
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination of arenes and phosphines by charge separated alkaline earth cations†
Lucia
Garcia
a,
Mathew D.
Anker
a,
Mary F.
Mahon
*a,
Laurent
Maron
*b and
Michael S.
Hill
 *a
*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@bath.ac.uk
bUniversité de Toulouse et CNRS, INSA, UPS, UMR 5215, LPCNO, 135 Avenue de Rangueil, F-31077 Toulouse, France
First published on 15th August 2018
Abstract
Generation of β-diketiminato group 2 cations, [(MeBDI)Ae]+ and [(t-BuBDI)Ae]+ (MeBDI = HC{(Me)CN-2,6-i-Pr2C6H3}2; t-BuBDI = HC{(t-Bu)CN-2,6-i-Pr2C6H3}2; Ae = Mg or Ca), in conjunction with the weakly coordinating anion, [Al{OC(CF3)3}4]−, allows the characterisation of charge separated alkaline earth η6-π adducts to toluene or benzene when crystallised from the arene solvents. Addition of 1,4-difluorobenzene to [(MeBDI)Mg]+ results in the isolation of [(MeBDI)Mg(1,4-F2C6H4)3]+ in which the fluorobenzene molecules coordinate via κ1-F–M interactions. Although DFT analysis indicates that the polyhapto arene binding to Mg is effectively electrostatic in origin, the interactions with Ca (Sr and Ba) are observed to invoke small but significant π overlap of the arene HOMOs with the alkaline earth valence nd orbitals. Reaction of triphenylphosphine with [(MeBDI)Mg]+ and [(t-BuBDI)Mg]+ in toluene solvent allows the isolation of the respective terminally coordinated magnesium-phosphine adducts. The resultant Mg–P bond lengths [2.5972(13), 2.6805(12) Å] are comparable to those previously observed in magnesium derivatives of terminal but formally anionic phosphide ligands, while the effectively electrostatic nature of the bonding is supported by DFT calculations.
Introduction
Non-covalent interactions between s-block cations and aromatic π systems play a pivotal role in the regulation of many biological phenomena.1,2 The importance of polyhapto arene-to-cation binding, for example during the transport of K+ and Ca2+ through membranes or to ensure the 3-dimensional integrity of many enzymes, has prompted intense interest in the nature of the interactions.3 A significant number of experimental and theoretical investigations have demonstrated that Ae2+ cations (Ae = Mg or Ca) can display affinities for benzene (e.g.I and II) and toluene in the gas phase as high as 300 kJ mol−1.3–12 These studies have also revealed that such interactions exert substantial electrostatic disruption of the arene π-electron density. More generally, Ae–π interactions have long been recognised to wield a significant influence over the structures and aggregation states adopted by a wide variety of organometallic, amido or phenoxy group 2 derivatives.13–21 In all such instances, however, the arene or π-substituent is a component of a more complex anion and benefits from the entropic advantage afforded by its intramolecular disposition. Despite their widespread biological significance, however, definitive structural characterisation of intermolecular interactions between Mg or Ca and neutral arenes has proven elusive, such that until very recently (vide infra) the only structurally authenticated examples of molecular species to feature intermolecular Ae–π interactions in the solid state were provided by Hanusa's bis(trimethylsilyl)butadiyne adduct of decamethylcalcocene (III)22 and various serendipitously crystallised benzene and toluene solvates of barium derivatives.23

In 2018, and while the current study was in progress, Harder and co-workers described the isolation of several cationic [(MeBDI)Ae]+ (MeBDI = HC{(Me)CN-2,6-i-Pr2C6H3}2; Ae = Mg or Ca) complexes as contact ion pairs of the [B(C6F5)4]− anion.38 These cations are evidently extremely potent Lewis acids coordinating benzene, 3-hexyne and even hexamethyldisiloxane.39 The benzene adducts, [(MeBDI)Mg·C6H6]+[B(C6F5)4]− (VI; shortest Mg⋯C distance = 2.367(2) Å) and [(MeBDI)Ca·C6H6]+[B(C6F5)4]− (VII, shortest Ca⋯C distance = 2.909(2) Å) were crystallographically characterised to display η3 and η6 binding, respectively, albeit both species also retain notably short Ae⋯F–C contacts to the weakly coordinating borate anions (VI, Mg⋯F 2.046(1); VII, Ca⋯F 2.453(1) Å).
Our own interest in these phenomena was prompted by our recent observation that the use of β-diketiminato calcium n-alkyls enables the unprecedented nucleophilic substitution of a C–H bond of benzene (Scheme 1).40 These reactions produce the n-alkyl benzenes through the generation of a calcium hydride such that rupture of the C–H bond occurs via an effective SN2 displacement of H−. Based on the experimental observation that the alkylation displays half order kinetics, it was proposed that the key step in this transformation was the generation of a monomeric and coordinatively unsaturated calcium alkyl. On this basis, density functional theory (DFT) calculations supported an ensuing mechanism of C–H activation which occurs through the assembly of a transition state (IV, Scheme 1) resembling a non-stabilized Meisenheimer complex. We deduced that the nucleophilic attack on the electron rich π-system was largely facilitated by the high degree of native charge separation between the calcium centre and the organic anion. In this manner, the mechanism of substitution is enabled by both the highly nucleophilic nature of the organic anion and η6 engagement of the π-electron density of benzene with the highly electrophilic calcium centre. This cooperative view of the reactivity implies that such Ae–π-arene interactions may not only provide an influence over structure and (bio)molecular function but also present a means to allow further unprecedented arene-centred transformations. Prior to Harder's very recent contributions,38,39 the only previously reported charge separated β-diketiminato alkaline earth cations are the ion pair systems, [(MeBDI)Ae(NC5H5)3]+[H2N{B(C6F5)3}]− (Ae = Ca, Sr), in which the coordination spheres of the electrophilic Ae centres are saturated by the addition of three N-donor pyridines.41 In this contribution, therefore, we provide an assessment of the potential of the truly charge separated β-diketiminato Mg and Ca cations to engage with arene π-systems and soft phosphine donors when generated in conjunction with Krossing's weakly nucleophilic aluminate anion, [Al{OC(CF3)3}4]−.42–44
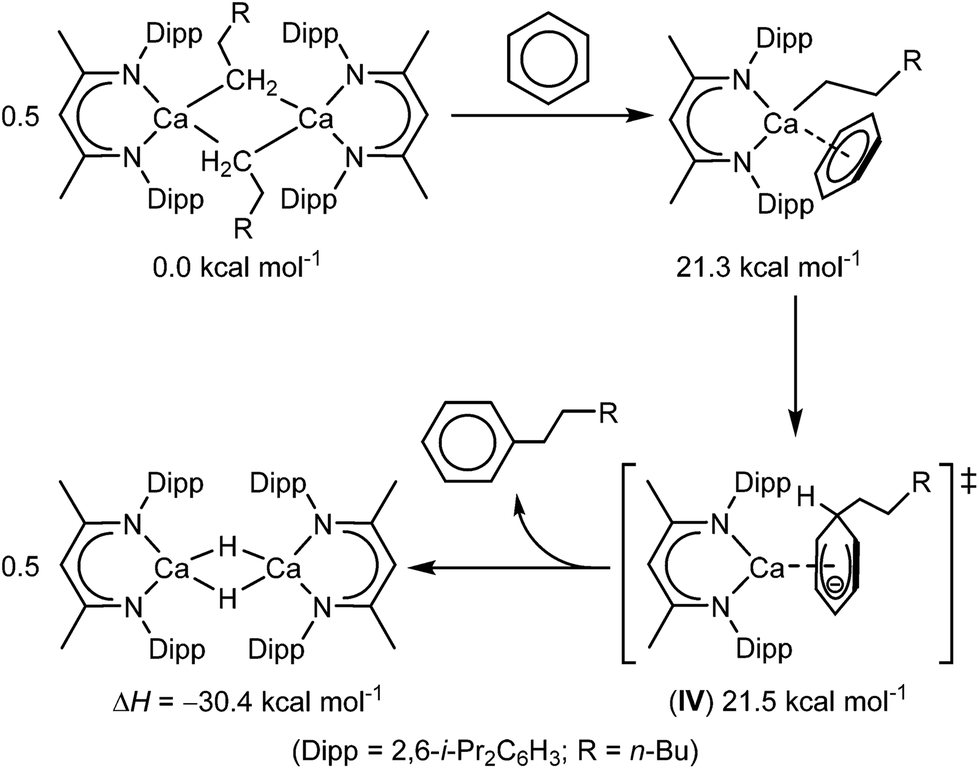 | ||
| Scheme 1 Calcium-mediated nucleophilic alkylation of benzene. | ||
Results and discussion
Arene adducts
