
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Energy transfer between Eu3+ and Nd3+ in near-infrared emitting β-triketonate coordination polymers†
Laura
Abad Galán
ab,
Alexandre N.
Sobolev
cd,
Brian W.
Skelton
c,
Eli
Zysman-Colman
 *b,
Mark I.
Ogden
*a and
Massimiliano
Massi
*a
*b,
Mark I.
Ogden
*a and
Massimiliano
Massi
*a
aSchool of Life and Molecular Science and Curtin Institute for Functional Molecules and Interfaces, Curtin University, Kent Street, Bentley 6102, WA, Australia. E-mail: m.massi@curtin.edu.au; m.ogden@curtin.edu.au
bOrganic Semiconductor Centre, EaStCHEM School of Chemistry, University of St Andrews, St Andrews, Fife KY16 9ST, UK. E-mail: eli.zysman-colman@st-andrews.ac.uk
cSchool of Molecular Sciences, M310, University of Western Australia, Perth 6009, WA, Australia
dCentre for Microscopy, Characterisation and Analysis, M310, University of the Western Australia, Perth 6009, WA, Australia
First published on 20th July 2018
Abstract
Isomorphous β-triketonate-based lanthanoid polymers containing tris(4-methylbenzoyl)methanide (mtbm) and Rb+ with formula {[Ln(Rb)(mtbm)4]2}n (Ln = Eu3+ and Nd3+) have been synthesised and structurally characterised. The photophysical properties for the Nd3+ complex presented relatively long lifetimes and high quantum yields in comparison with analogous β-diketonate complexes. Mixed lanthanoid complexes were also formed and their luminescence properties studied, with effective sensitisation of the 4F3/2 of Nd3+via the 5D0 of Eu3+, which is to the best of our knowledge the first example of Eu3+ to Nd3+ sensitisation in a structurally defined coordination complex or polymer.
Introduction
Much attention has been paid to materials incorporating trivalent lanthanoid cations due to their unique photophysical properties such as their line-like emission spectra and their long-lived excited state lifetimes as a result of intraconfigurational f–f transitions. In addition, their emission colours range from the UV to the near infrared (NIR), and it is exclusively characteristic of the specific lanthanoid cation (e.g. red emission from europium or green emission from terbium). The NIR region is of particular interest due to potential applications in a wide range of fields including night vision devices, telecommunication signalling and life science.1–6 Despite the listed advantages, trivalent lanthanoid cations cannot be directly excited with high efficiency, as intraconfigurational f–f transitions are parity and often spin forbidden. Therefore, π-conjugated ligands are routinely used as sensitisers, because of their greater efficiency in absorbing incident light with consequent energy transfer to lanthanoid excited states. This alternative pathway, which is termed antenna effect, is well established and is generally rationalised as energy transfer from the triplet state of the conjugated ligands, populated via intersystem crossing due to the strong spin–orbit coupling of the lanthanoid elements, to the excited energy levels of the lanthanoid cation.7,8Furthermore, in the design of emissive lanthanoid complexes it is also necessary to avoid high energy vibrations in close proximity to the metal centre, such as the presence of OH and NH bonds. The activation of vibrational modes of these bonds acts as an efficient quencher for lanthanoid excited states. In the case of NIR emission, which is of particular interest here, CH bonds can also become an efficient source of quenching.7
β-Diketones with aromatic substituents, such as dibenzoylmethane, have commonly been used as antenna ligands because of their good chelating properties and their ability to effectively sensitise the trivalent lanthanoid excited states, particularly in the solid state.9 Of the potential near-IR emitting systems, Nd3+ diketonate complexes have been studied in less detail than the Yb3+ and Er3+ compounds. Reported quantitative data (quantum yields, lifetimes) are very limited, despite the fact that a variety of Nd3+ β-diketonate complexes can be found in the literature over the last couple of decades.10–17 As for all of the near-IR emitting systems, the design of the Nd3+ β-diketonate complexes typically involves two main strategies to improve photophysical properties: (i) adjusting the triplet state energy of the antenna in order to optimise energy transfer to Nd3+ and (ii) minimising the nonradiative relaxation pathways.13,14
In extending the coordination chemistry of luminescent lanthanoid β-diketonate complexes, we have been recently exploring the use of β-triketonate molecules as antenna ligands for lanthanoids. These ligands have been found to support the formation of unique assemblies that display particularly enhanced ytterbium and erbium emission properties. Our previous studies with tris-benzoylmethane (tbmH) and tris(4-methylbenzoyl)methane (mtbmH) resulted in the isolation of tetranuclear assemblies and polymeric structures of formulation [Ln(Ae·HOEt)(tbm)4]2 (Ln3+ = Eu3+, Er3+, Yb3+/Ae+ = Na+, K+, Rb+)18,19 and {[Ln(Cs)(mtbm)4]2}n (Ln3+ = Eu3+ and Er3+),20 respectively. In contrast, our initial attempts to isolate the corresponding neodymium analogues were not successful and their photophysical properties remained unknown.
In this work, we report the successful extension of these studies to neodymium-containing assemblies using both tbmH and mtbmH ligands in the presence of RbOH and CsOH. The syntheses, crystal structures and emission properties of the resulting assemblies are reported.
Furthermore, since the structure of these complexes were found to be similar for the different lanthanoids, we have studied the formation of mixed assemblies for the purpose of investigating energy transfer processes or multiple emission from the same material. Excited states of a lanthanoid have been previously exploited to sensitise excited states of another lanthanoid.21–24 This approach is well established for certain pairings with NIR emitters, for example sensitisation of erbium luminescence via energy transfer from the 2F5/2 excited state of trivalent ytterbium,25–27 or ytterbium luminescence via visible emitters such as terbium or europium.28,29 In contrast, to our knowledge, neodymium sensitisation via other lanthanoids has not been reported in coordination complexes. Only three examples have been reported where Eu/Nd energy migration was used to determine the lanthanoid–lanthanoid distance following pure Förster mechanisms.30–32 However, these studies are focused on the quenching of the europium excited states but do not report any associated near-IR emission from the neodymium centres. This sensitisation process for neodymium emission has been seen in the case of Eu/Nd doped glasses,33,34 so should also be possible in the comparatively well-defined structure of a coordination complex. Indeed, this study presents the first example of a coordination complex with effective lanthanoid-lanthanoid energy transfer from the 5D0 of Eu3+ to the 4f* of Nd3+, leading to dual emission.
