
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDinuclear tethered pyridine, diimine complexes†‡
Wiebke Dammann,
Tabea Buban,
Carl Schiller and
Peter Burger *
*
Institut für Anorganische und Angewandte Chemie, Fachbereich Chemie, Universität Hamburg, Martin Luther King Platz 6, 20146 Hamburg, Germany. E-mail: burger@chemie.uni-hamburg.de
First published on 23rd July 2018
Abstract
The regioselective borylation of pyridine precursors, followed by Suzuki coupling with dihalogenated linker molecules, provides access to tethered ligands. Complexation with MX2 salts results in the formation of dinuclear metal compounds. Syntheses and crystal structures are reported along with a discussion on the rigidity/flexibility of these new ligand systems.
Introduction
Pyridine, diimine (PDI) ligands are nowadays ubiquitous in main group and transition metal chemistry and catalysis. Their popularity started almost two decades ago with the outstanding reports of Brookhart1 and Gibson2,3 on olefin polymerization with iron and cobalt PDI catalysts.4 The proven non-innocence5 of this class of ligands contributed further to their widespread use in inorganic chemistry. Therefore, it is surprising that there are only a few examples of covalently tethered PDI ligands, which would allow us to access dinuclear complexes.6 This is in contrast to other popular ligands, e.g. porphyrins, for which sophisticated synthetic strategies were developed to construct cofacial bimetallic complexes with a defined metal–metal distance for the bimolecular activation of O2 or the C–H-bond in methane.7 To the best of our knowledge, only the tethering modes shown in Fig. 1 have been reported in the literature along with their corresponding dinuclear transition metal complexes.6 | ||
| Fig. 1 Previously reported tethered PDI ligands. | ||
We have therefore set out to establish a general route to this type of ligand class. Herein, we report on the ligand syntheses and first examples of their corresponding dinuclear 3d transition metal complexes (Ni and Zn).
Our general goal was to couple the PDI ligands in the para position of the pyridine unit with suitable aromatic linkers a–d presented in Fig. 2. This gives a variety of ligands that allow access to metal complexes with well-defined metal–metal distances. In contrast, the use of the diphenylether spacer e leads to a less rigid ligand. Furthermore, the substituents of the ketimine group were varied to adjust their steric demand.
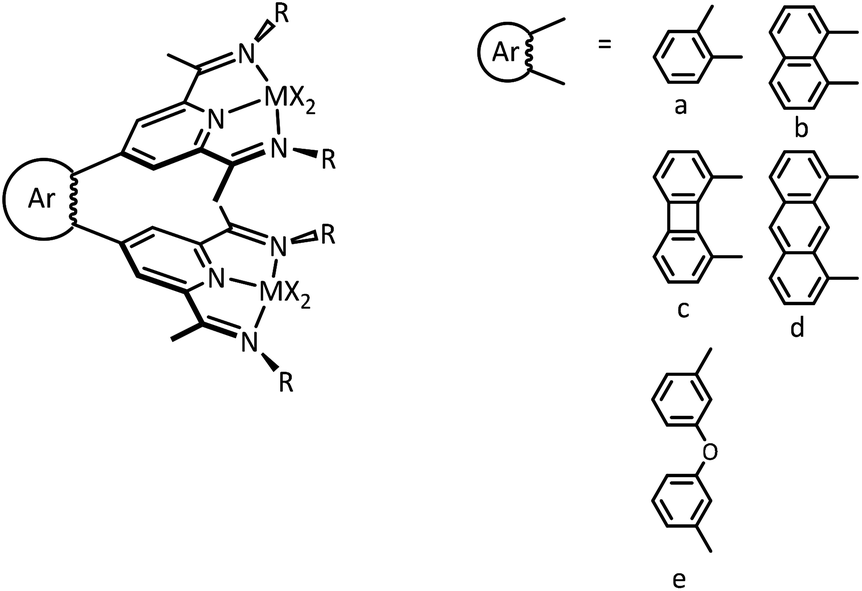 | ||
| Fig. 2 General motif of the tethered PDI complexes. Variations are accessible through changes of the bridging aromatic unit or the substituents at the ketimine group. | ||
Results and discussion
Ligand synthesis
The key to novel covalently tethered PDI ligands is the regioselective borylation of suitable precursors, followed by Suzuki coupling with the appropriate dihalogenated carbon linkers. Hence, the first desired compound is a para-borylated PDI ligand. An analogous terpyridine derivative was previously synthesized by Schwalbe et al. They described a classic Miyaura borylation of a 4-bromo-terpyridine derivative.8 In our case, regioselective bromination in the 4-position of the pyridine moiety is challenging. The scale-up of the reported three-step synthesis of 4-bromo-2,6-diacetylpyridine9 led to low yields and was deemed unsuitable. Therefore, a regioselective C–H bond activation followed by borylation was developed. To the best of our knowledge, the C–H bond activation step of pyridine-diimine ligands is not yet described in the literature. While this manuscript was completed, Chirik et al. reported on an adventitious borylation step in a PNP Co-complex.10 The unique feature of our organo-boron compounds is the wide range of accessible products gained through C–C bond formation.Both the ketal protected or diketimine pyridine derivatives 1,11 and 2, 33,12 allowed exclusive borylation with bis(pinacolato)diboron in the para-position of the pyridine ring (Scheme 1). It ought to be mentioned that the first reaction step has to be the formation of the catalyst derived from the iridium dimer [Ir(OMe)COD]2, 4,4′-di-tert-butyl-2,2′-dipyridine and bis(pinacolato)diboron. Otherwise, the formation of an unreactive iridium PDI complex is observed. In the literature, different reaction conditions for borylation are described.13 We have used the procedure reported by Steel et al. without further optimization.14
 | ||
| Scheme 1 Regioselective borylation of compounds 1–3. | ||
The borylated products 4–6 were obtained in good yields in an analytically pure form as white (4) or yellow (5 and 6) solids through direct crystallization from the reaction mixture. It is noteworthy that our attempts to accomplish borylation of the unprotected 2,6-diacetyl pyridine were unsuccessful, since a non-separable mixture of the starting materials and the desired product was obtained.
The successful regioselective borylation was demonstrated by the observation of a singlet in the 1H-NMR spectrum for the meta pyridine protons of compounds 4–6 (see the Experimental section). An exemplary molecular structure observed in the solid state is shown for compound 5 in Fig. 3.
 | ||
| Fig. 3 ORTEP plot of 5 (left) and ligand 12 (right) with anisotropic displacement parameters shown at the 50% probability level. Hydrogen atoms are omitted for clarity. The pinacol moiety of 5 is disordered and only one part is shown. Selected bond lengths for 5 (Å): N3a–C8a, 1.283(3); C8a–C7a, 1.496(4); C7a–N1a, 1.348(3); N1a–C3a, 1.345(3); C3a–C2a, 1.508(4); C2a–N2a, 1.272(3); Selected bond lengths 12 (Å): N3b–C8b, 1.274(2); C8b–C7b, 1.501(2); C7b–N1b, 1.342(2); N1b–C3b, 1.343(2); C3b–C2b, 1.501(2); C2b–N2b, 1.272(2). | ||
In the next step, compounds 4–6 were employed in Suzuki coupling reactions15 with the appropriate dihalogenated compound (Scheme 2). Using standard protocols16 with palladium catalysts (Pd(PPh3)4, Pd(dppf)Cl2 or Pd2dba3/XPhos), the desired C–C-coupled products 8–12 were obtained in good to excellent yields as yellow solids. The reaction conditions vary depending on the dihalogenated compound that is used as the aromatic linker (Scheme 2 and Table 1) and are adapted from the general optimized procedure.16,17
 | ||
| Scheme 2 Reaction conditions of the Suzuki coupling reactions of compounds 5 and 6. Compound 8: 1,2-dibromobenzene, Pd2dba2, XPhos, K3PO4, KF, dioxane/water, 110 °C, 3 h. Compound 9: 1,8-dibromobiphenylene, Pd(PPh3)4, K2CO3, THF, 90 °C, 2 d. Compound 10: 1,8-dichloroanthracene, Pd(PPh3)4, XPhos, KF, K3PO4, dioxane/water, 105 °C, 19 h. Compound 11: 1-bromo-3-(3-iodophenoxy)benzene, KF, K2CO3, Pd(dppf)Cl2, THF/water, 80 °C, 2 h. | ||
| Compound | Aromatic linker Ar | Substituent R | Yield |
|---|---|---|---|
| 8 | 1,2-Benzene | Mesityl | 93% |
| 9 | 1,8-Biphenylene | 2,6-Dimethyl-6-tert-butylphenyl | 65% |
| 10 | 1,8-Anthracene | Mesityl | 98% |
| 11 | 3,3′-Diphenylether | Mesityl | 67% |
| 12 | 1,8-Naphthalene | Propyl | 71% |
The numbering scheme with the used aromatic linker and the substituents of the imine group is presented in Table 1.