In a manner reminiscent of the recently described behaviour of compounds VI and VII, an initial reaction performed in toluene-d8 between [(BDI)Mgn-Bu] (1) and [Ph3C]+[Al{OC(CF3)3}4]− resulted in the immediate formation of two immiscible phases. This observation is characteristic of the generation of charge separated metalorganic species and consequent liquid clathrate formation.45,46 Although analysis of this mixture by 19F{1H} NMR spectroscopy provided a single sharp resonance at δ −74.9 ppm, in common with many previously reported examples of liquid clathrates, the resultant 1H and 13C{1H} NMR spectra were broad and largely uninformative. Similar reactions performed between the trityl aluminate and 1 in C6D6 or with the alternative magnesium organometallic, [(t-BuBDI)Mgn-Bu] (2) bearing a more sterically encumbered tert-butyl substituted β-diketiminate ligand in toluene-d8 provided similar observations and the formation of two phase systems. Although clathrate formation hindered definitive solution characterisation by NMR spectroscopy, slow diffusion of hexane into all three reaction solutions at room temperature provided colourless crystals of compounds 3, 4 and 5 suitable for single crystal X-ray analysis (Scheme 2). | ||
| Scheme 2 Synthesis of compounds 3, 4 and 5. | ||
Although isolated samples of compounds 3–5 provided NMR spectra similar to those of the reaction solution when redissolved in either C6D6 or toluene-d8, solution analysis in THF-d8 definitively established the bulk production of the cationic derivatives as their corresponding THF adducts. More significantly, the constitutions of compounds 3 and 4 in the solid state were identified by single crystal X-ray analysis as the respective toluene- and benzene-adducted magnesium ion pairs [(MeBDI)Mg(C6D5CD3)]+[Al{OC(CF3)3}4]− (3) and [(MeBDI)Mg(C6D6)]+[Al{OC(CF3)3}4]− (4) (Fig. 1). Although single, the crystals of compound 5 were very weakly diffracting and the refinement of the structural model was compromised by several factors, all of which contributed to high residuals. Nonetheless, the structural connectivity of 5 was unambiguously assigned (Fig. S10†) confirming it as the toluene-adducted magnesium ion pair derivative [(t-BuBDI)Mg(C6D5CD3)]+[Al{OC(CF3)3}4]−. Hence, the synthesis of compounds 3–5 (Scheme 2) is, strongly reminiscent of that described for both the recently reported compound VI, which was prepared by treatment of [(MeBDI)Mgi-Pr] with the trityl salt of [B(C6F5)4]−,38 and [(MeBDI)Mg{HB(C6F5)3}], obtained by a similar hydride abstraction reaction of [(MeBDI)Mg(NMe2BH2NMe2BH3)] with B(C6F5)3.47,48 Significantly, however, the borate anions present in both of these previously reported compounds each bind to the magnesium centres through the above noted Mg⋯FC interactions or a combination of B–H and ortho-C–F coordinative contacts, respectively.
 | ||
| Fig. 1 ORTEP representations of the Mg1-containing cationic components of (a) compound 3 and (b) compound 4 (30% probability ellipsoids). Hydrogen atoms have been removed for clarity. Selected bond lengths (Å) and angles (°): (3) Mg1–N1 1.9847(18), Mg1–N2 1.9915(18), Mg1–C30 2.520(3), Mg1–C31 2.607(3), Mg1–C32 2.741(3), Mg1–C33 2.679(3), Mg1–C34 2.574(3), Mg1–C35 2.517(3), C30–Mg1–C31 31.86(10), C30–Mg1–C32 55.13(9), C30–Mg1–C33 64.52(9), C30–Mg1–C34 56.48(10), C31–Mg1–C32 30.09(9), C31–Mg1–C33 53.73(10), C33–Mg1–C32 29.80(10), C34–Mg1–C31 64.85(10), C34–Mg1–C32 54.34(10), C34–Mg1–C33 30.60(10), C35–Mg1–C30 31.96(10), C35–Mg1–C31 56.10(10), C35–Mg1–C32 64.35(9), C35–Mg1–C33 54.94(10) C35–Mg1–C34 31.67(10); (4) Mg1–N1 1.981(3), Mg1–N2 1.971(3), Mg1–C30 2.582(5), Mg1–C31 2.541(5), Mg1–C32 2.507(6), Mg1–C33 2.554(5), Mg1–C34 2.593(6), Mg1–C35 2.611(6), C30–Mg1–C34 55.6(2), C30–Mg1–C35 31.1(2), C31–Mg1–C33 54.8(2), C31–Mg1–C34 65.0(2), C31–Mg1–C35 55.6(2). | ||
The most notable structural feature of compound 3 is the marginally asymmetric η6-coordination of the toluene molecule to the magnesium centre. Although a separation of 2.204 Å is observed between the magnesium centre and the centroid defined by the aromatic ring, the presence of the C32-bound toluene methyl substituent results in a range of Mg–C distances between 2.517(3) (Mg1–C35) and 2.741(3) Å (Mg1–C32). In contrast, the magnesium centre of compound 4 interacts much more symmetrically with the η6-bound benzene molecule providing Mg–C distances which lie in a narrow range between 2.507(6) (Mg1–C32) and 2.611(6) Å (Mg1–C35). Although all of these distances are significantly longer than the closest Mg–C contact observed in compound VI (2.367(2) Å),38 the arene binding to magnesium in this latter species is significantly perturbed by its interaction with the more coordinating [B(C6F5)4]− anion such that the overall hapticity is lowered to η3 and with the two other closest Mg–C contacts elongated to 2.686(2) and 2.810(2) Å. Although all of the Mg–C distances observed in compounds 3 and 4 are significantly longer than typical values (ca. 1.95–2.1 Å) calculated for interactions between Mg2+ ions and simple monocyclic arenes,6–11 it should be noted that these theoretical results were performed on gas phase dications rather than the monocationic and BDI-supported species described here. Notably, however, the experimentally deduced distances are commensurate with or are shorter than those calculated for the isoelectronic interaction between C6-arenes and either ‘naked’ Na+ or water-adducted sodium monocations.6
A further crystallisation of compound 3 performed in the presence of a stoichiometric excess of 1,4-difluorobenzene provided a further new ion paired magnesium complex, compound 6. Although the Mg⋯F–C interactions were too labile to be observed in solution by NMR spectroscopy, 6 was identified by single crystal X-ray analysis as [(MeBDI)Mg(1,4-F2C6H4)3]+[Al{OC(CF3)3}4]− in which three molecules of 1,4-difluorobenzene interact with the alkaline earth centre via κ1-F–M interactions (Fig. 2). Similar κ1 binding of fluorobenzenes is very uncommon and has only been observed previously in a handful of d0 transition metal complexes.49–53 1,4-Difluorobenzene, in particular, is very weakly coordinating and has more typically been observed to interact with metal-centres either through its constituent π-system54–58 or in the unique coordination polymer [{LiN(SiMe3)2}2·1,4-C6H4F2]∞ in which each 1,4-difluorobenzene bridges between two dimeric {LiN(SiMe3)2}2 units by κ1-Li⋯F interactions.59 Compound 6 is, thus, unique in providing the first observation of this κ1 coordination mode for 1,4-difluorobenzene. As expected, the Mg–F interactions [2.029(3)–2.100(3) Å] are significantly longer than those observed in the limited number of molecular magnesium fluorides that have been structurally characterized, for example Parkin's unique terminal fluoride, [{Tpt-Bu,Me}MgF] (Tpt-Bu,Me = tris(3-tert-butyl-5-pyrazolyl)hydroborate),60 [1.7977(11) Å] and those of the dinuclear complex [(MeBDI)Mg(μ-F)(THF)]2 [1.951(2) Å].61 The observation that the Mg–F interactions in 6 are effectively commensurate with those observed in the ion paired species VI [2.046(1) Å] and in several magnesium derivatives of the {HB(C6F5)3}− anion [ca. 2.08 Å] emphasises the highly electrophilic nature of the [(MeBDI)Mg]+ unit.48,62
