotic curve reaching ∼2.5 cm3 K mol−1, indicating significant population of the S = 3/2 excited state at high temperatures.7
If we ever hope to exploit the SCO properties of dpa-based tricobalt complexes, it is necessary to better understand the role of the ligand environment on the core geometry and magnetic behaviour. To this end, we have completed the series of halide adducts with the first examples of the fluorido and iodido complexes of [Co3(dpa)4X2], namely [Co3(dpa)4F2]·2CH2Cl2, (1·2CH2Cl2), [Co3(dpa)4I2]·C2H4Cl2 (4·C2H4Cl2) and [Co3(dpa)4I2]·Et2O (4·Et2O). During this work, we also obtained new solvates of the chlorido and bromido adducts, [Co3(dpa)4Cl2]·Et2O (2·Et2O) and [Co3(dpa)4Br2]·Et2O (3·Et2O). To contextualize their structural and magnetic temperature dependence, we have examined the magnetic data for a number of previously reported chlorido8 and bromido11 adducts, and report the previously-undetermined thermodynamic parameters characterizing their SCO behaviour.
Experimental section
Materials
All reactions were carried out under an inert atmosphere of argon or nitrogen using standard Schlenk and glove box techniques. Anhydrous CoCl2 from Fisher Chemicals was stored at 120 °C, NaI and AgBF4 from Alfa Aesar were dried under vacuum (10−3 mbar) for 12 h and stored in a glovebox, anhydrous tetrabutylammonium fluoride (TBAF) solution (1 M in THF) and CoBr2 from Sigma Aldrich were stored in a nitrogen atmosphere. Dichloromethane (CH2Cl2), diethyl ether (Et2O) and acetonitrile (MeCN) were purified using an Inert Technologies solvent purification system. Anhydrous dimethylformamide (DMF), n-hexane and 1,2-dichloroethane (C2H4Cl2) were purchased from Acros and degassed prior to use. [Co3(dpa)4Cl2]·nCH2Cl2 (n = 1, 2) was prepared according to the literature procedure.13Physical measurements
CHN elemental analyses were performed by the Service d'Analyse Elémentaire, UMR 7565, Université de Lorraine, France and by PLACAMAT, UMS 3626, Université de Bordeaux. IR spectra were measured on a Nicolet 6700 FT-IR using a Smart iTR accessory between 550 and 4000 cm−1.Crystallography
Single crystals suitable for X-ray diffraction were selected under immersion oil in ambient conditions and attached to a MiTeGen microloop. For structures at 350 K, the crystals were fixed to a pin using Apiezon AP101 grease. The crystals were mounted and centred in the X-ray beam using a video camera. Data collection was performed on a Bruker APEXII Quasar diffractometer with Mo Kα (λ = 0.71073 Å) radiation. The data were collected using a routine to survey reciprocal space, and were indexed by the APEX2 program.14 Data were reduced and integrated using SAINT+ (ref. 14) and an absorption correction was applied using SADABS.15 The structures were solved using direct methods and refined by least-squares refinement on F2 followed by difference Fourier synthesis.16 All hydrogen atoms were introduced at idealized positions and were allowed to ride on the neighbouring atoms with relative isotropic displacement coefficients. Crystal and refinement data are given in Tables S1–S5.† Cif files have been deposited with the Cambridge Structural Database CCDC 1865577–1865593, 1877180 and 1877181.Magnetic measurements
Magnetic measurements were carried out with an MPMS-XL Quantum Design SQUID magnetometer, working between 1.8 and 400 K with applied dc fields ranging from −7 to 7 T. Measurements were performed on freshly filtered polycrystalline samples of 1·2CH2Cl2 (22.97 mg), 2·Et2O (20.82 mg), 3·Et2O (22.10 mg), 4·C2H4Cl2 (17.94 mg) and 4·Et2O (17.43 mg) sealed in a polypropylene bag (3 × 0.5 × 0.02 cm). Prior to the experiments, the field-dependent magnetization was measured at 100 K in order to confirm the absence of any bulk ferromagnetic impurities. The magnetic data were corrected for the sample holder and the intrinsic diamagnetic contributions.Synthesis
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) , cm−1): 1602s, 1590s, 1546m, 1466w, 1450w, 1419m, 1360s, 1308s, 1281m, 1150s, 1053w, 1023s, 882m, 756m, 737m, 649w.
, cm−1): 1603s, 1591s, 1546m, 1468s, 1457s, 1416s, 1363s, 1313s, 1279m, 1251w, 1152s, 1104m, 1041w, 1018s, 932w, 884s, 779w, 762s, 740s, 639w, 565w.
, cm−1): 1603s, 1591s, 1546m, 1467s, 1456s, 1416s, 1362s, 1313s, 1279m, 1250w, 1164w, 1152s, 1104s, 1042w, 1018s, 964w, 933w, 885s, 843w, 761s, 739s, 647w, 563w.
, cm−1): 1603s, 1593s, 1547s, 1454s, 1418s, 1370s, 1313s, 1236w, 1162w, 1146m, 947w, 925m, 886s, 769m, 751m, 729m, 670m, 573w, 557w.
, cm−1): 1604s, 1591s, 1548m, 1455s, 1420s, 1368s, 1312s, 1167m, 1148s, 1107m, 1045w, 1017m, 1004w, 925w, 885m, 756s, 734s, 636s, 580w.
, cm−1): 1602s, 1590s, 1546m, 1466w, 1450w, 1419m, 1360s, 1308s, 1281m, 1150s, 1053w, 1023s, 882m, 756m, 737m, 649w.
, cm−1): 1603s, 1591s, 1546m, 1468s, 1457s, 1416s, 1363s, 1313s, 1279m, 1251w, 1152s, 1104m, 1041w, 1018s, 932w, 884s, 779w, 762s, 740s, 639w, 565w.
, cm−1): 1603s, 1591s, 1546m, 1467s, 1456s, 1416s, 1362s, 1313s, 1279m, 1250w, 1164w, 1152s, 1104s, 1042w, 1018s, 964w, 933w, 885s, 843w, 761s, 739s, 647w, 563w.
