
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stable Fe(III) phenoxyimines as selective and robust CO2/epoxide coupling catalysts†
Eszter
Fazekas
 ,
Gary S.
Nichol
,
Michael P.
Shaver
and
Jennifer A.
Garden
*
,
Gary S.
Nichol
,
Michael P.
Shaver
and
Jennifer A.
Garden
*
EaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: j.garden@ed.ac.uk
First published on 30th August 2018
Abstract
Three phenoxyimine Fe(III)Cl complexes bearing electronically diverse -Cl, -H or -tBu substituents in the ortho position were synthesised. X-ray crystallographic analysis of the complexes reveals mononuclear structures with pentacoordinate iron centres and trigonal bipyramidal geometries. All three complexes demonstrated excellent catalytic activities towards CO2/epoxide coupling to selectively form cyclic carbonates, with catalyst activity correlating with the electron withdrawing nature of the ortho-substituent (Cl > H > tBu) and thus the Lewis acidity of the metal centre. The chloro-substituted complex displayed remarkable activity in the synthesis of propylene carbonate from propylene oxide and CO2, reaching turnover frequencies (TOF) up to 760 h−1 in the presence of TBABr co-catalyst at 120 °C and 20 bars of CO2 pressure. Importantly, the catalyst is also very robust, functioning with high substrate loading, under air or in the presence of water. The substrate scope was successfully extended to other terminal epoxides including epichlorohydrin (TOF = 900 h−1) and to the more challenging internal epoxide, cyclohexene oxide (TOF = 80 h−1). These are amongst the highest TOF values reported for an iron CO2/epoxide coupling catalyst to date.
Introduction
Catalytic transformations of carbon dioxide (CO2) are an important strategy to valorise this waste gas and potentially reduce the environmental impact of its accumulation in the atmosphere. Epoxide coupling is an effective and atom efficient method to use this inexpensive, renewable C1 building block. Depending on the catalyst system and the reaction conditions used, polycarbonates and/or cyclic carbonates are formed; the latter are applied as high boiling solvents, ion-carriers for batteries, reagents for lignin functionalisation, fuel additives and precursors to polymer synthesis.1–6 The key to CO2/epoxide coupling is to combine high selectivity with high activity, as the synthesis of both cyclic carbonates and polycarbonates requires the catalyst system to overcome the significant activation energy barrier of these transformations.1,7,8Some of the most active catalyst systems comprise homogeneous complexes with a Lewis acidic metal centre (for epoxide coordination and activation) and a nucleophile (for ring opening of the epoxide). Ligand design has enabled the development of highly efficient catalyst systems, including mono- or bi-metallic ligand supported complexes with ammonium or phosphonium salt co-catalysts, or bifunctional catalysts which incorporate the onium salt into the ligand backbone.9–12 One popular ligand framework is the privileged salen ligand scaffold, which has supported many Lewis acidic complexes mediating a wide array of small and large molecule transformations (Scheme 1).13–15 While the ubiquitous salen ligands have shown some success in CO2/epoxide coupling, their bidentate phenoxyimine (half salen) analogues have been underexplored.16 Breaking the bridge between the phenoxy groups creates a more flexible coordination environment, enabling the balance between the steric hindrance and the accessibility of the metal centre to be better fine-tuned (Scheme 1). Previous findings in the field suggest that the coordination mode of the metal centre is a key feature in allowing the efficient conversion of sterically challenging internal epoxides to cyclic carbonates.1
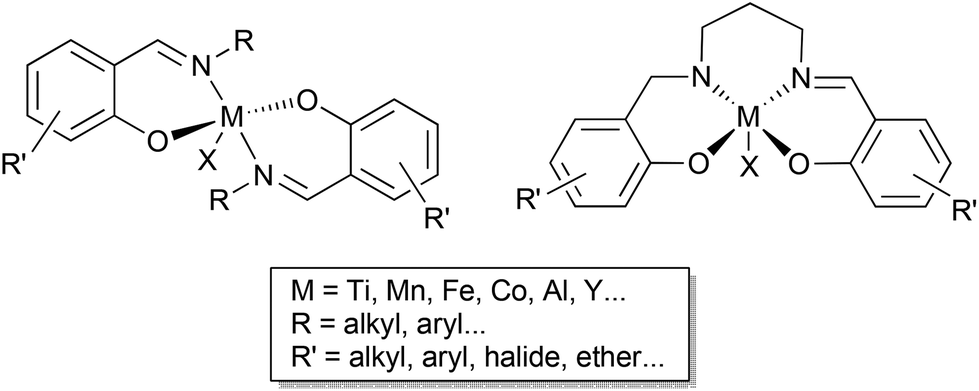 | ||
| Scheme 1 General structure of a bis(phenoxyimine) complex vs. a salen complex. | ||
We have had a longstanding interest in iron-mediated catalysis thanks to its abundance, low cost, and low toxicity.17,18 In particular, Fe3+ compounds offer a user-friendly route into catalysis, due to their air- and moisture-stability which enables convenient synthesis and handling. Only a few examples of mono- or bi-metallic Fe(III) complexes have been reported as catalysts for CO2/epoxide couplings and in most cases high catalyst loadings were required to achieve good conversions.16,19–27 Uniting these joint ligand and metal benefits, we targeted a series of phenoxyimine Fe(III) chloride complexes as robust and flexible catalysts for epoxide/CO2 coupling.
