
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing porphyrin photostability when locked in metal–organic frameworks†
Ghandi F.
Hassan
 ,
Nour
El Hoda Saad
,
Mohamad
Hmadeh
* and
Pierre
Karam
*
,
Nour
El Hoda Saad
,
Mohamad
Hmadeh
* and
Pierre
Karam
*
Chemistry Department, American University of Beirut, P.O. Box 11-0236, Riad El-Solh, 1107 2020, Beirut, Lebanon. E-mail: mohamad.hmadeh@aub.edu.lb; pierre.karam@aub.edu.lb; Fax: (+961) (1)365217
First published on 9th October 2018
Abstract
Porphyrins have been widely used in many optical devices given their unique photochemical properties. Their poor photostability has, however, limited their wide applications in bioimaging and biosensing schemes. Herein, we report the remarkable photostability enhancement of the porphyrin, carboxyphenyl porphyrin (TCPP-H2) when locked in a zirconium based metal–organic framework (MOF-525). Steady-state ensemble fluorescence spectroscopy experiments showed minimal changes (2%) in the recorded signal when MOF-525 was continuously illuminated as compared to a 16% decrease for free porphyrins. Single particle fluorescence imaging revealed bright microparticles with exceptional photostability and no-blinking within the experiment window. This study highlights the use of metal–organic frameworks for preparing photostable microstructures by leveraging on their unique self-assembly properties.
Introduction
The continuous rise of fluorescence microscopy as a fundamental method in many fields of science such as imaging and biosensing has been accompanied by a great demand for developing bright and photostable probes.1–5 Quantum dots have emerged as popular material for both fluorescence imaging as well as sensing.6,7 However, quantum dots have demonstrated some limitations; their blinking under continuous irradiation limits their application in fluorescence imaging, and their toxicity hinders their implementation in biosensing schemes.8,9 Consequently, new materials such as conjugated polymer nanoparticles have been recently developed to serve as bright and stable probes for both sensing and imaging.10–14 While photostability and brightness are drawing a lot of attention, little focus has been directed towards developing new probes that have a large separation between their excitation and emission wavelengths. Creating such a fluorophore will provide a better signal-to-noise ratio in fluorescence imaging microscopy, and in developing sensitive biosensing assays.15,16Porphyrins have many unique photophysical properties that have attracted significant attention directed towards optimizing them for their use in photodynamic therapy,17,18 dye-sensitized solar cells,19,20 and the fabrication of optoelectronic21–23 and photon up-conversion devices.24 Of special importance to fluorescence imaging, porphyrin emission has a large red shift from its maximum absorbance (ca. 200 nm), allowing for an efficient separation between the excitation wavelength and the collected emitted photons, thus achieving sensitive multicolor measurements. Despite this major advantage, there have been no widespread applications that integrate porphyrins into an imaging or a sensing scheme. This limitation is due to their tendency to aggregate in water,25 a phenomenon which influences their physical, chemical, and most importantly, their photophysical properties.26 When aggregated, porphyrin fluorescence brightness is compromised due to self-quenching.27
To overcome this impediment, many approaches were constructed. One necessitated solubilizing porphyrins using micelles or amphiphilic polymers, ensuring their deaggregation in water, and subsequently reducing the self-quenching pathways.28 Another approach was to engineer them onto nanostructured materials. For instance, when meso-tetrakis(1-methylpyridinium-4-yl)porphyrin chloride was adsorbed onto ZnO nanoparticles through electrostatic interactions, the symmetry of the porphyrin macrocycle increased which hindered the rotational relaxation of the meso unit and/or decreased the intramolecular charge transfer. This molecular structural change resulted in a six-fold enhancement in the fluorescence intensity. It is also believed that the increased rigidity of the porphyrin upon the physical adsorption also contributed to the fluorescence signal enhancement.29
With the issue of brightness tackled, photostability was another important parameter that had to be improved, especially for porphyrin-based probes, since they are specifically prone to fast degradation given their efficient production of singlet oxygen. For example, this photostability enhancement was achieved when porphyrins were incorporated inside the molecular sieve channels of AIPO4-5.30