Experimental
General procedures
All reagents and solvents were purchased from chemical suppliers and used as received without further purification. The ligand tribenzoylmethane (tbmH), was prepared as previously reported.31 Hydrated LnCl3 (Ln = Eu3+, Er3+, Yb3+) was prepared by the reaction of Ln2O3 with hydrochloric acid (5 M), followed by evaporation of the solvent under reduced pressure. Infrared spectra (IR) were recorded on solid-state samples using an attenuated total reflectance PerkinElmer Spectrum 100 FT-IR. IR spectra were recorded from 4000 to 650 cm−1; the intensities of the IR bands are reported as strong (s), medium (m), or weak (w), with broad (br) bands also specified. Melting points were determined using a BI Barnsted Electrothermal 9100 apparatus. Elemental analyses were obtained at Curtin University, Australia. Nuclear magnetic resonance (NMR) spectra were recorded using a Bruker Avance 400 spectrometer (400.1 MHz for 1H; 100 MHz for 13C) at 300 K. The data were acquired and processed by the Bruker TopSpin 3.1 software. All of the NMR spectra were calibrated to residual solvent signals.Selected equations
The values of the radiative lifetime (τR), and intrinsic quantum yield (ΦLnLn), can be calculated with the following equations.36 | (1) |
In eqn (1), the refractive index (n) of the solvent is used (assumed value of 1.5 in the solid state), the value 14.65 s−1 is the spontaneous emission probability of the 7F1 ← 5D0 transition reported previously. ITot is the total integration of the Eu3+ emission spectrum, and IMD is the integration of the 7F1 ← 5D0 transition.
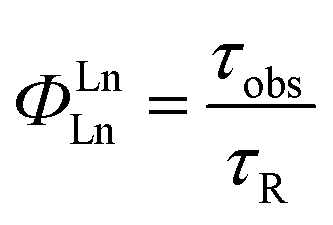 | (2) |
The sensitisation efficiency (ηsens) can be determined using eqn (3) below:
 | (3) |
The rate of energy transfer (KET) and quantum efficiency of energy transfer (ΦET) can be calculated according to the following equations:
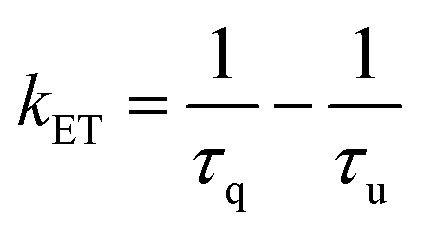 | (4) |
 | (5) |
In eqn (4) and (5), τq and τu are the 5D0 decay lifetime of Eu3+ in the presence or absence of the quencher (Nd3+), respectively.
For dipole–dipole exchange mechanisms or Förster the donor–acceptor distance (RDA) can be calculated following eqn (6):
 | (6) |
Photophysical measurements
Absorption spectra were recorded at room temperature using a PerkinElmer Lambda 35 UV/Vis spectrometer. Uncorrected steady-state emission and excitation spectra were recorded using an Edinburgh FLSP980-stm spectrometer equipped with a 450 W xenon arc lamp, double excitation and emission monochromators, a Peltier-cooled Hamamatsu R928P photomultiplier (185–850 nm) and a Hamamatsu R5509-42 photomultiplier for detection of NIR radiation (800–1400 nm). Emission and excitation spectra were corrected for source intensity (lamp and grating) and emission spectral response (detector and grating) by a calibration curve supplied with the instrument. Overall quantum yields (ΦLLn) were measured with the use of an integrating sphere coated with BenFlect.36 In the case of the NIR, overall quantum yields were measured using two different detectors and [Yb(phen)(tta)3] in toluene (ΦLLn = 1.6%),37 where tta is thenoyltrifluoroacetone, as reference to calibrate the set up according to the procedure previously reported by our group.9Excited-state decays (τ) were recorded on the same Edinburgh FLSP980-stm spectrometer using a microsecond flashlamp. The goodness of fit was assessed by minimising the reduced χ2 function and by visual inspection of the weighted residuals.
Synthesis
{[Eu(Rb)(mtbm)4]2}n. 36 mg, 21%. M.p. 267–269 °C. Elemental analysis calcd (%) for C200H168Rb2Eu2O24 (1.75·H2O): C, 68.53; H, 5.05; found: C, 68.53; H, 4.74. IR (ATR): ν = 2919 w, 1634 w, 1602 m, 1577 m, 1538 s, 1408 m, 1360 s, 1275 m, 1183 m, 1151 m, 1115 w, 1021 w, 899 s, 837 m, 7836 s, 721 m, 694 w.
{[Nd(Rb)(mtbm)4]2}n. 28 mg, 16%. M.p. 289–291 °C. Elemental analysis calcd (%) for C100H84RbNdO12 (1.5·H2O): C, 66.49; H, 5.30; found: C, 66.24; H, 4.93. IR (ATR): ν = 2920 w, 2164 w, 1634 m, 1602 m, 1574 m, 1529 s, 1405 m, 1342 s, 1273 m, 1184 m, 1151 m, 1112 w, 1034 w, 899 s, 825 m, 780 s, 763 s, 721 m.
