
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDinitrogen photoactivation: status quo and future perspectives
Vera
Krewald

Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: v.krewald@bath.ac.uk
First published on 3rd May 2018
Abstract
While the thermal activation of the dinitrogen molecule has been demonstrated in numerous examples with a variety of transition metals and ligand frameworks, the use of light to induce a weakening or splitting of the strong N–N bond is less well explored. Six complexes that bind N2 in a linear μ–η1:η1-end-on fashion between two transition metals are known to cleave dinitrogen after absorbing a photon and relaxing from an electronically excited state. This Perspective article reviews the molecular complexes known to be capable of dinitrogen photocleavage, and discusses mechanistic insights into the photoactivation process gained from experimental and computational studies. In an extension of previous hypotheses for pathways to dinitrogen photoactivation that would facilitate easy protonation of the μ-N atoms, a scheme is presented that may help to identify other complexes that could be capable of dinitrogen photoactivation.
Introduction
The first sentence of a review by Chatt and Leigh from 1972 reads: “Nitrogen fixation is topical and the subject of many detailed reviews”.1 Despite almost 50 years having passed since, this description of the field remains accurate to date, which should of course not be interpreted as a lack of progress. In fact, many milestones towards a better understanding of dinitrogen fixation in nature, in industry and in the development of novel catalysts in many laboratories have been achieved in the past decades: besides Nobel prizes to Fritz Haber in 1918 “for the synthesis of ammonia from its elements”2 and to Gerhard Ertl in 2007 “for his studies of chemical processes on solid surfaces”, in particular the Haber–Bosch process,3 the atomic-level description of the active site of nitrogenase4–6 and the first molecular complexes that cleave dinitrogen thermally7 or photochemically8 were achieved. Dinitrogen activation remains a challenging field of research, fuelled by the wish to ever better understand nitrogen fixation in nature, and the need to develop catalysts that can directly incorporate air-derived nitrogen atoms into useful chemicals. The key reasons why dinitrogen remains difficult to break and utilise are the same as recognised decades ago: a dissociation enthalpy of 944 kJ mol−1 due to its triple bond, an ionisation potential of 15.6 eV (cf. 14.0 eV for CO), a large HOMO–LUMO gap, and the lack of a dipole moment.1The industrial route to ammonia, the Haber–Bosch process, is energy efficient and well established with an annual production in 2015 that consumed ca. 1% of the world energy production.9,10 The overwhelming majority of the ammonia produced with the Haber–Bosch process is used to generate artificial fertiliser. While the question of whether the process could be replaced by an alternative that relies on molecular catalysts remains open, the Haber–Bosch process in its current form is not without problems. It consumes fossil hydrogen from natural gas, in quantities that amount to ca. 2% of the world annual production,9 so that it is overall unsustainable; however, this could probably be remedied once a water oxidation catalyst that is capable of producing renewable hydrogen in an industrial scale is found and established.11,12
The ongoing search for N2-to-NH3-converting molecular complexes has ramifications beyond any potential industrial use. First of all, there is scientific curiosity as to the atomic-level details of this process, and much to be learned from the chemical clarity and elegance of molecular catalysts capable of small molecule activation. Equally, the study of molecular complexes helps to unravel the mechanism of dinitrogen activation in nature.13–18 Besides biochemical, spectroscopic and computational efforts to shed light on this key reaction,14,19 synthetic complexes that mimic the molecular structure and electronics of the active site(s) have been of great utility.20 Despite significant advances in the understanding of the molecular composition and electronic structure of the nitrogen-fixing cofactor(s) in enzymes, the precise mechanism remains clouded.14,21–23
Most bulk and fine chemicals are currently sourced from fossil resources, including important N-heterocyclic compounds. Clearly, the economic pressure is currently not sufficiently high to prevent mankind from burning these precious resources. It is, however, equally clear that alternative approaches and methods will be required at some point in the future to maintain the ubiquity of synthetic materials that we have come to depend on. These approaches may include recycling of existing N-containing materials, the valorisation of natural sources such as proteins, and the incorporation of air-derived nitrogen atoms into valuable chemicals. The latter idea motivates the search for catalysts that can activate or split dinitrogen and subsequently form N–E bonds (E = main group element, e.g. C, H, Si).24,25
Two different targets of dinitrogen fixation can be distinguished: one that aims to produce ammonia, i.e. reduces nitrogen, and one that aims to produce N-incorporated products, in which the nitrogen atom may be formally oxidised or reduced. The requirements for catalysts that are capable of carrying out these reactions are of course not mutually exclusive, but they may differ, potentially affecting the metal identity, the metal oxidation states, their oxidative or reductive power, the hapticity of the ligands, and the need for internal acids or bases among many other chemical parameters.
