
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethyl camouflage in the ten-vertex closo-dicarbaborane(10) series. Isolation of closo-1,6-R2C2B8Me8 (R = H and Me) and their monosubstituted analogues†
Mario
Bakardjiev
a,
Oleg L.
Tok
a,
Aleš
Růžička
b,
Zdeňka
Růžičková
b,
Josef
Holub
a,
Drahomír
Hnyk
 a,
Zbyněk
Špalt
a,
Jindřich
Fanfrlík
c and
Bohumil
Štíbr
*a
a,
Zbyněk
Špalt
a,
Jindřich
Fanfrlík
c and
Bohumil
Štíbr
*a
aInstitute of Inorganic Chemistry, Academy of Sciences of the Czech Republic, Husinec-Řež 1001, Czech Republic
bDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic
cInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 2, 166 10 Prague 6, Czech Republic
First published on 23rd July 2018
Abstract
Reported are procedures leading to the first types of methyl camouflaged dicarbadecaboranes with fewer than eleven vertices. The compounds contain the closo-1,6-C2B8 scaffolding inside the egg-shaped hepta – decamethyl sheath, which imparts unusually high air and solvolytic stability to all of these compounds.
Introduction
The chemistry of polyalkylated twelve-vertex carboranes, compounds derived from closo [CB11H12]− and C2B10H12, were pioneered by Hawthorne's and Michl's groups.1,2 This class of carboranes represents a new genre in organic chemistry, “spherical hydrocarbons” whose icosahedral core is sheathed by a coating of alkyl substituents. In the dicarbaborane area pertinent to this work, the reaction of 1,2-C2B10H12 (o-carborane) or 1,7-C2B10H12 (m-carborane) with excess MeI in the presence of AlCl3 afforded octamethyl derivatives 1,2,3,6-H4C2B10Me8 and 1,2,3,7-H4C2B10Me8 in high yield;3 the two positively charged boron positions adjacent to both carbons remain unmethylated. Complete B-methylation was, however, achieved via methylation of p-carboranes 1,12-R2C2B10H10 (R = H, Me and Br) with MeOTf/HOTf at reflux for 20 h to cleanly afford the B-permethylated carboranes 1,12-R2C2B10Me10 in yields over 90%.4 These successful experiments, together with those achieved in the methylation of arachno-6,9-C2B8H14,5 prompted us to examine a similar methylation strategy in the ten-vertex closo-1,6-C2B8H10 carborane series in which, except for electrophilic halogenation,6 no work on B-substitution has so far been reported. In this work we would like to present synthetic studies on reactions of dicarbaboranes closo-1,6-R2C2B8H8 (1) (where R = H or Me) with already established methylation agents that lead to permethylation or, at least, to persubstitution on B-vertices.Results and discussion
Scheme 1 (left) demonstrates that the HOTf-catalyzed MeOTf methylation of the parent closo-1,6-H2C2B8H8 (1a) dicarbaborane in a Pyrex tube leads at different temperatures (110–175 °C) and varying periods of reaction time (48–120 h) to persubstitution in all B-positions to generate a mixture of the B-permethylated derivative closo-1,6-H2C2B8Me8 (2a) and the triflate closo-1,6-H2C2B8Me7-8-OTf (2b). The mixture was separated by LC on a silica gel support, using hexane and a 10% CH2Cl2-hexane mixture as the mobile phase. The yields of 2a and 2b varied in ranges 15–40% and 50–70%, respectively (for approximate yields ±5%, see Table 1 and for calculations on the reaction mechanisms, see ref. 7). The table suggests that raising temperatures over shorter reaction time seem to lend support to the formation of the B-permethylated 2a and 4a. | ||
