
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An investigation of the roles furan versus thiophene π-bridges play in donor–π-acceptor porphyrin based DSSCs†
Michele
Cariello‡
 a,
Saifaldeen M.
Abdalhadi‡
a,
Pankaj
Yadav
b,
Jean-David
Decoppet
b,
Shaik M.
Zakeeruddin
*b,
Michael
Grätzel
b,
Anders
Hagfeldt
c and
Graeme
Cooke
*a
a,
Saifaldeen M.
Abdalhadi‡
a,
Pankaj
Yadav
b,
Jean-David
Decoppet
b,
Shaik M.
Zakeeruddin
*b,
Michael
Grätzel
b,
Anders
Hagfeldt
c and
Graeme
Cooke
*a
aWestCHEM, School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: graeme.cooke@glasgow.ac.uk
bLaboratory of Photonics and Interfaces, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland. E-mail: shaik.zakeer@epfl.ch
cLaboratory of Photomolecular Science, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland
First published on 26th April 2018
Abstract
Dye-sensitized solar cells (DSSCs) continue to attract interest due to their lower cost production compared to silicon based solar cells and their improving power conversion efficiencies. Porphyrin-based sensitizers have become an important sub-class due to their strong absorption characteristics in the visible region, convenient modulation of properties through synthetic manipulation and class-leading power conversion efficiencies. In this article, we report the synthesis and characterization of two porphyrin-based dyes and their application as sensitizers in DSSCs. A thiophene and a furan moiety have been incorporated into the push–pull architecture as a π-bridge, allowing the systematic investigation of how these moieties influence the physical properties of the dyes and the performance of their resulting DSSCs. A significant difference in PCEs has been observed, with the furan containing dye (PorF, PCE = 4.5%) being more efficient than the thiophene-based analogue (PorT, PCE = 3.6%) in conjunction with the iodide/triiodide redox electrolyte.
Introduction
Photovoltaics (PV) offer an attractive renewable energy technology and there is a continued interest in this field due to the potentially inexhaustible energy source provided by sun.1 During the last twenty years, interest in dye-sensitised solar cells (DSSCs) as promising PV technology has increased, due to their ease of fabrication, relatively low cost and interesting design possibilities in terms of tunable transparency and colour. Since the development of the first working device in 1991 by Grätzel and O'Reagan,2 there has been a huge effort directed towards increasing the power conversion efficiency (PCE) of devices by optimising their components, with a particular emphasis being placed on the sensitiser, as it determines the light harvesting and absorption properties of the device. Ruthenium-based dyes, such as N719, have monopolised the early progress in this field, however, metal-free organic dyes have become increasingly attractive due to their lower cost, together with higher molar extinction coefficients, easier synthesis, purification and structural modification.3More recently, the development of porphyrin-based dyes has revitalised research in metal-incorporating dyes, due to the employment of less expensive metals (such as Zn, instead of Ru), their synthetically tunable optical properties and good photostability.4–6 In particular, the position of these bands, together with the redox properties,7,8 can be tuned through the insertion of a metal in the central cavity9 and through the substitution of the porphyrin periphery, which can be done by functionalising the β-positions and the meso-positions. While zinc is the most preferred metal due to its low-cost and ability to broaden the optical absorption of porphyrins,10 the types of substituent that could be added to the peripheral positions to tune the properties of porphyrin derivatives is enormous.