The targeted chemical space for products resulting from dinitrogen fixation is vast, however, the principal approach to activate or cleave the N–N triple bond relies on a catalyst that binds N2 and then weakens the N–N interaction so that the nitrogen atoms are primed to form new bonds. The energy required to enable this reaction sequence is usually derived thermally. A less explored route to activate or cleave dinitrogen is a photochemical process,26 and consequently, our current knowledge of this route is insufficient to systematically and strategically optimise dinitrogen photocleavage catalysts.
This perspective aims to review the field of molecular dinitrogen photoactivation, to identify shared principles among the complexes relevant to this reaction, and to indicate possible paths to a better understanding of the photophysical and photochemical processes. We begin with a brief summary of the nature of metal–nitrogen interactions and highlight a few examples of thermal dinitrogen fixing complexes and catalysts, before exploring in greater detail the photochemical dinitrogen activation systems.
Nitrogen binding in metal complexes
The nature of the interaction between dinitrogen and metals has been covered in many excellent reviews,27,28 and only a few key points are summarised here. Dinitrogen is generally not a good ligand: it does not have a dipole moment and is a weaker σ-donor and π-acceptor than isoelectronic CO.28,29 The strength and character of the nitrogen-metal interaction alters the nitrogen–nitrogen interaction, i.e. N2 is activated by depletion of electron density from its bonding orbitals (σ-donor) and donation into its antibonding orbitals (π-acceptor), Fig. 1a. The resulting overall N–N bond strength – equivalent to the degree of activation – can be determined from the N–N distance in crystal structures, or measured by the N–N stretching frequency, which can then be translated into an estimate for the N–N distance according to Badger's rule.28,30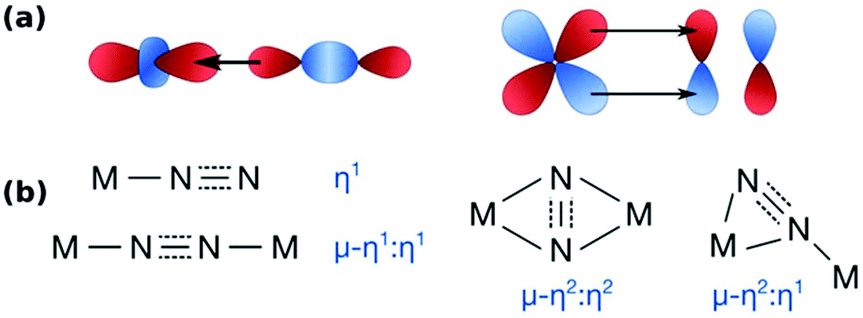 | ||
| Fig. 1 (a) Electronic interactions in end-on M–N–N complexes: σ-donor (left) and π-acceptor pathways (right). (b) Binding modes in mono- and di-nuclear metal complexes of dinitrogen; the μ–η1:η1-end-on configuration can be linear, cis or trans. | ||
Dinitrogen can bind η1-end-on, μ–η1:η1-end-on, μ–η2:η2-side-on, and μ–η1:η2-side-on; the η1-side-on motif remains unisolated (see Fig. 1b).27 Side-on vs. end-on coordination can be subtly influenced by the extent of nonbonded steric interactions.31 Electronically, dinitrogen is formally bound as N2, N2–, N22–, N23– and N24– with varying spin states, although in comparison with the more electron-accepting O2 molecule, the more reduced forms are formed with more strongly reducing metals.28
Thermal routes to dinitrogen activation
N2 is cleaved with elemental Li to yield Li3N, and complexes containing Cr, Mo, W, Fe and Ti in reaction mixtures with LiAlH4, RMgX, RLi, or R3Al have also been known to produce NH3 from N2 since at least the 1960s.32 To reductively cleave dinitrogen, six electrons are required, i.e. at least a d3–d3 configuration or equivalent in a transition metal dimer. The first report of a well-defined transition metal complex capable of spontaneous thermal dinitrogen splitting was by Laplaza and Cummins in 1995:7 a three-coordinate Mo(III) (d3) complex with three NRAr (R = Me3C, Ar = 3,5-C6H3Me2) ligands was shown to bind dinitrogen end-on, attach a second Mo(III) unit resulting in [(NRAr)3Mo(μ-N2)Mo(NRAr)3] with a linear Mo–N–N–Mo unit, Fig. 2a, and subsequently cleave the N–N bond at 30 °C. The transition state is thought to be of ‘zig-zag’-shape, see Fig. 2b. Chemically unassisted thermal dinitrogen cleavage reactions, i.e. without an added source of reducing equivalents, yielding a transition metal nitrido product remain rare, with only a few reported in the literature to date.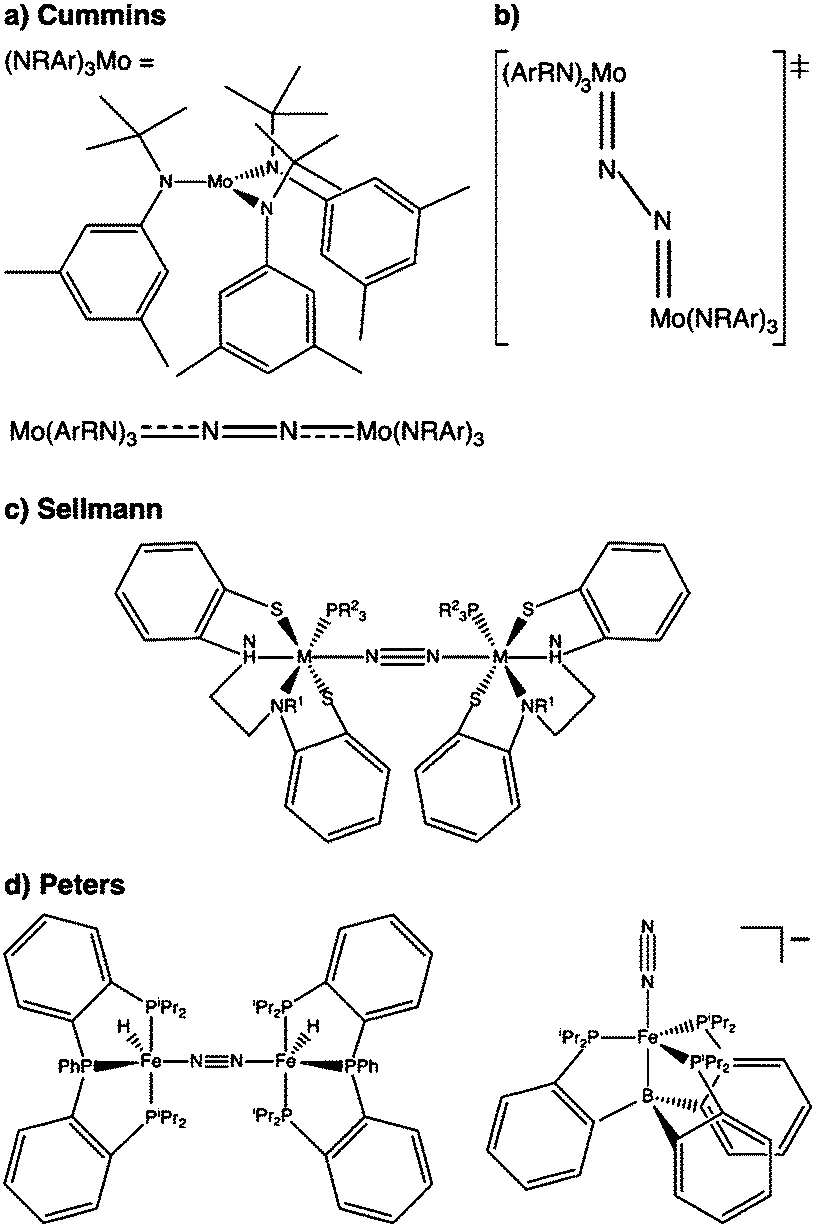 | ||
| Fig. 2 Thermal dinitrogen activation complexes: (a) Cummins, (b) zig-zag transition state relevant to the Cummins complex, (c) Sellmann, (d) Peters: left [P2P′PhFeH]2(μ-N2), right [P3BFe(N2)]−. | ||
One of the most famous molecular catalytic systems capable of activating dinitrogen was developed by Yandulov and Schrock,33,34 an extension of Schrock's earlier work on molybdenum complexes with triamidoamine ligands.35 The design principle that presumably leads to a closed catalytic cycle without significant decomposition or dimerisation of chemical intermediates is the formation of a pocket by the three 2,4,6-iPr3C6H2 rings attached to the amido-nitrogens of the ligand, in which the molybdenum nitrido unit is protected.
A different ligand design idea was pursued by Sellmann, with the aim of coupling proton and electron transfer steps:36–38 the ligand contains thiolate groups, which can be protonated and deprotonated relatively easily, Fig. 2c. Mononuclear iron complexes of the Sellmann ligand bind NH3 and N2H4, and N2H2 is found to bridge two iron centres stabilised by such a ligand.39 A ruthenium ion binds N2 end-on,40 and ruthenium dimers with bridging N2 and N2H2 were isolated and characterised as well.41
In recent years, Nishibayashi and coworkers have developed a series of complexes for dinitrogen activation that are based around PNP, PPP and PCP pincer-type ligands, which stabilise a range of transition metals (e.g. Mo, Cr, Fe, Co) and produce ammonia from N2.42–45
Many more complexes capable of activating and transforming dinitrogen have been synthesised and characterised, and a number of thorough reviews summarising the current state of the art are available.24,46–54
Photochemical routes to dinitrogen activation
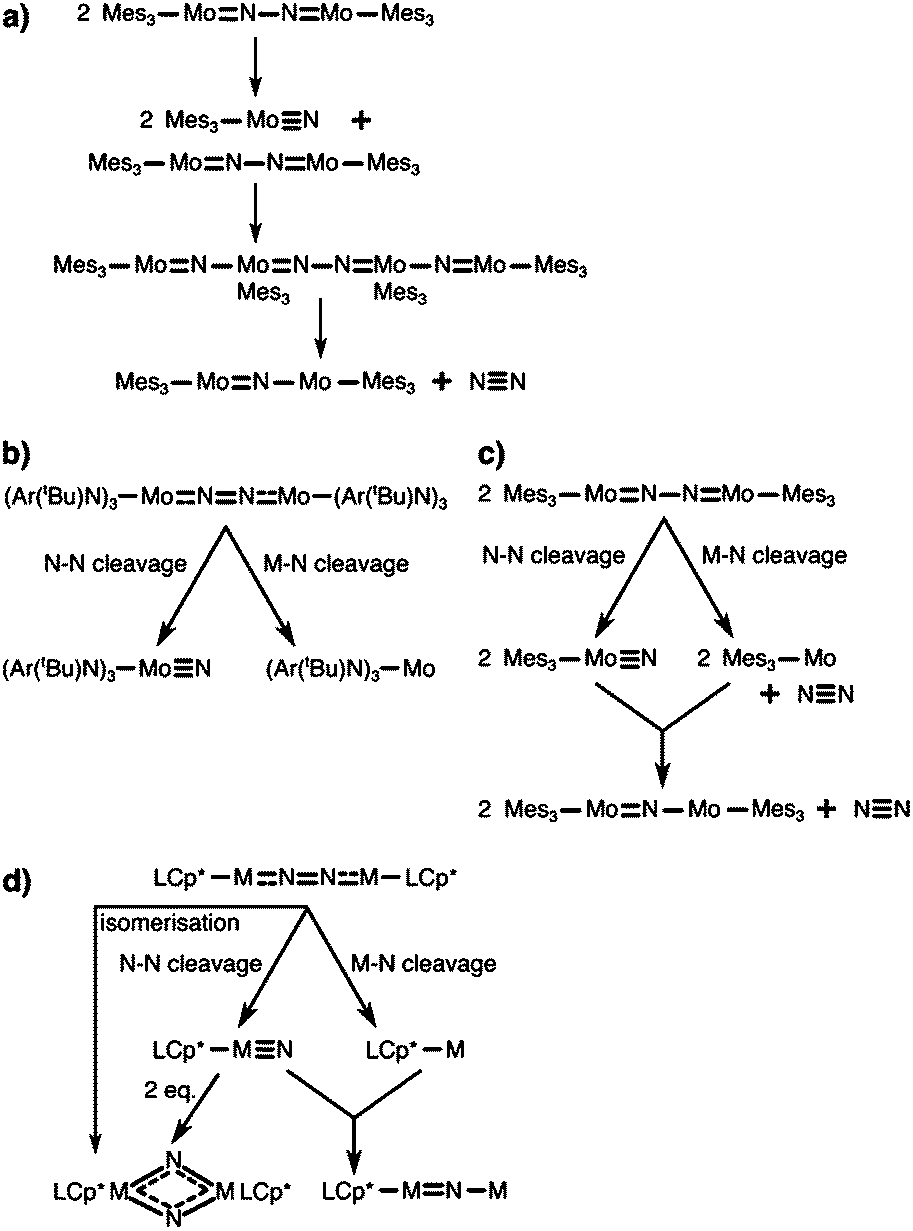