[Nd(Rb·HOEt)(tbm)4]2. 21 mg, 13%. M.p. 259–261 °C. Elemental analysis calcd (%) for C180H132Rb2Nd2O26: C, 68.19; H, 4.20; found: C, 67.70; H, 3.78. IR (ATR): ν = 3065 w, 1739 w, 1644 w, 1645 w, 1597 w, 1583 m, 1540 m, 1491 w, 1448 m, 1374 s, 1297 s, 1277 s, 1181 w, 1151 m, 1073 w, 1012 w, 897 s, 823 m, 779 w, 747 s.
Crystallography
Crystallographic data for the structures were collected at 100(2) K on an Oxford Diffraction Gemini or Xcalibur diffractometer using Mo Kα or Cu Kα radiation. Following absorption corrections and solution by direct methods, the structures were refined against F2 with full-matrix least-squares using the SHELX-2014 crystallographic package.38Unless stated below, anisotropic displacement parameters were employed for the non-hydrogen atoms. All hydrogen atoms were added at calculated positions and refined by use of a riding model with isotropic displacement parameters based on those of the parent atom.
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (No. 2), a = 14.9383(5), b = 15.9699(4), c = 17.9990(7) Å, α = 84.625(2), β = 74.799(3), γ = 88.086(2)°, V = 4125.3(2) Å3, Z = 1, Dc = 1.388 g cm−3, μ = 6.673 mm−1. F000 = 1770, Cu Kα radiation, λ = 1.54178 Å, 2θmax = 134.8°, 44
(No. 2), a = 14.9383(5), b = 15.9699(4), c = 17.9990(7) Å, α = 84.625(2), β = 74.799(3), γ = 88.086(2)°, V = 4125.3(2) Å3, Z = 1, Dc = 1.388 g cm−3, μ = 6.673 mm−1. F000 = 1770, Cu Kα radiation, λ = 1.54178 Å, 2θmax = 134.8°, 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 958 reflections collected, 14682unique (Rint = 0.0549). Final GooF = 1.059, R1 = 0.0386, wR2 = 0.0890, R indices based on 13164 reflections with I > 2σ(I), |Δρ|max = 0.67 e Å−3, 1054 parameters, 3 restraints. The water molecule hydrogen atoms were refined with geometries restrained to ideal values. CCDC 1829212.†
(no. 2), a = 14.9907(3), b = 15.9632(3), c = 17.9930(4) Å, α = 84.954(2), β = 74.674(2), γ = 88.124(2)°, V = 4136.30(15) Å3, Z = 1, Dc = 1.378 g cm−3, μ = 6.017 mm−1. F000 = 1764, Cu Kα radiation, λ = 1.54178 Å, 2θmax = 134.6°, 89010 reflections collected, 14738unique (Rint = 0.0665). Final GooF = 1.001, R1 = 0.0335, wR2 = 0.0787, R indices based on 13047 reflections with I > 2σ(I), |Δρ|max = 0.84 e Å−3, 1054 parameters, 9 restraints. The water molecule hydrogen atoms were refined with geometries restrained to ideal values. CCDC 1829213.†
(no. 2), a = 14.0539(2), b = 14.7835(3), c = 19.7708(4) Å, α = 99.829(2), β = 107.431(2), γ = 90.137(2)°, V = 3855.27(13) Å3, Z = 1, Dc = 1.405 g cm−3, μ = 1.367 mm−1. F000 = 1666, Mo Kα radiation, λ = 0.71073 Å, 2θmax = 64.7°, 84599 reflections collected, 25545unique (Rint = 0.0650). Final GooF = 1.002, R1 = 0.0496, wR2 = 0.0959, R indices based on 19518 reflections with I > 2σ (I), |Δρ|max = 1.1 e Å−3, 961 parameters, 13 restraints. One phenyl ring and two solvent ethanol molecules were modelled as being disordered over two sets of sites with occupancies constrained to 0.5 and with the non-hydrogen atoms refined with isotropic displacement parameters. Geometries of the disordered atoms were restrained to ideal values. CCDC 1829214.†
086 reflections collected, 11254unique (Rint = 0.0839). Final GooF = 1.069, R1 = 0.0486, wR2 = 0.1117, R indices based on 8726 reflections with I > 2σ(I), |Δρ|max = 1.7 e Å−3, 820 parameters, 17 restraints. The solvent was modelled as an ethanol molecule disordered about a crystallographic inversion centre. Geometries were restrained to ideal values. Water molecule hydrogen atoms were located and refined with geometries restrained to ideal values. CCDC 1829215.†
516 reflections collected, 4906 unique (Rint = 0.0436). Final GooF = 1.090, R1 = 0.0386, wR2 = 0.1095, R indices based on 4314 reflections with I > 2σ(I), |Δρ|max = 2.5 e Å−3, 349 parameters, 0 restraints. CCDC 1829216.†
616 reflections collected, 3671 unique (Rint = 0.0558). Final GooF = 1.037, R1 = 0.0455, wR2 = 0.1174, R indices based on 3361 reflections with I > 2σ(I), |Δρ|max = 2.4 e Å−3, 265 parameters, 0 restraints. CCDC 1829217.†
958 reflections collected, 14682unique (Rint = 0.0549). Final GooF = 1.059, R1 = 0.0386, wR2 = 0.0890, R indices based on 13164 reflections with I > 2σ(I), |Δρ|max = 0.67 e Å−3, 1054 parameters, 3 restraints. The water molecule hydrogen atoms were refined with geometries restrained to ideal values. CCDC 1829212.†