Metal–organic frameworks (MOFs) have recently emerged as a new class of crystalline extended materials that are formed by the self-assembly of metal clusters with polytopic organic linkers.31–36 Their high versatility, which is related to the wide range of building blocks from which they can be produced, has led to a wide variety of applications, including gas separation, gas storage, catalysis, purification and sensing.37–40 Because of the strong chemical bonding and higher coordination number, the Zr-based cluster, Zr6O4(OH)4(CO2)12, found in UiO-66 (Zr6O4(OH)4(BDC)6; BDC = terephthalate) is one of the most stable inorganic clusters.41,42 Thus, this inorganic secondary building unit (SBU) has been employed as a platform to build thermally and chemically stable MOFs that are critical for practical applications.43,44 Recently, MOFs combining zirconium clusters and porphyrin-based linkers have been synthesized and showed interesting physical and chemical properties that allowed them to be used in catalysis, light-harvesting and oxygen transportation.45
In this work, we argued that metal–organic frameworks are an ideal platform to assemble porphyrins into higher ordered structures that will ensure (1) disaggregation of porphyrins and (2) rigidification of their structures. This will most definitely lead to their fluorescence enhancement and to increased photostability. To this end, MOF-525 appeared to be a great choice for such a study.46 Indeed, this cubic structure has a ftw topology, each cube is composed of eight corner-sharing Zr6O4(OH)4 units and six face-sharing porphyrin units, where each porphyrin unit bridges four Zr6O4(OH)4 units. In addition, it exhibited excellent chemical stability in different solvents and under a wide pH range.47,48 As such, it was an ideal fluorescent porphyrin-based MOF to test, in the hope of exploring its photophysical properties that is of importance for future exploitation in imaging and sensing applications. To this end, we report the exceptional photostability of zirconium–porphyrin (TCPP-H2)-based MOF-525 when their fluorescence signal is measured at both the ensemble and the single particle level.
Results and discussion
MOF-525 cubic crystals were synthesized under conditions similar to those reported in the literature (see the Experimental section for more details).46 The crystallinity and phase purity of the sample were confirmed by powder X-ray diffraction (PXRD) analysis (Fig. 1). Scanning electron microscopy images (SEM) revealed the formation of homogeneous cuboctahedral crystals of 2–3 μm. The infra-red (IR) spectra of MOF-525 and free porphyrin linker were recorded and demonstrated that the free base porphyrin did not coordinate to Zr cations through the N of the pyrrole units (νNH-stretching = 3428 cm−1) (Fig. S1†). | ||
| Fig. 1 (A–C) SEM images of the prepared MOF-525, (D) crystal structure of MOF-525, (E) PXRD pattern of MOF-525 crystals compared to the simulated one, and (F) N2 isotherm of MOF-525 at 77 K. | ||
On the other hand, we observed that the free C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the ligand (1724 cm−1) was shifted to a lower wavenumber in the MOF spectrum (1406 cm−1) due to the coordination to the Zr cluster. In order to assess the porosity of the MOF-525 structure, the N2 adsorption/desorption isotherm of the activated sample was measured and then the Brunauer–Emmett–Teller (BET) method exhibited a surface area of 2600 m2 g−1 which is in agreement with the reported value.46
O stretching vibration of the ligand (1724 cm−1) was shifted to a lower wavenumber in the MOF spectrum (1406 cm−1) due to the coordination to the Zr cluster. In order to assess the porosity of the MOF-525 structure, the N2 adsorption/desorption isotherm of the activated sample was measured and then the Brunauer–Emmett–Teller (BET) method exhibited a surface area of 2600 m2 g−1 which is in agreement with the reported value.46