| Scheme 1 Synthesis of B-methyl derivatives of the closo-1,6-H2C2B8H8 family. | ||
| Subst. | °C | Time (h) | Yield 2a or 4a (%) | Yield 2b or 4b (%) |
|---|---|---|---|---|
| 1a | 110 | 120 | 20 | 70 |
| 1a | 130 | 90 | 25 | 65 |
| 1a | 175 | 48 | 40 | 50 |
| 1b | 110 | 120 | 15 | 70 |
| 1b | 130 | 90 | 15 | 75 |
| 1b | 175 | 48 | 40 | 40 |
An attempt at AlCl3-catalyzed methylation of 1a with neat MeI (Pyrex tube, 80 °C, 72 h) yielded, however, only the iodo compound closo-1,6-H2C2B8Me7-8-I (2c) (yield ∼75%) which was isolated and purified, similarly as in the preceding case, via LC chromatography. It is evident that the yields of the permethylated 2a are exacerbated by the formation of the 8-X substituted compounds 2b and 2c. As demonstrated on the reaction between 2a and HOTf in MeOTf (170 °C, ∼50 h, yield of 2b 96%), these substituted compounds are likely to be formed as a consequence of electrophilic H+-attack by HX evolved in the methylation reactions (X = OTf and I) at the most negative B8 site6 of 2a:
| {1,6-H2C2B8Me8 (2a) + HX → CH4 + 1,6-H2C2B8Me7-8-X (2b and 2c) | (1) |
It should be also noted that 2c can be also prepared in 81% yield by MeI/AlCl3 methylation of the “pre-iodinated” closo-1,6-H2C2B8H7-8-I (1c) derivative.6
Scheme 1 (right) also shows that the HOTf-catalyzed MeOTf methylation of the C-dimethylated dicarbaborane closo-1,6-Me2C2B8H8 (1b) (110 °C, 120 h) leads only to the octamethylated product closo-1,6-Me2C2B8Me6-2,3-H2 (3a, identified by 11B NMR, see Fig. S16†), in which boron positions between both cluster carbon vertexes remain unmethylated, which is probably due to combined sterical and electronic effects of the CMe groups. However, under forcing conditions (15 h, 175 °C), the methylation can be completed to form a mixture of the permethylated closo-1,6-Me2C2B8Me8 (4a) (yield ∼15%) with the triflate closo-1,6-Me2C2B8Me7-8-OTf (4b) (yield ∼75%) which was separated via LC in a 10% dichloromethane/hexane mixture. Nevertheless, the best yield of 4a (∼40%) was achieved at 175 °C over a period of 48 h (see Table 1). Unfortunately, an attempt at AlCl3-catalyzed methylation of 1b with neat MeI (80 °C, 72 h) yielded an inseparable mixture of at least three products, in which the prevailing iodo compound, closo-1,6-Me2C2B8Me7-8-I (4c), could be identified by 11B NMR spectroscopy (see Fig. S16†).
The structures of 8-OTf and 8-I-derivatives 2b and 2c were determined by an X-ray diffraction analysis (see Fig. 1 and 2). Their crystallographic parameters thus extend those reported previously for 1b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 and 1,6-H2C2B8H7-8-Br6 derivatives of 1. Both compounds adopt Archimedean antiprismatic geometry with apical distances generally shorter than intra- and interbelt separations, the C–B bond vectors being generally shorter than B–B bonds. The structures unambiguously confirm the B8-substitution along with persubstitution in all other B- positions of the cluster. The crystals