The concept of dinitrogen photoactivation holds the promise to use sunlight as the energy source for one of the most difficult small molecule activation reactions, while at the same time storing the energy in a useful form by generating valuable intermediates or chemicals. To the best of the author's knowledge, dinitrogen photoactivation and -cleavage was first discussed by Fischler and von Gustorf in 1975.55 They introduced the idea that particular electronic excitations would lead to distinct geometric and electronic changes of a dinitrogen molecule bound to a transition metal, which would in turn alter the chemical reactivity of this N2 entity.55 Specifically, they hypothesised for an end-on Fe–N2 complex that (i) d–d-transitions would depopulate an Fe–N π-orbital and fill an Fe–N σ*-orbital, thus weakening the Fe–N bond twofold and inducing its homolytic cleavage (Fig. 3, orange arrow); and (ii) LMCT transitions would populate an N–N π*-orbital, thus increasing the basicity of the nitrogen atoms and facilitating their protonation (Fig. 3, blue arrow).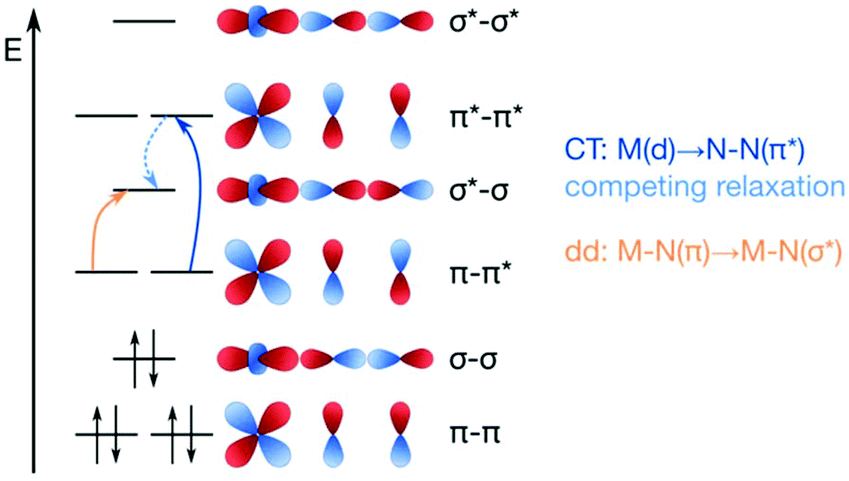 | ||
| Fig. 3 Two theoretical pathways relevant to dinitrogen photocleavage according to Fischler and von Gustorf for an Fe-η1-N2 complex (metal d-electrons not specified). Orange: M–N(π) to M–N(σ*) excitation; dark blue: M(d) to N–N(π*) excitation, shown here as M(d) is the M–N(π–π*) orbital; light blue: competing relaxation into M–N(σ*).55 | ||
The first experimental evidence for molecular dinitrogen photoactivation was published in 2001,8 followed by further examples in 2008,56 2010,57 201458 and 2015,31 alongside computational studies into plausible mechanisms in 200459 and 2018.60 In all of these examples, the molecular catalyst acts directly as the photosensitizer. Dinitrogen photoactivation has furthermore been shown to occur on surfaces, which was also studied computationally.61–66 A photochemical route to the activation or cleavage of dinitrogen is thus a well-founded concept that warrants further experimental and theoretical exploration.
In the following, we will discuss three categories of complexes relevant to dinitrogen photoactivation; those that (i) show increased turnover upon irradiation, (ii) yield metal-nitrido products, and (iii) directly incorporate N2-derived nitrogen atoms into molecules. It should be noted that there are also μ-N2 dimers that form from metal nitrido complexes, both thermally and under irradiation, which is an obviously undesired side reaction to any light-induced dinitrogen splitting reaction.58,67–73
Complexes that show increased turnover upon irradiation
The Peters group has shown that for two of their iron complexes the yield of ammonia increases upon irradiation of the reaction mixture with a Hg lamp.74 These complexes are a μ–η1:η1-bridged dimer, [P2P′PhFeH]2(μ-N2), and an η1-bound monomer, [P3BFe(N2)]−, Fig. 2d. The turnover numbers of N2 to NH3 with HBArF4 were significantly increased under Hg lamp irradiation but otherwise identical conditions. Mechanistic investigations support the idea that light-induced elimination of H2 might lead to P2P′PhFe0(N2) as a plausible photoproduct, which acts as the catalyst.74 A direct role of electronically excited states in the activation of the N–N bond was thus not shown conclusively.Complexes that yield metal nitrido products
The first complex for which the light-induced cleavage of a bridging dinitrogen was shown experimentally is [Mes3MoV(μ-N2)MoVMes3] (Mes: 2,4,6-Me3C6H2), Fig. 4a, in which the Mo–N and N–N interactions were characterised as double and single bonds, respectively.8 Notably, the complex is thermally stable, but cleaves the N–N bond upon irradiation with UV-light (λ = 365 nm). The final product in this reaction is the mono-μ-N complex [Mes3Mo(μ-N)MoMes3]. The authors suggest that this product forms upon reaction of the initial photocleavage product, the Lewis-basic [Mes3MoVI(N)], with unreacted (μ-N2) dimer. The product of two such coupling reactions would be [Mes3Mo(μ-N)MoMes3(μ-N2)MoMes3(μ-N)MoMes3], which upon expulsion of the central dinitrogen unit would form two mono-μ-N molybdenum dimers, Fig. 5a.8 The authors furthermore note that although there are structural similarities between [Mes3MoV(μ-N2)MoVMes3] and the [(NRAr)3Mo(μ-N2)Mo(NRAr)3] complex by Cummins and coworkers7 introduced above, the ligands in the latter have σ- and π-donor properties. This would explain the thermal stability of their complex in contrast to the thermally induced dinitrogen splitting activity of the Cummins complex.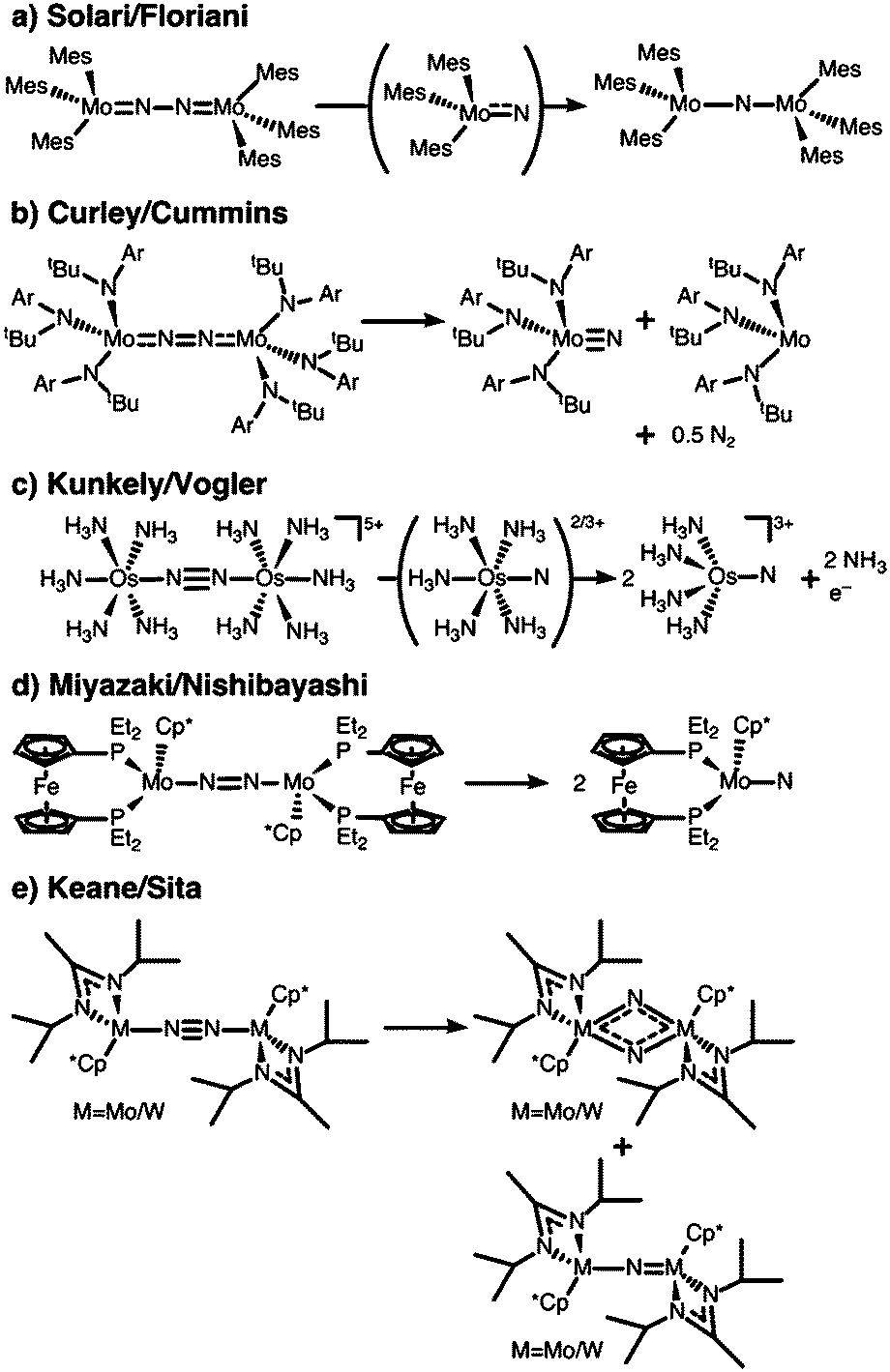 | ||
| Fig. 4 Complexes shown to be active in dinitrogen photoactivation: (a) Solari et al.;8 (b) Curley et al.;56 (c) Kunkely et al.;57 (d) Miyazaki et al.;58 (e) Keane et al.31 | ||
 | ||
| Fig. 5 Bifurcated reaction pathways in dinitrogen photocleavage and possible alternative routes to observed products. (a) Proposed reaction sequence to the observed mono-μ-N product of Solari et al.8 (b) Bifurcated photochemical routes according to Curley et al. for the Laplaza–Cummins system.56,75 (c) Proposed alternative mechanism to the mono-μ-N product of Solari et al. according to Curley et al.56 (d) Possible pathways to products observed in Keane et al.,31 photo-induced isomerisation of the μ-N2 complex to the bis-μ-N complex, dimerisation of two metal nitrido complexes after N–N photocleavage to the bis-μ-N complex, and formation of the mono-μ-N product analogous to (c). | ||
Interestingly, the Laplaza/Cummins complex capable of thermal dinitrogen splitting was later shown to also be capable of photochemical dinitrogen splitting.56,75 With a view to the first computational study of dinitrogen photoactivation (Reiher et al.,59vide infra), Cummins and coworkers had reasoned that the geometry of the excited state predicted by Reiher et al. resembled the well-known ‘zig-zag’ intermediate identified in computational studies of the thermal dinitrogen activation pathway, Fig. 2b. Indeed, irradiation of the intense visible absorption band (544 nm) of [(NRAr)3Mo(μ-N2)Mo(NRAr)3] with a Hg–Xe arc lamp (546 nm) at low temperatures to suppress the thermal cleavage process resulted in a bleach at 542 nm and an increased absorbance at ca. 430 nm.56 The authors observed the anticipated [(NRAr)3MoN] product from homolytic N2 splitting, as well as a [(NRAr)3Mo] monomer with a vacant coordination site.