Since the first report of a porphyrin-based sensitiser providing a PCE of 2.6%,11 significant effort has been directed towards understanding the role substituents play on modulating the device characteristics of porphyrin based DSSCs. Functionality attached to both β- and meso-positions have been investigated, with the latter typically providing the best performing dyes.12 However, the performance of this class of dyes is generally lower than that of the most efficient Ru-based dyes, due to their poor injection yields.13,14 To circumvent this issue, the development of meso-substituted porphyrin dyes featuring a D–π-A architecture represents a step forward regarding the performance of these molecules in DSSCs.15,16 In particular, substituting the porphyrin core with electron donating moieties and electron withdrawing units attached at opposite sides of the porphyrin ring, results in the generation of an intrinsic dipole moment which favours the electron injection of the dye into the conduction band of TiO2. Finally, the introduction of alkylated aromatic units17–19 at the two side meso-positions has led to a further improvement of performance, due to the minimisation of charge recombination arising from the dye aggregation.20,21 Thanks to these developments, the PCE of porphyrin-sensitised devices has grown rapidly and has provided the new record PCE of 13%.22 Nevertheless, despite the high efficiencies reported for some porphyrin dyes, the synthesis of unsymmetrical derivatives is problematic and generally results in low yielding multi-step procedures leading to the formation of several side-products.23,24
In this article, we report the synthesis of two porphyrin-based dyes (Fig. 1) and their employment as sensitisers in DSSCs. Both the molecules incorporate three triphenylamine (TPA) moieties as donor units and differ for the presence of either a thiophene or a furan ring in the π-bridge (A3B-type).12 The three bulky TPA units have been incorporated into the dyes in view of their combined electron-donating character and ability to negate dye aggregation on the TiO2 surface.12 Thiophene and furan residues have been incorporated into the π-bridge, allowing the role these heterocycles have in controlling device performance of this class of dyes. Thiophene and its derivatives have been extensively studied as π-spacers in sensitisers for DSSCs, due to their smaller resonance energy (29 kcal mol−1 for thiophene, 36 kcal mol−1 for benzene) which enhances the conjugation by lowering the energy required for charge transfer (CT).25,26 The employment of furan as π-spacer should further improve the dye performance, due to its lower aromatic energy (16 kcal mol−1). It has also been verified that furan shows comparable optical properties and charge carrier mobility with thiophene27 and better solubility when incorporated in polyconjugated systems.28 Moreover, the higher oxidation potential of furan should improve the hole location, hence the stability of the dye,29 and its PCE when compared to its thiophene analogue.27,30,31 However, previous work has shown that thiophene, rather than furan, when incorporated as π-bridges of porphyrin dyes results in better PCEs, presumably due to the more electron withdrawing character and/or improved interactions between the dye and TiO2 due to thiophene moiety.32–35