, cm−1): 1603s, 1593s, 1547s, 1454s, 1418s, 1370s, 1313s, 1236w, 1162w, 1146m, 947w, 925m, 886s, 769m, 751m, 729m, 670m, 573w, 557w.
, cm−1): 1604s, 1591s, 1548m, 1455s, 1420s, 1368s, 1312s, 1167m, 1148s, 1107m, 1045w, 1017m, 1004w, 925w, 885m, 756s, 734s, 636s, 580w.
Results
Synthesis
The fluorido adduct [Co3(dpa)4F2]·2CH2Cl2, 1·2CH2Cl2, was synthesized from [Co3(dpa)4(BF4)2], prepared in situ by combining AgBF4 and [Co3(dpa)4Cl2]13 in CH2Cl2 and stirring for several hours. The dark green solution was then filtered to remove AgCl and treated with tetrabutylammonium fluoride in THF, giving a deep orange solution, from which an orange solid crystallized rapidly as 1·2CH2Cl2 in 59% yield. Attempts to synthesize 1 directly from [Co3(dpa)4Cl2] using 2 eq. of AgF gave a mixture of [Co3(dpa)4F2] and [Co3(dpa)4Cl2] complexes, while 3 eq. yielded a mixture of products, including the oxidized 1[BF4]·4CH2Cl2, which was crystallographically characterized (Fig. S1, Tables S6 and S12†). Attempts to cleanly synthesize this latter compound by oxidation of 1·2CH2Cl2 with NOBF4 were unsuccessful.Compounds 2·Et2O and 3·Et2O were obtained from recrystallization of [Co3(dpa)4Cl2] and [Co3(dpa)4Br2],11 respectively, from DMF/Et2O.
The iodido adduct [Co3(dpa)4I2], 4, was obtained from treating [Co3(dpa)4Cl2] with a large excess of NaI in MeCN. After stirring overnight, the solvent was removed under reduced pressure and the reddish-brown solid was extracted with C2H4Cl2. When the solution was cooled to −15 °C, brown needles of 4·C2H4Cl2 were obtained in 56% yield. For 4·Et2O, the solid was first extracted with CH2Cl2 and filtered to remove excess NaI and NaCl. The CH2Cl2 was in turn evaporated under reduced pressure, the solid extracted with MeCN and layered with Et2O, yielding a crop of 4·Et2O as brown plates in 73% yield.
Crystal structures
Diffraction data for crystals of 1·2CH2Cl2, 2·Et2O, 3·Et2O, 4·Et2O and 4·C2H4Cl2 were collected at a minimum of three temperatures from 85 K. Diagrams of the structures at 85 K are represented in Fig. 1 and selected bond distances are given in Tables 1–3.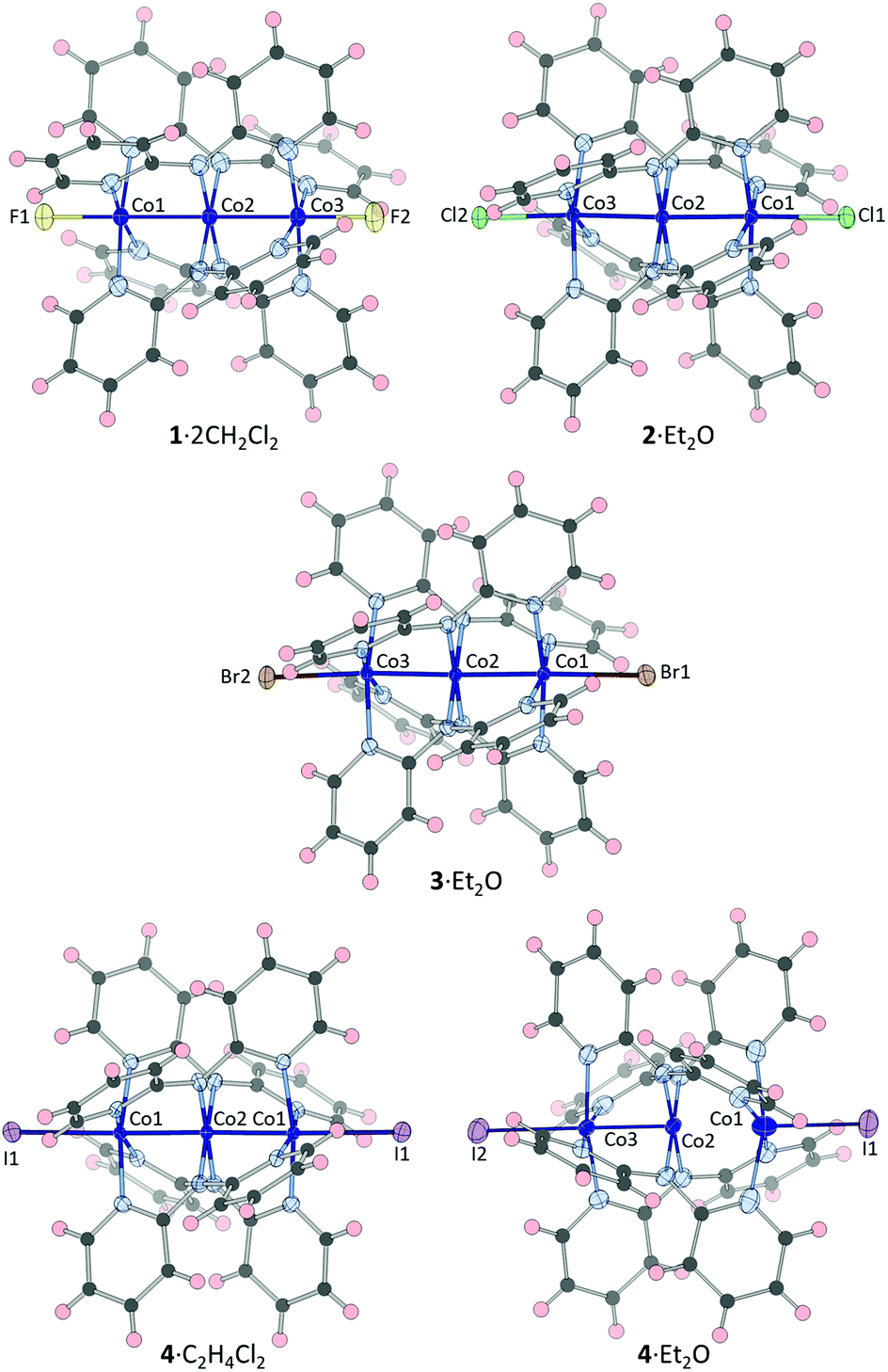 | ||
| Fig. 1 Thermal ellipsoid (50%) representations of the new structures from X-ray diffraction data at 85 K. Solvents of crystallization have been omitted. | ||
| T (K) | 85 | 120 | 250 |
|---|---|---|---|
| Co(1)–Co(2) | 2.3265(6) | 2.3257(6) | 2.3304(9) |
| Co(2)–Co(3) | 2.3156(6) | 2.3157(6) | 2.3153(9) |
| Co(1)–F(1) | 2.011(1) | 2.0084(19) | 2.016(3) |
| Co(3)–F(2) | 2.005(1) | 2.005(2) | 2.007(3) |
| T (K) | 85 | 120 | 298 | 350 |
|---|---|---|---|---|
| 2·Et2O | ||||
| Co(1)–Co(2) | 2.3323(2) | 2.3312(3) | 2.3752(5) | 2.3964(6) |
| Co(2)–Co(3) | 2.3209(2) | 2.3198(3) | 2.3299(5) | 2.3397(6) |
| Co(1)–Cl(1) | 2.4816(4) | 2.4823(4) | 2.4574(7) | 2.4350(9) |
| Co(3)–Cl(2) | 2.4430(3) | 2.4443(4) | 2.4471(7) | 2.4412(9) |
| 3·Et2O | ||||
| Co(1)–Co(2) | 2.3303(2) | 2.3295(2) | 2.3691(6) | 2.3862(7) |
| Co(2)–Co(3) | 2.3162(2) | 2.3162(2) | 2.3218(5) | 2.3308(7) |
| Co(1)–Br(1) | 2.6852(2) | 2.6890(2) | 2.6363(6) | 2.6096(7) |
| Co(3)–Br(2) | 2.6167(2) | 2.6205(2) | 2.6081(6) | 2.6002(7) |
| 4·Et2O | ||||
|---|---|---|---|---|
| T (K) | 85 | 170 | 240 | 298 |
| Co(1)–Co(2) | 2.4295(3) | 2.4504(8) | 2.4645(4) | 2.4680(15) |