(no. 2), a = 14.9907(3), b = 15.9632(3), c = 17.9930(4) Å, α = 84.954(2), β = 74.674(2), γ = 88.124(2)°, V = 4136.30(15) Å3, Z = 1, Dc = 1.378 g cm−3, μ = 6.017 mm−1. F000 = 1764, Cu Kα radiation, λ = 1.54178 Å, 2θmax = 134.6°, 89010 reflections collected, 14738unique (Rint = 0.0665). Final GooF = 1.001, R1 = 0.0335, wR2 = 0.0787, R indices based on 13047 reflections with I > 2σ(I), |Δρ|max = 0.84 e Å−3, 1054 parameters, 9 restraints. The water molecule hydrogen atoms were refined with geometries restrained to ideal values. CCDC 1829213.†
(no. 2), a = 14.0539(2), b = 14.7835(3), c = 19.7708(4) Å, α = 99.829(2), β = 107.431(2), γ = 90.137(2)°, V = 3855.27(13) Å3, Z = 1, Dc = 1.405 g cm−3, μ = 1.367 mm−1. F000 = 1666, Mo Kα radiation, λ = 0.71073 Å, 2θmax = 64.7°, 84599 reflections collected, 25545unique (Rint = 0.0650). Final GooF = 1.002, R1 = 0.0496, wR2 = 0.0959, R indices based on 19518 reflections with I > 2σ (I), |Δρ|max = 1.1 e Å−3, 961 parameters, 13 restraints. One phenyl ring and two solvent ethanol molecules were modelled as being disordered over two sets of sites with occupancies constrained to 0.5 and with the non-hydrogen atoms refined with isotropic displacement parameters. Geometries of the disordered atoms were restrained to ideal values. CCDC 1829214.†
086 reflections collected, 11254unique (Rint = 0.0839). Final GooF = 1.069, R1 = 0.0486, wR2 = 0.1117, R indices based on 8726 reflections with I > 2σ(I), |Δρ|max = 1.7 e Å−3, 820 parameters, 17 restraints. The solvent was modelled as an ethanol molecule disordered about a crystallographic inversion centre. Geometries were restrained to ideal values. Water molecule hydrogen atoms were located and refined with geometries restrained to ideal values. CCDC 1829215.†
516 reflections collected, 4906 unique (Rint = 0.0436). Final GooF = 1.090, R1 = 0.0386, wR2 = 0.1095, R indices based on 4314 reflections with I > 2σ(I), |Δρ|max = 2.5 e Å−3, 349 parameters, 0 restraints. CCDC 1829216.†
616 reflections collected, 3671 unique (Rint = 0.0558). Final GooF = 1.037, R1 = 0.0455, wR2 = 0.1174, R indices based on 3361 reflections with I > 2σ(I), |Δρ|max = 2.4 e Å−3, 265 parameters, 0 restraints. CCDC 1829217.†
Results and discussion
The tbmH and mtbmH molecules were synthesised according to the previously reported methodology.20,39 Following a similar procedure to that previously reported for the preparation of {[Ln(Cs)(tbm)4]2} (Ln3+ = Eu, Er, Yb) and {[Ln(Cs)(mtbm)4]2}n (Ln3+ = Eu, Er),20 one equivalent of hydrated LnCl3 (Ln3+ = Eu, Nd) was made to react with four equivalents of mtbmH and four equivalents of RbOH in ethanol. Slow evaporation of the solvent resulted in the formation of suitable crystals for X-ray diffraction, revealing the formation of coordination polymers with formula {[Ln(Rb)(mtbm)4]2}n where Ln3+ = Eu(1), Nd(2). The compositions of the isolated species were further confirmed by elemental analysis and IR spectroscopy. The resulting solids are isolated with variable degrees of solvation, which has been found previously for these Ln3+/Ae+ based complexes.19,20The Eu3+/Nd3+ mixed assemblies were synthesised in a similar fashion to the {[Ln(Rb)(mtbm)4]2}n, except for the use of mixtures of hydrated EuCl3 and NdCl3 in molar ratios of Nd3+ of 0.25 (3), 0.50 (4) and 0.75 (5).
Analogous syntheses were attempted with CsOH and NdCl3 in order to assess the effect of the different alkaline base in the mixed systems. However, only the cesium-containing coordination polymer [Cs(mtbm)]n was deposited (ESI†).20
When the same procedure was followed for the hydrated NdCl3 and tbmH with RbOH or CsOH, the formation of assemblies with formula [Nd(Rb)(tbm)4]2 and [Nd(Cs·2HOEt)(dbm)4]n was found, respectively. The [Nd(Rb)(tbm)4]2 (6) complex presents a similar structure to the previously reported tetranuclear assemblies.19 In contrast, the isolation of the [Nd(Cs·2HOEt)(dbm)4]n linear polymer shows the second example of a possible in situ retro-Claisen condensation reaction of tbmH in the presence of CsOH and hydrated NdCl3 resulting in the formation of a β-diketonate complex similar to previously reported examples (ESI†).20 The hypothesis that the triketonate ligands undergo a retro-Claisen condensation reaction under these reaction conditions is currently under investigation and the results will be presented elsewhere.
Finally, when the same procedure was attempted with YbCl3, a dimeric structure was crystallised with formula [Yb(mtbm)3(H2O)2]2 (ESI†). Due to difference in composition and symmetry of this structure in comparison with the polymeric species of complexes 1 and 2, Yb3+ was not further investigated for the purpose of this study.