After successfully preparing and characterizing MOF-525, we assessed its photophysical properties and the effect of structural assembly on its fluorescence signal. When comparing free porphyrins to MOF-525 in deionized water for optically matched solutions (Fig. S2†), a 14 time fluorescence enhancement was observed at 660 nm (Fig. 2). We believe that the deliberately positioned porphyrins in the metal–organic framework are significantly far to prevent self-quenching (20 Å). Indeed, when incremental amounts of NaCl were added to the solution of TCPP-H2 prepared in water, a fluorescence enhancement was observed (Fig. S3†). This enhancement could be interpreted by the disaggregation of porphyrins when the solution's ionic strength is increased. Similar results were observed when tetrakisphenyl porphyrins were modified with branched polyethylene glycol (PEG). The fluorescence intensity increased with the increase of the substitution from 2 to 4 for the same porphyrin concentration, suggesting that fully substituted PEG provides better solubility in water and avoids self-aggregation of the porphyrin core.49
 | ||
Fig. 2 Fluorescent emission of optically matched aggregated porphyrin prepared in water ((■) TCPP-H2) and porphyrin locked in a zirconium-based metal–organic framework (( ) MOF-525). ) MOF-525). | ||
However, the recorded fluorescence intensity slightly decreased when a MOF-525 solution was compared to an optically matched solution of deaggregated TCPP-H2 prepared in buffer. This might be the effect of the coordinating Zr4+ ions. Indeed, upon the addition of increasing concentrations of zirconium cations, we observed a quenching in the fluorescent emission and the Stern–Volmer plot showed a positive deviation indicating a favorable interaction between the free base porphyrin and the metal ion (Fig. S4†). Structural perturbation of porphyrins may also induce major changes in electronic and chemical properties. Upon comparison of planar porphyrins with the perturbed ones, the latter showed reduced quantum yields (up to 10 fold lower).50
To evaluate the intrinsic photostability of porphyrins assembled and locked in a MOF structure, steady-state fluorescence spectroscopy measurements were acquired for a solution of optically matched (Fig. S4†) free porphyrins and MOF-525, upon continuous excitation at 415 nm. Their emission was recorded at 645 nm (Fig. 3). The fluorescence trajectory for MOF 525 showed remarkable photostability with only a 2% decrease in intensity over the tested time interval (32 min). On the other hand, the fluorescence intensity of optically matched free porphyrins decreased by 16% under the same experimental conditions. To ensure that no structural degradation is inflicted on the tested MOF, powder X-ray diffraction (PXRD) patterns were recorded before and after photoexcitation and prove conclusively that the structure of the framework is maintained after photoexcitation (Fig. S5†).
 | ||
Fig. 3 Fluorescence intensity versus time trajectories of MOF-525 ( ) and TCPP-H2 (■) upon continuous excitation at 415 nm and the collection of emission intensity at 660 nm. The experiment was done in 10 mM HEPES buffer pH = 7.3 and 150 mM NaCl. ) and TCPP-H2 (■) upon continuous excitation at 415 nm and the collection of emission intensity at 660 nm. The experiment was done in 10 mM HEPES buffer pH = 7.3 and 150 mM NaCl. | ||
The observed ensemble photostability of MOF-525 might prove instrumental for cellular bioimaging and biosensing applications. To evaluate the photostability of MOF-525 at the single particle level, fluorescence microscopy imaging was acquired. MOF-525 crystals were imaged using an upright fluorescence microscope using a 40× objective with NA = 0.8 coupled to an excitation filter of 390–420 nm at a power, measured out of the objective, equal to 8 mW cm−2. The emission was collected using a 600 nm long-path filter, in the presence and the absence of an antifade solution with a time interval of 500 ms for 90 s (limit of our processor). Time–intensity trajectories were extracted using the ImageJ software and subsequently corrected for background signals. Individual traces showed no sign of blinking (Fig. S6†) at least within the time resolution of our experiment (ca. 500 ms). In the absence of an antifade solution, the trajectories showed an initial fast decrease in the fluorescence signal (within the first 7 seconds) equivalent to 16.8% of the original intensity (458 ± 221 a.u. N = 1302), and then remained stable throughout the experiment as shown in Fig. 4. We speculate that the quick intensity drop might be due to some un-coordinated and non-specifically adsorbed porphyrin units. In the presence of the antifade mixture, the intensity−time trajectories remained stable with no apparent decrease in the fluorescence intensity with an average initial intensity of 697 ± 105 a.u. (N = 1316), 52% higher than that of MOF-525 with no antifade. We were unable to acquire images for TCPP-H2 molecules given their weak brightness and limited photostability under the same experimental conditions as those for MOF-525.