| Co(2)–Co(3) | 2.2942(3) | 2.2928(8) | 2.2937(4) | 2.3047(14) |
| Co(1)–I(1) | 2.7728(3) | 2.7535(7) | 2.7449(3) | 2.7542(13) |
| Co(3)–I(2) | 2.8620(3) | 2.8620(7) | 2.8719(3) | 2.8702(13) |
| 4·C2H4Cl2 | |||
|---|---|---|---|
| T (K) | 85 | 120 | 298 |
| Co(1)–Co(2) | 2.3146(3) | 2.3111(4) | 2.3558(5) |
| Co(1)–I(1) | 2.8904(4) | 2.8925(4) | 2.8447(6) |
The fluorido adduct 1·2CH2Cl2 crystallizes in the space group C2/c with the tricobalt complex on a general position. The Co–Co distances are similar at 85 K, with a difference (ΔCo–Co) of only 0.0109(8) Å, and the Co–Co and Co–F distances remain essentially constant with increasing temperature (Table 1). In the crystal packing, the dichloromethane molecules interact with the axial fluorido ions through C–H⋯F hydrogen bonds, forming a one-dimensional motif (Fig. S2†). Notably, the ligand–solvent interactions are not identical on both sides of the complex, and the closer contact between the dichloromethane molecule and fluorido ion is associated with the slightly longer Co–Co distance. This compound has the unusual feature of having quite similar Co–Npyr and Co–Namide distances (Table S7†); all other known examples of tricobalt EMACs have much shorter Co–amide distances with respect to the Co–pyridine distances.
[Co3(dpa)4Cl2]·Et2O (2·Et2O) and [Co3(dpa)4Br2]·Et2O (3·Et2O) are isostructural, crystallizing in the space group P21/c, with almost equal Co–Co distances at 85 K (ΔCo–Co = 0.0114(3) and 0.0141(3) Å, respectively, Table 2). The slight asymmetry of the trimetallic core is associated with pairwise X⋯H–C interactions between neighbouring molecules in the crystal packing (Fig. S3†). The two complexes display the same temperature dependence; specifically, both the longer Co–Co distance and the longer Co–N distances increase with increasing temperature, while the other Co–Co and Co–N distances remain essentially the same (Tables 2, S8 and S9). However, this divergence does not hold with respect to the Co–Cl and Co–Br distances, which are more alike at 350 K (ΔCo–X = 0.006(1) and 0.009(1) Å) than at 85 K (ΔCo–X = 0.0386(5) and 0.0685(3) Å).
Two [Co3(dpa)4I2] solvates were crystallographically characterized. Compound 4·Et2O (C2/c) has highly dissimilar Co–Co distances at 85 K with ΔCo–Co = 0.1353(4) Å (Table 3). This asymmetry is associated with a short contact of ca. 3 Å between the iodido ligand coordinated to the cobalt ion participating in the long Co–Co distance and the ether solvent (Fig. S4†), reminiscent of the asymmetric solvent interactions observed in [Co3(dpa)4Cl2]·2CH2Cl2.10 The temperature dependence of 4·Et2O is characterized by an increase of the asymmetry due to a divergence of the Co–Co, Co–I and average terminal Co–N bond distances with increasing temperature. This asymmetrization implicates only one half of the molecule: the longer Co–Co and terminal Co–N (Table S10†) distances increase and the shorter Co–I distance decreases from 85 to 298 K, while the other distances do not change substantially. In this way, the bond distances in 4·Et2O are highly dissimilar at 298 K, although they are still far from convergence at 85 K. It should be noted that this is the same trend previously observed in the unsymmetric chlorido adduct 2·2CH2Cl2,7 although it is less pronounced in the iodido adduct.
