
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of the first Zn6-hexagon sandwich-tungstoantimonate via rearrangement of a non-lacunary Krebs-type polyoxotungstate†‡
Elias
Tanuhadi
 a,
Alexander
Roller
b,
Gerald
Giester
c,
Ioannis
Kampatsikas
a and
Annette
Rompel
*a
a,
Alexander
Roller
b,
Gerald
Giester
c,
Ioannis
Kampatsikas
a and
Annette
Rompel
*a
aUniversität Wien, Fakultät für Chemie, Institut für Biophysikalische Chemie, Althanstraße 14, 1090 Wien, Austria. E-mail: annette.rompel@univie.ac.at; Web: http://www.bpc.univie.ac.at
bUniversität Wien, Fakultät für Chemie, Zentrum für Röntgenstrukturanalyse, Währinger Straße 42, 1090 Wien, Austria
cUniversität Wien, Fakultät für Geowissenschaften, Geographie und Astronomie, Institut für Mineralogie und Kristallographie, Althanstraße 14, 1090 Wien, Austria
First published on 11th October 2018
Abstract
A novel synthetic pathway to obtain the first Zn6 hexagon tungstoantimonate [(Zn(H2O))6(B-α-SbW9O33)2]6− ([Zn6-α-SbW9] (1)) via rearrangement of a non-lacunary Krebs-POM precursor (C12N4H11)4K4[(Mn(H2O)3)2((Mn0.5W0.5)O2)2(B-β-SbW9O33)2] ([Mn-β-SbW9]) has been developed. Addition of ortho-phenylenediamine (opda) in order to optimize the synthesis of [Mn-β-SbW9] led to the crystallization of a novel Krebs-type Zn-POM (C12N4H11)4Na5[((Zn0.8W0.2)(H2O)3)2((Zn0.2W0.8)O2)2(B-β-SbW9O33)2] ([(Zn/W)2-β-SbW9] (2)) comprising four disordered Zn-centers after replacement of MnCl2 with ZnCl2. The compounds were characterized in the solid state by single-crystal and powder X-ray diffraction (XRD), IR spectroscopy, thermogravimetric analysis (TGA) and elemental analysis and in solution by UV-vis spectroscopy and ESI-mass spectrometry. The Krebs-POM archetype and the Zn6 hexagon tungstoantimonate have been investigated towards their interactions with Human Serum Albumin (HSA) as a model protein using SDS-PAGE and trypthophan fluorescence quenching.
Polyoxometalates (POMs)1 represent a large class of mostly anionic clusters, which are composed of metals in high oxidation states linked by oxygen atoms resulting in a vast variety of unique structures with possible applications in material science,2 photochemistry3 and biological chemistry4,5 including protein crystallography.6 Within the class of transition metal substituted POMs (TMSPs), the sandwich-type family represents the largest group. Sandwich-POMs generally are constructed by two lacunary species that are linked by heteroatoms. The family of sandwich POM compounds can be divided into further subtypes like the Krebs-7,8 and Hervé-9 type. Krebs-type POMs, which are constituted by two lone-pair containing β-Keggin lacunary fragments, e.g. [β-Sb(III)W9O33]9−,10 have shown considerable importance in the field of both homo- and heterogeneous catalysis in the past.11 The first representatives of this structural family, [M2(H2O)6(WO2)2(β-SbW9O33)2](14−2n)− (Mn+ = Fe3+, Co2+, Mn2+, Ni2+), were reported by Bösing et al. in 1997.10 Since then, a broad variety of Krebs-POM representatives has been reported, among them the tetra-substituted Krebs-polyanions, first synthesized and characterized by Kortz and co-workers in 2002.8 However, mixed transition metal substituted Krebs-type compounds remain rare. Most sandwich-type POMs contain two to five transition metals in the linking belt. Hexa-substituted analogues, however, especially the subclass of hexagon-sandwich polyoxotungstates, which comprise a hexagon-shaped linking-belt, have been reported scarcely, among them, Na4Zn2[Zn2(H2O)10(ZnCl)6(B-α-BiW9O33)2],12 [(Zn(Hen))6(B-α-AsW9O33)2]13 (en = ethylenediamine), [(CuCl)6(B-α-AsW9O33)2],14 [(MnCl)6(B-α-SbW9O33)2],14 [(CuCl)6(B-α-SbW9O33)2],14,15 and [(MnCl)6(B-α-AsW9O33)2].15 To the best of our knowledge, [(MnCl)6(B-α-SbW9O33)2] and [(CuCl)6(B-α-SbW9O33)2], published by Yamase et al.14,15 represent the only hexa-substituted sandwich POMs containing the [SbW9O33]9− building block.
