
Sterically demanding methoxy and methyl groups in ruthenium complexes lead to enhanced quantum yields for blue light triggered photodissociation†

Abstract
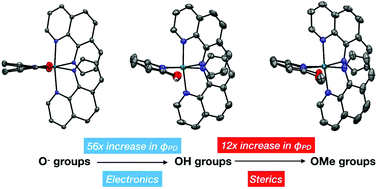
Ruthenium complexes containing a sterically congested metal center can serve as light activated prodrugs through photo-activated chemotherapy (PACT). In this work, we modified PACT agents containing 6,6′-dihydroxybipyridine (6,6′-dhbp) (Papish et al., Inorg. Chem., 2017, 56, 7519) by replacing it with a sterically bulky isoelectronic ligand, 6,6′-dimethoxybipyridine (6,6′-dmbp). The resulting complexes, [(phen)2Ru(6,6′-dmbp)]Cl2 (2OMe, phen = 1,10-phenanthroline) and [(dop)2Ru(6,6′-dmbp)]Cl2 (3OMe, dop = 2,3-dihydro-[1,4]dioxino[2,3-f][1,10]phenanthroline), have been fully characterized and display enhanced quantum yields for blue light triggered photodissociation of 0.024(6) and 0.0030(2), respectively. We have also synthesized 4OH = [(dmphen)2Ru(4,4′-dhbp)]Cl2 wherein dmphen = 2,9-dimethyl-1,10-phenanthroline and 4,4′-dhbp = 4,4′-dihydroxybipyridine. These ligands enhance steric bulk near the metal center and move the hydroxy groups further from the metal center, respectively. Complex 4OH displays a relatively low quantum yield of 0.0014(2). All of the new complexes (2OMe, 3OMe, 4OH) were tested in breast cancer cells (MDA-MB-231) and were non-toxic (IC50 > 100 μM). This has been interpreted in terms of unfavorable log(Do/w) values and furthermore photodissociation alone is insufficient for cytotoxicity. We also report the crystal structures of 4OH and 2OMe, the thermodynamic acidity of complex 4OH, and the redox potentials for all new complexes.
 Please wait while we load your content...
Please wait while we load your content...