Results and discussion
Three phenoxyimine ligand precursors were targeted, bearing ortho-substituents of H (L1, Scheme 2), tBu (L2) or Cl (L3). Accordingly, all three ligands were synthesised in excellent yields (95–99%) via the straightforward condensation of methyl amine and mono-substituted salicylaldehydes (Scheme 2).28 Confirming the synthesis of L1, L2 and L3, no aldehyde resonances were present in the 1H NMR spectra, and diagnostic imine resonances were observed (CDCl3 solvent, L1, δ = 8.33; L2, δ = 8.35; L3, δ = 8.30). Complexes C1–C3 were synthesised through deprotonation of pro-ligands L1–L3 using NaH (1.1 eq.) to form the corresponding Na-phenolates. Subsequent reaction with anhydrous FeCl3 (0.5 eq.) in THF solvent at room temperature gave an immediate colour change from yellow to dark purple, which indicated the formation of the bis-ligated Fe(III) chloride complexes C1–C3. After purification via filtration to remove residual NaCl, the products were isolated in high yields (74–98%, Scheme 2) as dark purple/brown solids. As the paramagnetically shifted 1H NMR spectra of C1–C3 provided limited information, the stoichiometry was confirmed via high resolution mass spectrometry and elemental analysis.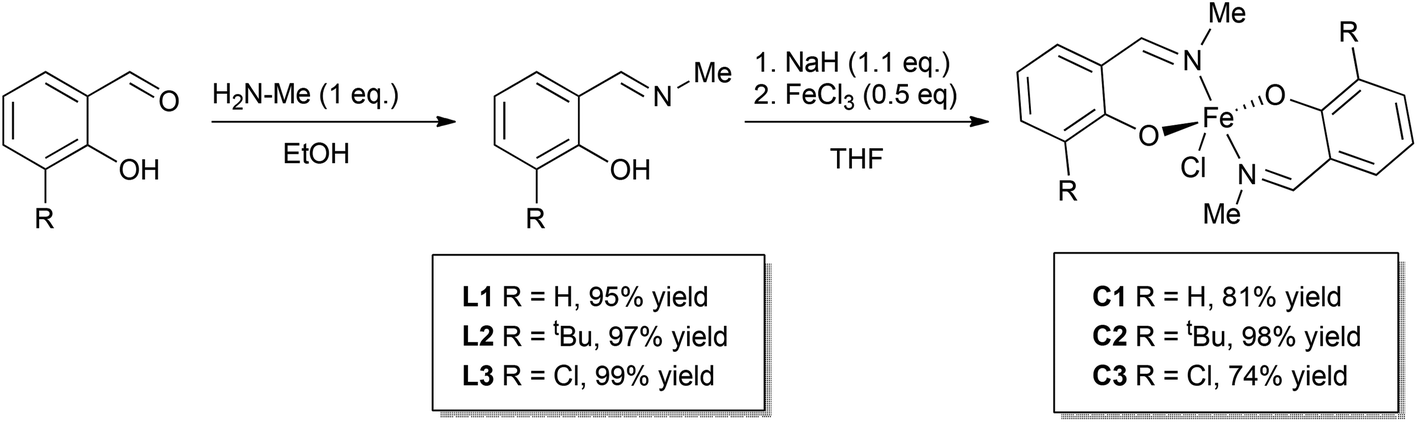 | ||
| Scheme 2 Synthesis of pro-ligands L1–L3 and Fe(III) phenoxyimine complexes C1–C3. | ||
The complexes proved to be oxygen- and moisture-stable, and single crystals suitable for X-ray diffraction were obtained under air. Crystals of complexes C1 and C3 were obtained via the slow evaporation of DCM, while crystals of C2 were obtained via the slow evaporation of toluene. Single crystal X-ray diffraction studies revealed that all three complexes have a mononuclear structure, with a pentacoordinate Fe(III) centre displaying a slightly distorted trigonal bipyramidal (TBP) geometry. In each case, the TBP character is high, as established using the factors of equatorial TBP (TBPe: C1 ≥ 99.9%; C2 ≥ 99.9%; C3 ≥ 99.9%) and axial TBP (TBPa: C1 ≥ 99.9%; C2 = 97.7%; C3 = 99.8%) proposed by Tamao and Ito (Fig. 1–3).29 For all three complexes, the axial N–Fe–N bond angle deviates slightly from linearity (N–Fe–N, C1, 177.44(5)°; C2, 169.7(2)°; C3, 174.92(7)°), while the sum of the equatorial O–Fe–Cl and O–Fe–O bond angles is 360° (C1, 360.00°; C2, 360.01°; C3, 360.01°). The steric bulk of the ortho-substituents notably influences the distortion of the TBP geometry at the Fe centre, with the sterically demanding tBu group leading to the greatest deviation from linearity for the N–Fe–N bond angle, and the lowest TBPa character. These ‘half salen’ frameworks significantly differ from Fe(III) salen complexes,30 as the phenoxyimine ligands are arranged such that both nitrogen and both oxygen atoms are in mutually trans positions. This difference in geometry arises from the lack of a bridge connecting the two phenoxy groups, enabling the phenoxyimine ligands to minimise the steric repulsion of the substituted