Importantly, this finding implies a bifurcated reaction pathway: ‘unproductive’ metal–nitrogen bond cleavage on one hand, and the desired nitrogen–nitrogen bond cleavage on the other, Fig. 5b. Based on their observations, Cummins and coworkers re-interpreted the findings by Solari et al.,8 suggesting that the final mono-μ-N complex [Mes3Mo(μ-N)MoMes3] might be formed upon coupling of a [Mes3Mo] fragment (resulting from Mo–N scission) and a [Mes3Mo(N)] fragment (resulting from N–N scission), Fig. 5c.56 Unlike for the Mes-substituted complex, the (NRAr)-substitution is too bulky to allow the formation of a mono-μ-N complex as was shown explicitly by Johnson et al.76
In 2010, Kunkely and Vogler investigated the propensity of the mixed-valent, valence-delocalised system [Os(II,III)2(μ-N2)(NH3)10]5+ for light-induced N–N bond splitting, Fig. 4e.57 They reasoned that its stability in aqueous solution, the intense colours of the starting compound and reaction product, and the rather unactivated N–N bond made for an ideal test case. Irradiation of a solution of [Os(II,III)2(μ-N2)(NH3)10]5+ at wavelengths shorter than 450 nm resulted in bleaching of its green colour and the formation of an osmium-nitrido product, [OsVI(N)(NH3)4]3+.57 It was also shown that intermittently, [OsVI(N)(NH3)5]3+ is formed and thus the authors inferred that a second immediate product must be [OsV(N)(NH3)5]2+ which would subsequently disproportionate.57 One ammonia ligand in each of the initial photoproducts is lost due to the strong trans-influence of the nitrido group.
Kunkely and Vogler furthermore carried out a similar experiment with [Os(III,III)2(μ-N2)(NH3)10]6+, which might yield directly the two desired Os(VI) nitrido complexes. However, this one-electron oxidised complex ‘vigorously’ expels dinitrogen upon irradiation of its aqueous solution, as it also occurs thermally at temperatures above 5 °C.57 These two structurally similar, but mechanistically disparate complexes thus showcase that a delicate electronic structure balance is required to achieve light-induced dinitrogen splitting. The osmium complexes are an example of the two reaction paths discussed above – metal–nitrogen bond scission and nitrogen–nitrogen bond scission – occurring in two closely related complexes.
Nishibayashi and coworkers isolated a series of complexes of the general formula [(depf)(Cp*)Mo(μ-N2)Mo(Cp*)(depf)]n+ (depf = 1,1′-bis(diethylphosphino)ferrocene), Fig. 4d, in which the bridging dinitrogen unit is formally [N2]4– (n = +2), [N2]3– (n = +1), [N2]2– (n = 0).58 Concomitant with an N–N bond length decrease as the charge increases, the calculated N–N Mayer bond order rises from 1.40 to 1.60 and 1.80.58 The most reduced complex in this series cleaves the N–N bond upon irradiation with visible light (λ > 400 nm) in benzene solution, yielding two Mo(IV) nitrido complexes, [(depf)(Cp*)Mo(N)]. Excitation with wavelengths above 580 nm did not induce a reaction. Interestingly, the monomeric photoproduct [(depf)(Cp*)Mo(N)], which does not carry significant N-based spin density according to DFT calculations, re-forms the dimeric structure upon chemical oxidation.58 The monomer can be protonated with 1,4-cyclohexadiene/FcBArF4 and [LutH][FcBArF4] (LutH = 2,6-lutidinium). Similarly to the case of the two osmium dimers discussed above, only this specific complex in the redox series is active in dinitrogen photocleavage: irradiation of [(depf)(Cp*)Mo(μ-N2)Mo(Cp*)(depf)]+/2+ was ineffective.
Complexes that incorporate N-atoms into molecules
A very interesting pair of complexes was published by Sita and coworkers in 2015:31 μ–η1:η1-dinitrogen-bridged Mo and W dimers that produce isocyanate derivatives from CO2 and R3ECl (E = C, Si, Ge) upon irradiation with light, Fig. 4e.31 The dimers are of the general formula [L(Cp*)M(μ-N2)M(Cp*)L] with M either Mo or W (L = N(iPr)C(Me)N(iPr)), and are thermally stable in hydrocarbon solution up to 100 °C.31 These complexes are analogues of the previously published Ta, Hf, Zr and Ti dimers with the same ligand framework,77,78 some of which can also be reacted further to form N–Si bonds.79 A molybdenum dimer with a less bulky ligand (L′ = N(Et)C(Ph)N(Et)) produces HN(SiMe3)2 in a three-step catalytic cycle from N2, Me3SiCl and a proton source with a formal overall Mo(II)/Mo(IV) redox process.80Upon irradiation of the Mo complex with a Rayonet carousel of a medium-pressure Hg lamp, several photoproducts appear to form based on the UV/vis spectral changes over time. In fact, two products could be crystallised in the case of Mo: a [L(Cp*)MoV(μ-N)2MoV(Cp*)L] dimer with a diamond-shaped core, and a mono-μ-N complex of the formula [L(Cp*)MoIII(μ-N)MoIV(Cp*)L]. Based on the latter, immediate analogies to the interpretation of the Floriani and Cummins complexes can be drawn: here, too, two competing photoactivation pathways may be at play, Mo–N and N–N cleavage, Fig. 5d. As an alternative route to the diamond-shaped dimer, one can imagine that two metal nitrido products dimerise to form the bis-μ-N complex, Fig. 5d. However, it is equally possible that the dimer with a diamond-shaped core is a true photoproduct from an isomerisation process. This means that photochemical pathways beyond the established ‘zig-zag’ structure must be considered and probed to fully unravel the processes in dinitrogen photocleavage. For the W complex, irradiation produces well-defined isosbestic points in the UV/vis spectrum. The photoproduct was assigned as the dimeric bis-μ-N-bridged species based on the 1H NMR spectrum; however it is unclear at this point whether this observation precludes the viability of both bond dissociation pathways for the W complex.
In summary, all experimentally characterised dinitrogen photoactivation catalysts share the same linear geometry of the M2(μ-N2) core. Importantly, there are indications that the dinitrogen photoactivation pathway is bifurcated in some systems, implying that M–N and N–N bond scission processes compete.