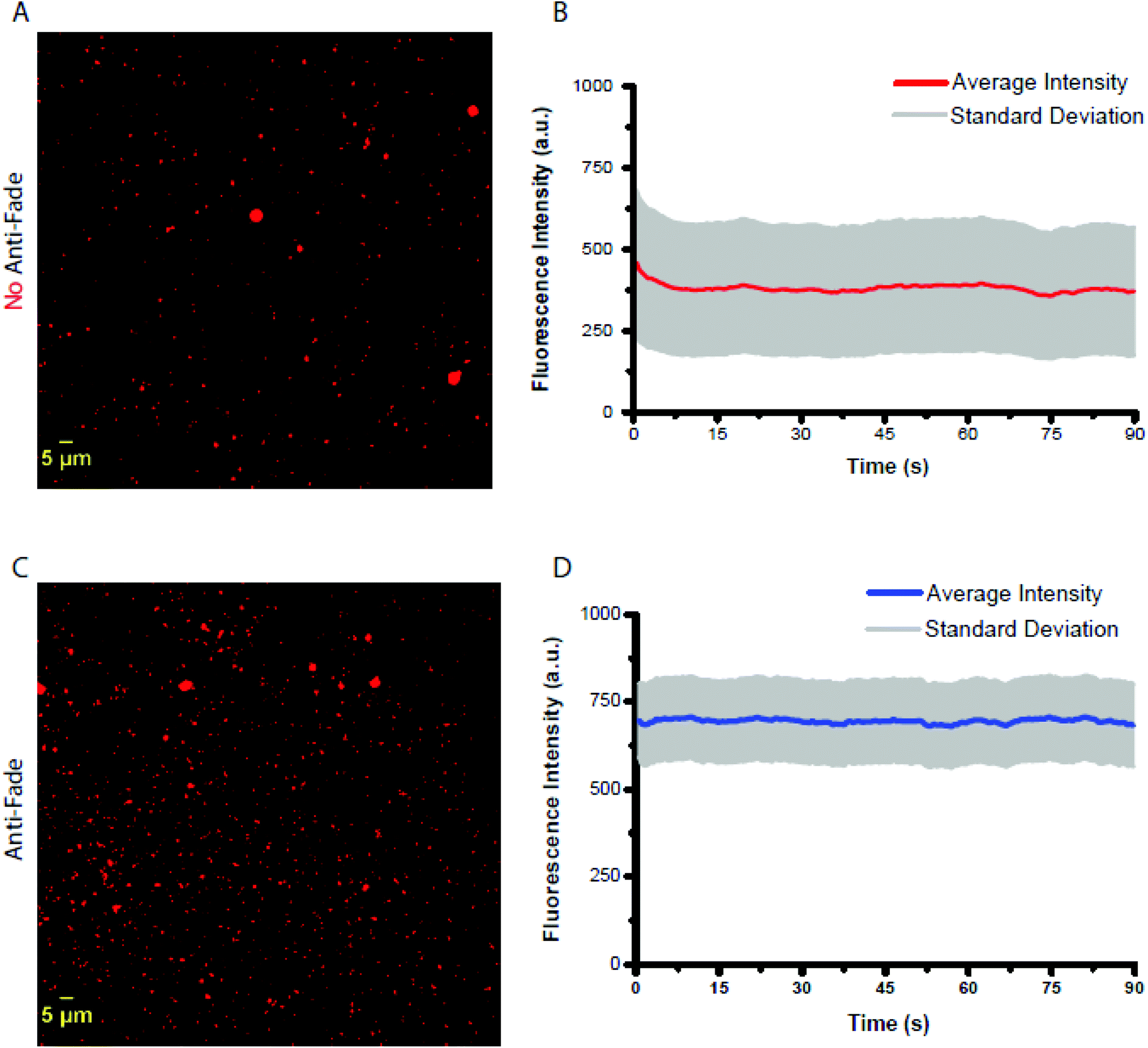 | ||
| Fig. 4 Upright fluorescent images of MOF-525 in (A and B) the presence and (C and D) the absence of anti-fade solution imaged using a 40× objective with NA = 0.8 coupled to an excitation filter of 390–420 nm at a power, measured out of the objective, equal to 8 mW cm−2. The emission was collected using a 600 nm long-path filter with a time interval of 500 ms for 90 s (limit of our processor). Average time–intensity trajectories were extracted using the ImageJ software and subsequently corrected for background signals. | ||
We believe that the MOF structure prompts the interplay of many factors leading to the observed photostability. Porphyrins come with high singlet-to-triplet conversion rates, and a long-lived triplet excited state that makes them prone for photodegradation in solutions by sensitizing oxygen; energy transfer from the excited triplet state to the ground state of triplet dioxygen (3O2) leads to the formation of singlet dioxygen (1O2) by spin inversion.51 Subsequently, the highly reactive singlet state attacks the porphyrin, predominantly in its meso-positions, resulting in its systematic photodegredation overtime.52,53 The rigid structure of the MOF might impose some structural constraints by locking the porphyrin in a planar structure. It has been previously reported that induced planarity in porphyrins reduces intersystem crossing.50,54 Porphyrin photostability was observed to be enhanced when closely packaged. It is believed that close neighboring porphyrin units enhance the process of triplet–triplet annihilation inducing a decrease in the production of singlet oxygen.30 Another factor might be introduced by the zirconium cation. Recently, Cosa et al. have highlighted the role of transition metal ions such as Ni2+ to quench the triplet excited state of organic dyes by triplet–triplet energy transfer to ligand field states in coordination complexes.55,56 Specifically, Ni2+ was reported to suppress the blinking and sensitization of singlet oxygen. As such, a dramatic improvement in the photophysics of red and green organic dyes was reported. Specifically to porphyrin, Cavaleiro et al. have observed enhanced photostability when porphyrins were complexed with copper when compared to them being free. They showed that the copper ion reduces the triplet lifetime by up to three orders of magnitude and subsequently reduces the production of singlet oxygen.57 Similarly, Ni2+ and Co2+ were shown to quench the triplet state of porphyrins.58 Zirconium (Zr4+) has also been reported to efficiently quench the triplet excited state cyclopentadiene complexes.59
As a result, we believe that a combination of structural configuration coupled with the physiochemical inhibition of intersystem crossing is responsible for the observed enhancement in the brightness and photostability of porphyrins.