Crystal structures
The structures of the two {[Ln(Rb)(mtbm)4]2}n (Ln3+ = Nd, Eu) complexes are isomorphous and structurally similar to the previously reported Cs-based polymers with formula {[Ln(Cs)(mtbm)4]2}n (Ln3+ = Eu, Er).20 See Table 1 for a list of bond lengths and angles. The units formed of two Ln3+, two Rb+ metal centres and eight mtbm− ligands are isomorphous to the previously reported tetranuclear assemblies.18 The Ln3+ is eight coordinated, with four mtbm− ligands coordinated by two of the O-keto atoms in a bidentate mode. In this case, the third O-keto of two of the ligands are linked to Rb+ cations forming the tetranuclear assembly and the polymer, respectively (Fig. 1). | ||
| Fig. 1 Representation of the X-ray structure of 2, {[Nd(Rb)(mtbm)4]2}n, where hydrogen atoms have been omitted for clarity. | ||
| 1 {[Eu(Rb)(mtbm)4]2}n | 2 [Nd(Rb)(mtbm)4]2}n | 6 [Nd(Rb·HOEt)(tbm)4]2 | |
|---|---|---|---|
| a Subsequent units. b Different chain. | |||
| Ln–O | 2.327(2)–2.405(2) | 2.363(2)–2.444(2) | 2.390(2)–2.450(2) |
| Ae–O | 2.816(2)–2.983(2) | 2.817(2)–2.989(2) | 2.822(2)–3.051(2) |
| Ae(1)–Ae(2) | 8.1196(5) | 8.1312(5) | 8.3053(6) |
| Ae(1)–Ae(2a) | 8.7992(5) | 8.8013(5) | – |
| Ln(1)–Ln(2) | 9.4915(5) | 9.5391(5) | 8.9836(5) |
| Ln(1)–Ln(2a) | 11.0901(6) | 11.0929(5) | 13.8915(6) |
| Ln(1)–Ae(1) | 4.0943(4) | 4.1044(3) | 4.1340(3) |
| Ln(1)–Ae(2) | 8.1849(5) | 8.8169(5) | 7.5993(6) |
| Ae(2)–Ln(1a) | 8.8145(5) | 8.2087(5) | — |
| Ln(1)–Ln(1b) | 14.9383(7) | 14.9907(5) | 14.0539(5) |
Here, a H2O molecule is found in the lattice with two hydrogen bonds formed with two keto O(22) and O(31). Intermolecular interactions between chains are present where the lanthanoid centres sit at distances longer than 14 Å (ESI†). The geometry of the eight coordinate Ln3+ is best described as triangular dodecahedron (ESI†).
The structure of the [Nd(Rb·HOEt)(tbm)4]2 is isomorphous to the previously published tetranuclear assemblies with Ln3+ = Eu3+, Er3+ and Yb3+.19 where the eight coordinated Nd3+ adopts a geometry best described as distorted triangular dodecahedron (Fig. 2).
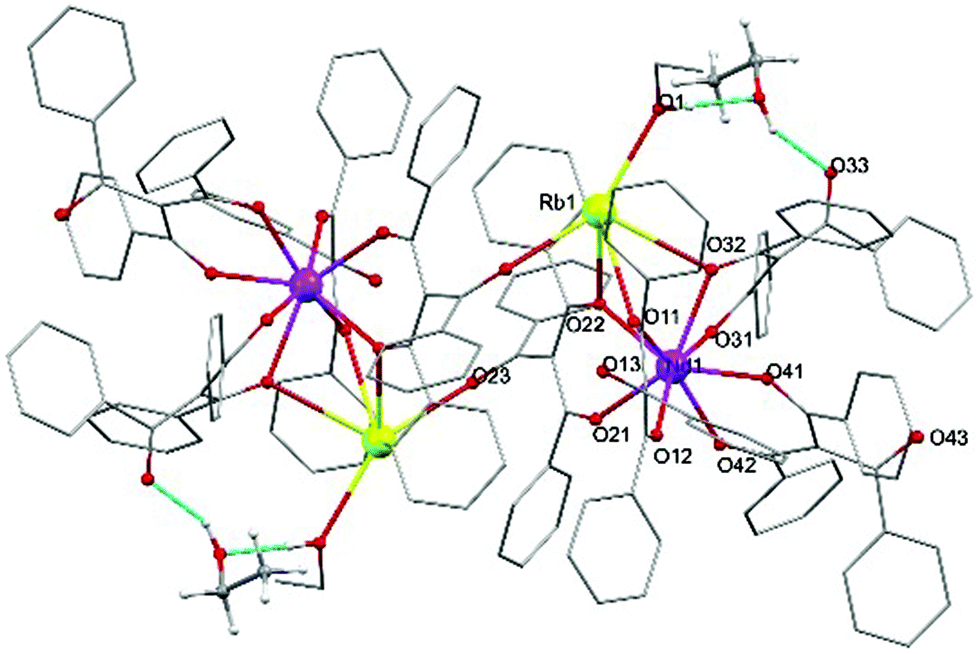 | ||
| Fig. 2 Representation of the X-ray structure of 6, [Nd(Rb)(tbm)4]2, where hydrogen atoms have been omitted for clarity except for those on the solvent EtOH molecule. | ||
Photophysical investigation
The photophysical data for complexes 1–6 including excited state lifetime decay (τobs), calculated radiative decay (τR), intrinsic photoluminescence quantum yield (ΦLnLn), overall photoluminescence quantum yield (ΦLnLn) and calculated sensitisation efficiency (ηsens), are reported in Table 2. The emission properties were recorded in the solid state due to the low stability of the complexes in polar solvents and poor solubility in nonpolar solvents, as previously demonstrated for analogous systems.18| Complex |
χ
Nd3+a |
λ
emb (nm) |
τ obs (μs) | τ r (ms) | Φ Ln Ln (%) |
Φ
L
Lnc (%) |
η sens | k ET (s−1) | τ ET (s) | Φ ET (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Solution phase compositions in the reaction mixture. b Emission spectra recorded using λexc = 350 nm. c Quantum yield measured with the use of an integrating sphere. d Literature value for Nd3+.16 | ||||||||||
| 1 {[Eu(Rb)(mtbm)4]2}n | 0 | 612 (Eu3+) | 507 | 0.86 | 59 | 31 | 52 | — | — | — |
| 2 {[Nd(Rb)(mtbm)4]2}n | 1 | 1060 (Nd3+) | 11 | 0.27d | 4.2 | 1.34 | 32 | — | — | — |
| 3 {[Eu1−xNdx (Rb)(mtbm)4]2}n | 0.25 | 612(Eu3+) | 335 | 0.681 | 49 | 17.5 | 35 | 1.0 × 103 | 9.87 × 10−4 | 34 |
| 1060 (Nd3+) | 8.7 | 0.27d | 3.1 | 0.23 | 7 | |||||
| 4 {[Eu1−xNdx (Rb)(mtbm)4]2}n | 0.5 | 612(Eu3+) | 183 | 0.46 | 40 | 6.55 | 16 | 3.5 × 103 | 2.86 × 10−4 | 64 |
| 1060 (Nd3+) | 11.0 | 0.27d | 4.1 | 0.74 | 18 | |||||
| 5 {[Eu1−xNdx (Rb)(mtbm)4]2}n | 0.75 | 612(Eu3+) | 143 | 0.54 | 27 | 1.44 | 5 | 5.0 × 103 | 1.99 × 10−4 | 72 |
| 1060 (Nd3+) | 8.7 | 0.27c | 3.2 | 0.44 | 14 | |||||
| 6 [Nd(Rb·HOEt)(tbm)4]2 | 1 | 1060 (Nd3+) | 8.8 | 0.27d | 3.3 | 0.58 | 17 | — | — | — |
As shown before, the energy of the mtbm and tbm triplet states (21140 cm−1 and 20704 cm−1)18,20 are sufficiently high to sensitise the 5D0 (∼17200 cm−1) of Eu3, the 2F5/2 (∼10200 cm−1) of Yb3+ and the 4I13/2 (∼6566 cm−1) of Er3+. Therefore, energy transfer to the 4F3/2 (∼11260 cm−1) state of Nd3+ is also expected. In fact, each emission spectrum shown herein is the result of an effective antenna effect, a conclusion that is supported by the broad excitation spectra which match with the absorption profile of the corresponding ligands.
