
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePost-synthetic modification of zirconium metal–organic frameworks by catalyst-free aza-Michael additions†
Harina
Amer Hamzah
 ,
Tom S.
Crickmore
,
Daniel
Rixson
and
Andrew D.
Burrows
*
,
Tom S.
Crickmore
,
Daniel
Rixson
and
Andrew D.
Burrows
*
Department of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: a.d.burrows@bath.ac.uk
First published on 26th September 2018
Abstract
The reactions of the zirconium MOF [Zr6O4(OH)4(bdc-NH2)6] (UiO-66-NH2, bdc-NH2 = 2-amino-1,4-benzenedicarboxylate) with the Michael acceptors acrylonitrile (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCN), acrylic acid (CH2CHCO2H), methyl acrylate (CH2CHCO2Me) and methyl vinyl ketone (CH2CHC(O)Me) led to post-synthetic modification of the MOF through C–N bond formation without loss of crystallinity. The reactions with acrylonitrile and acrylic acid go to completion, yielding [Zr6O4(OH)4(bdc-NHCH2CH2CN)6] (UiO-66-AN, 1) and [Zr6O4(OH)4(bdc-NHCH2CH2CO2H)6] (UiO-66-AA, 2) respectively, whereas those with methyl acrylate and methyl vinyl ketone are incomplete, yielding [Zr6O4(OH)4(bdc-NH2)0.66(bdc-NHCH2CH2CO2Me)5.34] (UiO-66-MA, 3) and [Zr6O4(OH)4(bdc-NH2)2.76(bdc-NHCH2CH2C(O)Me)3.24] (UiO-66-MVK, 4), respectively. The acrylonitrile-modified MOF UiO-66-AN undergoes further reaction with sodium azide in the presence of zinc(II) chloride in n-butanol to form the tetrazolate-modified MOF [Zr6O4(OH)4(bdc-NHCH2CH2CN)4.74(bdc-NHCH2CH2CN4H)1.26] (UiO-66-TZ, 5).
CHCN), acrylic acid (CH2CHCO2H), methyl acrylate (CH2CHCO2Me) and methyl vinyl ketone (CH2CHC(O)Me) led to post-synthetic modification of the MOF through C–N bond formation without loss of crystallinity. The reactions with acrylonitrile and acrylic acid go to completion, yielding [Zr6O4(OH)4(bdc-NHCH2CH2CN)6] (UiO-66-AN, 1) and [Zr6O4(OH)4(bdc-NHCH2CH2CO2H)6] (UiO-66-AA, 2) respectively, whereas those with methyl acrylate and methyl vinyl ketone are incomplete, yielding [Zr6O4(OH)4(bdc-NH2)0.66(bdc-NHCH2CH2CO2Me)5.34] (UiO-66-MA, 3) and [Zr6O4(OH)4(bdc-NH2)2.76(bdc-NHCH2CH2C(O)Me)3.24] (UiO-66-MVK, 4), respectively. The acrylonitrile-modified MOF UiO-66-AN undergoes further reaction with sodium azide in the presence of zinc(II) chloride in n-butanol to form the tetrazolate-modified MOF [Zr6O4(OH)4(bdc-NHCH2CH2CN)4.74(bdc-NHCH2CH2CN4H)1.26] (UiO-66-TZ, 5).