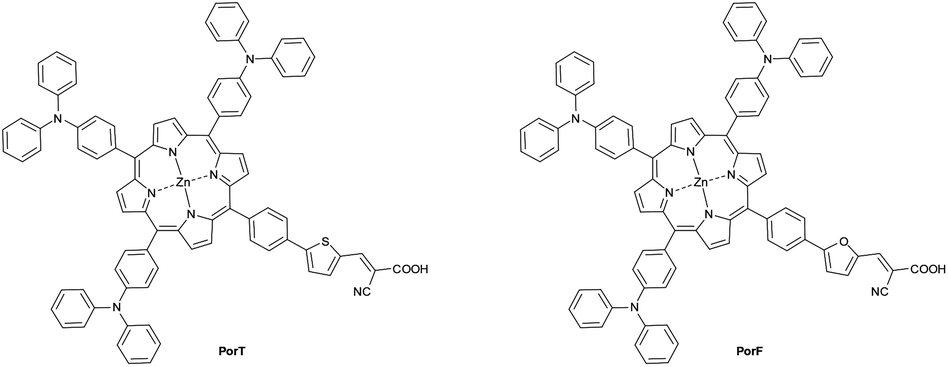 | ||
| Fig. 1 Chemical structure of dyes PorT and PorF. | ||
Results and discussion
Synthesis
Porphyrin based dyes PorT and PorF were synthesized according to Scheme 1. The synthesis of the central meso-substituted porphyrins was performed according to Lindsey's method36,37 (Scheme 1). 4-(Diphenylamino) benzaldehyde 1 and 4-bromobenzaldehyde 2 in 3.5/1 molar ratio were reacted with an excess of pyrrole to afford compound 3 in modest yield. This was then quantitatively converted to the Zn-based porphyrin 4, by reaction with zinc acetate. A Suzuki cross-coupling reaction between 4 and the boronic acids 5a and 5b, was carried out affording aldehydes 6a and 6b, respectively. These were then reacted with cyanoacetic acid through a Knoevenagel condensation to obtain the final dyes PorT and PorF.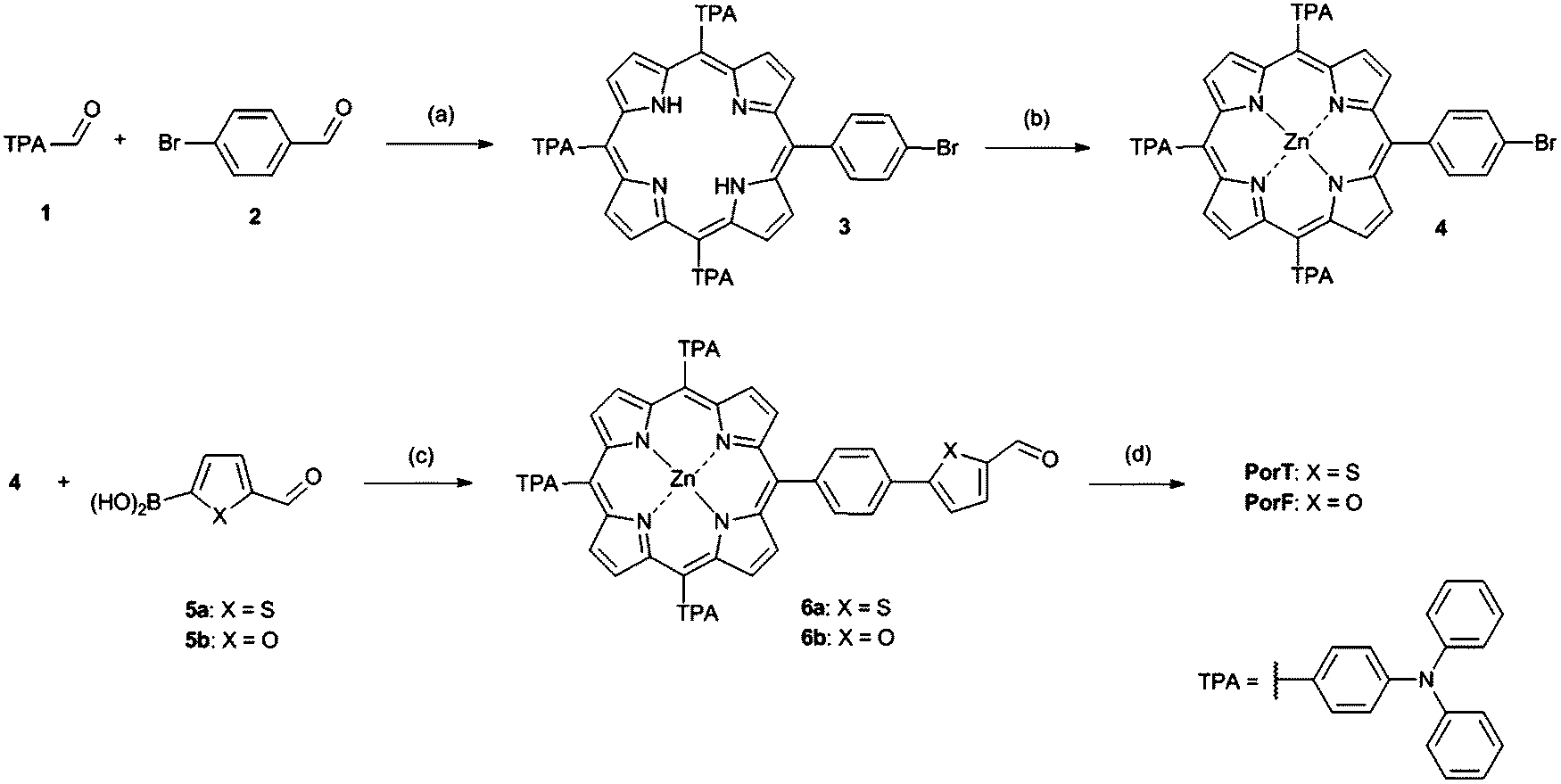 | ||
| Scheme 1 Synthesis of dyes PorT and PorF. Conditions: (a) Pyrrole, DDQ, BF3·O(C2H5)2, Et3N, DCM, r.t., 3 h; (b) Zn(OAc)2, MeOH, DCM, r.t., 18 h; (c) Pd(dppf)Cl2, K2CO3 (2 M), DME, 80 °C, 2 d; (d) cyanoacetic acid, Zn(OAc)2, THF, AcOH, 70 °C, 4 h. | ||
Optical and electrochemical properties
The UV-Vis absorption spectra of the two dyes recorded in solution (Fig. 2) show the typical behaviour of metalloporphyrins, consisting of an intense and sharp B-band and two weaker Q-bands.38 The former, at 438 nm for PorT and 426 nm for PorF, have an intensity of the order of 105 M−1 cm−1, whilst the latter, at 565 nm and 609 nm for PorT and 553 nm and 597 nm for PorF, are one order of magnitude weaker. The absorption maxima of PorT and PorF are at 650 nm and 625 nm, respectively, corresponding to optical bandgaps (EG,OPT) of 1.91 eV and 1.98 eV.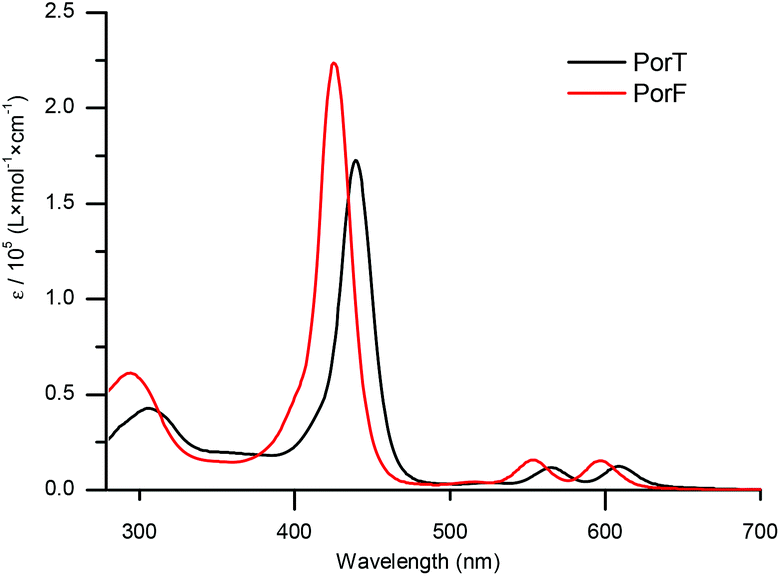 | ||
| Fig. 2 UV-vis absorption spectra of dyes PorT and PorF in DMF (C = 10−5 M). | ||
The solution-based electrochemical properties of both dyes were explored by cyclic voltammetry (CV, Fig. 3) and square wave voltammetry (SWV, ESI Fig. S1†). Both dyes undergo pseudo-reversible one-electron reduction and oxidation. SWV allowed us to estimate the ionization potential (IP), electron affinity (EA) and fundamental band gap (EG,F), respectively (Table 1). Both Ered and Eox of PorF are more positive than those of PorT, of about 100 mV (−1.72 and 0.31 V for PorT and −1.62 and 0.40 V for PorF), resulting in more negative energy values for IP and EA.
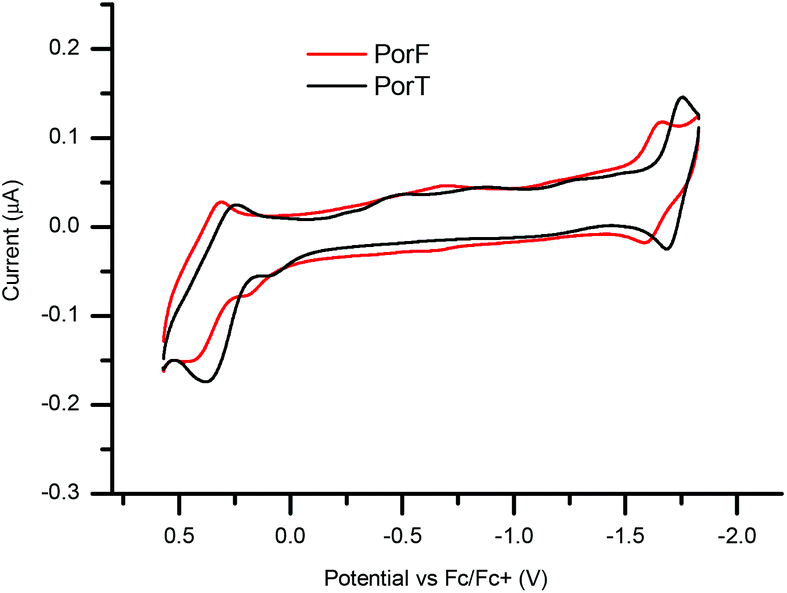 | ||
| Fig. 3 Cyclic voltammetry plots of dyes PorT and PorF in DMF (C = 10−3 M). | ||
| Dye |
E
G,OPT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a/eV a/eV |
I
Pb/eV (HOMOc/eV) |
E
Ab/eV (LUMOc/eV) |
E
G,Fd/eV |
|---|---|---|---|---|
| a Calculated using the equation: EG,OPT = 1240/λonset. b Estimated using the equations: IP (eV) = −4.8 − Eox and EA (eV) = −4.8 − Ered.44 c Values obtained by DFT calculation. d Calculated using the formula: EG,F = EA − IP. | ||||
| PorT | 1.9 | −5.1 (−5.13) | −3.1 (−3.07) | 2.0 |
| PorF | 2.0 | −5.2 (−5.14) | −3.2 (−3.02) | 2.0 |
Theoretical calculations