The central cobalt ion in 4·C2H4Cl2 is bisected by a 2-fold rotation axis, yielding crystallographically equivalent Co–Co, Co–X and terminal average Co–N distances. The complex in 4·C2H4Cl2 exhibits a non-negligible increase of 0.0412(6) Å in the Co–Co distance from 85 to 300 K. This modification is concomitant with an increase in the average terminal Co–N distance (Table S11†) and a decrease in the Co–I distance with increasing temperature. This structural temperature dependence has been previously observed in the likewise rigorously symmetric compounds 2·THF and 2·cyclohexane, both of which show a thermally-induced SCO.8 We will revisit the relationship between the structural temperature dependence and the SCO in the discussion section.
Temperature dependence of the magnetic susceptibility
Variable temperature dc magnetic susceptibility measurements were carried out on freshly-filtered polycrystalline samples of 1·2CH2Cl2, 2·Et2O, 3·Et2O, 4·Et2O and 4·C2H4Cl2 from 1.85 K to a maximum temperature dictated by the respective thermal stability of the compounds, as determined by verifying the reversibility of the curves above 300 K (Fig. 2). As shown in the χT vs. T plots, all compounds exhibit a low temperature plateau around 0.55 cm3 mol−1 K, corresponding to a doublet ground state with g values of ca. 2.4. The magnetic susceptibility data were fit to the ideal solution model (eqn (1)),17 with SLS = 1/2 and SHS = 3/2. The resulting thermodynamic parameters are given in Table 4. To complement these results, the thermodynamic parameters from a (re)fitting of the magnetic data for several previously-reported compounds have been included (χT vs. T plots are presented in Fig. S5 and S6†). As the value of χTHS could not always be determined from the experimental data and taking 4·C2H4Cl2 as a reference, the gHS value was fixed at 2.66 for all compounds except 4·Et2O, 4·C2H4Cl2 and 2·2CH2Cl2. The ΔH, T1/2 and gLS parameters were allowed to refine freely.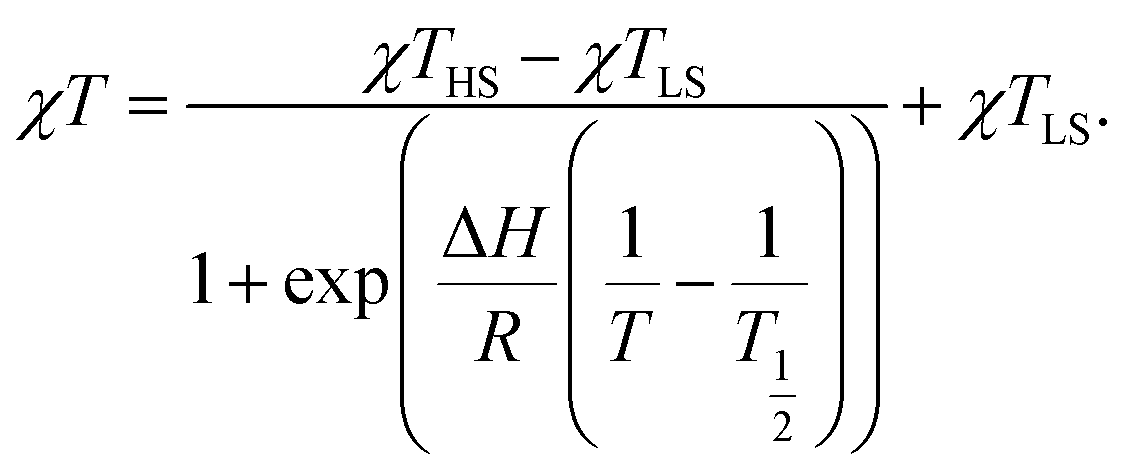 | (1) |
 | ||
| Fig. 2 χT versus T plots at 1000 Oe for the newly reported compounds, χ being the molar magnetic susceptibility defined as M/H. Lines are the fits to the ideal solution model (eqn (1)). | ||
| ΔH (kJ mol−1) | ΔS (J mol−1 K−1) | g LS | g HS | T 1/2 (K) | |
|---|---|---|---|---|---|
| a g HS was fixed in the fitting process. b Values from ref. 7. c Data from ref. 8. d Data from ref 11. n.r. = not reported. | |||||
| 1·2CH2Cl2 | 18.6(5) | 40.4 | 2.45(5) | 2.66a | 460(10) |
| 2·Et2O | 14.6(5) | 43.6 | 2.39(5) | 2.66a | 335(5) |
| 3·Et2O | 13.3(5) | 40.9 | 2.44(5) | 2.66a | 325(5) |
| 4·Et2O | 1.5(2) | 12.3 | 2.59(5) | 2.32(5) | 122(5) |
| 4·C2H4Cl2 | 9.5(5) | 36.5 | 2.45(5) | 2.66(5) | 260(5) |
| 2·CH2Cl2 | 18.0b | 54.7b | 2.35(2)b | 2.35(2)b | 329b |
| 16.0(5) | 44.8 | 2.39(5) | 2.66a | 357(5) | |
| 2·2CH2Cl2 | n.r.b | n.r.b | 3.21(2)b | 2.51(2)b | n.r.b |
| 1.6(2) | 8.3 | 3.37(5) | 2.76(5) | 193(5) | |
2·1.75C7H8·0.5C6H14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
6.6(5) | 28.7 | 2.41(5) | 2.66a | 230(5) |
| 2·C4H8Oc | 11.6(5) | 39.7 | 2.37(5) | 2.66a | 292(5) |
|
2·C6H6c |
12.6(5) | 43.6 | 2.36(5) | 2.66a | 289(5) |
|
2·C6H12c |
9.1(5) | 33.3 | 2.46(5) | 2.66a | 273(5) |
|
3·CH2Cl2d |
14.9(5) | 34.5 | 2.38(5) | 2.66a | 431(5) |
|
3·1.75C7H8·0.5C6H14d |
8.0(5) | 33.2 | 2.41(5) | 2.66a | 241(5) |
|
3·C6H12d |
8.2(5) | 40.4 | 2.49(5) | 2.66a | 203(5) |
Discussion
In this study, we wished to explore the relationship between the axial ligand, the molecular geometry and the spin-crossover properties of dpa-based tricobalt EMACs. We will approach these questions in succession, and first examine the role of the axial ligand on the molecular geometry. Based on the data presented here and found in the literature, there does not appear to be a correlation between the nature of the axial ligand and the molecular geometry for the chlorido, bromido and iodido adducts, which, at room temperature, range from rigorously symmetric to quite unsymmetric, depending on the solvent of crystallization. Concerning the fluorido adduct, so far only one example is known. In 1·2CH2Cl2, the difference in Co–Co distances, ΔCo–Co, is less than 0.05 Å, and this complex is therefore considered “symmetric”, or more precisely “quasi-symmetric”, as ΔCo–Co, while small, is nonzero.These results, taken together, are consistent with the observation that the geometry of complexes with weaker axial ligands is sensitive to crystal packing, while the presence of relatively strong field axial ligands favours symmetric or quasi-symmetric structures. Specifically, the symmetry in [Co3(dpa)4(CN)2]·CH2Cl2 is crystallographically enforced, while all the known –NCS− and N(CN)2− adducts have similar Co–Co distances, with the largest variation being 0.014 Å.9 An alternative hypothesis concerning the fluorido adduct is that the low polarizability of this anion does not lend itself to significant interactions with other moieties that can distort the {Co3} core. Undoubtedly, further examples of tricobalt complexes with axial fluorido ligands should be sought in an effort to clarify this observation.