Introduction
Metal–organic frameworks (MOFs) can be regarded as a new generation of porous materials1 and they continue to attract considerable attention for applications as wide-ranging as carbon capture,2 catalysis,3 drug delivery,4 and chemical weapon detoxification.5 Much of this interest derives from the enormous diversity of MOF structures, with variation of both the metal centres and organic linkers providing an effectively limitless number of possible materials with different pore sizes and shapes. Of particular interest for many applications is the potential for precise functionalisation of the pore walls, which is possible by using a linker ligand that contains an appropriate functional group capable of projecting into the pores. Unfortunately, many functional groups are intolerant to the synthetic conditions used to prepare MOFs, so direct reaction of a metal salt and the ligand containing the desired functionality often does not yield the anticipated product. Post-synthetic modification6,7 has emerged as a powerful tool for circumventing this issue, and in many cases, it provides the only route to including a particular functional group onto the framework of a MOF structure.A wide range of covalent post-synthetic modification reactions have been developed over recent years, including conversion of primary amines into amides,8 isocyanates,9 ureas,10 azides,11 β-amidoketones12 and secondary amines,13 aldehydes into hydrazones,14 azides into triazoles15 and bromides into nitriles,16 in addition to oxidation,17 reduction18 and Mannich reactions.19 Many of these post-synthetic modification reactions were developed on zinc MOFs, whose limited moisture stability places constraints on the reactions that can be utilised and the applications of the modified MOFs. The more recent extension of post-synthetic modification protocols to moisture- and acid-stable MOFs allows the use of reactions that would destroy more sensitive frameworks. Thus, for example, we previously reported that the amino groups in [Cr3O(F/OH)(H2O)2(bdc-NH2)3] (MIL-101(Cr)-NH2, bdc-NH2 = 2-amino-1,4-benzenedicarboxylate) could be transformed into diazonium ions, which could subsequently be converted into halides or azo dyes.20 Post-synthetic modifications on derivatives of the water-stable zirconium MOF [Zr6O4(OH)4(bdc)6] (UiO-66, bdc = 1,4-benzenedicarboxylate) have recently been reviewed by Marshall and Forgan.21
We are interested in developing new post-synthetic methods that allow different functional groups to be incorporated into stable MOFs. In this context, aza-Michael reactions provide a potential method of introducing a range of functionalities into MOF structures. The aza-Michael reaction is a conjugate addition in which a nitrogen nucleophile adds to the β-carbon of an electron-deficient alkene.22 The reaction is a versatile method for constructing new C–N bonds, and it generally requires a Lewis acid catalyst such as Bi(OTf)3 or SmI2 to proceed.23,24 More recent studies have shown that the aza-Michael reaction can take place without the aid of a catalyst. For example, Li and co-workers demonstrated that azoles react readily with a range of β,γ-unsaturated α-keto esters to afford new C–N bond adducts,25 whereas Legros, Crousse and co-workers reported that aza-Michael additions could progress in polar protic solvents without any promoting agent.26 In this report, they demonstrated that the choice of the solvent is crucial in determining the products obtained, with both the mono-adduct and the di-adduct accessible.
Encouraged by the precedence for catalyst-free Michael additions, we sought to assess whether such reactions could be employed in the post-synthetic modification of MOFs. To this end we have investigated the aza-Michael addition reactions between [Zr6O4(OH)4(bdc-NH2)6] (UiO-66-NH2) and acrylonitrile (CH2CHCN), acrylic acid (CH2CHCO2H), methyl acrylate (CH2CHCO2Me) and methyl vinyl ketone (CH2CHC(O)Me). UiO-66-NH2 was selected for study based on the high chemical stability of UiO-66 MOFs in different solvents and the relatively large pore apertures present (Fig. 1).27 This pore window size is crucial, as a MOF with insufficiently large apertures would not allow for diffusion of the reactants into the framework, which would restrict reactions to the external crystal surfaces. In this context, UiO-66-NH2 has an aperture size of 6.0 Å (ref. 27, 28) which is large enough for diffusion of small reactants into the pores. Furthermore, UiO-66 structures generally contain defects such as missing linkers29–31 and, as a consequence, reagents that are larger than the pore windows can, in practice, access reactive sites within the pores.
 | ||
| Fig. 1 (a) Part of the UiO-66 structure, with hydrogen atoms on the disordered hydroxide ligands in the Zr6O4(OH)4(O2C)6 secondary building units omitted for clarity. (b) Space-filling representation of a triangular window in UiO-66. The amino group in UiO-66-NH2 is disordered across the four positions of the benzene ring and has not been observed crystallographically. O, red; C, black; H, grey; Zr, green. | ||
Results and discussion
UiO-66-NH2 was synthesised using a previously reported procedure.32 The as-synthesised MOF was rinsed and soaked in MeOH for 3 days, replacing the solvent with fresh MeOH every 24 hours. Subsequently, the yellow microcrystalline material was collected via centrifugation and dried under reduced pressure. The reaction between UiO-66-NH2 and acrylonitrile was initially carried out under reflux for 24 h using a range of solvents. While a small degree of conversion (4–5%) was observed using the polar protic solvents hexafluoroisopropanol and 2,2,2-trifluoroethanol, the best conversion (38%) was found using water as the solvent. In all cases, the degree of conversion was obtained from the 1H NMR spectrum of the digested MOF. Most previous work32–35 has used hydrofluoric acid to digest UiO-66 and its derivatives. We found that UiO-66-NH2 and its derivatives were readily digested in ammonium fluoride solution, and have used NH4F/D2O as a digestion method throughout this work in order to avoid the hazards associated with HF, with DMSO-d6 added for 1H NMR analyses.19Lengthening the reaction time from 24 h to 5 days increased the extent of modification to 50%, though further increase of the reaction time did not lead to higher percentage conversions. However, complete conversion was achieved on leaving the reaction in an autoclave at 120 °C for 2 days to yield a product with the formula [Zr6O4(OH)4(bdc-NHCH2CH2CN)6], UiO-66-AN, 1 (Scheme 1). It is likely that the closed environment created by the autoclave prevents acrylonitrile vapour from being lost from the reaction vessel, and this combined with the higher temperature leads to full conversion.
