
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity studies of an imine-functionalised phosphaalkene; unusual electrostatic and supramolecular stabilisation of a σ2λ3-phosphorus motif via hydrogen bonding†
Daniel
Morales Salazar
 *,
Arvind Kumar
Gupta
and
Andreas
Orthaber
*
*,
Arvind Kumar
Gupta
and
Andreas
Orthaber
*
Molecular Inorganic Chemistry, Department of Chemistry - Ångström Laboratories, Uppsala University, Sweden. E-mail: andreas.orthaber@kemi.uu.se; daniel.salazar@kemi.uu.se
First published on 11th May 2018
Abstract
A P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C heavy-alkene analogue that is unreactive towards the addition of strong acids on its double-bond is presented; instead, a strategically located imine nitrogen on the periphery forms protonated adducts displaying hydrogen bonding interactions. These materials are significantly more stable than the parent species, demonstrating an unprecedented approach towards the stabilisation of a multiple-bonded heavy main group fragment, in this case, a phosphaalkene. An HCl adduct self-assembles with H2O into a dimeric network displaying a discrete quadrilateral hydrogen-bonded arrangement.
C heavy-alkene analogue that is unreactive towards the addition of strong acids on its double-bond is presented; instead, a strategically located imine nitrogen on the periphery forms protonated adducts displaying hydrogen bonding interactions. These materials are significantly more stable than the parent species, demonstrating an unprecedented approach towards the stabilisation of a multiple-bonded heavy main group fragment, in this case, a phosphaalkene. An HCl adduct self-assembles with H2O into a dimeric network displaying a discrete quadrilateral hydrogen-bonded arrangement.
The study of molecular systems incorporating multiple-bonded heavy p-block elements has challenged chemists for decades. The high reactivity exhibited by heavy multiple bonds (E
C, EE, E![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C; E: third to sixth-row element from group 13–16) renders these species increasingly interesting at the fundamental and applied level.1
C; E: third to sixth-row element from group 13–16) renders these species increasingly interesting at the fundamental and applied level.1
Early on, breaking of the double-bond-rule with reports on the synthesis of stable species containing PC, PN, SiC, SiSi, AsC, and other multiple bonds, were accompanied by studies with small molecules (e.g. HCl, H2O, O2, Br2, MeOH, MeI) to confirm the sensitivity and reactivity of the multiple bonds.2 For example, the addition of HX (X: Hal−, Nuc−) to PC compounds (Scheme 1) is an archetypical example showcasing their increased reactivity. For other species, similar pathways (hydrolysis, oxidation, addition) lead to decomposition products in most reactions with small molecules.3 In sporadic cases, a multiple-bonded fragment is kept upon reaction with small molecules via rearrangement,4 substitution/addition reactions,5 or due to increased stability of the unusual bonding by other means (e.g. aromaticity in phosphinine).6 For example, dicoordinate phosphorus species display increased stability to small molecules in some examples, which is expressed in the ability to avoid protonation or alkylation.5,7
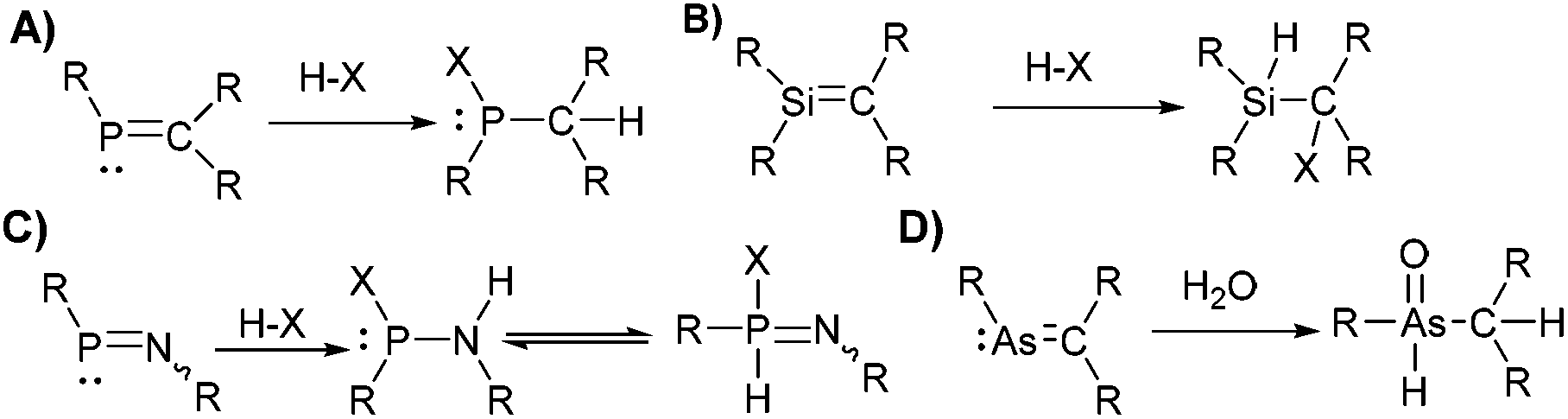 | ||
| Scheme 1 Selected reactivity of main group compounds with double bonding to a heavier element. Hydrogen halide addition on (A) PC (classically polarised) (B) SiC or SiSi bonds, (C) PN bonds, (D) and hydrolysis of AsC. | ||
The initially suggested instability of these species led to a naturally apparent dichotomy in which efforts have focused on either the stabilisation and isolation of the exotic motif as final materials8 (e.g. polymer chemistry and materials science), or their utilisation as highly-reactive synthons9 that can act as intermediates in driving unique chemical transformations (e.g. small-molecule activation,10 unique reactivity,11 catalysis,12 polymer-precursors,13 high-spin materials14). Regarding the former direction, finding ways to stabilise systems featuring multiple bonding to heavier elements are among the crucial goals in the modern main group community; increased stability to small molecules should broaden the scope of applying these fascinating materials in catalysis and optoelectronic materials.