To gain insight in electronic properties of the two dyes, density functional theory (DFT) calculations were performed using Gaussian 09.39 The optimized geometry shows a similar conformation for both the molecules, with the dihedral angles between the porphyrin and the appended benzene moieties of about 60°. Although this limits the conjugation throughout the molecule, a twisted structure could effectively decrease the dye aggregation by obstructing the formation of π-stacks among the porphyrin units.40 Corresponding molecular orbitals for PorT and PorF are shown in Fig. 4. The HOMO is delocalised throughout the TPA donors and the porphyrin ring, while the LUMO is spread over the acceptor arm. This good separation is normally not found in porphyrin dyes having the π-bridge co-planar with the porphyrin core,41,42 indicating that the absence of the intramolecular CT band is likely due to the twisted geometry of the molecule. Fig. 4 also shows that the spatial distribution of the LUMO+1 is confined on the porphyrin core, suggesting that an optical transition between the HOMO and the LUMO+1 would be more favourable. The estimated energy of the transition is 2.54 eV and 2.64 eV, for PorT and PorF, respectively, corresponding to a wavelength of 488 nm and 469 nm. The values are in good approximation with the experimental offset of the Soret bands (S0 → S2) of both the dyes. This suggests that the π-bridge does not have a significant role in the light absorption, which presumably occurs due to the presence of the porphyrin core. However, the π-bridge should play a significant role in the extraction of electrons upon dye excitation.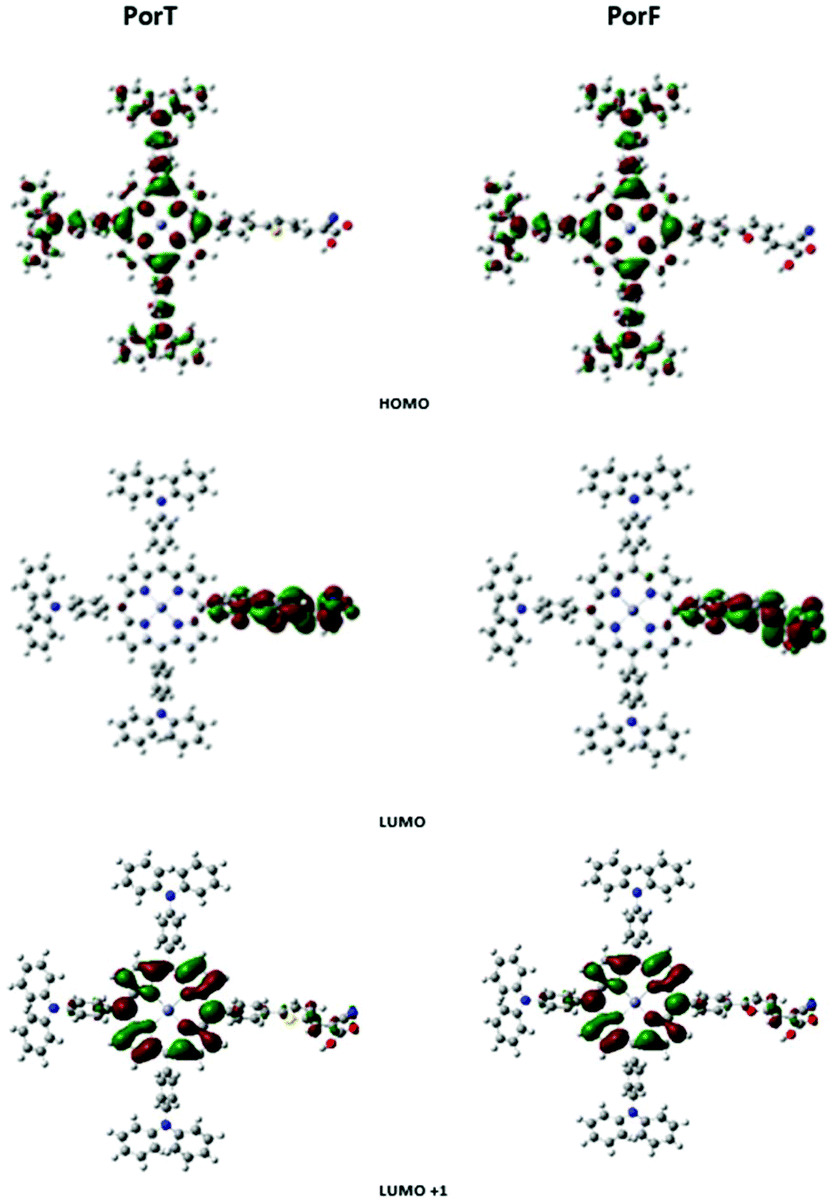 | ||
| Fig. 4 Frontier molecular orbital representation of PorT and PorF calculated by DFT. | ||
A summary of the properties of the two dyes is provided in Table 1. It is worth observing that the calculated properties are in good agreement with both the experimentally estimated ones and those calculated in previous work for similar molecules.43
Device performance
The above dyes were tested as sensitisers in DSSCs by applying cobalt and iodide/triiodide redox electrolytes on 4 μm + 4 μm double layer TiO2 films. The photovoltaic characteristics of these devices, A–D, measured at AM 1.5G irradiance (100 mW cm−2), are tabulated in Table 2. For dye PorF, Device A, the highest PCE obtained is 4.1% with a VOC of 0.700 V, a JSC of 7.7 mA cm−2 and a FF of 75%. In turn, device B exhibits VOC of 0.660 V, JSC of 8.9 mA cm−2, and PCE of 4.5%. Furthermore, employing the same cobalt and iodide/triiodide based redox electrolytes with PorT dye, labelled as devices C and D, respectively, the photovoltaic characteristics are tabulated in Table 2. The data show that having thiophene (PorT) closer to the surface of TiO2 enhances the recombination compared to furan (PorF) dye, which is reflected in the lower VOC of PorT dye with both electrolytes. To the best of our knowledge, these results are in contrast with related studies on porphyrin-based dyes. However, these results are in good agreement with several studies on metal-free push–pull dyes27,31,45 which showed an increase in VOC when the device is sensitised with a furan-based dye compared to its thiophene analogue. As suggested by the authors, this difference may be due to the enhanced suppression of the electron recombination between TiO2 and electrolyte.| Device | Dye | Electrolyte | J SC [mA cm−2] | V OC [mV] | FF (%) | PCE [%] |
|---|---|---|---|---|---|---|
| A | PorF | Co2+/Co3+ | 7.7 | 700 | 0.75 | 4.1 |
| B | PorF | I−/I3− | 8.9 | 660 | 0.71 | 4.5 |
| C | PorT | Co2+/Co3+ | 5.9 | 688 | 0.77 | 3.1 |
| D | PorT | I−/I3− | 8.1 | 606 | 0.74 | 3.6 |
| Ref | Y123 | I−/I3 | 12.7 | 749 | 0.76 | 7.3 |
| Ref | Y123 | Co2+/Co3+ | 12.7 | 808 | 0.78 | 8.0 |
