
Boron carbonyl complexes analogous to hydrocarbons
Jiaye
Jin  a
and
Mingfei
Zhou
*a
a
and
Mingfei
Zhou
*a
a
and
Mingfei
Zhou
*a
Abstract
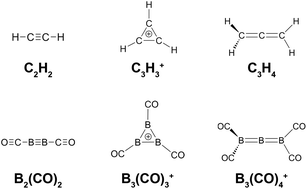
Recent studies on boron carbonyl complexes show their intriguing structural and bonding properties, enriching our knowledge on main group coordination chemistry. The isolobal relationships between BCO and CH and the more generally applicable CO/H− and B−/C analogies are employed to understand the structure and bonding of boron carbonyl complexes, bridging the boron carbonyl chemistry to the well-known hydrocarbon analogues.
- This article is part of the themed collection: 2018 Frontier and Perspective articles
 Please wait while we load your content...
Please wait while we load your content...