The solubilities of these ligands differ, depending on both the bridging unit and the ketimine substituent. While 8 and 11 are soluble in dichloromethane, THF and diethylether and even slightly soluble in n-hexane, the solubility of the other ligands (9, 10 and 12) is lower. Compound 9 is soluble in THF, while 10 is only slightly soluble in THF and dichloromethane and is insoluble in diethylether and n-hexane.
The analogous coupling with 1,8-diiodonaphthalene was not successful, which we attribute to the steric hindrance of the PDI moieties 5. Therefore, we planned on reducing the steric demand by introducing n-propyl-substituents. In the alternative synthetic route for 12 shown in Scheme 3, the Suzuki coupling of 4 was followed by the cleavage of the ketal protecting group to yield 7 in moderate yield. The condensation of 7 with n-propylamine led to ligand 12 in yields up to 71%. In contrast to 8–11, ligand 12 is obtained as a white solid and displays a higher solubility, e.g. in THF, dichloromethane and diethylether. An advantage of this synthetic route is the potential use of different amines for the condensation to access a wide variety of ligands with different steric demands.
 | ||
| Scheme 3 Suzuki coupling of compound 4 and consecutive condensation with n-propylamine to the imine. | ||
The 1H-NMR spectra of ligands 8–12 display a singlet with an integral of 4 for the meta pyridine protons. For the ketimine methyl groups, a singlet with an integral corresponding to 12 protons is observed. This indicates time averaged C2v-symmetry in solution for 8–12. Single crystal X-ray crystallographic data of the ligands clearly reveal lowering of the symmetry in the solid state.
The solid-state structure of 12 (Fig. 3) illustrates that the nitrogen atoms of the ketimine group are in E,E conformation. This is consistent with previously reported PDI ligands. The bond distances and angles are in the expected range.18 For the solid state structure of 8, see the ESI.‡
Complexation
In order to probe the propensity of this new type of ligand system, we turned to complexation reactions with water-free zinc and nickel dichloride (ZnCl2 and NiCl2) as well as zinc bistriflate (ZnOTf2, Scheme 4). The reaction conditions vary only slightly depending on the ligand and the used salts MX2. The zinc PDI complexes are obtained by stirring the appropriate ligand with a zinc salt at room temperature. The nickel complex 9-NiCl2 was synthesized at 90 °C to increase the solubility of the nickel dichloride starting material. | ||
| Scheme 4 Complexation with MX2. 8-ZnCl2: ZnCl2, dichloromethane, RT, 18 h. 9-NiCl2: NiCl2, THF/acetone, 90 °C, 24 h. 10-ZnOTf2: ZnOTf2, acetonitrile/dichloromethane, RT, 16 h. 11-ZnCl2: ZnCl2, THF, RT, 2 h; 12-ZnCl2: ZnCl2, dichloromethane, RT, 15 h. | ||
The zinc complexes are isolated as yellow (8-ZnCl2, 10-ZnOTf2, and 11-ZnCl2) and pale yellow (12-ZnCl2) solids with varying solubilities. While complexes 12-ZnCl2 and 10-ZnOTf2 are soluble in dichloromethane and slightly soluble in THF, compound 11-ZnCl2 is soluble in dichloromethane, but insoluble in THF. Compound 8-ZnCl2 is the least soluble complex and is insoluble in dichloromethane and THF. In DMF, acetonitrile and nitromethane low solubility was observed.
The 1H-NMR spectra of these complexes reveal a singlet with an integral of 4 protons for the meta protons of the pyridine units. Another singlet with an integral for 12 protons is observed for the resonance of the ketimine methyl groups (see the Experimental section). This observation points to time averaged C2v-symmetrical complexes in solution. Due to the low solubility of complex 8-ZnCl2, a full assignment of all 1H-NMR signals was not possible. Nevertheless, the 1H-NMR spectrum reveals interesting solvent dependent characteristics. Using acetonitrile, a singlet with an integral of 4 protons at 7.83 ppm is observed, which can be assigned to the isochronous protons of the phenyl linker. In DMF-d7, broad signals are observed for both aromatic and aliphatic protons. The origin of these broad signals is presently unknown. It has to be noted that the dielectric constants of both solvents are nearly the same.
The nickel complex 9-NiCl2 is obtained as a brown solid, which is soluble in THF and DMF. The broad signals and high dispersion of the 1H-NMR chemical shift support the expected paramagnetism of the dinickel system, which is corroborated by an effective magnetic moment spin only value of μeff = 4.33μB, indicative of two independent S = 1 nickel centers. According to μeff2 = ∑μi2, a slightly smaller value of 4μB is expected (for details see the ESI‡).
Complexation of 10 with ZnOTf2 leads to the formation of 10-ZnOTf2. The solid state structure (Fig. 4) shows a bond angle between the anthracene linker and the PDI unit of 124.3(4)° and the zinc atoms lie above the plane spanned by the three nitrogen atoms of the PDI moiety. This distortion can be explained by the large steric demand of the triflate ligands. Using a more flexible linker molecule (11-ZnCl2), a larger intramolecular zinc–zinc distance of 18.31 Å can be observed in the solid state structure (Fig. 4 and Table 2).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| Compound | Bond legth (Å) | Bond angle (°) | ||
|---|---|---|---|---|
| a The quality of the crystallographic data of complex 11-ZnCl2 is poor. While the constitution is confirmed, we refrained to include geometric parameters. | ||||
| 8-ZnCl2 | Zn1a–N1a | 2.108(2) | N2a–Zn1a–N3a | 142.92(8) |
| Zn1a–N2a | 2.250(2) | N1a–Zn1a–Cl1a | 104.38(6) | |
| Zn1a–N3a | 2.273(2) | N1a–Zn1a–Cl2a | 146.23(6) | |
| Zn1a–Cl1a | 2.282(1) | N1a–Zn1a–N2a | 72.62(8) | |
| Zn1a–Cl2a | 2.222(1) | Cl1a–Zn1a–Cl2a | 109.34(3) | |
| N3a–C2a | 1.276(3) | |||
| C8a–N2a | 1.281(3) | |||
| 9-NiCl2 | Ni1–N1b | 1.979(4) | N2b–Ni1–N3b | 146.57(16) |
| Ni1–N2b | 2.165(4) | N1b–Ni1–Cl1 | 93.19(14) | |
| Ni1–N3b | 2.128(4) | N1b–Ni1b–Cl2b | 161.75(14) | |
| Ni1–Cl1 | 2.264(2) | N1b–Ni1–N2b | 76.16(17) | |
| Ni1–Cl2 | 2.232(2) | Cl1–Ni1–Cl2 | 104.95(6) | |
| N3b–C8b | 1.270(6) | |||
| C7b–N1b | 1.331(6) | |||
| N1b–C3b | 1.325(6) | |||
| C3a–N1a | 1.339(6) | |||
| N1a–C7a | 1.329(6) | |||
| C2b–N2b | 1.274(6) | |||
| 10-ZnOTf2 | Zn1–N1bb | 2.029(4) | N2bb–Zn1–N3bb | 146.8(2) |
| Zn1–N2bb | 2.184(4) | N1bb–Zn1–O1bb | 149.4(2) | |
| Zn1–N3bb | 2.205(4) | N1bb–Zn1–O4bb | 101.1(1) | |
| Zn1–O1bb | 1.933(3) | N1bb–Zn1–N2bb | 75.6(2) | |
| Zn1–O4bb | 2.010(3) | O1bb–Zn1–O4bb | 108.9(2) | |
| N2bb–C8bb | 1.279(6) | |||
| N1bb–C7bb | 1.330(6) | |||
| N3bb–C2bb | 1.277(6) | |||
| N1bb–C3bb | 1.330(6) | |||
| 12-ZnCl2 | Zn1a–N1a | 2.105(2) | N2a–Zn1a–N3a | 145.18(6) |
| Zn1a–N2a | 2.321(2) | N1a–Zn1a–Cl1a | 139.50(4) | |
| Zn1a–N3a | 2.241(2) | N1a–Zn1a–Cl2a | 106.22(4) | |
| Zn1a–Cl1a | 2.2477(5) | N1a–Zn1a–N2a | 73.11(6) | |
| Zn1a–Cl2a | 2.2800(5) | Cl1a–Zn1a–Cl2a | 114.00(2) | |
| N2a–C2a | 1.282(2) | |||
| N1a–C7a | 1.338(2) | |||
| N1a–C3a | 1.339(2) | |||
| N3a–C8a | 1.279(2) | |||
 | ||
| Fig. 4 ORTEP plot of complexes 8-ZnCl2, 9-NiCl2, 10-Zn(OTf)2, 11-ZnCl2 and 12-ZnCl2 with anisotropic displacement parameters shown at the 50% probability level. Hydrogen atoms, co-crystallized solvent molecules and the mesityl substituents of the imine (except one carbon atom for each mesityl group) are omitted for clarity. | ||
Flexibility and rigidity of the ligand system
It is intended to employ the new ligand systems in the dinuclear activation of small molecules with square-planar group 9 transition metal complexes. A particular focus will be placed on systems with ketimine N-substituents with little steric demand as in ligand 12, e.g. propyl groups. In order to get a first impression of the accessible range of metal–metal distances, we therefore turned to DFT calculations for square-planar iridium chlorido systems with coordination number 4 rather than the 5-coordinate MX2 complexes reported herein (Fig. 5). We also focused on ligands with methyl imine substituents as realistic models for the N-propyl substituted ligands, e.g.12. These calculations were performed in two ways. For the first type of relaxed potential energy scans (PES), the metal–metal bond distance was varied under C2v-symmetry constraint (Fig. 6). Dispersion interactions were accounted for using Grimme's D3-corrections for the employed tpss functional. | ||
| Fig. 5 Relaxed potential energy scan for the variation of the iridium–iridium distance under C2v-symmetry constraint (left) for the iridium chlorido model system (right). | ||
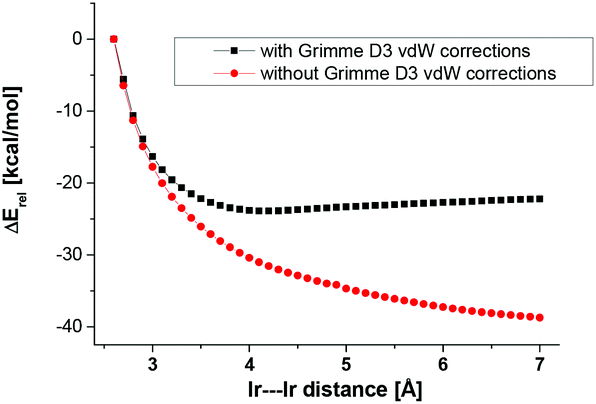 | ||
| Fig. 6 Potential energy scan with C2v-symmetry constraints of the phenyl bridged complex with and without vDW (Grimme D3) corrections. | ||
Overall, the potentials are rather flat in the metal–metal distance range of 3–7 Å. This illustrates the flexibility of these ligand systems to adjust to transition state geometries in bimolecular activation steps. This holds in particular for the phenyl bridged system, which displays a weakly pronounced minimum at 3.8 Å. The importance of the interannular van der Waals interactions is underlined in an exemplary fashion through the comparison of the DFT results with and without Grimme's D3 correction (Fig. 6).