The incident photon-to-current conversion efficiency (IPCE) spectrum of the devices is shown in Fig. 5a (inset). The IPCE spectra of both the devices A and B have peak maxima at 450 and 620 nm and the tail goes up to 700 nm. The IPCE spectrum reflects closely the absorption spectra of dyes. The integrated current of IPCE spectra of devices is close to the measured current. The difference in the HOMO level of these dyes with the cobalt electrolyte redox energy level is large enough to provide a driving force for the dye regeneration. Transient photovoltage characterisation under open circuit voltage was performed to understand the differences in the photovoltaic parameters of devices with two different electrolytes. The plot of charge vs. potential for the devices A and B shows similar behaviour but at the same charge density the VOC is higher for cobalt-based electrolyte (Fig. 5b) which is consistent with the difference in the energy level of these two electrolytes.
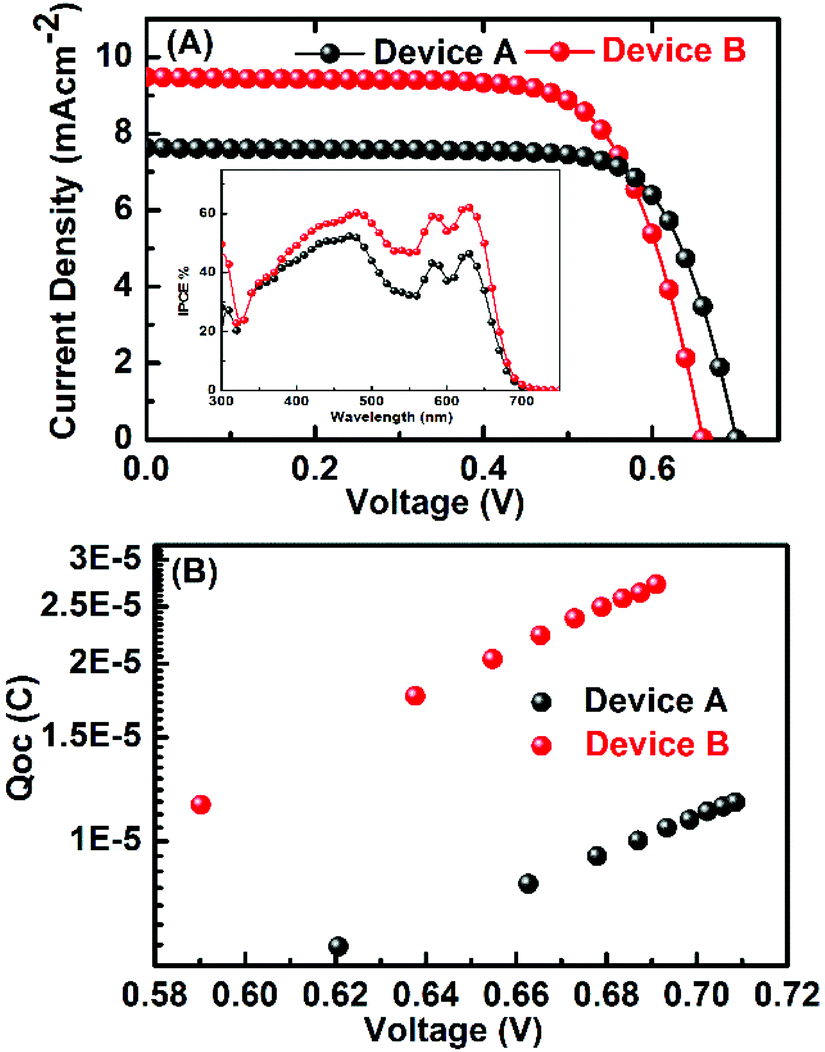 | ||
| Fig. 5 (a) Photocurrent density vs. voltage (J–V) curves of devices A and B with PorT under standard AM 1.5G illumination. The inset is the IPCE of devices. (b) Charge vs. VOC curves. | ||
Conclusions
In conclusion, two porphyrin dyes containing furan and thiophene as π-conjugated spacers have been synthesized. The systematic modification of the dyes has allowed us to investigate the role this has on the optical and redox properties of the dyes together with its influence on DSSC device performance. Modification of this type leads to a significant change in the dyes solution optical and redox properties, with PorT having a lower estimated optical band gap. PorF gave rise to the best performing DSSC devices in terms of power conversion efficiencies, which was independent of the electrolyte used. This work is in contrast with related studies that have indicated that thiophene-based bridges tend to give rise to superior power conversion efficiencies, and indicates that the role the structure of the π-conjugated spacer has in determining device characteristics of porphyrin based dyes is dependent upon the parent structure of the porphyrin.Experimental
General information
Chemicals were purchased from Sigma-Aldrich, TCI and Alfa Aesar, and were used without further purification. All reactions were run under an argon atmosphere. Solvents were purified using a PureSolv solvent purifier system. NMR spectra were performed with either a Bruker AVIII 400 MHz or a Bruker AVIII 500 MHz spectrometer and all reported chemical shifts are relative to TMS. Mass spectra were performed by the National Mass Spectroscopy Facility (NMSF), Swansea University (UK). UV-vis spectra were recorded on a PerkinElmer Lambda 25 instrument. Optically determined band gaps (EOPT) were estimated using the absorption edge of the longest wavelength absorption (λ) using EG,OPT (eV) = (1240/λ(nm)). Cyclic voltammetry measurements were undertaken using a CH Instruments 440A electrochemical analyser using a platinum working electrode, a platinum wire counter electrode and a silver wire pseudo-reference electrode. Ferrocene was used as an external standard and all redox couples are reported versus the ferrocene/ferrocenium (Fc/Fc+) redox couple, adjusted to 0.0 V. The solutions were prepared using dry DMF containing electrochemical grade tetrabutylammonium hexafluorophosphate (0.1 M) as the supporting electrolyte. The solutions were purged with nitrogen gas for 3 min prior to recording the electrochemical data.Theoretical calculations