The majority of POM compounds is prepared by a simple one-pot synthetic procedure, which follows mainly self-assembly mechanisms. These mechanisms are rather complex and most often lead to novel, yet unprecedented products, making the selective preparation of new hexagon sandwich POM com-pounds challenging. Therefore, the development of synthetic routes to design the desired clusters is of special interest. Herein, we report a novel multistep-synthesis, which includes the rearrangement of a Krebs-type manganese POM, [(Mn(H2O)3)2((Mn0.5W0.5)O2)2(B-β-SbW9O33)2] abbreviated [Mn-β-SbW9], in order to obtain the first Zn6-hexagon tungsto-antimonate [(Zn(H2O))6(B-α-SbW9O33)2]6− in the following termed [Zn6-α-SbW9] (1). On the way to synthesizing the target compound, we were able to obtain the literature known [Mn-β-SbW9] POM cluster more selectively and faster16 by using o-phenylenediamine (opda). In the presence of Zn2+ applying this optimization strategy selectively led to the crystallization of the novel Krebs-type zinc compound (C12N4H11)4Na5[((Zn0.8W0.2)(H2O)3)2((Zn0.2W0.8)O2)2(B-β-SbW9O33)2] ([(Zn/W)2-β-SbW9] (2)) composed of a linking belt with four disordered zinc centres. Replacement of Zn2+ with Cd2+ and decrease in pH yielded the novel mixed Krebs-type compound (TMA)2Na6H8[(Mn(H2O)3)2(CdCl0.8O1.2)2(B-β-SbW9O33)2] ([Cd2Mn2-β-SbW9] (3)), TMA = tetramethylammonium. As the interaction of Krebs-type POTs with proteins has never been investigated, the interaction of all three Krebs-type clusters [Mn-β-SbW9], [(Zn/W)2-β-SbW9] (2) and [Cd2Mn2-β-SbW9] (3) and the Zn6-hexagon sandwich-tungstoantimonate [Zn6-α-SbW9] (1) with Human Serum Albumin (HSA) as a model protein has been studied by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis and tryptophan fluorescence quenching.
The first step in the synthesis of [Zn6-α-SbW9] (1) (Fig. 1) was the preparation of the Krebs-POM precursor [Mn-β-SbW9].16 Upon addition of opda to a stirred reaction mixture of Na9[B-SbW9O33] and MnCl2, the in situ formed [(Mn(H2O)3)2((Mn0.5W0.5)O2)2(B-β-SbW9O33)2] catalyzed the oxidation reaction of opda to 2,3-diaminophenazine (2,3-DAP) (Scheme 1), which co-crystallized together with [Mn-β-SbW9] (Fig. S9‡). Addition of opda resulted in a significantly lower water solubility of [Mn-β-SbW9], which subsequently facilitated its crystallization process. X-ray-crystallographic studies on [Mn-β-SbW9] (CCDC number 1569737, Tables S6 and S7‡) revealed π–π-interactions between the aromatic 2,3-DAP systems. As a result, a more stabilized crystal packing was obtained (Fig. S9‡), which in turn led to an accelerated crystallization of [Mn-β-SbW9] upon cooling of the stirred solution to room temperature. It is worth noting that literature known synthetic protocols of [Mn-β-SbW9] lead to a mixture of the anions [Na3(H2O)6Mn3(μ-OAc)2(B-α-SbW9O33)2]11− and [Mn3W(H2O)8(B-β-SbW9O33)2]4−,17 whereas addition of opda selectively enables isolation of the Krebs-β-SbW9 anion (80% yield based on W). Besides SXRD, [Mn-β-SbW9] was characterized in the solid state by ATR-IR spectroscopy (Fig. S1‡). The same reaction procedure using ZnCl2 instead of MnCl2 led to the crystallization of the novel Krebs-type zinc compound [(Zn/W)2-β-SbW9] (2) with four zinc centres disordered with tungsten (CCDC number 1569739, Tables S8 and S9‡). However, the full d10-electron configuration of Zn2+ led to only little opda-conversion, resulting in a lower product yield of [(Zn/W)2-β-SbW9] (2) (12% yield based on W). As a matter of fact, addition of catalytic amounts of FeCl3,18 to overcome this problem, led to a higher opda-conversion resulting in an optimized yield of [(Zn/W)2-β-SbW9] (2) (70% yield based on W). Interestingly, omission of opda in the procedure leads to [Zn2(H2O)6(WO2)2(B-β-SbW9O33)2]10–19 not exhibiting (Zn/W) disorders as characterized by SXRD, ATR-IR spectroscopy (Fig. S2‡) and elemental analysis.