 | ||
| Scheme 1 Post-synthetic modification of UiO-66-NH2 on reaction with acrylonitrile. | ||
The 1H NMR spectrum for UiO-66-AN following digestion (Fig. S1†) shows triplets at δ 2.71 ppm and δ 3.42 ppm which correspond to the β- and α-protons to the secondary nitrogen of D2bdc-NHCH2CH2CN. The resonances at δ 6.95 (dd), 7.05 (d) and 7.65 (d) ppm correspond to the protons from the aromatic ring of D2bdc-NHCH2CH2CN. The addition of a second acrylonitrile molecule into UiO-66-AN to form the di-adduct with bdc-N(CH2CH2CN)2 substituents was ruled out through comparison of the integrals for the aromatic and aliphatic protons. Furthermore, the negative ion ESI mass spectrum of UiO-66-AN digested in NH4F/H2O confirms the presence of the singly deprotonated anion of H2bdc-NHCH2CH2CN at m/z = 233.0561 (predicted [M − H]− = 233.0562). The FTIR spectrum for UiO-66-AN (Fig. S2†) shows a broad peak at 3364 cm−1 which corresponds to ν(N–H) of the modified framework. The presence of ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) at 2161 cm−1 confirms the presence of the cyanoethyl groups. The high similarity between the powder X-ray diffraction (PXRD) pattern of UiO-66-AN and that of UiO-66-NH2 indicates that the structural integrity of the MOF was retained upon post-synthetic modification (Fig. S3†). This illustrates the robustness of the UiO-66 framework, as neither the post-synthetic modification reaction nor the high temperature employed has any effect on the crystallinity of the MOF. The greater intensity of the peak at 2θ 12° in the PXRD pattern compared with that in the simulated pattern is consistent with previous observations with UiO-66 type MOFs and is related to desolvation, with the calculated pattern based on the activated framework.
N) at 2161 cm−1 confirms the presence of the cyanoethyl groups. The high similarity between the powder X-ray diffraction (PXRD) pattern of UiO-66-AN and that of UiO-66-NH2 indicates that the structural integrity of the MOF was retained upon post-synthetic modification (Fig. S3†). This illustrates the robustness of the UiO-66 framework, as neither the post-synthetic modification reaction nor the high temperature employed has any effect on the crystallinity of the MOF. The greater intensity of the peak at 2θ 12° in the PXRD pattern compared with that in the simulated pattern is consistent with previous observations with UiO-66 type MOFs and is related to desolvation, with the calculated pattern based on the activated framework.
Following this optimisation of the reaction conditions, the aza-Michael reaction was repeated using acrylic acid, methyl acrylate, methyl vinyl ketone and vinyl phosphonic acid as Michael acceptors. Successful conversions were observed in the cases of acrylic acid, methyl acrylate and methyl vinyl ketone, as summarised in Scheme 2. Reactions with vinyl phosphonic acid yielded amorphous powders as evidenced by PXRD and were not explored further.
 | ||
| Scheme 2 Post-synthetic modification of UiO-66-NH2 on reaction with acrylic acid, methyl acrylate and methyl vinyl ketone. | ||
For the reaction with acrylic acid, the 1H NMR spectrum of the NH4F/D2O-digested product UiO-66-AA (Fig. S4†) showed peaks at δ 6.94 (dd), 7.09 (d) and 7.54 (d) ppm, corresponding to the aromatic protons of D2bdc-NHCH2CH2CO2D. Two sets of triplets at δ 2.34 and 3.23 ppm were also observed and the absence of peaks corresponding to D2bdc-NH2 confirmed full conversion to [Zr6O4(OH)4(bdc-NHCH2CH2CO2H)6] 2. PXRD (Fig. S5†) revealed that this product has retained crystallinity and is structurally similar to UiO-66-NH2. Despite the success of these small scale reactions, efforts to scale up the synthesis of 2 proved difficult. 1H NMR spectra carried out on the digested products of larger scale reactions were more complicated than those of the small scale reactions, showing the presence of D2bdc-NHCH2CH2CO2D alongside D2bdc-N(CH2CH2CO2D)2 and D2bdc-NH2, with the presence of the doubly-substituted product confirmed by ESI-MS (m/z 326.09, [M + H]+). For this reason, thermal analyses and gas adsorption studies were not carried out on 2.