The deconvolution of packing and electronic effects on the core geometry is unfortunately complicated by the lack of an isostructural series for the four halide analogues to date. While the chlorido and bromido adducts are often isostructural,8,11 the fluorido and the iodido adducts do not follow the same pattern. For example, 1·2CH2Cl2 (C2/c) is not isostructural with 2·2CH2Cl2 (I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) ), and while 2·Et2O and 3·Et2O (P21/c) are isostructural, the iodido adduct 4·Et2O (C2/c) is not. We can conclude that obtaining an isostructural series is not a trivial task, and may indeed be impossible, due to the varying sizes of the axial ligands under consideration. Nonetheless, for the isostructural pairs 2/3·Et2O and 2/3·1.75toluene·0.5hexane, the core symmetries and temperature dependences within each pair are comparable. Likewise, their spin crossover properties are similar, with almost identical T1/2 values, ∼330 K for the diethyl ether solvates and ∼235 K for the toluene/hexane solvates (Table 4). This observation suggests that the SCO behaviour in the chlorido and bromido adducts is principally a result of molecular geometry and crystal packing, and that the similar ligand field of the Cl− and Br− anions does not allow us to differentiate any electronic effect for these axial ligands.
), and while 2·Et2O and 3·Et2O (P21/c) are isostructural, the iodido adduct 4·Et2O (C2/c) is not. We can conclude that obtaining an isostructural series is not a trivial task, and may indeed be impossible, due to the varying sizes of the axial ligands under consideration. Nonetheless, for the isostructural pairs 2/3·Et2O and 2/3·1.75toluene·0.5hexane, the core symmetries and temperature dependences within each pair are comparable. Likewise, their spin crossover properties are similar, with almost identical T1/2 values, ∼330 K for the diethyl ether solvates and ∼235 K for the toluene/hexane solvates (Table 4). This observation suggests that the SCO behaviour in the chlorido and bromido adducts is principally a result of molecular geometry and crystal packing, and that the similar ligand field of the Cl− and Br− anions does not allow us to differentiate any electronic effect for these axial ligands.
Indeed, the important influence of the molecular geometry on the spin-crossover properties has been previously observed in the disparate behaviour of the respective symmetric and unsymmetric complexes 2·CH2Cl2 and 2·2CH2Cl2.7 We now have a second comparative example in the form of two iodido complexes, where the dichloroethane solvate is rigorously symmetric and the diethyl ether solvate is unsymmetric. From Fig. 3, which collects the T1/2 values for all measured compounds, it can be seen that the T1/2 values for the unsymmetric 2·2CH2Cl2 and 4·Et2O (in red) are significantly lower than those of their more symmetric analogues.
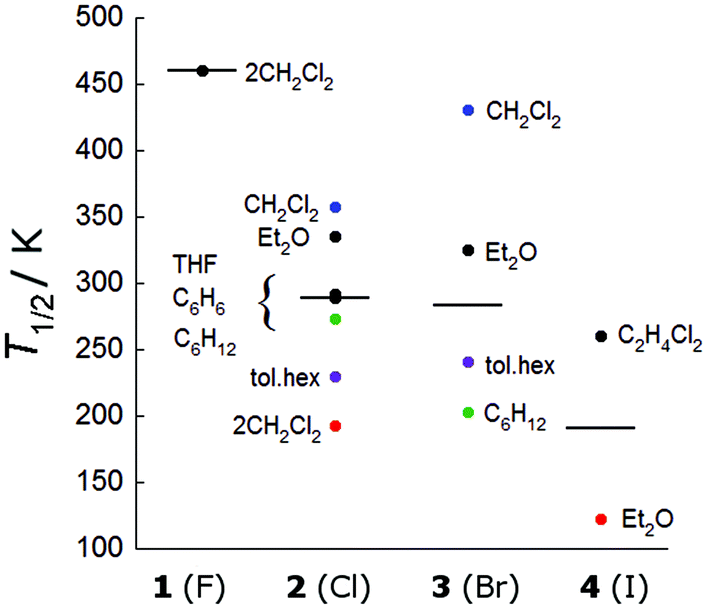 | ||
| Fig. 3 Distribution of T1/2 values for all reported compounds. Black lines represent the median and coloured dots are discussed in the text. | ||
While there is considerable overlap between the series, the T1/2 values generally follow the trend F− > Cl− ≈ Br− > I−. The impact of the fluorido ligands in stabilizing the S = 1/2 spin state has been previously seen in a series of one-dimensional polymers of alternating [(Co3(dpa)4)]2+ and MF62− units, which also demonstrated very high spin-crossover temperatures (T1/2 > 400 K).18,19 It should also be mentioned that the structure of 1·2CH2Cl2 does not display any significant geometric changes up to 250 K, a temperature where the compound remains fully in the low spin state, suggesting that our assumption that the changes in geometry are indeed representative of the spin-crossover phenomenon is reasonable.