imine ligands. These complexes bear similar structural motifs to those observed with related phenoxyimine Fe(III)–chloride complexes, including those with bulky substituents on the imine groups.16,31–37 The bond metrics are comparable to reported examples of structurally characterised phenoxyimine Fe(III) complexes,16 with Fe–N bond lengths ranging from 2.092(2) to 2.111(1) Å and Fe–O bond lengths ranging from 1.8757(9) to 1.889(2) Å.33,34,38
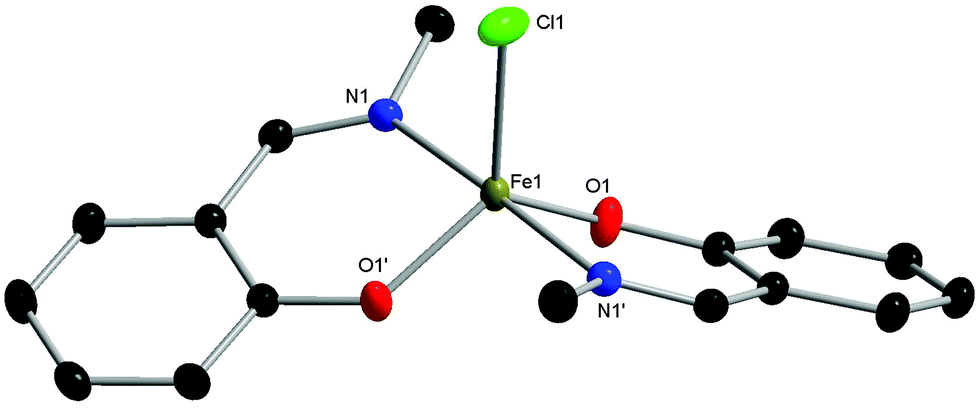 | ||
| Fig. 1 Molecular structure of C1 with ellipsoids set at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å): Fe1–Cl1 2.2713(5), Fe1–O1 1.8758(9), Fe1–O1′ 1.8757(9), Fe1–N1 2.111(1), Fe1–N1′ 2.111(1). Selected bond angles (°): O1–Fe1–Cl1 119.50(3), O1′–Fe1–Cl1 119.50(3), O1′–Fe1–O1 121.00(6), O1′–Fe1–N1′ 90.29(4), O1–Fe1–N1′ 88.46(4), O1–Fe1–N1 90.28(4), O1′–Fe1–N1 88.46(4), N1′–Fe1–Cl1 91.28(3), N1–Fe1–Cl1 91.28(3), N1–Fe1–N1′ 177.44(5). | ||
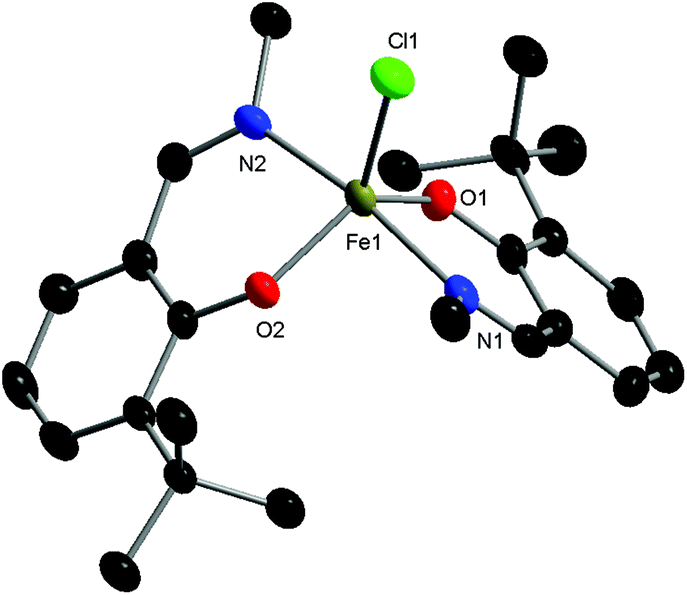 | ||
| Fig. 2 Molecular structure of C2 with ellipsoids set at the 50% probability level. Hydrogen atoms and co-crystallised solvent have been omitted for clarity. Selected bond lengths (Å): Fe1–Cl1 2.245(2), Fe1–O1 1.880(5), Fe1–O2 1.881(4), Fe1–N1 2.103(6), Fe1–N2 2.108(6). Selected bond angles (°): O1–Fe1–Cl1 116.1(2), O2–Fe1–Cl1 119.4(2), O2–Fe1–O1 124.6(2), N1–Fe1–Cl1 93.6(2), N1–Fe1–O1 87.5(2), N1–Fe1–O2 87.5(2), N2–Fe1–Cl1 96.8(2), N2–Fe1–O1 88.5(2), N2–Fe1–O2 86.9(2), N2–Fe1–N1 169.7(2). | ||
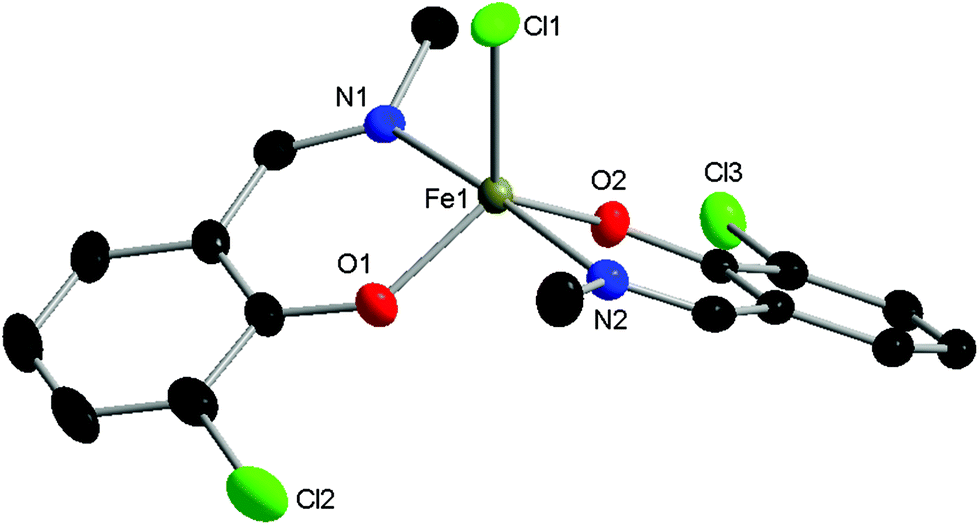 | ||
| Fig. 3 Molecular structure of C3 with ellipsoids set at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å): Fe1–Cl1 2.2517(6), Fe1–O1 1.871(2), Fe1–N1 2.092(2), Fe1–O2 1.889(2), Fe1–N2 2.104(2). Selected bond angles (°): O1–Fe1–N1 88.64(7), O1–Fe1–Cl1 117.12(5), O1–Fe1–O2 121.78(7), O1–Fe1–N2 89.43(7), N1–Fe1–Cl1 92.25(5), N1–Fe1–N2 174.92(7), O2–Fe1–N1 89.00(7), O2–Fe1–Cl1 121.11(5), O2–Fe1–N2 88.02(7), N2–Fe1–Cl1 92.81(5). | ||
The C–O bond lengths of complexes C1, C2 and C3 are short [1.328(1) Å, 1.318(8) Å and 1.321(3) Å, respectively] in comparison to the related protonated ligand bearing a naphthalene substituent on the imine N [C–O, 1.354(2) Å].39 These short C–O bonds are indicative of resonance delocalisation of the anionic charge on the phenoxide ligand in complexes C1–C3. Furthermore, the C–C bond lengths of the (O–)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–C(N) scaffold within C1–C3 lie between the expected bond lengths for C(aromatic)C(aromatic) double bonds and C(aromatic)–C single bonds (1.40 and 1.52 Å, respectively),40 suggestive of resonance delocalisation through the phenoxyimine moiety. This resonance delocalisation is most pronounced for the electron donating tBu-substituted complex C2 [C1–C6, 1.434(9); C6–C7, 1.43(1) Å], followed by the unsubstituted CH analogue C1 [C1–C6, 1.417(2); C6–C7, 1.447(2) Å] and the electron withdrawing Cl analogue C3 [C1–C6, 1.411(3); C6–C7, 1.450(3) Å].