For the reaction with methyl acrylate, the 1H NMR spectrum of the NH4F/D2O-digested product (Fig. S6†) showed peaks at δ 6.88 (d), 7.03 (s) and 7.61 (d) ppm, corresponding to the aromatic protons of D2bdc-NHCH2CH2CO2Me, indicating that the post-synthetic modification was successful. Furthermore, two sets of triplets at δ 2.27 and 3.21 ppm can be seen in the aliphatic region, which correspond to the β- and α-protons to the secondary nitrogen, respectively. By comparing the integrals of the aromatic protons for D2bdc-NHCH2CH2CO2Me with those for D2bdc-NH2, also present, the degree of conversion can be calculated as 89%. This gives an average formula for UiO-66-MA of [Zr6O4(OH)4(bdc-NH2)0.66(bdc-NHCH2CH2CO2Me)5.34] 3. The negative ion ESI mass spectrum of NH4F/H2O-digested UiO-66-MA confirmed the presence of singly deprotonated anion of H2bdc-NHCH2CH2CO2Me at m/z = 266.0661 (predicted [M − H]− = 266.0665). The FTIR spectrum showed a weak peak at 3364 cm−1 (Fig. S7†), which corresponds to ν(N–H). The presence of ester groups on the modified framework was confirmed by observation of ν(CO) at 1712 cm−1. PXRD (Fig. S8†) revealed that UiO-66-MA was crystalline, and the similarities between its powder pattern and that for UiO-66-NH2 confirmed that the framework remained intact and retained its crystallinity upon the aza-Michael reaction.
The reaction with methyl vinyl ketone was undertaken using the same conditions as those with acrylonitrile and methyl acrylate. The 1H NMR spectrum of the digested product (Fig. S9†) confirmed that reaction had occurred, with new signals in both the aromatic and aliphatic regions. Specifically, signals at δ 6.90 (d), 7.05 (s) and 7.63 (d) ppm correspond to the aromatic protons in D2bdc-NHCH2CH2C(O)Me, whereas triplets at δ 2.72 and 3.27 ppm correspond to the β- and α-protons relative to the secondary nitrogen atom. Based on the aromatic signals for D2bdc-NH2 and D2bdc-NHCH2CH2C(O)Me, it can be deduced that the reaction proceeded with 54% conversion. The formula for UiO-66-MVK can therefore be expressed as [Zr6O4(OH)4(bdc-NH2)2.76(bdc-NHCH2CH2C(O)Me)3.24] 4. The FTIR spectrum of the solid sample shows a broad peak at 3367 cm−1 (Fig. S10†) which corresponds to ν(N–H). The presence of ketone functional groups on the modified framework was confirmed by observation of ν(CO) at 1710 cm−1. PXRD (Fig. S11†) revealed that UiO-66-MVK has the same gross structure as UiO-66-NH2, showing that, as with the aza-Michael additions using acrylonitrile, acrylic acid and methyl acrylate, post-synthetic modification does not affect the crystallinity of the MOF.
The results from the post-synthetic aza-Michael reactions are summarised in Table 1. The degree of conversion from primary amine to the aza-Michael product is related to the electron withdrawing power of the acceptor used, with the nitrile group in acrylonitrile being more electron withdrawing than the ester and ketone groups in methyl acrylate and methyl vinyl ketone, respectively.