of 2a and 4a were not suitable for crystallographic studies and their molecular geometries were therefore optimized at the MP2/TZVP level (Fig. 3 and 4). The optimization revealed that the comparable B–B and B–Me bonding vectors are very similar to those found crystallographically for 2b and 2c. The computation has also led to a good agreement between theoretical and experimental δ(11B) values for 2a and 4a (max. deviation less than 3 ppm), for the individual values see ESI.†
8 and 1,6-H2C2B8H7-8-Br6 derivatives of 1. Both compounds adopt Archimedean antiprismatic geometry with apical distances generally shorter than intra- and interbelt separations, the C–B bond vectors being generally shorter than B–B bonds. The structures unambiguously confirm the B8-substitution along with persubstitution in all other B- positions of the cluster. The crystals of 2a and 4a were not suitable for crystallographic studies and their molecular geometries were therefore optimized at the MP2/TZVP level (Fig. 3 and 4). The optimization revealed that the comparable B–B and B–Me bonding vectors are very similar to those found crystallographically for 2b and 2c. The computation has also led to a good agreement between theoretical and experimental δ(11B) values for 2a and 4a (max. deviation less than 3 ppm), for the individual values see ESI.†
 | ||
| Fig. 1 ORTEP (30% probability level) representation of the molecular structure of closo-1,6-H2C2B8Me7-8-OTf (2b). Selected bond lengths (Å): O1–B8 1.473(3), C1–B2 1.594(4), C1–B4 1.606(4), B2–B3 1.888(4), B4–B5 1.867(4), B5–B2 1.839(4), C6–B2 1.744(4), C6–B7 1.772(4), C6–B10 1.621(4), B3–B7 1.808(4), B4–B8 1.829(4), B2–B9 1.810(4), B7–B8 1.826(4), B7–B10 1.728(4), B9–B10 1.728(4), B2–C2 1.569(4), B10–C10 1.571(4); angles (°): O1–B8–B4 112.3(2), O1–B8–B7 103.6(2), B2–C1–B3 72.8(2), B3–C1–B4 110.8(2), B2–B3–B4 89.9(2), B2–C6–B3 65.3(2), C6–B7–B8 84.6(2), B9–C6–B7 97.4(2). | ||
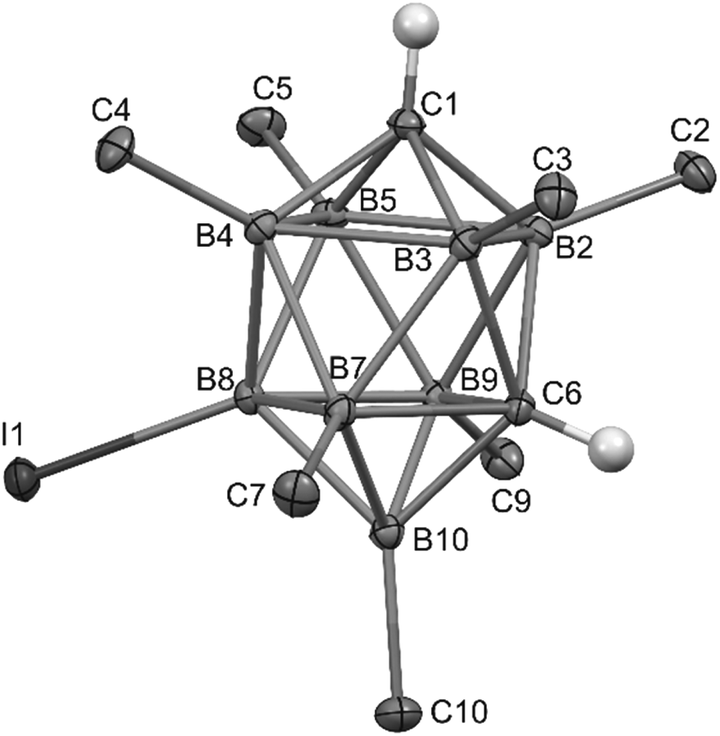 | ||
| Fig. 2 ORTEP (40% probability level) representation of the molecular structure of closo-1,6-H2C2B8Me7-8-I (2c). Selected bond lengths (Å): I1–B8 2.176(3), C1–B2 1.596(4), C1–B4 1.615(4), B2–B3 1.896(4), B4–B5 1.879(4), B5–B2 1.851(4), C6–B2 1.746(4), C6–B7 1.774(4), C6–B10 1.620(4), B3–B7 1.807(4), B4–B8 1.828(4), B2–B9 1.806(4), B7–B8 1.821(4), B7–B10 1.724(4), B9–B10 1.720(4), B2–C2 1.569(4), B10–C10 1.564(3); angles (°): I1–B8–B4 119.3(2), I1–B8–B7 129.0(2), B2–C1–B3 72.9(2), B3–C1–B5 111.1(2), B2–B3–B4 89.6(2), B2–C6–B3 65.7(2), C6–B7–B8 85.4(2), B9–C6–B7 96.4(2). | ||
 | ||
| Fig. 3 Molecular model of closo-1,6-H2C2B8Me8 (2a) as obtained from the MP2/TZVP optimization. B2–B3 is the longest vector in the molecule, viz 1.889 Å. The other selected internal coordinates are (distances in Å, angles in °): C1–B2 1.593, C1–B4 1.610, B4–B5 1.854, B5–B2 1.843, C6–B2 1.736, C6–B7 1.764, C6–B10 1.623, B3–B7 1.799, B4–B8 1.832, B2–B9 1.800, B7–B8 1.845, B7–B10 1.707, B9–B10 1.707, B2–C1–B3 72.7, B3–C1–B5 110.2, B2–B3–B4 89.5, B2–C6–B3 65.9, C6–B7–B8 85.4. | ||
 | ||