C–C(N) scaffold within C1–C3 lie between the expected bond lengths for C(aromatic)C(aromatic) double bonds and C(aromatic)–C single bonds (1.40 and 1.52 Å, respectively),40 suggestive of resonance delocalisation through the phenoxyimine moiety. This resonance delocalisation is most pronounced for the electron donating tBu-substituted complex C2 [C1–C6, 1.434(9); C6–C7, 1.43(1) Å], followed by the unsubstituted CH analogue C1 [C1–C6, 1.417(2); C6–C7, 1.447(2) Å] and the electron withdrawing Cl analogue C3 [C1–C6, 1.411(3); C6–C7, 1.450(3) Å].
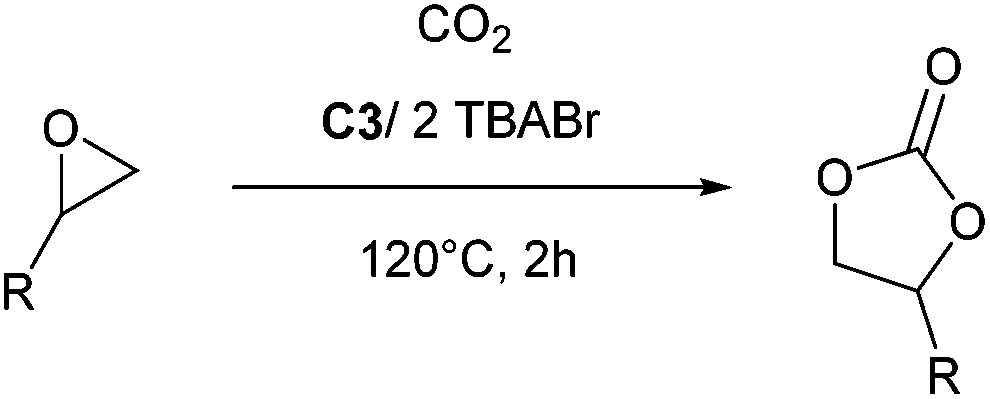
To test the catalytic activity of complexes C1–C3 towards CO2/epoxide coupling, propylene oxide (PO) was selected as a benchmark substrate, as it has previously been studied with other iron catalysts.16 Initially, complex C2 was tested under solvent free conditions using 0.1 mol% catalyst loading with 0.1 mol% tetrabutylammonium iodide (TBAI) as a co-catalyst (vs. PO), using 20 bar pressure of CO2 at 120 °C. 1H and 13C NMR studies of the crude mixture revealed that the catalysts were selective towards the formation of cyclic propylene carbonate (Fig. S8†).41 Under these conditions, C2 successfully achieved 99% conversion to cyclic propylene carbonate in 12 hours (Table 1, entry 2). Control reactions testing only the Fe-complex C2 (entry 4) or only co-catalyst TBAI (entry 5) showed that both components individually display low activity, however, their combination displayed a synergistic effect with a significantly higher conversion achieved (cf. entries 1 and 3). A systematic optimisation of the reaction conditions was subsequently performed. Catalyst C2 displayed tolerance towards an increased substrate loading of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2000 ([Fe]:[PO]), reaching a high TOF value of 400 h−1 (Table 1, entry 6). Doubling the co-catalyst ratio from 1 to 2 equivalents (vs. catalyst C2) further improved the turnover frequency of the catalyst system, from 400 h−1 to 480 h−1 (Table 1, entries 6 and 8, respectively). Finally, tetrabutylammonium bromide (TBABr) was investigated as a co-catalyst, which resulted in a modest improvement in the catalytic activity (entry 12, TOF = 510 h−1). Kinetic studies were performed and confirmed a linear, first-order relationship between substrate conversion and time, highlighting the catalyst stability under the reaction conditions tested (Fig. 4). All three Fe(III) phenoxyimine complexes were subsequently screened for CO2/PO coupling using the optimised reaction conditions (Table 1, entries 11–13).
:2000 ([Fe]:[PO]), reaching a high TOF value of 400 h−1 (Table 1, entry 6). Doubling the co-catalyst ratio from 1 to 2 equivalents (vs. catalyst C2) further improved the turnover frequency of the catalyst system, from 400 h−1 to 480 h−1 (Table 1, entries 6 and 8, respectively). Finally, tetrabutylammonium bromide (TBABr) was investigated as a co-catalyst, which resulted in a modest improvement in the catalytic activity (entry 12, TOF = 510 h−1). Kinetic studies were performed and confirmed a linear, first-order relationship between substrate conversion and time, highlighting the catalyst stability under the reaction conditions tested (Fig. 4). All three Fe(III) phenoxyimine complexes were subsequently screened for CO2/PO coupling using the optimised reaction conditions (Table 1, entries 11–13).