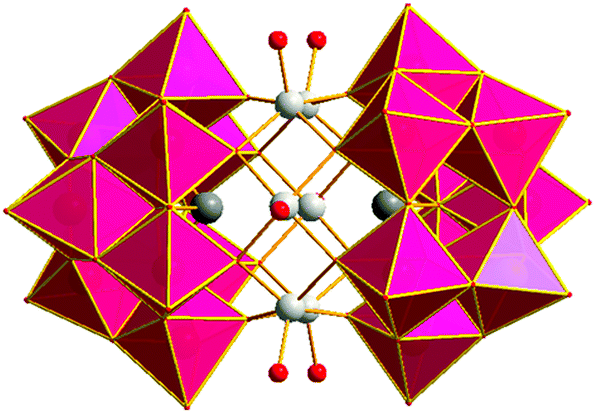 | ||
| Fig. 1 Polyhedral representation of [Zn6-α-SbW9] (1). Colour legend: WO6, pink; Sb, dark grey; Zn, silver; Ot, red. | ||
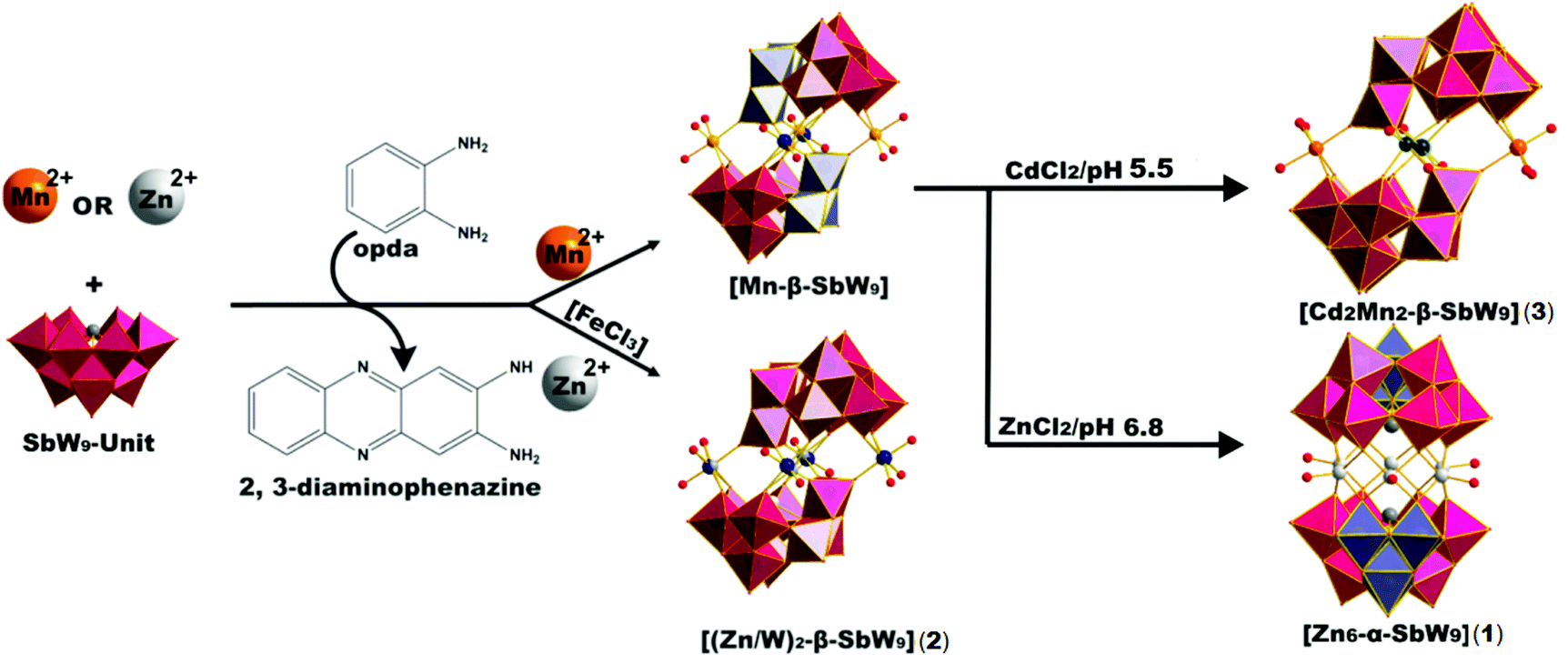 | ||
| Scheme 1 Structure and synthesis of [Zn6-α-SbW9] (1) and [(Zn/W)2-β-SbW9] (2). The synthesis starts from the [SbW9] unit and the transition metal (TM) of choice. [Mn-β-SbW9] was used as precursor for the synthesis of the title compound, which was obtained by exchange of the manganese linker cations with zinc cations and β/α-isomerization of the precursor. Counter cations are omitted for clarity. Color legend: WO6, pink octahedra; 60° rotated WO6, ice blue octahedra; Sb, dark grey spheres; O, dark red spheres; Zn, light grey spheres; Mn, orange spheres; Cd, dark green spheres, W in the linking belt, dark blue spheres. Opda – o-phenylenediamine; 2,3-DAP – 2,3-diaminophenazine, ACN – acetonitrile. | ||
Attempts to prepare [Zn6-α-SbW9] (1) by reacting Na9[B-α-SbW9O33] with ZnCl2 were not successful. Considering the synthesis of [(CuCl)6(B-α-AsW9O33)2]12−, which only worked by preparing the non-lacunary [{EuH2O}2(B-α-AsW9O33)2]12− polyanion to replace the europium-linker cations in the linking belt with a Cu6-hexagon in a subsequent reaction step, we decided to use the Krebs-POM [Mn-β-SbW9] as a non-lacunary precursor to prepare the Zn6-hexagon sandwich target compound [Zn6-α-SbW9] (1). Regarding the decreased water solubility of [Mn-β-SbW9], the second reaction step (Scheme 1) was performed by using a solvent mixture of acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water = (1:4). After addition of an excess of ZnCl2 to the dissolved [Mn-β-SbW9] precursor, the reaction solution was refluxed for 40 min at pH 6.8. An aqueous solution of SbW9 contains alpha and beta species in equilibrium. Based on SXRD studies performed by Krebs and co-workers10 on isolated SbW9 containing species at different pH values, the stable pH range of the [α-SbW9]-moiety is known to be between 6.0 and 7.5. Lower pH values result in a higher tendency of the SbW9 unit to undergo further condensation. This increased tendency further results in the generation of the [β-SbW9]-isomer, which proves to be stable in the pH range of 4.5 to 5.5 indicated by the formation of the literature known Zn2-β-SbW9-Krebs-type analogue19 at pH values lower than 6.0 and formation of [Cd2Mn2-β-SbW9] (3) upon replacing ZnCl2 with CdCl2 at pH 5.5 (Scheme 1). Filtration of the brownish reaction mixture, after dropwise addition of a tetramethylammonium chloride (TMACl) solution, gave a yellowish solution, which was left to evaporate for one week to give a mixture of red crystal plates (TMA)(C12N4H11)5[(Zn(H2O))6(B-α-SbW9O33)2] ((TMA)(C12N4H11)5[Zn6-α-SbW9] (1)) (CCDC 1569738, Tables S10 and S11‡) and colourless block shaped crystals (TMA)6[(Zn(H2O))6(B-α-SbW9O33)2] ((TMA)6[Zn6-α-SbW9] (1)) (CCDC 1576366, Tables S12 and S13.‡ Replacement of zinc chloride with cadmium chloride at pH 6.8 to yield the cadmium containing hexagon analogue, resulted in precipitates, which despite all our efforts could not be identified.