Next we probed the ligand flexibility through relaxed potential energy scans without symmetry constraints. The inspection of the PES for the biphenylene bridged model in C2v- and C1-symmetry clearly reveals that the symmetry constraint imposes an energetic increase of at least 10 kcal mol−1 (Fig. 6).
Inspection of Fig. 7 reveals two distinct minima at 2.63 and 5.21 Å. The minimum at the shorter metal–metal distance corresponds to the μ-chlorido-bridged structure presented in Fig. 8.
 | ||
| Fig. 7 DFT (PBE-D3) relaxed potential energy scan with C2v- and without symmetry for the phenylenyl bridged complex. | ||
 | ||
Fig. 8 Front and side views of the minimum structure (Ir–Ir distance: 2.63 Å;  C, C,  N, N,  Cl, Cl,  Ir). Ir). | ||
The Ir–μ-chlorido distances of 2.496 and 2.491 Å are nearly identical indicating a symmetrical μ-chlorido bridging unit, which displays an acute Ir–Cl–Ir angle of 63.5°. The remaining Ir–Cl bond to the terminal chlorido ligand has a significantly shorter bond length of 2.387 Å. This is yet longer than the Ir–Cl bonds of 2.308 Å observed in the other minimum of the PES at an Ir⋯Ir distance of 5.21 Å, which corresponds to the unbridged molecular structure presented in Fig. 9.
 | ||
Fig. 9 Front and side views corresponding to the minimum structure (Ir⋯Ir distance: 5.21 Å;  C, C,  N, N,  Cl, Cl,  Ir). Ir). | ||
The PES presented in Fig. 7 displays another weakly pronounced minimum at an Ir⋯Ir distance of 3.48 Å, which is presumably stabilized by interannular vDW interactions of the PDI ligand. The corresponding molecular structure is presented in Fig. 10.
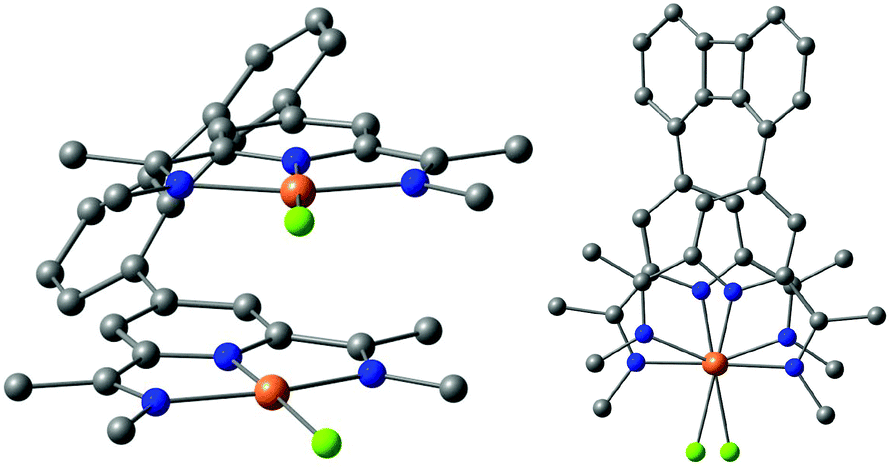 | ||
Fig. 10 Front and top views corresponding to the minimum structure at 3.48 Å ( C, C,  N, N,  Cl, Cl,  Ir). Ir). | ||
Two energy minima at short and long Ir⋯Ir distances of ca. 2.65 Å and in the range of 4.8 to 5.8 Å were also observed for the other bridging units (see Table 3 and the ESI‡ for details). For the phenyl and naphthyl systems, however, rather than an additional minimum at intermediate distances, we noted transition states for the interconversion of the μ-chlorido bridged to the unbridged structures (for details see the ESI‡). This is shown in an exemplary fashion for the system with the phenyl linker in Fig. 11, which displays a small barrier of 9 kcal mol−1 (naphthyl linker: 7 kcal mol−1) at the DFT PBE-D3 level.
 | ||
Fig. 11 Transition state for the conversion of the μ-Cl-bridged and unbridged iridium system with the phenyl linker (νimag = −44 cm−1). The Ir–Ir distance in Å and the scaled vectors are presented ( C, C,  N, N,  Cl, Cl,  Ir). Ir). | ||
| Compound | Aromatic linker Ar | Substituent R | M–M distance [Å] |
|---|---|---|---|
| 8-ZnCl2 | 1,2-Benzene | Mesityl | 7.76 |
| 9-NiCl2 | 1,8-Biphenylene | 2,6-Dimethyl-6-tert-butylphenyl | 7.17 |
| 10-ZnOTf2 | 1,8-Anthracene | Mesityl | 8.39 |
| 11-ZnCl2 | 3,3′-Diphenylether | Mesityl | 18.31 |
| 12-ZnCl2 | 1,8-Naphthalene | Propyl | 5.88 |
The μ-chlorido-bridged structure is an unexpected structural motif. In order to get more accurate insights into the thermodynamic situation, we also performed DLPNO-CCSD(T) single point calculations for the DFT optimized minimum structures. The DFT and CCSD(T) results qualitatively agree and clearly reveal the unbridged structure as a global minimum with a slightly stronger preference according to the CCSD(T) calculation (Table 4). In particular, for the biphenylene backbone, the μ-chlorido bridged structure should be thermally accessible. We will follow up on this issue including a description of the detailed electronic structure and spectroscopic properties of the μ-chlorido bridged complexes once we have the experimental results.
