
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrocycles containing 1,1′-ferrocenyldiselenolato ligands on group 4 metallocenes†
Clara
Sanchez-Perez
ab,
Caroline E.
Knapp
 b,
Minna M.
Karjalainen
a,
Raija
Oilunkaniemi
a,
Claire J.
Carmalt
*b and
Risto S.
Laitinen
*a
b,
Minna M.
Karjalainen
a,
Raija
Oilunkaniemi
a,
Claire J.
Carmalt
*b and
Risto S.
Laitinen
*a
aLaboratory of Inorganic Chemistry, Environmental and Chemical Engineering, University of Oulu, P. O. Box 3000, FI-90014 Oulu, Finland
bDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: c.j.carmalt@ucl.ac.uk
First published on 12th March 2018
Abstract
Macrocyclic [Fe(η5-C5H4Se)2M(η5-C5H4R)2]2 [M = Ti (1), Zr (2), Hf (3), R = H; and M = Zr (4), Hf (5), R = tBu] were prepared and characterized by 77Se NMR spectroscopy and the crystal structures of 1–3 and 5 were determined by single-crystal X-ray diffraction. The crystal structure of 4 is known and the complex is isomorphous with 5. 1–5 form mutually similar macrocyclic tetranuclear complexes in which the alternating Fe(C5H4Se)2 and M(C5H4R)2 centers are linked by selenium bridges. The thermogravimetric analysis (TGA) of 1–3 under a helium atmosphere indicated that the complexes undergo a two-step decomposition upon heating. The final products were identified using powder X-ray diffraction as FexMSe2, indicating their potential as single-source precursors for functional materials.
Introduction
Transition metal dichalcogenides (TMDs) have been widely studied over the years due to their ability to intercalate species with apparent implications in catalytic and biological processes1–3 and biosensors.4 Due to their ability to intercalate species with substantial alteration of their electronic structure but minimum changes in their crystal structure, they became widely used as a new generation of semiconductors5–7 and battery systems.8 In particular, transition metal diselenides and ditellurides have exhibited band gaps lying within the UV-visible region, which makes them promising candidates for solar cell energy devices.9 Conventional methods for preparation of binary and ternary transition metal dichalcogenides (TMDs) require heating of high purity elements at high temperatures for extensive periods, involving several homogenizing steps making their synthesis long and expensive.10–12 The decomposition of metal chalcogenido complexes to metal chalcogenides takes place at significantly lower temperatures, providing high purity materials required for electronic applications in somewhat shorter timeframes. In recent years, the chemistry of organo-derivatives of selenium and tellurium has been widely developed after the discovery that these materials may be used as precursors for semiconducting binary metal chalcogenides.13 Extension of this area to include various metallocene dichalcogenides has been possible because of the development of equipment for air-sensitive synthesis. Alkoxide and thiolate groups as ligands have been widely studied in the development of metal-chalcogenide precursors in comparison with lesser known homologous complexes containing heavier elements. Selenium and tellurium analogues of common sulphur-containing precursors are often unknown or not readily available, and their metal-chalcogenolates are generally non-volatile, polymeric compounds.14Attempts to improve the stability of these compounds have yielded several complexes of the type [(η5-C5H4R)2M(SeR)2], [(η5-C5H4R)2MSe2R′′] and the bulkier [(η5-C5H4R)2M(E{SiMe3})2] (M = group 4 element).13 These studies show that there is increasing stability down the group, where the highest reactivity is observed when M = Ti.
Traditional synthetic routes for aryl chalcogenides involve the use of chalcogenols, but due to their toxicity, synthetic routes involving dilithium salts are preferentially used nowadays.13,15 Special interest was focussed on zirconocene dichalcogenides although high temperatures were required for the complex formation.3,15–17 Access to titanium and hafnium analogues through transmetallation decreased their overall yields dramatically to 40%,18 and therefore the use of dilithium salts of diselenolate ligands with metallocene dichlorides was a good compromise for simple synthetic design and achieving high yields.19 Few examples are found in the literature involving sulfur and tellurium analogue species.20,21 An important strategy to increase the stability and control the reactivity of metallocene dichalcogenides is the choice of appropriate ligands surrounding the transition metal.22
Aryl substituents bonded to metallocene dichalcogenides have been shown to increase the molecular stability in comparison with alkyl fragments.16 Also, the choice of flexible organic groups in the selenolato ligands are shown to allow maximum interaction between the d-orbitals of the metal and the p-orbitals of the chalcogen atoms.23,24
A lot of research has been focused on the field of single-source precursors for binary transition metal selenides; however ternary systems are still only achievable through multi-source precursor synthesis.12 Intercalation of group 4 metal dichalcogenides with first row transition metals creates an overlap of the M–M 3d electronic states, which is predicted to tune their optical properties.25 As these materials are potentially interesting for photovoltaic applications, the development of precursors for their deposition is long overdue. To the best of our knowledge, we are reporting the first class of single-source precursors for iron-doped group 4 transition metal dichalcogenides.