| Michael acceptor | % Conversion | Product |
|---|---|---|
| a Small scale reaction. | ||
| CH2CHCN |
100 | [Zr6O4(OH)4(bdc-NHCH2CH2CN)6] 1 (UiO-66-AN) |
| CH2CHCO2H |
100a | [Zr6O4(OH)4(bdc-NHCH2CH2CO2H)6] 2 (UiO-66-AA) |
| CH2CHCO2Me |
89 | [Zr6O4(OH)4(bdc-NH2)0.66(bdc-NHCH2CH2CO2Me)5.34] 3 (UiO-66-MA) |
| CH2CHC(O)Me |
54 | [Zr6O4(OH)4(bdc-NH2)2.76(bdc-NHCH2CH2C(O)Me)3.24] 4 (UiO-66-MVK) |
In order to access whether the nitrile groups in UiO-66-AN can be transformed into other functional groups, further post-synthetic modification reactions were assessed. Initial results suggested that the most promising of these was the reaction of UiO-66-AN with sodium azide in the presence of zinc(II) chloride, which was anticipated to convert the nitrile groups into tetrazoles. This reaction was found to be very dependent on the solvent used, with no conversion observed using water, ethanol, n-propanol, DMF or acetonitrile. In contrast, a reaction was observed when n-butanol was used as the solvent (Scheme 3).
 | ||
| Scheme 3 Post-synthetic modification of UiO-66-AN on reaction with sodium azide in the presence of zinc(II) chloride and n-butanol. | ||
The successful conversion of nitrile groups to tetrazoles was confirmed by 1H NMR spectroscopy of the digested product (Fig. S12†). New sets of peaks (δ 6.92d, 7.08s and 7.61d ppm) were observed in the aromatic region, in addition to the peaks which correspond to the aromatic protons of D2bdc-NHCH2CH2CN. The new peaks correspond to the protons from the benzene ring of D2bdc-NHCH2CH2CN4H. A triplet due to the β-protons to the secondary nitrogen of D2bdc-NHCH2CH2CN4H is clearly visible at δ 3.0 ppm, though the triplet of the neighbouring α-protons is only partly visible due to overlapping with the more dominant triplet resulting from D2bdc-NHCH2CH2CN. The percentage conversion from nitrile to tetrazole was estimated on the basis of the integrals to be approximately 21%, which gives an formula for UiO-66-TZ as [Zr6O4(OH)4(bdc-NHCH2CH2CN)4.74(bdc-NHCH2CH2CN4H)1.26] 5. The negative ion ESI mass spectrum of the digested product confirmed the singly deprotonated anion of H2bdc-NHCH2CH2CN4H at m/z = 276.0727 (predicted [M − H]− = 276.0733).
In order to investigate the structural integrity of the PSM product, PXRD was carried out on the solid sample UiO-66-TZ. The sample was extensively rinsed with n-BuOH prior to the PXRD measurement and the pattern of the n-BuOH-rinsed sample shows additional peaks at 2θ 31.5°, 34.5°, 36.0° and 47.5° in addition to those corresponding to the UiO-66 framework (Fig. S13†). These additional peaks do not derive from unreacted reagents, and they were removed by washing the sample with water. Reactions were undertaken using different times and catalyst loadings, but conversion could not be raised above 21%. This is likely to be a consequence of pore blocking by the relatively large ethyltetrazole groups, which is consistent with the nitrogen adsorption data (vide infra).
The thermal stability of the post-synthetically modified products 1 and 3–5 was assessed by thermogravimetric analysis (TGA) (Fig. S14†). The samples were rinsed with water prior to the TGA measurements. An initial mass loss of 2–17% was seen in all cases between 50 and 120 °C. This can be attributed to the loss of included water molecules, and the different amounts reflect the remaining pore space following the modification reactions. A second small (∼3%) and gradual mass loss (120 °C–440 °C) may be attributed to residual DMF solvent in the pores and/or the dehydroxylation of the Zr6O4(OH)4 nodes. The UiO-66 series of MOFs are known to undergo complete dehydroxylation reactions below 300 °C.34 Normally, a plateau region in a TGA profile represents the range of temperature that can be used to produce the activated form of a MOF. Due to the gradual mass loss in the 120 °C–440 °C region, 120 °C was chosen as the activation temperature, which is consistent with that used in previous studies on UiO-66 derivatives.36–38 The mass loss above 440 °C corresponds to decomposition of the framework, and is similar to previous reports for UiO-66-NH2 and related MOFs.32,39 These results implies that post-synthetic modification does not alter the thermal stability of UiO-66-NH2.