:water = (1:4). After addition of an excess of ZnCl2 to the dissolved [Mn-β-SbW9] precursor, the reaction solution was refluxed for 40 min at pH 6.8. An aqueous solution of SbW9 contains alpha and beta species in equilibrium. Based on SXRD studies performed by Krebs and co-workers10 on isolated SbW9 containing species at different pH values, the stable pH range of the [α-SbW9]-moiety is known to be between 6.0 and 7.5. Lower pH values result in a higher tendency of the SbW9 unit to undergo further condensation. This increased tendency further results in the generation of the [β-SbW9]-isomer, which proves to be stable in the pH range of 4.5 to 5.5 indicated by the formation of the literature known Zn2-β-SbW9-Krebs-type analogue19 at pH values lower than 6.0 and formation of [Cd2Mn2-β-SbW9] (3) upon replacing ZnCl2 with CdCl2 at pH 5.5 (Scheme 1). Filtration of the brownish reaction mixture, after dropwise addition of a tetramethylammonium chloride (TMACl) solution, gave a yellowish solution, which was left to evaporate for one week to give a mixture of red crystal plates (TMA)(C12N4H11)5[(Zn(H2O))6(B-α-SbW9O33)2] ((TMA)(C12N4H11)5[Zn6-α-SbW9] (1)) (CCDC 1569738, Tables S10 and S11‡) and colourless block shaped crystals (TMA)6[(Zn(H2O))6(B-α-SbW9O33)2] ((TMA)6[Zn6-α-SbW9] (1)) (CCDC 1576366, Tables S12 and S13.‡ Replacement of zinc chloride with cadmium chloride at pH 6.8 to yield the cadmium containing hexagon analogue, resulted in precipitates, which despite all our efforts could not be identified.
Interestingly, XRD studies of both crystal fractions revealed the same Zn6-hexagon anion in both crystal fractions, with the 2,3-DAP cation lacking in the colourless block shaped crystals, which indicates the complete rearrangement of the Krebs-type precursor [Mn-β-SbW9] to [Zn6-α-SbW9] (1). By using the inorganic di-substituted counterpart Na10[Mn2(H2O)6(WO2)2(B-β-SbW9O33)2]·40H2O19 as a precursor, (TMA)6[Zn6-α-SbW9] (1) could be obtained as a pure crystalline phase in lower yields (12%). The same reaction procedure, replacing ZnCl2 with CdCl2 in an acetate buffer (pH = 5.5) led to the formerly mentioned dominance of the beta isomer resulting in the novel mixed substituted Krebs-POM compound [Cd2Mn2-β-SbW9] (3) (CCDC 1849600, Tables S14 and S15‡) (Scheme 1).
(TMA)6[Zn6-α-SbW9] (1) was additionally characterized in the solid state by ATR-IR spectroscopy (Fig. S3 and S4‡), PXRD (Fig. S13‡) and elemental analysis. SXRD-analysis showed that the four polyanions, [Mn-β-SbW9], [(Zn/W)2-β-SbW9] (2), [Cd2Mn2-β-SbW9] (3) and (TMA)(C12N4H11)5[Zn6-α-SbW9] (1), crystallize in a triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , whereas (TMA)6[Zn6-α-SbW9] (1) belongs to the monoclinic space group C2/m.
, whereas (TMA)6[Zn6-α-SbW9] (1) belongs to the monoclinic space group C2/m.