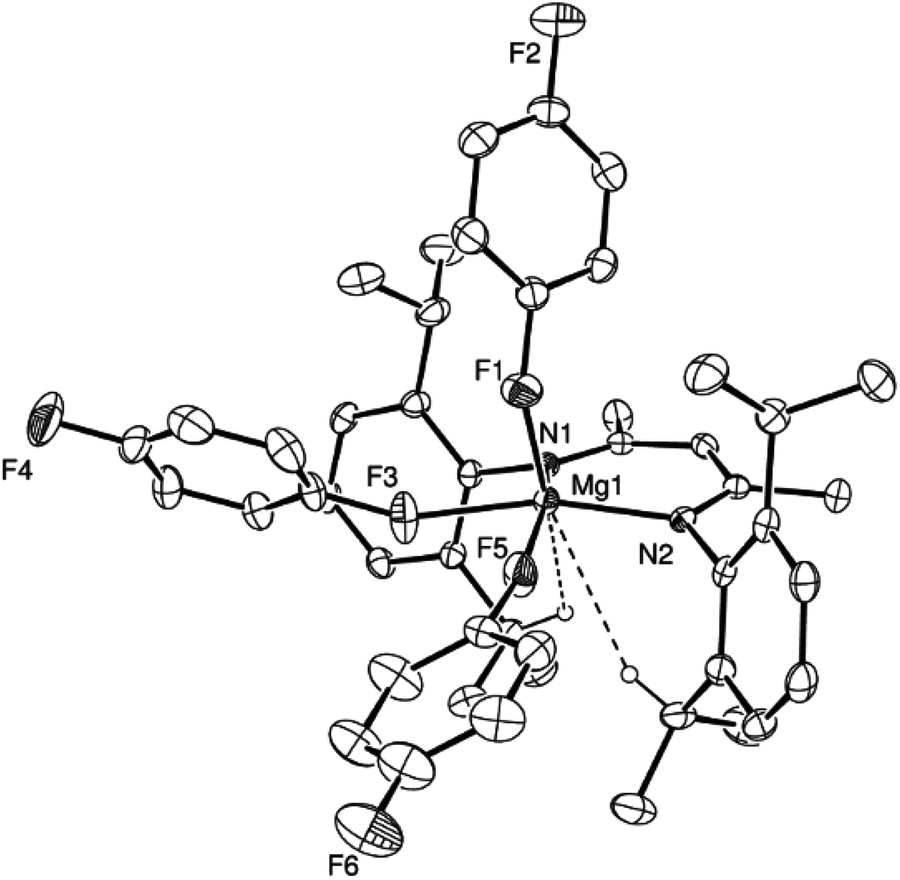 | ||
| Fig. 2 ORTEP representation of the cationic component of compound 6 (30% probability ellipsoids). Hydrogen atoms, apart from H15 and H27 and the second disordered component of the 1,4-difluorobenzene based on F5 have been omitted for clarity. Selected bond lengths (Å) and angles (°): Mg1–N1 2.008(3), Mg1–N2 2.015(3), Mg1–F1 2.029(3), Mg1–F3 2.100(3), Mg1–F5 2.073(4), N1–Mg1–N2 96.92(13), N1–Mg1–F5 161.7(3), N2–Mg1–F1 102.94(13), N2–Mg1–F3 164.48(14), N1–Mg1–F1 111.84(13), N1–Mg1–F3 92.40(13), F1–Mg1–F3 84.90(13), F1–Mg1–F5 81.4(3), N2–Mg1–F1 102.94(13), N2–Mg1–F3 164.48(14), N2–Mg1–F5 92.1(3), F5–Mg1–F3 75.7(3). | ||
Compound VII was synthesized through reaction of [(MeBDI)H2]+[B(C6F5)4]− and [Ca(p-t-Bu-benzyl)2]. A further reaction between [(MeBDI)CaN(SiMe3)2] and [Ph3C]+[Al{OC(CF3)3}4]− performed in C6D6 also provided two immiscible phases, which, after slow diffusion of hexane into the reaction mixture, provided the target calcium cation, compound 7 as a crop of colourless crystals. After decantation, analysis of the remaining soluble components evidenced the production of a single organic side-product, compound 8, which displayed a series of resonances characteristic of phenyl dearomatization in its 1H NMR spectrum. Although compound 7 formally contains the same [(MeBDI)Ca(C6D6)]+ cation as compound VII, like the magnesium-benzene adduct (3), the absence of a coordinating anion ensures that the arene π-system interacts symmetrically with the calcium centre via Ca–C interactions of ca. 2.93 Å (Fig. 3). These distances are comparable to those observed in VII despite the higher level of charge separation enabled by the use of the less coordinating aluminate anion. Reactions between trityl derivatives of weakly coordinating anions and sterically demanding organophosphines have previously been observed to result in nucleophilic attack at a position para to the central trityl carbon and generation of cylcohexadienyl derivatives.63 A similar process was inferred to have provided compound 8, which was confirmed as the product of bis(trimethylsilyl)amide para-phenyl addition by its independent synthesis through reaction of KN(SiMe3)2 and the trityl aluminate. The course of the reaction to provide compounds 7 and 8 was, thus, deduced to proceed as shown in Scheme 3.
 | ||
| Fig. 3 ORTEP representation of the cationic component of compound 7 (30% probability ellipsoids). Hydrogen atoms have been removed for clarity. Selected bond lengths (Å) and angles (°): Ca1–N1 2.239(3), Ca1–C16 2.932(5), Ca1–C17 2.935(5), Ca1–C18 2.932(5), N1′–Ca1–N1 76.26(14), N1–Ca1–C16 139.39(18), N1′–Ca1–C16 129.46(17), N1–Ca1–C17 166.05(17), N1′–Ca1–C17 114.84(14), N1–Ca1–C18 155.76(18), N1′–Ca1–C18 118.74(16). Symmetry operations used to generate equivalent atoms ′ 1 − x, +y, 1 − z. | ||
 | ||
| Scheme 3 Synthesis of compounds 7 and 8. | ||
Calculations were carried out on compound 7 as well as on the congeneric series of [(MeBDI)Ae(C7H8)]+ heavier alkaline earth-toluene cations (Ae = Mg, Ca, Sr, Ba). The geometries were optimised at the DFT level (B3PW91) including dispersion effects (D3BJ), a methodology which has previously proven its ability to describe subtle interactions in alkaline earth metal complexes.64 The optimised geometry of complex 7 (opt. 7) corresponded closely with the experimentally observed structure and provided individual Ca–C(benzene) distances of 2.95 Å (versus ∼2.93 Å experimentally). This further demonstrates the appropriateness of the computational approach. Analysis of the bonding within opt. 7, indicated that the arene–Ca interaction (HOMO−9) is, primarily, a consequence of significant π donation from the aromatic benzene HOMO into an empty 3d orbital at the calcium centre (Fig. 4). The consequent disruption to the π system can be quantified by analysing the Natural Bonding Orbital (NBO) for the benzene fragment. The total Natural charge of this fragment is +0.13, indicating a depletion of density that is mainly derived from the π system (average occupancy of 1.64e).
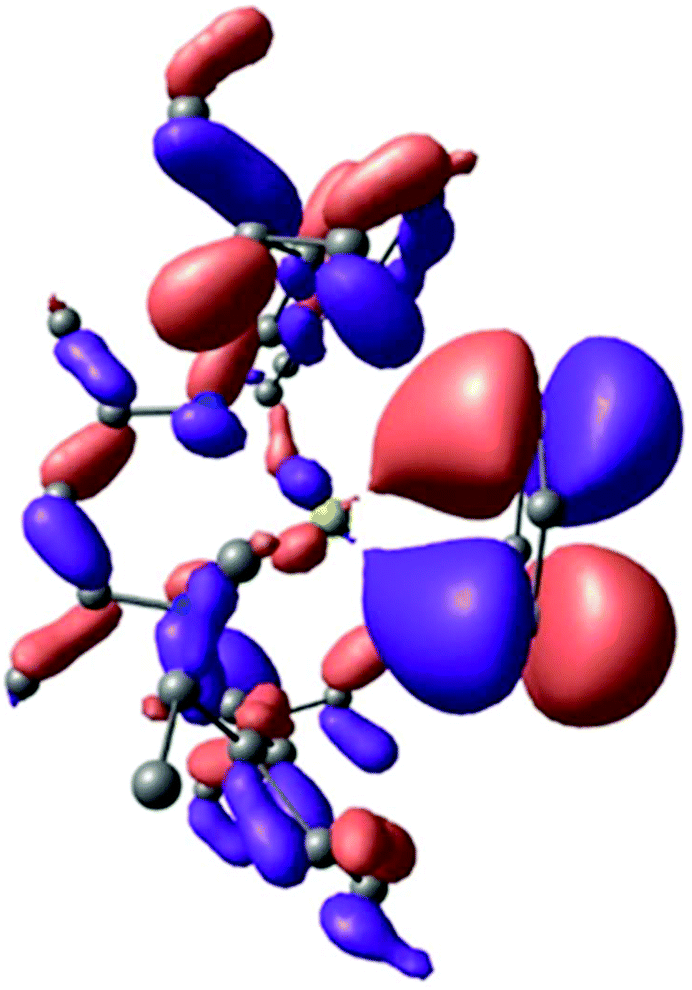 | ||
| Fig. 4 HOMO−9 molecular orbital of opt. 7 indicating the ligand to metal π-donation. | ||
A similar study was carried out on the Ae–toluene cations. While the interaction with Mg was found to be largely electrostatic in origin (Fig. S29a†), the HOMO−8 of all three heavier alkaline earth cations, [(MeBDI)Ae(C7H8)]+ (Ae = Ca, Sr, Ba), exhibited a similar π-symmetric interaction between the toluene π-system and a d orbital of the alkaline earth centre (Fig. S29b†). The percentage of the d contribution in this HOMO−8 orbital was found to decrease from 8.5% (Ca) to 5.3% (Sr) and 2.9% (Ba) in line with the expected increase of d orbital energy from the 3rd to the 5th period, mainly because of relativistic effects. Although the invocation of (n − 1)d orbital participation in the bonding of heavier alkaline earth elements has been somewhat controversial,65 their radial maxima have been calculated to lie in the same regions as the outermost core (n − 1)p orbitals.66 Notably, backbonding interactions from the 5d orbitals of barium have also very recently been implicated in the red shifted CO vibrations which result from co-condensation of laser ablated barium atoms with CO/Ne mixtures.67 Despite the significant variation in d orbital participation, NBO analysis of all three [(MeBDI)Ae(C7H8)]+ (Ae = Ca, Sr, Ba) cations indicated that the total Natural charge of the toluene fragment is +0.11, consistent with minor but potentially significant depletion of π-electron density. Although these results contrast with those of Harder and co-workers, a different functional was used in the two cases and it may be anticipated that all three heavier alkaline earth centres may be similarly implemented to activate arenes toward otherwise unfavourable nucleophilic attack.