| Bridge | Phenyl | Biphenylenyl | Naphthyl | Anthracenyl |
|---|---|---|---|---|
| Preference for unbridged complex DFT values in parentheses [kcal mol−1] | −11.0 (−8.9) | −2.8 (−2.5) | −7.0 (−1.6) | −6.2 (−0.8) |
| M⋯M distance μ-Cl-bridged [Å] | 2.65 | 2.63 | 2.63 | 2.63 |
| M⋯M distance unbridged [Å] | 5.82 | 5.21 | 4.83 | 5.65 |
Conclusions and outlook
Herein, we reported synthetic routes for para-pyridine tethered PDI ligands and the corresponding divalent 3d-transition metal complexes. Currently, we are investigating the activation of small molecules and the magnetic interaction of dinuclear group 9 and 12 transition metal PDI complexes, which will be reported in due course.Experimental section
Materials and methods
Manipulations involving air- and moisture-sensitive compounds were conducted under an atmosphere of nitrogen using standard Schlenk techniques or in a glovebox filled with a nitrogen atmosphere. THF, dioxane and MTBE were distilled before use from sodium/benzophenone. Absolute dichloromethane, n-hexane and n-pentane were taken from an MBraun solvent purification system. Deuterated THF was dried over sodium benzophenone ketyl and stored under nitrogen. Compound 1,11 1,8-dibromobiphenylene,19 1,8-dichloroanthracene20 and 1,8-diiodonaphthalene21 were synthesized following published procedures. All other chemicals were purchased from commercial sources and used as received.Instrumentation
1H- and 13C-NMR spectra were recorded using Bruker Fourier 300 MHz, Bruker AVANCE 400 MHz, Bruker AVANCE I 400 MHz, Bruker DRX500 MHz or Bruker AVANCE III 600 MHz NMR spectrometers. All spectra were recorded at 298 K. 1H-NMR and 13C-NMR chemical shifts are reported in parts per million (ppm). Abbreviations used in the description of the NMR data are as follows: br, broad; s, singlet; d, doublet; t, triplet; q, quartet; and m, multiplet. MALDI mass spectra were recorded on a MALDI TOF/TOF Bruker UltrafleXtreme Smartbeam II Laser using an anthracene matrix. ESI mass spectra were recorded using an Agilent 6224 ESI-TOF or Bruker maXis ESI-Q-TOF spectrometer. Elemental analyses were performed using an Elementar Vario ELIII or EuroVector/Hekatech EuroEA elemental analyzer. X-ray data were collected on a Bruker AXS SMART APEX II diffractometer with Mo Kα radiation at 100 K and a SuperNova Oxford diffractometer with Mo Kα or Cu Kα radiation.X-ray crystallography
Compounds 8-ZnCl2 and 10-Zn(OTf2) displayed strongly disordered co-crystallized solvent molecules, which could not be refined and were treated using the PLATON SQEEZE procedure/program.22 Details of the crystallographic data collection are summarized in the ESI.‡Computations
The calculations were carried out on a local and the “Hummel” computing cluster of the University of Hamburg computing center (RRZ). DFT calculations were performed with ver. 7.1 and 7.2 of the parallelized Turbomole program package.23 For the potential energy scans, def-SV(P) basis sets were employed for the C, H, N and Cl atoms, while a def2-TZVP basis24 with the corresponding Stuttgart-Dresden pseudopotential ECP-60-MWB was utilized for iridium.25 The TPSS functional with Grimme's D3-van der Waals corrections with the Becke–Johnson damping scheme was employed for the PES.26 The PBE-D3 functional showed good agreement with the results from the DLPNO-CCSD(T) method and was employed for all other calculations.27 The RI-DFT method was used with the corresponding RIJ-auxiliary basis. The relaxed potential energy scans were performed with a shell script of the Turbomole program package. The observed minima in the PES were fully optimized without geometry or symmetry constraints employing def2-TZVP basis sets for all atoms and Stuttgart-Dresden ECP-60-MWB pseudopotentials for Ir. The minima were confirmed by the absence of imaginary frequencies in the analytic calculations of the second derivatives. Geometries optimized at the PBE-D3 level (ridft) with the large def2-TZVP basis set were used for single point DLPNO-CCSD(T) calculations with the Orca 4.01 program system.28 For the latter, the RIJCOSX approximation was used for the RHF calculations in combination with def2-TZVPP and the corresponding def2-TZVPP auxiliary basis and a Stuttgart-Dresden ECP-60-MWB pseudopotential for Ir. The DLPNO accuracy parameter was left at the default value (NormalPNO). The T1 diagnostics are in the range of 0.013–0.014 thus revealing no deviation from the single determinantal reference solution.291-Iodo-3′-bromodiphenyl ether
500 mg (2.49 mmol) 3-bromophenylboronic acid, 547 mg (2.49 mmol) 3-iodophenol, 136 mg (0.749 mmol) Cu(OAc)2 and 1 g molecular sieve 4 Å were evacuated three times and suspended in 10 mL dry dichloromethane. The mixture was cooled to 0 °C and 1.7 mL (13 mmol) dry NEt3 was added. Afterwards, a balloon with O2 was attached and the flask was purged with O2. The reaction mixture was allowed to warm to room temperature overnight. The solids were filtered off and the solvent of the filtrate was removed in vacuo to give a brown oil. The crude product was purified by column chromatography with petroleum ether as the eluent. Upon drying under high vacuum, 468 mg (1.25 mmol, 50%) of a colorless oil were obtained.

1 H-NMR (600 MHz, CD2Cl2) δ [ppm] = 7.51 (ddd, J = 7.8, 1.6, 1.0 Hz, 1H, H4′′), 7.40 (dd, J = 2.4, 1.6 Hz, 1H, H2′′), 7.29 (ddd, J = 8.0, 1.9, 1.0 Hz, 1H, H3′), 7.24 (t, J = 8.1 Hz, 1H, H5′), 7.19 (t, J = 2.1 Hz, 1H, H2′), 7.10 (t, J = 8.0 Hz, 1H, H5′′), 7.02 (ddd, J = 8.3, 2.4, 1.0 Hz, 1H, H6′′), 6.97 (ddd, J = 8.1, 2.4, 1.0 Hz, 1H, H6′). 13C-NMR (150 MHz, CD2Cl2) δ [ppm] = 158.1 (C3′), 157.6 (C3′′), 133.5 (C4′′), 131.8 (5′′), 131.6 (C5′), 128.6 (C2′′), 127.3 (C4′), 123.4 (C1′), 122.6 (C2′), 119.0 (6′′), 118.1 (C6′), 94.8 (C1′′). Elemental analysis: Calc.: C, 38.43; H, 2.15. Found: C, 38.73; H, 2.24.
4
Bis(pinacolato)diboron (8.3 g, 33 mmol), 4,4-di-tert-butyl-2,2′-bipyridine (107 mg, 400 μmol) and [Ir(OMe)COD]2 (265 mg, 400 μmol) were dissolved in 450 mL anhydrous n-hexane and stirred for one hour at room temperature. 1 (7.05 g, 29.8 mmol) was added to the red solution and the mixture was heated to reflux for 4.5 hours under stirring. The mixture was allowed to cool to RT and the solvent was reduced to 200 mL. The resulting white precipitate was collected by filtration and washed with cold n-pentane. After recrystallisation from n-hexane, the product was obtained as a white solid (9.44 g, 25.0 mmol, 84%).
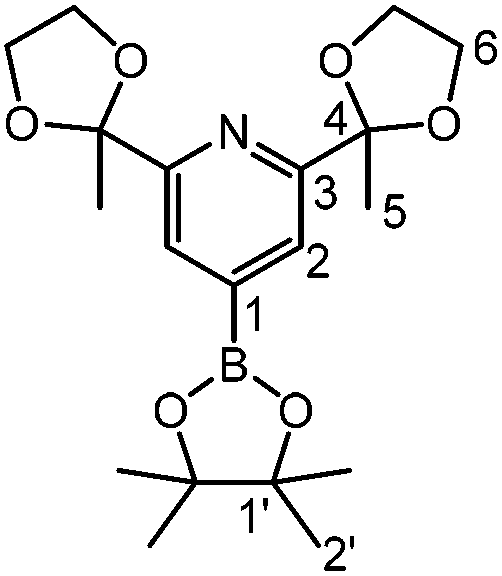
1 H-NMR (300 MHz, CDCl3): δ [ppm] = 7.80 (s, 2H, H2); 4.11–3.93 (m, 8H, H6); 1.76 (s, 6H, H5); 1.34 (s, 12H, H2′). 13C-NMR (75 MHz, CDCl3): δ [ppm] = 159.6 (C3); 123.8 (C2); 109.0 (C4); 84.6 (C1′); 25.0 (C2′); 24.9 (C5). ESI-MS: m/z = 378.2 [M]+. Elemental analysis: Calc.: C, 60.49; H, 7.48; N, 3.71. Found: C, 60.57; H, 7.41; N, 3.67.
5
53 mg (80 μmol) [Ir(OMe)COD]2, 703 mg (2.77 mmol) bis(pinacolato)diboron and 73 mg (0.27 mmol) 4,4′-di-tert-butyl-2,2′-dipyridine were dissolved in 50 mL anhydrous MTBE and stirred at room temperature until the solution turned deep red. Then 1.07 g (2.68 mmol) 2 were added and the solution was refluxed for 1 h at 85 °C. The solution was allowed to cool to RT, upon which a yellow solid precipitated. The latter was collected by filtration, washed with MTBE and dried in vacuo to yield 5 as an analytically pure yellow solid (785 mg, 1.50 mmol, 56%).