A few compounds show anomalous behaviour and deserve further comment. The crystallographically symmetrical (ΔCo–Co = 0) 3·CH2Cl2 has an exceptionally high T1/2 value compared to its congener 2·CH2Cl2 (431 K vs. 357 K, blue dots in Fig. 3). Although the two complexes are isostructural near room temperature, 2·CH2Cl2 undergoes a phase change which breaks the equivalence of the Co–Co distances at lower temperatures. But this does not explain the unusual temperature dependence observed in 3·CH2Cl2. Unlike the other rigorously symmetric compounds 2·THF, 2·C6H12, 3·C6H12 and 4·C2H4Cl2 where the Co–X distance decreases with increasing temperature (Table 5), in 3·CH2Cl2, the Co–X distance significantly increases with temperature. The reason for this uncharacteristic temperature dependence is not clear from the packing diagrams, but is likely related to the unusual T1/2 value, and theoretical calculations may help elucidate this behaviour.
| 2·THF | 2·C6H12 | 3·C6H12 | 3·CH2Cl2 | 4·C2H4Cl2 | |
|---|---|---|---|---|---|
| δT (K) | 120–295 | 213–295 | 110–298 | 111–240 | 85–298 |
| Co(1)–Co(2) | 0.037(1) | 0.031(1) | 0.064(1) | 0.007(1) | 0.041(1) |
| Co(1)–X(1) | −0.017(1) | −0.014(1) | −0.054(1) | 0.043(1) | −0.046(1) |
| Co(1)–Navg | 0.038(3) | 0.031(4) | 0.060(3) | 0.015(9) | 0.042(3) |
| Co(2)–Navg | 0.009(3) | 0.009(4) | 0.011(3) | 0.005(9) | 0.006(3) |
| T 1/2 (K) | 292(5) | 273(5) | 203(5) | 431(5) | 260(5) |
| Ref. | 8 | 11 | This work | ||
Remaining within the crystallographically symmetric series, we note that 3·C6H12 displays a particularly low T1/2 value compared to the chlorido analogue 2·C6H12 (203 K vs. 273 K, green dots in Fig. 3). These complexes are not isostructural and 2·C6H12 undergoes a phase transition between 120 and 213 K, which is clearly observable in the χT vs. T plot (Fig. S7†). Nonetheless, they follow the same temperature dependence and both compounds begin their spin-crossover around 100 K, but the conversion in 2·C6H12 is broader than in 3·C6H12 (Fig. S7†). Comparing the structures at 120 (2·C6H12) and 110 K (3·C6H12), we note that two short contacts of 2.28 and 2.30 Å are found between the complexes in 3·C6H12, while only one comparable contact (2.26 Å) is found in 2·C6H12. An increased cooperativity between trinuclear complexes in 3·C6H12 may thus explain its more “abrupt” thermal conversion to the high spin state.20
We now turn our attention to compounds where the symmetry of the metal core is not crystallographically enforced (Table 6). For these compounds, excluding 1·2CH2Cl2 and 4·Et2O, for which the T1/2 values are likely influenced by the electronic effect of the axial ligands, the T1/2 values range from 193 to 357 K. As might be expected based on the low T1/2 values displayed by the unsymmetric compounds 2·2CH2Cl2 and 4·Et2O, there is a correlation between SCO temperature and the asymmetry of the complex in the chlorido and bromide series. For example, if we compare ΔCo–Co values from crystallographic data collected around room temperature, we find that large differences in Co–Co distances are associated with lower T1/2 values (Fig. 4). The linear fit is slightly better when the averages of the two ΔCo–Co values for the two molecules in the asymmetric unit of 2·1.75toluene·0.5hexane (0.161 and 0.130 Å) and 3·1.75toluene·0.5hexane (0.157 and 0.109 Å) are used (Fig. S8†). Compound 2·C6H6 has a lower than expected T1/2 for being a quite symmetrical compound (0.025 Å at 316 K). But here again, the temperature dependence for 2·C6H6 is atypical, in that both the Co–Co and terminal Co–N distances expand with increasing temperature, mirroring the behaviour for the crystallographically symmetric compounds. Interestingly, the χT vs. T curve is almost superimposable on that of the crystallographically symmetric 2·C4H8O.8
 | ||
| Fig. 4 Correlation between T1/2 and the differences in Co–Co bond distances for structures obtained close to room temperature (295–316 K). | ||
| 1·2CH2Cl2 | 2·Et2Oa | 3·Et2Oa | |
|---|---|---|---|
| δT (K) | 85–250 | 85–298 | 85–298 |
| Co(1)–Co(2) | 0.004(1) | 0.043(1) | 0.039(1) |
| Co(2)–Co(3) | 0.000(1) | 0.009(1) | 0.006(1) |
| Co(1)–X(1) | 0.005(3) | −0.024(1) | −0.049(1) |
| Co(3)–X(2) | 0.002(3) | 0.004(1) | −0.009(1) |