Mechanistic aspects in thermal and photochemical dinitrogen activation
Many papers and reviews have been dedicated to the question of how thermal dinitrogen activation proceeds on the molecular level,81–87 most notably the Chatt88 and Schrock89 cycles, which cannot be repeated here. We focus below on mechanistic aspects of the families of complexes that are capable of thermal and photochemical dinitrogen activation, and review available computational studies on dinitrogen photofixation.A computational study by Reiher et al.59 investigated the second scenario put forward by Fischler and von Gustorf (Fig. 3), namely an electronic excitation that leads to an excited state with an altered electronic and geometric structure in which the μ-N can be protonated more easily. The individual steps involved were laid out as (i) coordination of N2 without a requirement for bond weakening, (ii) formation of a well-defined excited state that activates the bound N2 by populating an antibonding orbital, e.g. a π* orbital, Fig. 6a, and (iii) protonation of the thus formed diazenoid structure and subsequent reduction; whether the protons are already stored in the complex before the activation step or the protonation takes place after light activation is open.59 With such a dinitrogen reduction and protonation scenario in mind, Sellmann-type complexes of Fe and Ru (see above) were chosen as models for this theoretical study, in which the thiolate ligand can be protonated and thus act as an ‘internal acid’.
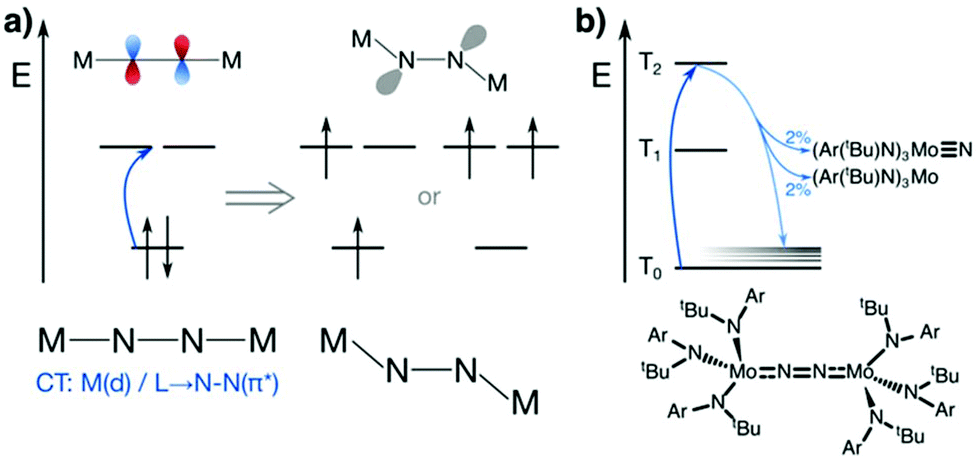 | ||
| Fig. 6 (a) Photoactivation scheme proposed by Reiher et al.: formation of an excited state in which the LUMO of N–N π* character is singly or doubly occupied (total spin unspecified), resulting in an excited state geometry that has lone pairs at the μ-N atoms which facilitate protonation. (b) Photochemical pathways seen by Huss et al. for the Laplaza–Cummins complex: excitation into the second excited triplet state (dark blue), rapid internal conversion and vibrational relaxation (light blue) with 2% cleavage of M–N and 2% cleavage of N–N bonds. | ||
Using DFT, TD-DFT and MP2 methods, it was found that the LUMO of the linear M–N–N–M core is dominated by N–N π* character, and that the geometry resulting from its occupation has bent M–N–N units (ca. 131–147°) akin to the ‘zig-zag’ transition state in thermal dinitrogen activation, Fig. 2b. The bridging nitrogen atoms begin to form lone pairs in the excited state, Fig. 6a, which was shown to facilitate their protonation by the internal acids.59 The same geometric distortion as in the structure with a doubly occupied LUMO was found for an excited triplet state, implying that several types of excitation may in principle be relevant to dinitrogen photoactivation. The character of the orbitals involved in the activation process in this study are dominated by the M2N2 core, regardless of the metal and the protonation state of the ligand. There is a striking resemblance to many other complexes of similar geometry for which electronic structure analyses have since been published, e.g. for several of the Nishibayashi systems in which the HOMO is of M–N π* and N–N π character.58,90 Of course, other complexes are known to have different HOMO and LUMO character, e.g. the Laplaza–Cummins system in which the HOMO is of M–N π and N–N π* and the LUMO is of M–N π* and N–N π character, as characterised by DFT later.56 As noted in Reiher et al.,59 the vertical excitation into a N2 π*orbital may be a universal process that enables dinitrogen activation and subsequent protonation for many linear M2-μ-N2 complexes – provided of course they have an appropriate electronic structure.
Theoretical studies on the mechanism of thermal dinitrogen splitting with the Laplaza/Cummins complex have shown that the steric bulk of the ligands is important for stabilising the [(NRAr)3Mo(N2)] adduct, and that the energetics of the reaction are sensitive to the ligand and metal properties.91 The [(NRAr)3Mo(μ-N2)Mo(NRAr)3] dimer is a triplet in its electronic ground state according to magnetic susceptibility measurements,56 although computational studies indicate that ligand rotations may result in a favourable singlet ground state depending on the nature of R (tBu or iPr).92 Several independent studies have found that a zig-zag transition state, i.e. substantial deviation from a linear Mo–N–N–Mo core with Mo–N–N angles of ca. 130° (Fig. 2b), is formed en route to cleaving the N–N bond.91,93 This structure has a significantly elongated N–N separation of ca. 1.56 Å compared with 1.20 Å in the ground state, and undergoes N–N scission.91 The importance of a careful design of the metal environment was emphasised early by Morokuma and coworkers: a Mo dimer with an amine ligand trans to the μ-N2 bridge does not split N2,† likely due to the competing σ-donation effects of the amine with the N2 ligand.
The Laplaza/Cummins system is also the only one for which the photophysical and photochemical processes underpinning dinitrogen photoactivation have been investigated experimentally.75 Having observed two photoproducts, indicative of a bifurcated reaction path (see above),56 a subsequent time-resolved spectroscopy study75 set out to map the processes at greater time resolution (60 fs). The initial excitation at 2.3 eV primarily results in the population of the second excited triplet state from the triplet ground state, shifting electron density from an orbital with Mo–N π and N–N π* character to one with predominant Mo–N π* and N–N π character according to DFT calculations. The excited state decays rapidly via internal conversion (IC) to the ground state (300 fs), followed by vibrational cooling (<1 ps), and the dissociation takes place before complete vibrational cooling is achieved, Fig. 6b.75 The authors rationalise the rapid deactivation of the excited state not with a dissociative excited state that returns to the ground state via solvent cage recombination, but rather with direct internal conversion to a vibrationally excited ground state. This assessment is based on the excited state not having the σ* character and high yields usually seen for such impulsive bond breaking processes, as well as the nearly equal amounts of N–N and Mo–N cleavage products observed. If the excited state did indeed break the Mo–N bond and recombined to form the dimer in the solvent cage, equal yields of N–N and Mo–N cleavage products would only be consistent with equal barriers for these two bond cleavage processes – whereas in fact it is known that N–N cleavage is associated with higher barriers in the ground state than Mo–N cleavage. The authors thus consider the most likely scenario that bond cleavage and vibrational energy relaxation compete once the system has reached the ground state via IC.75 Relaxation to the ground state is dominant, and Mo–N and N–N bond cleavage processes each make up ca. 2% of the dissociation channels, Fig. 6b. The fact that equal amounts of Mo–N and N–N bond dissociation products are observed is presumably due to the vibrational energy distribution during IC, which does not follow the statistics expected from thermal barriers. This inherent bias towards N–N cleavage rather than Mo–N cleavage is exciting, as it implies that modifications of the system that impede vibrational relaxation and dissociation into reactants would improve the overall yield of the Mo–N product.75