The crystal structure of [(Zn/W)2-β-SbW9] (2) shows two [β-SbW9]-lacunary species linked by four zinc centres. All four zinc centres exhibit a disorder with tungsten resulting in the typical Krebs-type structure with idealized C2h symmetry (Fig. S10‡). Regarding the synthetic conditions of [(Zn/W)2-β-SbW9] (2), which include the use of an acidic buffer (pH = 4.8), the disorder with tungsten is in accordance with the results for the disordered alpha-arsenotungstate compounds observed at lower pH values, reported by Kortz et al. in 2001.20 Zinc and tungsten show inverse occupancy ratios at the inner (Zn:W = 20:80) and outer (Zn:W = 80:20) site of the linking belt. The zinc centres exhibit a distorted octahedral coordination environment with three H2O ligands bound to the peripheral zinc positions and Zn–O bond lengths ranging from 1.664(2) at the inner site of the belt to 2.237(1) Å between the peripheral zinc centres and the H2O ligands (Fig. S11‡). As for the architecture of [Cd2Mn2-β-SbW9] (3), SXRD studies revealed two [β-SbW9]-lacunary species linked by a belt comprising two Cd2+ centres at the inner positions, whereas the peripheral sites of the belt are occupied by two Mn2+ metal centres. All four metal centres exhibit a distorted octahedral coordination environment with Cd–O bond lengths ranging from 1.962(1) to 2.105(2) Å and Mn–(OH2) distances between 2.164(1) and 2.209(2) Å. The crystal structure of [Zn6-α-SbW9] (1), (Fig. 1) presents a polyanion with idealized D3d symmetry consisting of two [(Zn(H2O))3(B-β-SbW9O33)] building blocks related by an inversion centre (Fig. S11A‡). Both building blocks are formed by rearrangement of the precursor [β-SbW9] moiety to the alpha isomer upon 60° rotation of a W3O13 unit. Occupation of the three vacant sites with the corresponding number of Zn2+ results in the [(Zn(H2O))3(B-α-SbW9O33)] building block comprising three zinc centres arranged in a triangular shape with Zn–Zn distances ranging from 3.089 to 3.114 Å and a bond angle of 119.49°. Each zinc ion inside the belt shows distorted square-pyramidal coordination geometry with four oxygen atoms belonging to two [SbW9O33] clusters and a terminal H2O-ligand (Ot) (Fig. S11B‡). The Zn–O distances vary between 2.025 and 2.062 Å with Ot–Zn–O bond angles from 102.061° to 109.783°.
The number of water molecules in the novel compounds [(Zn/W)2-β-SbW9] (2)·13H2O, [Cd2Mn2-β-SbW9] (3)·51.5H2O and (TMA)6[(Zn(H2O))6(B-α-SbW9O33)2] (1)·37.5H2O was determined using thermogravimetric analysis (TGA) (Fig. S6–S8 and Tables S2–S4‡). The five polyanions were investigated by UV-vis spectroscopy (Fig. S15 and Table S16‡). The spectra are characterized by an absorption maximum at 272 nm attributed to the pπ(Ob) → dπ*(W) ligand-to-metal charge-transfer typical for the Keggin-type framework.21 A second absorption maximum in the spectra of the 2,3-DAP-containing polyanions22 was observed around 425 nm corresponding to aromatic transitions. ESI-MS was performed on (TMA)(C12N4H11)5[(Zn(H2O))6(B-α-SbW9O33)2] (1). The major peaks observed show the [(Zn(H2O))6(B-α-SbW9O33)2]6− moiety and a series of various tungsten related species based on the polyanion (Fig. S16 and Table S17‡).
The interactions of the peripheral Lewis acid metal centres in the compounds [Mn-β-SbW9], [(Zn/W)2-β-SbW9] (2), [Cd2Mn2-β-SbW9] (3) and (TMA)6[(Zn(H2O))6(B-α-SbW9O33)2] (1) with Human Serum Albumin (HSA) as a model protein were investigated to assess whether sandwich POM compounds show any proteolytic character in solution(s) with proteins, as has been reported for other POM archetypes.23 SDS-PAGE (Fig. S17‡) was applied in 10 mM NaOAc buffer pH 5.5 (Fig. S17G–N,‡ for [Mn-β-SbW9], (2), (1) and (3)) and in 25% DMSO solution (Fig. S17A–F,‡ for [Mn-β-SbW9], (2) and (3)) respectively, to ensure a relaxed conformation of the model protein. The results reveal that all four compounds show no hydrolytic activity as indicated by intact HSA bands at 66 kDa even at 65 °C and 100-fold excess of each sandwich POM.