Density functional theory (DFT) calculations were performed using Gaussian 09, revision D.01.39 Global minimum states were confirmed by absence from imaginary frequencies under ground-state geometry optimization followed by vibrational frequency calculations. All calculations were conducted with Becke's three-parameter hybrid and Lee–Yang–Parr's gradient corrected correlation (B3LYP) functional, 6-311G(d,p) basis set for H, C, N, O, S and Lanl2DZ for Zn, under vacuum.Device fabrication and testing
State-of-the-art double layer mesoporous TiO2 layer (4 μm thickness of 20 nm particle (DSL 18NR-T, DYESOL) plus 4 μm thickness of 400 nm light scattering particles (HPW-400NRD, CCIC)) were deposited on FTO conducting glass (Solar-4 mm, Nippon Sheet Glass Co, Ltd). The double layer TiO2 film was sensitized by immersing it into a solution of the PorF and PorT dyes (0.025 mM) and chenodeoxycholic acid (1.25 mM) in a tert-butanol/acetonitrile mixture (1:1 v/v) for 13 h at room temperature. A platinized FTO conducting glass (LOF TECH 7, Pilkington) was used as a counter electrode. The composition of the iodide based electrolyte (Z960) is 1.0 M 1,3-dimethylimidazolium iodide, 0.5 M tert-butylpyridine, 0.03 M iodine, 0.05 M LiI, 0.1 M GuNCS in acetonitrile:valeronitrile (85:15 v/v) and the composition of the cobalt electrolyte is 0.22 M [Co(bpy)3](TFSI)2, 0.05 M [Co(bpy)3](TFSI)3, 0.1 M LiClO4, and 0.2 M tert-butylpyridine in acetonitrile. Devices A and B were fabricated by using PorF with cobalt electrolyte and iodide/triiodide redox electrolyte, respectively. Devices C and D were fabricated by using PorT with cobalt electrolyte and iodide/triiodide redox electrolyte, respectively. An antireflection film (ARCTOP, Mihama Co.,) was attached on the photoanode side.
For photovoltaic measurements of the DSSCs, a solar simulator equipped with a 450 W xenon light source (Osram XBO 450) with a filter (Schott 113) was employed, whose power was regulated to the AM 1.5 solar standard by using a reference Si photodiode equipped with a colour-matched filter (KG-3, Schott) to reduce the mismatch in the region of 350–750 nm between the simulated light and AM 1.5 to less than 4%. The measurement-settling time between applying a voltage and measuring a current density for the J–V characterization of DSSCs was fixed to 80 ms with a Keithley model 2400 digital source meter. The photocurrent action spectra were measured with an Incident Photon-to-Current Conversion Efficiency (IPCE) test system. The modulation frequency used was about 2 Hz and light from a 300 W xenon lamp (ILC Technology, USA) was focused through a computer controlled Gemini-180 double monochromator (John Yvon Ltd, UK). A white light bias was used to bring the total light intensity on the device under testing closer to operating conditions. A light mask was used on the DSSCs, so the illuminated active area of DSCs was fixed to 0.159 cm2.
Synthesis
:DCM, 3:2), affording 3 (0.483 g, 15%) as a dark green powder. m.p. 212–214 °C; δH (500 MHz, CDCl3, TMS) 9.01 (m, 6H), 8.83 (d, J 4.7, 2H), 8.13–8.04 (m, 8H), 7.91 (d, J 8.3, 2H), 7.50–7.44 (m, 6H), 7.44–7.37 (m, 24H), 7.19–7.11 (m, 6H), −2.71 (s, 2H); δC (125 MHz, CDCl3, TMS) 148.0, 147.8, 141.5, 136.2, 136.1, 135.9, 135.8, 135.8, 130.07, 129.7, 125.1, 125.1, 125.1, 125.0, 123.5, 123.5, 122.6, 121.5, 121.5, 120.7, 120.4, 118.3; HRMS (NSI) m/z calcd for C80H57BrN7 [M + 2H]2+: 597.6963; found 597.6956.