Conclusion
This work presents a simple way to enhance the photostability of porphyrins, when assembled in a metal–organic framework. The MOF microparticles were photostable when tested at the ensemble and the single particle level. We believe these microparticles would be of great importance in the field of bioimaging by allowing the staining and subsequently sensing rare sub-cell population. In addition, porphyrin used in solar cells will benefit from brighter and more photostable sensitizers.Materials and methods
Materials
4,4′,4′′,4′′′-(Porphine-5,10,15,20-tetrayl)tetrakis(benzoic acid) and zirconyl chloride octahydrate in addition to all the other reagents and solvents, were purchased from Sigma-Aldrich and used without further purification. The infrared spectroscopy (IR) spectra were recorded on a FT-IR spectrometer Thermo-Nicolet working in the transmittance mode, in the 450–3950 cm−1 range. Thermogravimetric analysis (TGA) was performed with Netzsch TG 209 F1 Libra apparatus. The analyses were carried out in a N2 flow from 30 to 800 °C at a heating rate of 3 K min−1. Powder X-ray diffraction (PXRD) patterns were collected using a Bruker D8 advance X-ray diffractometer (Bruker AXS GmbH, Karlsruhe, Germany) at 40 kV and 40 mA (1600 W) using Cu Kα radiation (k = 1.5418 Å). Scanning electron microscopy (SEM) was performed using a MIRA3 TESCAN electron microscope where the samples were first coated with a thin layer (10 nm) of gold. Nitrogen sorption measurements were carried out at 77 K. Prior to the measurements; the samples were activated under dynamic vacuum at 120 °C for 48 hours.MOF-525 synthesis
MOF-525 Zr6(OH)4O4(C48N4O8H26)3 Zirconyl chloride octahydrate (12.5 mg, 0.037 mmol) was added to N,N-dimethylformamide (DMF, 10 mL) and then was sonicated for thirty minutes.46 After sonication, tetrakis(4-carboxyphenyl)porphyrin (2.5 mg, 0.037 mmol) was added and was sonicated again for ten minutes. Acetic acid (2.5 mL) was then added to the solution. The solution was placed in a 20 mL scintillation vial and heated in an oven at 65 °C for three days. The crystals were then washed with DMF (5 × 10 mL) over a three-hour period. The DMF was removed and replaced by acetone (5 × 30 mL) over a five-day period. The collected crystals of MOF-525 were heated at 120 °C under dynamic vacuum (30 mTorr) for 48 h, in order to evacuate the pores.Steady-state spectroscopy
UV-Vis spectra for optically matched porphyrin and MOF-525 solutions were acquired using a Jasco V-570 spectrophotometer in either water or a buffer solution of 10 mM HEPES (pH = 7.3) and 150 mM NaCl. Fluorescence spectra were recorded with a Thermo Scientific Lumina Fluorescence Spectrometer upon excitation at 415 nm and the emission was collected between 600 nm and 800 nm with a cell holder temperature maintained at 20 °C, and under constant stirring at 600 rpm.Single particle imaging
A solution of porphyrin or MOF-525 was placed on a glass slide and left to dry. Twenty microliters of a ProLong gold antifade (P36930) solution were added and covered immediately with a coverslip. The samples were imaged using an upright fluorescence microscope using a 40× objective with NA = 0.8 coupled to an excitation filter of 390–420 nm at a power, measured out of the objective, equal to 8 mW cm−2. The emission was collected using a 600 nm long-path filter, in the presence and the absence of an antifade with a time interval of 500 ms for 90 s (limit of our processor).Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge the funding provided by the American University of Beirut Research Board (#103009) and the K. Shair Central Research Science Laboratory.References
- M. Fernández-Suárez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929 CrossRef PubMed.
- K. M. Dean and A. E. Palmer, Nat. Chem. Biol., 2014, 10, 512 CrossRef CAS PubMed.
- S. A. McKinney, C. S. Murphy, K. L. Hazelwood, M. W. Davidson and L. L. Looger, Nat. Methods, 2009, 6, 131 CrossRef CAS PubMed.
- N. Panchuk-Voloshina, R. P. Haugland, J. Bishop-Stewart, M. K. Bhalgat, P. J. Millard, F. Mao, W.-Y. Leung and R. P. Haugland, J. Histochem. Cytochem., 1999, 47, 1179–1188 CrossRef CAS PubMed.
- Y. Niko, P. Didier, Y. Mely, G.-I. Konishi and A. S. Klymchenko, Sci. Rep., 2016, 6, 18870 CrossRef CAS PubMed.
- X. Gao, L. Yang, J. A. Petros, F. F. Marshall, J. W. Simons and S. Nie, Curr. Opin. Biotechnol., 2005, 16, 63–72 CrossRef CAS PubMed.
- K. D. Wegner and N. Hildebrandt, Chem. Soc. Rev., 2015, 44, 4792–4834 RSC.
- A. L. Efros and D. J. Nesbitt, Nat. Nanotechnol., 2016, 11, 661 CrossRef CAS PubMed.
- Y. Wang, R. Hu, G. Lin, I. Roy and K.-T. Yong, ACS Appl. Mater. Interfaces, 2013, 5, 2786–2799 CrossRef CAS PubMed.