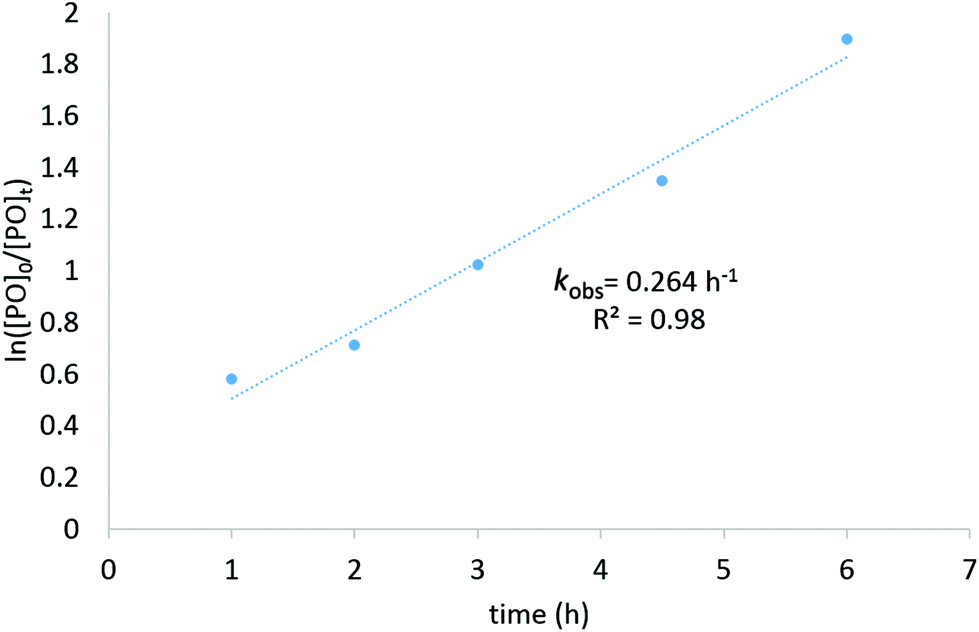 | ||
| Fig. 4 Kinetic plot for the synthesis of propylene carbonate using C2 with 2000 equivalents of substrate. | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Complex | t (h) | [PO]/[Fe] | Co-cat. | [Co-cat]/[Fe] | Conv. (%) | TON | TOF (h−1) |
| Conditions: 100 ml stainless steel autoclaves, 20 bar CO2 pressure, 120 °C, neat. Conversion was determined using 1H NMR spectra of crude reaction mixtures.a The reaction was carried out without an iron complex, 2 equivalents of TBAI were added per 1000 equivalents of epoxide.b 100 equivalents H2O/[Fe] was added.c The reaction was carried out under air.d The reaction was carried out using unpurified propylene oxide (99%). | ||||||||
| 1 | C2 | 24 | 1000 | TBAI | 1 | 99 | 990 | 41 |
| 2 | C2 | 12 | 1000 | TBAI | 1 | 99 | 990 | 83 |
| 3 | C2 | 2 | 1000 | TBAI | 1 | 56 | 560 | 280 |
| 4 | C2 | 24 | 1000 | — | 0 | 13 | 130 | 5 |
| 5a | — | 2 | 1000 | TBAI | 2 | 15 | — | — |
| 6 | C2 | 2 | 2000 | TBAI | 1 | 40 | 800 | 400 |
| 7 | C2 | 2 | 1000 | TBAI | 2 | 91 | 910 | 455 |
| 8 | C2 | 2 | 2000 | TBAI | 2 | 48 | 960 | 480 |
| 9 | C1 | 2 | 2000 | TBAI | 2 | 48 | 960 | 480 |
| 10 | C3 | 2 | 2000 | TBAI | 2 | 69 | 1380 | 690 |
| 11 | C1 | 2 | 2000 | TBABr | 2 | 53 | 1060 | 530 |
| 12 | C2 | 2 | 2000 | TBABr | 2 | 51 | 1020 | 510 |
| 13 | C3 | 2 | 2000 | TBABr | 2 | 76 | 1520 | 760 |
| 14b | C3 | 2 | 2000 | TBABr | 2 | 43 | 860 | 430 |
| 15c | C3 | 2 | 2000 | TBABr | 2 | 65 | 1300 | 650 |
| 16 | C3 | 2 | 10000 |
TBABr | 2 | 11 | 1100 | 550 |
| 17 | C3 | 26 | 10000 |
TBABr | 2 | 63 | 6300 | 242 |
| 18d | C3 | 24 | 10000 |
TBABr | 2 | 51 | 5100 | 213 |
The reactivity trends fall in line with the electron withdrawing/donating ability of the ortho-substituent, with the electron withdrawing Cl group (C3) giving the highest catalytic activity (TOF = 760 h−1, entry 13), followed by the ortho-H analogue C1 (TOF = 530 h−1, entry 11) and the electron donating tBu substituent C2 (TOF = 510 h−1, entry 12). Comparing C1–C3 to other Fe(III) catalysts known for this transformation is rather complex due to the range of different conditions used, as shown by specific catalytic comparators provided in the ESI (Table S2†). However, the high catalytic activity of ortho-chloro-substituted C3 is particularly notable, as it is amongst the fastest reported Fe-based catalyst systems for cyclic carbonate formation. It seems likely that the presence of electron withdrawing chloro- groups increases the Lewis acidity of the Fe centre, facilitating epoxide coordination to promote nucleophilic attack and ring-opening.42 These findings conform to previous studies of cobalt(III) amidoamine ligands, which showed a similar trend; complexes bearing electron withdrawing chloro- or nitro- substituents gave significantly enhanced catalyst activities in comparison to the methyl substituted analogues.43
In addition to their high catalytic activity, it is particularly noteworthy that C1, C2 and C3 are all air-stable complexes. When the coupling reaction was set up under air, ortho-chloro catalyst C3 still displayed high activities towards cyclic propylene carbonate formation, with only a minor reduction in the TOF value observed (entry 13, TOF = 760 h−1; entry 15, TOF = 650 h−1). Furthermore, catalyst system C3 also displayed some tolerance towards water; the addition of 100 equivalents of water (vs. catalyst C3) still gave high TOF values of 430 h−1 (entry 14). Catalyst C3 is noteworthy as it demonstrates high catalytic activities even at low catalyst loadings of 0.01 mol% (1:10000 [Fe]:[PO]), displaying a TOF of up to 550 h−1 after 2 hours (entry 16), slowing only after 8 hours (Fig. S12†). The robustness of C3 was further demonstrated when high conversion was maintained using 10000 equivalents of unpurified (wet) PO (entry 18). The water tolerance of catalyst C3 was supported by FT-IR spectroscopic studies (refer to ESI† for further details).