In order to investigate the porosity of the products, N2 adsorption measurements were carried out on 1 and 3–5 at 77 K. Prior to the measurements, the samples were activated at 120 °C for 12 h. 1H NMR analysis of the digested activated samples confirmed that no solvent molecules were present upon activation. Furthermore, the PXRD patterns of the activated samples were similar to those prior to activation, indicating that the activation conditions did not alter the crystallinity of the MOFs. The N2 adsorption isotherms for UiO-66-NH2 and 1 and 3–5 are shown in Fig. 2, and the calculated BET surface areas for these MOFs are given in Table 2.
 | ||
| Fig. 2 Adsorption and desorption of N2 by 1 and 3–5 at 77 K in comparison to UiO-66-NH2. | ||
| Compound | S BET (m2 g−1) |
|---|---|
| [Zr6O4(OH)4(bdc-NH2)6], UiO-66-NH2 | 1041 |
| UiO-66-AN, 1 | 452 |
| UiO-66-MA, 3 | 392 |
| UiO-66-MVK, 4 | 527 |
| UiO-66-TZ, 5 | 52 |
The BET surface area for UiO-66-NH2 obtained in this work is similar to values reported in the literature for this material, which range from 778 to 1293 m2 g−1.32,39–42 The derivatives 1, 3 and 4 exhibited Type I isotherms with each product showing a lower degree of porosity than UiO-66-NH2, which is consistent with the mass increase caused by the tag groups in the pores which also reduce the space available for guest inclusion. The percentage conversion of the post-synthetic modification reaction also has an impact on the BET surface area and, in general, the higher the percentage conversion, the lower the surface area observed. This accounts for the higher value of SBET for 4 in comparison to 1 and 3. The effect of pore blocking with large introduced substituents is especially apparent in the essentially non-porous nature of 5, which shows a Type II isotherm.
The CO2 isotherms of 1 and 3–5 at 273 K were also recorded, and are shown in Fig. 3. The CO2 uptakes are lower than that for UiO-66-NH2, which is likely to be related to the pore blocking effect of the tag groups as this limits diffusion of CO2 molecules into the pores. Furthermore, the absence of the primary amine group in the pores may contribute to the lower CO2 uptake. It is notable than the CO2 uptake for 1 is higher than that for 3. This is likely to be due to the relative sizes of the tag groups. Unexpectedly, the CO2 loading of 5 is somewhat higher than that for 4 despite the former being non-porous to nitrogen. One possible reasoning for this observation is the strong interaction of CO2 molecules with the tetrazole moieties. MOFs with nitrogen-rich environments such as those with tetrazole- and triazole-functionalities have been previously reported to have high affinity towards CO2,43,44 and in the case of 5 much of this may occur on external surfaces.
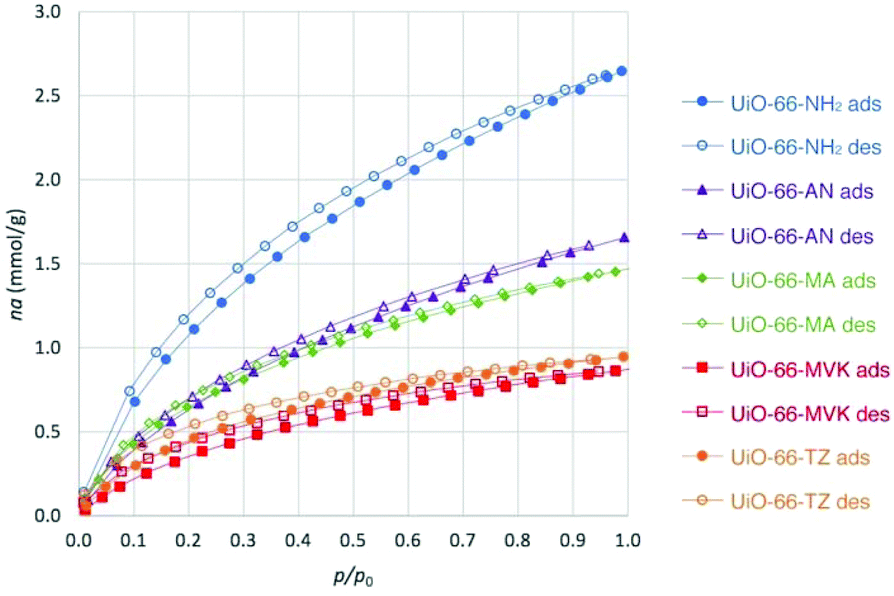 | ||
| Fig. 3 Adsorption and desorption of CO2 by 1 and 3–5 at 273 K in comparison to UiO-66-NH2. | ||
Conclusions
A new versatile, single-step post-synthetic modification reaction for MOFs has been developed. In this process, the primary amino group in UiO-66-NH2 is converted into a range of secondary amines bearing different functional groups via a catalyst-free aza-Michael addition. The reaction was achieved by reacting UiO-66-NH2 with four different alkenes containing electron withdrawing groups, CH2CHR (R = CN, CO2H, CO2CH3, C(O)CH3). The degrees of conversion were governed by the strength of the electron withdrawing groups attached to the alkenes with the stronger electron withdrawing groups leading to higher conversion. Following optimisation, the highest conversion (100%) was achieved whilst using either acrylonitrile or acrylic acid, with incomplete reactions for methyl acrylate (89%) and methyl vinyl ketone (54%). A second post-synthetic modification was successfully performed on UiO-66-AN in which the MOF was treated with NaN3 and ZnCl2 catalyst in n-BuOH. This reaction resulted in the partial conversion of the nitrile groups into tetrazoles.