- C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086–3109 CrossRef CAS PubMed.
- C. Wu, T. Schneider, M. Zeigler, J. Yu, P. G. Schiro, D. R. Burnham, J. D. McNeill and D. T. Chiu, J. Am. Chem. Soc., 2010, 132, 15410–15417 CrossRef CAS PubMed.
- S. Wang, J. W. Ryan, A. Singh, J. G. Beirne, E. Palomares and G. Redmond, Langmuir, 2016, 32, 329–337 CrossRef CAS PubMed.
- W. Sun, S. Hayden, Y. Jin, Y. Rong, J. Yu, F. Ye, Y.-H. Chan, M. Zeigler, C. Wu and D. T. Chiu, Nanoscale, 2012, 4, 7246–7249 RSC.
- B. Sun, B. Zhao, D. Wang, Y. Wang, Q. Tang, S. Zhu, B. Yang and H. Sun, Nanoscale, 2016, 8, 9837–9841 RSC.
- N. C. Shaner, P. A. Steinbach and R. Y. Tsien, Nat. Methods, 2005, 2, 905 CrossRef CAS PubMed.
- W. T. Mason, Fluorescent and luminescent probes for biological activity: a practical guide to technology for quantitative real-time analysis, Elsevier, 1999 Search PubMed.
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC.
- Y. Zhou, X. Liang and Z. Dai, Nanoscale, 2016, 8, 12394–12405 RSC.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242 CrossRef CAS PubMed.
- A. Aziz, A. R. Ruiz-Salvador, N. C. Hernández, S. Calero, S. Hamad and R. Grau-Crespo, J. Mater. Chem. A, 2017, 5, 11894–11904 RSC.
- A. Ambroise, R. W. Wagner, P. D. Rao, J. A. Riggs, P. Hascoat, J. R. Diers, J. Seth, R. K. Lammi, D. F. Bocian and D. Holten, Chem. Mater., 2001, 13, 1023–1034 CrossRef CAS.
- R. Dong, Y. Bo, G. Tong, Y. Zhou, X. Zhu and Y. Lu, Nanoscale, 2014, 6, 4544–4550 RSC.
- Y. Tian, C. M. Beavers, T. Busani, K. E. Martin, J. L. Jacobsen, B. Q. Mercado, B. S. Swartzentruber, F. van Swol, C. J. Medforth and J. A. Shelnutt, Nanoscale, 2012, 4, 1695–1700 RSC.
- A. Shalav, B. Richards and M. Green, Sol. Energy Mater. Sol. Cells, 2007, 91, 829–842 CrossRef CAS.
- S. B. Brown, M. Shillcock and P. Jones, Biochem. J., 1976, 153, 279–285 CrossRef CAS PubMed.
- R. Redmond, E. J. Land and T. Truscott, in Methods in porphyrin photosensitization, Springer, 1985, pp. 293–302 Search PubMed.
- Q. Liu, H. Zhou, J. Zhu, Y. Yang, X. Liu, D. Wang, X. Zhang and L. Zhuo, Mater. Sci. Eng., C, 2013, 33, 4944–4951 CrossRef CAS PubMed.
- X. Dong, C. Wei, L. Lu, T. Liu and F. Lv, Mater. Sci. Eng., C, 2016, 61, 214–219 CrossRef CAS PubMed.
- S. M. Aly, M. Eita, J. I. Khan, E. Alarousu and O. F. Mohammed, J. Phys. Chem. C, 2014, 118, 12154–12161 CrossRef CAS.
- D. Wo, A. K. Sobbi and O. Franke, Zeolites, 1995, 15, 540–550 CrossRef.
- S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2014, 5, 32–51 RSC.
- H. Wang, J. Xu, D. S. Zhang, Q. Chen, R. M. Wen, Z. Chang and X. H. Bu, Angew. Chem., Int. Ed., 2015, 54, 5966–5970 CrossRef CAS PubMed.