Phosphine adducts
Treatment of two-phase solutions of compounds 3 and 5 in toluene-d8 with stoichiometric equivalents of Ph3P induced minor but significant downfield shifts in the resultant 31P{1H} NMR spectra in comparison to the resonant frequency of the pure phosphine (δ −5.30 ppm). This solution-based evidence for the generation of the respective magnesium phosphine coordination complexes 9 (δ −4.94 ppm) and 10 (δ −4.61 ppm) (Scheme 4) was confirmed by the isolation of colourless crystals of both compounds by slow diffusion of hexane into the reaction mixtures. The results of the respective single crystal X-ray diffraction analyses are shown in Fig. 5a and b and establish that the monodentate phosphine coordinates in a terminal fashion to the magnesium centres in both complexes. | ||
| Scheme 4 Synthesis of compounds 9 and 10. | ||
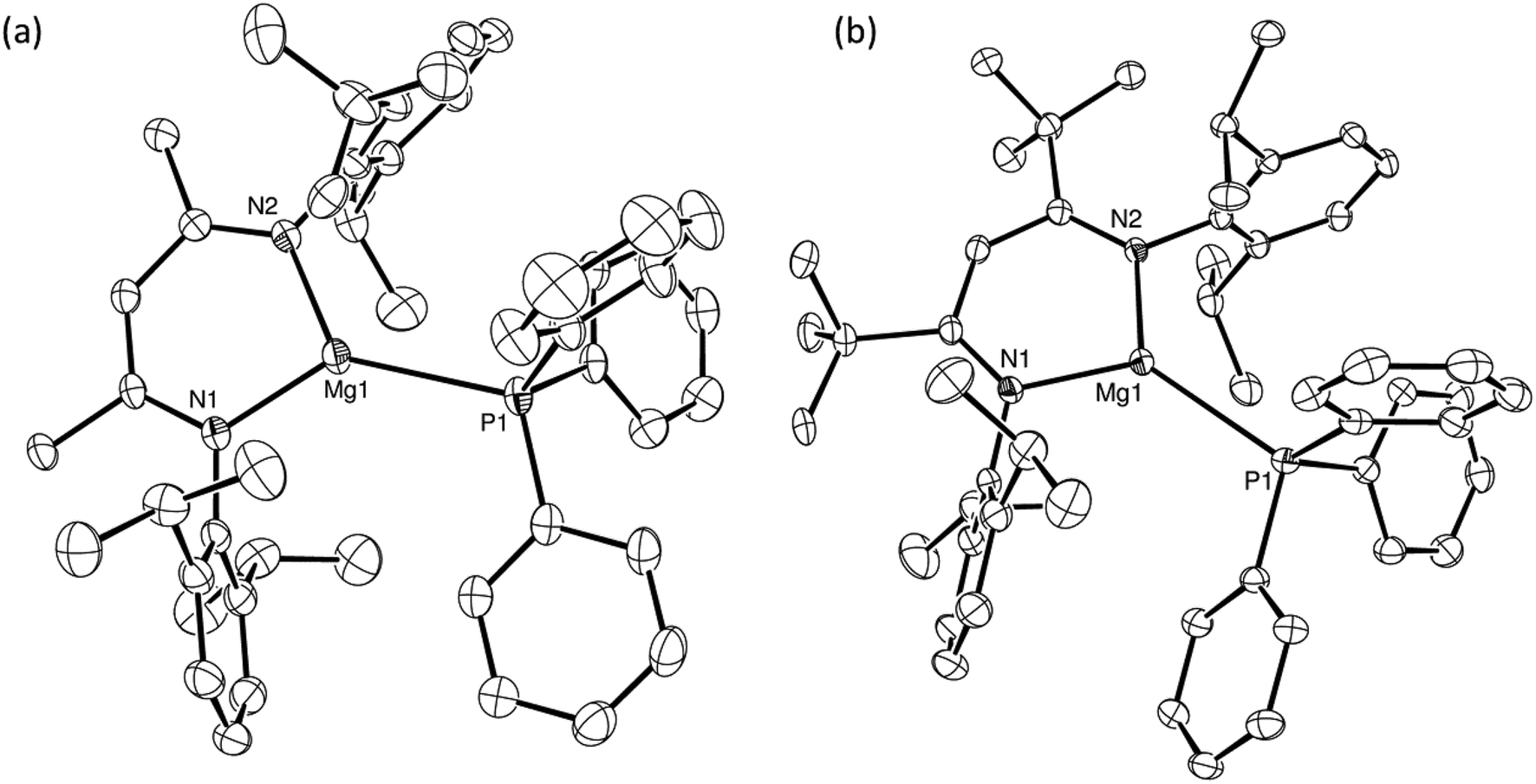 | ||
| Fig. 5 ORTEP representations (25% probability ellipsoids) of the cationic components in (a) compound 9 and (b) compound 10. Hydrogen atoms throughout, plus the minor disordered components of the carbon atoms in the PPh3 ligand of compound 9 are omitted for clarity. Selected bond lengths (Å) and angles (°): 9: P1–Mg1 2.5972(13), Mg1–N1 1.977(3), Mg1–N2 1.983(3), N1–Mg1–P1 134.47(10), N2–Mg1–P1 127.44(9), N1–Mg1–N2 97.51(12); 10: P1–Mg1 2.6805(12), Mg1–N1 1.989(2), Mg1–N2 1.988(2), N1–Mg1–P1 130.37(8), N2–Mg1–P1 130.96(7), N2–Mg1–N1 98.25(10). | ||
The magnesium centres of both compounds 9 and 10 are effectively co-planar with the delocalised β-diketiminate ligands, lying only 0.064 and 0.078 Å out of the respective least squares planes defined by the C3N2 chelate. Replacement of the methyl groups of the supporting ligand of 9 by the bulkier tert-butyl substituents of 10 induces only minor adjustments to the distorted trigonal planar magnesium coordination geometry. The consequently increased steric constraints imposed on the coordination environment of 10, however, are reflected in a significant elongation of the Mg–P distance from 2.5972(13) Å to 2.6805(12) Å. Consistent with the formal positive charges and the lower coordination numbers of the magnesium cations, however, both of these bond lengths are notably shorter than those observed in compounds IV [2.770(1), 2.761(1) Å]36 and V [2.65(1), 2.66(1) Å]37 and are more typical of the Mg–P separations reported for terminal primary and secondary magnesium phosphide derivatives such as [Mg(PHPh)2(TMEDA)] [2.592(5), 2.587(5) Å]25 and [(MeBDI)MgPPh2(THF)] [2.531(3) Å],35 the latter of which comprises the identical β-diketiminate ligand as that employed in the synthesis of compound 9.
Density functional theory (DFT) calculations (B3PW91) were carried out to assess the nature of the Mg–P interaction of compound 10. The optimised geometry of opt. 10 accurately replicates experimentally observed structure, providing a Mg–P distance of 2.65 Å. Analysis of the magnesium to phosphorus bonding (HOMO−5, Fig. 6) indicated that the interaction may be considered as largely electrostatic in origin through what is an effective polarisation of the phosphine lone pair to the [(t-BuBDI)Mg]+ cation.