| Co(1)–Navg | −0.001(3) | 0.041(2) | 0.041(3) |
| Co(2)–Navg | −0.002(3) | 0.006(2) | 0.010(3) |
| Co(3)–Navg | −0.001(3) | 0.014(2) | 0.012(3) |
| T 1/2 (K) | 460(10) | 335(5) | 325(5) |
| Ref. | This work | This work | This work |
| 2·1.75tol·0.5hex | 3·1.75tol·0.5hex | |||
|---|---|---|---|---|
| δT(K) | 90–298b | 110–295b | ||
| Co(1)–Co(2) | 0.105(2) | 0.122(2) | 0.080(2) | 0.117(2) |
| Co(2)–Co(3) | 0.000(2) | −0.002(2) | 0.003(2) | 0.006(2) |
| Co(1)–X(1) | −0.080(4) | −0.075(4) | −0.080(2) | −0.120(1) |
| Co(3)–X(2) | 0.000(4) | −0.014(4) | 0.006(1) | 0.020(1) |
| Co(1)–Navg | 0.097(10) | 0.111(10) | 0.075(7) | 0.110(6) |
| Co(2)–Navg | 0.009(10) | 0.007(10) | 0.009(6) | 0.013(6) |
| Co(3)–Navg | −0.003(10) | 0.006(11) | 0.003(6) | 0.003(6) |
| T 1/2 (K) | 230(5) | 241(5) | ||
| Ref. | 8 | 11 | ||
| Compound | 2·C6H6c | 2·2CH2Cl2 | 4·Et2O | |
|---|---|---|---|---|
| δT(K) | 170–316 | 133–298 | 85–298 | |
| a Values only given to 298 K for better comparison with other complexes. b Two molecules in the asymmetric unit. c Two molecules in the asymmetric unit between 170–260 K and one molecule at 316 K, due to a phase transition. The values were obtained by taking the difference between the distances for the unique molecule at 316 K and the two independent molecules at 170 K. | ||||
| Co(1)–Co(2) | 0.039(1) | 0.038(2) | 0.031(1) | 0.039(2) |
| Co(2)–Co(3) | 0.028(1) | 0.023(2) | 0.004(1) | 0.011(1) |
| Co(1)–X(1) | −0.029(2) | −0.040(2) | −0.028(3) | −0.019(1) |
| Co(3)–X(2) | 0.003(2) | −0.018(2) | 0.007(2) | −0.008(1) |
| Co(1)–Navg | 0.028(6) | 0.032(7) | 0.027(6) | 0.039(6) |
| Co(2)–Navg | 0.001(6) | 0.004(7) | 0.008(6) | 0.006(5) |
| Co(3)–Navg | 0.023(6) | 0.027(8) | 0.004(6) | 0.008(5) |
| T 1/2 (K) | 289(5) | 193(5) | 122(5) | |
| Ref | 8 | 7 | This work | |
Notably, the isostructural compounds 2·1.75toluene·0.5 hexane and 3·1.75toluene·0.5hexane demonstrate T1/2 values (violet dots in Fig. 3) intermediate between the quasi-symmetric and unsymmetric complexes. These complexes exhibit a marked temperature dependence, transforming from almost symmetric to unsymmetric within the measured temperature range. This observation, as well as the relationship between ΔCo–Co and T1/2 described above, suggests that the distinction between “(quasi)-symmetric” and “unsymmetric” compounds may not be very meaningful. Rather, it seems reasonable, based on observed trends, that the compounds in Table 6 would theoretically all be quite symmetric in the low temperature limit and unsymmetric in the high temperature limit, as exemplified by 2/3·1.75toluene·0.5hexane, where the transformation happens to occur within a measurable temperature range.
To explore this idea, we plotted previously published structural data for unsymmetric 2·2CH2Cl2 and the new data from (quasi)-symmetric 2·Et2O in Fig. 5. The two data sets have been aligned (requiring an offset of 0.02 Å) to emphasize the point that the trends for 2·Et2O are simply an extrapolation of those for 2·2CH2Cl2. Interestingly, the crossing of the two independent Co–Cl distances, due to the gradual elongation of Co(1)–Cl(1) with decreasing temperature, is replicated in both compounds. The similarity of the temperature dependence not only suggests that the same states are implicated in the spin-crossover for both the (quasi)-symmetric and unsymmetric compounds, but provides experimental evidence for the shallow 2B potential energy state proposed by Pantazis et al.,21 which links the symmetric 2A low spin state with the unsymmetric 4B high spin state and accounts for the gradual geometric changes previously observed for 2·2CH2Cl2. In this view, the location of the complex on the 2B surface at a given temperature determines both the T1/2 value and the asymmetry of the core, a relationship which is consistent with experimental evidence for crystallographically unconstrained compounds. This can be contrasted with the temperature dependence of the crystallographically symmetrical complexes, which implies the existence of a low-lying symmetrical quartet state, which is not present in the current theoretical picture. Experimental and theoretical work on rigorously symmetrical complexes is underway.