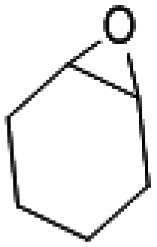
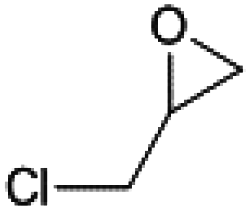
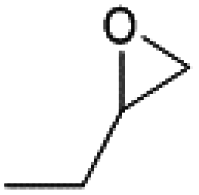
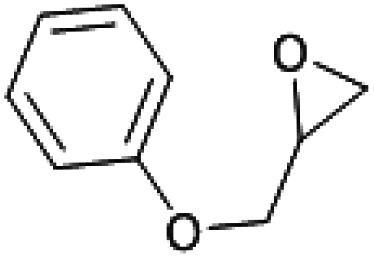
The substrate scope of catalyst C3 towards CO2/epoxide coupling was investigated by testing a range of epoxides, including cyclohexene oxide (CHO), epichlorohydrin, 1,2-epoxybutane and 1,2-epoxy-3-phenoxypropane (Table 2). Catalyst C3 demonstrated tolerance towards a broad substrate scope, successfully converting all five substrates tested to the corresponding cyclic carbonates. For the terminal epoxides, the catalyst activity falls in line with the nature of the epoxide substituent, with the presence of electron withdrawing substituents facilitating the epoxide ring opening. Accordingly, the highest conversion was observed for epichlorohydrin (TOF = 900 h−1, Table 2, entry 3), and the lowest conversion (of a terminal epoxide) was achieved for butylene oxide (TOF = 500 h−1, entry 4). In agreement with previously observed trends,1 the lowest conversions were observed for the most sterically hindered internal epoxide, cyclohexene oxide, as expected due to the formation of a strained bicyclic carbonate product. Through extending the reaction time to 48 hours, high conversion of CHO to cyclohexene carbonate (CHC) was achieved (74%, Table S1,† entry 3). This is indeed surprising, as catalysts which selectively give CHC in high yields are still rare, as many catalyst systems yield a mixture of poly(cyclohexene carbonate) (PCHC) and CHC.1,20,44,45 Importantly, catalyst C3 exclusively formed cis-CHC at high conversions, which is quite unusual as the formation of the trans-isomer often occurs through the thermodynamically favourable back-biting of a poly(cyclohexene carbonate) chain. However, resonances corresponding to trans-CHC (3.9 ppm) were absent from the 1H NMR spectra, and IR spectroscopy and mass spectrometry (EI and MALDI) studies of the product mixture confirmed the lack of polymer formation (Fig. S13–15†).1,20 The formation of cis-CHC occurs through a double inversion pathway, which can be favoured through the addition of excess co-catalyst.20,46–50
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Substrate loading | Conv. (%) | TON | TOF (h−1) |
| Conditions: 100 ml stainless steel autoclaves, 20 bar CO2 pressure, 120 °C, 2 hours, neat. Conversion was determined using 1H NMR spectra of crude reaction mixtures. | |||||
| 1 |
|
2000 | 76 | 1520 | 760 |
| 2 |
|
2000 | 8 | 160 | 80 |
| 3 |
|
2000 | 90 | 1800 | 900 |
| 4 |
|
2000 | 50 | 1000 | 500 |
| 5 |
|
1500 | 69 | 1035 | 518 |
It has previously been proposed that the metal geometry is of key importance for the conversion of sterically congested internal epoxides to the corresponding cyclic carbonates, with complexes bearing a trigonal bipyramidal geometry around the metal centre typically showing greater success.1,51 Complexes C1–C3 all display a distorted trigonal bipyramidal geometry, in contrast to the square pyramidal geometries often observed with Fe(III) salen complexes.52–55 The flexible coordination modes available when using phenoxyimine ligands may present an advantage over the well-established salen analogues. These findings suggest that phenoxyimine ligand supported metal complexes have significant potential for a broad scope of CO2/epoxide coupling reactions.
Conclusions
In conclusion, three new iron(III) phenoxyimine complexes were synthesised and tested in CO2/epoxide coupling reactions. All three earth-abundant, air-tolerant complexes display excellent performance in the selective catalytic coupling of CO2/epoxides, when activated by the presence of a tetrabutylammonium bromide or iodide co-catalyst. These results highlight that for this class of catalysts, the activity is influenced by the presence of electron donating or electron withdrawing ortho-substituents on the phenoxyimine ligand. The highest catalyst activities were achieved using the electron withdrawing chloro-substituted complex C3, with TOF values up to 900 h−1. Importantly, the complexes are very robust, tolerating the presence of both air and water, and very flexible, selectively forming the cis isomer of cyclohexene carbonate from the sterically challenging secondary epoxide. These iron catalyst precursors offer significant potential as stable, robust and selective catalysts for the valorisation of carbon dioxide.Experimental
General procedure for ligand precursors L1–L3
Equimolar amounts of a salicylaldehyde (salicylaldehyde, 3-tert-butylsalicylaldehyde or 3-chlorosalicylaldehyde) and methylamine (40% w/w solution in water) were dissolved in ethanol (0.2 M) and stirred at room temperature for 16 hours. The resulting yellow solutions were dried over MgSO4, filtered, and the solvents were subsequently evaporated in vacuo. L1 was obtained as an orange oil, whilst L2 and L3 were further purified via recrystallisation from ethanol, to produce L2 as green crystals and L3 as yellow crystals. Characterisation data for L1 was in agreement with reported values in the literature.56
Data for
L2: (1.86 g, 97%) 1H NMR (500 MHz, CDCl3) δ 14.11 (s, 1H, OH), 8.35 (s, 1H, HCN), 7.31 (d, J = 7.7 Hz, 1H, ArH), 7.10 (d, J = 7.6 Hz, 1H, ArH), 6.80 (t, J = 7.6 Hz, 1H, ArH), 3.48 (d, J = 1.5 Hz, 3H, NCH3), 1.44 (s, 9H, CCH3). 13C NMR (126 MHz, CDCl3) δ 166.89 (CN), 160.51 (C-OH), 137.43, 129.32, 129.13, 118.75, 117.63 (Ar-C), 45.72 (N-CH3), 34.83 (CCH3), 29.32 (CCH3). HRMS (EI): m/z [M]+ 191.1319 calculated [M]+ 191.1310.