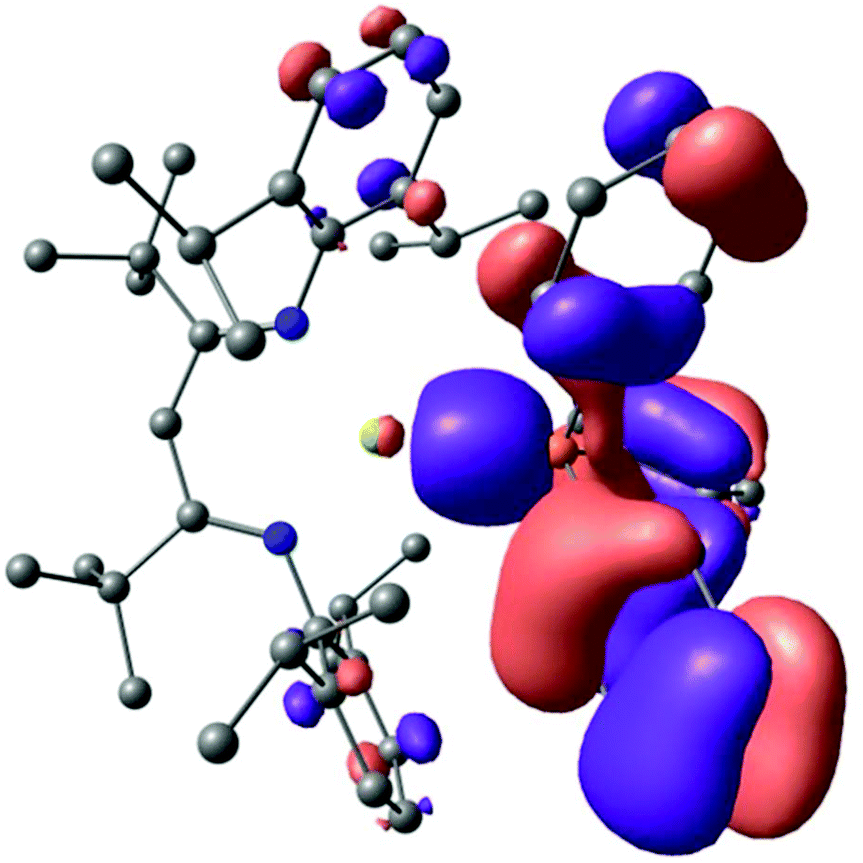 | ||
| Fig. 6 HOMO−5 molecular orbital of opt. 10 highlighting the polarisation of the phosphine lone pair electron density to magnesium. | ||
Conclusions
In conclusion, we have shown that generation of β-diketiminato magnesium and calcium cations in conjunction with the weakly coordinating [Al{OC(CF3)3}4]− anion in benzene or toluene, allows the isolation of completely charge separated η6 adducts in which the polyhapto coordination of the arene ligand is unperturbed by any interaction with the anion. DFT calculations implicate minor but potentially significant 3d orbital participation in the binding to calcium. The magnesium cations react with PPh3 to allow the isolation of the first molecular species in which magnesium is coordinated by a monodentate phosphine base. While the Mg–P binding is effectively electrostatic, it has not escaped our attention that the combination of the hard electrophilic magnesium cations of compounds 9 and 10 with the softer basic PPh3 is reminiscent of the combinations of p-block derivatives that can give rise to so-called ‘frustrated’ Lewis pair reactivity with small molecule substrates. We are, thus, continuing investigate the implications of these observations and to explore the properties of these outwardly simple highly electrophilic molecules.Experimental section
All manipulations were carried out using standard Schlenk line and glovebox techniques under an inert atmosphere of argon. NMR experiments were conducted in J. Young tap NMR tubes made up and sealed in a Glovebox. NMR spectra were collected at 298 K on an Agilent ProPulse spectrometer operating at 500 MHz (1H), 126 MHz (13C), 125.8 MHz (31P) and 470 MHz (19F) and referenced relative to residual solvent resonances. Microanalysis were performed by Mr S. Boyer of London Metropolitan Enterprises. C6D6, tol-d8 and THF-d8 were purchased from Fluorochem Ltd and Sigma-Aldrich Ltd and dried over molten potassium before distilling under argon and storing over molecular sieves in the glovebox. 1,4-Difluorobenzene and triphenylphosphine (PPh3) were purchased from Sigma-Aldrich Ltd and triphenylcarbenium tetrakis(perfluoro-tert-butoxy)aluminate ([Ph3C][Al(OC(CF3)3)4]) from Ionic Liquids Technologies GmbH and used without further purification. [(MeBDI)MgnBu], [(t-BuBDI)MgnBu] and [(MeBDI)CaN(SiMe3)2] (MeBDI = HC{(Me)CN(Dipp)}2, t-BuBDI = HC{(tBu)CN(Dipp)}2, Dipp = 2,6-iPr2C6H3) were synthesised by literature procedures.68–70Synthesis of [(MeBDI)Mg(C6D5CD3)][Al(OC(CF3)3)4] (3)
[(MeBDI)MgnBu] (30 mg, 0.060 mmol) and [Ph3C][Al(OC(CF3)3)4] (73 mg, 0.060 mmol) were dissolved in toluene-d8 (0.5 mL) in a sealed J. Young NMR tube, with the appearance of two immiscible phases which were left overnight at room temperature. Colourless crystals of 3 suitable for X-ray analysis were formed by slow diffusion of hexane into the reaction mixture. Yield: 78 mg, 90%. 1H NMR (500 MHz, tol-d8, 298 K): Isomer A (50%): δ 7.15–6.93 (6H, CH Dipp), 4.55 (s broad, 1H, CH{C(Me)NDipp}2), 2.21 (hept broad, 3JHH = 6.4 Hz, 4H, CH(CH3)2), 1.09 (s broad, 6H, CH{C(CH3)NDipp}2), 1.01 (d broad, 3JHH = 6.2 Hz, 12H, CH(CH3)2), 0.85 ppm (d broad, 3JHH = 5.9 Hz, 12H, CH(CH3)2); Isomer B (50%): δ 7.37–6.71 (6H, CH Dipp), 4.55 (s, 1H, CH{C(Me)NDipp}2), 2.27 (hept, 3JHH = 6.9 Hz, 4H, CH(CH3)2), 1.31 (s, 6H, CH{C(CH3)NDipp}2), 1.08 (d, 3JHH = 6.9 Hz, 12H, CH(CH3)2), 0.89 ppm (d, 3JHH = 6.7 Hz, 12H, CH(CH3)2). 19F{1H} NMR (470 MHz, tol-d8, 298 K): Isomer A (50%): δ −75.09 ppm (s); Isomer B (50%): δ −74.90 ppm (s). Elemental analysis (%). Found: C 37.83, H 3.20, N 2.21. Calculated for C45H41AlF36O4MgN2 ([(MeBDI)Mg][Al(OC(CF3)3)4]): C 38.36, H 2.93, N 1.99.Synthesis of [(MeBDI)Mg(C4D8O)][Al(OC(CF3)3)4] (3·THF)
[(MeBDI)Mg(C6D5CD3)][Al(OC(CF3)3)4] (30 mg, 0.020 mmol) was dissolved in THF-d8 (0.5 mL) in a sealed J. Young NMR tube. The solvent was evaporated to dryness and the residue washed with hexane (3 × 5 mL) to afford a pale yellow solid. Yield: 29 mg, 98%. 1H NMR (500 MHz, THF-d8, 298 K): δ 7.29–7.05 (6H, CH Dipp), 5.16 (s, 1H, CH{C(Me)NDipp}2), 3.05 (hept, 3JHH = 6.9 Hz, 4H, CH(CH3)2), 1.82 (s, 6H, CH{C(CH3)NDipp}2), 1.24 ppm (d, 3JHH = 6.9 Hz, 24H, CH(CH3)2). 13C{1H} NMR (126 MHz, THF-d8, 298 K): δ 172.7 (s, NC(Me)), 144.0 (s, C–iPr), 143.0 (s, N–C Dipp), 127.3 (s, p-CH Dipp), 125.2 (s, m-CH Dipp), 123.6 (s, p-CH Dipp), 96.0 (s, CH{C(Me)NDipp}2), 29.3 (s, CH(CH3)2), 25.2 (s, CH3 Dipp), 24.6 (s, CH3 Dipp), 24.5 ppm (s, CH–CH3 Dipp). 19F{1H} NMR (470 MHz, THF-d8, 298 K): δ −74.03 ppm (s). Elemental analysis (%). Found: C 39.32, H 3.37, N 1.98. Calculated for C49H49AlF36O5MgN2: C 39.73, H 3.33, N 1.89.Synthesis of [(MeBDI)Mg(C6D6)][Al(OC(CF3)3)4] (4)
[(MeBDI)MgnBu] (30 mg, 0.060 mmol) and [Ph3C][Al(OC(CF3)3)4] (73 mg, 0.060 mmol) were dissolved in C6D6 (0.5 mL) in a sealed J. Young NMR tube, with the appearance of two immiscible phases which were left overnight at room temperature. Colourless crystals of 4 suitable for X-ray analysis were formed by slow diffusion of hexane into the reaction mixture. Yield: 81 mg, 91%. 1H NMR (500 MHz, C6D6, 298 K): Isomer A (70%): δ 7.37–6.71 (6H, CH Dipp), 4.55 (s, 1H, CH{C(Me)NDipp}2), 2.17 (hept, 3JHH = 7.4 Hz, 4H, CH(CH3)2), 1.29 (s, 6H, CH{C(CH3)NDipp}2), 0.98 (d, 3JHH = 6.3 Hz, 12H, CH(CH3)2), 0.84 ppm (d, 3JHH = 6.4 Hz, 12H, CH(CH3)2); Isomer B (30%): δ 7.37–6.71 (6H, CH Dip), 4.53 (broad, 1H, CH{C(Me)NDipp}2), 2.24 (broad, 4H, CH(CH3)2), 1.29–1.27 (broad, 6H, CH{C(CH3)NDipp}2), 1.07–1.03 (broad, 12H, CH(CH3)2), 0.89–0.86 ppm (broad, 12H, CH(CH3)2). 19F{1H} NMR (470 MHz, C6D6, 298 K): Isomer A (70%): δ −74.83 ppm (s); Isomer B (30%): δ −75.08 ppm (s). Elemental analysis (%). Found: C 37.93, H 3.10, N 2.11. Calculated for C45H41AlF36O4MgN2 ([(MeBDI)Mg][Al(OC(CF3)3)4]): C 38.36, H 2.93, N 1.99.Synthesis of [(t-BuBDI)Mg(C6D5CD3)][Al(OC(CF3)3)4] (5)