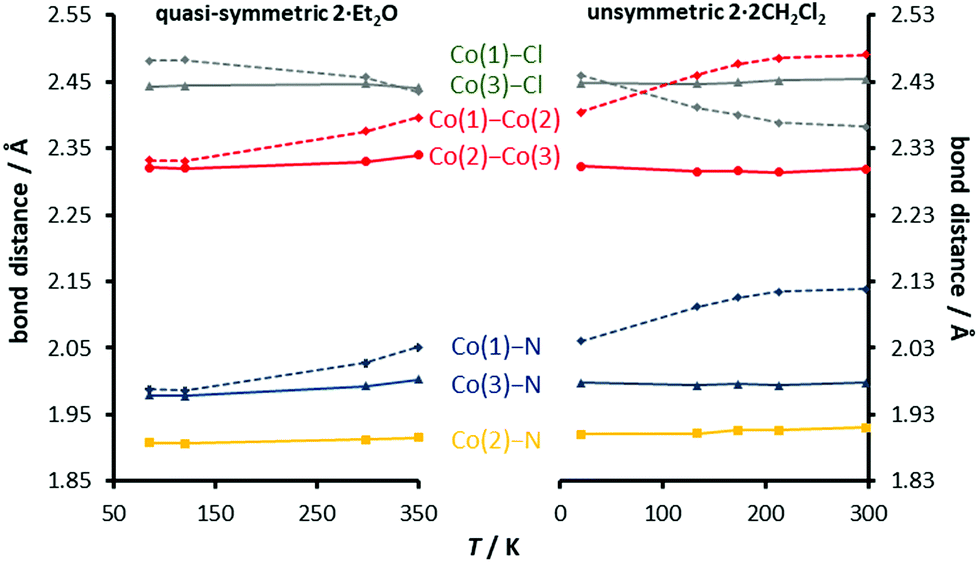 | ||
| Fig. 5 Comparison of the structural data from X-ray crystallography on two distinct crystals, the quasi-symmetric and unsymmetric forms of [Co3dpa4Cl2]. | ||
Conclusions
The synthesis of fluorido and iodido adducts of tricobalt paddlewheel complexes has been accomplished for the first time, allowing a more comprehensive investigation of the influence of the axial ligands on their core geometry and spin-crossover properties. Although the relationship between axial ligands, geometry and the spin-crossover properties is far from simple, several conclusions can be derived. While there is quite a bit of overlap in the T1/2 values among the four halide series, the presence of iodido ligands tends to stabilize the high spin state compared to their congeners, while fluorido ligands favour a much higher T1/2 value. The structural geometry of complexes with I−, Br− and Cl− ligands appears to be mainly a function of the crystal packing, while the only example of a fluorido compound is quite symmetric. Higher asymmetry in the tricobalt core is associated with destabilization of the low spin state, as exemplified by lower T1/2 values, suggesting a continuum of core geometries consistent with the previous theoretical calculations.These conclusions are offered with the caveat that the picture could easily evolve with the discovery of new halide adducts, and work is currently focused on efforts to obtain genuinely unsymmetric examples in the fluorido and bromido series.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the CNRS, the University of Bordeaux, the Conseil Régional de la Nouvelle Aquitaine and the France-Canada Research Fund (doctoral bursary for A. S.). LRF thanks the Ministry of Science, Innovation and Universities (Spain) for Grant MAT2015-68200-C2-1-P with funding FEDER. The authors thank the GdR MCM-2 (Magnétisme et Commutation Moléculaires) and the MOLSPIN COST action CA15128. The authors thank S. Exiga and P. Rosa for technical assistance and F. A. Cotton, C. A. Murillo, L. M. Daniels and K. R. Dunbar for their historical contributions, upon which much of the current analysis is based.Notes and references
- J. F. Berry in Multiple Bonds between Metal Atoms, ed. F. A. Cotton, C. A. Murillo and R. A. Walton, Springer, USA, 2005, pp. 669–706 Search PubMed.
- M. Majumdar and J. K. Bera, in Macromolecules Containing Metal and Metal-Like Elements, ed. A. Abd-El Aziz, C. E. Carraher, C. U. Pittman and M. Zeldin, John Wiley & Sons, Inc., 2009, vol. 9, pp. 182–253 Search PubMed.
- S.-A. Hua, M.-C. Cheng, C.-h. Chen and S.-M. Peng, Eur. J. Inorg. Chem., 2015, 2015, 2510 CrossRef CAS.
- F. A. Cotton, L. M. Daniels, C. A. Murillo and I. J. Pascual, J. Am. Chem. Soc., 1997, 119, 10223 CrossRef CAS.
- J. F. Berry, F. A. Cotton, T. Lu, C. A. Murillo, B. K. Roberts and X. Wang, J. Am. Chem. Soc., 2004, 126, 7082 CrossRef CAS PubMed.
- M. Spivak, V. Arcisauskaite, X. Lopez, J. E. McGrady and C. de Graaf, Dalton Trans., 2017, 46, 6202 RSC.
- R. Clérac, F. A. Cotton, L. M. Daniels, K. R. Dunbar, K. Kirschbaum, C. A. Murillo, A. A. Pinkerton, A. J. Schultz and X. Wang, J. Am. Chem. Soc., 2000, 122, 6226 CrossRef.
- R. Clérac, F. A. Cotton, L. M. Daniels, K. R. Dunbar, C. A. Murillo and X. Wang, Inorg. Chem., 2001, 40, 1256 CrossRef.
- F. A. Cotton, C. A. Murillo and X. Wang, Inorg. Chem., 1999, 38, 6294 CrossRef CAS PubMed.
- R. D. Poulsen, J. Overgaard, A. Schulman, C. Østergaard, C. A. Murillo, M. A. Spackman and B. B. Iversen, J. Am. Chem. Soc., 2009, 131, 7580 CrossRef CAS PubMed.
- R. Clérac, F. A. Cotton, L. M. Daniels, K. R. Dunbar, C. A. Murillo and X. Wang, J. Chem. Soc., Dalton Trans., 2001, 386 RSC.
- R. Clérac, F. A. Cotton, S. P. Jeffery, C. A. Murillo and X. Wang, Inorg. Chem., 2001, 40, 1265 CrossRef.
- F. A. Cotton, L. M. Daniels, G. T. Jordan IV and C. A. Murillo, J. Am. Chem. Soc., 1997, 119, 10377 CrossRef CAS.
- Bruker APEX2, SAINT+, Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- Bruker SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2001 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. Kahn, in Molecular Magnetism, VCH Publishers, New York, 1993, p. 59 Search PubMed.
- V. Bulicanu, K. S. Pedersen, M. Rouzières, J. Bendix, P. Dechambenoit, R. Clérac and E. A. Hillard, Chem. Commun., 2015, 51, 17748 RSC.
- M. Cortijo, V. Bulicanu, K. S. Pedersen, M. Rouzières, J. Bendix, R. Clérac and E. A. Hillard, Eur. J. Inorg. Chem., 2018, 320 CrossRef CAS.
- A. Hauser, J. Jeftic, H. Romstedt, R. Hinek and H. Spiering, Coord. Chem. Rev., 1999, 190/192, 471 CrossRef.
- D. A. Pantazis and J. E. McGrady, J. Am. Chem. Soc., 2006, 128, 4128 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Thermal ellipsoid plot of 1[BF4]·4CH2Cl2, diagrams showing intramolecular interactions for 1·2CH2Cl2, 3·Et2O and 4·Et2O, crystal data and refinement and selected bond distance and angles tables for newly reported compounds, χT vs. T plots and fits for previously reported compounds. CCDC 1865577–1865593 and 1877180–1877181. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt03833c |
| ‡ The compound 2·Et2O was reported as a communication to the Cambridge Structural Database by M. Shatruk, RefCode GOJRII. |
| This journal is © The Royal Society of Chemistry 2018 |