:DCM, 1:4), affording 6b (75.8 mg, 75%) as a purple solid. m.p. 230–233 °C; δH (400 MHz, CDCl3, TMS) 9.76 (s, 1H), 9.13 (m, 6H), 8.98 (d, J 4.7, 2H), 8.33 (d, J 8.4, 2H), 8.23 (d, J 8.3, 2H), 8.13–8.06 (m, 6H), 7.47 (m, 7H), 7.42 (m, 24H), 7.18–7.10 (m, 7H); δC (100 MHz, CDCl3, TMS) 177.5, 159.8, 152.6, 150.8, 150.6, 150.6, 149.9, 148.1, 148.1, 147.6, 144.8, 136.8, 136.7, 135.6, 135.6, 135.3, 132.5, 132.4, 132.3, 132.3, 131.7, 129.7, 128.3, 125.0, 125.0, 123.8, 123.4, 121.6, 121.5, 121.5, 121.4, 119.8, 108.4; HRMS (MALDI) m/z calcd for C85H57N7O2Zn [M]+˙: 1271.3865; found 1271.3872.
:1, v:v). Ammonium acetate (29.9 mg, 0.388 mmol) and zinc acetate dihydrate (68.0 mg, 0.310 mmol) were added and the resulting mixture was stirred for 4 h at 70 °C under N2. The mixture was allowed to cool to r.t. and water (50 mL) was added. After stirring for further 10 min at r.t., the precipitate was filtered off, washed with water (50 mL) and dried under vacuum. The crude compound was purified by column chromatography (SiO2, DCM:MeOH, 9:1) affording PorT (0.100 g, 95%) as a dark green solid. m.p. 264–267 °C; δH (400 MHz, d6-DMSO, TMS, 80 °C) 8.96–8.90 (m, 6H), 8.31–8.25 (m, 3H), 8.19–8.15 (m, 2H), 8.12–8.04 (m, 6H), 7.50–7.43 (m, 12H), 7.41–7.34 (m, 18H), 7.21–7.14 (m, 6H); HRMS (NSI) m/z calcd for C88H59N8O2SZn [M + H]+: 1355.3768; found 1355.3770.
:1, v:v). Ammonium acetate (30.3 mg, 0.392 mmol) and zinc acetate dihydrate (68.9 mg, 0.314 mmol) were added and the resulting mixture was stirred for 4 h at 70 °C under N2. The mixture was allowed to cool to r.t. and water (50 mL) was added. After stirring for further 10 min at r.t., the precipitate was filtered off, washed with water (50 mL) and dried under vacuum. The crude compound was purified by column chromatography (SiO2, DCM:MeOH, 9:1) affording PorF (97.9 mg, 93%) as a dark green solid. m.p. 230–233 °C; δH (400 MHz, d6-DMSO, TMS) 8.97–8.93 (m, 6H), 8.87 (d, J 4.6, 2H), 8.30 (s, 4H), 8.11–8.06 (m, 6H), 7.90 (s, 1H), 7.55 (d, J 3.6, 1H), 7.50–7.45 (m, 12H), 7.40–7.35 (m, 18H), 7.20–7.15 (m, 6H); HRMS (NSI) m/z calcd for C88H59N8O3Zn [M − H]−: 1337.3851; found 1337.3866.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
GC and MC thank the EPSRC for funding (EP/E036244/1, EP/J500434/1). GC thanks the National Mass Spectroscopy Facility (NMSF), Swansea University (UK). S. M. A. acknowledges the Ministry of Higher Education and Scientific Research in Iraq and Kufa University for funding and support. We acknowledge Swiss National Science Foundation for financial support with the project entitled as “Fundamental studies of mesoscopic devices for solar energy conversion” with project number 20020_169695. P. Y. acknowledges the support from Swiss Government Excellence postdoctoral fellowship.Notes and references
- J. Gong, K. Sumathy, Q. Qiao and Z. Zhou, Ren. Sust. Energ. Rev., 2017, 68(1), 234–246 CrossRef CAS.
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef.
- M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC.
- M. G. Walter, A. B. Rudine and C. C. Wamser, J. Porphyrins Phthalocyanines, 2010, 14, 759–792 CrossRef CAS.
- A. Harriman, J. Chem. Soc., Faraday Trans. 2, 1981, 77, 1281–1291 RSC.
- P. K. Goldberg, T. J. Pundsack and K. E. Splan, J. Phys. Chem. A, 2011, 115, 10452–10460 CrossRef CAS PubMed.
- J. Feng, Y. Jiao, W. Ma, M. K. Nazeeruddin, M. Grätzel and S. Meng, J. Phys. Chem. C, 2013, 117, 3772–3778 CAS.
- A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed.
- F. Arkan and M. Izadyar, Mater. Chem. Phys., 2017, 196, 142–152 CrossRef CAS.
- T. D. Santos, A. Morandeira, S. Koops, A. J. Mozer, G. Tsekouras, Y. Dong, P. Wagner, G. Wallace, J. C. Earles, K. C. Gordon, D. Officer and J. R. Durrant, J. Phys. Chem. C, 2010, 114, 3276–3279 Search PubMed.
- A. Kay and M. Graetzel, J. Phys. Chem., 1993, 97, 6272–6277 CrossRef CAS.
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330–12396 CrossRef CAS PubMed.