[(t-BuBDI)MgnBu] (30 mg, 0.051 mmol) and [Ph3C][Al(OC(CF3)3)4] (62 mg, 0.051 mmol) were dissolved in toluene-d8 (0.5 mL) in a sealed J. Young NMR tube, with the appearance of two immiscible phases that were left overnight at room temperature. Storage of this mixture at room temperature with a third layer of hexane yielded colourless crystals of 5 suitable for crystallographic analysis. Yield: 65 mg, 80%. 1H NMR (500 MHz, C6D6, 298 K): δ 7.05 (6H, CH Dipp), 5.19 (s, 1H, CH{C(tBu)NDipp}2), 3.30 (hept, 3JHH = 6.9 Hz, 4H, CH(CH3)2), 1.32 (d, 3JHH = 5.1 Hz, 30H, CH(CH3)2 and C–CH3), 0.96 ppm (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2). 19F{1H} NMR (470 MHz, tol-d8, 298 K): δ −74.17 ppm (s). Elemental analysis (%). Found: C 44.61, H 4.08, N 1.76. Calculated for C58H61AlF36O4MgN2: C 43.94, H 3.88, N 1.77.Synthesis of [(t-BuBDI)Mg(C4D8O)][Al(OC(CF3)3)4] (5·THF)
In a sealed J. Young NMR tube, [(t-BuBDI)Mg(C6D5CD3)][Al(OC(CF3)3)4] (30 mg, 0.019 mmol) was dissolved in THF-d8 (0.5 mL). The solvent was evaporated to dryness and the residue washed with hexane (3 × 5 mL) to afford a pale yellow solid. Yield: 28 mg, 96%. 1H NMR (500 MHz, THF-d8, 298 K): δ 7.22–7.16 (5H, m,p-CH Dipp), 7.06 (t, 3JHH = 7.8 Hz, 1H, p-CH Dipp), 5.65 (s, 1H, CH{C(tBu)NDipp}2), 3.12 (hept, 3JHH = 6.8 Hz, 4H, CH(CH3)2), 1.39 (d, 3JHH = 6.7 Hz, 12H, CH(CH3)2), 1.23 (d, 3JHH = 6.9 Hz, 12H, CH(CH3)2), 1.21 ppm (s, 18H, C–CH3). 13C{1H} NMR (126 MHz, THF-d8, 298 K): δ 181.5 (s, NC(tBu)), 145.4 (s, N–C Dipp), 142.5 (s, C–iPr), 126.8 (s, p-CH Dipp), 125.4 (s, m-CH Dipp), 123.4 (s, p-CH Dipp), 97.4 (s, CH{C(tBu)NDipp}2), 45.7 (s, CtBu), 33.3 (s, CH3tBu), 29.1 (s, CH(CH3)2), 26.6 (s, CH3 Dipp), 24.7 ppm (s, CH3 Dipp). 19F{1H} NMR (470 MHz, THF-d8, 298 K): δ −79.98 ppm (s). Elemental analysis (%). Found: C 42.11, H 4.03, N 1.76. Calculated for C55H61AlF36O5MgN2: C 42.20, H 3.93, N 1.79.Synthesis of [(MeBDI)Mg(C6D4F2)3][Al(OC(CF3)3)4] (6)
[(MeBDI)MgnBu] (30 mg, 0.060 mmol) and [Ph3C][Al(OC(CF3)3)4] (73 mg, 0.060 mmol) were dissolved in 1,4-difluorobenzene (0.5 mL) with the appearance of two immiscible phases and the resulting mixture vigourously stirred for one minute and left overnight at room temperature. Colourless crystals of 6 suitable for X-ray crystallography were grown by slow diffusion of hexane into the reaction mixture. Yield: 94 mg, 89%. 19F{1H} NMR (470 MHz, C6D6, 298 K): δ −74.85 (s), 119.58 (q) ppm. Elemental analysis (%). Found: C 43.53, H 3.08, N 1.66. Calculated for C63H53AlF42O4MgN2: C 43.21, H 3.05, N 1.60.Synthesis of [(MeBDI)Ca(C6D6)][Al(OC(CF3)3)4] (7)
[(MeBDI)CaN(SiMe3)2] (30 mg, 0.049 mmol) and [Ph3C][Al(OC(CF3)3)4] (59 mg, 0.049 mmol) were dissolved in C6D6 (0.5 mL) in a sealed J. Young NMR tube, with the appearance of two immiscible phases. Colourless crystals of 7 suitable for X-ray analysis were formed by slow diffusion of hexane into the reaction mixture. Yield: 59 mg, 76%. 19F{1H} NMR (470 MHz, C6D6, 298 K): δ −74.80 ppm (s). Elemental analysis (%). Found: C 37.45, H 2.87, N 2.20. Calculated for C45H41AlCaF36N2O4 ([(BDI)Ca][Al(OC(CF3)3)4]): C 37.30, H 2.85, N 1.93.Synthesis of [(MeBDI)Ca(C4D8O)3][Al(OC(CF3)3)4] (7·THF)
[(MeBDI)Ca(C6D6)][Al(OC(CF3)3)4] (30 mg, 0.049 mmol) was dissolved in THF-d8 (0.5 mL) in a sealed J. Young NMR tube. Colourless crystals of 7·THF suitable for X-ray analysis were formed by slow diffusion of hexane into the reaction mixture. Yield: 29 mg, 94%. 1H NMR (500 MHz, THF-d8, 298 K): δ 7.23 (d, 3JHH = 7.7 Hz, 4H, o,m-CH Dipp), 7.15 (t, 3JHH = 7.7 Hz, 2H, p-CH Dipp), 4.98 (s, 1H, CH{C(Me)NDipp}2), 3.10 (hept, 3JHH = 6.9 Hz, 4H, CH(CH3)2), 1.73 (s, 6H, CH{C(CH3)NDipp}2), 1.24 ppm (2d, 3JHH = 6.9 Hz, 24H, CH(CH3)2). 13C{1H} NMR (126 MHz, THF-d8, 298 K): δ 168.0 (s, NC(Me)), 147.5 (s, N–C Dipp), 142.6 (s, C–iPr), 125.9 (s, p-CH Dipp), 125.0 (s, o,m-CH Dipp), 95.6 (s, CH{C(Me)NDipp}2), 29.3 (s, CH(CH3)2), 25.1 (s, CH{C(CH3)NDipp}2), 25.0 (s, CH3 Dipp), 24.7 ppm (s, CH3 Dipp). 19F{1H} NMR (470 MHz, THF-d8, 298 K): δ −74.02 ppm (s). Elemental analysis (%). Found: C 38.83, H 3.27, N 2.01. Calculated for C49H49AlCaF36O5N2 ([(BDI)Ca(C4D8O)][Al(OC(CF3)3)4]): C 39.32, H 3.30, N 1.87.Synthesis of 8
[Ph3C][Al(OC(CF3)3)4] (91 mg, 0.075 mmol) and KN(SiMe3)2 (15 mg, 0.075 mmol) were dissolved in C6D6 (0.5 mL) in a sealed J. Young NMR tube and vigorously stirred for 1 minute, forming a pink solution. The solvent was evaporated to dryness and the resulting residue extracted in hexane and filtered. 1H NMR (400 MHz, C6D6, 298 K): δ 7.27–7.23 (4H, o-CH Ph), 7.14–7.09 (4H, m-CH Ph), 7.07–7.02 (2H, p-CH Ph), 6.61 (dd, 3JHH = 10.2, 5JHH = 2.5 Hz, 2H, HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCHN), 5.73 (dd, 3JHH = 10.2, 4JHH = 3.8 Hz, 2H, HC–CHN), 4.54 (3JHH = 10.3, 2.5 Hz, 1H, HC–N), 0.20 ppm (s, 18H, CH3). 13C{1H} NMR (126 MHz, C6D6, 298 K): δ 142.3 (s, Cipso Ph), 140.4 (s, CPh2), 135.5 (s, C–CHN), 131.2 (s, o-CH Ph), 128.4 (s, m-CH Ph), 127.5 (s, p-CH Ph), 126.5 (s, CC–CHN), 53.2 (s, HCN), 3.3 ppm (broad, SiCH3). 29Si{1H} NMR (99.4 MHz, C6D6, 298 K): δ 6.21 ppm.