| Fig. 4 Molecular model of closo-1,6-Me2C2B8Me8 (4a) as obtained from the MP2/TZVP optimization. B2–B3 is the longest vector in the molecule, viz 1.886 Å. The other selected internal coordinates are (distances in Å, angles in °): C1–B2 1.599, C1–B4 1.620, B4–B5 1.851, B5–B2 1.841, C6–B2 1.750, C6–B7 1.799, C6–B10 1.625, B3–B7 1.796, B4–B8 1.837, B2–B9 1.797, B7–B8 1.853, B7–B10 1.705, B9–B10 1.705, B2–C1–B3 72.2, B3–C1–B5 109.1, B2–B3–B4 89.5, B2–C6–B3 65.2, C6–B7–B8 87.1. | ||
The HF/cc-pVTZ calculations of the electrostatic potential (ESP) surface show that the parent 1a has hydridic BH vertices which can form dihydrogen bonds,9 whereas the hydridic B-bound hydrogens in 2a and 4a are replaced by methyl groups of amphiphilic character.10 The hydrogen atoms of the methyl groups have positive ESP surface and the exo-skeletal carbon atoms have negative ESP surface (see Fig. 5). From the viewpoint of electron transmission, the Me groups behave as weak electron acceptors, which might be surprising, as it is opposed to toluene, xylene, hexamethylbenzene etc. However, the electron transmission just follows the established electronegativity concept that reflects the fact that exo-skeletal substituents are bonded via classical 2-centre 2-electron bonds.11 This agrees with the concept elaborated by Viñas et al. for a 12-vertex closo system.12 Conceivably, the whole 11B NMR spectra of 2a and 4a are significantly paramagnetically deshielded to high frequencies (i.e. downfield shifted) with respect to that of 1a; for the computed 11B NMR spectrum of the latter see ESI.†
 | ||
| Fig. 5 Computed (HF/cc-pVTZ) electrostatic potential (ESP) surface for (A) 1a, (B) 2a and (C) 4a. The color range of the ESP in kcal mol−1. | ||
The structures of all compounds isolated in this study are also in excellent agreement with the results of multinuclear 11B, [11B-11B]-COSY,131H, and 13C NMR measurements that led to complete assignments of individual cage BMe, BX and CH units. Graphical inter-comparisons of the 11B NMR spectra of all compounds isolated are in Fig. S15 and S16.† The 11B and 11B-{1H}-decoupled NMR spectra of all B-methylated isomers isolated are identical, exhibiting only singlet resonances, which unambiguously points to persubstitution in all B-vertexes. All the 11B NMR spectra of the Cs-symmetry (a slight gear effect of the Me groups) 2–4 derivatives, exhibiting 1:1:2:2:2 patterns of singlets, are in agreement with the presence of the C1–C6–B10 plane.
The 13C and 1H NMR spectra of compounds of type 2 and 4 are consistent with the presence of two different CH or CMe and cluster units in positions 1 and 6 (reading upfield) and six or seven, mostly well resolved, Me groups resonating in the high-field area (see comparative Fig. S17 and S18†). Due to the Me-substitution, all the 11B and 13C resonances in the spectra of the B-permethylated compounds 2 are deshielded when compared to those of the B-unsubstituted counterparts of structure 1, while the corresponding 1H-resonances of cage CH units are shielded. Similar features can be also traced in the spectra of 8-X compounds with differences attributed to electronic effects induced by OTf and I substituents. The 19F NMR spectra of the triflato compounds 2b and 4b are consistent with the presence of one CF3 group (see Fig. S17 and S18†).
