
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the affinity of catechols and salicylic acids towards Al(III): characterization of Al–chelator interactions†
Gabriele
Dalla Torre
 ab,
Jon I.
Mujika
a,
Elena
Formoso
a,
Eduard
Matito
ac,
Maria J.
Ramos
b and
Xabier
Lopez
*a
ab,
Jon I.
Mujika
a,
Elena
Formoso
a,
Eduard
Matito
ac,
Maria J.
Ramos
b and
Xabier
Lopez
*a
aKimika Fakultatea, Euskal Herriko Unibertsitatea UPV/EHU, and Donostia International Physics Center (DIPC), P.K. 1072, 20080 Donostia, Euskadi, Spain. E-mail: xabier.lopez@ehu.es
bUCIBIO/REQUIMTE, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, Rua do Campo Alegre, s/n, Porto, Portugal
cIKERBASQUE, Basque Foundation for Science, 48013 Bilbao, Euskadi, Spain
First published on 11th June 2018
Abstract
Due to aluminum's controversial role in neurotoxicity, the goal of chelation therapy, the removal of the toxic metal ion or attenuation of its toxicity by transforming it into less toxic compounds, has attracted considerable interest in the past years. In the present paper we present, validate and apply a state-of-the-art theoretical protocol suitable for the characterization of the interactions between a chelating agent and Al(III). In particular, we employ a cluster-continuum approach based on Density Functional Theory calculations to evaluate the binding affinity of aluminum for a set of two important families of aromatic chelators: salicylic acids and catechols. Our protocol shows very good qualitative agreement between the computed binding affinities and available experimental stability constants (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β) values for 1:1, 1:2 and 1:3 complexes. Then, we have investigated the nature of the Al–O bond in an enlarged dataset of 27 complexes of 1:1 stoichiometry, by means of the QTAIM and Energy Decomposition Analysis (EDA). Quite interestingly, we have found that although the Al–O interaction is mainly electrostatic, there is a small but significant degree of covalency that explains the modulation of binding affinities in both families of compounds by the addition of electron donating (CH3, OCH3) or withdrawing (NO2, CF3) substituents. The role of aromaticity and the mechanisms of action of the different functional groups were also evaluated. Finally, we have analyzed the competition between Al(III) and proton toward the binding of these chelators, giving a rationalization of the different trends found experimentally between logβ and the amount of free aluminum in solution in the presence of a given ligand (p[Al]). In summary, we propose a validated and comprehensive computational protocol that can provide a valuable help toward the design and tuning of new efficient aluminum chelators.
β) values for 1:1, 1:2 and 1:3 complexes. Then, we have investigated the nature of the Al–O bond in an enlarged dataset of 27 complexes of 1:1 stoichiometry, by means of the QTAIM and Energy Decomposition Analysis (EDA). Quite interestingly, we have found that although the Al–O interaction is mainly electrostatic, there is a small but significant degree of covalency that explains the modulation of binding affinities in both families of compounds by the addition of electron donating (CH3, OCH3) or withdrawing (NO2, CF3) substituents. The role of aromaticity and the mechanisms of action of the different functional groups were also evaluated. Finally, we have analyzed the competition between Al(III) and proton toward the binding of these chelators, giving a rationalization of the different trends found experimentally between logβ and the amount of free aluminum in solution in the presence of a given ligand (p[Al]). In summary, we propose a validated and comprehensive computational protocol that can provide a valuable help toward the design and tuning of new efficient aluminum chelators.
Introduction
Aluminum is the third most abundant element in the Earth's crust, following oxygen and silicon. However, complex but effective geochemistry has prevented its solubilization,1,2 allowing biological systems to evolve in the absence of this abundant metal. Nonetheless, in the past century, human intervention has made aluminum bioavailable in a myriad different ways, and as a consequence, important trace amounts of this element are found in the human body. The introduction of a nonessential element into biological cycles has raised justified concerns about its biological effects and potential toxicity,3–5 and the scientific literature on the adverse health effects of aluminum is extensive.6 Although the exact mechanisms of aluminum toxicity are not well understood at the atomic level, there is increasing evidence that aluminum promotes oxidative stress,7–9 inhibits the normal function of several enzymes (such as hexokinase,10 glutamate dehydrogenase,11–13etc.), interferes with several key cell metabolism cycles,14–16 and alters the structure and chemistry of important metabolites12 and cofactors.17 Aluminum is also considered a neurotoxic element.18 Early studies supported this hypothesis, linking aluminum and the formation of neurofibrillary tangles (NFT).19 In fact, both experimental and theoretical studies have underlined the ability of aluminum to bind to phosphorylated peptides20,21 and to promote the hyperphosphorylation of normal neurofilaments.22 In addition, the ability of aluminum to contribute to Aβ-amyloid aggregation has been recently demonstrated,23,24 and growing evidence links aluminum to be a decisive contributing factor in Alzheimer's disease.25–27In this controversial context, the quest for chelating agents that could be an effective treatment for aluminum-related disorders has attracted considerable interest.28–32 In particular, catechols and salicylic acids have emerged as very promising building blocks for the design of effective aluminum chelators, because they constitute two of the strongest bidentate aluminum binding species.33 The reason for that relies on the fact that Al(III) is a hard Lewis acid (and the hardest trivalent metal), and therefore it prefers to coordinate to hard Lewis bases such as phenoxide and carboxylate.33 Moreover, the interaction of aluminum with such functional groups is supposed to be mainly electrostatic in nature.30 Due to this inherent affinity, it is not surprising that the biochemistry of important neurotransmitters like catecholamines is highly affected by the presence of aluminum.34–36 It has been shown that aluminum affects the signaling process mediated by these neurotransmitters,37 it alters their content in animal models,34 and it interferes with enzymatic activities that involve these neurotransmitters.38,39 Because of their strong binding affinity, both catechols and salicylic acids have been extensively studied28,30,40,41 in the framework of aluminum chelation therapy with the aim of finding improved and aluminum-specific chelators by tuning their chemical environment with different substituents. The efficiency of low molecular mass aluminum–chelator complexes has been studied by means of several experimental techniques, such as potentiometric titrations, UV/Vis spectroscopy, 1H NMR and ESI-MS.40–44 Nevertheless, the effects mediated by the inclusion of different substituents in the molecule and how they may modulate the binding affinity toward aluminum are still not well understood.41,43 In this sense, the understanding of the effect of electron withdrawing groups (EWGs) and electron donating groups (EDGs), the role played by aromaticity in these chelators, the rationalization of complex stability, and the specific nature of the Al–O bonds is of paramount importance to guide the quest for improved aluminum chelating agents. However, often this relevant information cannot be deduced directly from experimental procedures alone.
The use of state-of-the-art theoretical methods can provide valuable insights into the properties of these systems, as demonstrated elsewhere.45–47 In the present work, we present a comprehensive computational protocol to investigate the behavior of different chelating agents interacting with Al(III). Validation with respect to available experimental data is also performed. Then, the validated protocol is applied to the characterization of the substituent effects and the bonding nature of various aluminum–chelator complexes, as well as their aromatic-related properties, in order to provide a thorough rationalization of the behavior of these chelators. We have considered two main families of chelating agents, salicylic acids and catechols, bearing electron donating groups (EDGs, methyl and methoxy) and electron withdrawing groups (EWGs, nitro and trifluoromethyl) placed at different positions along the aromatic ring and in different quantities (see Fig. 1 and Table 2). These substituents were chosen since they exert opposite effects through different mechanisms of action (resonance and/or induction). Our results demonstrate that although the Al–O bond is mainly of an ionic nature, as it corresponds to a hard metal ion, the trend in the stability for these complexes is mainly determined by covalent dative interactions. We also analyze how Al(III)/proton competition modulates the properties of these chelators.
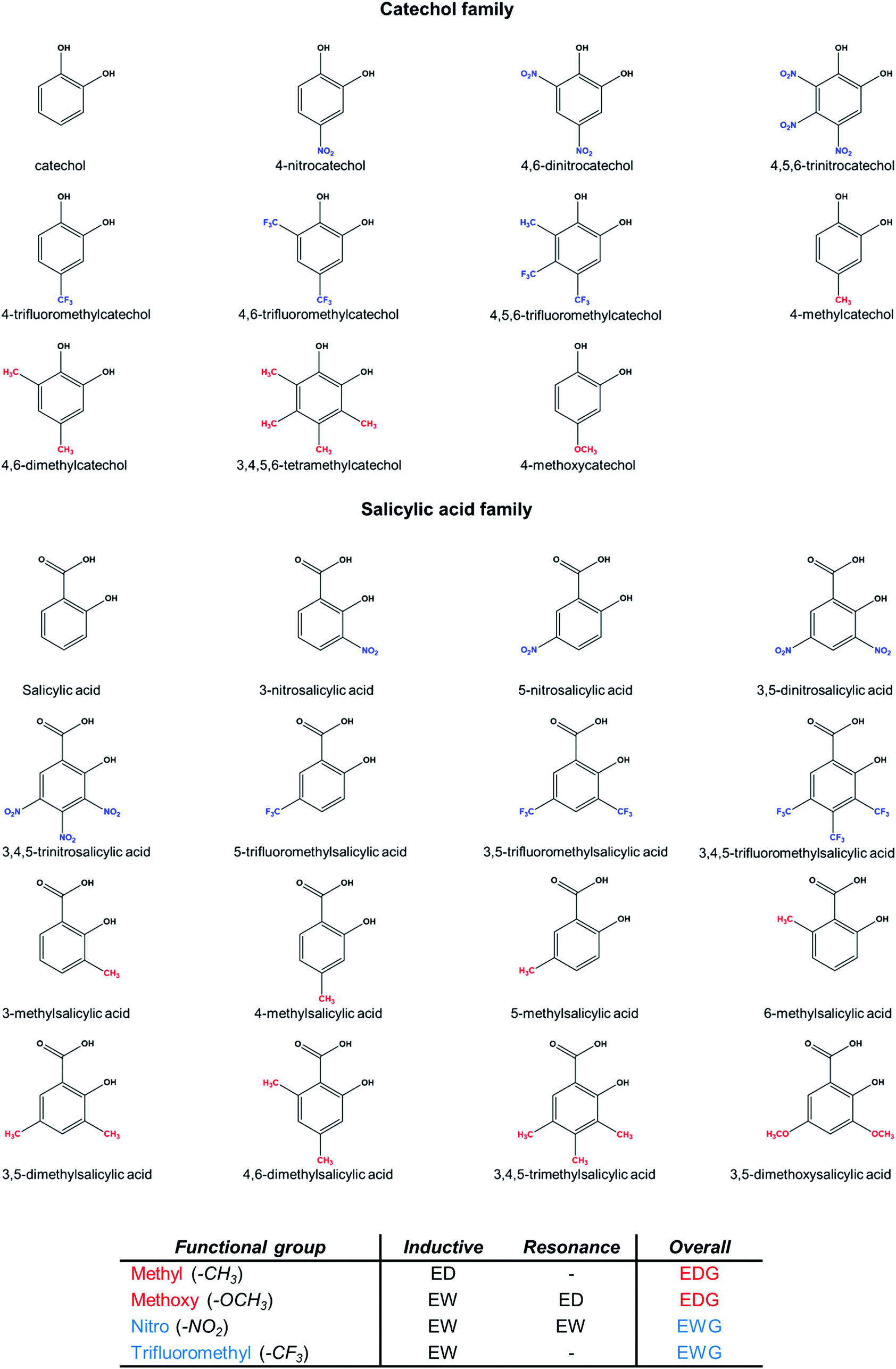 | ||
| Fig. 1 Summary of the two families of chelators with the four different substituents considered in this work: methyl and methoxy (EDGs), nitro and trifluoromethyl (EWGs). The different mechanisms of action of the substituents are also summarized. | ||
Methods
The computational protocol that we developed for the investigation of different Al(III)–chelator complexes is outlined in Fig. 2 and presented in the following sections. For the sake of simplicity, we provide only a schematic overview of the whole protocol; therefore, for the full theory and technical details of each methodology we redirect the reader to the specific computational details section in the ESI.†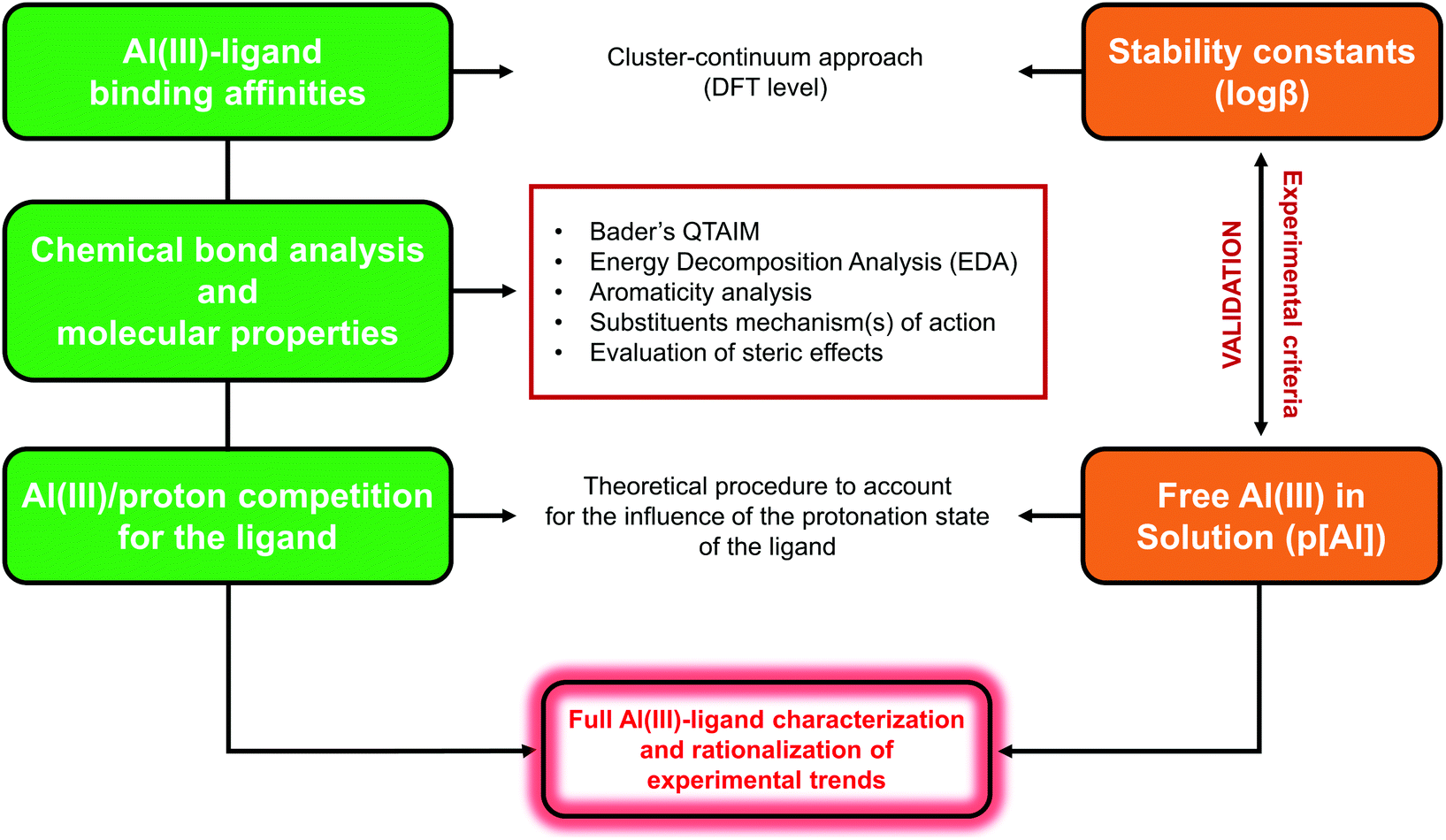 | ||
| Fig. 2 Schematic representation of the theoretical protocol developed in this work. | ||
Definition of binding affinities: cluster-continuum approach
In order to investigate the thermodynamics of all Al(III)–chelator complexes in aqueous solution, we utilized the so-called cluster-continuum approach,48–50 with the first-coordination shell of aluminum surrounded by explicit water molecules in an octahedral fashion and the effects of the remaining solvent considered with a continuum dielectric model (see Fig. 4). Optimization and single point calculations were performed at the B3LYP-D3(BJ)/6-311++G(3df,2p)//B3LYP-D3(BJ)/6-31++G(d,p) level using the integral equation formalism variant (IEFPCM) solvation model.51 Such a choice was made since it has been shown that the introduction of dispersion corrections on both geometry and single point energy calculations improve the overall results.52 In general, the addition of methods that properly take into account dispersion energies in DFT has been proven to improve the precision of computed non-covalent interactions.53,54 For results with different density functionals and the MP2 method and their evaluation versus experimental stability constants see ESI Tables S1–S5 and Fig. S1–S2.†We characterized bidentate Al–Lig complexes with 1:1, 1:2 and the 1:3 stoichiometry following the ligand substitution reaction shown in (1):
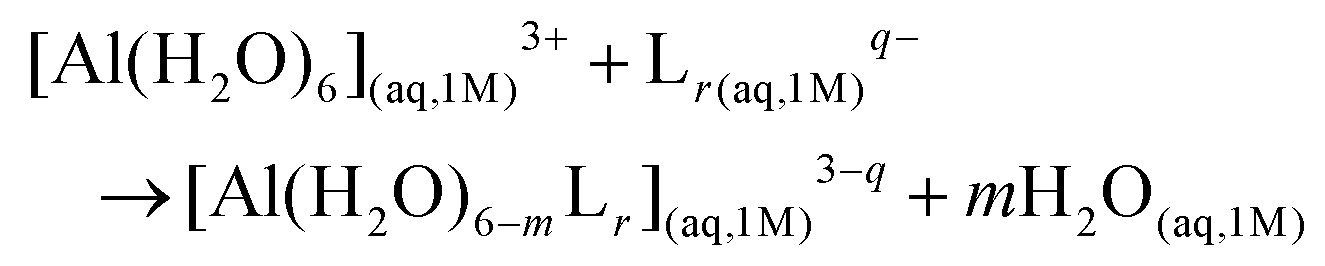 | (1) |
:1, 1:2 and 1:3 complexes, respectively. Notice that we consider the ligand in its unprotonated form, which is the state considered when evaluating experimental log(β) (the validation of binding affinities section).
The enthalpy in solution corresponding to the binding of the ligand to Al(III) is therefore calculated as:
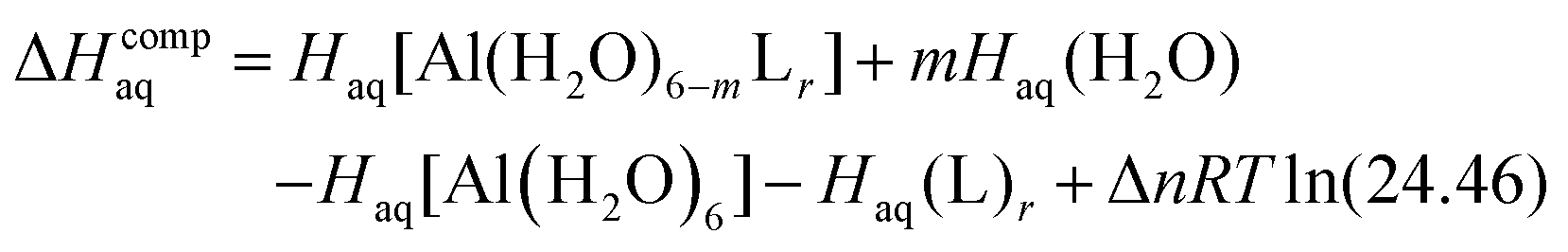 | (2) |
Since the enthalpies are determined using an ideal gas at 1 atm as the standard state, the last term in eqn (2) corresponds to the volume change due to the transformation from 1 atm to 1 M in solution, where Δn refers to the change in the number of species in the reaction.55 In a similar way, the free energy of the complexes is determined as:
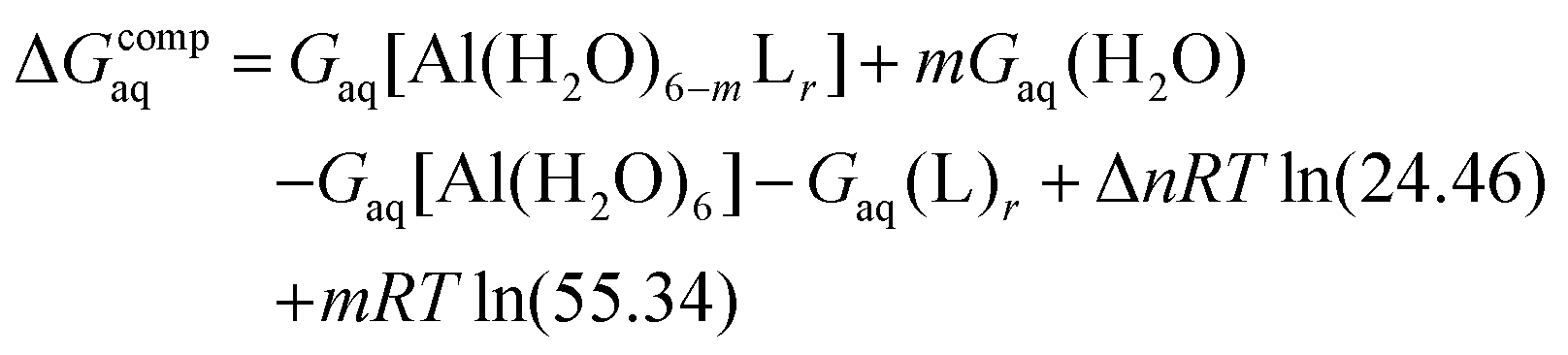 | (3) |
The validation of binding energies with respect to experimental stability constants (i.e. logβ) is thoroughly discussed in the validation of binding affinities section.
Chemical bond analysis and evaluation of molecular properties
At the second stage, to provide a quantitative and qualitative characterization of the interactions arising in these Al(III)–chelator complexes, as well as to unveil the effect of different substituents (EDGs and EWGs) toward complex stability, we have employed several state-of-the-art computational techniques summarized as follows:• Quantum Theory Of Atoms In Molecules (QTAIM): Bader's theory56 allows the classification of the nature of a given bond according to the characteristics of its Bond Critical Point (BCP), such as the electron density at the BCP ρ(rBCP), the Laplacian of the electron density ∇2ρ(rBCP) and the total energy density H(rBCP). Delocalization Indices (D.I.AB) provide a mean of the average number of electron pairs shared between two atoms A and B.
• Energy Decomposition Analysis (EDA): The EDA scheme by Morokuma57 and Ziegler and Rauk58 decomposes the total interaction energy (ΔEint) between two molecules into three main components, that is, an electrostatic interaction term (ΔEelstat), an orbital interaction term (ΔEoi) and a Pauli repulsion term (ΔEPauli). Therefore, the EDA scheme allows the measurement and quantification of the electrostatic and covalent effects that may arise in a given complex.
• Aromaticity analysis: The analysis of the aromaticity of a molecule according to the Iring59 and MCI60 aromatic descriptors is useful to compare the overall aromatic character of a given ligand with respect to a reference (i.e. benzene for an aromatic compound, cyclohexane for a non-aromatic one); moreover, it is possible to analyze the effects that the addition of substituents of different nature may have on the aromatic-based properties (like resonance) of the ligand, so as to provide a rationale for their mechanism of action.
• Evaluation of possible steric effects: Steric hindrances may take place between two or more functional groups placed close to one another; accordingly, it is important to evaluate the change in stability due to repulsive phenomena upon the addition of bulky functional groups.
Aluminum ion and proton competition
Finally, we developed a strategy to account for the influence of the protonation constants of a given ligand toward the stability of the complex with Al(III), and, therefore, for the competition between Al(III) and the proton for the ligand. The latter aspect is particularly important upon addition of substituents because different functional groups lead to different effects against ligand's pKa, modulating the overall basicity/acidity of the chelator.40,41,61 The whole strategy is presented and discussed in the proton and aluminum ion competition section, and its evaluation and validation with respect to the experimental p[Al] criteria is also provided.Results and discussion
Validation of binding affinities
Experimentally, chelation affinity is usually measured using two different criteria: p[M] and logβ (cumulative stability constant).62p[M] is defined as the negative logarithm of the concentration of the free metal in solution, calculated for total [ligand] = 10−5 M and total [metal] = 10−6 M at pH 7.4, usually calculated from data at 25 °C and 0.1 M ionic strength.63 This criteria is usually useful when comparing different chelators, as p[M] takes into account the effects of ligand protonation and denticity, so that it can provide a general and qualitative insight about the chelation properties of the molecule in solution.62–64 On the other hand, stability constants (logβabc) can be expressed as aM + bL + cH ⇌ MaLbHc, where M is the metal, L is the ligand (in its unprotonated form) and H stands for a given protonation state. In other words, this is a measure of the strength of the interaction between the metal and the ligand that form the complex.
In order to validate our approach, theoretical binding energies of 1:1, 1:2 and 1:3 complexes were evaluated with respect to the available experimental logβ and p[Al] values taken from ref. 40. At this stage, those complexes with available experimental data were included, namely: catechol, 4-nitrocatechol, salicylic acid, 3-nitrosalicylic acid, 5-nitrosalicylic acid and 3,5-dinitrosalicylic acid. Optimized geometries are shown in Fig. 4, and the ΔGcompaq and ΔHcompaq values reported in Table 1 along with experimental data.
:1, 1:2 and 1:3 Al–ligand complexes with available experimental log(β) and p[Al] data, taken from ref. 40
| Stoichiometry | Ligand | Theoretical | Experimental | |||
|---|---|---|---|---|---|---|
| ΔHcompaq | ΔGcompaq | log(β) | p[Al] | |||
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 Complexes :1 Complexes
|
Catecholates | Catecholate | −88.4 | −91.4 | 16.3 | 10.1 |
| 4-Nitrocatecholate | −71.6 | −75.8 | 13.3 | 14.2 | ||
| Salicylates | Salicylate | −76.9 | −78.7 | 13.3 | 8.2 | |
| 3-Nitrosalicylate | −64.2 | −66.7 | 9.5 | 8.7 | ||
| 5-Nitrosalicylate | −63.3 | −65.4 | 9.3 | 8.4 | ||
| 3,5-Dinitrosalicylate | −55.0 | −57.1 | 6.9 | 9.1 | ||
|
1:2 Complexes
|
Catecholates | Catecholate | −151.8 | −157.7 | 31.7 | 10.1 |
| 4-Nitrocatecholate | −124.0 | −130.8 | 24.8 | 14.2 | ||
| Salicylates | Salicylate | −130.8 | −136.9 | 24.2 | 8.2 | |
| 3-Nitrosalicylate | −110.6 | −114.6 | 17.7 | 8.7 | ||
| 5-Nitrosalicylate | −109.2 | −114.2 | 17.7 | 8.4 | ||
| 3,5-Dinitrosalicylate | −95.4 | −101.0 | 13.3 | 9.1 | ||
|
1:3 Complexes
|
Catecholates | Catecholate | −183.1 | −191.7 | 41.1 | 10.1 |
| 4-Nitrocatecholate | −154.8 | −164.6 | 33.7 | 14.2 | ||
| Salicylates | Salicylate | −160.6 | −167.5 | 32.1 | 8.2 | |
| 3-Nitrosalicylate | −139.8 | −145.8 | 23.7 | 8.7 | ||
| 5-Nitrosalicylate | −137.8 | −141.7 | 23.7 | 8.4 | ||
| 3,5-Dinitrosalicylate | −125.8 | −127.9 | 18.5 | 9.1 | ||
| Total correlation coefficients | 0.9692 | 0.1235 | ||||
As we can see in Fig. 3, our theoretical protocol is able to describe the relative affinity for this set of molecules. Indeed, theoretical ΔGcompaq shows the same trends as the experimental logβ for all stoichiometries, with a total correlation coefficient of 0.9692. On the other hand, if we analyze the trends observed for p[Al] (Table 1), we can see that these trends are not the same order as for logβ and ΔGcompaq. Notice that p[Al] does not depend on the stoichiometry (Table 1); indeed, it is often used as an indirect ligand affinity indicator.64 However, the observed discrepancies between p[Al] and logβ are due to the fact that, as previously mentioned, p[Al] depends not only on the stability of the complexes, but also on other factors like metal/proton competition, the number of different Al(III)–chelator species present at pH 7.4 and the denticity of the chelator.62,64 This denotes the limits of using p[M] alone, as a unique measure of complex stability. A more detailed discussion about p[Al] is provided in the proton and aluminum ion competition section.
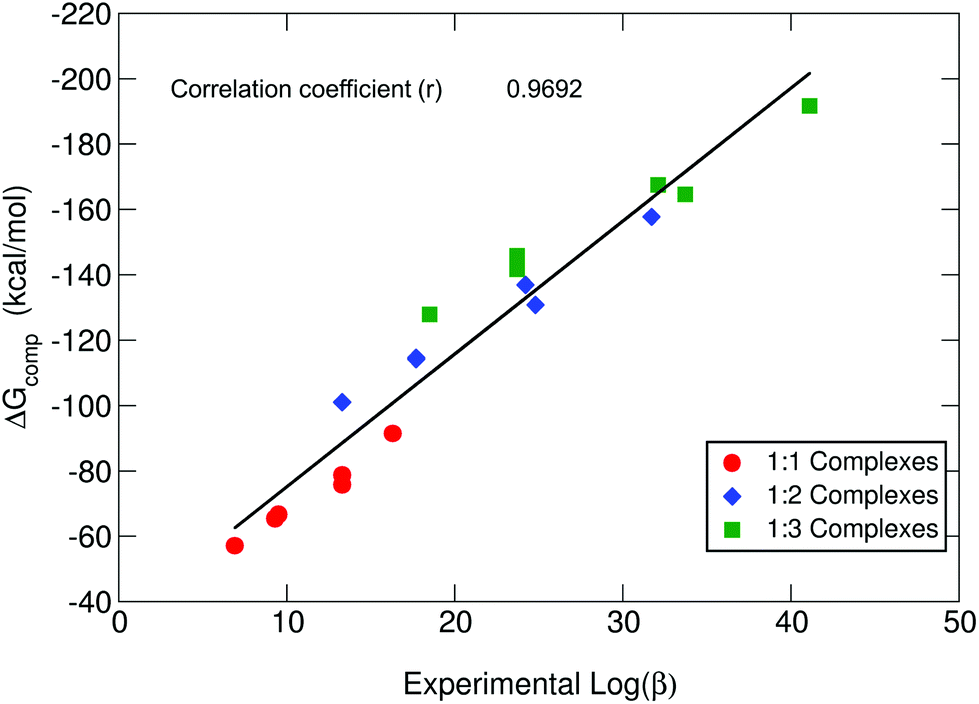 | ||
| Fig. 3 B3LYP-D3(BJ) binding energies (ΔGcompaq) versus experimental stability constants (logβ).40 | ||
Optimized geometries for 1:1, 1:2 and 1:3 complexes can be found in Fig. 4. In all complexes the ligands interact bidentately with aluminum. Since Al(III) is always hexacoordinated, the remaining coordination sites are filled with water molecules. In 1:1 and 1:2 complexes, aluminum is always placed coplanar to the aromatic rings. In 1:2 complexes, the two ligands are not fully coplanar as they are slightly tilted towards one another (deviation dihedrals in the 8.0–11.0 degree range, Fig. 4). In the case of 1:3 complexes, whereas the catechol family still retains the coplanarity (Fig. 4) of aluminum with respect to the aromatic rings, salicylic acid complexes show slight distortions, due to π–π stacking interactions that arise between the adjacent aromatic rings.
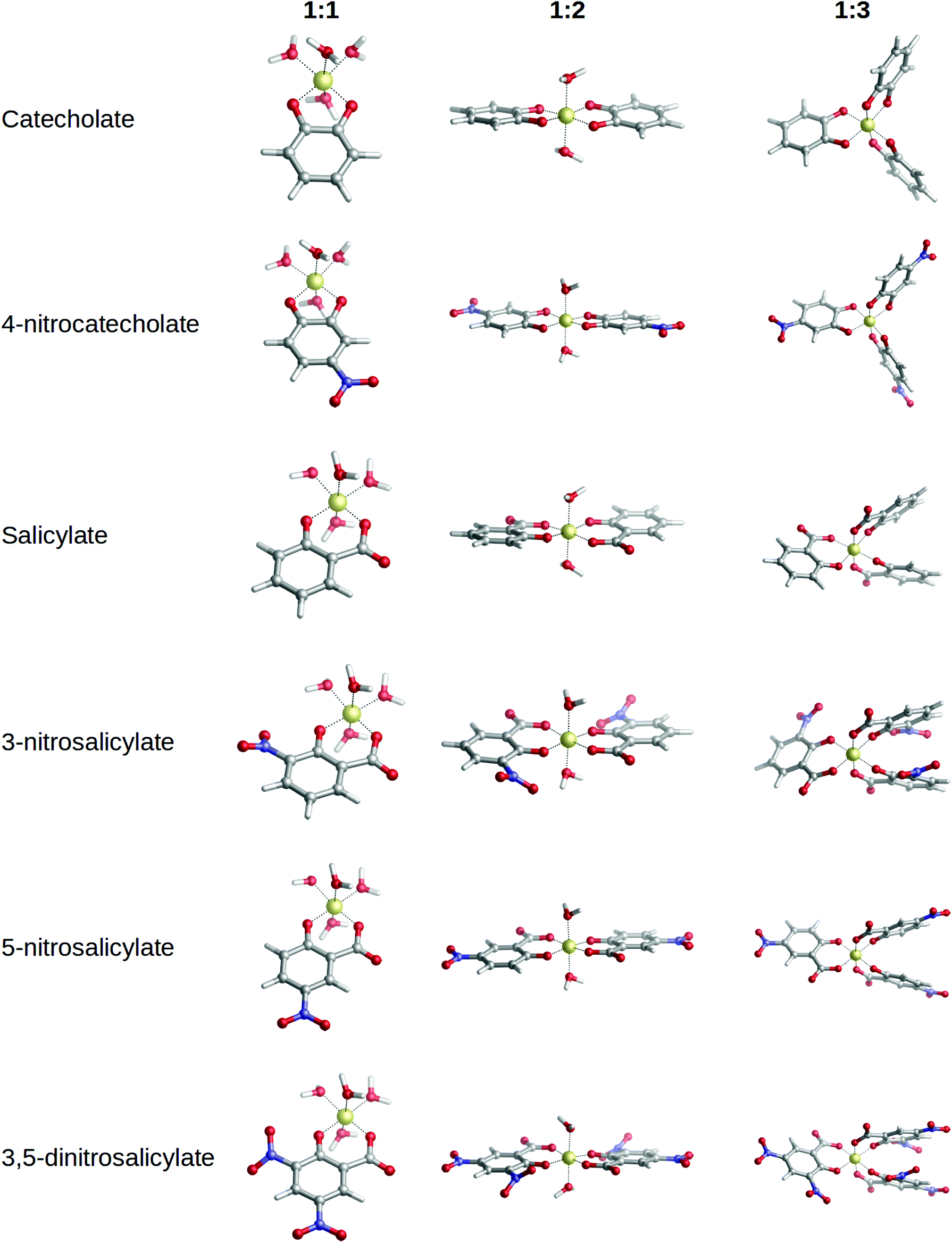 | ||
| Fig. 4 Optimized geometries of 1:1, 1:2 and 1:3 Al(III)–chelator complexes used to validate the theoretical protocol: catechol, 4-nitrocatechol, salicylic acid, 3-nitrosalicylic acid, 5-nitrosalicylic acid, 3,5-dinitrosalicylic acid. | ||
Finally, we would like to point out that we repeated our calculations with other dispersion corrected DFT functionals, as well as at the MP2 level of theory (see ESI Tables S1–S5 and Fig. S1 and S2†), finding a good agreement between all different methods and B3LYP-D3(BJ) binding energies, which further validates our approach.
Modulation of the binding affinities by electron donating and withdrawing groups
Once our theoretical binding energies were validated with respect to available stability constants, and taking into account that the relative affinities are not affected by the different stoichiometries, we focus on 1:1 complexes and enlarge our dataset of possible chelators by considering the four types of substituents presented in Fig. 1: methyl and methoxy (EDGs), and nitro and trifluoromethyl (EWGs). These substituents were placed at different positions of the catecholate/salicylate rings, and in different quantities. In this way, a total of 27 complexes were considered (1:1 complexes of Fig. 4 and 5). Results can be found in Table 2.
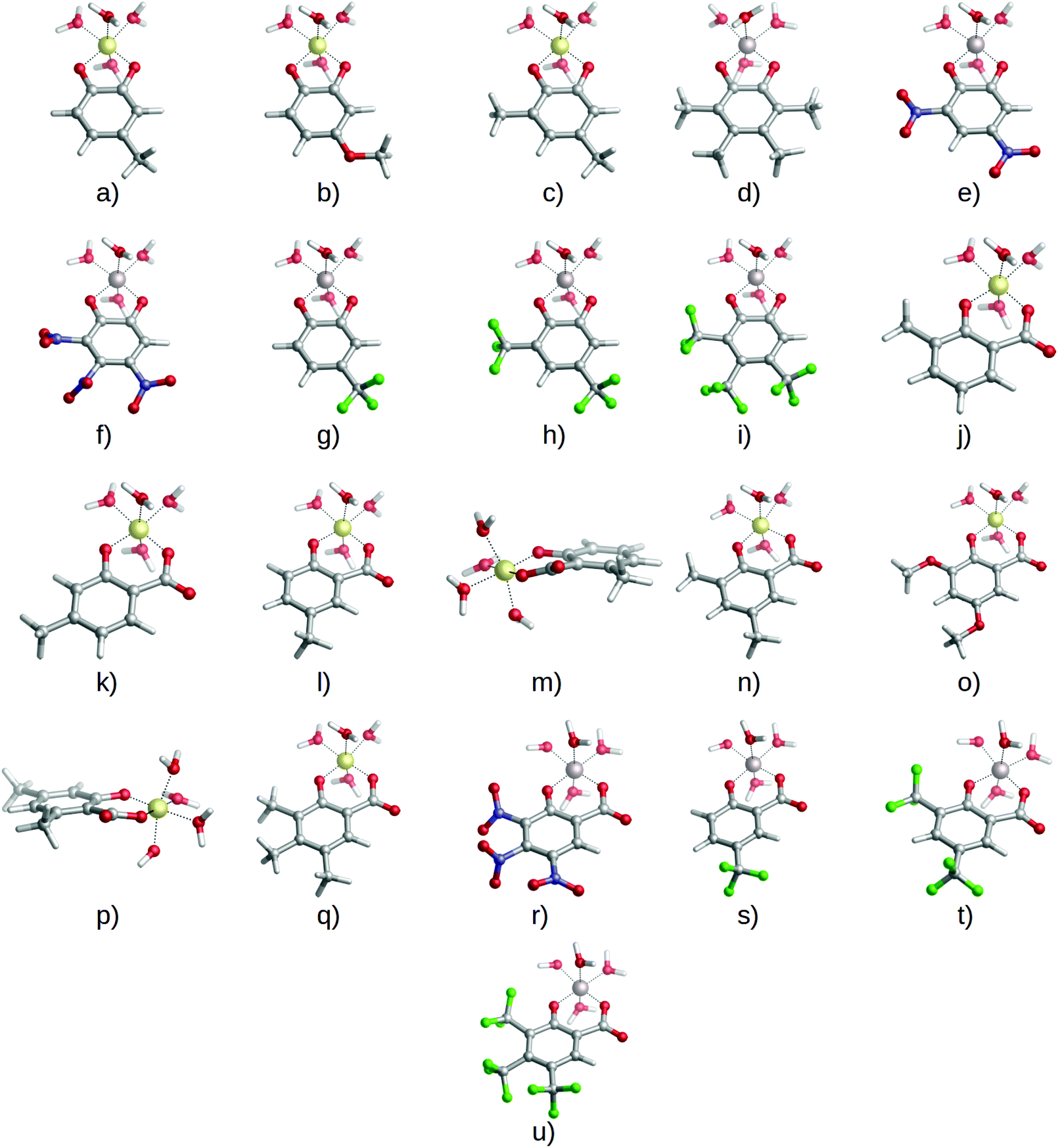 | ||
| Fig. 5 Optimized geometries of 1:1 complexes between aluminum and catecholates or salicylates bearing the four different substituents: (a) 4-methylcatecholate, (b) 4-methoxycatecholate, (c) 4,6-dimethylcatecholate, (d) 3,4,5,6-tetramethylcatecholate, (e) 4,6-dinitrocatecholate, (f) 4,5,6-trinitrocatecholate, (g) 4-trifluoromethylcatecholate, (h) 4,6-trifluoromethylcatecholate, (i) 4,5,6-trifluoromethylcatecholate, (j) 3-methylsalicylate, (k) 4-methylsalicylate, (l) 5-methylsalicylate, (m) 6-methylsalicylate, (n) 3,5-dimethylsalicylate, (o) 3.5-dimethoxysalicylate, (p) 4,6-dimethylsalicylate, (q) 3,4,5-trimethylsalicylate, (r) 3,4,5-trinitrosalicylate, (s) 5-trifluoromethylsalicylate, (t) 3,5-trifluoromethylsalicylate, and (u) 3,4,5-trifluoromethylsalicyate. | ||
:1 complexes (compounds are shown in Fig. 1) considering the whole dataset of compounds bearing different substituents
| Ligand | ΔHcompaq | ΔGcompaq |
|---|---|---|
| Catecholates | ||
| Catecholate | −88.4 | −91.4 |
| Electron withdrawing groups | ||
| 4-Nitrocatecholate | −71.6 | −75.8 |
| 4,6-Dinitrocatecholate | −62.0 | −65.9 |
| 4,5,6-Trinitrocatecholate | −52.8 | −56.4 |
| 4-Trifluoromethylcatecholate | −81.4 | −87.1 |
| 4,6-Trifluoromethylcatecholate | −75.5 | −78.6 |
| 4,5,6-Trifluoromethylcatecholate | −70.1 | −74.0 |
| Electron donating groups | ||
| 4-Methylcatecholate | −89.5 | −93.3 |
| 4,6-Dimethylcatecholate | −91.6 | −95.4 |
| 3,4,5,6-Tetramethylcatecholate | −97.2 | −101.2 |
| 4-Methoxycatecholate | −89.6 | −92.8 |
| Salicylates | ||
| Salicylate | −76.9 | −78.7 |
| Electron withdrawing groups | ||
| 3-Nitrosalicylate | −64.2 | −66.7 |
| 5-Nitrosalicylate | −63.3 | −65.4 |
| 3,5-Dinitrosalicylate | −57.1 | −55.0 |
| 3,4,5-Trinitrosalicylate | −50.5 | −52.4 |
| 5-Trifluoromethylsalicylate | −70.3 | −75.3 |
| 3,5-Trifluoromethylsalicylate | −65.5 | −67.6 |
| 3,4,5-Trifluoromethylsalicylate | −61.8 | −64.2 |
| Electron donating groups | ||
| 3-Methylsalicylate | −79.2 | −82.5 |
| 4-Methylsalicylate | −79.3 | −82.3 |
| 5-Methylsalicylate | −79.3 | −82.9 |
| 6-Methylsalicylate | −74.9 | −77.2 |
| 3,5-Dimethylsalicylate | −80.7 | −83.6 |
| 4,6-Dimethylsalicylate | −76.4 | −79.4 |
| 3,4,5-Trimethylsalicilate | −82.0 | −85.3 |
| 3,5-Dimethoxysalicylate | −79.5 | −82.3 |
Our results show that the inclusion of methyl and methoxy groups leads to larger binding energies when compared with the unsubstituted compounds of both families, whereas the inclusion of nitro and trifluoromethyl groups leads to lower affinities. The destabilizing effect of the inclusion of a nitro group is larger than the destabilizing effect of a trifluoromethyl group and, moreover, larger than the stabilizing effect of the inclusion of both methyl/methoxy groups. This can be qualitatively explained in terms of inductive and resonance effects (see Fig. 1): nitro is an EWG by both inductive and resonance effects, whereas trifluoromethyl is an EWG only by induction. Moreover, methoxy shows contrary effects that partially compensate, i.e. an electron withdrawing effect by induction and a donating one by resonance. Finally, methyl is electron donating only by the inductive effect. As we will see in the role of aromaticity section, resonance effect dominates over inductive effect and methoxy has an overall electron donating behavior. Our results are consistent with the hypothesis by Nurchi et al.40 in that the decrease in the stability constants caused by the nitro substituent was due to a mixture of inductive/resonance effects. On the other hand, in a more recent paper, Nurchi et al.41 also pointed to the increase in the stability of complexes formed by methoxysalicylic acids and aluminum, although the origin of the enhancement of ligand affinity by methoxy substituents was not deeply analyzed. Another interesting feature that can be observed from our calculated binding affinities is the additive character of the substituent effects: the higher the number of the substituents, the stronger their modulation of binding affinities. On the other hand, the specific position of the substituent in the aromatic ring does not lead to significant differences in the stability of both families of chelators (Table 2). These latter findings are in agreement with those reported in the literature, considering the stability constants of differently substituted (EWGs or EDGs) salicylic acids.40,41
In order to rationalize the opposite behavior of the two different types of substituents and to obtain a more detailed picture about the change in the electronic structure of these complexes, we proceeded to characterize the nature of the Al–O interactions by means of the QTAIM theory and Energy Decomposition Analysis (EDA).
Chemical bond analysis of Al–chelator interactions
:1 complexes are shown in the ESI Table S6.† The values of the electron density at all Al–O bond critical points, ρ(rBCP), are rather small; such a situation has been reported in the literature as a typical feature for metal-containing systems.65–67 Interestingly, there is very good correlation between ρ(rBCP) and binding affinities (ΔGcompaq): the higher the value of ρ(rBCP), the stronger the affinity (see ESI Fig. S3†). In addition, we find positive values of ∇2ρ(rBCP), and small but negative values of the energy densities at the bond critical points H(rBCP), consistently for all Al–O BCPs. Positive values of ∇2ρ(rBCP) and H(rBCP) are indicative of closed-shell interactions (i.e. ionic or electrostatic bonds), while negative values for both quantities indicate the presence of shared (covalent) interactions. The mixed situation present in our results, previously reported for bonds involving metals,65 suggests that, although the Al–O bonds are mainly of ionic nature, there is also a small degree of covalency that could play a significant role. It is worth emphasizing that also both the Laplacians and the energy densities at all Al–O BCPs show a strong correlation with binding energies (ESI Fig. S4 and S5,† respectively). To further investigate these findings, we decided to calculate the delocalization indices (D.I.) for all the Al–O bonds. These indices are quantities integrated in the whole volumes of the respective atom basins, and therefore they give a more global and reliable picture of a bond interaction than the analysis based on the properties of a single point in space like the bond critical point.
β when available) binding affinities. It is also important to note that values of aluminum localization indices (average number of electrons localized on a given atom) are stable among different compounds, suggesting that no charge transfer takes place in these aluminum–chelator complexes.
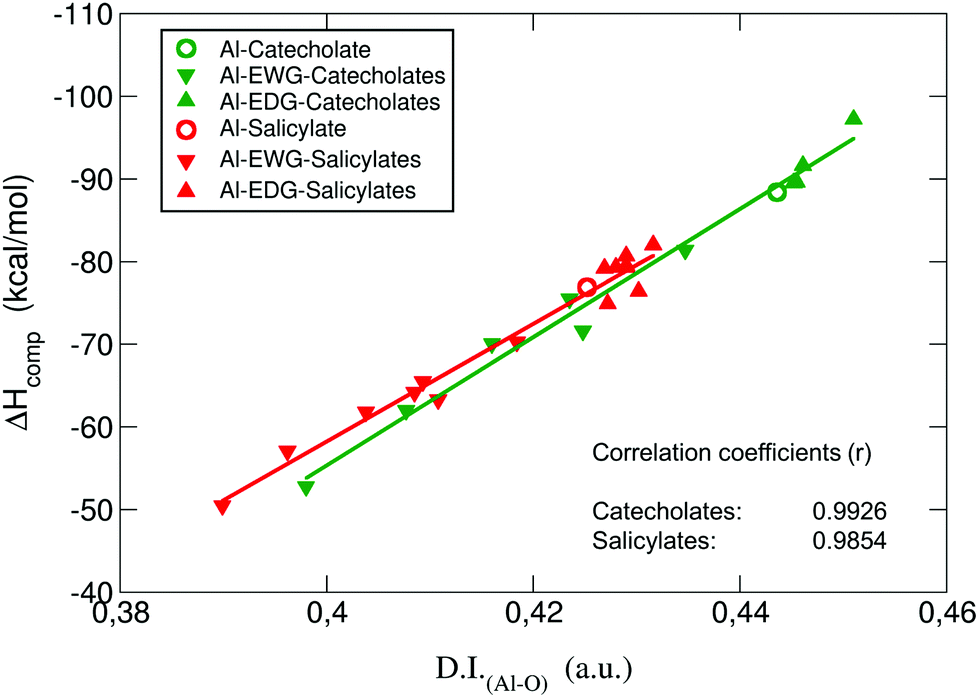 | ||
| Fig. 6 Binding enthalpies ΔHcompaq in kcal mol−1versus the sum of the two Al–O delocalization indices (D.I.Al–O) in a.u. for all complexes. | ||
Finally, we would also like to highlight the effect that substituents have in the modulation of atomic charges at the oxygen atoms coordinated to aluminum. In general, methyl and methoxy groups tend to increase the negative charges at those oxygen atoms, whereas the presence of nitro and trifluoromethyl groups lead to lower negative charges in both families (see the ESI Table S7†). Quite interestingly, high electron delocalizations from the lone pairs of the two oxygens to the 3s and 3p orbitals of aluminum were assessed by means of the Natural Bond Orbital approach. According to these latter findings, we can rationalize such small covalent character as a dative interaction between the two oxygen donors and the formally empty orbitals of the metal.
To further investigate the relative contributions of the electrostatic and covalent components of these Al–O bonds, we performed the Energy Decomposition Analysis (EDA) of all compounds.
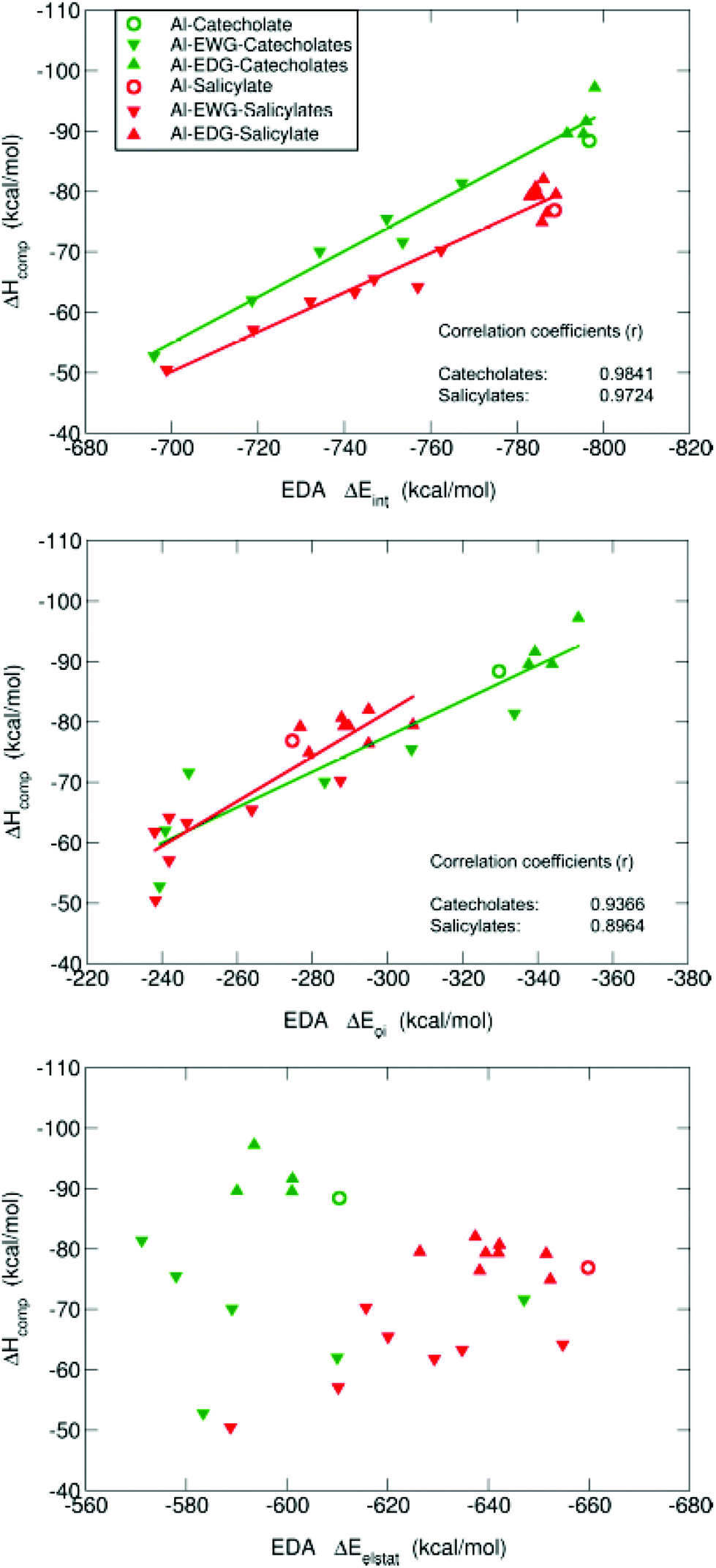 | ||
| Fig. 7 Representation of the binding enthalpies (ΔHcompaq) versus three components of the Energy Decomposition Analysis: (i) total interaction energies (top diagram), (ii) orbital interaction term (middle diagram) and (iii) electrostatic energy term (bottom diagram) for all the 11 aluminum–catecholate and 16 aluminum–salicylate compounds. All energies are in kcal mol−1. | ||
In summary, in agreement with QTAIM analysis, the tuning of the covalency of the Al–O bonds by the different EWG/EDG substituents modulates the differential affinities towards aluminum shown by these chelators. In this sense, the introduction of nitro and trifluoromethyl groups in the catecholate and salicylate rings leads to smaller absolute values of ΔEoi, and this decrease is significantly larger for the former than for the latter. On the other hand, methyl and methoxy substituents lead to larger orbital interactions.
The role of aromaticity
The aromatic character of the ligands could be important to transmit the substituent effects. As can be seen in Fig. 1, the four substituents provide different effects: nitro is an EWG by both inductive and resonance effects, trifluoromethyl is an EWG only by inductive effect, methoxy shows opposite effects that partially compensate, (electron withdrawing by induction and electron donating by resonance), and finally methyl is an EDG only by the inductive effect. Therefore, we chose to investigate how the aromaticity of the ligands changes upon aluminum binding and introduction of substituents. We investigate the aromatic character of all complexes according to the Iring and MCI aromatic descriptors69 (see specific computational details in the ESI and Table S9†). Both give similar trends and we will focus our discussion on Iring indices. Benzene is used as reference for an aromatic compound, while cyclohexane for a non-aromatic one. As expected, both catechol and salicylic acid show lower aromatic characters than the pure benzene ring. Upon deprotonation, both catecholate and salicylate display a sizable reduction of the aromatic character to 0.0235 and 0.0306 a.u., respectively. Notice, however, that upon aluminum binding the values are restored to 0.0345 and 0.0351 a.u. Thus, in terms of aromaticity, the interaction with aluminum recovers the values obtained for the original protonated catechol and salicylic acid. For the rest of the discussion, we will focus on Al-bound complexes, taking as reference the corresponding unsubstituted Al-catecholate/salicylate complex.In both families of chelators, the addition of substituents, independently of the electron donating/withdrawing nature, decreases the aromatic character of the complexes. Moreover, such a decrease in aromaticity follows a clear trend depending on the number of substituents that are added, so that the higher the number of substituents, the lower the aromatic character of the corresponding complex. Interestingly, substituents with a mechanism of action mediated by resonance (nitro and methoxy) show a larger decrease of aromaticity than those that work through inductive effect (methyl and trifluoromethyl). The lowest aromatic character is observed for tri-nitro-substituted compounds (4,5,6-trinitrocatecholate and 3,4,5-trinitrosalicylate), with values of 0.0207 and 0.0249 a.u., respectively. Regarding the electron-donating substituents, methoxy leads to lower aromaticity indices than methyl, because in aromatic molecules resonance effect dominates over inductive effect and methoxy has an overall electron donating behavior.
One may ask, as partially hypothesized by Dean et al.42 for similar pyridine-based aluminum chelators, whether the aromatic character of a chelating agent is one of the main factors contributing to the different stabilities of the Al–chelator complexes. Clearly, our calculations point` to a negative answer. Both EWGs and EDGs decrease the aromatic character of the compounds, but in the latter case there is an increase in the affinity towards aluminum. Thus, aromaticity does not play a direct role in the stabilization of these aluminum–chelator complexes.
Nevertheless, the role of aromaticity is critical to modulate the mechanism of action of the substituents through resonance. In order to analyze this aspect, we calculated the binding energies of a series of non-aromatic 4-R-1,2-dihydroxy-cyclohexanes (see Fig. 8), and evaluated the changes in the binding affinities towards aluminum caused by the introduction of the four substituents listed in Fig. 1. The results are summarized in Fig. 8, where we depict the relative binding energies ΔΔGcompaq of each complex with respect to the unsubstituted chelator in each case. We can see important differences in ΔΔGcompaq between aromatic and non-aromatic compounds: while in the case of non-aromatic chelators the range of ΔΔGcompaq expands from −0.6 kcal mol−1 to 6.4 kcal mol−1, in the case of the aromatic catecholates ΔΔGcompaq expands to a much larger range, from −1.9 kcal mol−1 to 15.7 kcal mol−1. This is indicative of a larger sensitivity of aromatic chelators towards substituent effects. Notice for instance, the large increase in ΔΔGcompaq when considering the nitro group, 6.4 kcal mol−1 (non-aromatic chelator) versus 15.7 kcal mol−1 (aromatic chelator); clearly, this difference demonstrates that when the resonance transmission mechanism of the substituent is absent, the nitro group loses some of its electron-withdrawing character, partially maintained by the inductive-based one. Methoxy is a very significant case: while in the case of the aromatic chelator –OCH3 leads to stabilizing effects (−1.4 kcal mol−1), in the case of the non-aromatic compound it leads to a destabilizing effect (0.8 kcal mol−1). This is due to the fact that –OCH3 acts as an EDG by resonance, but as an EWG by inductive effect. Accordingly, when resonance is absent like in 4-methoxy-1,2-dihydroxy-cyclohexane, the inductive-based electron withdrawing mechanism is the only one working.
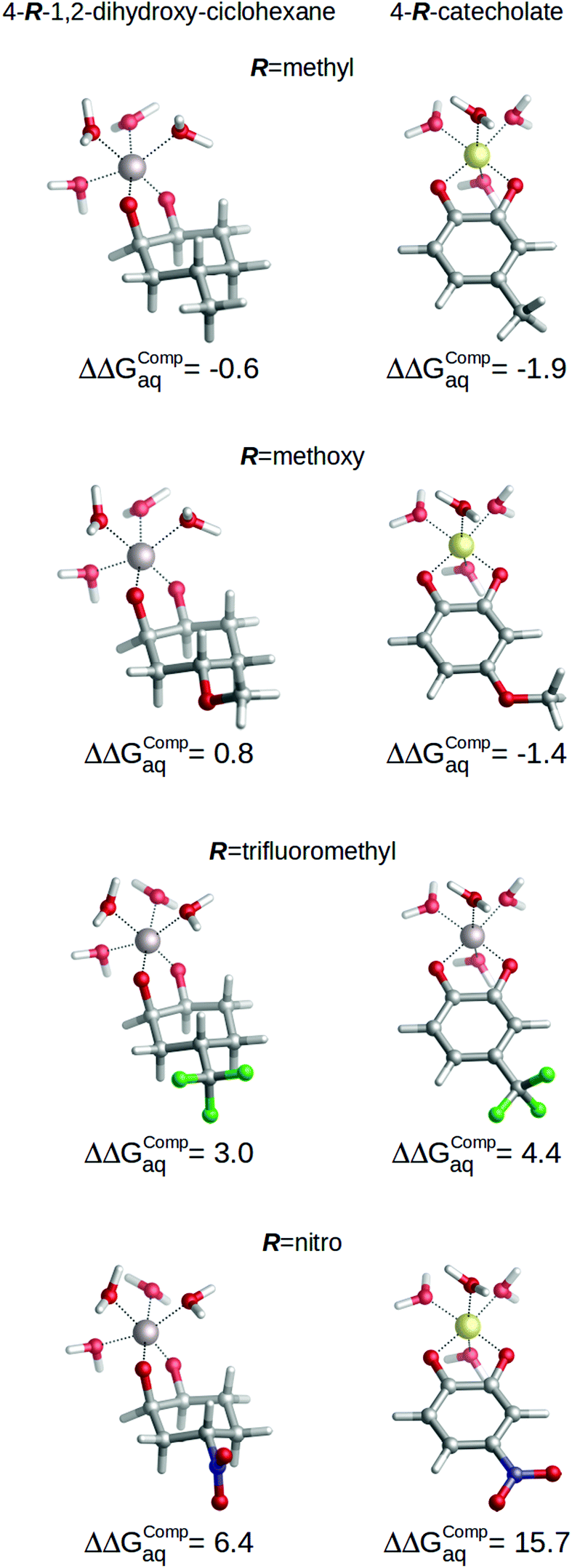 | ||
| Fig. 8 Relative binding energies (ΔΔGCompaq) of non-aromatic (4-R-1,2-dihydroxy-cyclohexane) and aromatic (4-R-catecholate) chelators calculated with respect to their unsubstituted counterparts. R can be methyl, methoxy, nitro or trifluoromethyl. All energies are in kcal mol−1. | ||
In summary, although the introduction of electron donating/withdrawing substituents in both catecholate and salicylate families of chelators reduce the aromaticity of the compounds, the complexes still retain enough aromatic character to permit the transmission of substituent effects by a combination of both resonance and inductive mechanisms. This is a key factor in tuning the covalent character of the Al–O interactions.
Proton and aluminum ion competition
So far, we have not considered the possible competition between Al(III) and proton(s) for ligand binding. In other words, the overall performance of a chelator at a given pH will be dictated not only by the stability of the corresponding aluminum–ligand complex, but also by the deprotonation capacity of a ligand at a certain pH. As a result, the experimental trends in logβ and p[Al] to characterize the performance of a given chelator can differ62,64 (see Table 1). For instance, catechol shows a p[Al] value of 10.1, and the introduction of a nitro electron-withdrawing group raises this value up to 14.2, indicating better chelation properties.40 However, as we have seen, 4-nitro-catecholate shows a lower logβ value than the unsubstituted catecholate (see Table 1). Conversely, in the case of salicylic acids, Nurchi et al.41 reported that methoxy-substituted salicylic acids (an overall electron-donating group) show significant higher p[Al] values (9.6/10.2 for ortho/para methoxy-salicylic acid) than the unsubstituted one (8.2), while the introduction of a nitro group provokes only a moderate variation of p[Al], (8.4/8.7 for ortho/para nitro-salicylic acid). What is the reason for these differential trends between salicylic acids and catechols, and between logβ and p[Al] in catechols? We have to take into account that whereas logβ is a measure of the stability of the complex with respect to the dissociation of unprotonated ligands, p[Al] takes into account additional factors, like proton/metal ion competition. Another important difference is that logβ is specific for each stoichiometry, while different stoichiometries and denticities contribute to a given value of p[Al].28,64
In order to account for proton/metal ion competition in our calculations, we have evaluated the relative proton affinities of the different ligands, and combine them with the relative aluminum affinities. The procedure is as follows: we evaluate the relative proton affinities of the ligands with a given functional group with respect to the unsubstituted catechol and salicylic acid, by the estimation of the following ΔΔGnH(Lsubs−2) reaction energy:
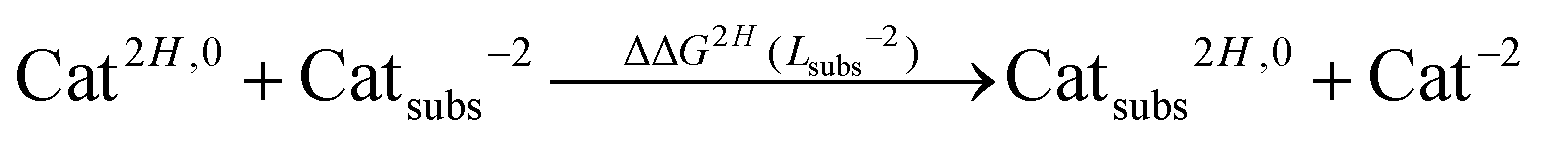 | (4) |
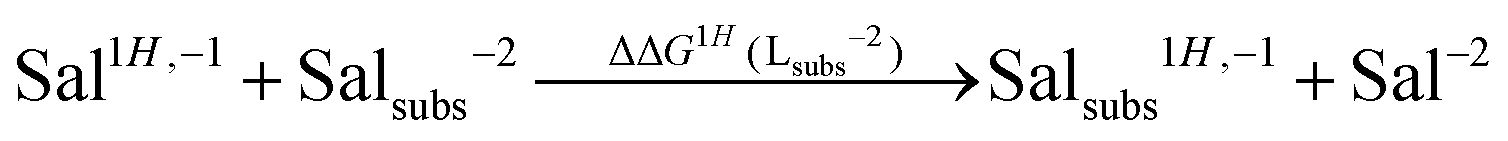 | (5) |
As one can see in these equations, there is an important difference between catechols and salicylic acids. The pKa values of catechols (see Table 3 and ref. 40) are such that at neutral pH both chelating positions are likely to be protonated and, therefore, Al(III) binding has to compete with the removal of two protons from the ligand. However, the first pKa1 of salicylic acid is so low (see Table 3 and ref. 40 and 41) that at neutral pH the carboxylic group is undoubtedly unprotonated; accordingly, the binding of the aluminum ion only involves the removal of the hydroxyl proton. Besides, we define a relative aluminum affinity of a given ligand in each family of compounds with respect to the unsubstituted ligand, using the ΔGcompaq values of Table 2, namely:
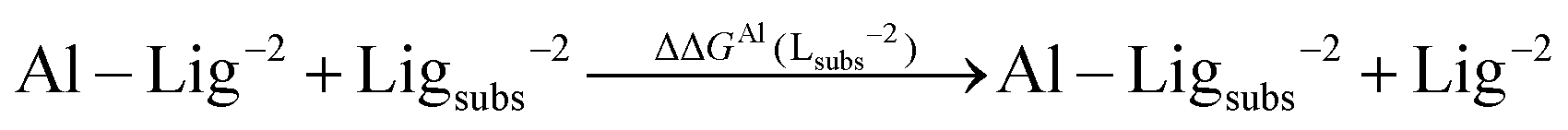 | (6) |
| ΔΔGAl(Lsubs−2) = ΔGcompaq(Al–Lsubs−2) − ΔGcompaq(Al–L−2) | (7) |
| ΔΔGAl(Al–LsubsnH) = ΔΔGAl(Al–Lsubs−2) − ΔΔGnH(Lsubs−2) | (8) |
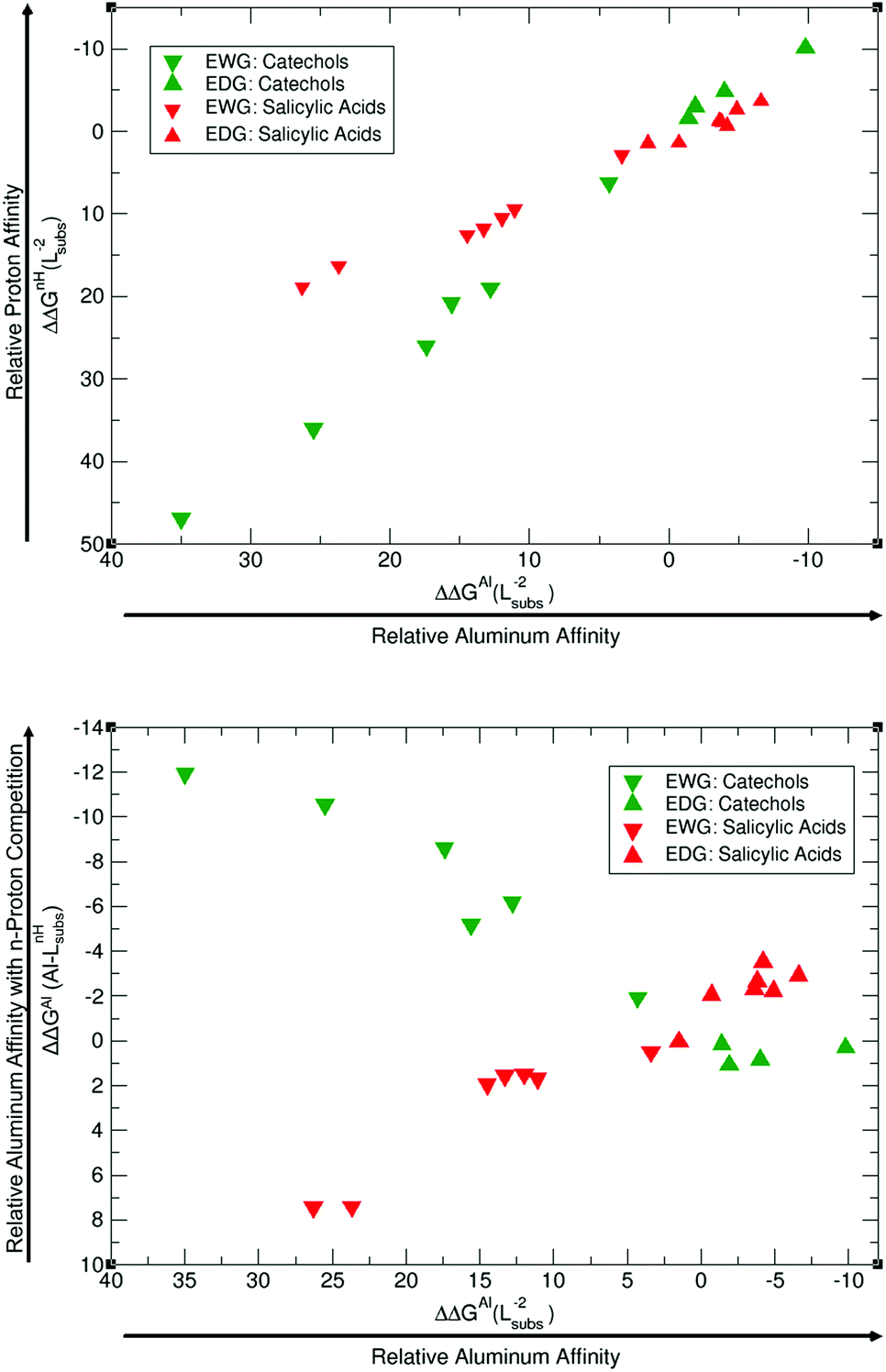 | ||
| Fig. 9 Proton versus aluminum competition analysed as: (a) ΔΔGnH(Lsubs−2) vs. ΔΔGAl(Lsubs−2) (top diagram) and (b) ΔΔGAl(Al–LsubsnH) vs. ΔΔGAl(Lsubs−2) (bottom diagram). Terms defined in the body text. All relative affinities are in kcal mol−1. | ||
| Ligand | ΔΔHsubs2H | ΔΔGsubs2H | pKa1 (exp) | pKa2 (exp) |
|---|---|---|---|---|
| Cat2H,0 + Catsubs−2 → Catsubs2H,0 + Cat−2 | ||||
| Catechol | 0.0 | 0.0 | 9.2 | 14.3 |
| Electron withdrawing groups | ||||
| 4-Nitrocatechol | 21.3 | 20.8 | 6.6 | 10.7 |
| 4,6-Dinitrocatechol | 36.0 | 36.0 | ||
| 4,5,6-Trinitrocatechol | 47.6 | 46.9 | ||
| 4-Trifluoromethylcatechol | 9.3 | 6.2 | ||
| 4,6-Trifluoromethylcatechol | 18.7 | 19.0 | ||
| 4,5,6-Trifluoromethylcatechol | 26.4 | 26.0 | ||
| Electron donating groups | ||||
| 4-Methylcatechol | −1.6 | −3.0 | ||
| 4,6-Dimethylcatechol | −3.9 | −4.8 | ||
| 3,4,5,6-Tetramethylcatechol | −9.1 | −10.1 | ||
| 4-Methoxycatechol | −1.6 | −1.6 | ||
| Sal1H,−1 + Salsubs−2 → Salsubs1H,−1 + Sal−2 | ||||
| Salicylic acid | 0.0 | 0.0 | 3.1 | 13.6 |
| Electron withdrawing groups | ||||
| 3-Nitrosalicylic acid | 10.8 | 10.5 | 1.5 | 9.9 |
| 5-Nitrosalicylic acid | 11.4 | 11.8 | 1.7 | 10.0 |
| 3,5-Dinitrosalicylic acid | 16.0 | 16.3 | −0.1 | 7.0 |
| 3,4,5-Trinitrosalicylic acid | 18.9 | 18.9 | ||
| 5-Trifluoromethylsalicylic acid | 5.2 | 2.9 | ||
| 3,5-Trifluoromethylsalicylic acid | 9.7 | 9.4 | ||
| 3,4,5-Trifluoromethylsalicylic acid | 12.7 | 12.6 | ||
| Electron donating groups | ||||
| 3-Methylsalicylic acid | −0.9 | −1.1 | ||
| 4-Methylsalicylic acid | −1.0 | −1.3 | ||
| 5-Methylsalicylic acid | −1.1 | −0.7 | ||
| 6-Methylsalicylic acid | 2.5 | 1.5 | ||
| 3,5-Dimethylsalicylic acid | −2.4 | −2.7 | ||
| 4,6-Dimethylsalicylic acid | 1.5 | 1.4 | ||
| 3,4,5-Trimethylsalicylic acid | −3.4 | −3.7 | ||
| 3,5-Dimethoxysalicylic acid | −2.1 | −1.3 | ||
As one can see in Fig. 9A, EDGs lie at the top-right side of the diagram, whereas EWGs are at the bottom-left side, manifesting that those ligands that have the largest affinities for aluminum also display the largest affinities for protons. This is the case for both catechols and salicylic acids, but with an important difference. Catechols span a wider range of relative proton affinities than salicylic acids, a fact mainly attributed to the fact that two protons are removed in catechol and only one in salicylic acids. In order to estimate the aluminum relative binding affinity in the presence of protonated ligands (Fig. 9B), we have to combine these two relative proton/aluminum affinities according to eqn (8), to yield ΔΔGAl(Al–Lsubs2H) (displayed in the y-axis of Fig. 9B). Our data clearly show an inverse trend between ΔΔGAl(Al–Lsubs2H) and ΔΔGAl(Al–Lsubs−2) for catechols, but not for salicylic acids. Our results for catechols suggest that the introduction of EWGs leads to a better Al(III) chelation performance upon competition with the removal of two protons, and this corresponds to the previously described experimental increase of p[Al] with nitro-substitution (Tables 1 and 3). In the case of salicylic acids, since we are only removing one proton upon aluminum binding, relative aluminum affinity is still the overall leading factor in chelator binding, and now it is the introduction of an EDG that clearly improves the performance of the chelator. This is again in agreement with the clear increase in the experimental p[Al] of salicylic acids upon the introduction of methoxy groups.41 In summary, our results demonstrate that in the competition between aluminum binding and deprotonation, the latter factor dominates when the binding of Al(III) requires the removal of two protons from the ligand, whereas the former is dominant if only one proton has to be removed, in agreement with the experimental results for catechols and salicylic acids, respectively.40,41
To complete our analysis, we provide a possible explanation of how the introduction of EWGs (i.e. nitro) in salicylic acids lead to similar albeit a bit higher p[Al] values.40 Our data for 1:1 complexes show a moderate decrease in ΔΔGAl(Al–Lsubs1H) for both 3- and 5-nitro-substitution (namely 1.5 kcal mol−1), which in principle should point to a lower value of p[Al]. One aspect should be remarked in this regard: the experimental values of p[Al] don't take into account only 1:1 aluminum–ligand stoichiometry, but different stoichiometrical complexes like 1:2 and 1:3. Therefore, we recalculated the differential binding free energies for 1:2 and 1:3 stoichiometries of the single nitro-substituted salicylic acids of Table 1, namely,
| ΔΔGAl(Al–[Lsubs1H]n) = ΔΔGAl(Al–[Lsubs−2]n) + n × ΔΔGsubs1H | (9) |
:1 complexes, we obtain values for ΔΔGAl(Al–[Lsubs1H]1) of 1.5 kcal mol−1 (3-nitro), 1.5 kcal mol−1 (5-nitro); for 1:2 complexes: 1.3 kcal mol−1 (3-nitro), −0.9 kcal mol−1 (5-nitro); finally, for 1:3 complexes, we found −9.8 kcal mol−1 (3-nitro), −9.6 kcal mol−1 (5-nitro). Thus, we can observe how the stoichiometry is an additional contributing factor in the modulation of the aluminum ion/proton competition for ligand binding, with higher stoichiometries favoring those substituents that lead to a more favorable deprotonation (i.e. EWGs), albeit lower interaction with aluminum itself. The overall result is that, for 1:3 complexes, the introduction of EWGs promotes their chelation to aluminum because of the lower protonation energies (i.e. lower protonation constants, Table 3), and would lead to higher p[Al] values; the case of 1:2 complexes lies in between 1:1 and 1:3-related behavior. The fact that the experimental data point only to a moderate increase in p[Al] upon nitro introduction in salicylic acids suggests that different stoichiometries with opposite effects are contributing to these values, and thus there is a partial cancelation and compensation of their effects. Finally, it is important to mention that both families of compounds, when protonated, form an intramolecular hydrogen bond between the two phenolates (catechol) and phenolate and carboxylate (salicylic acid), which is a further factor contributing to the modulation of the aluminum/proton competition.
Tuning the molecule: the role of substituents
The present paper provides the most complete and thorough theoretical study of the interaction of aluminum with catechols and salicylic acids chelating agents done so far. We have identified and rationalized important factors affecting ligand binding to aluminum, which are crucial to design new chelators of increased affinity. Namely, we have characterized the strength of the aluminum–ligand interactions, interpreted the binding strengths in terms of the electrostatic/covalent nature of the Al–O bonds, unveiled the role played by the aromaticity of these chelators, rationalized the modulation of the stability through addition of substituents and, finally, determined how aluminum/proton competition affects the overall activity of a chelator.In this sense, our calculations demonstrate that although the bond is mainly electrostatic in nature, as it corresponds to a hard metal, the fine tuning of the stability in both families of chelators is mediated through the modulation of the covalent character of the Al–O bonds. This covalent character can be classified as a dative bond from the lone pair of the oxygens to the 3s, 3p valence shell of Al(III). The increase in the dative Al–O bond character through the introduction of EDGs leads to complexes of higher stability, whereas EWGs lead to complexes of lower stability, in agreement with the experimental trends of logβ (Table 1). Such a picture is also coherent with the Pearson's Hard and Soft Acids and Bases (HSAB) principle;70 indeed, the two phenolate groups of catechol are harder Lewis bases than the carboxylate one of salicylic acid, because of the intrinsic resonance of the COO− moiety, and therefore the former are expected to show higher affinity for hard Lewis acids such as Al(III). This is quite interesting considering that the salicylate family shows, overall, a higher electrostatic interaction (three negatively charged oxygens) than the catecholate family,61 as shown by EDA results (see Table S8†); nevertheless, the catecholate family has a higher affinity for the trivalent metal, a fact that can be related to the more covalent Al–O bond as revealed by both QTAIM and EDA results and summarized in Fig. S6.†
We have also determined the role that the aromatic nature of these two families of chelators plays in the metal–ligand complexes. Aromaticity is only slightly affected upon aluminum binding, being more sensible to the introduction of EDG/EWG substituents in the ring. In both salicylates and catecholates, the introduction of both electron donating/withdrawing substituents leads to a lower aromatic character. Nevertheless, a significant degree of aromaticity is maintained in all complexes, which is pivotal to modulate and transmit some of the resonance-based substituent effects.
We should remark that, although the covalent character is the main driving factor in the modulation of the affinity toward aluminum for these two families of chelators, other factors can also affect the observed stability. For instance, in some of the complexes we found steric hindrances that put them out of the general trend in binding energy (see Table 2). Indeed, 6-methylsalicylate has a lower stability when compared with unsubstituted salicylic acid, despite the presence of an EDG, which should enhance its binding affinity. The optimized geometry for that compound shows that the six-membered ring formed by aluminum, the carboxylate and the enolate groups is slightly distorted from full planarity (by 16.8° and 20.9°), suggesting a steric repulsion between methyl at the 6 position and the carboxylate group (compound ‘m’ in Fig. 5). Such a situation leads to a decrease in binding affinity (Table 2). Moreover, if we consider 4,6-dimethylsalicylate (compound ‘p’ in Fig. 5), we can see that the addition of a second methyl in position 4 partially recovers the stability and planarity of the complex (12.6° and 15.6°) when compared with salicylate (Table 2), because of the electron donating effect that counterbalances the steric repulsion of the methyl in position 6. However, the recovered stability is still not as high as for another di-substituted compound like 3,5-dimethylsalicylate (0.1° and 0.1°), where no steric effects are present (compound ‘n’ in Fig. 5). This situation was also hypothesized by Dean et al.43 for similar compounds. It is clear that, when considering new strategies toward the improvement and design of new Al(III) chelating agents, one should carefully consider possible repulsive phenomena. Regarding proton/aluminum ion competition, we have been able to reproduce the inverse trends in ligand affinity when comparing logβ and p[Al] values for catechols (Table 1), and to explain how the introduction of an electron withdrawing group in catechols, but electron donating group in salicylic acids, enhances the chelation properties of the ligands upon competition with protonation. Taking into account that the metal/proton competition for ligand binding is critical to determine the performance of a given ligand in chelation therapy, as established by Hider et al.,62 complex stability is also important in order to compete with other endogenous ligands (like citrate) in an open biological environment.71 Moreover, if the stability of the Al(III)–chelator complex is too weak, then the metal may prefer to form the very stable [Al(OH)4]− hydroxo complex.61,72 We have found that those substituents that favor aluminum binding (in terms of logβ) also favor protonation.40,41,62 The overall effect is a balance between the Al(III)–ligand complex stability and the competition with H+. In this sense, EWGs, by lowering the affinity toward aluminum, also favor deprotonation (by lowering the protonation constants of the ligand), and this latter factor is the dominant one at a pH in which aluminum competes with two protons for ligand binding. Conversely, when only one proton has to be removed, like in salicylic acids at weakly acidic or neutral pH, the nature of the dominant factor shifts to aluminum complex stability. Other factors such as stoichiometry of the complex can also contribute to the proton/aluminum ion competition toward a given ligand. Our results suggest that higher stoichiometries favor deprotonation as a leading factor in the overall performance of a given chelator.
Conclusions
In the present work, we have developed, validated and applied a state-of-the-art theoretical protocol suitable for the investigation of two families of bidentate Al(III) chelating agents (catechols and salicylic acids). Trends in binding affinities show very good agreement with respect to available experimental data. We have rationalized our results analyzing the nature of the Al–O bonds, finding that the covalent part of a mainly ionic Al–O interaction is the driving force in the fine-tuning of the stability of these complexes. Such a covalent character is modulated by the opposite effect of the substituents: methyl and methoxy groups increase this covalency, leading to higher affinities, whereas the nitro and trifluoromethy groups decrease the covalent component leading to lower binding affinities. We have also determined how the overall performance of a chelator depends critically on the metal/proton competition toward ligand binding. In summary, the present work establishes a reliable and transferable theoretical protocol aimed to test the behavior of metal organic chelators, which would help in the future design and tuning of novel chelating agents of increased efficacy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been funded by the Spanish MINECO/FEDER Projects CTQ2015-67608-P (X. L.), CTQ2014-52525-P (E. M.), EUIN2017-88605 (E. M.) and the Basque Country Consolidated Group Project No. IT588-13. The authors would like to thank the technical and human support provided by the SGI/IZO (SGIker) of the UPV/EHU. Financial support comes as well from the European Commission, Horizon 2020 Research and Innovation Programme (grant agreement N 642294 TCCM). This work received financial support from the European Union (FEDER funds POCI/01/0145/FEDER/007728) and National Funds (Fundação para a Ciência e Tecnologia and Ministério da Educação e Ciência–FCT/MEC) UID/MULTI/04378/2013, under the Partnership Agreement PT2020; NORTE-01-0145-FEDER-000024, supported by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF).References
- C. Exley, J. Inorg. Biochem., 2003, 97, 1–7 CrossRef PubMed.
- J. Beardmore, X. Lopez, J. I. Mujika and C. Exley, Sci. Rep., 2016, 6, 30913 CrossRef PubMed.
- R. B. Martin, Acc. Chem. Res., 1994, 27, 204–210 CrossRef.
- R. A. Yokel and P. J. McNamara, Pharmacol. Toxicol., 2001, 88, 159–167 CrossRef.
- C. A. Shaw and L. Tomljenovic, Immunol. Res., 2013, 56, 304–316 CrossRef PubMed.
- C. C. Willhite, N. A. Karyakina, R. A. Yokel, N. Yenugadhati, T. M. Wisniewski, I. M. F. Arnold, F. Momoli and D. Krewski, Crit. Rev. Toxicol., 2014, 44(Suppl 4), 1–80 CrossRef PubMed.
- C. Exley, Free Radical Biol. Med., 2004, 36, 380–387 CrossRef PubMed.
- J. I. Mujika, F. Ruipérez, I. Infante, J. M. Ugalde, C. Exley and X. Lopez, J. Phys. Chem. A, 2011, 115, 6717–6723 CrossRef PubMed.
- F. Ruipérez, J. I. Mujika, J. M. Ugalde, C. Exley and X. Lopez, J. Inorg. Biochem., 2012, 117, 118–123 CrossRef PubMed.
- C. Exley, N. C. Price and J. D. Birchall, J. Inorg. Biochem., 1994, 54, 297–304 CrossRef PubMed.
- S. J. Yang, J. W. Huh, J. E. Lee, S. Y. Choi, T. U. Kim and S. W. Cho, Cell. Mol. Life Sci., 2003, 60, 2538–2546 CrossRef PubMed.
- J. I. Mujika, J. M. Ugalde and X. Lopez, J. Phys. Chem. B, 2014, 118, 6680–6686 CrossRef PubMed.
- P. Zatta, E. Lain and C. Cagnolini, Eur. J. Biochem., 2000, 267, 3049–3055 CrossRef PubMed.
- J. Lemire, C. Auger, R. Mailloux and V. D. Appanna, J. Neurosci. Res., 2014, 92, 464–475 CrossRef PubMed.
- J. Lemire, R. Mailloux, S. Puiseux-Dao and V. D. Appanna, J. Neurosci. Res., 2009, 87, 1474–1483 CrossRef PubMed.
- R. J. Mailloux, R. Hamel and V. D. Appanna, J. Biochem. Mol. Toxicol., 2006, 20, 198–208 CrossRef PubMed.
- E. Formoso, J. I. Mujika, S. J. Grabowski and X. Lopez, J. Inorg. Biochem., 2015, 152, 139–146 CrossRef PubMed.
- V. Kumar and K. D. Gill, Arch. Toxicol., 2009, 83, 965–978 CrossRef PubMed.
- I. Klatzo, H. Wisniewski and E. Streicher, J. Neuropathol. Exp. Neurol., 1965, 24, 187–199 CrossRef PubMed.
- R. Grande-Aztatzi, J. M. Mercero and J. M. Ugalde, Phys. Chem. Chem. Phys., 2016, 18, 11879–11884 RSC.
- T. Kiss, K. Gajda-Schrantz and P. F. Zatta, Neurodegenerative Diseases and Metal Ions, John Wiley & Sons, Ltd, 2006, ch. 13, pp. 371–393 Search PubMed.
- M. Abdel-ghany, A. Khalek and D. Shalloway, J. Biol. Chem., 1993, 268, 11976–11981 Search PubMed.
- C. Exley, Coord. Chem. Rev., 2012, 256, 2142–2146 CrossRef.
- J. I. Mujika, J. Rodríguez-Guerra Pedregal, X. Lopez, J. M. Ugalde, L. Rodríguez-Santiago, M. Sodupe and J.-D. Maréchal, Chem. Sci., 2017, 8, 5041–5049 RSC.
- L. Tomljenovic, J. Alzheimers Dis., 2011, 23, 567–598 Search PubMed.
- C. Exley, Front. Neurol., 2014, 5, 1–6 Search PubMed.
- V. B. Gupta, S. Anitha, M. L. Hegde, L. Zecca, R. M. Garruto, R. Ravid, S. K. Shankar, R. Stein, P. Shanmugavelu and K. S. Jagannatha Rao, Cell. Mol. Life Sci., 2005, 62, 143–158 CrossRef PubMed.
- G. Crisponi, V. M. Nurchi, V. Bertolasi, M. Remelli and G. Faa, Coord. Chem. Rev., 2012, 256, 89–104 CrossRef.
- T. Kiss, J. Inorg. Biochem., 2013, 128, 156–163 CrossRef PubMed.
- R. A. Yokel, Coord. Chem. Rev., 2002, 228, 97–113 CrossRef.
- G. Crisponi, A. Dean, V. Di Marco, J. I. Lachowicz, V. M. Nurchi, M. Remelli and A. Tapparo, Anal. Bioanal. Chem., 2013, 405, 585–601 CrossRef PubMed.
- G. Crisponi, V. M. Nurchi, J. I. Lachowicz, M. Crespo-Alonso, M. A. Zoroddu and M. Peana, Coord. Chem. Rev., 2015, 284, 278–285 CrossRef.
- A. E. Martell, R. D. Hancock, R. M. Smith and R. J. Motekaitis, Coord. Chem. Rev., 1996, 149, 311–328 CrossRef.
- T. Kaur, R. K. Bijarnia and B. Nehru, Drug Chem. Toxicol., 2009, 32, 215–221 CrossRef PubMed.
- T. Kiss, I. Sovago and R. B. Martin, J. Am. Chem. Soc., 1989, 111, 3611–3614 CrossRef.
- T. Kiss, Arch. Gerontol. Geriatr., 1995, 21, 99–112 CrossRef PubMed.
- A. C. Miu and O. Benga, J. Alzheimers Dis., 2006, 10, 179–201 Search PubMed.
- A. J. Cross, T. J. Crow, E. K. Perry, R. H. Perry, G. Blessed and B. E. Tomlinson, Br. Med. J. (Clin. Res. Ed.), 1981, 282, 93–94 CrossRef.
- M. Milanese, M. I. Lkhayat and P. Zatta, J. Trace Elem. Med. Biol., 2001, 15, 139–141 CrossRef.
- V. M. Nurchi, T. Pivetta, J. I. Lachowicz and G. Crisponi, J. Inorg. Biochem., 2009, 103, 227–236 CrossRef PubMed.
- V. M. Nurchi, M. Crespo-Alonso, L. Toso, J. I. Lachowicz, G. Crisponi, G. Alberti, R. Biesuz, A. Dominguez-Martin, J. Niclos-Gutierrez, J. M. Gonzalez-Perez and M. A. Zoroddu, J. Inorg. Biochem., 2013, 128, 174–182 CrossRef PubMed.
- A. Dean, E. Sija, E. Zsigo, M. G. Ferlin, D. Marton, V. Gandin, C. Marzano, D. Badocco, P. Pastore, A. Venzo, R. Bertani, T. Kiss and V. Di Marco, Eur. J. Inorg. Chem., 2013, 2013, 1310–1319 CrossRef.
- A. Dean, M. G. Ferlin, P. Brun, I. Castagliuolo, R. A. Yokel, A. Venzo, G. Giorgio Bombi and V. B. Di Marco, Inorg. Chim. Acta, 2011, 373, 179–186 CrossRef.
- V. M. Nurchi, G. Crisponi, J. I. Lachowicz, S. Murgia, T. Pivetta, M. Remelli, A. Rescigno, J. Niclos-Gutierrez, J. M. Gonzalez-Perez, A. Dominguez-Martin, A. Castineiras and Z. Szewczuk, J. Inorg. Biochem., 2010, 104, 560–569 CrossRef PubMed.
- O. Gutten and L. Rulisek, Inorg. Chem., 2013, 52, 10347–10355 CrossRef PubMed.
- Q. Y. Wu, Y. T. Song, L. Ji, C. Z. Wang, Z. F. Chai and W. Q. Shi, Phys. Chem. Chem. Phys., 2017, 19, 26969–26979 RSC.
- J. H. Lan, W. Q. Shi, L. Y. Yuan, J. Li, Y. L. Zhao and Z. F. Chai, Coord. Chem. Rev., 2012, 256, 1406–1417 CrossRef.
- J. R. Pliego and J. M. Riveros, J. Phys. Chem. A, 2001, 105, 7241–7247 CrossRef.
- D. Riccardi, H. B. Guo, J. M. Parks, B. Gu, L. Liang and J. C. Smith, J. Chem. Theory Comput., 2013, 9, 555–569 CrossRef PubMed.
- J. I. Mujika, E. Rezabal, J. M. Mercero, F. Ruipérez, D. Costa, J. M. Ugalde and X. Lopez, Comput. Struct. Biotechnol. J., 2014, 9, e201403002 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef PubMed.
- R. Lonsdale, J. N. Harvey and A. J. Mulholland, J. Chem. Theory Comput., 2012, 8, 4637–4645 CrossRef PubMed.
- W. Hujo and S. Grimme, J. Chem. Theory Comput., 2013, 9, 308–315 CrossRef PubMed.
- R. Sedlak, T. Janowski, M. Pitonak, J. Rezac, P. Pulay and P. Hobza, J. Chem. Theory Comput., 2013, 9, 3364–3374 CrossRef PubMed.
- J. Alí-Torres, L. Rodríguez-Santiago and M. Sodupe, Phys. Chem. Chem. Phys., 2011, 13, 7852–7861 RSC.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford Univ. Press, Oxford, 1990 Search PubMed.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef.
- T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef.
- M. Giambiagi, M. S. de Giambiagi, C. D. dos Santos Silva and A. P. de Figueiredo, Phys. Chem. Chem. Phys., 2000, 2, 3381–3392 RSC.
- P. Bultinck, R. Ponec and S. Van Damme, J. Phys. Org. Chem., 2005, 18, 706–718 CrossRef.
- T. Kiss, K. Atkari, M. Jezowska-bojczuk and P. Decock, J. Coord. Chem., 1993, 29, 81–96 CrossRef.
- R. Hider, Thalassemia Rep., 2014, 4, 19–27 Search PubMed.
- G. Crisponi and M. Remelli, Coord. Chem. Rev., 2008, 252, 1225–1240 CrossRef.
- C. Bazzicalupi, A. Bianchi, C. Giorgi, M. P. Clares and E. Garcia-Espana, Coord. Chem. Rev., 2012, 256, 13–27 CrossRef.
- G. Gervasio, R. Bianchi and D. Marabello, Chem. Phys. Lett., 2004, 387, 481–484 CrossRef.
- T. S. Koritsanszky and P. Coppens, Chem. Rev., 2001, 101, 1583–1628 CrossRef PubMed.
- B. Silvi and C. Gatti, J. Phys. Chem. A, 2000, 104, 947–953 CrossRef.
- E. Matito, M. Solà, P. Salvador and M. Duran, Faraday Discuss., 2007, 135, 325–345 RSC.
- F. Feixas, E. Matito, J. Poater and M. Solà, Chem. Soc. Rev., 2015, 44, 6434–6451 RSC.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef.
- R. Milacic, S. Murko and J. Scancar, J. Inorg. Biochem., 2009, 103, 1504–1513 CrossRef PubMed.
- P. Rubini, A. Lakatos, D. Champmartin and T. Kiss, Coord. Chem. Rev., 2002, 228, 137–152 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C8DT01341A |
| This journal is © The Royal Society of Chemistry 2018 |