The emission spectrum of {[Eu(Rb)(mtbm)4]2}n (1) shows the characteristic Eu3+ emission bands attributed to the 7FJ ← 5D0 (J = 0–6) region 580–820 nm (Fig. 3).40,41 The 7F0 ← 5D0 transition is strictly forbidden by the selection rules and is only observable for low symmetry complexes. The magnetic dipole-allowed band (7F1 ← 5D0) is split into two sublevels inherent to tetragonal crystal fields. This is in agreement with the splitting of the hypersensitive band (7F2 ← 5D0) in four sublevels. The splitting of the main transitions is in accordance with the shape analysis, which suggests that the local symmetry of the Eu3+ cation is best described as a distorted triangular dodecahedron.
 | ||
| Fig. 3 Normalised emission plot for {[Eu(Rb)(mtbm)4]2}n (red trace) and {[Nd(Rb)(mtbm)4]2}n (black trace) in the solid state, with excitation wavelength at 350 nm. Inset: highlight of the splitting of the magnetic dipole transition for the Eu3+ complex. | ||
The excited state decay was satisfactorily fitted as a monoexponential function, giving a value of observable lifetime (τobs) of 507 μs. From the emission spectrum, the radiative decay (τr) was calculated to be 0.86 ms. With an integrating sphere, the overall quantum yield (ΦLLn) was measured as 31%. From these data, the intrinsic quantum yield (ΦLnLn) as ratio τobs/τr could be calculated to be 59% with a sensitisation efficiency of 52%.
These data are of the same order as the previously reported {[Eu(Cs)(mtbm)4]2}n,20 showing that the exchange in the alkaline base has little impact on the photophysical properties.
The emission spectrum of {[Nd(Rb)(mtbm)4]2}n (2) shows the characteristic Nd3+ emission bands from the 4IJ ← 4F3/2 (J = 9/2, 11/2, 13/2) with maxima at 910, 1060 and 1350 nm respectively (Fig. 3).42 These bands are structured as a consequence of the crystal field effect from the ligands. The excited state decay was measured to be 11 μs after deconvolution from instrumental response. This value of τobs is relatively high in comparison to the previously reported β-diketonate compounds21,28 and of the same order of magnitude as highly conjugated systems where the triplet state is lowered in energy to better match the emissive lanthanoid excited state energy.14,43
Although it is known that the radiative decay for Nd3+ ranges from 0.2 to 0.5 ms,4 a standard value of 0.27 ms is generally accepted for the Nd3+ complexes in the solid state.16 The intrinsic quantum yield can therefore be estimated to 4.2%. The overall quantum yield, using an integrating sphere following previously reported procedure for the use of two different detectors,9 was found to be 1.34%, with a sensitisation efficiency of 32%. These data highlight that reducing non-radiative decays due to the removal of the C–H bond is an effective way to enhance the photophysical properties of the Nd3+ emitters.
As the structures for Eu3+ and Nd3+ are isomorphous, {[Ln(Rb)(mtbm)4]2}n, mixtures of both lanthanoids were prepared (3–5) in order to investigate sensitisation of the 4F3/2 of Nd3+via the 5D0 of Eu3+ (Fig. 4).