Data for
L3: (1.10 g, 99%) 1H NMR (500 MHz, CDCl3) δ 14.51 (s, 1H, OH), 8.29 (s, 1H, HCN), 7.39 (d, J = 7.9 Hz, 1H, ArH), 7.14 (d, J = 7.7 Hz, 1H, ArH), 6.77 (t, J = 7.8 Hz, 1H, ArH), 3.49 (d, J = 1.5 Hz, 3H, CH3). 13C NMR (126 MHz, CDCl3) δ 165.8 (CN), 158.8 (C-OH), 132.7, 129.6, 122.2, 119.3, 118.1 (Ar-C) 44.9 (CH3). HRMS (EI): m/z [M]+ 169.0315 calculated [M]+ 169.0294.
General procedure for complexes C1–C3
To a solution of pro-ligand (L1–L3) in THF, 1.1 equivalents of NaH was gradually added and the mixture was stirred at ambient temperature for 1 hour. A solution of anhydrous FeCl3 (0.5 eq.) in THF was added dropwise to afford a dark coloured mixture that was stirred for a further 16 hours at room temperature. Solvents were evaporated in vacuo and the crude product was taken up in toluene. The NaCl by-product was removed by filtration through Celite and the filtrate was dried in vacuo. The crude products were washed three times with hexane to afford dark purple-brown powders. Single crystals suitable for X-ray diffraction analysis were obtained via slow evaporation of DCM or toluene.Data for C1 (0.42 g, 81%) HRMS (EI): m/z [M]+ 359.0205 calculated [M]+ 359.0250 elemental analysis calculated for C16H16ClFeN2O2: C, 53.4; H, 4.5; N, 7.8. Found: C, 53.3; H, 4.65; N, 7.6.
Data for C2 (2.11 g, 98%) HRMS (EI): m/z [M]+ 471.1523 calculated [M]+ 471.1502 elemental analysis calculated for C24H32FeN2O2: C, 61.1; H, 6.84; N, 5.9. Found: C, 60.2; H, 6.2; N, 6.0.
Data for C3 (0.49 g, 74%) HRMS (EI): m/z [M]+ 426.9474 calculated [M]+ 426.9475 elemental analysis calculated for C16H14Cl3FeN2O2: C, 44.85; H, 3.3; N, 6.5. Found: C, 44.7; H, 3.35; N, 6.5.
General procedure for the synthesis of cyclic carbonates from CO2 and epoxides
Reactions were carried out in 100 mL stainless steel autoclaves equipped with a magnetic stirrer, heating mantle (controlled by a thermocouple) and a pressure gauge. Complex C1, C2 or C3 (10.0 μmol) and co-catalyst (TBAI or TBABr) were placed into the autoclave and purged with argon. The relevant epoxide (2000 eq.) was added and the autoclave was subsequently pressurised with 20 bar of CO2. The autoclave was heated to 120 °C for 24 hours, then rapidly cooled in an ice bath for 1 hour and slowly vented. An aliquot of the crude reaction mixture was immediately taken up in CDCl3 and analysed via1H NMR spectroscopy, to determine epoxide conversion via integration of product resonances against starting material resonances (refer to ESI† for further details).Crystallography
Single crystal X-ray diffraction data were collected on an Rigaku Oxford Diffraction SuperNova diffractometer fitted with an Atlas CCD detector with Mo-Kα radiation (λ = 0.7107 Å) or Cu-Kα radiation (λ = 1.5418 Å). Crystals were mounted under Paratone on MiTeGen loops. The structures were solved by direct methods using SHELXS or SHELXT and refined by full-matrix least-squares on F2 using SHELXL interfaced through Olex2.57,58 Molecular graphics for all structures were generated using Diamond.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge the University of Edinburgh for funding.References
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- H. Büttner, L. Longwitz, J. Steinbauer, C. Wulf and T. Werner, Top. Curr. Chem., 2017, 375, 50 CrossRef PubMed.
- D. C. Webster and A. L. Crain, Prog. Org. Coat., 2000, 40, 275–282 CrossRef.
- H. Zhang, H.-B. Liu and J.-M. Yue, Chem. Rev., 2014, 114, 883–898 CrossRef PubMed.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- A. Duval and L. Avérous, ACS Sustainable Chem. Eng., 2017, 5, 7334–7343 CrossRef.
- H. Sun and D. Zhang, J. Phys. Chem. A, 2007, 111, 8036–8043 CrossRef PubMed.
- C.-H. Guo, H.-S. Wu, X.-M. Zhang, J.-Y. Song and X. Zhang, J. Phys. Chem. A, 2009, 113, 6710–6723 CrossRef PubMed.
- M. Chihiro, T. Tomoya, O. Kanae and E. Tadashi, Angew. Chem., Int. Ed., 2015, 54, 134–138 CrossRef PubMed.
- C. J. Whiteoak, N. Kielland, V. Laserna, F. Castro-Gomez, E. Martin, E. C. Escudero-Adan, C. Bo and A. W. Kleij, Chemistry, 2014, 20, 2264–2275 CrossRef PubMed.
- H. Vignesh Babu and K. Muralidharan, Dalton Trans., 2013, 42, 1238–1248 RSC.
- J. Qin, P. Wang, Q. Li, Y. Zhang, D. Yuan and Y. Yao, Chem. Commun., 2014, 50, 10952–10955 RSC.
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC.
- A. W. Kleij, Eur. J. Inorg. Chem., 2009, 193–205 CrossRef.
- J. A. Castro-Osma, M. North and X. Wu, Chem. – Eur. J., 2016, 22, 2011–2017 CrossRef PubMed.
- F. A. Al-Qaisi, N. Genjang, M. Nieger and T. Repo, Inorg. Chim. Acta, 2016, 442, 81–85 CrossRef.
- K. Zhu, M. P. Shaver and S. P. Thomas, Chem. Sci., 2016, 7, 3031–3035 RSC.
- K. Zhu, J. Dunne, M. P. Shaver and S. P. Thomas, ACS Catal., 2017, 7, 2353–2356 CrossRef.