The spectra of the permethylated compounds 2a and 4a are exemplified in Fig. 6 and 7 along with signal assignments, for selection of other key NMR measurements see ESI (Fig. S1–S14†).
 | ||
| Fig. 6 190.2 MHz 11B, 600 MHz 1H, and 150.9 MHz 13C NMR spectra of 1,6-H2C2B8Me8 (2a) (CDCl3). | ||
 | ||
| Fig. 7 190.2 MHz 11B, 600 MHz 1H, and 150.9 MHz 13C NMR spectra of 1,6-Me2C2B8Me8 (4a) (CDCl3). | ||
Of extreme importance for further developments in the field of B-clusters containing less than twelve cage vertices is the question of their resistance to various cage-degradation agents. According to our experience, the ten-vertex dicarbaboranes are most sensitive to the degradation with alcohols. In order to estimate the effect of the Me-sheath, we have tested the behaviour of the 1a/2a pair to ethanolysis in the presence of a ten-fold excess of EtOH than required by eqn (2) and (3):
| 1,2-H2C2B8H8 + 24EtOH → 8B(OEt)3 + C2H6 + 14H2 | (2) |
| 1,2-H2C2B8Me8 + 16EtOH → 8MeB(OEt)2 + C2H6 + 6H2 | (3) |
The progress of the reactions could be easily monitored by integrated 11B NMR and a two-day treatment with EtOH had no effect to 2a, while the unprotected 1a was completely degraded to give B(OEt)3 over a period of ∼10 h.
Experimental
Materials and methods
All the reactions were carried out under argon atmosphere. Dichloromethane and hexane were dried over CaH2 and freshly distilled before use. Other conventional chemicals were of reagent or analytical grade and were used as purchased. NMR spectroscopy was performed at 400 and 600 Mz (for 1H), inclusive of standard [11B-11B]-COSY13 experiments (all theoretical cross-peaks were observed) leading to complete assignments of all resonances to individual cage B-vertexes. Chemical shifts are given in ppm to high-frequency (low field) of Ξ = 32.083971 MHz (nominally F3B·OEt2 in CDCl3) for 11B (quoted ± 0.5 ppm), Ξ = 25.144 MHz for 13C (quoted ± 0.5 ppm), and Ξ = 100 MHz for 1H (quoted ± 0.05 ppm), Ξ is defined as in ref. 14 and the solvent resonances were used as internal secondary standards. The starting carboranes 1 and 1c were prepared according to the reported methods.6Dimethylation of closo-1,6-C2B8H10 (1a) on carbon vertexes
A solution of 1a (120 mg, 1 mmol) in dry Et2O (ca. 10–20 ml) was cooled to −78 °C and then treated dropwise with 2.5 M LiBu (solution in hexane) (1 ml, 2.5 mmol) under stirring. The off-white slurry of the Li+ salt was stirred for additional 1 h prior to dropwise addition of methyl triflate (MeOTf, m.w. 164.1) (410 mg, 2.5 mmol) under cooling down in an dry-ice bath. The mixture was then left stirring for additional 2 h at room temperature. After adding 5% hydrochloric acid (10 ml) under repeated cooling and shaking, the Et2O layer was separated and evaporated to provide closo-1,6-Me2C2B8H8 (1b) (140 mg, 93%) on sublimation. The purity was assessed by 11B NMR spectroscopy (see Fig. S6 and S7†).closo-1,6-R2C2B8Me8 (2a and 4a) and closo-1,6-R2C2B8Me7-8-OTf (2b and 4b) (where R = H or Me)
A solution of carboranes 1a or 1b (reaction scale ∼1.5 mmol) in neat MeOTf (10 ml) was treated with three drops of HOTf and the mixture was heated at different temperatures for varying times in a thick-walled reaction vessel equipped with a Teflon screw cap. In each experiment were the volatiles evaporated and the residue extracted with hexane and filtered through a silica gel plug. The filtrate was then treated with silica gel (∼5 g) and the hexane evaporated. The residual material was mounted onto the top of a column packed with silica gel (∼2.5 × 20 cm) which was then eluted with hexane, followed by a 15% CH2Cl2/hexane mixture to collect two main fractions of RF (anal., hexane) ∼0.6 and 0.10 which were identified by 11B NMR and isolated via solvent evaporation as compounds 2a, 2b, 4a, and 4b. Further purification can be achieved by vacuum sublimation at bath temperature ∼130 °C. The isolated yields of individual compounds are in Table 1.For 2a: MS (ESI−): m/z (max.) calcd 233.28, found 233.25; for C10H26B8 (m.w. 232.80) calcd 51.59% C, 11.26% H; found 50.79% C, 11.05% H. For 2b MS (ESI−): m/z (max.) calcd 367.21, found 367.30; for C10H23B8O3SF3 (m.w. 366.84) calcd 32.74% C, 6.32% H; found 32.30% C, 6.11% H. For 4a: MS (ESI−): m/z (max.) calcd 261.32, found 261.30; for C12H30B8 (m.w. 260.85) calcd 55.25% C, 11.59% H; found 54.80% C, 11.05% H. For 4b MS (ESI−): m/z (max.) calcd 395.24, found 395.25; for C12H27B8O3SF3 (m.w. 394.89) calcd 36.50% C, 6.89% H; found 36.10% C, 6.58% H.
closo-1,6-H2C2B8Me7-8-OTf (2b) from closo-1,6-H2C2B8Me8 (2a)
A solution of compound 2a (40 mg, 0.17 mmol) in MeOTf (2 ml) was treated with HOTf (204 mg, 1.36 mmol) and heated at 170 °C for ∼50 h in a sealed glass ampoule. The volatiles were then evaporated, the residue dissolved in hexane and filtered through a silica gel plug. The hexane was then evaporated to give 60 mg (96%) of a white crystals which were identified as 2b as in the preceding experiment.closo-1,6-H2C2B8Me7-8-I (2c)
(a) From1a: A solution of carborane 1a (122 mg, 1.0 mmol) in neat MeI (10 ml) was heated in a thick-walled reaction vessel equipped with a Teflon screw cap on addition of anhydrous AlCl3 (ca. 14 mg, 0.14 mmol) at 80 °C for 72 hours. The volatiles were evaporated and the residue extracted with hexane. The extract was then treated with silica gel (∼5 g) and the hexane evaporated. The residual material was mounted atop of a column packed with silica gel (∼2.5 × 20 cm) which was then eluted with 10% CH2Cl2 in hexane to collect the main fractions of RF (anal., hexane) ∼0.2 which was isolated via solvent evaporation and identified by 11B NMR as compound 2c (269 mg, 78%). Further purification can be achieved by vacuum sublimation at bath temperature ∼130 °C. For 2c: MS (ESI−): m/z (max.) calcd 345.16, found 345.12; for C9H23B8I (m.w. 344.67) calcd 31.36% C, 6.73% H; found 30.47% C, 6.51% H. An analogous experiment with 1b gave a mixture of at least three compounds (350 mg, assessed by 11B NMR) that could not be separated via LC. (b) From1c: A solution of the iodo carborane 1c (51 mg, 0.21 mmol) in neat MeI (3 ml) was heated in a thick-walled reaction vessel equipped with a Teflon screw cap on addition of anhydrous AlCl3 (ca. 20 mg, 0.2 mmol) at 80 °C for 72 hours. Further work-up as in the preceding experiment led to the isolation of 59 mg (81%) of 2c as a single product which was identified by 11B NMR spectroscopy.NMR test of resistance toward ethanolysis for closo-1,6-C2B8H10 (1a) and closo-1,6-H2C2B8Me8 (2a)
Compounds 1a (25 mg, 0.2 mmol) and 2a (25 mg, 0.1 mmol) were dissolved in ethanol (2 ml) In a NMR tube and both solutions were monitored via integrated 11B NMR at ambient temperature. After ∼10 h exposure the signals of 1a have completely disappeared (see Fig. S15†) to form B(OEt)3, while those of 2a did not change their intensity during 48 h (no formation of B(OEt3) was detected).Computational details
Magnetic shielding was calculated using the GIAO-MP2 method incorporated into Gaussian0915 utilizing the IGLO-II basis with the MP2/TZVP geometry and frozen core electrons. Electrostatic potentials were computed at the HF/cc-pVDZ level using Gaussian09 and Molekel4.316 programs. It has recently been shown that this basis set size is sufficient for these purposes.17X-ray crystallography
The X-ray data for the triflato and iodo derivatives 2b and 2c (colourless crystals by slow evaporation of a hexane solution) were collected at 150(2)K with a Bruker D8-Venture diffractometer equipped with Mo (Mo/Kα radiation; λ = 0.71073 Å) microfocus X-ray (IμS) source, Photon CMOS detector and Oxford Cryosystems cooling device was used for data collection. The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm. Data were corrected for absorption effects using the Multi-Scan method (SADABS). Obtained data were treated by XT-version 2014/5 and SHELXL-2014/7 software implemented in APEX3 v2016.5-0 (Bruker AXS) system.18 Hydrogen atoms were mostly localized on a difference Fourier map, however to ensure uniformity of treatment of crystal, all hydrogen were recalculated into idealized positions (riding model) and assigned temperature factors Hiso(H) = 1.2Ueq (pivot atom) or of 1.5Ueq (methyl). H atoms in methyl groups were placed with C–H distances of 0.96 while the hydrogen atoms of the C–H in the carborane cage were assigned according to the maxima on the difference Fourier map. Rint = ∑|Fo2 − Fo,mean2|/∑Fo2, S = [∑(w(Fo2 − Fc2)2)/(Ndiffrs − Nparams)]½ for all data, R(F) = ∑||Fo| − |Fc||/∑|Fo|for observed data, wR(F2) = [∑(w(Fo2 − Fc2)2)/(∑w(Fo2)2)]½ for all data. Crystallographic data for structural analysis have been deposited with the Cambridge Crystallographic Data Centre, CCDC no. 1573271 and 1573272† for 2b and 2c, respectively. Crystallographic data for 2b: C10H23B8F3O3S, M = 366.82, orthorhombic, P212121, a = 10.2153(5), b = 12.7603(7), c = 15.3713(9) Å, β = 90°, Z = 4, V = 2003.65(19) Å3, Dc = 1.216 g cm−3, μ = 0.193 mm−1, Tmin/Tmax = 0.3997/0.7455; −12 ≤ h ≤ 13, −14 ≤ k ≤ 16, −19 ≤ l ≤ 19; 17134 reflections measured (θmax = 27.12°), 4409 independent (Rint = 0.0291), 4098 with I > 2σ(I), 241 parameters, S = 1.046, R1 (obs. data) = 0.0394, wR2 (all data) = 0.1050; max., min. residual electron density = 0.185, −0.306 e Å−3. Flack parameter 0.02(2). Crystallographic data for 2c: C9H23B8I, M = 344.65, monoclinic, P21/n, a = 7.7088(4), b = 13.5358(7), c = 16.1895(9) Å, β = 102.909(2) °, Z = 4, V = 1646.59(15) Å3, Dc = 1.390 g cm−3, μ = 1.919 mm−1, Tmin/Tmax = 0.5122/0.745; −9 ≤ h ≤ 9, −17 ≤ k ≤ 17, −20 ≤ l ≤ 20; 28878 reflections measured (θmax = 27.12°), 3635 independent (Rint = 0.0263), 3116 with I > 2σ(I), 178 parameters, S = 1.182, R1 (obs. data) = 0.0257, wR2 (all data) = 0.0811; max., min. residual electron density = 0.472, −1.252 e Å−3.
Conclusions
The work is the initial venture into the area of permethylated chemistry of closed cages with fewer than eleven vertices. It was demonstrated that all B-positions in the closo-1,6-R2C2B8H8 (1) framework can be furnished with methyl substituents via electrophilic reactions with MeOTf and/or MeI reagents. In contrast to its 12-vertex 1,7-R2C2B10H10 analogue,3 all the B-positions, inclusive of those between carbon vertexes, can be methylated. The reactions result in permethylated derivatives of 1a which can be, in fact, viewed as rigid, egg-shaped hydrocarbons with a carborane core inside (see Fig. 5). It should be underlined that such structural arrangements impart an exceptionally high air and solvolytic stability to the otherwise moderately stable compounds 1 by creating a protective methyl sheath around the most sensitive boron section of the molecule.5 For example, stability tests have proved that compound 2a can be quantitatively recovered from ethanolic solution after a 48 h exposure to solvolysis, while the unprotected 1a is decomposed to B(OEt)3 over ∼10 h ethanolysis, as assessed by 11B NMR measurements. Furthermore, from experience we know that solid 2a is stable in air for at least two weeks without any noticeable degradation to B(OH)3. This straightforward enhancement of stability actually brings positive expectations for smaller-cage B-chemistry that was practically abandoned for air-instability as one of the reasons; one might therefore expect new developments in the area of highly alkylated compounds with less than twelve vertexes in the near future. Moreover, Me-substituted derivatives thus far isolated can be used as “Me-labels” for designed syntheses in specific areas of carborane chemistry, for example in metal-insertion, cluster-rearrangement or cluster-degradation reactions; relevant experiments are being in progress in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Grant Agency of the Czech Republic (project no. 16-01618S).Notes and references
- For review, see: J. J. Rockwell, A. Herzog, T. Peymann, C. B. Knobler and M. F. Hawthorne, Curr. Sci., 2000, 78, 405–409 Search PubMed.