Fluorescence quenching as a well-established method to investigate ligand–protein interactions has been applied to investigate the interactions of HSA/BSA and different POM archetypes, including the Keggin,24 Wells-Dawson25 and wheel-shape-structured26 POM architectures. Herein, fluorescence quenching was applied to gain more insight into the interaction between the sandwich POM archetype and the fluorophore tryptophan of HSA for the first time. All four fluorescence spectra of HSA show a maximum at 313 nm, which decreases with increased POM concentration indicating the binding of POM to HSA (Fig. S18–S21‡). Furthermore, a clear hypsochromic shift of the maximum to lower wavelengths upon increased POM concentration indicates a decrease in polarity within the immediate tryptophan environment. It is worth noting that all POMs form a 1:1 complex with the protein, except for [Cd2Mn2-β-SbW9] (3), which showed the strongest interaction with HSA resulting in 1 to 2 POM molecules binding to the protein (Table S18‡). This could be explained both by electrostatic and structural factors, as [(Mn(H2O)3)2(CdCl0.8O1.2)2(B-β-SbW9O33)2]16− [Cd2Mn2-β-SbW9] (3) shows the highest negative charge relative to the other Krebs-POMs [(Mn(H2O)3)2((Mn0.5W0.5)O2)2(B-β-SbW9O33)2]8− ([Mn-β-SbW9]) and [((Zn0.8W0.2)(H2O)3)2((Zn0.2W0.8)O2)2(B-β-SbW9O33)2]9− ([(Zn/W)2-β-SbW9] (2)), whereas the title compound [(Zn(H2O))6(B-α-SbW9O33)2]6− ([Zn6-α-SbW9] (1)), which shows a stronger binding compared to [(Mn(H2O)3)2((Mn0.5W0.5)O2)2(B-β-SbW9O33)2]8− ([Mn-β-SbW9]), comprises more accessible metal centres. These results suggest non-proteolytic POM protein interactions of the sandwich-POM archetype, which, potentially could be used as an additive in POM assisted protein crystallography.6
Conclusions
In conclusion, the synthetic pathway presented in this paper may open novel reaction routes for the synthesis of new hexagon-POM structures. The potential of the Krebs-POM archetype as a non-lacunary synthetic precursor and its interactions with HSA as a model protein have been explored.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Austrian Science Fund (FWF): P27534 and the University of Vienna. We are grateful to Ing. Peter Unteregger for his support with ESI-MS measurements, Mass Spectrometry Centre, University of Vienna, Ao.Univ.-Prof. Christian Lengauer, Institut für Mineralogie und Kristallographie for TGA measurements. We acknowledge Assoc.-Prof. Andreas Limbeck, Institute of Chemical Technologies and Analytics, TU Wien for ICP-OES investigations. Lastly, the authors wish to thank Nadiia Gumerova, PhD, Dipl.-Ing. Matthias Pretzler, Dr Joscha Breibeck and Emir Al-Sayed, MSc for valuable discussions concerning this work.Notes and references
- M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer-Verlag, Berlin, 1983 Search PubMed.
- K. Kastner, A. J. Kibler, E. Karjalainen, J. A. Fernandes, V. Sans and G. N. Newton, J. Mater. Chem. A, 2017, 5, 11577–11581 RSC.
- K. Suzuki, F. Tang, Y. Kikukawa, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2014, 21, 5356–5360 CrossRef PubMed.
- A. Bijelic, M. Aureliano and A. Rompel, Chem. Commun., 2018, 54, 1153–1169 RSC.
- A. Bijelic, M. Aureliano and A. Rompel, Angew. Chem., Int. Ed. DOI:10.1002/anie.201803868; (Angew. Chem.), DOI:10.1002/ange.201803868, in press.