We anticipate that the aza-Michael process described is likely to be readily extended to other MOF systems that possess high stability towards H2O and alcohols. Current work is seeking to investigate this in addition to expanding the range of alkenes that can be used as Michael acceptors in this process. Furthermore, we are studying additional post-synthetic modification reactions on UiO-66-AN and investigating the acid behaviour of UiO-66-AA.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The University of Bath and Majlis Amanah Rakyat (MARA) are thanked for a PhD studentship (to HAH).Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- J. Yu, L.-H. Xie, J.-R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674 CrossRef CAS PubMed.
- L. Zhu, X.-Q. Liu, H.-L. Jiang and L.-B. Sun, Chem. Rev., 2017, 117, 8129 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232 CrossRef CAS PubMed.
- N. S. Bobbitt, M. L. Mendonca, A. J. Howarth, T. Islamoglu, J. T. Hupp, O. K. Farha and R. Q. Snurr, Chem. Soc. Rev., 2017, 46, 3357 RSC.
- S. M. Cohen, Chem. Rev., 2012, 112, 970 CrossRef CAS PubMed.
- A. D. Burrows, in Postsynthetic Modification of Metal-Organic Frameworks in Metal-Organic Framework Materials, ed. L. R. MacGillivray and C. M. Lukehart, Wiley, 2014, p. 195 Search PubMed.
- K. K. Tanabe, Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2008, 130, 8508 CrossRef CAS PubMed.
- C. Volkringer and S. M. Cohen, Angew. Chem., Int. Ed., 2010, 49, 4644 CrossRef CAS PubMed.
- E. Dugan, Z. Wang, M. Okamura, A. Medina and S. M. Cohen, Chem. Commun., 2008, 3366 RSC.
- M. Savonnet, D. Bazer-Bachi, N. Bats, J. Perez-Pellitero, E. Jeanneau, V. Lecocq, C. Pinel and D. Farrusseng, J. Am. Chem. Soc., 2010, 132, 4518 CrossRef CAS PubMed.
- W. J. Gee, L. K. Cadman, H. Amer Hamzah, M. F. Mahon, P. R. Raithby and A. D. Burrows, Inorg. Chem., 2016, 55, 10839 CrossRef CAS PubMed.
- A. D. Burrows and L. L. Keenan, CrystEngComm, 2012, 14, 4112 RSC.
- A. D. Burrows, C. G. Frost, M. F. Mahon and C. Richardson, Angew. Chem., Int. Ed., 2008, 47, 8482 CrossRef CAS PubMed.
- Y. Goto, H. Sato, S. Shinkai and K. Sada, J. Am. Chem. Soc., 2008, 130, 14354 CrossRef CAS PubMed.
- M. Kim, S. J. Garibay and S. M. Cohen, Inorg. Chem., 2011, 50, 729 CrossRef CAS PubMed.
- A. D. Burrows, C. G. Frost, M. F. Mahon and C. Richardson, Chem. Commun., 2009, 4218 RSC.
- W. Morris, C. J. Doonan, H. Furukawa, R. Banerjee and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 12626 CrossRef CAS PubMed.
- H. Amer Hamzah, W. J. Gee, P. R. Raithby, S. J. Teat, M. F. Mahon and A. D. Burrows, Chem. – Eur. J., 2018, 24, 11094 CrossRef CAS PubMed.
- D. Jiang, L. L. Keenan, A. D. Burrows and K. J. Edler, Chem. Commun., 2012, 48, 12053 RSC.