Sita and coworkers recently noted that the photochemical reactions of the tungsten and molybdenum complexes ([L(Cp*)M(μ-N2)M(Cp*)L], L = N(iPr)C(Me)N(iPr)) are slow and suffer from poor efficiency and atom economy. An additional obstacle to further developing this chemistry was the unknown mechanism of the photochemical pathway.94 Having identified a correlation between the ligand size and the degree of N2 activation in Hf and Ta complexes, they reduced the steric bulk of the ligand (N(iPr)C(Me)N(iPr) to N(Et)C(Ph)N(Et)) and found that, as expected, the Mo and W complexes switched to thermal dinitrogen cleavage.94 Along the thermal route, the bis-μ-N-bridged MoV dimer [Cp*LMoV(μ-N)2MoVLCp*] with L = N(Et)C(Ph)N(Et) is found, the analogue of which with L = N(iPr)C(Me)N(iPr) has been crystallised from the photochemical dinitrogen cleavage reaction (see Fig. 4). The isomerisation coordinate of a simplified model of the analogous Ta dimer was suggested to involve an end-on/side-on bridging core, M2(μ–η2:η1-N2), and a side-on/side-on bridging dimer, M2(μ–η2:η2-N2).93,95 It is currently unclear whether the first intermediate with M2(μ–η2:η1-N2) is also relevant to the thermal isomerisation of the molybdenum dimer, although the otherwise similar chemistry of the Sita series certainly suggests that this may be the case. This would then open the question of whether such a side-on/end-on intermediate is also reached photochemically when irradiating ([L(Cp*)M(μ-N2)M(Cp*)L], L = N(iPr)C(Me)N(iPr)), as opposed to a ‘zig-zag’-intermediate. Clearly, these are not the only possibilities, an obvious alternative being direct photoisomerisation into the M2(μ-N)2 core, which would be consistent with the observation of an irradiation induced change in coordination mode from end-on to side-on in isolated TiN2 complexes.96
Computationally, the characterisation of photochemical reaction coordinates for transition metal complexes is highly challenging. The first computational study on a complex that is known to be active in dinitrogen photoactivation focused on the [Os(II,III)2(μ-N2)(NH3)10]5+ dimer by Kunkely and Vogler.60 Using multireference CASPT2/CASSCF methods considering spin–orbit coupling, we characterised the electronic ground state and excited doublet and quartet states at the equilibrium geometry, and the evolution of the electronic structure of the twelve lowest-lying states along the N–N dissociation coordinate. It is clear from a purely MO-based picture that a molecular orbital of N–N σ* character would be the one that is energetically most affected along the dissociation coordinate, Fig. 7. Indeed, a stepwise increase in N–N distance resulted in drastic changes of the electronic structure, in particular related to electronic states in which an orbital of σ character between osmium and μ-N atoms and σ* character between the two μ-N bridges becomes first singly, then doubly occupied. Additionally, the in silico description of the N–N separation process allows for a quantitative evaluation of electronic structure descriptors such as bond orders and relative orbital occupation numbers.60 Clearly, more research is needed to fully map other dissociation coordinates and competing processes. However, it is of note for the following discussion that in our computational analysis of this osmium dimer a significant number of doubly excited determinants were seen below the experimentally determined energy that induces N–N scission.60
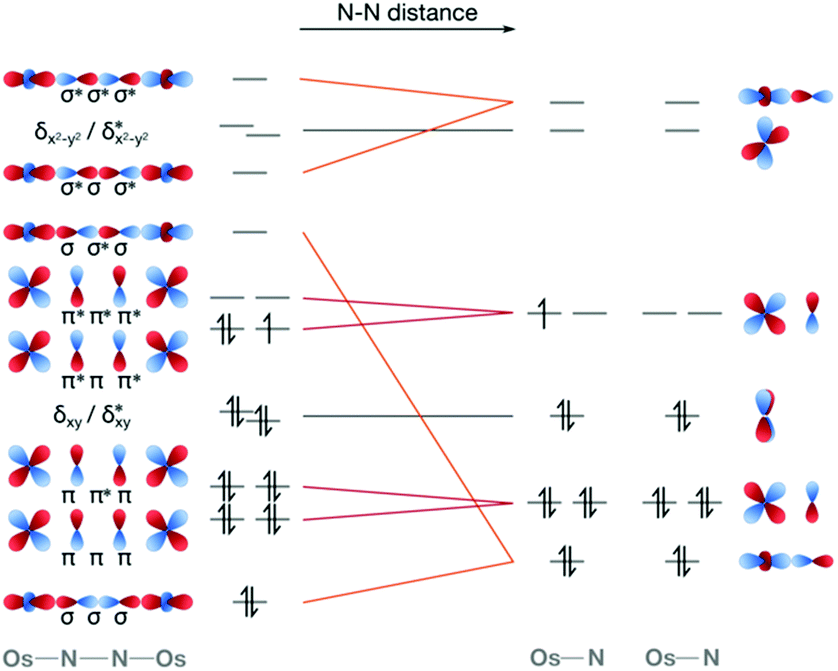 | ||
| Fig. 7 Molecular orbital scheme of the linear dinitrogen cleavage coordinate from a Os(II,III)2(μ-N)2 core with octahedrally coordinated metals as in [Os(II,III)2(μ-N2)(NH3)10]5+ to two independent OsN monomers. | ||
Having presented an overview of the currently available experimental and computational insights into dinitrogen photoactivation, we can now differentiate two scenarios for light-induced activation of the N–N triple bond and suggest types of excitation that would most likely facilitate the implementation of these scenarios. Firstly, in the scenario of Reiher et al. the protonation of the μ-N bridges requires an activated bridge with an onset of lone pair formation. This may be achieved with excitations into N–N π* orbitals (Fig. 8, left). When aiming at full dinitrogen photocleavage, the excitation should probably aim at orbitals of N–N σ* character, which will most likely lie higher in energy than the π* orbitals as shown schematically on the right of Fig. 8. Alternatively, an excited state with a different type of acceptor orbital may lead to a geometry in which an MO of significant N–N σ* character is lowered sufficiently in energy to allow its occupation, which would be a more indirect route to dinitrogen photocleavage.
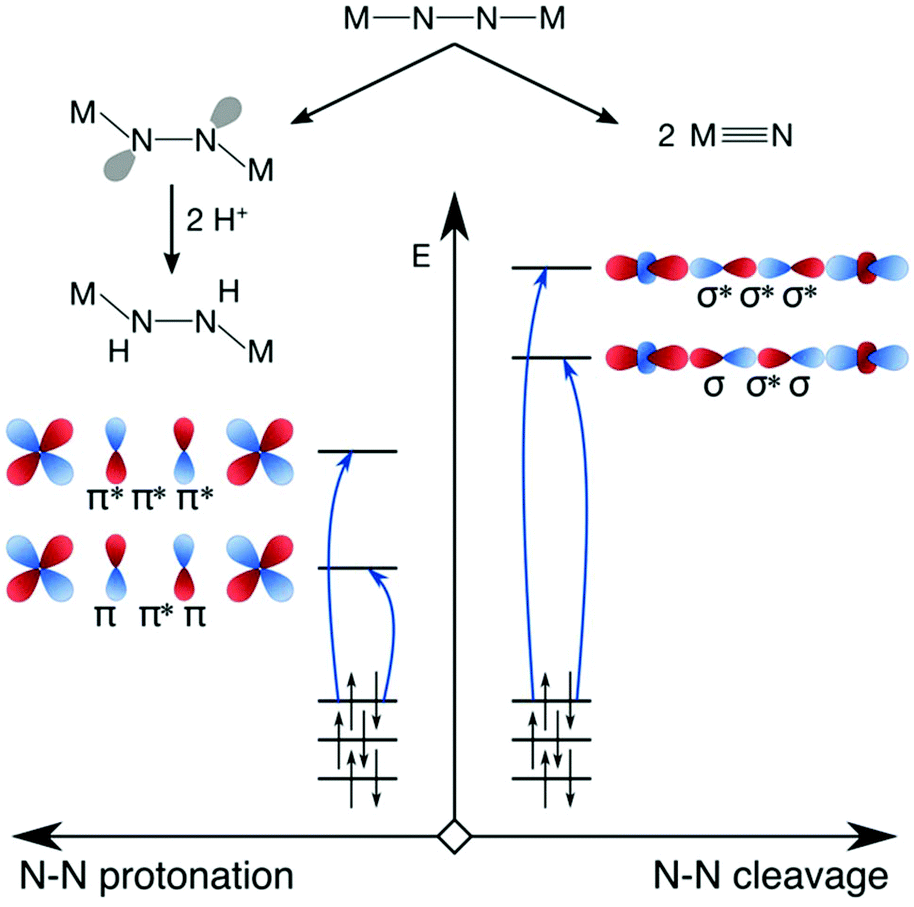 | ||
| Fig. 8 Hypothetical scenarios for dinitrogen photoactivation that would lead to μ-N protonation (left) and N–N scission (right). The character of the donor orbitals is discussed in the text. | ||
For either scenario, one may also consider which type of orbital the electron(s) should ideally originate from to maximise the degree of activation. In the first scenario, distortion of the molecule into a zig-zag geometry can be achieved by a charge-transfer excitation from a ligand-based orbital as shown by Reiher et al. It can also be hypothesized that an excitation from a N–N π orbital might be effective, as it would weaken the N–N interaction twofold. With the N–N σ MO as the dominant remaining bonding interaction between the N–N bridges, rotations about this axis might be less hindered than in the ground state. For full N–N photoscission into two metal nitrido complexes, the most effective donor orbital would likely be one of N–N bonding character (σ or π), as this would deplete a bonding orbital of electrons while populating an antibonding orbital.
These considerations are based on a rather simplified one-electron picture that considers the ground state molecular orbitals, rather than the actual excited state which may of course have significant multireference character. Furthermore, any coupling between close-lying excited states, population transfer during the dynamic relaxation processes and vibrational relaxation are not taken into account, meaning that other types of excitation may lead to either scenario if dynamic processes induce particular distortions or vibrations that lower the above-mentioned virtual orbitals such that their occupation becomes possible. Nevertheless, the chemically intuitive ground state molecular orbital point of view may help in identifying other systems that might be amenable to either of the dinitrogen photoactivation coordinates. For instance, if particularly bright and/or isolated bands in the UV-vis spectrum of a compound are due to excitations that are dominated by configurations in which such orbitals are singly or doubly occupied, the candidate may be a good test case for dinitrogen photoactivation.