- (a) A. Bijelic and A. Rompel, Coord. Chem. Rev., 2015, 299, 22–38 CrossRef CAS PubMed; (b) A. Bijelic and A. Rompel, Acc. Chem. Res., 2017, 50, 1441–1448 CrossRef CAS PubMed.
- I. Loose, E. Droste, M. Bösing, H. Pohlmann, M. H. Dickman, C. Rosu, M. T. Pope and B. Krebs, Inorg. Chem., 1999, 38, 2688–2694 CrossRef CAS.
- U. Kortz, M. G. Savelieff, B. S. Bassil, B. Keita and L. Nadjo, Inorg. Chem., 2002, 41, 783–789 CrossRef CAS PubMed.
- L.-H. Bi, M. Reicke, U. Kortz, B. Keita, L. Nadjo and R. J. Clark, Inorg. Chem., 2004, 43, 3915–3920 CrossRef CAS PubMed.
- M. Bösing, I. Loose, H. Pohlmann and B. Krebs, Chem. – Eur. J., 1997, 3, 1232–1237 CrossRef.
- M. Bösing, A. Nöh, I. Loose and B. Krebs, J. Am. Chem. Soc., 1998, 120, 7252–7259 CrossRef.
- H. Tan, Z. Zhang, D. Liu, Y. Qi, E. Wang and Y. Li, J. Cluster Sci., 2008, 19, 543–550 CrossRef CAS.
- J. Y. Niu, X. Ma, J. W. Zhao, P. T. Ma, C. Z. Zhang and J. P. Wang, CrystEngComm, 2011, 13, 4834–4837 RSC.
- T. Yamase, K. Fukaya, H. Nojiri and Y. Ohshima, Inorg. Chem., 2006, 45, 7698–7704 CrossRef CAS PubMed.
- T. Yamase, H. Ishikawa, A. Hiroko, K. Fukaya, H. Nojiri and H. Takeuchi, Inorg. Chem., 2012, 51, 4606–4619 CrossRef CAS PubMed.
- B. W. Chen, W. L. Chen, Y. G. Li and E. B. Wang, J. Cluster Sci., 2011, 22, 73–85 CrossRef CAS.
- J. P. Wang, P. T. Ma, J. Li, H. Y. Niu and J. Y. Niu, Chem. – Asian J., 2008, 3, 822–833 CrossRef CAS PubMed.
- A. Abdolmaleki and S. M. Ahmadi, Can. J. Chem., 2011, 89, 1202–1206 CrossRef CAS.
- M. Piepenbrink, E. M. Limanski and B. Krebs, Z. Anorg. Allg. Chem., 2002, 628, 1187–1191 CrossRef CAS.
- U. Kortz, N. K. Al-Kassem, M. G. Savelieff, N. A. Al Kadi and M. Sadakane, Inorg. Chem., 2001, 40, 4742–4749 CrossRef CAS PubMed.
- L. Bi, B. Li, L. Wu and Y. Bao, Inorg. Chim. Acta, 2009, 362, 3309–3313 CrossRef CAS.
- P. Zhou, H. Liu, S. Chen, L. Lucia and H. Zhan, Molbank, 2011, 2011, M730 CrossRef.
- K. Stroobants, E. Moelants, H. G. T. Ly, P. Proost, K. Bartik and T. N. Parac-Vogt, Chem. – Eur. J., 2013, 19, 2848–2858 CrossRef CAS PubMed.
- V. Goovaerts, K. Stroobants, G. Absilis and T. N. Parac-Vogt, Phys. Chem. Chem. Phys., 2013, 15, 18378–18387 RSC.
- G. Zhang, B. Keita, C. T. Craescu, S. Miron, P. de Olivera and L. Nadjo, Biomacromolecules, 2008, 9, 812–817 CrossRef CAS PubMed.
- G. Zhang, B. Keita, J. C. Brochon, P. de Oliveira, L. Nadjo, C. T. Craescu and S. Miron, J. Phys. Chem. B, 2007, 111, 1809–1814 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Dr Dr h. c. Bernt Krebs on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1576366, 1849600 and 1569737–1569739. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt02787k |
| This journal is © The Royal Society of Chemistry 2018 |