- R. J. Marshall and R. S. Forgan, Eur. J. Inorg. Chem., 2016, 2016, 4310 CrossRef CAS.
- J. L. Vicario, D. Badía, L. Carrillo, J. Etxebarria, E. Reyes and N. Ruiz, Org. Prep. Proced. Int., 2005, 37, 513 CrossRef CAS.
- R. Varala, M. M. Alam and S. R. Adapa, Synlett, 2003, 720 CAS.
- I. Reboule, R. Gil and J. Collin, Tetrahedron Lett., 2005, 46, 7761 CrossRef CAS.
- J. Wang, P.-F. Li, S. H. Chan, A. S. C. Chan and F. Y. Kwong, Tetrahedron Lett., 2012, 53, 2887 CrossRef CAS.
- K. De, J. Legros, B. Crousse and D. Bonnet-Delpon, J. Org. Chem., 2009, 74, 6260 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef PubMed.
- Q. Yang, H. Jobic, F. Salles, D. Kolokolov, V. Guillerm, C. Serre and G. Maurin, Chem. – Eur. J., 2011, 17, 8882 CrossRef CAS PubMed.
- C. A. Trickett, K. J. Gagnon, S. Lee, F. Gándara, H.-B. Bürgi and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 11162 CrossRef CAS PubMed.
- C. L. Hobday, R. J. Marshall, C. F. Murphie, J. Sotelo, T. Richards, D. R. Allan, T. Düren, F.-X. Coudert, R. S. Forgan, C. A. Morrison, S. A. Moggach and T. D. Bennett, Angew. Chem., Int. Ed., 2016, 55, 2401 CrossRef CAS PubMed.
- S. Øien, D. Wragg, H. Reinsch, S. Svelle, S. Bordiga, C. Lamberti and K. P. Lillerud, Cryst. Growth Des., 2014, 14, 5370 CrossRef.
- S. J. Garibay and S. M. Cohen, Chem. Commun., 2010, 46, 7700 RSC.
- M. Kandiah, S. Usseglio, S. Svelle, U. Olsbye, K. P. Lillerud and M. Tilset, J. Mater. Chem., 2010, 20, 9848 RSC.
- R. Ameloot, M. Aubrey, B. M. Wiers, A. P. Gómora-Figueroa, S. N. Patel, N. P. Balsara and J. R. Long, Chem. – Eur. J., 2013, 19, 5533 CrossRef CAS PubMed.
- C. O. Audu, H. G. T. Nguyen, C.-Y. Chang, M. J. Katz, L. Mao, O. K. Farha, J. T. Hupp and S. T. Nguyen, Chem. Sci., 2016, 7, 6492 RSC.
- Z. Hu, K. Zhang, M. Zhang, Z. Guo, J. Jiang and D. Zhao, ChemSusChem, 2014, 7, 2791 CrossRef CAS PubMed.
- A. Kronast, S. Eckstein, P. T. Altenbuchner, K. Hindelang, S. I. Vagin and B. Rieger, Chem. – Eur. J., 2016, 22, 12800 CrossRef CAS PubMed.
- C. Wang, X. Liu, J. P. Chen and K. Li, Sci. Rep., 2015, 5, 16613 CrossRef CAS PubMed.
- S. M. Chavan, G. C. Shearer, S. Svelle, U. Olsbye, F. Bonino, J. Ethiraj, K. P. Lillerud and S. Bordiga, Inorg. Chem., 2014, 53, 9509 CrossRef CAS PubMed.
- D. Sun, Y. Fu, W. Liu, L. Ye, D. Wang, L. Yang, X. Fu and Z. Li, Chem. – Eur. J., 2013, 19, 14279 CrossRef CAS PubMed.
- J. B. DeCoste, M. A. Browe, G. W. Wagner, J. A. Rossin and G. W. Peterson, Chem. Commun., 2015, 51, 12474 RSC.
- H. Saleem, U. Rafique and R. P. Davies, Microporous Mesoporous Mater., 2016, 221, 238 CrossRef CAS.
- D.-M. Chen, J.-Y. Tian, M. Chen, C.-S. Liu and M. Du, ACS Appl. Mater. Interfaces, 2016, 8, 18043 CrossRef CAS PubMed.
- S. Seth, G. Savitha and J. N. Moorthy, Inorg. Chem., 2015, 54, 6829 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses and characterisation of products. See DOI: 10.1039/c8dt03312a |
| This journal is © The Royal Society of Chemistry 2018 |