 | ||
| Fig. 4 Energy level diagram and energy transfer occurring for the mixed complexes {[Eu1−xNdx (Rb)(mtbm)4]2}n. | ||
The emission spectra of the mixed complexes show the characteristic emission bands from the 7FJ ← 5D0 (J = 0–6) of Eu3+ in the visible region (580–820 nm) and the 4IJ ← 4F3/2 (J = 9/2, 11/2, 13/2) Nd3+ bands in the NIR region (850–1400 nm) with identical splitting in comparison with the pure complexes 1 and 2, respectively. This suggests that the structure is preserved with the mixed lanthanoid polymers. The intensity of the Nd3+ emission bands increases when the molar ratio of Nd3+ is higher (Fig. 5). The lifetime of the excited state of Eu3+ is shortened as the amount of Nd3+ increases, from 507 μs for 1 (where Nd3+ is absent) to 335 μs, 183 μs and 143 μs for 3, 4 and 5, respectively. From these numbers, the highest energy transfer quantum efficiency can be calculated to be 72% for complex 5. Overall quantum yields were measured, finding decreasing values for Eu3+ of 17.5%, 6.55% and 1.44% for complexes 3–5, respectively (Table 2). In the case of Nd3+, both lifetime and overall quantum yield values seem to be reduced by the presence of Eu3+. These results indicate possible quenching of the 4F3/2 of Nd3+ by the 7FJ of Eu3+ as previously suggested in the literature.34 However, the ratio of this energy transfer was calculated to be only 20%, which is perhaps the main difference from previous Eu/Nd coordination compounds where no neodymium emission was reported.30–32
 | ||
| Fig. 5 Nd3+ emission plot for complex 2 (black trace), 3 (red trace), 4 (blue trace) and 5 (green trace) with excitation wavelength at 350 nm normalised at 612 nm (Eu3+ emission). | ||
Typically, energy transfer between lanthanoid centres is considered limited for distances longer than 9 Å because of slow energy migration.44 In fact, if a purely dipole–dipole exchange mechanism is considered, the donor–acceptor distance can be calculated to be 7.7 Å following eqn (6), for a quantum efficiency of energy transfer (ΦET) of 0.72 for complex 5. However, in our system and when considering one polymeric chain, the shortest distance between two lanthanoid centres is 9.5 Å. Therefore, the sensitisation to *f states of Nd3+ from the 5D0 of Eu3+ for complexes 3–5 seems not to be a pure Förster mechanism, and a ligand-mediated Dexter mechanism may have some contribution.45,46
As a control experiment, equimolar mechanically-ground mixtures of 1 and 2 were studied. The lifetime of the 5D0 of Eu3+ was found to be 356 μs, shorter than the pure complex 1 (τobs = 507 μs) and longer with respect to the solution-phase mixed equimolar complex 4 (τobs = 507 μs). These data suggest that there is energy transfer between chains occurring at 30% of efficiency. Taking into consideration the long Ln–Ln distances between chains (∼15 Å) based on the crystal structure, the energy transfer process may occur via intermolecular interactions (ESI†).
Finally, the emission spectrum of [Nd(Rb·HOEt)(tbm)4]2 (6) shows the three characteristic Nd3+ bands from the 4IJ ← 4F3/2 (J = 9/2, 11/2, 13/2) similarly to the {[Nd(Rb)(mtbm)4]2}n (1) (ESI†). The values of lifetime (τobs), intrinsic quantum yield (ΦLnLn) and overall quantum yield (ΦLLn) were found to be 8.85 μs, 3.3% and 0.58%, respectively. The main difference with complex 2 arises from a lower overall quantum yield, maintaining the values of lifetime and intrinsic quantum yields, which suggests that the sensitisation process from tbm to the 4f* accepting states of Nd3+ is not as efficient as in the mtbm based complexes.
Conclusions
In this report the study of β-triketonate based lanthanoid complexes has been extended to Nd3+, presenting new examples of tetranuclear assemblies ([Nd(Rb·HOEt)(tbm)4]2) and coordination polymers with formula {[Nd(Rb)(mtbm)4]2}n. The fact that isomorphous structures were found for the mtbm and Eu3+ ({[Nd(Rb)(mtbm)4]2}n), opened up the possibility to synthesise mixed lanthanoid complexes with the aim of achieving f–f energy transfer. Indeed, an example of a mixed mtbm based lanthanoid coordination polymer with efficient sensitisation from the 5D0 of Eu3+ to the 4F3/2 of Nd3+ was formulated. The emission studies of the pure and mixed complexes show particularly good photophysical properties in the case of Nd3+via both mechanisms: standard antenna and f–f sensitisation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was partially supported by the Australian Research Council's Discovery Projects funding scheme (project DP170101895), and a Royal Society International Exchanges Grant. E. Z.-C. thanks the EPSRC (EP/M02105X/1) for support. L. A. G thanks Curtin University for the Australian Postgraduate Award. The authors acknowledge access to the facilities at the Centre for Microscopy, Characterisation and Analysis, University of Western Australia.Notes and references
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- J.-C. G. Bünzli, Luminescence Bioimaging with Lanthanide Complexes, 2014, vol. 1st Search PubMed.
- J.-C. G. Bünzli and S. V. Eliseeva, J. Rare Earths, 2010, 28, 824–842 CrossRef.
- J.-C. G. Bünzli and S. V. Eliseeva, Photophysics of Lanthanoid Coordination Compounds, 2013, vol. 8 Search PubMed.
- V. V. Utochnikova, A. Grishko, A. Vashchenko, A. Goloveshkin, A. Averin and N. Kuzmina, Eur. J. Inorg. Chem., 2017, 1–6 Search PubMed.
- S. V. Eliseeva and J.-C. G. Bünzli, New J. Chem., 2011, 35, 1165 RSC.
- J.-C. G. Bünzli and S. V. Eliseeva, Springer Ser. Fluoresc., 2011, 1–45 Search PubMed.
- S. Cotton, Lanthanide and Actinide Chemistry, 2005 Search PubMed.
- L. Abad Galán, B. L. Reid, S. Stagni, A. N. Sobolev, B. W. Skelton, E. G. Moore, G. S. Hanan, E. Zysman-Colman, M. I. Ogden and M. Massi, Dalton Trans., 2018, 47, 7956–7964 RSC.
- O. Sun, T. Gao, J. Sun, G. Li, H. Li, H. Xu, C. Wang and P. Yan, CrystEngComm, 2014, 16, 10460–10468 RSC.
- N. M. Shavaleev, S. J. A. Pope, Z. R. Bell and M. D. Ward, Dalton Trans., 2003, 0, 808–814 RSC.
- G. Zucchi, O. Maury, P. Thuéry and M. Ephritikhine, Inorg. Chem., 2008, 47, 10398–10406 CrossRef PubMed.
- W. Li, J. Li, H. Li, P. Yan, G. Hou and G. Li, J. Lumin., 2014, 146, 205–210 CrossRef.
- L. Yang, Z. Gong, D. Nie, B. Lou, Z. Bian, M. Guan, C. Huang, H. J. Lee and W. P. Baik, New J. Chem., 2006, 30, 791 RSC.
- M. Iwamuro, Y. Wada, T. Kitamura and N. Nakashima, Phys. Chem. Chem. Phys., 2000, 2, 2291–2296 RSC.