In here, we explore the reactions of the imine containing phosphaalkene (1)15 with a variety of strong acids (Scheme 2). Despite being a sensitive compound under atmospheric conditions, when 1 reacts with hydrogen chloride (HCl), trifluoromethanesulfonic acid (HOTf), and hexafluorophosphoric acid (HPF6(aq.)) under inert conditions, the PC bond remains unchanged. The resulting protonated derivatives are significantly stabilised in comparison to 1 as demonstrated by simple decomposition experiments followed via31P-NMR spectroscopy. Remarkably, reacting 1 and HCl with subsequent exposure to the atmosphere, leads to a hydrogen-bonded dimer (2·H2O) as verified by X-ray diffraction, FTIR, NMR and elemental analysis, offering an example of a multiple bonded heavier main group compound (RPCR2) involved in supramolecular chemistry via hydrogen bonding (i.e. HB) interactions.16
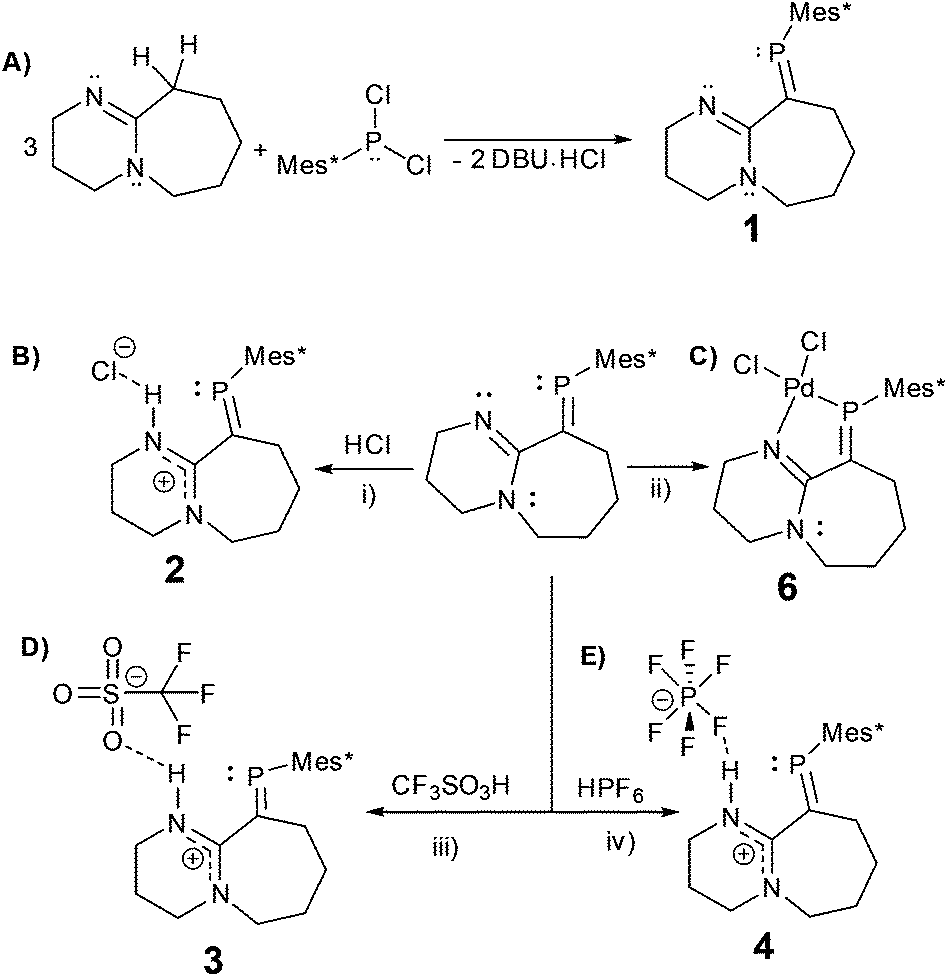 | ||
| Scheme 2 Simplified synthetic procedures: (A) 1; THF, −78 °C (B) 2; (i) anhydrous HCl (2 M, THF), THF, −78 °C. (C) 6; (ii) [Pd(MeCN)2Cl2], DCM, rt. (D) 3; (iii) CF3SO3H (neat), MS (3 Å), THF, −78 °C. (E) 4; (iv) HPF6 (60% aq.), MS (3 Å), THF, −78 °C. | ||
The reaction of DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) with Mes*PCl2 (Mes* = 2,4,6-tri-tert-butylphenyl) leads to phosphaalkene 1, previously reported based on spectroscopic data.17 Despite potential stabilisation of 1via π-conjugation and charge delocalisation, (Fig. S1†), the PC bond is highly air/moisture sensitive, and decomposition products are spectroscopically visible after a few minutes of exposure to atmospheric conditions (Fig. S2†).
After several attempts, single-crystals of 1 were obtained by diffusion of hexanes into dichloromethane (DCM) under inert conditions (Fig. 1). The crystal structure of 1 displays bond lengths reflecting P1C3, N1C2 double bonds and N2–C2, C2–C3 single bonds, the latter is somewhat long for two singly bonded Csp2 centres (1.494(4) Å), thus the bicyclic torsional strain frustrates the possibility to achieve electronic delocalization across the PC to NC–N pathway. The PC bond length (1.676(3) Å) and low-field 31P-NMR (δ = 254 ppm) indicate 1 is a “classically polarised” phosphaalkene (Pδ+Cδ−)18 with a proximal, but non-conjugated imine functionality. The π-accepting nature of the sp2 hybridised P and N atoms as well as their proximity renders 1 interesting as an ancillary ligand as reported for other systems (vide infra). The reactivity of 1 with anhydrous HCl was tested, and a downfield shift of ∼60 ppm in the 31P-NMR spectrum was observed (Fig. S2†); such perturbation is not related to bond length changes (vide infra) on the environment around the phosphorus. We were unable to obtain single crystals of 2 under inert conditions, but only when left crystallising under ambient conditions, resulting in the hydrated form 2·H2O (Fig. 2A). Structural analysis of 2·H2O shows the formation of a dimeric hydrochloride hydrate salt, which self-assembled from 2 + H2O. The crystallisation was attempted twice again with slight variations of the conditions, and yet, the same crystal structure was obtained, which indicated the motif is energetically preferential. The reproducibility is interesting from a crystal-engineering perspective where the desire for the rational design of systems leading to discrete structures prevails.19 The molecular structure of 2·H2O was solved in the monoclinic space group C2/c. Heavy atoms were refined anisotropically, and hydrogen atoms were placed at geometric positions with fixed C–H distances, except for the N–H and water hydrogen atoms, which were located on the electron difference map. The asymmetric unit consists of the cationic fragment [1 + H]+, as well as a hydrogen bonded chloride anion and a water molecule.
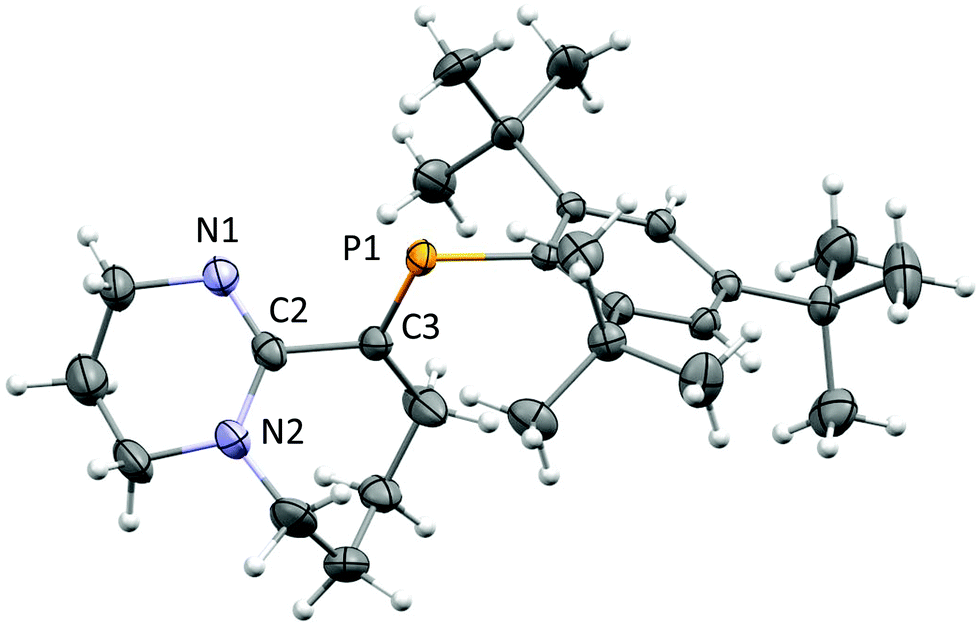 | ||
| Fig. 1 ORTEP representation of 1.20 Ellipsoids are plotted at 50% probability levels. Selected interatomic distances (Å): P1C3, 1.676(3). N1C2, 1.285(4). N2–C2, 1.370(4). C2–C3, 1.496(4). See Table S1.† | ||
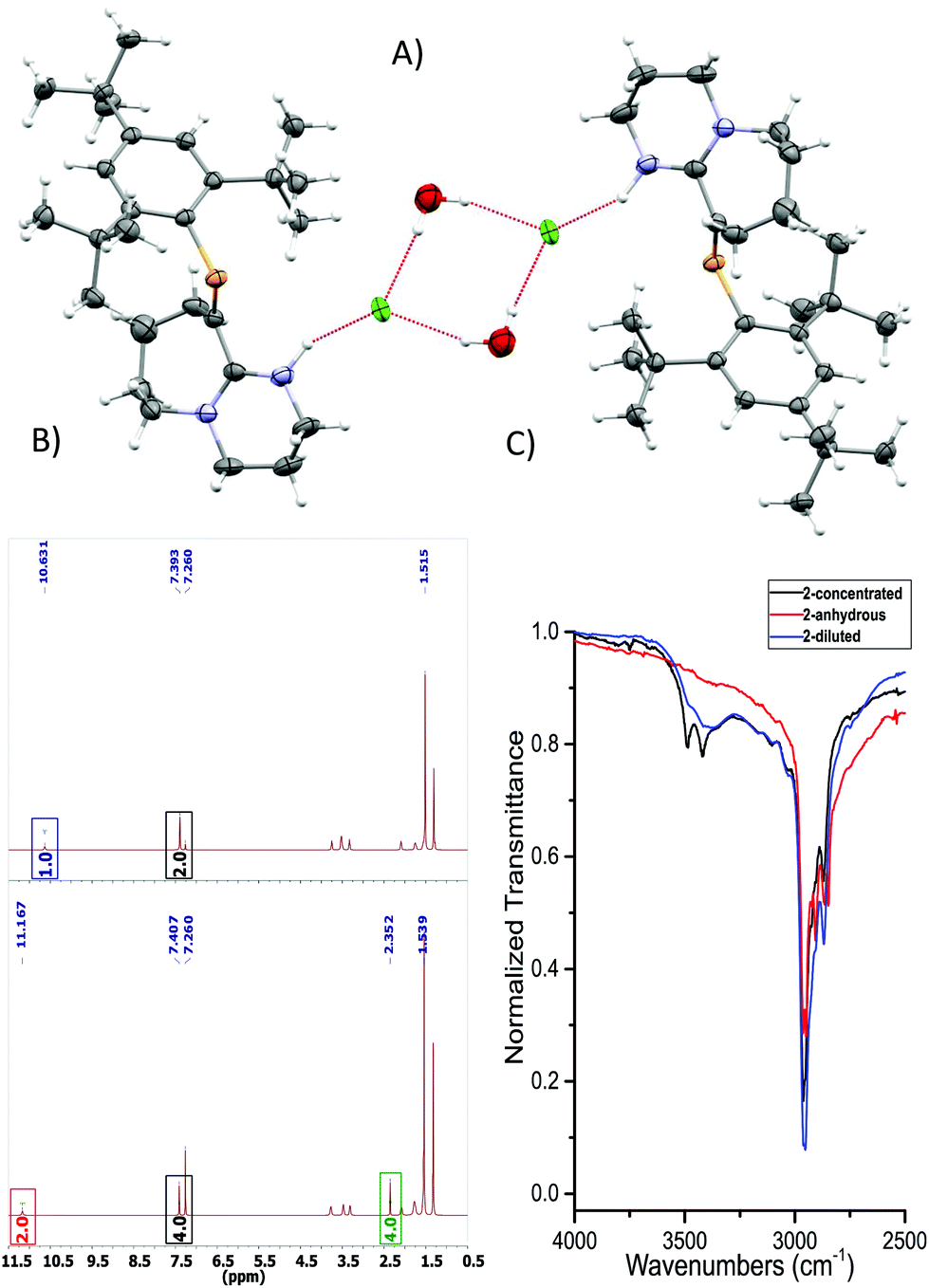 | ||
| Fig. 2 (A) ORTEP representation of 2·H2O. Ellipsoids are plotted at 50% probability levels. (B) 1H-NMR spectra of 2 and 2 + H2O. Top: Anhydrous. Bottom: H2O titrated 2. Red and blue box: N–H signal, green box: H2O signal shifted due to the presence of HB. Bottom integrals assume a dimeric species. (C) FTIR: –OH region expansion of 2 and 2 + H2O experiments (ESI/Fig. S6†). | ||
As a hydrogen-bond network, (Fig. 2A & S3†) the quadrilateral centred ring (∼0.11 nm2) with the two peripheral iminium hydrogen donors is leading to a significant stabilisation as rationalised by the repeated crystallisations. A search on the Cambridge Structural Database (CSD) of the Cl⋯(H2O)2⋯Cl arrangement without constraints, yielded 1512 hits; restricting the search to the isolated dimeric motif observed in 2·H2O (a ring; with two chloride anions as HBA from three donors each, and two H2O molecules as doubly HBD each) gave no results. A few isostructural arrangements, but with slightly different hydrogen bond coordination numbers on the HB network exist (see Fig. S3†). The discrete nature of the central HB motif in 2·H2O epitomises the small-limit of such water/chloride hydrogen-bonded cluster, in this case, embedded in a hydrophobic environment.21 Regardless of the interpretation, the study of hydrate and chloride containing rings and other topological arrangements is fundamentally important due to the ubiquitous nature of HB interactions involving water and halides.22 We hypothesise that the addition of a water molecule via hydrolysis to an otherwise reactive PC double bond as seen in 1, is attenuated due to the strategic location of the iminium–chloride pair (2), which leads to hydrogen bonding interactions (2·H2O); further stabilisation via HB networks is favoured over decomposition of the functional PC centre, explaining the increased stability of both 2 (anhydrous) and 2·H2O over 1 (see Fig. S4†). Substantial changes in the atomic orbital contributions to the FMO densities of 1vs.2/2·H2O were ruled out by DFT calculations. Protonation of an imine over reaction at the multiple bonded heavier group 15 element was only recently reported for the first time, however not altering the stability of the phosphinine in this case.6b
An insignificant elongation of the PC bond to 1.679(3) Å (ref. 23) from 1 to 2·H2O contrasts with the N1C2–N2 motif, which switches from a localised double/single bond towards a delocalized cationic situation with 1.325(4) and 1.318(4) Å bond lengths, respectively. Despite the significant separation of N2 and Cl1 from P1 (>3 Å), partial π-bond delocalisation and other weak electrostatic interactions such as London dispersion may assist in stabilising HB interactions (Fig. S5†). Solution NMR experiments were performed (pages S18–S22†) to demonstrate the prevalence of HB in 2 and 2 + H2O (not exclusively of the dimer). For example, titration of 2 (Fig. 2B) with a stoichiometric amount of deoxygenated water, resulted in downfield shifts of the N–H and H2O signals strongly suggesting that a hydrogen bonded equilibrium is cooperatively established. Additional experiments (e.g. concentration dependency) in which specific experimental conditions led to expected chemical shift changes of the N–H, and the appearance of minor species in 31P-NMR from complex equilibria (ion-pairs, hydrogen-bonded adducts, and others), validated the presence of HB in solution (see ESI†). The same type of evidence has been consistently used to demonstrate the presence of HB in the past.24
The HB situation of 2 was further studied via infrared spectroscopy (Fig. 3C and Fig. S6 and S7†). The anhydrous form of 2 (red curve) lacks any –OH stretches at >3300 cm−1. On the other hand, measurements obtained from a dilute sample of 2 in DCM (0.5% H2O), shows the arising of a discrete band at ∼3384 cm−1, which corresponds to a HB –OH vibrational mode (blue curve). A three-fold concentration increase of 2 (i.e. more likely to participate in HB) to yield an approximate equimolar 2/H2O mixture, led to the black spectrum featuring two, well-resolved discrete bands at 3490 and 3421 cm−1. These vibrations occur at approximately 250 cm−1 lower wavenumbers than the –OH stretches of free H2O in CCl4, indicating HB interactions. The two –OH resonances separated by only 69 cm−1 from the other reliably confirm the self-assembly of the dimeric motif in solution (Fig. S6 and S7† for details)25 and correlate well with solid-state IR-data. Theoretically, QTAIM molecular graphs (Bader analysis) verifies the HB interactions of this cluster (Fig. S8†).26 Examples of hydrogen bonding in compounds containing unsaturated/multiple bonded heavy main group motifs are rare.16a,c,27
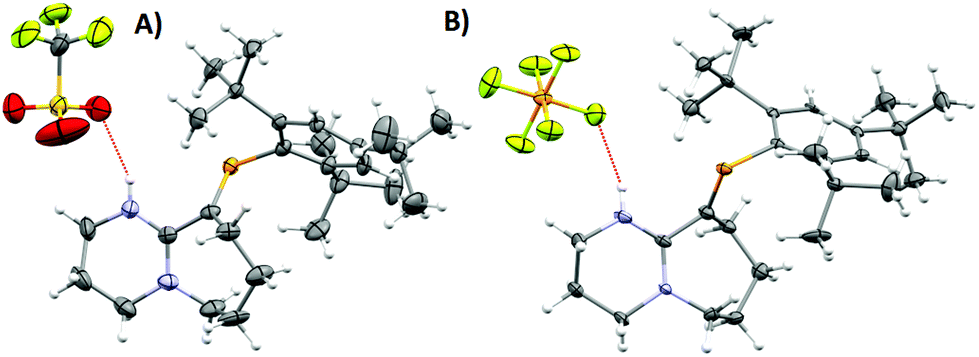 | ||
| Fig. 3 ORTEP representations of (A) 3 (B) 4 (only one of the two independent molecules is displayed; see the asymmetric unit in Fig. S11†). Ellipsoids are plotted at 50% probability levels. | ||
Intrigued by the stability of 1 after the reaction with HCl, its reactivity with TfOH (previously used to yield phosphenium cations from phosphaalkenes)28 and HPF6 (60% aq.), acids orders of magnitude stronger than HCl, were tested. The 31P-NMR spectra of 3 and 4 exhibited low-field shifts typical of phosphaalkenes >300 ppm and were confirmed crystallographically (Fig. 3). Dicoordinate phosphenium cations also exhibit strongly deshielded 31P NMR resonances and a Δδ31P of 70 ppm and higher compared to their corresponding parent phosphaalkenes;29 as stated before for 1 (Fig. S1†), a resonance structure with a cationic phosphorus centre is plausible. However, the observed geometric parameters in the solid-state structures of 2·H2O, 3, and 4 (vide infra) suggest that the charge delocalisation mainly occurs via the nitrogen atoms (Fig. S8†). Despite the pronounced effect on the 31P chemical shifts for all adducts, we believe that the observed species are typical of customarily polarised phosphaalkene units. The solid-state structures of 3 and 4 (Table S1†) show similar geometric parameters for the PC unit in comparison to 2·H2O. The N1–C2–N2 motif displays similar bond lengths, whereas the PC distances are slightly longer in 3 (1.689(4) Å) and 4 (1.695(5)/1.688(5) Å) in comparison to 2·H2O and 1. In both cases, strong HB interactions between the cationic iminium and the anion are observed.30 The donor–acceptor distances are in the range of 2.932(7)–3.097(3) Å, thus significantly shorter than in the DBU·HCl adduct (5) (3.156(6) Å), (Table S2†). DBU salts together with other adduct derivatives and its parent compound DBU are widely relevant in various areas such as catalysis, ionic liquids, organic synthesis, and others,31 however, the single crystal structure of this organic hydrochloride salt was reported for the first time (Fig. S9†). These hydrogen bonds are fundamentally interesting because they lie at the Brønsted acid/base and HB interface.32
Additionally, single crystals of the Pd(II) complex (6) from the reaction of 1 with [PdCl2(MeCN)2] were obtained (Fig. S10†); as previously observed, the resulting PC bond length shortened upon metal coordination (1.648(7) Å),33 and as a result of the formation of the square planar complex, the torsion angle between the PC and NC is reduced (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12.3(7)°) with respect to the other species (52–57°) (Table S1†). The relative donor strengths of the N and the P binding sites are reflected by the Pd–Cl distances of 2.296(2) and 2.335(2) Å to the trans-chlorides, respectively suggesting stronger P binding.
:12.3(7)°) with respect to the other species (52–57°) (Table S1†). The relative donor strengths of the N and the P binding sites are reflected by the Pd–Cl distances of 2.296(2) and 2.335(2) Å to the trans-chlorides, respectively suggesting stronger P binding.
In summary, a broad set of fascinating PC containing systems obtained from the reactions of 1 with very strong acids and a Pd(II) precursor (1, 2·H2O, 3, 4, 6), as well as the structurally unreported hydrochloride salt of DBU (5), are presented. The protonated derivatives are significantly stabilised towards decomposition of their phosphaalkene in comparison to 1, proving a vital role of both the anionic fragments and of HB interactions in suppressing undesired processes. The HCl adduct self-assembles into a dimeric species selectively, and exhibits a novel HB type of quadrilateral arrangement stabilised by two HB donor-only water molecules and two triply HB accepting chloride anions as the smallest water–chloride cluster of that type. The potential use of these systems as supramolecular precursors, in catalysis, and other applications may be envisioned. The introduction of this concept incentivises the use of rational design towards compounds that incorporate formidable multiple bonding to heavier p-block elements by strategically positioning saviour atoms on the periphery of the main group scaffold.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Swedish research council (Vetenskapsrådet 2017-03727), the EU COST action on Smart Inorganic Polymers (SIPs), and the Olle-Engkvist foundation for their support.Notes and references
- (a) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef PubMed; (b) R. West, Angew. Chem., Int. Ed. Engl., 1987, 26, 1201–1211 CrossRef; (c) J. I. Bates, J. Dugal-Tessier and D. P. Gates, Dalton Trans., 2010, 39, 3151–3159 RSC; (d) C. Präsang and D. Scheschkewitz, Chem. Soc. Rev., 2016, 45, 900–921 RSC.
- (a) J. Heinicke and A. Tzschach, Phosphorus Sulfur Relat. Elem., 1984, 20, 347–356 CrossRef; (b) R. Appel, F. Knoll and I. Ruppert, Angew. Chem., Int. Ed. Engl., 1981, 20, 731–744 CrossRef; (c) R. Appel, G. Maier, H. P. Reisenauer and A. Westerhaus, Angew. Chem., Int. Ed. Engl., 1981, 20, 197–197 CrossRef; (d) A. H. Cowley and S. K. Mehrotra, J. Am. Chem. Soc., 1983, 105, 2074–2075 CrossRef; (e) D. Gudat, M. Nieger and E. Niecke, J. Chem. Soc., Dalton Trans., 1989, 693–700 RSC; (f) T. C. Klebach, R. Lourens and F. Bickelhaupt, J. Am. Chem. Soc., 1978, 100, 4886–4888 CrossRef; (g) K. Sugamata, T. Sasamori and N. Tokitoh, Chem. Lett., 2013, 43, 95–96 CrossRef; (h) J. I. Bates and D. P. Gates, J. Am. Chem. Soc., 2006, 128, 15998–15999 CrossRef PubMed.
- (a) T. C. Klebach, R. Lourens and F. Bickelhaupt, J. Am. Chem. Soc., 1978, 100, 4886–4888 CrossRef; (b) T. A. Van der Knaap, T. C. Klebach, R. Lourens, M. Vos and F. Bickelhaupt, J. Am. Chem. Soc., 1983, 105, 4026–4032 CrossRef; (c) M. J. Fink, D. J. De Young, R. West and J. Michl, J. Am. Chem. Soc., 1983, 105, 1070–1071 CrossRef; (d) P. Rosa, C. Gouverd, G. Bernardinelli, T. Berclaz and M. Geoffroy, J. Phys. Chem. A, 2003, 107, 4883–4892 CrossRef; (e) R. West, Polyhedron, 2002, 21, 467–472 CrossRef; (f) L. Weber, S. Buchwald, A. Rühlicke, H.-G. Stammler and B. Neumann, Z. Anorg. Allg. Chem., 1993, 619, 934–942 CrossRef; (g) V. K. Greenacre, I. J. Day and I. R. Crossley, Organometallics, 2017, 36, 435–442 CrossRef; (h) C. Moser, M. Nieger and R. Pietschnig, Organometallics, 2006, 25, 2667–2672 CrossRef; (i) O. C. Presly, T. J. Davin, M. Green, R. J. Kilby, S. M. Mansell, J. E. McGrady and C. A. Russell, Eur. J. Inorg. Chem., 2008, 2008, 4511–4515 CrossRef.
- M. Driess, H. Pritzkow and M. Sander, Angew. Chem., Int. Ed. Engl., 1993, 32, 283–285 CrossRef.
- E. Niecke and D. Gudat, Angew. Chem., Int. Ed. Engl., 1991, 30, 217–237 CrossRef.
- (a) Y. Zhang, F. S. Tham, J. F. Nixon, C. Taylor, J. C. Green and C. A. Reed, Angew. Chem., Int. Ed., 2008, 47, 3801–3804 CrossRef PubMed; (b) H. Huang, G. Tao, Z. Wei, J. Hou, M.-c. Wang, Z. Duan and F. Mathey, Organometallics, 2018, 37, 464–468 CrossRef.
- S. Loss, C. Widauer and H. Grützmacher, Angew. Chem., Int. Ed., 1999, 38, 3329–3331 CrossRef PubMed.
- (a) M. Kobayashi, N. Hayakawa, T. Matsuo, B. Li, T. Fukunaga, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, J. Am. Chem. Soc., 2016, 138, 758–761 CrossRef PubMed; (b) K. Tamao, M. Kobayashi, T. Matsuo, S. Furukawa and H. Tsuji, Chem. Commun., 2012, 48, 1030–1032 RSC; (c) K. Duck, B. W. Rawe, M. R. Scott and D. P. Gates, Angew. Chem., Int. Ed., 2017, 56, 9507–9511 CrossRef PubMed; (d) R. C. Smith and J. D. Protasiewicz, J. Am. Chem. Soc., 2004, 126, 2268–2269 CrossRef PubMed; (e) D. Morales Salazar, E. Mijangos, S. Pullen, M. Gao and A. Orthaber, Chem. Commun., 2017, 53, 1120–1123 RSC.
- (a) R. J. Gilliard, D. Heift, Z. Benkő, J. M. Keiser, A. L. Rheingold, H. Grützmacher and J. D. Protasiewicz, Dalton Trans., 2018, 47, 666–669 RSC; (b) G.-L. Hou, B. Chen, W. J. Transue, Z. Yang, H. Grützmacher, M. Driess, C. C. Cummins, W. T. Borden and X.-B. Wang, J. Am. Chem. Soc., 2017, 139, 8922–8930 CrossRef PubMed; (c) A. Hinz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2016, 55, 15515–15519 CrossRef PubMed.
- (a) Y. Peng, B. D. Ellis, X. Wang, J. C. Fettinger and P. P. Power, Science, 2009, 325, 1668–1670 CrossRef PubMed; (b) L. E. Longobardi, C. A. Russell, M. Green, N. S. Townsend, K. Wang, A. J. Holmes, S. B. Duckett, J. E. McGrady and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 13453–13457 CrossRef PubMed.
- P. K. Majhi, K. C. Chow, T. H. Hsieh, E. G. Bowes, G. Schnakenburg, P. Kennepohl, R. Streubel and D. P. Gates, Chem. Commun., 2016, 52, 998–1001 RSC.
- (a) S. Ito, T. Shinozaki and K. Mikami, Eur. J. Org. Chem., 2017, 6889–6900 CrossRef; (b) H.-o. Taguchi, D. Sasaki, K. Takeuchi, S. Tsujimoto, T. Matsuo, H. Tanaka, K. Yoshizawa and F. Ozawa, Organometallics, 2016, 35, 1526–1533 CrossRef; (c) A. J. Arduengo, J. C. Calabrese, A. H. Cowley, H. V. R. Dias, J. R. Goerlich, W. J. Marshall and B. Riegel, Inorg. Chem., 1997, 36, 2151–2158 CrossRef PubMed.
- B. W. Rawe, C. M. Brown, M. R. MacKinnon, B. O. Patrick, G. J. Bodwell and D. P. Gates, Organometallics, 2017, 36, 2520–2526 CrossRef.
- (a) X. Pan, X. Wang, Z. Zhang and X. Wang, Dalton Trans., 2015, 44, 15099–15102 RSC; (b) G. Tan, S. Li, S. Chen, Y. Sui, Y. Zhao and X. Wang, J. Am. Chem. Soc., 2016, 138, 6735–6738 CrossRef PubMed; (c) G. Tan, J. Li, L. Zhang, C. Chen, Y. Zhao, X. Wang, Y. Song, Y.-Q. Zhang and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 12741–12745 CrossRef PubMed.
- R. Appel and L. Krieger, J. Organomet. Chem., 1988, 354, 309–312 CrossRef.
- (a) Y. Mao, K. M. H. Lim, Y. Li, R. Ganguly and F. Mathey, Organometallics, 2013, 32, 3562–3565 CrossRef; (b) G. Becker, Z. Anorg. Allg. Chem., 1981, 480, 38–48 CrossRef; (c) G. Becker, M. Rössler and G. Uhl, Z. Anorg. Allg. Chem., 1982, 495, 73–88 CrossRef.
- R. Appel and L. Krieger, J. Organomet. Chem., 1988, 354, 309–312 CrossRef.
- L. Weber, Eur. J. Inorg. Chem., 2000, 2000, 2425–2441 CrossRef.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565–565 CrossRef.
- (a) J. H. Loehlin and A. Kvick, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 3488–3490 CrossRef; (b) M. N. Hoque, A. Basu and G. Das, Cryst. Growth Des., 2012, 12, 2153–2157 CrossRef; (c) A. Basu and G. Das, Chem. Commun., 2013, 49, 3997–3999 RSC; (d) D.-X. Wang, S.-X. Fa, Y. Liu, B.-Y. Hou and M.-X. Wang, Chem. Commun., 2012, 48, 11458–11460 RSC.
- (a) G. A. Jeffrey, Crystallogr. Rev., 2003, 9, 135–176 CrossRef; (b) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef; (c) Q. He, P. Tu and J. L. Sessler, Chem, 2018, 4, 46–93 CrossRef PubMed; (d) J. E. Combariza, N. R. Kestner and J. Jortner, J. Chem. Phys., 1994, 100, 2851–2864 CrossRef; (e) N. Agmon, J. Phys. Chem. A, 1998, 102, 192–199 CrossRef; (f) W. H. Thompson and J. T. Hynes, J. Am. Chem. Soc., 2000, 122, 6278–6286 CrossRef; (g) J. N. Moorthy, R. Natarajan and P. Venugopalan, Angew. Chem., Int. Ed., 2002, 41, 3417–3420 CrossRef.
- (a) D. Nauroozi and A. Orthaber, Eur. J. Inorg. Chem., 2016, 2016, 709–717 CrossRef; (b) S. Shah, T. Concolino, A. L. Rheingold and J. D. Protasiewicz, Inorg. Chem., 2000, 39, 3860–3867 CrossRef PubMed.
- (a) P. L. Wash, E. Maverick, J. Chiefari and D. A. Lightner, J. Am. Chem. Soc., 1997, 119, 3802–3806 CrossRef; (b) C. Reichardt and T. Welton, in Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, pp. 7–64 Search PubMed; (c) M. Fleischmann, D. Drettwan, E. Sugiono, M. Rueping and R. M. Gschwind, Angew. Chem., Int. Ed., 2011, 50, 6364–6369 CrossRef PubMed; (d) E. E. Simanek, M. I. M. Wazeer, J. P. Mathias and G. M. Whitesides, J. Org. Chem., 1994, 59, 4904–4909 CrossRef; (e) R. Wyler, J. de Mendoza and J. Rebek, Angew. Chem., Int. Ed. Engl., 1993, 32, 1699–1701 CrossRef.
- A. Maeda, J. Sasaki, Y. Shichida and T. Yoshizawa, Biochemistry, 1992, 31, 462–467 CrossRef PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef.
- (a) G. Becker, Z. Anorg. Allg. Chem., 1977, 430, 66–76 CrossRef; (b) S. G. Ruf, J. Dietz and M. Regitz, Tetrahedron, 2000, 56, 6259–6267 CrossRef; (c) X. Chen, S. Alidori, F. Puschmann Florian, G. Santiso-Quinones, Z. Benkő, Z. Li, G. Becker, H. F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 1641–1645 CrossRef PubMed; (d) R. Suter, Y. Mei, M. Baker, Z. Benkő, Z. Li and H. Grützmacher, Angew. Chem., Int. Ed., 2016, 56, 1356–1360 CrossRef PubMed.
- (a) D. Gudat, E. Niecke, B. Krebs and M. Dartmann, Chimia, 1985, 39, 277–279 Search PubMed; (b) J. I. Bates and D. P. Gates, Chem. – Eur. J., 2012, 18, 1674–1683 CrossRef PubMed; (c) L. Weber, S. Kleinebekel and T. Haase, Z. Anorg. Allg. Chem., 2000, 626, 1857–1862 CrossRef.
- (a) A. H. Cowley, M. C. Cushner and J. S. Szobota, J. Am. Chem. Soc., 1978, 100, 7784–7786 CrossRef; (b) N. Burford, T. S. Cameron, J. A. Clyburne, K. Eichele, K. N. Robertson, S. Sereda, R. E. Wasylishen and W. A. Whitla, Inorg. Chem., 1996, 35, 5460–5467 CrossRef PubMed; (c) D. Gudat, Coord. Chem. Rev., 1997, 163, 71–106 CrossRef.
- (a) M. S. Miran, H. Kinoshita, T. Yasuda, M. A. B. H. Susan and M. Watanabe, Chem. Commun., 2011, 47, 12676–12678 RSC; (b) T. S. Teets, J. A. Labinger and J. E. Bercaw, Organometallics, 2013, 32, 5530–5545 CrossRef.
- (a) M. Pschenitza, S. Meister and B. Rieger, Chem. Commun., 2018, 54, 3323–3326 RSC; (b) Z. Z. Yang, L. N. He, C. X. Miao and S. Chanfreau, Adv. Synth. Catal., 2010, 352, 2233–2240 CrossRef; (c) P. G. Jessop, D. J. Heldebrant, X. Li, C. A. Eckert and C. L. Liotta, Nature, 2005, 436, 1102–1102 CrossRef PubMed.
- P. Gilli, L. Pretto, V. Bertolasi and G. Gilli, Acc. Chem. Res., 2009, 42, 33–44 CrossRef PubMed.
- (a) K. Takeuchi, H.-o. Taguchi, I. Tanigawa, S. Tsujimoto, T. Matsuo, H. Tanaka, K. Yoshizawa and F. Ozawa, Angew. Chem., Int. Ed., 2016, 55, 15347–15350 CrossRef PubMed; (b) S. Ikeda, F. Ohhata, M. Miyoshi, R. Tanaka, T. Minami, F. Ozawa and M. Yoshifuji, Angew. Chem., Int. Ed., 2000, 39, 4512–4513 CrossRef; (c) M. Klein, C. Albrecht, G. Schnakenburg and R. Streubel, Organometallics, 2013, 32, 4938–4943 CrossRef; (d) J. Dugal-Tessier, G. R. Dake and D. P. Gates, Angew. Chem., Int. Ed., 2008, 47, 8064–8067 CrossRef PubMed; (e) P. Le Floch, Coord. Chem. Rev., 2006, 250, 627–681 CrossRef; (f) S. C. Serin, F. S. Pick, G. R. Dake and D. P. Gates, Inorg. Chem., 2016, 55, 6670–6678 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1828164–1828169. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt01607k |
| This journal is © The Royal Society of Chemistry 2018 |