Results and discussion
The syntheses of [Fe(η5-C5H4E)2M(η5-C5H4R)2] (E = S, M = Ti, R = H; E = Se, M = Zr, R = H, tBu; M = Hf, R = tBu) have been reported,26–28 but the only crystallographically characterized complex is the formally dimeric macrocycle [Fe(η5-C5H4Se)2Zr(η5-C5H4tBu)2]2.28 In this contribution, a convenient tandem reaction involving chemically stabilized selenolates, purification and structural and spectroscopic characterization of the series of complexes [Fe(η5-C5H4Se)2M(η5-C5H4R)2]2 (M = Ti, R = H (1), M = Zr, R = H (2); M = Hf, R = H (3), tBu (5)) are reported. [Fe(η5-C5H4Se)2Zr(η5-C5H4tBu)2]2 (4) was also prepared and characterized by 77Se NMR spectroscopy. Complexes 1–3 have been studied as potentially suitable single-source precursors for FexMSe2 materials (0 < x < 1; M = Ti, Zr, Hf) upon thermal decomposition. As isolation of complexes 4 and 5 involved complicated and time-consuming air sensitive column chromatography, these complexes were not investigated as suitable precursors for CVD.Lithiation of ferrocene in hexane with tetramethylethylenediamine (TMEDA) proceeded with further addition of selenium powder to form Li2[Fe(η5-SeC5H4)2].29 The cyclic complexes [Fe(η5-C5H4Se)2M(η5-C5H4R)2]2 [R = H, M = Ti (1), Zr (2), Hf (3); R = tBu, M = Zr (4), Hf (5)] were isolated from the equimolar reaction of Li2[Fe(η5-SeC5H4)2] with the respective metallocene dichloride (Scheme 1), with high yields of complexes 1–3 (84% (1), 74% (2), and 69% (3)). The molecular structures of [Fe(η5-C5H4Se)2M(η5-C5H5)2]2 [M = Ti (1), Zr (2), Hf (3)] are shown in Fig. 1, left and that of the related complex [Fe(η5-C5H4Se)2Hf(η5-C5H4tBu)2]2 (5) is shown in Fig. 1, right. Selected bond lengths and angles are given in Table 1. Crystallographic parameters of complexes 1–3 and 5 are listed in Table 2.
 | ||
| Scheme 1 General synthesis of 1–5. | ||
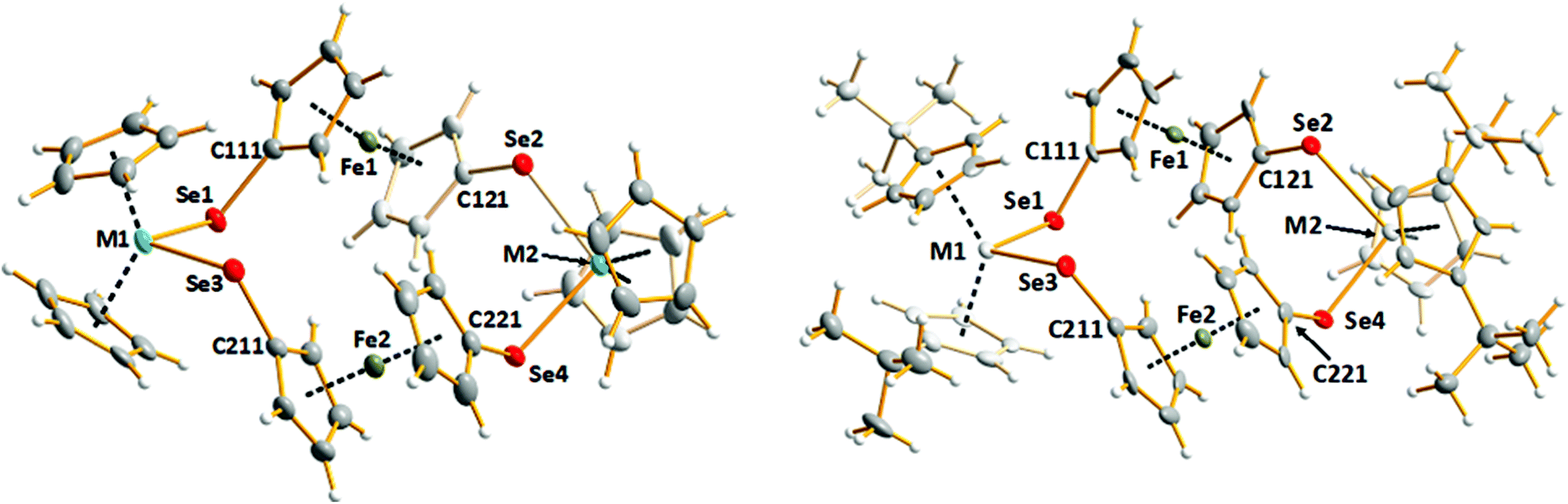 | ||
| Fig. 1 Molecular structures of [Fe(η5-C5H4Se)2M(η5-C5H5)2]2 [M = Ti (1), Zr (2), Hf (3)] (left) and [Fe(η5-C5H4Se)2Hf(η5-C5H4tBu)2]2 (5) (right) including the numbering of central atoms. Thermal ellipsoids are displayed at a 50% probability level. | ||
| 1 M = Ti | 2 M = Zr | 3 M = Hf | 5 M = Hf | |
|---|---|---|---|---|
| Lengths (Å) | ||||
| M1–Se1 | 2.5471(11) | 2.6434(11) | 2.6219(5) | 2.6438(11) |
| M1–Se3 | 2.5242(12) | 2.6396(12) | 2.6145(5) | 2.6180(10) |
| M2–Se2 | 2.5414(11) | 2.6284(15) | 2.6197(5) | 2.6368(11) |
| M2–Se4 | 2.5517(10) | 2.6521(14) | 2.6301(5) | 2.6253(10) |
| Se1–C111 | 1.907(4) | 1.918(7) | 1.918(4) | 1.923(8) |
| Se2–C121 | 1.907(4) | 1.907(7) | 1.913(4) | 1.902(8) |
| Se3–C211 | 1.902(4) | 1.908(7) | 1.912(5) | 1.906(9) |
| Se4–C221 | 1.904(5) | 1.921(7) | 1.916(5) | 1.894(9) |
| Angles (°) | ||||
| Se1–M1–Se3 | 97.80(4) | 102.52(4) | 102.947(16) | 98.53(3) |