- K. Nakano, K. Kobayashi, T. Ohkawara, H. Imoto and K. Nozaki, J. Am. Chem. Soc., 2013, 135, 8456–8459 CrossRef PubMed.
- A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC.
- C. J. Whiteoak, B. Gjoka, E. Martin, M. M. Belmonte, E. C. Escudero-Adán, C. Zonta, G. Licini and A. W. Kleij, Inorg. Chem., 2012, 51, 10639–10649 CrossRef PubMed.
- M. A. Fuchs, T. A. Zevaco, E. Ember, O. Walter, I. Held, E. Dinjus and M. Doring, Dalton Trans., 2013, 42, 5322–5329 RSC.
- A. Buonerba, A. De Nisi, A. Grassi, S. Milione, C. Capacchione, S. Vagin and B. Rieger, Catal. Sci. Technol., 2015, 5, 118–123 RSC.
- M. İkiz, E. İspir, E. Aytar, M. Ulusoy, Ş. Karabuğa, M. Aslantaş and Ö. Çelik, New J. Chem., 2015, 39, 7786–7796 RSC.
- C. J. Whiteoak, E. Martin, E. Escudero-Adán and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2233–2239 CrossRef.
- F. Della Monica, S. V. C. Vummaleti, A. Buonerba, A. D. Nisi, M. Monari, S. Milione, A. Grassi, L. Cavallo and C. Capacchione, Adv. Synth. Catal., 2016, 358, 3231–3243 CrossRef.
- F. Della Monica, B. Maity, T. Pehl, A. Buonerba, A. De Nisi, M. Monari, A. Grassi, B. Rieger, L. Cavallo and C. Capacchione, ACS Catal., 2018, 8, 6882–6893 CrossRef.
- F. M. García-Valle, R. Estivill, C. Gallegos, T. Cuenca, M. E. G. Mosquera, V. Tabernero and J. Cano, Organometallics, 2015, 34, 477–487 CrossRef.
- K. Tamao, T. Hayashi, Y. Ito and M. Shiro, Organometallics, 1992, 11, 2099–2114 CrossRef.
- R. Duan, C. Hu, X. Li, X. Pang, Z. Sun, X. Chen and X. Wang, Macromolecules, 2017, 9188–9195 CrossRef.
- J. E. Davies and B. M. Gatehouse, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 3641–3645 CrossRef.
- C. J. Magurany and C. E. Strouse, Inorg. Chem., 1982, 21, 2348–2350 CrossRef.
- J. M. Becker, J. Barker, G. J. Clarkson, R. van Gorkum, G. K. Johal, R. I. Walton and P. Scott, Dalton Trans., 2010, 39, 2309–2326 RSC.
- M. Amini, M. M. Haghdoost, M. Bagherzadeh, A. Ellern and L. Keith Woo, Polyhedron, 2013, 61, 94–98 CrossRef.
- Y. Elerman and H. Paulus, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 1971–1973 CrossRef.
- M. Amini, A. Arab, P. G. Derakhshandeh, M. Bagherzadeh, A. Ellern and L. K. Woo, Spectrochim. Acta, Part A, 2014, 133, 432–438 CrossRef PubMed.
- A. Mojtaba, B. Mina, D.-H. Sepideh, E. Arkady and W. L. Keith, Z. Anorg. Allg. Chem., 2014, 640, 385–389 CrossRef.
- J. A. Bertrand, J. L. Breece and P. G. Eller, Inorg. Chem., 1974, 13, 125–131 CrossRef.
- R. Jamjah, M. Nekoomanesh, R. Zahedi, G. Zohuri, F. Afshar Taromi and B. Notash, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, 522 CrossRef PubMed.
- G. S. Manku, Theoretical principles of inorganic chemistry, Tata McGraw-Hill, New Delhi, 1980 Search PubMed.
- L. Hongchun and N. Yongsheng, Polym. Adv. Technol., 2016, 27, 1191–1194 CrossRef.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef PubMed.
- P. Ramidi, N. Gerasimchuk, Y. Gartia, C. M. Felton and A. Ghosh, Dalton Trans., 2013, 42, 13151–13160 RSC.
- D. J. Darensbourg and A. D. Yeung, Macromolecules, 2013, 46, 83–95 CrossRef.
- M. Taherimehr, S. M. Al-Amsyar, C. J. Whiteoak, A. W. Kleij and P. P. Pescarmona, Green Chem., 2013, 15, 3083–3090 RSC.
- V. Laserna, G. Fiorani, C. J. Whiteoak, E. Martin, E. Escudero-Adán and A. W. Kleij, Angew. Chem., Int. Ed., 2014, 53, 10416–10419 CrossRef PubMed.
- J. Langanke, L. Greiner and W. Leitner, Green Chem., 2013, 15, 1173–1182 RSC.
- C. J. Whiteoak, E. Martin, E. Escudero-Adán and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2233–2239 CrossRef.
- F. Castro-Gómez, G. Salassa, A. W. Kleij and C. Bo, Chem. – Eur. J., 2013, 19, 6289–6298 CrossRef PubMed.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef PubMed.
- C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adán, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228–1231 CrossRef PubMed.
- L. D. Wickramasinghe, S. Mazumder, K. K. Kpogo, R. J. Staples, H. B. Schlegel and C. N. Verani, Chem. – Eur. J., 2016, 22, 10786–10790 CrossRef PubMed.
- Z. Chu, W. Huang, L. Wang and S. Gou, Polyhedron, 2008, 27, 1079–1092 CrossRef.
- A. Karahan, R. Kurtaran, Y. Yahsi, E. Gungor and H. Kara, J. Struct. Chem., 2016, 57, 731–736 CrossRef.
- B. Chen, D. S. Contreras, Y. L. Clancy and F. R. Fronczek, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, 732–734 CrossRef.
- P. A. Nikitina, A. S. Peregudov, T. Y. Koldaeva, L. G. Kuz'mina, E. I. Adiulin, I. I. Tkach and V. P. Perevalov, Tetrahedron, 2015, 71, 5217–5228 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra and single crystal X-ray diffraction data. CCDC 1855808–1855810. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt02919a |
| This journal is © The Royal Society of Chemistry 2018 |