1 H-NMR (300 MHz, THF-d8): δ [ppm] = 8.81 (s, 2H, H2), 6.86 (s, 4H, H9), 2.26 (s, 6H, H11), 2.21 (s, 6H, H5), 1.98 (s, 12H, H8), 1.37 (s, 12H, H2′). 13C-NMR (75 MHz, THF-d8): δ [ppm] = 167.9 (C4), 155.6 (C3), 147.7 (C7), 132.64 (C10), 129.4 (C9), 128.0 (C2), 125.7 (C6), 85.6 (C1′), 25.3 (C2′), 21.0 (C11), 18.2 (C8), 16.6 (C5). MALDI-MS: m/z = 532.3 [M]+; 508.2 [M − CH3]+. Elemental analysis: Calc: C, 75.49; N, 8.11; H, 8.00. Found: C, 75.71; N, 8.03; H, 8.09.
6
56 mg (85 μmol) [Ir(OMe)COD]2, 564 mg (2.22 mmol) bis(pinacolato)-diboron and 43 mg (0.16 mmol) 4,4′-di-tert-butyl-2,2′-dipyridine were dissolved in 50 mL absolute MTBE and stirred at room temperature. After the solution turned deep red, 1.02 g (2.11 mmol) 3 were added and the reaction mixture was refluxed at 85 °C for 20 h. Upon cooling to RT, a yellow precipitate formed, which was collected by filtration, washed with cold MTBE and dried in vacuo to yield a yellow solid (975 mg 1.60 mmol, 76%).

1 H-NMR (300 MHz, THF-d8): δ [ppm] = 8.81 (s, 2H, H2), 7.09 (s, 4H, H9), 2.22 (s, 6H, H5), 2.02 (s, 6H, H8), 1.37 (s, 12H, H2′), 1.32 (s, 18H, H12). 13C-NMR (75 MHz, THF-d8): δ [ppm] = 167.8 (C4), 155.6 (C3), 147.7 (C10), 146.2 (C7), 139.9 (C1), 128.0 (C2), 125.7 (C9), 125.3 (C6) 85.6 (C1′), 32.2 (C12), 25.3 (C2′), 27.5 (C11), 18.6 (C8), 16.7 (C5). MALDI-MS: m/z = 607.4 [M˙+]. Elemental analysis: Calc: C, 76.75; H, 8.97; N, 7.06. Found: C, 77.08; H, 8.96; N, 6.91.
7
4 (7.81 g, 19.0 mmol), 1,8-diiodonaphthalene (3.29 g, 8.65 mmol), K2CO3 (5.98 g, 43.3 mmol), Pd(PPh3)4 (200 mg, 173 μmol) and KF (1.10 g, 19.0 mmol) were placed in a Schlenk tube equipped with a J-Young high vacuum Teflon valve. 50 mL degassed THF and 2 mL degassed water were added and the reaction mixture was stirred overnight at 100 °C. After filtration through a glass frit, the filtrate was evaporated to dryness under high vacuum. The resulting solid was dissolved in 100 mL dichloromethane and washed with water (3 × 70 mL). The solvent of the organic phase was removed in vacuo and the resulting brown oil was redissolved in 100 mL acetone. After the addition of 3 drops of concentrated HCl (37%), the mixture was heated under reflux overnight. Upon cooling to room temperature, a white precipitate was formed, which was collected by filtration. The solid was washed with 50 mL acetone and dried under vacuum to yield a white solid (1.99 g, 4.42 mmol, 51%).

1 H-NMR (300 MHz, CDCl3): δ [ppm] = 8.08 (dd, 3JH9–H8 = 8.22 Hz, 4JH9–H7 = 1.34 Hz, 2H, H4′); 7.71 (s, 4H, H2); 7.63 (dd, 3JH8–H7 = 7.09 Hz, 3JH8–H9 8.22 Hz, 2H, H3′); 7.43 (dd, 3JH7–H8 = 7.09 Hz, 4JH7–H9 = 1.34 Hz, 2H, H2′); 2.65 (s, 12H, H5). 13C-NMR (75 MHz, CDCl3): δ [ppm] = 198.4 (C4); 152.9 (C1); 151.9 (C3); 135.4 (Cq); 135.2 (Cq); 131.3 (C2′); 131.0 (C4′); 128.1 (Cq); 125.9 (C3′); 125.5 (C2); 25.5 (C5). Elemental analysis: Calc: C, 74.65; H, 4.92; N, 6.22. Found: C, 74.54; H, 4.95; N, 6.16.
8
5 (100 mg, 191 μmol), KF (38 mg, 0.27 mmol), K3PO4 (116 mg, 546 μmol), Pd2dba3 (0.8 mg, 0.9 μmol) and XPhos (0.4 mg, 0.9 μmol) were placed in a Schlenk tube equipped with a J-Young high vacuum Teflon valve. 6 mL degassed dioxane and 1 mL degassed water were added and the resulting yellow solution was stirred at 110 °C for 3 h. After cooling to room temperature, the solvent was removed in vacuo. The residue was dissolved in a mixture of 7 mL dichloromethane and 7 mL water. The phases were separated and the aqueous phase was extracted with dichloromethane (3 × 7 mL). The combined organic phases were washed with water (3 × 7 mL) and dried over anhydrous Na2SO4. The solvent was removed in vacuo and the crude product was recrystallized from n-hexane to yield the product as yellow crystals (74 mg, 85 μmol, 93%). Crystals suitable for X-ray analysis were grown from a concentrated n-hexane solution.
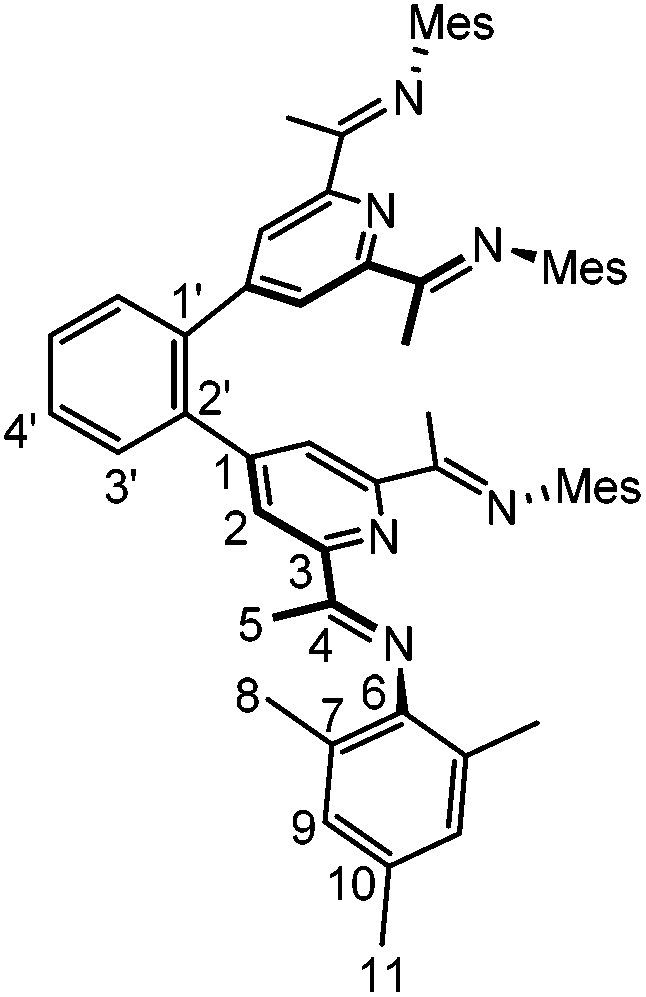
1 H-NMR (400 MHz, CD2Cl2): δ [ppm] = 8.32 (s, 4H, H2); 7.73–7.69 (m, 2H, H3′); 7.61–7.57 (m, 2H, H4′); 6.83 (s, 8H, H9); 2.26 (s, 12H, H11); 2.12 (s, 12H, H5); 1.81 (s, 24H, H8). 13C-NMR (100 MHz, CD2Cl2): δ [ppm] = 167.4 (C4); 155.7 (C3); 149.8 (C1); 146.8 (C6); 138.9 (C1′); 132.5 (C10); 131.0 (C3′); 129.7 (C4′); 128.9 (C9); 125.5 (C7); 123.9 (C2); 21.0 (C11); 18.0 (C8); 16.5 (C5). MALDI-MS: m/z = 868.5 [M]+. Elemental analysis: Calc: C, 82.91; H, 7.42; N, 9.67. Found: C, 82.99; H, 7.49; N, 9.16.
9
94 mg (0.16 mmol) 6, 24 mg (77 μmol) 1,8-dibromobiphenylene and 10 mg (8.6 μmol) Pd(PPh3)4 were dissolved in 20 mL absolute THF and placed in a Schlenk tube equipped with a J-Young high vacuum Teflon valve. To this solution, 123 mg (0.888 mmol) K2CO3 were added and the resulting suspension was stirred at 90 °C for 49 h. After cooling to room temperature, the reaction mixture was filtered and the filtrate was mixed with 250 mL methanol. The solvent was reduced in vacuo until the product precipitated. The solid was collected and washed with cold methanol. The yellow solid was obtained in a yield of 65% (56 mg, 50 μmol).