| Se2–M2–Se4 | 99.40(4) | 100.98(5) | 101.800(16) | 98.37(3) |
| 1·3CH3C6H5 | 2·(CH2)4O | 3·1.90CH2Cl2 | 5 | |
|---|---|---|---|---|
| a I ≥ 2σ(I). b R 1 = ∑||Fo| − |Fc||/∑|Fo|, wR2 = [∑w(Fo2 − Fc2)2/∑wFo4]1/2. | ||||
| Empirical formula | C57H52.50Fe2Se4Ti2 | C44H44OFe2Se4Zr2 | C41.91H39.81Cl3.81Fe2Hf2Se4 | C56H68Fe2Hf2Se4 |
| Relative molecular mass | 1262.84 | 1198.77 | 1462.90 | 1525.62 |
| Radiation (Å) | Mo-Kα, 0.7173 | Mo-Kα, 0.7173 | Cu-Kα, 1.5418 | Mo-Kα, 0.7173 |
| Crystal system | Triclinic | Triclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/c | P21/c |
| a (Å) | 11.413(2) | 11.542(2) | 11.648(2) | 13.866(3) |
| b (Å) | 14.514(3) | 14.304(3) | 23.329(5) | 20.452(4) |
| c (Å) | 15.645(3) | 14.484(3) | 15.788(3) | 18.464(4) |
| α (°) | 96.90(3) | 99.53(3) | ||
| β (°) | 101.22(3) | 109.08(3) | 94.372(3) | 90.62(3) |
| γ (°) | 90.15(3) | 108.16(3) | ||
| V (Å3) | 2522.7(9) | 2050.0(9) | 4277.5(14) | 5235.7(18) |
| T (K) | 120(2) | 120(2) | 155(2) | 120(2) |
| Z | 2 | 2 | 4 | 4 |
| F(000) | 1257 | 1160 | 2752 | 2944 |
| D calc. (g cm−3) | 1.662 | 1.942 | 2.272 | 1.935 |
| μ(Mo-Kα) (mm−1) | 3.792 | 4.773 | 7.312 | |
| μ(Cu-Kα) (mm−1) | 20.334 | |||
| Crystal size (mm) | 0.16 × 0.13 × 0.12 | 0.20 × 0.18 × 0.15 | 0.05 × 0.05 × 0.05 | 0.22 × 0.10 × 0.08 |
| θ range (°) | 2.92–26.00 | 3.01–26.00 | 3.36–73.42 | 2.94–26.00 |
| No. of reflns. collected | 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 338 338 |
21767 |
71499 |
34621 |
| No. of unique reflns. | 9831 | 7696 | 8598 | 10147 |
| No. of observed reflns.a | 8290 | 6357 | 8227 | 8528 |
| No. of parameters/restraints | 584/2 | 479/0 | 496/9 | 580/0 |
| R INT | 0.0826 | 0.1038 | 0.0500 | 0.1344 |
|
R
1a,b |
0.0488 | 0.0626 | 0.0326 | 0.0586 |
| wR2a,b |
0.1208 | 0.0773 | 0.0829 | 0.1427 |
| R 1 (all data)b | 0.0603 | 0.1559 | 0.0344 | 0.0718 |
| wR2 (all data)b | 0.1291 | 0.1675 | 0.0840 | 0.1526 |
| GOF on F2 | 1.027 | 1.061 | 1.059 | 1.095 |
| Δρmax,min (e Å−3) | 0.906, −0.722 | 1.278, −1.210 | 1.893, −1.480 | 2.336, −1.201 |
All five complexes 1–5 form similar macrocyclic tetranuclear complexes in which the alternating Fe(C5H4Se)2 and M(C5H4R)2 centres are linked by selenium bridges. The conformation of each ring molecule is remarkably similar, as evidenced by the selected dihedral angles shown in ESI Table S1.† The Ti–Se bond length range is 2.5242(12)–2.5517(10) Å in 1, the Zr–Se bond length range is 2.6284(15)–2.6525(14) Å in 2, and the Hf–Se bond length ranges are 2.6145(5)–2.6301(5) and 2.6180(10)–2.6438(11) Å in 3 and 5, respectively. The M–Se bond lengths in related titanocene, zirconocene, and hafnocene complexes are consistent, as shown by the illustrative examples for Ti–Se [2.5327(10)–2.6039(7) Å],30–34 Zr–Se [2.628(2)–2.6696(17)],20,28,34,35 and Hf–Se [2.6113(5)–2.649(3)].34 The