Conclusions
This contribution was aimed at summarising the current status of research into dinitrogen photoactivation. The six complexes known to be capable of dinitrogen photoactivation share as a common characteristic of a linear M–N–N–M core, in which the metal ions are tetrahedrally or octahedrally coordinated. There is direct evidence for a bifurcated reaction pathway in one of these complexes, resulting in products due to the desired N–N as well as the unproductive M–N bond cleavage. The reaction products of other dinitrogen photoactivation experiments indicate that bifurcated reaction pathways may be a common feature of this reaction. The main challenge appears to be the suppression of undesired side reactions while maximising the yield of metal-nitrido or NRx products. To date, two computational studies have begun to explore the electronic structures of the ground and excited states of complexes that are relevant to dinitrogen photoactivation. Compared with thermal dinitrogen activation, however, this line of research is relatively unexplored territory, and detailed experimental studies on all photochemical and photophysical processes would certainly be desirable to further advance our understanding of the underlying mechanism(s).While acknowledging that the excited state dynamics of the entire dinitrogen photoactivation process will inevitably be very challenging to resolve, an analysis of the known dinitrogen photocleavage catalysts has highlighted features that may help to identify more complexes susceptible to light-induced N2 activation: if electronic excitations into N–N antibonding orbitals are associated with high spectral intensities, irradiation of these bands may lead to an excited state distortion of ‘zig-zag’ shape and facilitate protonation of the μ-N atoms in case of N–N π* character, while for N–N σ* character the excited state may decay into metal nitrido complexes. From a computational point of view, the steps toward unravelling the functional principles of photochemical dinitrogen activation include the identification of relevant excited states, mapping possible dissociation pathways, and studying the dynamic dissociation processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out with a 50th Anniversary Prize Fellowship from the University of Bath and was initially supported by a H2020 Marie-S.-Curie Individual Fellowship of the European Commission, call H2020-MSCA-IF-2015, proposal number 703860, title: Solar Dinitrogen Activation, acronym: SolarAct. The COST action CM1305 “Explicit Control Over Spin-states in Technology and Biochemistry (ECOSTBio)” is gratefully acknowledged for network support.Notes and references
- J. Chatt and G. J. Leigh, Chem. Soc. Rev., 1972, 1, 121–144 RSC.
- F. Haber, in Fünf Vorträge aus den Jahren 1920–1923, 1924, vol. 1, pp. 1–24 Search PubMed.
- G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS PubMed.
- O. Einsle, F. A. Tezcan, S. L. Andrade, B. Schmid, M. Yoshida, J. B. Howard and D. C. Rees, Science, 2002, 297, 1696–1700 CrossRef CAS PubMed.
- K. M. Lancaster, M. Roemelt, P. Ettenhuber, Y. Hu, M. W. Ribbe, F. Neese, U. Bergmann and S. DeBeer, Science, 2011, 334, 974–977 CrossRef CAS PubMed.
- T. Spatzal, M. Aksoyoglu, L. Zhang, S. L. Andrade, E. Schleicher, S. Weber, D. C. Rees and O. Einsle, Science, 2011, 334, 940 CrossRef CAS PubMed.
- C. E. Laplaza and C. C. Cummins, Science, 1995, 268, 861–863 CAS.
- E. Solari, C. Da Silva, B. Iacono, J. Hesschenbrouck, C. Rizzoli, R. Scopelliti and C. Floriani, Angew. Chem., Int. Ed., 2001, 40, 3907–3909 CrossRef CAS PubMed.
- N. Cherkasov, A. O. Ibhadon and P. Fitzpatrick, Chem. Eng. Process. Process Intensif., 2015, 90, 24–33 CrossRef CAS.
- R. Schlögl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef PubMed.
- S. Paul, N. Cox and D. A. Pantazis, Inorg. Chem., 2017, 56, 3875–3888 CrossRef CAS PubMed.
- V. Krewald, M. Retegan and D. A. Pantazis, Top. Curr. Chem., 2016, 371, 23–48 CrossRef CAS PubMed.
- I. Djurdjevic, O. Einsle and L. Decamps, Chem. – Asian J., 2017, 12, 1447–1455 CrossRef CAS PubMed.
- C. C. Lee, M. W. Ribbe and Y. Hu, Met. Ions Life Sci., 2014, 14, 147–176 CAS.
- B. M. Hoffman, D. Lukoyanov, Z. Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- I. Klopsch, M. Finger, C. Würtele, B. Milde, D. B. Werz and S. Schneider, J. Am. Chem. Soc., 2014, 136, 6881–6883 CrossRef CAS PubMed.
- M. G. Scheibel, J. Abbenseth, M. Kinauer, F. W. Heinemann, C. Wurtele, B. de Bruin and S. Schneider, Inorg. Chem., 2015, 54, 9290–9302 CrossRef CAS PubMed.
- G. A. Silantyev, M. Förster, B. Schluschaß, J. Abbenseth, C. Würtele, C. Volkmann, M. C. Holthausen and S. Schneider, Angew. Chem., Int. Ed., 2017, 56, 5872–5876 CrossRef CAS PubMed.
- R. Bjornsson, F. Neese, R. R. Schrock, O. Einsle and S. DeBeer, J. Biol. Inorg. Chem., 2015, 20, 447–460 CrossRef CAS PubMed.
- I. Dance, Dalton Trans., 2010, 39, 2972–2983 RSC.
- B. M. Barney, H. I. Lee, P. C. Dos Santos, B. M. Hoffman, D. R. Dean and L. C. Seefeldt, Dalton Trans., 2006, 2277–2284 RSC.
- B. M. Hoffman, D. Lukoyanov, D. R. Dean and L. C. Seefeldt, Acc. Chem. Res., 2013, 46, 587–595 CrossRef CAS PubMed.
- Y. Hu and M. W. Ribbe, Angew. Chem., Int. Ed., 2016, 55, 8216–8226 CrossRef CAS PubMed.
- R. J. Burford and M. D. Fryzuk, Nat. Rev. Chem., 2017, 1, 0026 CrossRef.
- J. M. Smith, in Progress in Inorganic Chemistry, John Wiley & Sons, Inc., 2014, vol. 58, pp. 417–470 Search PubMed.
- C. Rebreyend and B. de Bruin, Angew. Chem., Int. Ed., 2015, 54, 42–44 CrossRef CAS PubMed.
- B. Peigne and G. Aullon, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, 71, 369–386 CAS.
- P. L. Holland, Dalton Trans., 2010, 39, 5415–5425 RSC.
- M. D. Fryzuk and S. A. Johnson, Coord. Chem. Rev., 2000, 200–202, 379–409 CrossRef CAS.
- R. M. Badger, J. Chem. Phys., 1934, 2, 128–131 CrossRef CAS.
- A. J. Keane, W. S. Farrell, B. L. Yonke, P. Y. Zavalij and L. R. Sita, Angew. Chem., Int. Ed., 2015, 54, 10220–10224 CrossRef CAS PubMed.
- M. E. Vol'Pin and V. B. Shur, Nature, 1966, 209, 1236–1236 CrossRef.
- D. V. Yandulov and R. R. Schrock, J. Am. Chem. Soc., 2002, 124, 6252–6253 CrossRef CAS PubMed.
- D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76–78 CrossRef CAS PubMed.
- M. Kol, R. R. Schrock, R. Kempe and W. M. Davis, J. Am. Chem. Soc., 1994, 116, 4382–4390 CrossRef CAS.
- D. Sellmann and J. Sutter, J. Biol. Inorg. Chem., 1996, 1, 587–593 CrossRef CAS.
- D. Sellmann and J. Sutter, Acc. Chem. Res., 1997, 30, 460–469 CrossRef CAS.
- D. Sellmann, J. Utz, N. Blum and F. W. Heinemann, Coord. Chem. Rev., 1999, 190–192, 607–627 CrossRef CAS.
- D. Sellmann, W. Soglowek, F. Knoch and M. Moll, Angew. Chem., Int. Ed. Engl., 1989, 28, 1271–1272 CrossRef.
- D. Sellmann, B. Hautsch, A. Rösler and F. W. Heinemann, Angew. Chem., Int. Ed., 2001, 40, 1505–1507 CrossRef CAS PubMed.
- D. Sellmann, A. Hille, A. Rosler, F. W. Heinemann, M. Moll, G. Brehm, S. Schneider, M. Reiher, B. A. Hess and W. Bauer, Chem. – Eur. J., 2004, 10, 819–830 CrossRef CAS PubMed.
- Y. Nishibayashi, Dalton Trans., 2012, 41, 7447–7453 RSC.