- M. K. Nazeeruddin, R. Humphry-Baker, D. L. Officer, W. M. Campbell, A. K. Burrell and M. Grätzel, Langmuir, 2004, 20, 6514–6517 CrossRef CAS PubMed.
- S. Cherian and C. C. Wamser, J. Phys. Chem. B, 2000, 104, 3624–3629 CrossRef CAS.
- M. S. Kang, S. H. Kang, S. G. Kim, I. T. Choi, J. H. Ryu, M. J. Ju, D. Cho, J. Y. Lee and H. K. Kim, Chem. Commun., 2012, 48, 9349–9351 RSC.
- R. B. Ambre, G.-F. Chang, M. R. Zanwar, C.-F. Yao, E. W.-G. Diau and C.-H. Hung, Chem. – Asian J., 2013, 8, 2144–2153 CrossRef CAS PubMed.
- T. Higashino, K. Kawamoto, K. Sugiura, Y. Fujimori, Y. Tsuji, K. Kurotobi, S. Ito and H. Imahori, ACS Appl. Mater. Interfaces, 2016, 8, 15379–15390 CAS.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- C.-L. Wang, J.-Y. Hu, C.-H. Wu, H.-H. Kuo, Y.-C. Chang, Z.-J. Lan, H.-P. Wu, E. Wei-Guang Diau and C.-Y. Lin, Energy Environ. Sci., 2014, 7, 1392–1396 CAS.
- V. Villari, P. Mineo, E. Scamporrino and N. Micali, RSC Adv., 2012, 2, 12989–12998 RSC.
- N. C. Maiti, S. Mazumdar and N. Periasamy, J. Phys. Chem. B, 1998, 102, 1528–1538 CrossRef CAS.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242 CrossRef CAS PubMed.
- S. J. Lee, C. D. Malliakas, M. G. Kanatzidis, J. T. Hupp and S. T. Nguyen, Adv. Mater., 2008, 20, 3543–3549 CrossRef CAS.
- K. E. Splan and J. T. Hupp, Langmuir, 2004, 20, 10560–10566 CrossRef CAS PubMed.
- S. Sharma, N. Zamoshchik and M. Bendikov, Isr. J. Chem., 2014, 54, 712–722 CrossRef CAS.
- J. H. Delcamp, Y. Shi, J.-H. Yum, T. Sajoto, E. Dell'Orto, S. Barlow, M. K. Nazeeruddin, S. R. Marder and M. Grätzel, Chem. – Eur. J., 2013, 19, 1819–1827 CrossRef CAS PubMed.
- S. Qu, B. Wang, F. Guo, J. Li, W. Wu, C. Kong, Y. Long and J. Hua, Dyes Pigm., 2012, 92, 1384–1393 CrossRef CAS.
- O. Gidron and M. Bendikov, Angew. Chem., Int. Ed., 2014, 53, 2546–2555 CrossRef CAS PubMed.
- P. Shen, X. Liu, S. Jiang, Y. Huang, L. Yi, B. Zhao and S. Tan, Org. Electron., 2011, 12, 1992–2002 CrossRef CAS.
- R. Li, X. Lv, D. Shi, D. Zhou, Y. Cheng, G. Zhang and P. Wang, J. Phys. Chem. C, 2009, 113, 7469–7479 CAS.
- J. Jia, Y. Zhang, P. Xue, P. Zhang, X. Zhao, B. Liu and R. Lu, Dyes Pigm., 2013, 96, 407–413 CrossRef CAS.
- H. Jia, X. Ju, M. Zhang, Z. Ju and H. Zheng, Phys. Chem. Chem. Phys., 2015, 17, 16334–16340 RSC.
- N. V. Krishna, J. V. S. Krishna, S. P. Singh, L. Giribabu, L. Han, I. Bedja, R. K. Gupta and A. Islam, J. Phys. Chem. C, 2017, 121, 6464–6477 CAS.
- M. Sreenivasu, A. Suzuki, M. Adachi, C. V. Kumar, B. Srikanth, S. Rajendar, D. Rambabu, R. S. Kumar, P. Mallesham, N. V. B. Rao, M. S. Kumar and P. Y. Reddy, Chem. – Eur. J., 2014, 20, 14074–14083 CrossRef CAS PubMed.
- S. Eu, S. Hayashi, T. Umeyama, A. Oguro, M. Kawasaki, N. Kadota, Y. Matano and H. Imahori, J. Phys. Chem. C, 2007, 111, 3528–3537 CAS.
- J. S. Lindsey, H. C. Hsu and I. C. Schreiman, Tetrahedron Lett., 1986, 27, 4969–4970 CrossRef CAS.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827–836 CrossRef CAS.
- J. Karolczak, D. Kowalska, A. Lukaszewicz, A. Maciejewski and R. P. Steer, J. Phys. Chem. A, 2004, 108, 4570–4575 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- I. Beletskaya, V. S. Tyurin, A. Y. Tsivadze, R. Guilard and C. Stern, Chem. Rev., 2009, 109, 1659–1713 CrossRef CAS PubMed.
- S. Arrechea, J. N. Clifford, L. Pellejà, A. Aljarilla, P. de la Cruz, E. Palomares and F. Langa, Dyes Pigm., 2016, 126, 147–153 CrossRef CAS.
- A. Aljarilla, J. N. Clifford, L. Pelleja, A. Moncho, S. Arrechea, P. d. l. Cruz, F. Langa and E. Palomares, J. Mater. Chem. A, 2013, 1, 13640–13647 CAS.
- S. Karthikeyan and J. Y. Lee, J. Phys. Chem. A, 2013, 117, 10973–10979 CrossRef CAS PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- J. T. Lin, P.-C. Chen, Y.-S. Yen, Y.-C. Hsu, H.-H. Chou and M.-C. P. Yeh, Org. Lett., 2009, 11, 97–100 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8dt00413g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |