
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulating p-hydroxycinnamate behavior as a ditopic linker or photoacid in copper(II) complexes with an auxiliary pyridine ligand†
Joan
Soldevila-Sanmartín
 a,
Teresa
Calvet
b,
Merce
Font-Bardia
c,
Concepción
Domingo
d,
José A.
Ayllón
*a and
Josefina
Pons
*a
a,
Teresa
Calvet
b,
Merce
Font-Bardia
c,
Concepción
Domingo
d,
José A.
Ayllón
*a and
Josefina
Pons
*a
aDepartament de Química, Universitat Autònoma de Barcelona, 08193-Bellaterra, Barcelona, Spain. E-mail: JoseAntonio.Ayllon@uab.es; Josefina.Pons@uab.es
bMineralogia, Petrologia i Geologia Aplicada, Universitat de Barcelona, Martí i Franquès s/n, 08028-Barcelona, Spain
cUnitat de Difracció de Raig-X, Centres Científics i Tecnològics de la Universitat de Barcelona (CCiTUB), Universitat de Barcelona, Solé i Sabarís, 1-3, 08028-Barcelona, Spain
dInstituto de Ciencia de los Materiales de Barcelona (CSIC), Campus UAB, 08193 Bellaterra, Spain
First published on 10th April 2018
Abstract
The reaction of copper(II) acetate monohydrate with p-hydroxycinnamic acid (HpOHcinn) and different pyridine derivatives (4-tert-butylpyridine, 4-tBupy; 4-acetylpyridine, 4-Acpy; 3-phenylpyridine, 3-Phpy; 4-phenylpyridine, 4-Phpy) was essayed in methanol solvent at room temperature. The crystal structures of the resulting compounds were elucidated. Their analysis shows that the choice of pyridine ligands determines different coordination modes of the pOHcinn ligand and the Cu(II) coordination, nuclearity and geometry. The pOHcinn acts as a monodentate carboxylate ligand in combination with 4-tBupy or 4-Phpy, yielding monomers and dimers, associated by hydrogen bonds into supramolecular networks in which the phenol group plays a key role. Conversely, in combination with 4-Acpy or 3-Phpy, the phenol group coordinates directly to the Cu(II), acting as a ditopic ligand and yielding 2D coordination polymers. The compound containing 3-Phpy shows interesting MeOH–H2O reversible exchange behavior. Not only has the pyridine auxiliary ligand had a tremendous effect on the coordination mode of pOHcinn, but also its reactivity is influenced. Particularly, in the case of the compound containing 4-Phpy, it undergoes a photoinduced process, in which the phenol group deprotonates and coordinates to Cu(II) as a phenoxy ligand. This yields a coordination polymer in which two different dimers alternate, bridged by the resulting pOcinn ligand. The magneto-structural correlation of this compound is also discussed.
Introduction
The need to cope with the world's energetic demands while preserving our environment has spurred the search for new functional materials. In this regard, it is remarkable the great amount of interest shown in porous materials over the last decade. Thus, Metal–Organic Frameworks (MOFs) and Covalent Organic Frameworks (COFs) have been investigated extensively due to the possibility of tailoring their structure to meet specific demands,1,2 some of them being on the doorstep of industrial application.3 Recently, a new class of porous materials, based in supramolecular lattices stabilized only by weak intermolecular forces, specially hydrogen bonds, has been gaining attention.4–7 They could present several interesting properties that outperform those of MOFs, such as wet-processing, low energy regeneration and mild reaction conditions. Despite being held via weak supramolecular forces, correct selection of the number and position of the intermolecular interactions can yield highly stable materials.8–10 These materials could be built from pure organic subunits, and also from discrete coordination complexes. In this context, p-hydroxybenzoic acid (HpOHBz) is an interesting ligand, which includes a phenol functional group occupying an opposite position with respect to the carboxylic group. In complexes containing this ligand, the phenol group usually participates in the generation of supramolecular networks via H-bonds.11,12 However, it is less common that this –OH group coordinates with metal sites forming 2D coordination polymers.13Recently, our group has shown that, in the [Cu(pOHBz)2(dPy)2] (dPy = 4-Phpy, 4-Bzpy, 3-PhPy) family of compounds, small changes in the nature and position of the pyridine substituent govern the different roles of the phenol group of the pOHBz ligand: it can be involved in hydrogen-bond interactions that lead to the formation of supramolecular networks, or it can be bonded to the copper cation, yielding 2D coordination polymers. These previous results clearly suggest the potential of organic–inorganic building blocks with peripheral phenol groups for the preparation of both supramolecular and covalent networks.14 Some of these compounds show incipient porosity. Hence, in light of the obtained results it was suggested that using ligands with longer distances between the carboxylate and the phenol functionalities could result in an increase in the overall empty space, an already successful strategy.15 The interest into phenol containing ligands is also being fueled by the widespread occurrence of tyrosyl radicals in metalloproteins with diverse catalytic activity and the achievement of similar model compounds that show catalytic activity.16
In this work, we selected (E)-3-(4-hydroxyphenyl)-2-propenoic acid (para-hydroxycinnamic acid, HpOHcinn) for the formation of compounds with channels or other cavities, considering that it is quasi-planar and has a rigid structure. HpOHcinn coordination compounds using Co(II),17 Cd(II),18 Mn(II),19 Pb(II),20 Zn(II),15,21–24 and lanthanides25–27 as metal centers have been previously described. Focusing on Cu(II) coordination compounds, to our knowledge five crystal structures have been described in the literature.18,28–31 Furthermore, all these examples show the great variability of coordination modes allowed by this ligand, such as monodentate, bidentate chelate or bidentate bridge and several have channels occupied by solvent molecules. As a result, monomers, dimers, tetramers and coordination polymers can be synthesized. However, in none of the reported structures the –OH moiety coordinates to a metal center. Moreover, this work continues studying the effects of the different auxiliary pyridine ligands in the coordination of the phenol and carboxylate groups. For this purpose, in combination with HpOHcinn, four pyridines with different sizes and functional groups were chosen: 4-terbutylpyridine (4-tBupy), 4-acetylpyridine (4-Acpy), 3-phenylpyridine (3-Phpy) and 4-phenylpyridine (4-Phpy).
Experimental section
Materials and methods
Elemental analyses (EA) of C, H and N were carried out on a Euro Vector 3100 instrument. Fourier transformed infrared (FTIR) spectra were recorded on a Tensor 27 (Bruker) spectrometer, equipped with an attenuated total reflectance (ATR) accessory model MKII Golden Gate with a diamond window in the range 4000–600 cm−1. Powder X-ray diffraction (PXRD) patterns were measured with a Siemens D5000 apparatus using the CuKα radiation. Patterns were recorded from 2θ = 5 to 50°, with a step scan of 0.02° counting for 1 s at each step. Magnetic measurements from 5 to 300 K were carried out with a Quantum Design MPMS-5S SQUID susceptometer using a 100 Oe field.
Synthesis procedures
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/CN)ar, 1531m, 1511s, 1438w δ(CC/CN)ar, 1421w, 1387s νs(COO−), 1277s, 1220s δ(OH), 1167s, 1071m δ(C–H)ar, ip, 1033w, 983s, 876w, 828s δ(C–H)ar, oop, 758m δ(C–H)ar, oop, 684br.
O), 1635m, 1603s νas(COO−), 1534s ν(CC/CN)ar, 1508s, 1441w δ(CC/CN)ar, 1414s, 1385s νs(COO−), 1355s, 1290w, 1258s, 1213s, 1168s, 1104w, 1058m δ(C–H)ar, ip, 1025w, 841s, 817s δ(C–H)ar, oop, 724s δ(C–H)ar, oop.
C/CN)ar, 1529m, 1509s, 1478m, 1454m δ(CC/CN)ar, 1374s νs(COO−), 1288m, 1221s δ(OH), 1168s, 1104w, 1045m δ(C–H)ar, ip, 990m, 934w, 875w, 832s δ(C–H)ar, oop, 771s δ(C–H)ar, oop, 751m, 695s δ(C–H)ar, oop.
C/CN)ar, 1548m, 1509s, 1439m δ(CC/CN)ar, 1365br, νs(COO−), 1237s δ(OH), 1168s, 1072m δ(C–H)ar, ip, 982w, 831s δ(C–H)ar, oop, 761s, 728m δ(C–H)ar, oop, 689m δ(C–H)ar, oop.
C/CN)ar, 1542s νas(COO−), 1502s, 1447m δ(CC/CN)ar, 1417w, 1386s νs(COO−), 1361s νs(COO−), 1285s, 1235s δ(OH), 1168s, 1102w, 1071w δ(C–H)ar, ip, 1046w, 982m, 862s, 831s δ(C–H)ar, oop, 762s, 728s δ(C–H)ar, oop, 689s δ(C–H)ar, oop.
C/CN)ar, 1531m, 1511s, 1438w δ(CC/CN)ar, 1421w, 1387s νs(COO−), 1277s, 1220s δ(OH), 1167s, 1071m δ(C–H)ar, ip, 1033w, 983s, 876w, 828s δ(C–H)ar, oop, 758m δ(C–H)ar, oop, 684br.
O), 1635m, 1603s νas(COO−), 1534s ν(CC/CN)ar, 1508s, 1441w δ(CC/CN)ar, 1414s, 1385s νs(COO−), 1355s, 1290w, 1258s, 1213s, 1168s, 1104w, 1058m δ(C–H)ar, ip, 1025w, 841s, 817s δ(C–H)ar, oop, 724s δ(C–H)ar, oop.
C/CN)ar, 1529m, 1509s, 1478m, 1454m δ(CC/CN)ar, 1374s νs(COO−), 1288m, 1221s δ(OH), 1168s, 1104w, 1045m δ(C–H)ar, ip, 990m, 934w, 875w, 832s δ(C–H)ar, oop, 771s δ(C–H)ar, oop, 751m, 695s δ(C–H)ar, oop.
C/CN)ar, 1548m, 1509s, 1439m δ(CC/CN)ar, 1365br, νs(COO−), 1237s δ(OH), 1168s, 1072m δ(C–H)ar, ip, 982w, 831s δ(C–H)ar, oop, 761s, 728m δ(C–H)ar, oop, 689m δ(C–H)ar, oop.
C/CN)ar, 1542s νas(COO−), 1502s, 1447m δ(CC/CN)ar, 1417w, 1386s νs(COO−), 1361s νs(COO−), 1285s, 1235s δ(OH), 1168s, 1102w, 1071w δ(C–H)ar, ip, 1046w, 982m, 862s, 831s δ(C–H)ar, oop, 762s, 728s δ(C–H)ar, oop, 689s δ(C–H)ar, oop.
The variation of the magnetization with temperature of 0.1052 g of 5 (0.085 mmol) in a 100 Oe field was measured. The calculated diamagnetic contribution of this compound was found to be 7.02 × 10−4 cm3 mol−1 using Pascal's constants.32
X-ray crystal structures
The X-ray intensity data were measured on a D8 Venture system equipped with a multilayer mono-chromate and a Mo microfocus (λ = 0.71073 Å). For compounds 1–3, a blue prism-like specimen was used for the X-ray crystallographic analysis. For compound 4, a blue needle specimen was used for the X-ray crystallographic analysis. For compound 5, a translucent blue-green specimen was used for the X-ray crystallographic analysis.Frames were integrated with the Bruker SAINT Software package using a narrow-frame algorithm. The structures were solved using the Bruker SHELXTL Software, packaged and refined using SHELXL (version-2018/3).33 Data were corrected for absorption effects using the multi-scan method (SADABS, version 2008/1). Crystal data and additional details of structure refinement for compounds 1–5 are included in the ESI (Tables S1 and S2†). Molecular graphics were generated with the program Mercury 3.9.34,35 Color codes for all molecular graphics are: orange (Cu), blue (N), red (O), grey (C), white (H).
The crystal quality of compound 4 was poor but an X-ray diffraction analysis was carried out, which clearly revealed the structure of the complex.
Results and discussion
Synthesis and general characterization
The reaction of copper acetate monohydrate (Cu(Ac)2·H2O) with (E)-3-(4-hydroxyphenyl)-2-propenoic acid (p-hydroxycinnamic acid, HpOHcinn) and different pyridine derivatives (dPy = 4-tBupy, 4-Acpy, 3-Phpy, 4-Phpy) was essayed using MeOH as solvent with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:4 for Cu:HpOHcinn:dPy, and at room temperature and atmospheric pressure. Pyridine derivatives were added in excess to neutralize the MeCOOH byproduct. The tested reactions yielded four different compounds: [Cu(μ-pOHcinn)2(4-tBupy)2(H2O)][Cu(μ-pOHcinn)2(4-tBupy)2(H2O)2] (1), [Cu(μ-pOHcinn)2(4-Acpy)2]n (2), {[Cu(μ-pOHcinn)2(3-Phpy)2]n·2MeOH}n (3) and [Cu(pOHcinn)2(4-Phpy)2]2[Cu(pOHcinn)2(4-Phpy)2]·1.5MeOH·2H2O (4) (Scheme 1). When a suspension of 4 in its mother liquor is exposed to sunlight, compound [Cu2(pOHcinn)2(trans-4-Phpy)4(μ-p-Ocinn)2Cu2(cis-4-Phpy)4]n (5) is obtained (Scheme 2), in which the phenol groups of half of the initial pOHCinn are deprotonated. Photoinduced intermolecular excited-state proton transfer (ESPT) reactions are ubiquitous in chemistry and biology.36,37 Ligands that contain a proton donating group are also prone to be involved in these processes, since an additional excited state perturbation can occur where the electron density shift, associated with light absorption, significantly influences the acidity of the ligand.16,38 Phenols are expected to be more acidic in the excited state than in the ground state.39,40
:2:4 for Cu:HpOHcinn:dPy, and at room temperature and atmospheric pressure. Pyridine derivatives were added in excess to neutralize the MeCOOH byproduct. The tested reactions yielded four different compounds: [Cu(μ-pOHcinn)2(4-tBupy)2(H2O)][Cu(μ-pOHcinn)2(4-tBupy)2(H2O)2] (1), [Cu(μ-pOHcinn)2(4-Acpy)2]n (2), {[Cu(μ-pOHcinn)2(3-Phpy)2]n·2MeOH}n (3) and [Cu(pOHcinn)2(4-Phpy)2]2[Cu(pOHcinn)2(4-Phpy)2]·1.5MeOH·2H2O (4) (Scheme 1). When a suspension of 4 in its mother liquor is exposed to sunlight, compound [Cu2(pOHcinn)2(trans-4-Phpy)4(μ-p-Ocinn)2Cu2(cis-4-Phpy)4]n (5) is obtained (Scheme 2), in which the phenol groups of half of the initial pOHCinn are deprotonated. Photoinduced intermolecular excited-state proton transfer (ESPT) reactions are ubiquitous in chemistry and biology.36,37 Ligands that contain a proton donating group are also prone to be involved in these processes, since an additional excited state perturbation can occur where the electron density shift, associated with light absorption, significantly influences the acidity of the ligand.16,38 Phenols are expected to be more acidic in the excited state than in the ground state.39,40
 | ||
| Scheme 1 Reactions carried out in this work. Isolated and characterized products are shown with their numbering scheme. | ||
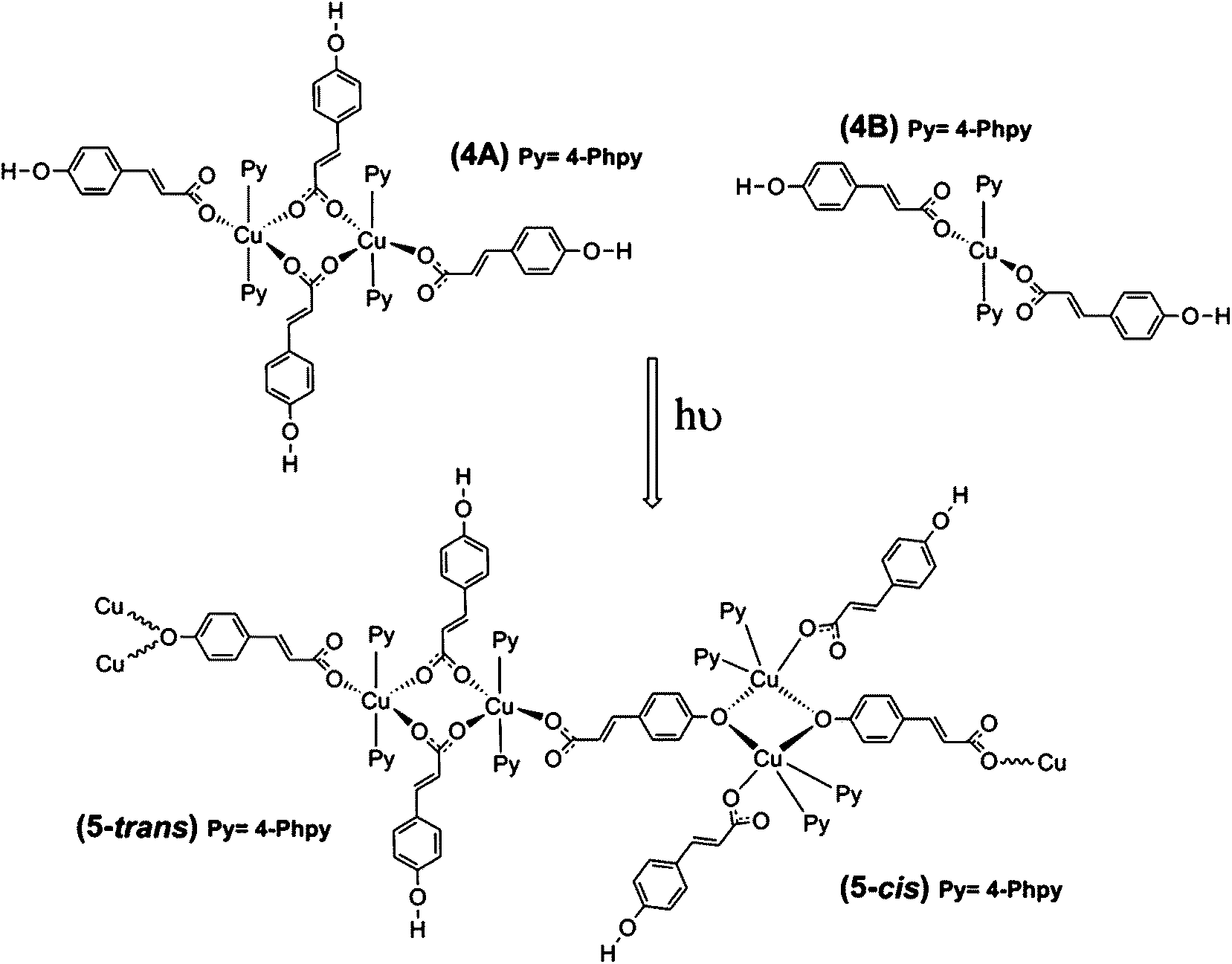 | ||
| Scheme 2 Transformation of compound 4 into compound 5via sunlight radiation. | ||
Products 1–5 were characterized via PXRD, Elemental Analyses (EA), ATR-FTIR spectroscopy and single crystal X-ray diffraction. Phase purity of the bulk samples for compounds 1, 2, 4 and 5 was confirmed via PXRD (ESI: Fig. S1–S4†). Compound 3, however, suffers a structural change when exposed to an open atmosphere; therefore, its PXRD patterns only match the one calculated from the solved structure when the sample is maintained under methanol vapors (see below). Again, EA data were also consistent with the elucidated single crystal X-ray structures of studied compounds, except for 3. ATR-FTIR spectra of the five compounds confirm the presence of the organic ligands used in the synthesis, including bands assignable to dPy and pOHcinn anion (ESI: Fig. S5–S9†). The region between 3500 and 3000 cm−1 shows several broad bands due the presence of ν(OH) and ν(CH)ar bands, hindering the possibility of extracting information on the role of the phenol group. However, in 3 (ESI: Fig. S10†) the disappearance of the band at 3396 cm−1 and the formation of a band at 3570 cm−1 were observed, which is consistent with the substitution of MeOH for H2O molecules in this compound. Bands assignable to carboxylate groups provide key information about their coordination mode.41,42 The difference measured between νas(COO−) and νs(COO−) is in the range of Δ = 218 cm−1–233 cm−1 for 1–5, indicating a monodentate coordination mode.41 In 4 and 5, other bands attributable to νas(COO−) and νs(COO−) appear, possibly indicating other coordination modes.
Molecular and extended structure
It was possible to obtain single crystals for all reported compounds. In most of the cases, they were obtained from the initial reaction mixture after partial solvent evaporation (1–3), or by cooling the solution in the fridge (4). Single crystals of 5 were obtained from a suspension of 4 in methanol under sunlight. All of them were of good quality, allowing the elucidation of the X-ray crystal structure. The obtained crystal structures of 1–5 show great flexibility in the coordination mode of the pOHcinn ligand (Scheme 3), which leads to the formation of monomers, dimers and polymers with different coordination environments and geometries.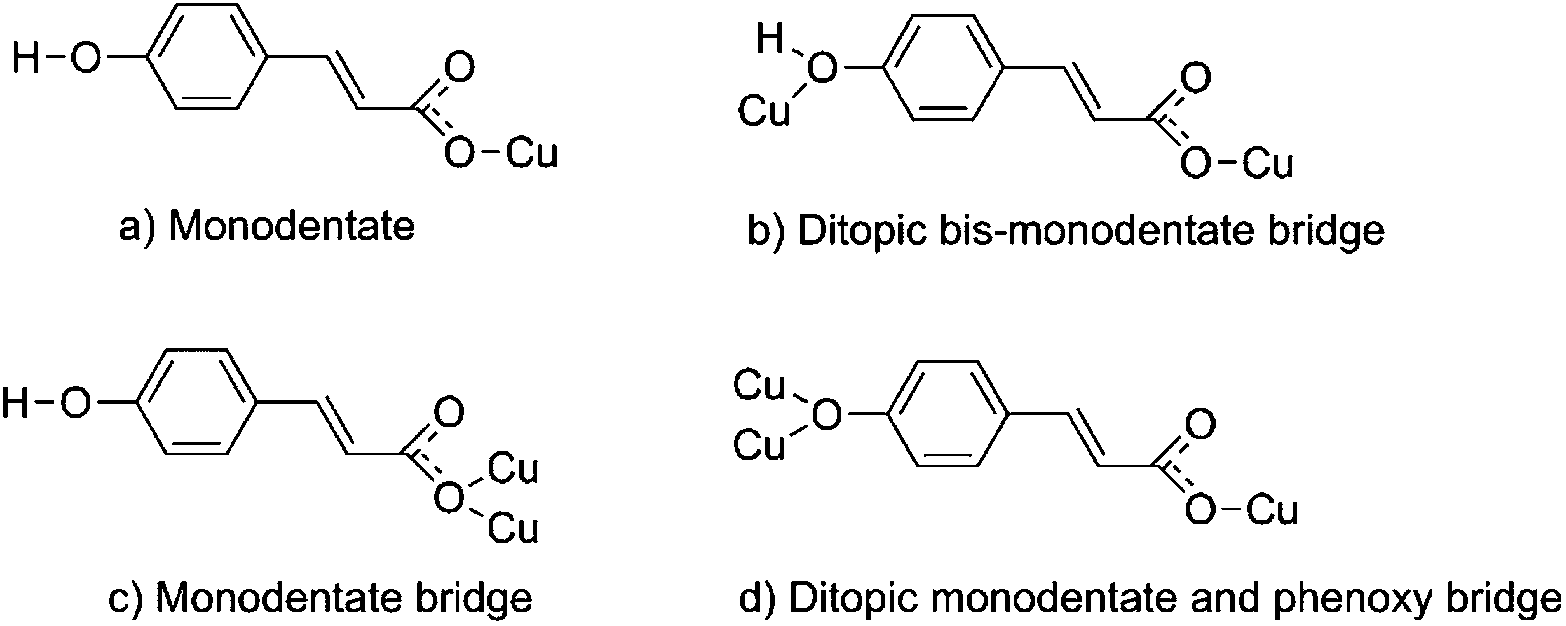 | ||
| Scheme 3 Coordination modes of the p-hydroxycinnamate ligand found in the presented structures. Note that the phenol group in coordination mode (d) is deprotonated. | ||
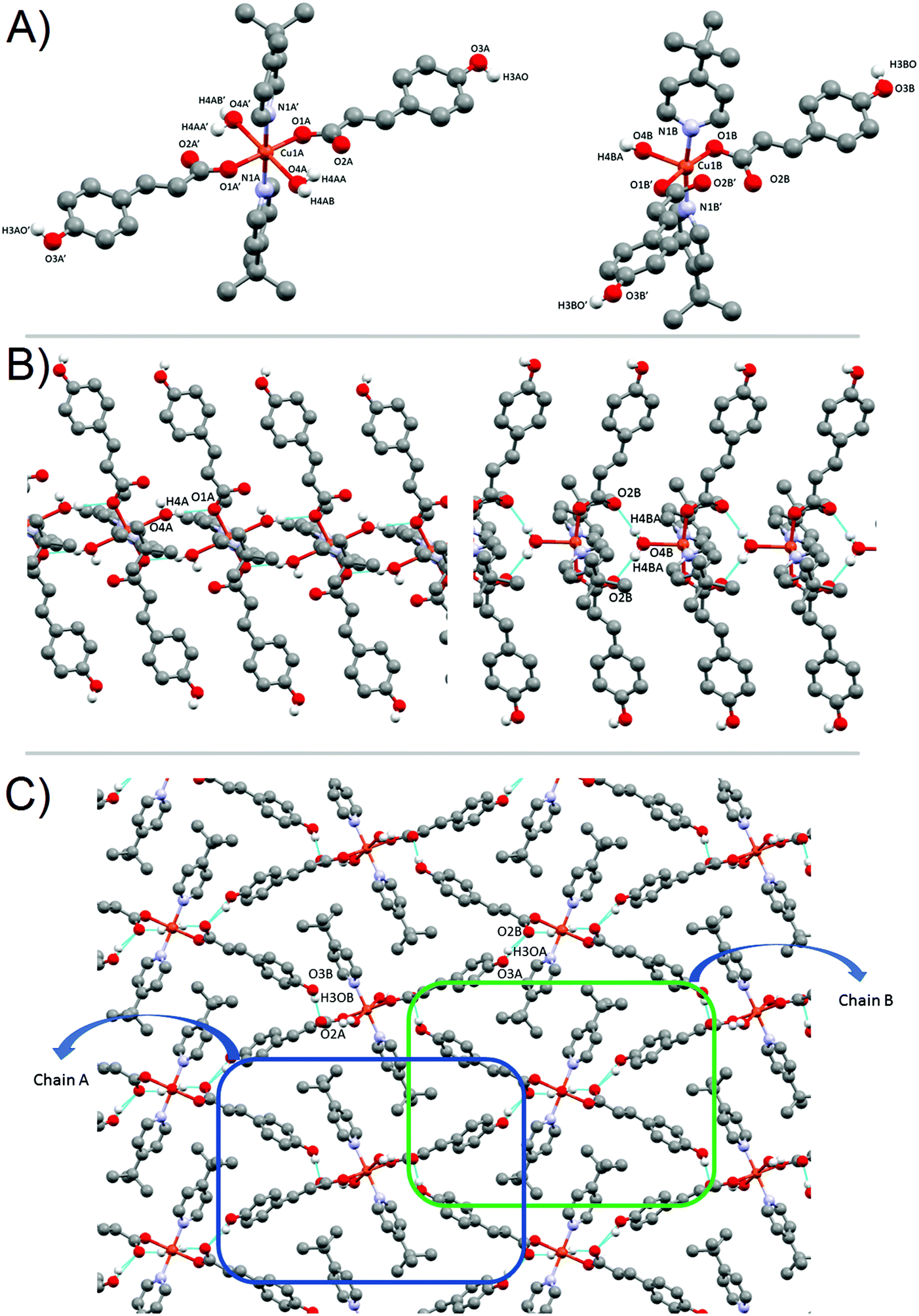 | ||
| Fig. 1 (A) Monomers 1A (left) and 1B (right), and their corresponding numbering scheme for relevant atoms. Only hydrogen atoms participating in H-bonds are shown. (b) View of the H-bond interaction in each individual chain (A left, B right). View along the c axis. (c) 3D stacking of monomers 1A and 1B. View along the b axis. | ||
For 1A, the basal plane is defined by two trans-coordinate 4-tBupy (Cu1A–N1B 2.019(3) Å) and two trans-coordinate monodentate pOHcinn moieties (Cu1A–O1A 1.982(3) Å). The apical positions are occupied by two weakly coordinating H2O molecules (Cu1A–O4A 2.440(3) Å). Bond angles are in the range of 85.12(9)–94.88(9)°. For 1B, 4-tBupy and pOHcinn ligands form the basal plane (Cu1B–N1B 2.005(3) Å, Cu1B–O1B 1.968(2) Å) and the H2O ligand lies in the apical position (Cu1B–O4B 2.208(4) Å). The bond angles lie in the range of 87.83(12)–94.82(9)° and in the range of 170.36(17)°–171.74(15)°. All these distances and angles are in good agreement with related Cu(II) carboxylate-pyridine compounds.14,44,45 Relevant distances and angles are summarized in Table 1.
| Molecule 1A | |||
| Bond lengths (Å) | |||
| Cu1A–O1A | 1.982(3) | Cu1A–O4A | 2.440(3) |
| Cu1A–N1A | 2.019(3) | ||
| Bond ngles (°) | |||
| O1A–Cu1A–O1A#1 | 180 | N1A–Cu1–O4A#1 | 87.35(11) |
| O1A–Cu1A–N1A#1 | 89.72(12) | O1A–Cu1A–O4A | 85.12(9) |
| O1A–Cu1A–N1A | 90.28(12) | N1A–Cu1A–O4A | 92.65(11) |
| N1A#1–Cu1A–N1A | 180 | O4A#1–Cu1A–O4A | 180 |
| O1A–Cu1A–O4A#1 | 94.88(9) | ||
| #1 −x + 1/2, −y + 3/2, −z + 1 | |||
| Molecule 1B | |||
| Bond lengths (Å) | |||
| Cu1B–O1B | 1.968(2) | Cu1B–O4B | 2.208(4) |
| Cu1B–N1B | 2.005(3) | ||
| Bond angles (°) | |||
| O1B–Cu1B–O1B#2 | 171.74(15) | N1B#2–Cu1B–N1B | 170.36(17) |
| O1B–Cu1B–N1B#2 | 91.48(12) | O1B–Cu1B–O4B | 94.13(8) |
| O1B–Cu1B–N1B | 87.83(12) | N1B–Cu1B–O4B | 94.82(8) |
| #2 −x + 1, y, −z + 1/2 | |||
The presence of two different monomeric units results in a supramolecular structure dominated by two distinct motifs: two different supramolecular 1-D chains along the b axis. Each chain is formed exclusively by the linkage via H-bonds of similar monomeric units; that is, a chain containing only 1A subunits and a different chain containing only 1B subunits. Similar motifs have been reported before in our group and in the literature.44,46 Each monomer 1A has a symmetric quadruple H-bond, involving the coordinated O1A from the pOHcinn ligand and H4AB of the coordinated H2O molecule (Fig. 1B). Monomer 1B, on the other hand, forms an asymmetric double H-bond. In this monomer, the involved O2B is the uncoordinated oxygen from the pOHcinn ligand and H4BA of the coordinated H2O molecule (Fig. 1B). Other interesting distances are between intrachain and interchain Cu(II), which are Cu1A⋯Cu1A 5.999 Å, Cu1B⋯Cu1B 5.999 Å and Cu1A⋯Cu1B 14.137 Å. Compared to similar 1D chain like structures, the interchain Cu⋯Cu distance is shorter when the carboxylate group belongs to acetate (around 8.2 Å)44,46 or cinnamate (13.091 Å).45 These chains are held together due to the role of the pOHcinn ligand. Its uncoordinated oxygen atoms of the carboxylate groups (O2A and O2B) form H-bonds with the –OH groups (H3AO and H3BO) of the different monomer. Thus, each monomer 1A is linked to four monomers 1Bvia the interaction of O2A with H3BO and each monomer 1B is linked to four monomers 1Avia the interaction of O2B with H3AO (Fig. 1C). It is noteworthy that O2B also participates in the H-bonding of the proper B chain, thus playing a central structural role. Relevant H-bond interactions are summarized in Table 2.
| D–H⋯A | D–H | H–D⋯A | D–H⋯A | |
|---|---|---|---|---|
| i [1/2 − x, 1/2 − y, 1 − z], ii [1 − x, −1 + y,1/2 − z], iii [x, 1 − y, 1/2 + z]. | ||||
| 1 | ||||
| O4A–H4AB⋯O1Ai | 2.08(1) | 0.837(15) | 2.917(3) | 178(5) |
| O4B–H4BA⋯O2Bii | 1.86(2) | 0.84(2) | 2.867(4) | 166(3) |
| O3B–H3OB⋯O2A | 1.90(4) | 0.84(4) | 2.715(4) | 163(4) |
| O3A–H3OA⋯O2Biii | 1.92 | 0.84 | 2.716(3) | 158 |
 | ||
| Fig. 2 (A) Polymers 2 (top) and 3 (down): their corresponding numbering scheme for relevant atoms. Only phenolic hydrogens are shown. (B) Polymeric 2D layers of compounds 2 (left) and 3 (right). | ||
Their basal plane is defined by two of the oxygen atoms provided by the carboxylate groups of two different pOHcinn ligands coordinating in a monodentate fashion (Cu–O1 1.9280(9) Å for 2 and 1.9640(12) Å for 3) and two nitrogen atoms corresponding to two pyridine ligands (4-Acpy) (2) or 3-Phpy (3) (Cu–N1 2.0521(11) Å for 2 and 1.9975(14) Å for 3). The apical positions are occupied by two phenolic groups from two different pOHcinn ligands (Cu–O3 2.5951(12) Å in 2 and 2.4925(13) in 3). Bond angles are in the range of 82.88(4)°–91.73(4)° for 2 and 89.07(5)–95.05(6) for 3. Relevant distances and angles for 2 and 3 are summarized in Table 3. The four pOHcinn anions bridge the four closer copper atoms in the (101) plane (2) or (½ 0 ½) (3) acting as a ditopic bis-monodentante bridging ligand (Scheme 3) via one oxygen from the carboxylate group and one oxygen of the phenol functionality, both from pOHcinn, forming 2D polymeric layers. Therefore, each POHcinn ligand is shared by two metal centers. This configuration has also been reported in the [Cu(pOHBz)2(4-Bzpy)2]n polymer with similar distances, except the Cu–Ophenol distance (2.7449(19) Å), which is longer than that in either 2 and 3.14 For 2 and 3, the uncoordinated oxygen O2 forms an intramolecular H-bond H3A, which is the hydrogen of the phenol group of an adjacent POHcinn ligand (Fig. 2A and B). This hydrogen bond defines a sort of 6-member ring [Cu–O1–C1–O2⋯H3–O3] reinforcing the layered structure. In 3, O2 also forms a second weaker hydrogen bond with a MeOH molecule.
| 2 | |||
| Bond lengths (Å) | |||
| Cu1–O1 | 1.9280(9) | Cu1–O3#1 | 2.5951(12) |
| Cu1–N1 | 2.0521(11) | ||
| Bond angles (°) | |||
| O1–Cu1–O1#1 | 180 | N1–Cu1–O3#1 | 82.88(4) |
| O1–Cu1–N1#1 | 90.58(4) | N1–Cu1–O3#2 | 97.13(4) |
| O1–Cu1–N1 | 89.42(4) | O1–Cu1–O3#1 | 91.73(4) |
| N1#1–Cu1–N1 | 180 | O1–Cu1–O3#2 | 88.27(4) |
| O3#1–Cu1–O3 | 180 | ||
| #1 1/2 + x, 1/2 − y, −1/2 + z #2 1/2 − x, 1/2 + y, 3/2 − z | |||
| 3 | |||
| Bond lengths (Å) | |||
| Cu1–O1 | 1.9640(12) | Cu1–N1 | 1.9975(14) |
| Cu1–O1#1 | 1.9640(12) | Cu1–O3#2 | 2.4925(13) |
| Bond angles (°) | |||
| O1–Cu1–O1#1 | 180 | N1–Cu1–O3#3 | 84.95(5) |
| O1–Cu1–N1#1 | 90.93(5) | N1–Cu1–O3#2 | 95.05(6) |
| O1–Cu1–N1 | 89.07(5) | O1–Cu1–O3#2 | 86.51(6) |
| N1#1–Cu1–N1 | 180 | O1–Cu1–O3#3 | 93.50(5) |
| O3#1–Cu1–O3 | 180 | ||
| #1 −x, −y + 1, −z + 1 #2 1/2 − x, 1/2 + y, 1/2 − z #3 −1/2 + x, 1/2 − y, 1/2 + z | |||
For 2 and 3 compounds, each monomer is linked to four other monomers, defining 2D-layers (Fig. 2B). The Cu(O1)2(N1)2 plane tilts 67.51° in 2 and 87.25° in 3 with respect to each other and in alternate fashion for each bonded monomer, while the carboxylate groups (O1–C1–O2) from the pOHcinn are almost coplanar with their benzene rings (8.13° in 2 and 19.66° in 3). This arrangement results in pOHcinn bridges being situated closer to the mean plane defined by the Cu(II) ions, while 4-Acpy or 3-Pypy ligands stand out, enabling them to interleave with pyridine derived ligands of the neighboring 2D layers (Fig. 3A). This supramolecular arrangement is similar to the one found in [Cu(μ-pOHBz)2(4-Phpy)2] and [Cu(μ-pOHBz)2(4-Bzpy)2]n.14 This allows the formation of H-bonds that hold the layers together in a 3D supramolecular structure. In 2, the carbonyl group of 4-Acpy is responsible for these interaction, forming an H-bond with the hydrogen on the para position of the neighboring 4-Acpy ligand. In 3, a different supramolecular force is holding the layers together; an H-π bonding between H12 of the 3-Phpy and the benzene ring of the pOHcinn ligand (Fig. 3B). Relevant H-bond interactions are summarized in Table 4.
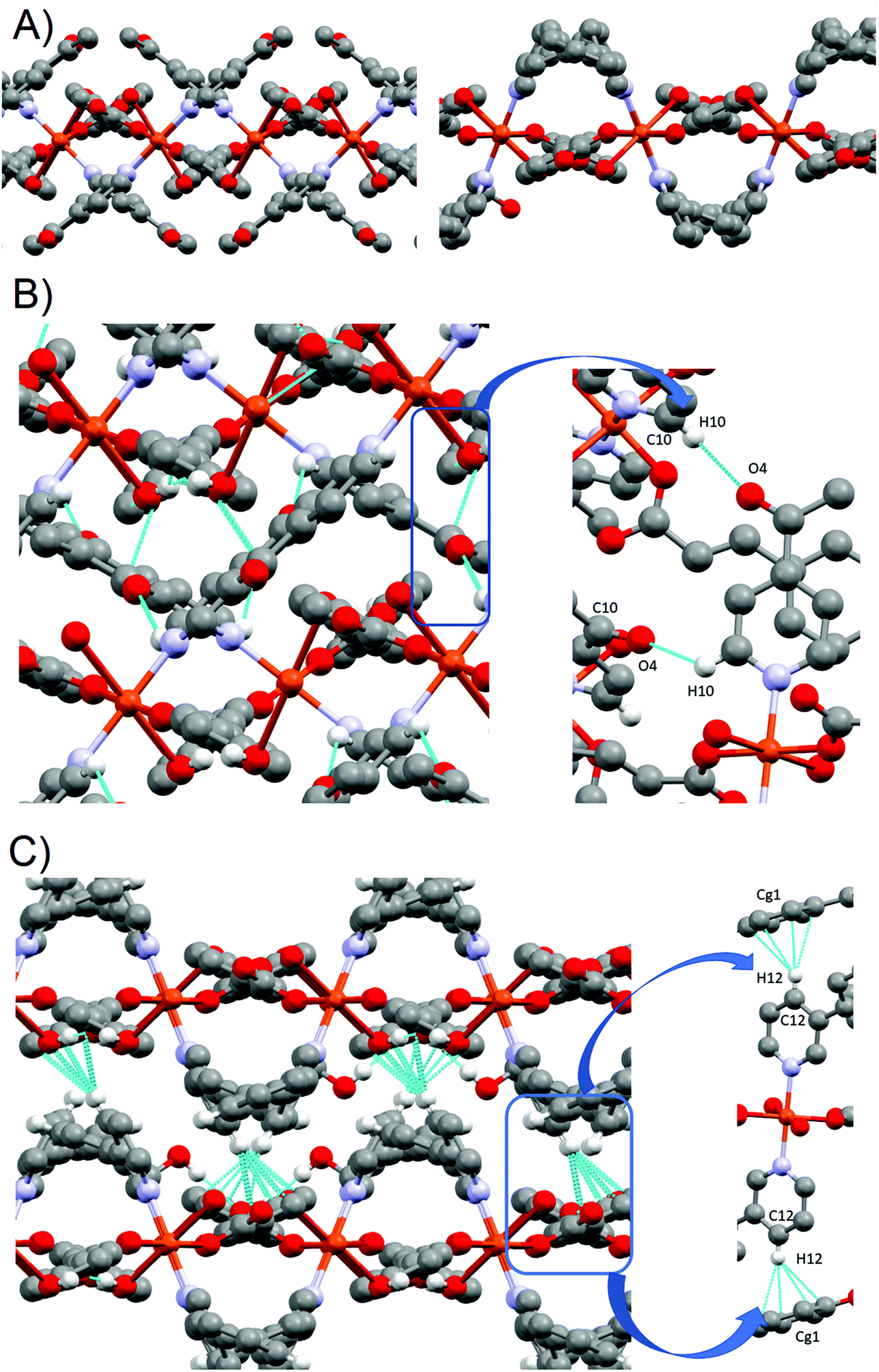 | ||
| Fig. 3 (A) Perpendicular cut of the 2D layers for 2 (left) and 3 (right). (B) H-bond interactions forming the supramolecular structure of 2 (top-left). Details of the main H-bonding holding the layer together (top-right). H-bond interactions forming the supramolecular structure of 3 (bottom-left). Details of the main H-bonding holding the layer together (bottom-right). | ||
| D–H⋯A | D–H | H–D⋯A | D–H⋯A | |
|---|---|---|---|---|
| Cg1 = C4–C5–C6–C7–C8–C9, i [1/2 + x, 1/2 − y, −1/2 + z]. | ||||
| 2 | ||||
| O3–H3A⋯O2 | 1.806(15) | 0.797(16) | 2.5637(15) | 158.3(15) |
| C10–H10⋯O4 | 2.476 | 0.950 | 3.0941(15) | 123 |
| 3 | ||||
| O3–H3A⋯O2i | 1.68(2) | 0.89(2) | 2.542(2) | 163(2) |
| O4–H4O⋯O2 | 1.96(3) | 0.83(3) | 2.771(2) | 165(2) |
| C12–H12⋯Cg1 | 2.467 | 0.950 | 3.355 | 155.65 |
Compound 2 shows a compact packing, facilitated by the orientation of the 4-Acpy ligand with respect to the 2D network. On the contrary, in 3, the larger size of the 3-Phpy ligand results in the phenyl substituents being orientated outwards, leaving tridimensional elongated cavities filled with MeOH molecules. These cavities are connected through choke points defining zig-zag channels (Fig. 4A). These channels occupy a total volume of approximately 10.6% (197.99 Å3, calculated using a probe radius of 1.2 Å).47 This different behavior can be related to the shape of the auxiliary pyridine ligands preventing close packing of the layers, as 4-Acpy results in an interlayer Cu⋯Cu distance of 8.434 Å (2), whereas for 3-Phpy this distance is 15.201 Å (3). Furthermore, the free space in 3 is much larger than the reported 4.2% for the [Cu(μ-pOHBz)2(4-Bzpy)2]n polymer.14 However, this result shows that not only a longer spacer between the phenol and the carboxylate functional groups is required for increasing the free space, but also functional groups that prevent the stacking of the layers from obstructing the cavities.
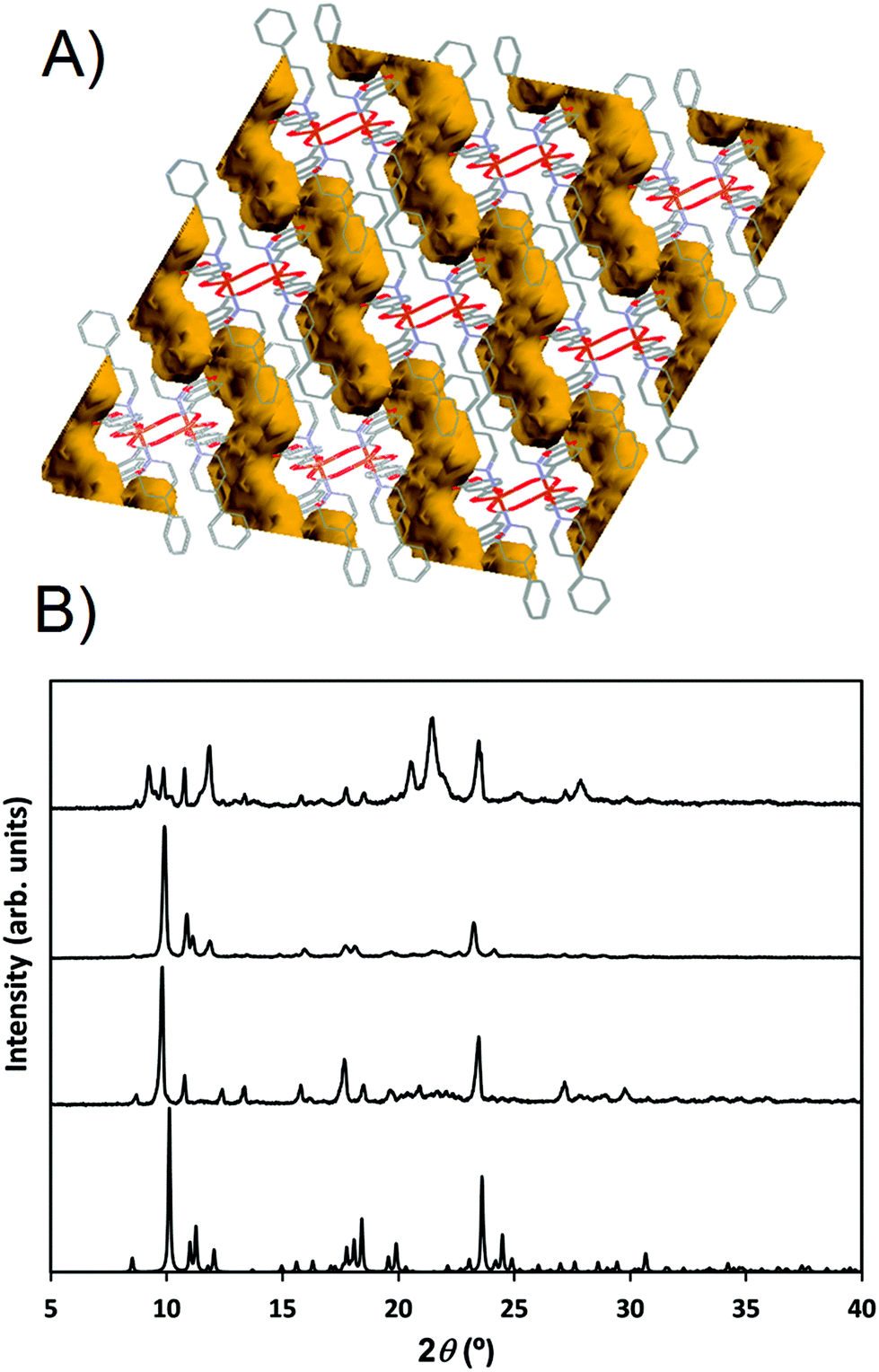 | ||
| Fig. 4 (A) Schematic representation of the channels occupied by the solvent molecules in 3. (b) PXRD pattern from 3 simulated from the resolved structure (single crystal XRD measured at 100 K, bottom). PXRD diffractogram of 3 measured at room temperature after exposure to air (middle-bottom). PXRD diffractogram of 3 measured at room temperature after exposure to a saturated MeOH atmosphere (middle-top). PXRD of the remaining powder after vacuum exposure of 3 (top). | ||
When exposed to air, the compound changes from dark blue crystals to pale blue powder. Furthermore, XRD analyses reveal that this process is accompanied by a structural change (Fig. 4B). FTIR-ATR spectra also confirm this structural change (ESI: Fig. S10†). EA of the pale blue powder suggests that MeOH molecules are substituted by two H2O molecules. Therefore, a MeOH–H2O exchange process occurs, in spite of the choke point, evidencing some structure flexibility. PXRD analysis shows that the process can be reversed by exposing the powdered sample to a MeOH saturated atmosphere, exchanging H2O molecules for MeOH molecules. The XRD pattern of the sample in contact with methanol vapors matches the simulated from the crystal structure data (Fig. 4). Therefore, it is clear that compound 3 is a 2D-MOF, which interchange water and methanol molecules in a reversible manner. However, trials to measure the specific surface area by studying N2 adsorption–desorption failed due to the collapse of the structure during the degasification step. EA performed after vacuum exposure confirms that all solvent molecules have been removed. This suggests that solvent molecules are necessary to stabilize the porous supramolecular net, and that full removal of the solvents causes irreversible collapse of the channels. PXRD performed after vacuum exposure denotes that a different structure is formed (Fig. 4B).
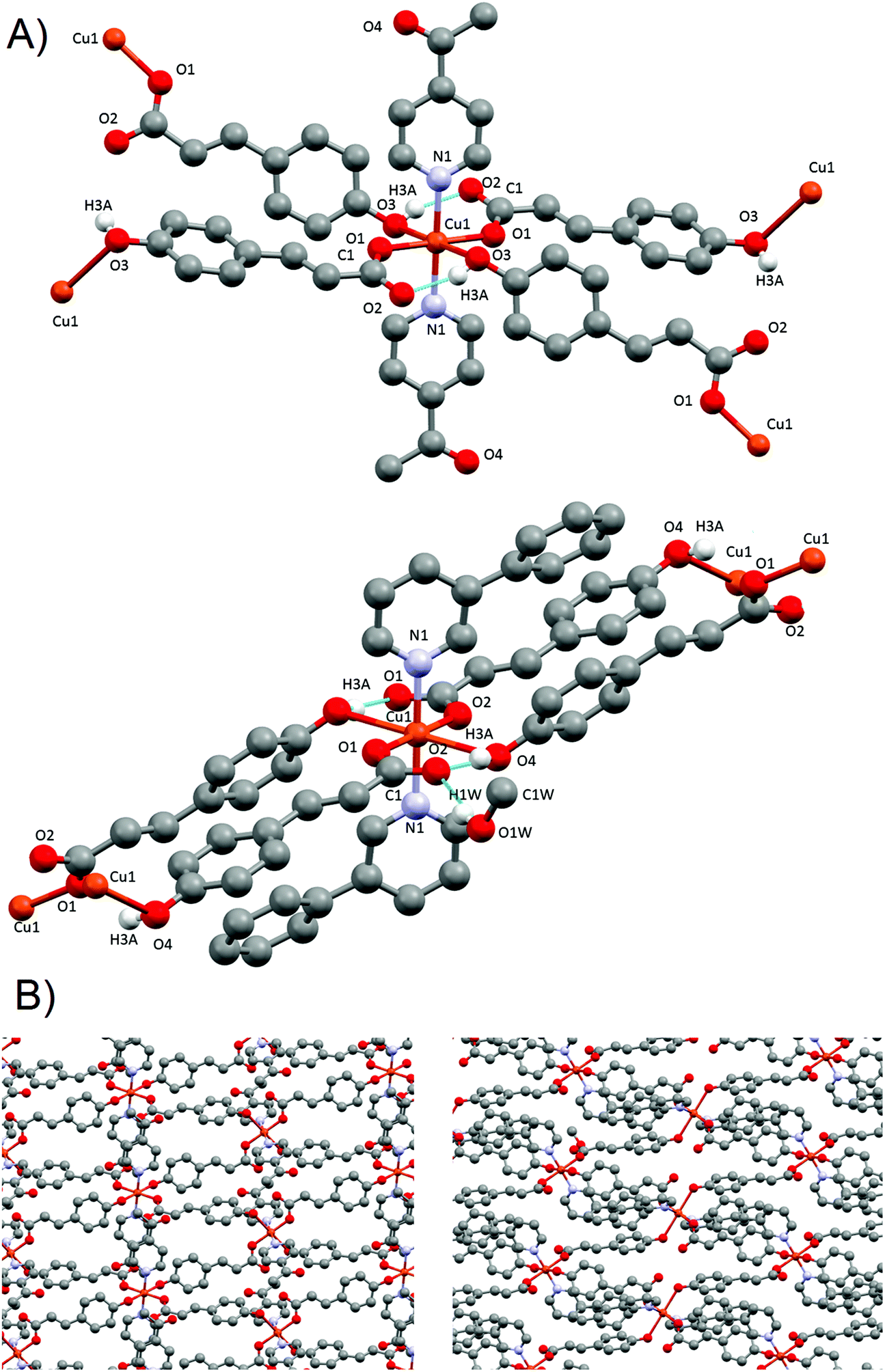
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group and has a complex structure containing two crystallographic independent units, a dimer [Cu(pOHcinn)2(4-Phpy)2]2 (4A) and a monomer [Cu(pOHcinn)2(4-Phpy)2] (4B) and included solvent molecules (Fig. 5A).
space group and has a complex structure containing two crystallographic independent units, a dimer [Cu(pOHcinn)2(4-Phpy)2]2 (4A) and a monomer [Cu(pOHcinn)2(4-Phpy)2] (4B) and included solvent molecules (Fig. 5A).
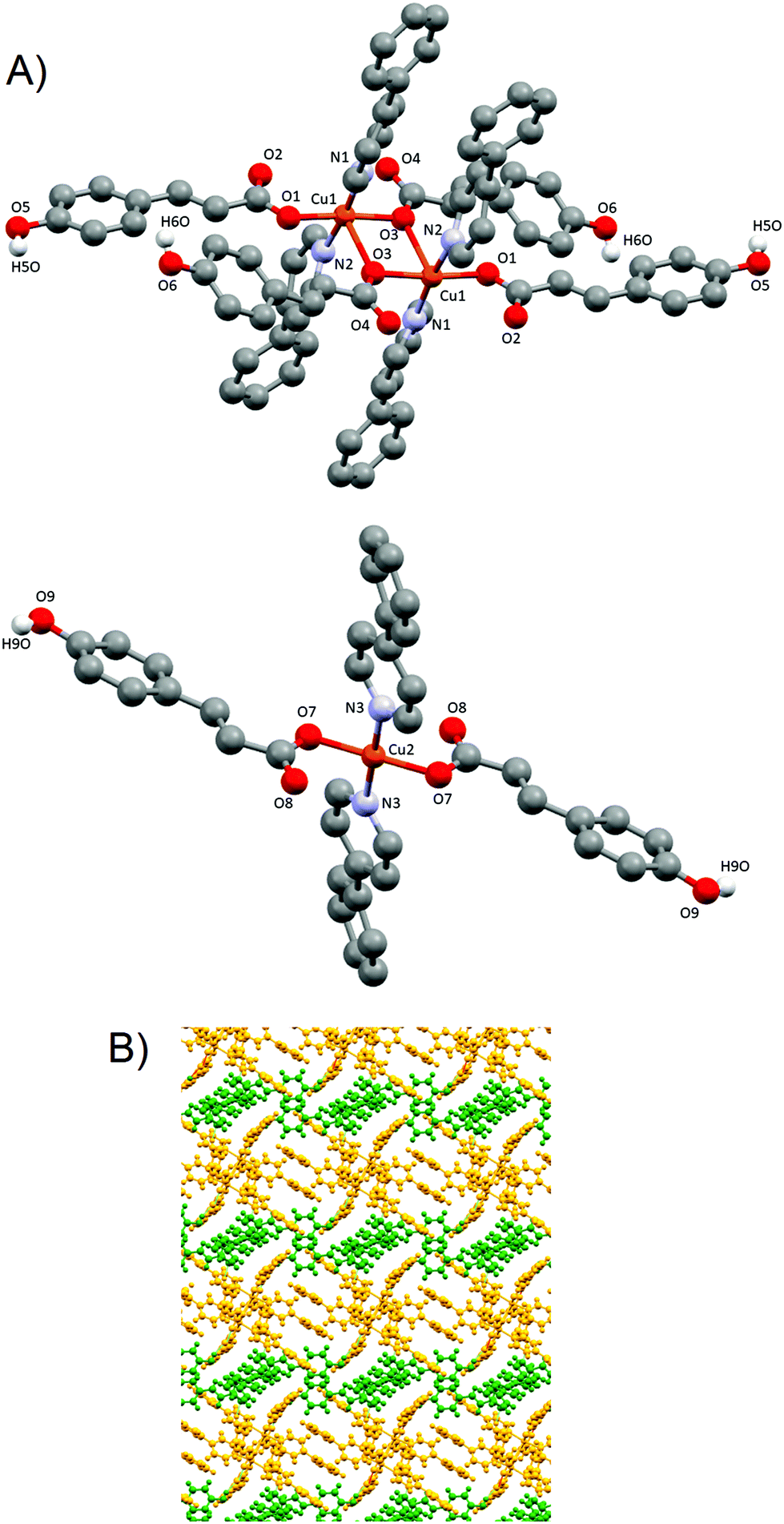 | ||
| Fig. 5 (A) Dimer 4A (top) and monomer 4B (down), and their corresponding numbering scheme for relevant atoms. Only phenolic hydrogens are shown. (B) Perpendicular view of the 2D layer formed by dimer 4A (yellow) and monomer 4B (green). | ||
Dimer 4A shows Cu(II) cations with a slightly distorted square pyramidal coordination geometry (τ = 0.028)43 and a [CuO3N2] core with the three oxygen atoms provided by three carboxylate groups from different pOHcinn ligands and two nitrogen atoms provided by two different 4-Phpy ligands. The basal plane is defined by one pOHcinn acting as a monodentate ligand (Cu1–O1 1.937(4) Å), another pOHcinn acting as a ditopic bis-monodentate bridged ligand (Cu–O3 1.967(3) Å) and the two nitrogen atoms of the 4-Phpy ligands (Cu1–N1 2.016(4) Å, Cu1–N2 2.002(4) Å). The apical position is occupied by one oxygen of a pOHcinn acting as a ditopic bis-monodentate bridging ligand (Cu1–O3#1 2.378(3) Å) (Scheme 3). The Cu⋯Cu separation is 3.417 Å. The bond angles are in the range of 76.70(13)°–94.87(15)° and 171.17(17)°–172.88(16)°. The smallest bond angle of 76.70(13)° is due to the rigid nature of the Cu1–O3–Cu1–O3 ring. The morphology of this compound is notably similar to the closely related [Cu(cinn)2(β-pic)2] compound (cinn = cinnamate, β-pic = β-picoline) described in the literature.34 The bond lengths and angles are in the range of similar compounds.45,48–50
Monomer 4B, on the other hand, presents a square planar geometry with a [CuO2N2] core, with two monodentate pOHcinn and two 4-Phpy ligands in trans positions (Cu2–O7 1.934(4) Å, Cu2–N3 2.004(4)). The bond angles are in the range of 89.52(17)°–90.48(17)°. Relevant distances and angles are provided in Table 5.
| Molecule 4A | |||
| Bond lengths (Å) | |||
| Cu1–O1 | 1.937(4) | Cu1–N2 | 2.002(4) |
| Cu1–O3 | 1.967(3) | Cu1–N1 | 2.016(4) |
| Cu1–O3#1 | 2.378(3) | Cu1⋯Cu1 | 3.417 |
| Bond angles (°) | |||
| O1–Cu1–O3 | 172.88(16) | O1–Cu1–O3#1 | 96.19(15) |
| O1–Cu1–N2 | 89.88(18) | O3–Cu1–O3#1 | 76.70(13) |
| O3–Cu1–N2 | 90.25(16) | N1–Cu1–O3#1 | 94.87(15) |
| O1–Cu1–N1 | 89.30(18) | N2–Cu1–O3#1 | 93.96(15) |
| O3–Cu1–N1 | 91.64(16) | N1–Cu1–N2 | 171.17(17) |
| #1 −x + 1, −y + 1, −z + 1 | |||
| Molecule 4B | |||
| Bond lengths (Å) | |||
| Cu2–O7 | 1.934(4) | Cu2–N3 | 2.004(4) |
| Cu2–O7#2 | 1.934(4) | Cu2–N3#2 | 2.004(4) |
| Bond angles (°) | |||
| O7–Cu2–O7#2 | 180.0 | O7–Cu2–N3 | 89.52(17) |
| O7–Cu2–N3#2 | 90.48(17) | O7#2–Cu2–N3 | 90.48(17) |
| O7#2–Cu2–N3#2 | 89.52(17) | N3–Cu3–N3#2 | 180.0 |
| #2 −x, −y + 1, −z | |||
The presence of occluded solvent molecules (MeOH and H2O), non-coordinating oxygen atoms of carboxylate from monodentate pOHcinn ligands and phenolic alcohols allows for the formation of 2D supramolecular layers. These layers can be described as alternating parallel threads of dimers 4A and monomers 4B (Fig. 5B). Each dimer interacts with two similar dimers due to the mediation of four H2O molecules, forming a symmetric double bridge. This double bridge involves a phenolic OH group (O5–H5O⋯O4 W) and a free oxygen of a monodentate pOHcinn (O4W–H4WB⋯O4, Fig. 6A).
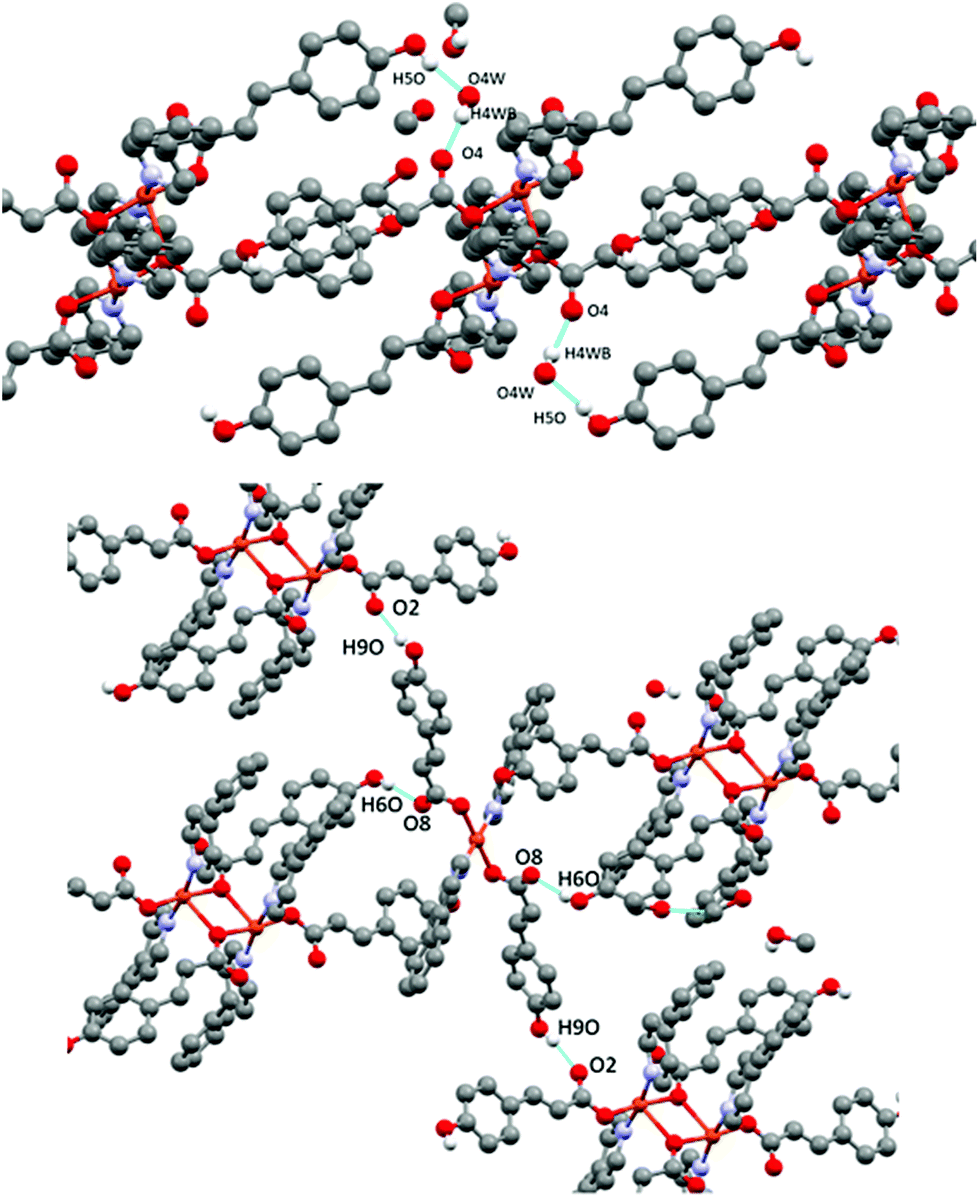 | ||
| Fig. 6 H-bond interactions forming the supramolecular structure of 4. Dimer centred view (top) and monomer centred view (bottom). | ||
Furthermore, each dimer interacts with two monomers, via a non-coordinating oxygen of the carboxylate from a monodentate pOHcinn ligand of the monomer and the remaining phenolic hydrogen (O6–H6O⋯O8) and another free oxygen of a monodentate pOHcinn ligand of the dimer and the remaining phenolic hydrogen of the monomer (O9–H9O⋯O2) (Fig. 6B). The intralayer Cu1⋯Cu2 distance is 10.563 Å, whereas the interlayer Cu1⋯Cu1 and Cu2⋯Cu2 distance is 12.956 Å. Relevant H-bond interactions are summarized in Table 6. The fact that in this compound solvent molecules occupy non interconnected cavities (12.9%, 370.42 Å3, using a probe radius of 1.2 Å)47 (Fig. 7) explains its stability compared with compound 3.
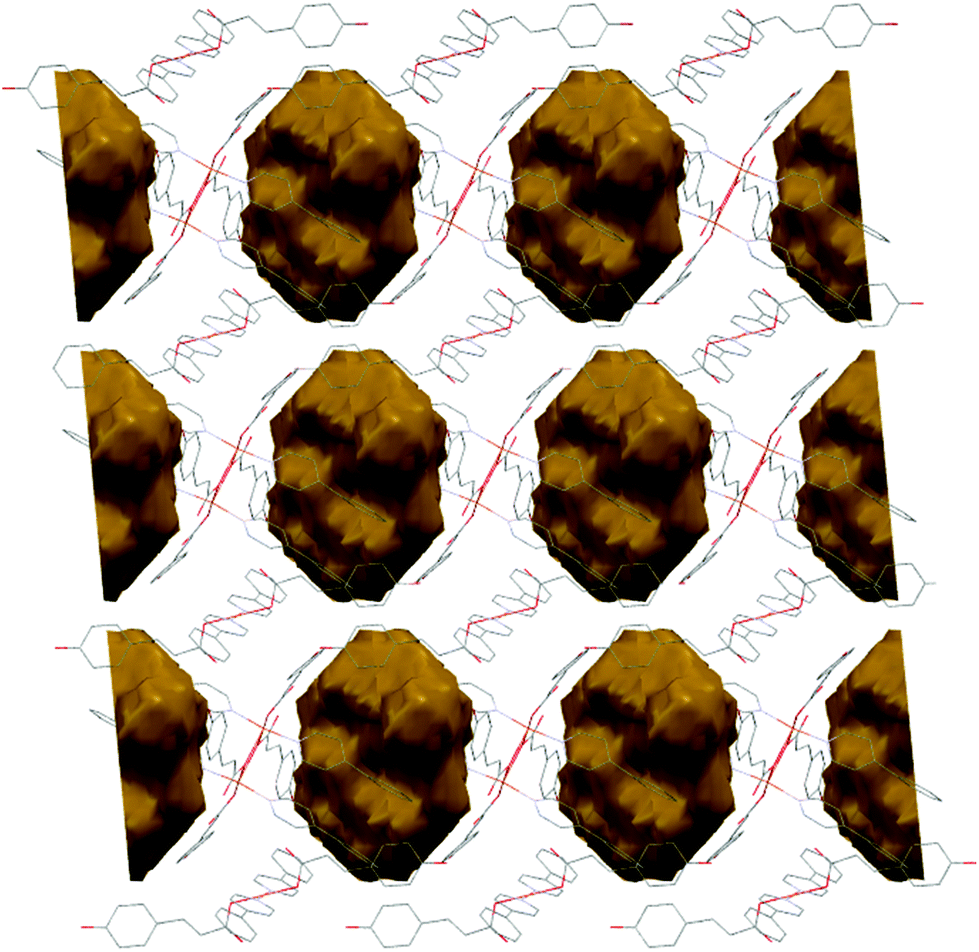 | ||
| Fig. 7 Schematic representation of the cavities occupied by solvent molecules in 4. | ||
| D–H⋯A | D–H | H–D⋯A | D–H⋯A | |
|---|---|---|---|---|
| i [2 − x,1 − y,1 − z], ii [1 − x, −y, 1 − z], iii [1 − x, 1 − y, 1 − z]. | ||||
| 4 | ||||
| O5–H5O⋯O1Wi | 1.88 | 0.82 | 2.641(8) | 161(8) |
| O9–H90⋯O2ii | 1.81 | 0.82 | 2.591(6) | 160 |
| O6–H6O⋯O8 | 1.83 | 0.82 | 2.617(7) | 160 |
| O1W–H1WB⋯O4iii | 1.82(5) | 0.83(4) | 2.624(8) | 161(8) |
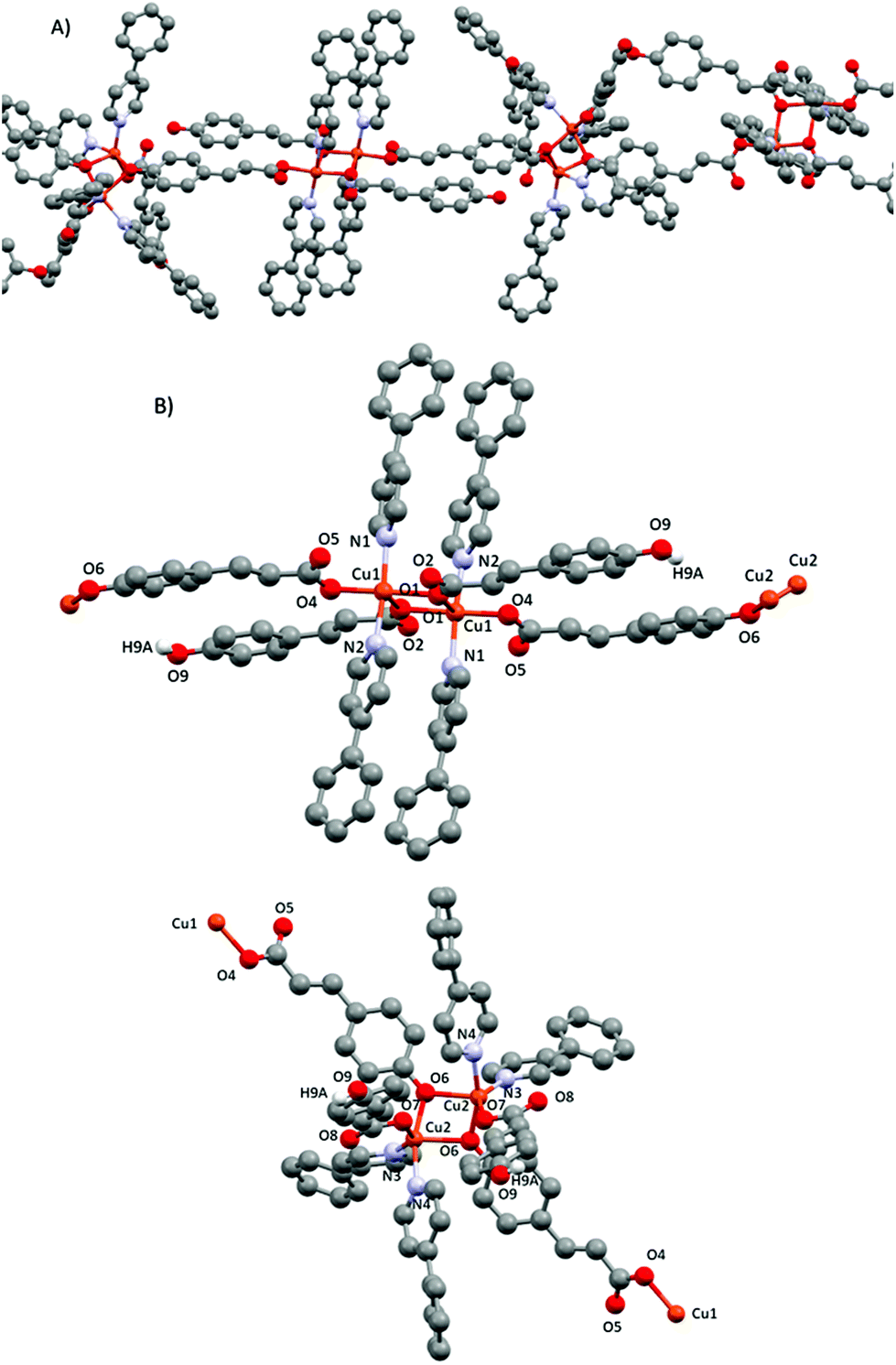 | ||
| Fig. 8 (A) Polymeric 1D chain of compound 5. (B) Corresponding dimeric subunits 5-trans (top) and 5-cis (down) with their corresponding numbering scheme. Only phenolic hydrogens are shown. | ||
In subunit 5-cis, the two Cu(II) cations are also penta-coordinated, with the pyridines being in a cis position. Thus, the coordination geometry is a distorted square pyramid (τ = 0.343).43 The basal plane is formed by one phenol oxygen of the linking pOcinn ligand bridging two Cu(II) atoms (Cu2–O6 1.9402(15) Å), one oxygen of a carboxylate from the monodentate pOHcinn ligand (Cu2–O7 2.0016(15) Å) and two nitrogen atoms of two 4-Phpy (Cu2–N3 2.0484(19) Å, Cu2–N4 1.9975(18) Å). The apical position is occupied by another phenol oxygen of the linking pOcinn ligand bridging two Cu(II) atoms (Cu2–O6 1.9402(15) and Cu2–O6#1 2.2521(15) Å). Relevant distances and angles are summarized in Table 7.
| #1 −x + 3/2, −y + 3/2, −z + 1 #2 −x, y, −z + 1/2. | |||
|---|---|---|---|
| 5 | |||
| Bond lengths (Å) | |||
| Cu1–O1 | 1.9755(15) | Cu2–O6 | 1.9402(15) |
| Cu1–O4 | 1.9365(15) | Cu2–O7 | 2.0016(15) |
| Cu1–N1 | 2.0166(19) | Cu2–N3 | 2.0484(19) |
| Cu1–N2 | 2.0246(19) | Cu2–N4 | 1.9975(18) |
| Cu1–O1#1 | 2.3438(16) | Cu2–O6#1 | 2.2521(15) |
| Cu1⋯Cu1 | 3.3888(9) | Cu2⋯Cu2 | 3.0127(9) |
| Bond angles (°) | |||
| O1–Cu1–O4 | 171.58(7) | O6–Cu2–N4 | 163.74(7) |
| O1–Cu1–N2 | 93.47(7) | O6–Cu2–O7 | 90.26(6) |
| O1–Cu1–N1 | 90.82(7) | O7–Cu2–N4 | 88.96(7) |
| O4–Cu1–N1 | 88.10(7) | O6–Cu2–N3 | 93.36(7) |
| O4–Cu1–N2 | 88.44(7) | N4–Cu2–N3 | 96.99(8) |
| O1–Cu1–O1#1 | 76.96(6) | O7–Cu2–N3 | 143.15(7) |
| O4–Cu1–O1#1 | 94.73(6) | O6–Cu2–O6#2 | 74.98(6) |
| N1–Cu1–O1#1 | 92.22(7) | N4–Cu2–O6#2 | 91.01(6) |
| N2–Cu1–O1#1 | 94.06(7) | O6#2–Cu2–O7 | 117.46(6) |
| N1–Cu1–N2 | 173.06(8) | N3–Cu2–O6#2 | 98.83(7) |
| N4–Cu2–Cu2#2 | 115.40(5) | ||
| N3–Cu2–Cu2#2 | 123.22(5) | ||
| O6–Cu2–Cu2#2 | 48.36(4) | ||
| O7–Cu2–Cu2#2 | 85.45(5) | ||
| O6#2–Cu2–Cu2#2 | 40.08(4) | ||
To summarize the differences between the two subunits, it is worth remarking the following: the most important of them is the fact that in subunit 5-trans all oxygen atoms are of the carboxylic groups of the pOHcinn ligand, whereas in subunit 5-cis they coordinate via carboxylic and phenolate groups. Furthermore, the relative position of their ligands is also different. Subunit 5-trans has a trans configuration, forming a sort of flat Cu1–O1–Cu1–O1 4-member ring with 4-Phpy and having pOHcinn ligands perpendicular (89.27°) and parallel (12.44°) to it, respectively, and a Cu1⋯Cu1 distance of 3.389 Å. The subunit 5-cis, although maintaining pOcinn ligands in the trans position, has the 4-Phpy ligands in the cis position (their pyridine rings defining a 96.99° angle between their centroids) and pOHcinn ligands acting as monodentate are also in cis positions. The Cu2–O6–Cu2–O6 ring has a V-like shape with a shorter Cu2⋯Cu2 distance of 3.013 Å. The two dimers lie at an intrachain Cu1⋯Cu2 distance of 11.470 Å.
The resulting 1-D polymeric structure could be described as a block copolymer with an ABAB configuration. These chains run along the (½ 0 ½) direction with an S-like shape. These chains are stacked in parallel via an interaction between a non-coordinated phenolic hydrogen and a free oxygen of a carboxylic acid (O9–H9A⋯O5), forming a 3D supramolecular structure. This supramolecular structure is further reinforced via weaker double C–H⋯O interactions (C41–H41⋯O1 and C42–H42⋯O1) involving hydrogen atoms in the pyridine ring of 4-Phpy and oxygen atoms of the carboxylate groups (Fig. 9). This arrangement results in an interchain Cu⋯Cu distance of 14.436 Å. Relevant supramolecular forces are summarized in Table 8.
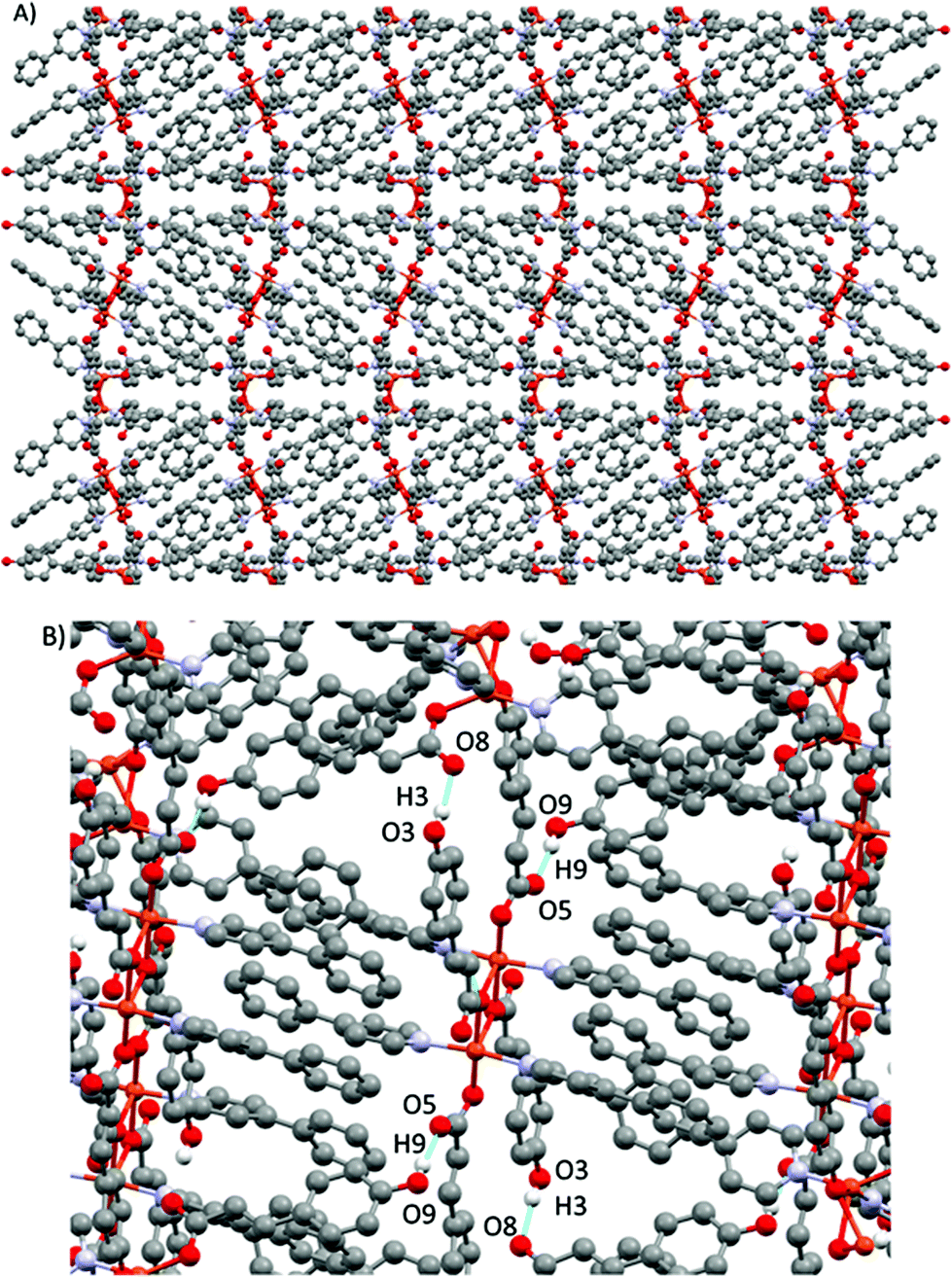 | ||
| Fig. 9 (A) View of the 3D supramolecular structure in 5. (B) Details of the supramolecular interactions in 5. | ||
| D–H⋯A | D–H | D–H⋯A | D–H⋯A | |
|---|---|---|---|---|
| I[−1/2 + x, −1/2 + y, z]. | ||||
| O9–H9A⋯O5I | 1.81(2) | 0.82(3) | 2.617(2) | 171.9(16) |
| C41–H41⋯O2 | 2.44 | 0.95 | 2.958(3) | 115 |
| C42–H42⋯O2 | 2.42 | 0.95 | 2.937(3) | 114 |
 | ||
| Fig. 10 Thermal variation of χpT for 5. The solid red line is the best fit to the proposed model (see text). | ||
In the first approximation, Cu(II) coupling through the pOcinn ligand was assumed negligible (shortest interdimer distance 11.866 Å). Therefore, the magnetic behavior of 5 must be considered as the addition of two independent dimers using a slightly modified Bleany–Bowers equation.50,51 The adjustment yielded a negligible ρ value of 0.01%. Therefore, this term was removed for the sake of simplification. The resulting equation was:
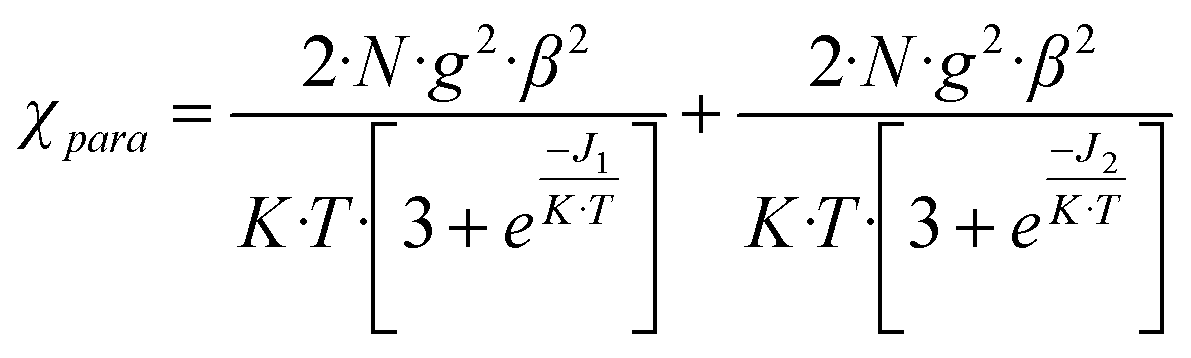
The model reproduces satisfactorily the magnetic properties in the range of 0 to 303 K with the parameters (g = 2.16; J1(cm−1) = −75.10; J2(cm−1) = −1.74; H = −JSiSi+1).
It has been known since longtime that Cu(II) dimers bridged by double phenoxy groups show a great variability of J values ranging from ferromagnetic to strong antiferromagnetic.52 This variability has prompted a strong debate about which are the magneto-structural correlations. The consensus is that five of them are key: the Cu⋯Cu distance, Cu–O–Cu angle, Cu–O distance, dihedral angle between the two coordination planes and planarity of the bonds around the coordinating atom.52,53 This is also applied in Cu(II) dimers bridged by phenoxy groups.54–56 Most of the Cu2O2 core magnetic pathways including phenolic oxygen atoms are antiferromagnetic,53,55–58 but a few of them are ferromagnetic.59 Taking this into account, the J1 value could be assigned to the 5-cis subunit, as its value is in the range of other phenoxy-bridged Cu(II) dimers.59 Its moderately antiferromagnetic interaction can be explained due the fact that the −2J value decreases with lower angles up to the crossover point of 77°.54 However, this parameter alone cannot explain this weak interaction, as J = −75.10 cm−1 is lower than expected with a Cu–O–Cu angle value of 91.57°. The weak antiferromagnetic interaction could also be due to the V shape of 5-cis, where O–Cu–O coordination planes having a dihedral angle of 51.24°. This morphology lowers the antiferromagnetic interaction due to the loss of orbital overlap.59
The other exchange magnetic pathway, J2 = −1.74 cm−1, which also shows an antiferromagnetic interaction, has a Cu2O2 core too, but including carboxylate bridges. Those pathways are well known and have been thoroughly studied.60 However, in 5-trans the bridging oxygen atoms are not both in the equatorial plane; in fact, one is in the equatorial plane and the other in the apical position. Reports of this combination for Cu(II) dimers are scarce and no general trends have been found yet.48–50 However, the consensus is that, although these types of core can be ferromagnetic or antiferromagnetic, they always show a weak interaction which agrees with the reported values here.48–50
Conclusions
A family of five Cu(II) coordination compounds including HpOHcinn acid and pyridine derivatives, 4-tBupy, 4-Acpy, 3-Phpy and 4-Phpy, is presented here. The analysis of their crystal structures shows how the choice of the pyridine ligand determines different coordination modes of the pOHcinn ligand, as well as the Cu(II) coordination, nuclearity and geometry. The pOHcinn acts as a monodentate carboxylate ligand in 1 and 4, yielding monomers and dimers, and the phenol group playing a key role in the hydrogen bond networks that determined the extended structure of both compounds. In contrast, when pOHcinn acts as a ditopic ligand, it leads to the formation of 2D coordination polymers, as observed in 2 and 3. The increase of the distance between the phenol and the carboxylate functionalities in the pOHcinn ligand is significant, compared to that with the previously studied pOHBz ligand, resulting in an overall increase of the available empty space for certain compounds. Compound 3 shows promising results, as it exchanges MeOH for H2O and vice versa, when exposed to different atmospheres, suffering a structural change in the process. This exchange is reversible, although full removal of solvents gives a non-porous material. Besides the effect of the pyridine substituents on the coordination mode of pOHcinn, the choice of the pyridine also determines its susceptibility to undergo a photoinduced process. Probably due to an increase in the acidity of the phenol group, half of the pOHcinn ligands in 4 are deprotonated when exposed to sunlight, generating a dianionic ligand that also acts as a linker between metal centers, resulting in the formation of polymer 5. This coordination polymer behaves magnetically as the sum of two independent dimers, due to a long interdimer distance (11.866 Å). One of the dimers has a J1 value of −75.10 cm−1 and is assigned to the 5-cis subunit. This value is somewhat lower than the reported values of similar dimers, which can be explained by other structural factors such as the shape and Cu⋯Cu distance. The other dimer has a value of J2 = −1.74 cm−1. Compared to other monodentate carboxylate bridged dimers, this value is low. This can be explained by the structural particularity mentioned before, that is, the fact of having one bridge in an equatorial position and one bridge in the axial position.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially financed by the Spanish National Plan of Research (projects CTQ2014-56324, CTQ2017-83632 and MAT2015-65756-R) and by the Generalitat de Catalunya (project 2014SGR377). C. D./ICMAB acknowledges financial support from the Spanish MEC, through the “Severo Ochoa” Programme for Centres of Excellence in R&D (SEV-2015-0496) J. S. also acknowledges the Universitat Autònoma de Barcelona for his pre-doctoral grant.References
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- P. Silva, S. M. F. Vilela, J. P. C. Tomé and F. A. Almeida Paz, Chem. Soc. Rev., 2015, 44, 6774–6803 RSC.
- G. Maurin, C. Serre, A. Cooper and G. Férey, Chem. Soc. Rev., 2017, 46, 3104–3107 RSC.
- Y. He, S. Xiang and B. Chen, J. Am. Chem. Soc., 2011, 133, 14570–14573 CrossRef CAS PubMed.
- H. Wang, H. Wu, J. Kan, G. Chang, Z.-Z. Yao, B. Li, W. Zhou, S. Xiang, J. Cong-Gui Zhao and B. Chen, J. Mater. Chem. A, 2017, 5, 8292–8296 CAS.
- A. Karmakar, R. Illathvalappil, B. Anothumakkool, A. Sen, P. Samanta, A. V. Desai, S. Kurungot and S. K. Ghosh, Angew. Chem., Int. Ed., 2016, 55, 10667–10671 CrossRef CAS PubMed.
- F. Hu, C. Liu, M. Wu, J. Pang, F. Jiang, D. Yuan and M. Hong, Angew. Chem., Int. Ed., 2017, 56, 2101–2104 CrossRef CAS PubMed.
- W. Yang, J. Wang, H. Wang, Z. Bao, J. C.-G. Zhao and B. Chen, Cryst. Growth Des., 2017, 17, 6132–6137 CAS.
- Z. Ju, G. Liu, Y.-S. Chen, D. Yuan and B. Chen, Chem. – Eur. J., 2017, 23, 4774–4777 CrossRef CAS PubMed.
- H. Wahl, D. A. Haynes and T. le Roex, Cryst. Growth Des., 2017, 17, 4377–4383 CAS.
- R. P. Sharma, A. Singh, A. Saini, P. Venugopalan, A. Molinari and V. Ferretti, J. Mol. Struct., 2009, 923, 78–84 CrossRef CAS.
- S. Gomathi and P. T. Muthiah, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 1498–1502 CAS.
- F. Valach, M. Dunaj-Jurčo, M. Melnik and N. N. Hoang, Z. Kristallogr., 1994, 209, 267–270 CAS.
- M. Sanchez-Sala, J. Pons, Á. Álvarez-Larena, C. Domingo and J. A. Ayllón, ChemistrySelect, 2017, 2, 11574–11580 CrossRef CAS.
- K. F. White, B. F. Abrahams, R. Babarao, A. D. Dharma, T. A. Hudson, H. E. Maynard-Casely and R. Robson, Chem. – Eur. J., 2015, 21, 18057–18061 CrossRef CAS PubMed.
- H. Görner, S. Khanra, T. Weyhermüller and P. Chaudhuri, J. Phys. Chem. A, 2006, 110, 2587–2594 CrossRef PubMed.
- X.-Y. Fan, K. Li, X.-C. Huang, T. Sun, K.-T. Wang, R.-R. Yun and H.-L. Wu, Z. Kristallogr. – New Cryst. Struct., 2009, 224, 59–61 CAS.
- Y. Z. Jia-Yuan Mao, H.-X. Fang, Q.-F. Xu, Q.-X. Zhou and J.-M. Lu, Chin. J. Inorg. Chem., 2008, 24, 1046–1050 Search PubMed.
- H.-L. Wu, W.-K. Dong and X.-L. Wang, Z. Kristallogr. – New Cryst. Struct., 2007, 222, 36–38 CAS.
- X. Qing-Feng, Z. Qiu-Xuan, L. Jian-Mei, X. Xue-Wei and Z. Yong, J. Solid State Chem., 2007, 180, 207–212 CrossRef.
- M. Kalinowska, L. Mazur, J. Piekut, Z. Rzączyńska, B. Laderiere and W. Lewandowski, J. Coord. Chem., 2013, 66, 334–344 CrossRef CAS.
- L. Chen, Z. Kristallogr. – New Cryst. Struct., 2009, 224, 565–566 CAS.
- V. Zeleňák, I. Císařová and P. Llewellyn, Inorg. Chem. Commun., 2007, 10, 27–32 CrossRef.
- L. Chen, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m807–m807 CAS.
- G. B. Deacon, M. Forsyth, P. C. Junk, S. G. Leary and W. W. Lee, Z. Anorg. Allg. Chem., 2009, 635, 833–839 CrossRef CAS.
- H. Li and C. W. Hu, J. Solid State Chem., 2004, 177, 4501–4507 CrossRef CAS.
- J. Yan, Y. Guo, H. Li, X. Sun and Z. Wang, J. Mol. Struct., 2008, 891, 298–304 CrossRef CAS.
- A. Mukherjee, M. K. Saha, M. Nethaji and A. R. Chakravarty, New J. Chem., 2005, 29, 596 RSC.
- S. Gupta, A. Mukherjee, M. Nethaji and A. R. Chakravarty, Polyhedron, 2005, 24, 1922–1928 CrossRef CAS.
- H.-L. Wu, J.-G. Liu, P. Liu, W.-B. Lv, B. Qi and X.-K. Ma, J. Coord. Chem., 2008, 61, 1027–1035 CrossRef CAS.
- A. Mukherjee, M. K. Saha, M. Nethaji and A. R. Chakravarty, Chem. Commun., 2004, 475, 716 RSC.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Van De Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. Van De Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- P. K. Singh and A. Awasthi, ChemPhysChem, 2017, 198–207 Search PubMed.
- N. Agmon, J. Phys. Chem. A, 2005, 109, 13–35 CrossRef CAS PubMed.
- A. Das, T. Banerjee and K. Hanson, Chem. Commun., 2016, 52, 1350–1353 RSC.
- M. Kuss-Petermann and O. S. Wenger, J. Phys. Chem. A, 2013, 117, 5726–5733 CrossRef CAS PubMed.
- R. M. O'Donnell, R. N. Sampaio, G. Li, P. G. Johansson, C. L. Ward and G. J. Meyer, J. Am. Chem. Soc., 2016, 138, 3891–3903 CrossRef PubMed.
- G. Gliemann, Ber. Bunsenges. Phys. Chem., 1978, 82, 1263–1263 CrossRef.
- G. Deacon, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- R. Kruszynski, A. Adamczyk, J. Radwańska-Doczekalska and T. Bartczak, J. Coord. Chem., 2002, 55, 1209–1217 CrossRef CAS.
- R. P. Sharma, A. Saini, P. Venugopalan, J. Jezierska and V. Ferretti, Inorg. Chem. Commun., 2012, 20, 209–213 CrossRef CAS.
- J. Soldevila-Sanmartín, J. A. Ayllón, T. Calvet, M. Font-Bardia and J. Pons, Polyhedron, 2017, 135, 36–40 CrossRef.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- J. Pasán, J. Sanchiz, C. Ruiz-Pérez, F. Lloret and M. Julve, New J. Chem., 2003, 27, 1557–1562 RSC.
- R. Baldomá, M. Monfort, J. Ribas, X. Solans and M. A. Maestro, Inorg. Chem., 2006, 45, 8144–8155 CrossRef PubMed.
- R. Bikas, P. Aleshkevych, H. Hosseini-Monfared, J. Sanchiz, R. Szymczak and T. Lis, Dalton Trans., 2015, 44, 1782–1789 RSC.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451–465 CrossRef CAS.
- V. H. Crawford, H. W. Richardson, J. R. Wasson, D. J. Hodgson and W. E. Hatfield, Inorg. Chem., 1976, 15, 2107–2110 CrossRef CAS.
- G. Dutta, R. K. Debnath, A. Kalita, P. Kumar, M. Sarma, R. B. Shankar and B. Mondal, Polyhedron, 2011, 30, 293–298 CrossRef CAS.
- L. K. Thompson, S. K. Mandal, S. S. Tandon, J. N. Bridson and M. K. Park, Inorg. Chem., 1996, 35, 3117–3125 CrossRef CAS PubMed.
- A. Biswas, M. G. B. Drew, J. Ribas, C. Diaz and A. Ghosh, Eur. J. Inorg. Chem., 2011, 2405–2412 CrossRef CAS.
- A. Biswas, M. G. B. Drew, J. Ribas, C. Diaz and A. Ghosh, Inorg. Chim. Acta, 2011, 379, 28–33 CrossRef CAS.
- N. Novoa, F. Justaud, P. Hamon, T. Roisnel, O. Cador, B. Le Guennic, C. Manzur, D. Carrillo and J.-R. Hamon, Polyhedron, 2015, 86, 81–88 CrossRef CAS.
- Q. R. Cheng, H. Zhou, Z. Q. Pan, G. Y. Liao and Z. G. Xu, Polyhedron, 2014, 81, 668–674 CrossRef CAS.
- P. Chaudhuri, R. Wagner and T. Weyhermüller, Inorg. Chem., 2007, 46, 5134–5136 CrossRef CAS PubMed.
- G. Psomas, C. P. Raptopoulou, L. Iordanidis, C. Dendrinou-Samara, V. Tangoulis and D. P. Kessissoglou, Inorg. Chem., 2000, 39, 3042–3048 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables with l crystallographic data, and PXRD patterns and ATR-FTIR spectra figures. CCDC 1820975–1820979. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt00645h |
| This journal is © The Royal Society of Chemistry 2018 |