- (a) B. T. King, Z. Janoušek, B. Grüner, M. Trammell, B. C. Noll and J. Michl, J. Am. Chem. Soc., 1996, 118, 3313–3314 CrossRef; (b) For review, see: S. Körbe, P. J. Schreiber and J. Michl, Chem. Rev., 2006, 106, 5208–5249 CrossRef PubMed.
- A. Herzog, A. Maderna, G. N. Harakas, C. B. Knobler and M. F. Hawthorne, Chem. – Eur. J., 1999, 5, 1212–1217 CrossRef.
- W. Jiang, C. B. Knobler, M. D. Mortimer and M. F. Hawthorne, Angew. Chem., Int. Ed. Engl., 1995, 34, 1332–1334 CrossRef.
- M. Bakardjiev, B. Štíbr, J. Holub, O. L. Tok, P. Švec, Z. Růžičková and A. Růžička, Inorg. Chem., 2016, 55, 7068–7074 CrossRef PubMed.
- M. Bakardjiev, B. Štíbr, J. Holub, Z. Padělková and A. Růžička, Organometallics, 2015, 34, 450–454 CrossRef.
- J. Kaleta, A. Akdag, R. Crespo, M.-C. Piqueras and J. Michl, ChemPlusChem, 2013, 78, 1174–1183 CrossRef.
- T. F. Koetzle, F. E. Scarbrough and W. N. Lipscomb, Inorg. Chem., 1970, 9, 2279–2285 CrossRef.
- J. Fanfrlík, M. Lepšík, D. Hořínek, Z. Havlas and P. Hobza, ChemPhysChem, 2006, 7, 1100–1105 CrossRef PubMed.
- (a) C. Esterhuysen, A. Hesselmann and T. Clark, ChemPhysChem, 2017, 18, 772–784 CrossRef PubMed; (b) J. Fanfrlík, A. Pecina, J. Řezáč, R. Sedlák, D. Hnyk, M. Lepšík and P. Hobza, Phys. Chem. Chem. Phys., 2017, 19, 18194–18200 RSC.
- In contrast, in the closo-1,6-C2B8 skeleton the C1 and C6 atoms participate in five multicenter bondings of 3c2e and 4c2e types, which results in in their positive charges as opposed to classical electronegativity complex. See: P. Melichar, D. Hnyk and J. Fanfrlík, Phys. Chem. Chem. Phys., 2018, 20, 4666–4675 RSC . Positive charges of endo-carbons in closo systems were also verified experimentally: D. Hnyk, V. Všetečka, L. Drož and O. Exner, Collect. Czech. Chem. Commun., 2001, 66, 1375–1379 CrossRef.
- F. Teixidor, G. Barberà, A. Vaca, R. Kivekäs, R. Sillanpää, J. Oliva and C. Viñas, J. Am. Chem. Soc., 2005, 127, 10158–10159 CrossRef PubMed.
- W. C. Hutton, T. L. Venable and R. N. Grimes, J. Am. Chem. Soc., 1984, 106, 29–37 CrossRef.
- W. A. McFarlane, Proc. R. Soc. London, Ser. A, 1968, 306, 175–199 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, 2009.
- (a) MOLEKEL 4.3: P. Flükiger, H. P. Lüthi, S. Portmann and J. Weber, Swiss Center for Scientific Computing, Manno (Switzerland), 2000 Search PubMed; (b) S. Portmann and H. P. Lüthi, Molekel: Chimia, 2007, 28, 555 Search PubMed.
- K. E. Riley, K.-A. Tran, P. Lane, J. S. Murray and P. Politzer, J. Comput. Sci., 2016, 17, 273 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data and computed 11B NMR, cartesian geometries. CCDC 1573271 and 1573272. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt02586j |
| This journal is © The Royal Society of Chemistry 2018 |