CHCHN), 5.73 (dd, 3JHH = 10.2, 4JHH = 3.8 Hz, 2H, HC–CHN), 4.54 (3JHH = 10.3, 2.5 Hz, 1H, HC–N), 0.20 ppm (s, 18H, CH3). 13C{1H} NMR (126 MHz, C6D6, 298 K): δ 142.3 (s, Cipso Ph), 140.4 (s, CPh2), 135.5 (s, C–CHN), 131.2 (s, o-CH Ph), 128.4 (s, m-CH Ph), 127.5 (s, p-CH Ph), 126.5 (s, CC–CHN), 53.2 (s, HCN), 3.3 ppm (broad, SiCH3). 29Si{1H} NMR (99.4 MHz, C6D6, 298 K): δ 6.21 ppm.
Synthesis of [(MeBDI)MgPPh3][Al(OC(CF3)3)4] (9)
In a sealed J. Young NMR tube, [(MeBDI)Mg(C6D5CD3)][Al(OC(CF3)3)4] (81 mg, 0.054 mmol) was dissolved in toluene-d8 (0.5 mL) and vigorously stirred for 1 minute with the appearance of two immiscible phases. The resulting mixture was subsequently treated with one equivalent of triphenylphosphine (14 mg, 0.054 mmol) and vigorously stirred for another minute. Colourless crystals of 9 suitable for X-ray analysis were formed by slow diffusion of hexane into the reaction mixture. Yield: 66 mg, 74%. 1H NMR (500 MHz, tol-d8, 298 K): δ 7.14–6.89 (21H, CH Dipp and PPh3), 4.95 (s, 1H, CH{C(Me)NDipp}2), 2.71 (hept, 3JHH = 6.9 Hz, 4H, CH(CH3)2), 1.51 (s, 6H, CH{C(CH3)NDipp}2), 1.04 (d, 3JHH = 6.9 Hz, 12H, CH(CH3)2), 0.62 ppm (d, 3JHH = 6.9 Hz, 12H, CH(CH3)2). 13C{1H} NMR (126 MHz, tol-d8, 298 K): δ 173.4 (s, NC(Me)), 144.4 (s, C–iPr), 142.0 (s, N–CMe), 141.8 (s, N–C Dipp), 133.8–125.2 (CH Dipp and Ph), 97.4 (s, CH{C(Me)NDipp}2), 29.3 (s, CH(CH3)2), 24.5 (s, CH3 Dipp), 23.7 (s, CH–CH3 Dipp), 23.1 ppm (s, CH3 Dipp). 31P{1H} NMR (125.8 MHz, tol-d8, 298 K): δ −4.9 ppm. 19F{1H} NMR (470 MHz, tol-d8, 298 K): δ −74.84 ppm. Elemental analysis (%). Found: C 44.97, H 3.49, N 1.63. Calculated for C63H56AlF36O4MgN2P: C 45.27, H 3.38, N 1.68.Synthesis of [(t-BuBDI)MgPPh3][Al(OC(CF3)3)4] (10)
[(t-BuBDI)Mg(C6D5CD3)][Al(OC(CF3)3)4] (82 mg, 0.052 mmol) was dissolved in toluene-d8 (0.5 mL) in a sealed J. Young NMR tube and vigorously stirred for 1 minute, with the appearance of two immiscible phases. The resulting mixture was subsequently treated with one equivalent of triphenylphosphine (14 mg, 0.052 mmol) and vigorously stirred for another minute. Colourless crystals of 10 suitable for X-ray crystallography were grown by slow diffusion of hexane into the reaction mixture. Yield: 70 mg, 77%. 31P{1H} NMR (125.8 MHz, tol-d8, 298 K): −4.6 ppm. 19F{1H} NMR (470 MHz, tol-d8, 298 K): δ −74.84 ppm. Elemental analysis (%). Found: C 46.61, H 4.08, N 1.76. Calculated for C69H68AlF36O4MgN2P: C 47.21, H 3.90, N 1.60.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank the EPSRC (UK) for funding this research (grant number: EP/N014456/1).References
- D. A. Dougherty, Science, 1996, 271, 163–168 CrossRef PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2013, 113, 2100–2138 CrossRef PubMed.
- S. E. Rodriguez-Cruz and E. R. Williams, J. Am. Soc. Mass Spectrom., 2001, 12, 250–257 CrossRef PubMed.
- S. Tsuzuki, T. Uchimaru and M. Mikami, J. Phys. Chem. A, 2003, 107, 10414–10418 CrossRef.
- A. S. Reddy and G. N. Sastry, J. Phys. Chem. A, 2005, 109, 8893–8903 CrossRef PubMed.
- A. S. Reddy, H. Zipse and G. N. Sastry, J. Phys. Chem. B, 2007, 111, 11546–11553 CrossRef PubMed.
- T. Rocha-Rinza and J. Hernandez-Trujillo, Chem. Phys. Lett., 2006, 422, 36–40 CrossRef.
- T. C. Dinadayalane, A. Hassan and J. Leszczynski, J. Mol. Struct., 2010, 976, 320–323 CrossRef.
- T. C. Dinadayalane, A. Hassan and J. Leszczynski, Theor. Chem. Acc., 2012, 131, 1131 Search PubMed.
- A. Mirchi, N. Sizochenko, T. Dinadayalane and J. Leszczynski, J. Phys. Chem. A, 2017, 121, 8927–8938 CrossRef PubMed.
- P. Kadlubanski, K. Calderon-Mojica, W. A. Rodriguez, D. Majumdar, S. Roszak and J. Leszczynski, J. Phys. Chem. A, 2013, 117, 7989–8000 CrossRef PubMed.
- C. Loh, S. Seupel, H. Gorls, S. Krieck and M. Westerhausen, Organometallics, 2014, 33, 1480–1491 CrossRef.
- C. Loh, S. Seupel, A. Koch, H. Gorls, S. Krieck and M. Westerhausen, Dalton Trans., 2014, 43, 14440–14449 RSC.
- S. C. Rosca, C. Dinoi, E. Caytan, V. Dorcet, M. Etienne, J. F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2016, 22, 6505–6509 CrossRef PubMed.
- S. C. Rosca, E. Caytan, V. Dorcet, T. Roisnel, J. F. Carpentier and Y. Sarazin, Organometallics, 2017, 36, 1269–1277 CrossRef.
- W. D. Buchanan, D. G. Allis and K. Ruhlandt-Senge, Chem. Commun., 2010, 46, 4449–4465 RSC.
- S. C. Rosca, V. Dorcet, T. Roisnel, J. F. Carpentier and Y. Sarazin, Dalton Trans., 2017, 46, 14785–14794 RSC.
- S. C. Rosca, V. Dorcet, J. F. Carpentier and Y. Sarazin, Inorg. Chim. Acta, 2018, 475, 59–64 CrossRef.
- H. Schumann, S. Schutte, H. J. Kroth and D. Lentz, Angew. Chem., Int. Ed., 2004, 43, 6208–6211 CrossRef PubMed.
- B. Freitag, H. Elsen, J. Pahl, G. Ballmann, A. Herrera, R. Dorta and S. Harder, Organometallics, 2017, 36, 1860–1866 CrossRef.