- T. M. George, S. Varughese and M. L. P. Reddy, RSC Adv., 2016, 6, 69509–69520 RSC.
- B. Li, H. Li, P. Chen, W. Sun, C. Wang, T. Gao and P. Yan, Dalton Trans., 2016, 45, 11459–11470 RSC.
- B. L. Reid, S. Stagni, J. M. Malicka, M. Cocchi, G. S. Hanan, M. I. Ogden and M. Massi, Chem. Commun., 2014, 50, 11580–11582 RSC.
- B. L. Reid, S. Stagni, J. M. Malicka, M. Cocchi, A. N. Sobolev, B. W. Skelton, E. G. Moore, G. S. Hanan, M. I. Ogden and M. Massi, Chem. – Eur. J., 2015, 21, 18354–18363 CrossRef PubMed.
- L. Abad Galán, B. L. Reid, S. Stagni, A. N. Sobolev, B. W. Skelton, M. Cocchi, J. M. Malicka, E. Zysman-colman, E. G. Moore, M. I. Ogden and M. Massi, Inorg. Chem., 2017, 56, 8975–8985 CrossRef PubMed.
- X. Rao, T. Song, J. Gao, Y. Cui, Y. Yang, C. Wu, B. Chen and G. Qian, J. Am. Chem. Soc., 2013, 135, 15559–15564 CrossRef PubMed.
- D. T. De Lill, A. De Bettencourt-Dias and C. L. Cahill, Inorg. Chem., 2007, 46, 3960–3965 CrossRef PubMed.
- S. Freslon, Y. Luo, G. Calvez, C. Daiguebonne, O. Guillou, K. Bernot, V. Michel and X. Fan, Inorg. Chem., 2014, 53, 1217–1228 CrossRef PubMed.
- F. Artizzu, A. Serpe, L. Marchiò, M. Saba, A. Mura, M. L. Mercuri, G. Bongiovanni, P. Deplano and F. Quochi, J. Mater. Chem. C, 2015, 3, 11524–11530 RSC.
- L. Song, Q. Wang, D. Tang, X. Liu and Z. Zhen, New J. Chem., 2007, 31, 506–511 RSC.
- S. Hinojosa, M. A. Meneses-Nava, O. Barbosa-García, L. A. Díaz-Torres, M. A. Santoyo and J. F. Mosiño, J. Lumin., 2003, 102–103, 694–698 CrossRef.
- F. Artizzu, F. Quochi, L. Marchiò, E. Sessini, M. Saba, A. Serpe, A. Mura, M. L. Mercuri, G. Bongiovanni and P. Deplano, J. Phys. Chem. Lett., 2013, 4, 3062–3066 CrossRef.
- S. Faulkner and S. J. A. Pope, J. Am. Chem. Soc., 2003, 125, 10526–10527 CrossRef PubMed.
- L. Zhou, P. A. Tanner, W. Zhou, Y. Ai, L. Ning, M. M. Wu and H. Liang, Angew. Chem., Int. Ed., 2017, 56, 10357–10361 CrossRef PubMed.
- J.-C. G. Bünzli and F. Ihringer, Inorg. Chim. Acta, 1996, 246, 195–205 CrossRef.
- C. M. Andolina and J. R. Morrow, Eur. J. Inorg. Chem., 2010, 2011, 154–164 CrossRef.
- J. J. Lessmann and W. D. W. Horrocks, Inorg. Chem., 2000, 39, 3114–3124 CrossRef PubMed.
- L. G. Van Uitert, E. F. Dearborn and J. J. Rubin, J. Chem. Phys., 1967, 46, 3551–3555 CrossRef.
- E. J. Sharp, M. J. Weber and G. Cleek, J. Appl. Phys., 1970, 41, 364–369 CrossRef.
- W. D. W. Horrocks, M. J. Rhee, A. Peter Snyder and D. R. Sudnick, J. Am. Chem. Soc., 1980, 102, 3650–3652 CrossRef.
- J. C. De Mello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef.
- M. P. Tsvirko, S. B. Meshkova, V. Y. Venchikov, Z. M. Topilova and D. V. Bol'shoi, Opt. Spectrosc., 2001, 90, 669–673 CrossRef.
- G. M. Sheldrick, Acta Crystallogr. Sect. C Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- B. L. Reid, S. Stagni, J. M. Malicka, M. Cocchi, G. S. Hanan, M. I. Ogden and M. Massi, Chem. Commun., 2014, 50, 11580–11582 RSC.
- A. M. Hilder, P. C. Junk, U. H. Kynast and M. M. Lezhnina, J. Photochem. Photobiol. A Chem., 2008, 202, 10–20 CrossRef.
- M. Hilder, M. Lezhnina, M. L. Cole, C. M. Forsyth, P. C. Junk and U. H. Kynast, J. Photochem. Photobiol. A Chem., 2011, 217, 76–86 CrossRef.
- L. Hu, J. Zhang, Q. Yin, P. Li and K. Du, Opt. Commun., 2014, 324, 26–29 CrossRef.
- S. I. Klink, G. A. Hebbink, L. Grave, P. G. B. Oude Alink, F. C. J. M. van Veggel and M. H. V. Werts, J. Phys. Chem. A, 2002, 106, 3681–3689 CrossRef.
- S. Omagari, T. Nakanishi, Y. Hirai, Y. Kitagawa, T. Seki, K. Fushimi, H. Ito and Y. Hasegawa, Eur. J. Inorg. Chem., 2017, 1–8 Search PubMed.
- A. Zam, S. V. Eliseeva, L. Guønøe, H. Nozary, S. Petoud and C. Piguet, Chem. – Eur. J., 2014, 20, 12172–12182 CrossRef PubMed.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1829212, 1829213, 1829214, 1829215, 1829216 and 1829217. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt02499e |
| This journal is © The Royal Society of Chemistry 2018 |