1 H-NMR (300 MHz, THF-d8): δ [ppm] = 8.11 (s, 4H, H2), 7.70 (m, 4H, H2′/H3′), 7.00 (s, 8H, H9), 6.87 (d, 2H, 3JH4′–H3′ = Hz, H4′), 1.99 (s, 12H, H5), 1.89 (s, 24H, H8), 1.29 (s, 36H, H12). 13C-NMR (75 MHz, THF-d8): δ [ppm] = 167.5 (C3), 157.3 (C4), 152.8 (C6′), 149.3 (C5′), 147.5 (C1′), 147.2 (C1), 146.2 (C10), 131.6 (C7), 131.1 (C3′), 130.6 (C2′), 125.8 (C6), 125.7 (C9), 121.8 (C2), 117.9 (C4′), 34.8 (C11), 32.1 (C12), 18.9 (C8), 17.3 (C5). MALDI-MS: m/z = 1110.7 [M]+; 1095.06 [M − CH3]+. Elemental analysis: Calc: C, 83.99; H, 8.32; N, 7.35. Found: C, 84.13; H, 8.33; N, 7.55.
10
1,8-Dichloroanthracene (504 mg, 2.04 mmol), 5 (2.23 g, 4.26 mmol), XPhos (13 mg, 28 μmol), Pd(PPh3)4 (26 mg, 22 μmol), KF (357 mg, 6.14 mmol) and K3PO4 (2.62 g, 12.3 mmol) were dissolved in 24 mL dioxane and 2 mL water. The reaction mixture was stirred at 105 °C for 19 h, upon which a yellow precipitate formed. After cooling to room temperature, the solids were collected by filtration, washed with 20 mL water, diethylether and tetrahydrofuran and dried in vacuo. 1.94 g (2.00 mmol, 98%) of a yellow powder was obtained.

1 H-NMR (600 MHz, CDCl3): δ [ppm] = 9.25 (s, 1H, H7′), 8.65 (s, 4H, H6′), 8.65 (s, 1H, H2), 8.12 (dd, 2H, J = 7.72 Hz, H2′), 7.62–7.56 (m, 4H, H3′/H4′), 6.84 (s, 8H, H9), 2.28 (s, 12H, H11), 2.21 (s, 12H, H5), 1.89 (s, 24H, H8). 13C-NMR (150 MHz, CDCl3): δ [ppm] = 167.2 (C3), 155.7 (C4), 149.7 (Canthracene), 146.3 (C6), 137.7 (C1′), 132.3 (Cmesityl), 132.1 (Canthracene), 129.7 (C2′), 129.5 (C1) 129.4 (Canthracene), 128.8 (C6′), 128.6 (C9), 125.5 (Canthracene), 125.3 (Cmesityl), 123.2 (C2), 120.8 (C7′), 20.9 (C11), 18.1 (C8), 16.7 (C5). MALDI-MS: m/z = 968.6 [M]+; 953.5 [M − CH3]+. Elemental analysis: Calc: C, 83.83; H, 7.07; N, 8.86. Found: C, 84.26; H, 7.07; N, 8.67.
11
650 mg (1.73 mmol) iodo-3′-bromodiphenyl ether, 1.91 g (3.64 mmol) 5, 1.44 g (10.4 mmol) K2CO3, 302 mg (5.20 mmol) KF and 40 mg (0.05 mmol) Pd(dppf)Cl2 were dissolved in 10 mL of degassed THF and 1 mL of degassed water was added. The reaction mixture was heated at 80 °C for 2 h. After cooling to RT, the organic phase was separated and the aqueous phase was extracted with 10 mL THF. The combined organic phases were reduced in vacuo and after the addition of 50 mL methanol, a yellow solid precipitated. The solid was collected by filtration and dried in vacuo. 1.12 g (1.17 mmol, 67%) of a yellow powder was obtained.

1 H-NMR (CD2Cl2, 500 MHz) δ [ppm] = 8.70 (s, 4H, H2), 7.63 (dt, J = 7.7, 1.3 Hz, 2H, H6′), 7.59 (t, J = 2.1 Hz, 2H, H2′), 7.50 (t, J = 8.0 Hz, 2H, H5′), 7.14 (ddd, J = 8.3, 2.4, 0.9 Hz, 2H, H4′), 6.89 (s, 8H, H9), 2.28 (s, 12H, H11), 2.23 (s, 12H, H5), 1.99 (s, 24H, H8). 13C-NMR (125 MHz, CD2Cl2) δ [ppm] = 167.9 (C4), 158.4 (C3′), 156.6 (C3), 149.0 (C1), 146.9 (C6), 141.0 (C1′), 132.7 (C10), 131.1 (C5′), 129.0 (C9), 125.7 (C7), 123.1 (C2′), 120.4 (C2), 119.9 (C4′), 118.6 (C6′), 21.0 (C11), 18.8 (C8), 16.9 (C5). Elemental analysis: Calc: C, 82.43; H, 7.13; N, 8.74. Found: C, 82.11; H, 7.22; N, 8.58.
12
7 (600 mg, 1.33 mmol) and catalytic amounts of para-toluenesulfonic acid were suspended in n-propylamine (3.0 mL, 36 mmol) and stirred at 45 °C for two days. The solvent was removed in vacuo. After recrystallisation from n-hexane, a white solid was obtained (582 mg, 0.947 mmol, 71%). Crystals suitable for X-ray analysis were grown from a concentrated THF/n-hexane solution at −35 °C.

1 H-NMR (600 MHz, CDCl3): δ [ppm] = 7.98 (dd, 2H, H4′); 7.68 (s, 4H, H2); 7.56 (dd, 2H, H3′); 7.49 (dd, 2H; H2′); 3.39–3.34 (m, 8H, H6); 2.24 (s, 12H, H5); 1.69 (m, 8H, H7); 0.98 (t, 12H, H8). 13C-NMR (150 MHz, CDCl3): δ [ppm] = 165.8 (C4); 155.1 (C3); 151.2 (C1); 137.8 (C1′); 135.3 (C5′); 131.2 (C2′); 129.8 (C4′); 128.8 (C6′); 125.4 (C3′); 122.0 (C2); 54.5 (C6); 24.2 (C7); 13.7 (C5); 12.4 (C8). Elemental analysis: Calc: C, 78.14; H, 8.20; N, 13.67. Found: C, 78.13; H, 8.23; N, 13.69.
8-ZnCl2
To a yellow solution of 12 (400 mg, 460 μmol) in 5 mL dichloromethane ZnCl2 (138 mg, 1.01 mmol) was added. The suspension was stirred at RT for 18 h. The solvent of the resulting yellow suspension was removed in vacuo and the resulting solid was washed with 20 mL water to yield a yellow solid (407 mg, 357 μmol, 89%). Crystals suitable for X-ray analysis were grown by slow diffusion of methanol into a dichloromethane solution.

1 H-NMR (300 MHz, CD3CN): δ [ppm] = 8.30 (s, 4H, H2); 7.83 (s, 4H, H3′/4′); 6.91 (s, 8H, H9); 2.27 (s); 2.16 (s); 2.09 (s, H24, H8). 1H-NMR (400 MHz, DMF-d7): δ [ppm] = 8.61 (br s, 4H), 7.88 (br s, 2H), 6.91 (s, 8H), 2.26 (s, 24H), 2.04 (br s, 24H). Elemental analysis: Calc: C, 63.12; H, 5.65; N, 7.36. Found: C, 62.64; H, 5.83; N, 7.25.
9-NiCl2
To a solution of 128 g (115 μmol) 9 in a mixture of 3 mL absolute THF and 3 mL absolute acetone, a suspension of 40 mg (0.31 mmol) nickel(II)chloride in 2 mL acetone was added. The reaction mixture was stirred at 90 °C in a Schlenk tube equipped with a J-Young high vacuum valve for 24 h. Upon cooling to RT, the resulting suspension was filtered and the filtrate was dried in vacuo. The brown solid was recrystallized from DMF to yield yellow crystals of 9-NiCl2 (51 mg, 37 μmol, 32%). Crystals suitable for X-ray analysis were grown from a THF solution. The magnetic moment was determined by variable temperature vibrating sample magnetometry (VSM, for details see the ESI‡).

1 H-NMR (THF-d8, 300 MHz): δ [ppm] = (w1/2) = 71 (71 Hz), 15.11 (30 Hz), 13.4 (185 Hz), 8.77 (24 Hz), 8.36 (24 Hz), 8.10 (7 Hz), 7.93 (11 Hz), 7.45 (14 Hz), 7.00 (10 Hz), 6.37 (18 Hz), 5.81 (62 Hz), 2.43 (8 Hz), 2.00 (8 Hz), 1.89 (7 Hz), 1.34 (7 Hz), 1.30 (6 Hz), 0.66 (46 Hz). MALDI-MS: m/z = 1205.74 [M − NiCl3]+, 1168.765 [M − NiCl4]+. Elemental analysis: Calc: C, 67.89; H, 6.72; N, 6.02. Found: C, 68.24; H, 6.76; N, 6.12.
10-Zn(OTf)2
100 mg (103 μmol) of 10 and 75 mg (0.21 mmol) of ZnOTf2 were suspended in a mixture of 10 mL acetonitrile and 5 mL dichloromethane. After 16 h, the solvent of the orange solution was removed in vacuo. The orange crude product was recrystallized twice from dichloromethane and diethylether to yield 151 mg (88 μmol, 86%) of yellow needles. Crystals suitable for X-ray analysis were grown from a dichloromethane solution overlaid with diethylether.
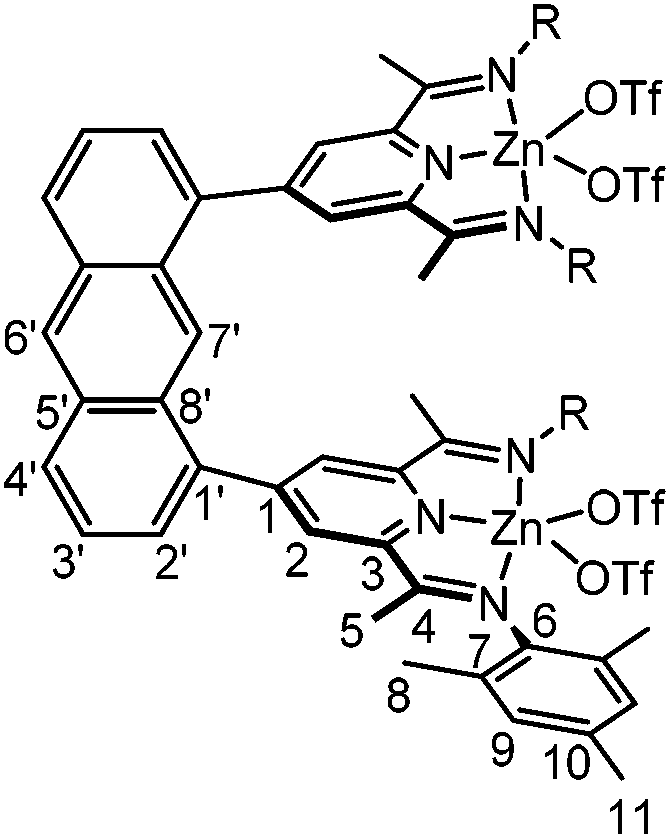
1 H-NMR (600 MHz, CD2Cl2): δ [ppm] = 8.85 (s, 1H, H7′); 8.67 (s, 1H, H6′); 8.45 (s, 4H, H2); 8.33 (d, 2H, H2′); 7.71 (t, 2H, H3′); 7.54 (d, 2H, H4′); 6.95 (s, 8H, H9); 2.33 (s, 12H, H5); 2.31 (s, 12H, H11); 2.02 (s, 24H, H8). 13C-NMR (150 MHz, CD2Cl2): δ [ppm] = 166.8 (C3); 159.8 (C5′); 147.5 (C4); 141.0 (C6); 136.8 (C10); 136.2 (C1); 132.6 (C8′); 131.8 (C2′); 131.5 (C4′); 130.1 (C7′); 129.9 (C9); 128.8 (C7); 128.4 (C2); 126.0 (C3′); 121.3 (C6′); 21.1 (C11); 18.5 (C8); 17.3 (C5). Elemental analysis: Calc: C, 64.25; H, 5.65; N, 6.81. Found: C, 63.75; H, 5.85; N, 6.45.
11-ZnCl2
100 mg (104 μmol) of 16 and 28 mg of ZnCl2 (0.21 mmol) were stirred in 10 mL THF. After 20 min, a yellow precipitate formed. After additional 2 h, the solvent was decanted and the yellow solid that remained was washed with 10 mL THF and dried in vacuo. 95 mg (77 μmol, 74%) of the product were obtained as a yellow powder in an analytically pure form. Crystals suitable for X-ray analysis were grown from a concentrated acetonitrile solution.
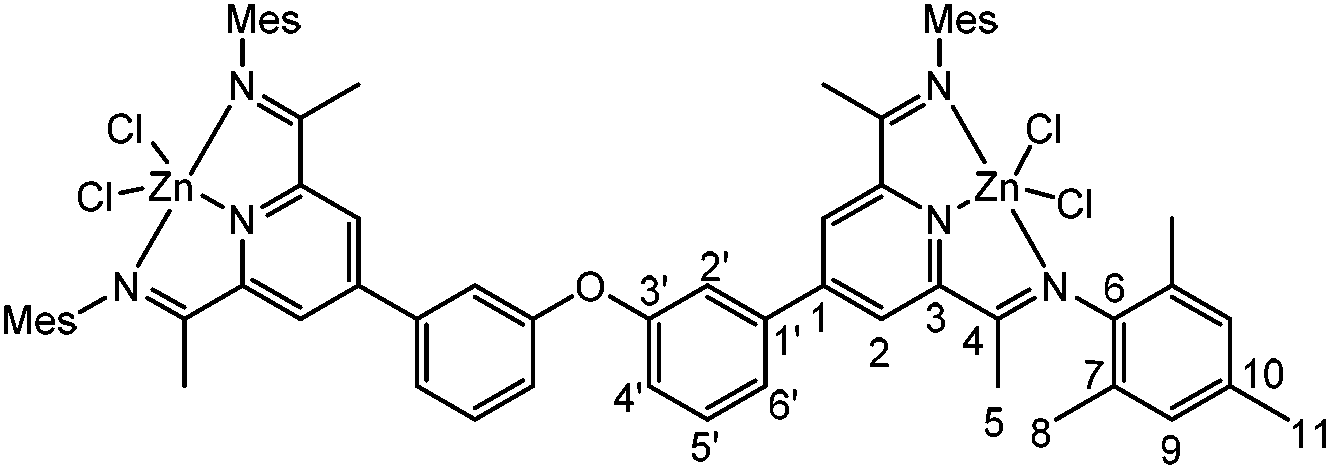
1 H-NMR (600 MHz, CD2Cl2) δ [ppm] = 8.39 (s, 4H, H2), 7.68–7.63 (m, 4H, H6′ + H2′), 7.60 (t, J = 2 Hz, 2H, H5′), 7.30 (ddd, J = 8 Hz, 2.4 Hz, 1.2 Hz, 2H, H4′) 6.95 (s, 8H, H9), 2.36 (s, 12H, H11) 2.30 (s, 12H, H5), 2.20 (s, 24H, H8). 13C-NMR (150 MHz, CD2Cl2) δ [ppm] = 164.8 (C4), 158.4 (C3′), 156.1 (C1), 150.0 (C3), 142.9 (C6), 138.8 (C1′), 135.6 (C10), 132.1 (C5′), 129.6 (C9), 128.6 (C7), 124.9 (C2), 123.7 (C2′), 121.5 (C4′), 119.5 (C6′), 21.1 (C11), 19.1 (C8), 17.4 (C5). Elemental analysis: Calc: C, 64.25; H, 5.65; N, 6.81. Found: C, 63.75; H, 5.85; N, 6.45.
12-ZnCl2
A solution of 3 (95 mg, 0.15 mmol) in 2 mL dichloromethane was added to a suspension of ZnCl2 (44 mg, 0.32 μmol) in 3 mL dichloromethane. After stirring at RT for 15 h, the pale yellow suspension was filtered. The solvent of the filtrate was evaporated in vacuo to yield a pale yellow solid (104 mg, 117 μmol, 76%). Crystals suitable for X-ray analysis were grown by slow diffusion of n-hexane into a THF solution.

1 H-NMR (300 MHz, CD2Cl2): δ [ppm] = 8.22 (d, 2H, H4′); 7.78 (dd, 2H, H3′); 7.74 (s, 4H, H2); 7.58 (d, 2H, H2′); 3.95–3.60 (m, 8H, H6); 2.29 (s, 12H, H5); 1.88 (m, 8H, H7); 0.96 (t, 12H, H7). 13C-NMR (75 MHz, CD2Cl2): δ [ppm] = 161.1 (C4); 159.2 (C1); 149.0 (C3); 136.0 (Cquart); 134.5 (C3′); 133.6 (C2′); 132.2 (C4′); 128.1 (Cq); 127.2 (Cq); 124.8 (C2); 54.4 (C6); 23.2 (C7); 15.3 (C5); 12.4 (C8). Elemental analysis: Calc: C, 54.14; H, 5.68; N, 9.47. Found: C, 53.59; H, 5.81; N, 9.21.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Max Völker for the crystallographic data collection and Uli Behrens for assistance with the refinement of disordered structures. Funding by Deutsche Forschungsgemeinschaft through SFB 668 “Magnetismus vom Einzelatom zur Nanostruktur” is gratefully acknowledged.References
- (a) B. L. Small and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 7143–7144 CrossRef; (b) B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049–4050 CrossRef; (c) L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef; (d) S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1204 CrossRef PubMed.