- R. A. Williams and T. P. Hanusa, J. Am. Chem. Soc., 1990, 112, 2454–2455 CrossRef.
- L. Bonomo, E. Solari, R. Scopelliti and C. Floriani, Chem. – Eur. J., 2001, 7, 1322–1332 CrossRef.
- H. Lehmkuhl, K. Mehler, R. Benn, A. Rufinska and C. Kruger, Chem. Ber., 1986, 119, 1054–1069 CrossRef.
- E. Hey, L. M. Engelhardt, C. L. Raston and A. H. White, Angew. Chem., Int. Ed. Engl., 1987, 26, 81–82 CrossRef.
- M. Westerhausen and W. Schwarz, Z. Anorg. Allg. Chem., 1994, 620, 304–308 CrossRef.
- M. Westerhausen, M. H. Digeser, H. Noth, T. Seifert and A. Pfitzner, J. Am. Chem. Soc., 1998, 120, 6722–6725 CrossRef.
- M. Westerhausen, M. Krofta and A. Pfitzner, Inorg. Chem., 1999, 38, 598 CrossRef PubMed.
- M. Westerhausen, M. H. Digeser, B. Wieneke, H. Noth and J. Knizek, Eur. J. Inorg. Chem., 1998, 517–521 CrossRef.
- S. Blair, K. Izod and W. Clegg, Inorg. Chem., 2002, 41, 3886–3893 CrossRef PubMed.
- S. Blair, K. Izod, W. Clegg and R. W. Harrington, Eur. J. Inorg. Chem., 2003, 3319–3324 CrossRef.
- M. Gartner, H. Gorls and M. Westerhausen, Inorg. Chem., 2008, 47, 1397–1405 CrossRef PubMed.
- B. M. Day and M. P. Coles, Eur. J. Inorg. Chem., 2010, 5471–5477 CrossRef.
- M. Arrowsmith, M. S. Hill, A. L. Johnson, G. Kociok-Kohn and M. F. Mahon, Angew. Chem., Int. Ed., 2015, 54, 7882–7885 CrossRef PubMed.
- M. J. Taylor, M. P. Coles and J. R. Fulton, Aust. J. Chem., 2015, 68, 635–640 Search PubMed.
- A. Pape, M. Lutz and G. Muller, Angew. Chem., Int. Ed. Engl., 1994, 33, 2281–2284 CrossRef.
- D. E. Gindelberger and J. Arnold, Inorg. Chem., 1994, 33, 6293–6299 CrossRef.
- J. Pahl, S. Brand, H. Elsen and S. Harder, Chem. Commun., 2018, 54, 8685–8688 RSC.
- J. Pahl, H. Elsen, A. Friedrich and S. Harder, Chem. Commun., 2018, 54, 7846–7849 RSC.
- A. S. S. Wilson, M. S. Hill, M. F. Mahon, C. Dinoi and L. Maron, Science, 2017, 358, 1168–1171 CrossRef PubMed.
- B. Liu, V. Dorcet, L. Maron, J. F. Carpentier and Y. Sarazin, Eur. J. Inorg. Chem., 2012, 3023–3031 CrossRef.
- I. Krossing and A. Reisinger, Coord. Chem. Rev., 2006, 250, 2721–2744 CrossRef.
- A. B. A. Rupp and I. Krossing, Acc. Chem. Res., 2015, 48, 2537–2546 CrossRef PubMed.
- T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789–899 RSC.
- J. L. Atwood and J. D. Atwood, Adv. Chem. Ser., 1976, 112–127 CrossRef.
- M. S. Hill, G. Kociok-Koehn and D. J. MacDougall, Inorg. Chem., 2011, 50, 5234–5241 CrossRef PubMed.
- M. D. Anker, M. Arrowsmith, P. Bellham, M. S. Hill, G. Kociok-Kohn, D. J. Liptrot, M. F. Mahon and C. Weetman, Chem. Sci., 2014, 5, 2826–2830 RSC.
- M. D. Anker, M. Arrowsmith, R. L. Arrowsmith, M. S. Hill and M. F. Mahon, Inorg. Chem., 2017, 56, 5976–5983 CrossRef PubMed.
- M. W. Bouwkamp, J. de Wolf, I. D. Morales, J. Gercama, A. Meetsma, S. I. Troyanov, B. Hessen and J. H. Teuben, J. Am. Chem. Soc., 2002, 124, 12956–12957 CrossRef PubMed.
- M. W. Bouwkamp, P. H. M. Budzelaar, J. Gercama, I. D. Morales, J. de Wolf, A. Meetsma, S. I. Troyanov, J. H. Teuben and B. Hessen, J. Am. Chem. Soc., 2005, 127, 14310–14319 CrossRef PubMed.
- F. Basuli, H. Aneetha, J. C. Huffman and D. J. Mindiola, J. Am. Chem. Soc., 2005, 127, 17992–17993 CrossRef PubMed.
- S. D. Pike, M. R. Crimmin and A. B. Chaplin, Chem. Commun., 2017, 53, 3615–3633 RSC.
- O. Eisenstein, J. Milani and R. N. Perutz, Chem. Rev., 2017, 117, 8710–8753 CrossRef PubMed.
- L. J. Radonovich, E. C. Zuerner, H. F. Efner and K. J. Klabunde, Inorg. Chem., 1976, 15, 2976–2981 CrossRef.
- A. K. Renfrew, A. D. Phillips, E. Tapavicza, R. Scopelliti, U. Rothlisberger and P. J. Dyson, Organometallics, 2009, 28, 5061–5071 CrossRef.
- B. R. Jagirdar, K. J. Klabunde, R. Palmer, L. J. Radonovich and T. Williams, Inorg. Chim. Acta, 1996, 250, 317–326 CrossRef.
- A. Khaleel and K. J. Klabunde, Inorg. Chem., 1996, 35, 3223–3227 CrossRef PubMed.
- M. Carrasco, N. Curado, E. Alvarez, C. Maya, R. Peloso, M. L. Poveda, A. Rodriguez, E. Ruiz, S. Alvarez and E. Carmona, Chem. – Eur. J., 2014, 20, 6092–6102 CrossRef PubMed.
- P. G. Williard and Q. Y. Liu, J. Org. Chem., 1994, 59, 1596–1597 CrossRef.
- M. Rauch, S. Ruccolo, J. P. Mester, Y. Rong and G. Parkin, Chem. Sci., 2016, 7, 142–149 RSC.
- H. J. Hao, H. W. Roesky, Y. Q. Ding, C. M. Cui, M. Schormann, H. G. Schmidt, M. Noltemeyer and B. Zemva, J. Fluorine Chem., 2002, 115, 143–147 CrossRef.
- N. L. Lampland, A. Pindwal, S. R. Neal, S. Schlauderaff, A. Ellern and A. D. Sadow, Chem. Sci., 2015, 6, 6901–6907 RSC.
- L. Cabrera, G. C. Welch, J. D. Masuda, P. R. Wei and D. W. Stephan, Inorg. Chim. Acta, 2006, 359, 3066–3071 CrossRef.
- C. N. de Bruin-Dickason, T. Sutcliffe, C. A. Lamsfus, G. B. Deacon, L. Maron and C. Jones, Chem. Commun., 2018, 54, 786–789 RSC.
- M. Kaupp, Angew. Chem., Int. Ed., 2001, 40, 3534–3565 CrossRef PubMed.
- G. Jeung, J. P. Daudey and J. P. Malrieu, Chem. Phys. Lett., 1983, 98, 433–438 CrossRef.
- X. Wu, L. L. Zhao, D. D. Jiang, I. Fernandez, R. Berger, M. F. Zhou and G. Frenking, Angew. Chem., Int. Ed., 2018, 57, 3974–3980 CrossRef PubMed.
- A. P. Dove, V. C. Gibson, P. Hormnirun, E. L. Marshall, J. A. Segal, A. J. P. White and D. J. Williams, Dalton Trans., 2003, 3088–3097 RSC.
- V. Balasanthiran, M. H. Chisholm, K. Choojun, C. B. Durr and P. M. Wambua, Polyhedron, 2016, 103, 235–240 CrossRef.
- M. R. Crimmin, M. S. Hill, P. B. Hitchcock and M. F. Mahon, New J. Chem., 2010, 34, 1572–1578 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the synthetic procedures and relevant NMR spectra, details of the X-ray analyses of compounds 3, 4, 6, 7, 7·THF, 9 and 10, details of the computational analysis and coordinates of the optimised structures (pdf). Crystallographic data have been deposited with the CCDC. CCDC 1838032 (3), 1853497 (4), 1838033 (6), 1838034 (7), 1838035 (7·THF), 1853498 (9) and 1853499 (10). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt03124j |
| This journal is © The Royal Society of Chemistry 2018 |