- K. Arashiba, E. Kinoshita, S. Kuriyama, A. Eizawa, K. Nakajima, H. Tanaka, K. Yoshizawa and Y. Nishibayashi, J. Am. Chem. Soc., 2015, 137, 5666–5669 CrossRef CAS PubMed.
- S. Kuriyama, K. Arashiba, K. Nakajima, H. Tanaka, K. Yoshizawa and Y. Nishibayashi, Eur. J. Inorg. Chem., 2016, 2016, 4856–4861 CrossRef.
- A. Eizawa, K. Arashiba, H. Tanaka, S. Kuriyama, Y. Matsuo, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2017, 8, 14874 CrossRef CAS PubMed.
- Y. Roux, C. Duboc and M. Gennari, ChemPhysChem, 2017, 18, 2606–2617 CrossRef CAS PubMed.
- M. Holscher and W. Leitner, Chem. – Eur. J., 2017, 23, 11992–12003 CrossRef PubMed.
- M. J. Bezdek and P. J. Chirik, Angew. Chem., Int. Ed., 2016, 55, 7892–7896 CrossRef CAS PubMed.
- R. J. Burford, A. Yeo and M. D. Fryzuk, Coord. Chem. Rev., 2017, 334, 84–99 CrossRef CAS.
- G. P. Connor and P. L. Holland, Catal. Today, 2017, 286, 21–40 CrossRef CAS PubMed.
- A. Eizawa and Y. Nishibayashi, in Nitrogen Fixation, Springer Verlag, 2017, 60(10), pp. 153–169 Search PubMed.
- S. Kuriyama and Y. Nishibayashi, in Nitrogen Fixation, Springer Verlag, 2017, 60(5), 215–234 Search PubMed.
- F. Tuczek, in Molybdenum and Tungsten Enzymes, Royal Society of Chemistry, 2016, vol. 6, pp. 223– 274 Search PubMed.
- H. Tanaka and K. Yoshizawa, in Nitrogen Fixation, Springer Verlag, 2017, 60(7), 171–196 Search PubMed.
- I. Fischler and E. K. von Gustorf, Naturwissenschaften, 1975, 62, 63–70 CrossRef CAS.
- J. J. Curley, T. R. Cook, S. Y. Reece, P. Müller and C. C. Cummins, J. Am. Chem. Soc., 2008, 130, 9394–9405 CrossRef CAS PubMed.
- H. Kunkely and A. Vogler, Angew. Chem., Int. Ed., 2010, 49, 1591–1593 CrossRef CAS PubMed.
- T. Miyazaki, H. Tanaka, Y. Tanabe, M. Yuki, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Angew. Chem., Int. Ed., 2014, 53, 11488–11492 CrossRef CAS PubMed.
- M. Reiher, B. Kirchner, J. Hutter, D. Sellmann and B. A. Hess, Chem. – Eur. J., 2004, 10, 4443–4453 CrossRef CAS PubMed.
- V. Krewald and L. Gonzalez, Chem. – Eur. J., 2018, 24, 5112–5123 CrossRef CAS PubMed.
- A. J. Medford and M. C. Hatzell, ACS Catal., 2017, 7, 2624–2643 CrossRef CAS.
- K. Hoshino, M. Inui, T. Kitamura and H. Kokado, Angew. Chem., Int. Ed., 2000, 39, 2509–2512 CrossRef CAS PubMed.
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS PubMed.
- T. Oshikiri, K. Ueno and H. Misawa, Angew. Chem., 2016, 55, 3942–3946 CrossRef CAS PubMed.
- Y. Hao, X. Dong, S. Zhai, H. Ma, X. Wang and X. Zhang, Chem. – Eur. J., 2016, 22, 18722–18728 CrossRef CAS PubMed.
- J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2017, 139, 4390–4398 CrossRef CAS PubMed.
- C.-M. Che, H.-W. Lam, W.-F. Tong, T.-F. Lai and T.-C. Lau, J. Chem. Soc., Chem. Commun., 1989, 1883–1884 RSC.
- D. C. Ware and H. Taube, Inorg. Chem., 1991, 30, 4605–4610 CrossRef CAS.
- H.-W. Lam, C.-M. Che and K.-Y. Wong, J. Chem. Soc., Dalton Trans., 1992, 1411–1416 RSC.
- O. Krahe, E. Bill and F. Neese, Angew. Chem., Int. Ed., 2014, 53, 8727–8731 CrossRef CAS PubMed.
- M. G. Scheibel, Y. Wu, A. C. Stuckl, L. Krause, E. Carl, D. Stalke, B. de Bruin and S. Schneider, J. Am. Chem. Soc., 2013, 135, 17719–17722 CrossRef CAS PubMed.
- M. G. Scheibel, B. Askevold, F. W. Heinemann, E. J. Reijerse, B. de Bruin and S. Schneider, Nat. Chem., 2012, 4, 552–558 CrossRef CAS PubMed.
- Y. Gloaguen, C. Rebreyend, M. Lutz, P. Kumar, M. Huber, J. I. van der Vlugt, S. Schneider and B. de Bruin, Angew. Chem., Int. Ed., 2014, 53, 6814–6818 CrossRef CAS PubMed.
- T. M. Buscagan, P. H. Oyala and J. C. Peters, Angew. Chem., Int. Ed., 2017, 56, 6921–6926 CrossRef CAS PubMed.
- A. S. Huss, J. J. Curley, C. C. Cummins and D. A. Blank, J. Phys. Chem. B, 2013, 117, 1429–1436 CrossRef CAS PubMed.
- M. J. A. Johnson, P. M. Lee, A. L. Odom, W. M. Davis and C. C. Cummins, Angew. Chem., Int. Ed. Engl., 1997, 36, 87–91 CrossRef CAS.
- M. Hirotsu, P. P. Fontaine, P. Y. Zavalij and L. R. Sita, J. Am. Chem. Soc., 2007, 129, 12690–12692 CrossRef CAS PubMed.
- M. Hirotsu, P. P. Fontaine, A. Epshteyn, P. Y. Zavalij and L. R. Sita, J. Am. Chem. Soc., 2007, 129, 9284–9285 CrossRef CAS PubMed.
- B. L. Yonke, A. J. Keane, P. Y. Zavalij and L. R. Sita, Organometallics, 2011, 31, 345–355 CrossRef.
- L. M. Duman and L. R. Sita, J. Am. Chem. Soc., 2017, 139, 17241–17244 CrossRef CAS PubMed.
- H. J. Himmel and M. Reiher, Angew. Chem., Int. Ed., 2006, 45, 6264–6288 CrossRef CAS PubMed.
- G. C. Stephan, C. Sivasankar, F. Studt and F. Tuczek, Chem. – Eur. J., 2008, 14, 644–652 CrossRef CAS PubMed.
- J. L. Crossland and D. R. Tyler, Coord. Chem. Rev., 2010, 254, 1883–1894 CrossRef CAS.
- N. Khoenkhoen, B. de Bruin, J. N. H. Reek and W. I. Dzik, Eur. J. Inorg. Chem., 2015, 2015, 567–598 CrossRef CAS.
- H. Tanaka, Y. Nishibayashi and K. Yoshizawa, Acc. Chem. Res., 2016, 49, 987–995 CrossRef CAS PubMed.
- F. Studt and F. Tuczek, Angew. Chem., Int. Ed., 2005, 44, 5639–5642 CrossRef CAS PubMed.
- J. Rittle and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 3161–3170 CrossRef CAS PubMed.
- C. J. Pickett, JBIC, J. Biol. Inorg. Chem., 1996, 1, 601–606 CrossRef CAS.
- R. R. Schrock, Acc. Chem. Res., 2005, 38, 955–962 CrossRef CAS PubMed.
- H. Tanaka, K. Arashiba, S. Kuriyama, A. Sasada, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2014, 5, 3737 Search PubMed.
- G. Christian, J. Driver and R. Stranger, Faraday Discuss., 2003, 124, 331–341 RSC.
- G. Christian, R. Stranger, B. F. Yates and D. C. Graham, Dalton Trans., 2005, 962–968 RSC.
- Q. Cui, D. G. Musaev, M. Svensson, S. Sieber and K. Morokuma, J. Am. Chem. Soc., 1995, 117, 12366–12367 CrossRef CAS.
- L. M. Duman, W. S. Farrell, P. Y. Zavalij and L. R. Sita, J. Am. Chem. Soc., 2016, 138, 14856–14859 CrossRef CAS PubMed.
- W. Zhang, Y. Tang, M. Lei, K. Morokuma and D. G. Musaev, Inorg. Chem., 2011, 50, 9481–9490 CrossRef CAS PubMed.
- M. Chen, G. Wang and M. Zhou, Chem. Phys. Lett., 2005, 409, 70–74 CrossRef CAS.
Footnote |
| † Dinitrogen splitting with this molybdenum dimer, [(tBuMe2SiNCH2CH2)3Mo(μ-N2)Mo(tBuMe2SiNCH2CH2)3], is computed to be endothermic by 10 kcal mol−1.93 |
| This journal is © The Royal Society of Chemistry 2018 |