- W. Jiang, J. Yang, Y.-Y. Liu, S.-Y. Song and J.-F. Ma, Inorg. Chem., 2017, 56, 3036–3043 CrossRef CAS PubMed.
- K. Adil, Y. Belmabkhout, R. S. Pillai, A. Cadiau, P. M. Bhatt, A. H. Assen, G. Maurin and M. Eddaoudi, Chem. Soc. Rev., 2017, 46, 3402–3430 RSC.
- J. A. Mason, J. Oktawiec, M. K. Taylor, M. R. Hudson, J. Rodriguez, J. E. Bachman, M. I. Gonzalez, A. Cervellino, A. Guagliardi and C. M. Brown, Nature, 2015, 527, 357 CrossRef CAS PubMed.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mat., 2017, 2, 17045 CrossRef CAS.
- H. Atallah, M. E. Mahmoud, A. Jelle, A. Lough and M. Hmadeh, Dalton Trans., 2018, 47, 799–806 RSC.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- C. A. Trickett, K. J. Gagnon, S. Lee, F. Gándara, H. B. Bürgi and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 11162–11167 CrossRef CAS PubMed.
- B. Mortada, T. A. Matar, A. Sakaya, H. Atallah, Z. Kara Ali, P. Karam and M. Hmadeh, Inorg. Chem., 2017, 56, 4739–4744 CrossRef PubMed.
- U. S. Arrozi, H. W. Wijaya, A. Patah and Y. Permana, Appl. Catal., A, 2015, 506, 77–84 CrossRef CAS.
- J. Park, Q. Jiang, D. Feng, L. Mao and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 3518–3525 CrossRef CAS PubMed.
- W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS PubMed.
- W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS PubMed.
- H.-L. Jiang, D. Feng, K. Wang, Z.-Y. Gu, Z. Wei, Y.-P. Chen and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 13934–13938 CrossRef CAS PubMed.
- W. J. Kim, M. S. Kang, H. K. Kim, Y. Kim, T. Chang, T. Ohulchanskyy, P. N. Prasad and K.-S. Lee, J. Nanosci. Nanotechnol., 2009, 9, 7130–7135 CAS.
- S. Gentemann, C. J. Medforth, T. P. Forsyth, D. J. Nurco, K. M. Smith, J. Fajer and D. Holten, J. Am. Chem. Soc., 1994, 116, 7363–7368 CrossRef CAS.
- A. K. Sobbi, D. Wohrle and D. Schlettwein, J. Chem. Soc., Perkin Trans. 2, 1993, 481–488, 10.1039/P29930000481.
- K. Smith, S. B. Brown, R. F. Troxler and J. J. Lai, Photochem. Photobiol., 1982, 36, 147–152 CrossRef CAS PubMed.
- T. Matsuura, K. Inoue, A. Ranade and I. Saito, Photochem. Photobiol., 1980, 31, 23–26 CrossRef CAS.
- S. Tsuchiya, Chem. Phys. Lett., 1990, 169, 608–610 CrossRef CAS.
- V. Glembockyte, J. Lin and G. Cosa, J. Phys. Chem. B, 2016, 120, 11923–11929 CrossRef CAS PubMed.
- V. Glembockyte, R. Lincoln and G. Cosa, J. Am. Chem. Soc., 2015, 137, 1116–1122 CrossRef CAS PubMed.
- J. A. S. Cavaleiro, H. Görner, P. S. S. Lacerda, J. G. MacDonald, G. Mark, M. G. P. M. S. Neves, R. S. Nohr, H.-P. Schuchmann, C. von Sonntag and A. C. Tomé, J. Photochem. Photobiol., A, 2001, 144, 131–140 CrossRef CAS.
- H. Linschitz and L. Pekkarinen, J. Am. Chem. Soc., 1960, 82(10), 2407–2411 CrossRef.
- G. Loukova, V. Smirnov and S. Starodubova, Russ. Chem. Bull., 2007, 56, 35–39 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8dt03638a |
| This journal is © The Royal Society of Chemistry 2018 |