- J. P. Britovsek, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. J. McTavish, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 7, 849–850 RSC.
- G. J. P. Britovsek, M. Bruce, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. Mastroianni, S. J. McTavish, C. Redshaw, G. A. Solan, S. Strömberg, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 8728–8740 CrossRef.
- G. H. Zohuri, S. M. Seyedi, R. Sandaroos, S. Damavandi and A. Mohammadi, Catal. Lett., 2010, 140, 160–166 CrossRef.
- (a) B. de Bruin, E. Bill, E. Bothe, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2000, 2936–2947 CrossRef; (b) P. H. M. Budzelaar, B. de Bruin and A. W. Gal, Inorg. Chem., 2001, 4649–4655 CrossRef.
- (a) J. O. Cabral, M. F. Cabral, M. G. Drew, F. S. Esho, O. Haas and S. M. Nelson, J. Chem. Soc., Chem. Commun., 1982, 1066–1067 RSC; (b) P. Barbaro, C. Bianchini, G. Giambastiani, I. G. Rios, A. Meli, W. Oberhauser, A. M. Segarra, L. Sorace and A. Toti, Organometallics, 2007, 26, 4639–4651 CrossRef; (c) J. Scott, S. Gambarotta, I. Korobkov and P. H. M. Budzelaar, J. Am. Chem. Soc., 2005, 127, 13019–13029 CrossRef PubMed; (d) R. W. Stotz and R. C. Stoufer, J. Chem. Soc. D, 1970, 1682–1683 RSC; (e) V. C. Gibson, S. McTavish, C. Redshaw, G. A. Solan, A. J. P. White and D. J. Williams, Dalton Trans., 2003, 221–226 RSC; (f) H. Sugiyama, I. Korobkov, S. Gambarotta, A. Möller and P. H. M. Budzelaar, Inorg. Chem., 2004, 43, 5771–5779 CrossRef PubMed; (g) H. Sugiyama, G. Aharonian, S. Gambarotta, G. P. A. Yap and P. H. M. Budzelaar, J. Am. Chem. Soc., 2002, 124, 12268–12274 CrossRef PubMed; (h) J. Scott, S. Gambarotta and I. Korobkov, Can. J. Chem., 2005, 279–285 CrossRef; (i) C. C. H. Atienza, C. Milsmann, S. P. Semproni, Z. R. Turner and P. J. Chirik, Inorg. Chem., 2013, 52, 5403–5417 CrossRef PubMed; (j) D. Lieb, F. C. Friedel, M. Yawer, A. Zahl, M. M. Khusniyarov, F. W. Heinemann and I. Ivanović-Burmazović, Inorg. Chem., 2013, 52, 222–236 CrossRef PubMed; (k) P. Cui, Q. Wang, S. P. McCollom, B. C. Manor, P. J. Carroll and N. C. Tomson, Angew. Chem., Int. Ed., 2017, 56, 15979–15983 CrossRef PubMed; (l) S. Takano, Y. Takeuchi, D. Takeuchi and K. Osakada, Chem. Lett., 2014, 43, 465–467 CrossRef; (m) J. W. Beattie, D. J. SantaLucia, D. S. White and S. Groysman, Inorg. Chim. Acta, 2017, 460, 8–16 CrossRef; G. Bombieri, F. Benetollo, W. T. Hawkins, A. Polo and L. M. Vallarino, Polyhedron, 1989, 8, 1923–1931 Search PubMed; A. W. A. Bligh, N. Choi, W. J. Cummis, E. G. Evangorou, J. D. Kelly and M. McPartlin, J. Chem. Soc., Dalton Trans., 1994, 3369–3376 Search PubMed.
- (a) X.-X. Zhang and B. B. Wayland, J. Am. Chem. Soc., 1994, 116, 7897–7898 CrossRef; (b) X.-X. Zhang, G. F. Parks and B. B. Wayland, J. Am. Chem. Soc., 1997, 119, 7938–7944 CrossRef; (c) X.-X. Zhang and B. B. Wayland, Inorg. Chem., 2000, 39, 5318–5325 CrossRef PubMed; (d) A. E. Sherry and B. B. Wayland, J. Am. Chem. Soc., 1990, 112, 1259–1261 CrossRef; (e) C. J. Chang, Z.-H. Loh, C. Shi, F. C. Anson and D. G. Nocera, J. Am. Chem. Soc., 2004, 126, 10013–10020 CrossRef PubMed; (f) Y. Deng, C. J. Chang and D. G. Nocera, J. Am. Chem. Soc., 2000, 122, 410–411 CrossRef; (g) J. P. Collman, C. M. Elliot, T. R. Halbert and B. S. Tovrog, Proc. Natl. Acad. Sci. U. S. A., 1977, 18–22 CrossRef; (h) J. P. Collman, F. C. Anson, C. E. Barnes, C. S. Bencosme, T. Geiger, E. R. Evitt, R. P. Kreh, K. Meier and R. B. Pettman, J. Am. Chem. Soc., 1983, 2694–2699 CrossRef; (i) G. Pognon, J. A. Wytko and J. Weiss, Org. Lett., 2007, 9, 785–788 CrossRef PubMed.
- M. Schwalbe, R. Metzinger, T. S. Teets and D. G. Nocera, Chemistry, 2012, 18, 15449–15458 CrossRef PubMed.
- (a) A. de Bettencourt-Dias, S. Viswanathan and A. Rollett, J. Am. Chem. Soc., 2007, 129, 15436–15437 CrossRef PubMed; (b) P. V. Ivchenko, I. E. Nifant'ev and I. V. Buslov, Tetrahedron Lett., 2013, 217–219 CrossRef.
- J. V. Obligacion and P. J. Chirik, ACS Catal., 2017, 7, 4366–4371 CrossRef PubMed.
- R. Breslow and S. Singh, Bioorg. Chem., 1988, 408–417 CrossRef.
- V. C. Gibson, B. S. Kimberley and P. J. Maddox, Iron, cobalt, manganese, and ruthenium complex polymerization catalysts, their manufactures and use in polymerizing olefins, PatentWO0015646, 2000 Search PubMed.
- S. M. Preshlock, B. Ghaffari, P. E. Maligres, S. W. Krska, R. E. Maleczka and M. R. Smith, J. Am. Chem. Soc., 2013, 135, 7572–7582 CrossRef PubMed.
- P. Harrisson, J. Morris, T. B. Marder and P. G. Steel, Org. Lett., 2009, 11, 3586–3589 CrossRef PubMed.
- (a) N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 3437–3440 CrossRef; (b) N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866–867 RSC.
- I. Maluenda and O. Navarro, Molecules, 2015, 20, 7528–7557 CrossRef PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 2457–2483 CrossRef.
- (a) P. R. Meehan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 888–890 CrossRef; (b) S. M. Boyt and A. B. Chaplin, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70(Pt 1), o73 CrossRef PubMed.
- H. O. House, D. G. Koepsell and W. J. Campbell, J. Org. Chem., 1972, 37, 1003–1011 CrossRef.
- Y. Koga, M. Kamo, T. Matsumoto and K. Matsubara, Eur. J. Inorg. Chem., 2011, 2011, 2869–2878 CrossRef.
- H. O. House, J. A. Hrabi and D. VanDerveer, J. Org. Chem., 1986, 51, 921–929 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 71, 9–18 CrossRef PubMed.
- TURBOMOLE V7.2 2017 & TURBOMOLE V7.1 2016, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef . (part of the TURBOMOLE basis set library).
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 123, 154101 CrossRef PubMed; S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; S. Grimme, L. Ehrlich and J. Goerigk, Comput. Chem., 2011, 32, 1456–1465 CrossRef PubMed.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef PubMed.
- F. Neese, ORCA – an Ab Initio, Density Functional and Semiempirical Program Package, V. 4.0.1, MPI für Chemische Energiekonversion, Mülheim a.d. Ruhr, Germany, 2017 Search PubMed.
- For T1, values up to 0.04 are accepted. T. J. Lee and P. R. Taylor, Int. J. Quantum Chem., 1989, 36, 199–207 CrossRef.
Footnotes |
| † Dedicated to Professor Felix Tuczek on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1845860, 1845398, 1845399, 1845400, 1845401, 1845402, 1846110 and 1856618. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt02347f |
| This journal is © The Royal Society of Chemistry 2018 |