coordination polyhedron around the transition metal atom defined by the cyclopentadienyl (Cp) ring centroids and the selenium atoms is a distorted tetrahedron of nearly C2v symmetry. The range of M–Se–C angles for complexes 1–3 and 5 spans 104–108°, giving rise to a variety of Se–M–Se angles of 98–103°. Complexes 1–3 were crystallized in different solvents. In every case the solvent plays an important role in the lattice, as shown in Fig. S1–S3 in the ESI.† The discrete complexes form quasi-2D layers, which are separated by layers of solvent molecules. Interestingly, [Fe(η5-C5H4Se)2Hf(η5-C5H4tBu)2]2 (5) crystallizes without the lattice solvent. In this case, the complexes form a three-dimensional network (see Fig. S4 in the ESI†) with weak H⋯Se hydrogen bonds. The shortest contacts are in the range 2.9598(4)–3.1648(5). The related [Fe(η5-C5H4Se)2Zr(η5-C5H4tBu)2]2 (4) is isomorphic with 5 and exhibits a similar hydrogen bonding network.28
Complexes 1–5 are air/moisture sensitive and unstable in solution to varying degrees; however, they are stable in the solid state under an argon atmosphere. Ferrocenylselenolate complexes of zirconium and hafnium were shown to be far less reactive towards air and could even be handled in air for some time, without decomposition noticeable in the 77Se NMR spectrum in solution. This fact may be rationalized on the basis of the corresponding higher redox potentials of these metals relative to titanium.13 Their enhanced stability compared to that of ferrocenylselenolate complexes of titanium goes in accord with the hard and soft acid and base (HSAB) theory. It has also been reported that the steric bulk of the substituent in the Cp ring also plays a role in the relative stabilities of the complexes.21 Due to the low stability of the species in solution, all spectroscopic measurements were made using freshly prepared solutions. Chemical shifts were in good agreement with those reported for structurally similar complexes.17,34,36,37
Selenium resonances are largely sensitive to a change in the transition metal. Changing the transition metal from Ti to Zr has a shielding effect of −400 ppm (δ = 963.0 ppm (1), 558.0 ppm (2)), which is substantially larger than that recorded previously using a phenyl substituent (Δδ = −170 ppm).36 On the other hand, the shielding effect from Zr to Hf (δ = 444.8 ppm (3)) is practically the same for structurally comparable complexes (Δδ = −125 ppm). This effect adds to the number of examples proving that titanium has markedly different chemistry from zirconium and hafnium.
Complexes 1–3 exhibit a marked trend in 77Se NMR shift; however in comparison, substitution on the metallocene moiety has little effect on the shift. For example, substitution of H in 2 and 3 for a tBu group in 4 and 5 yields virtually the same shift (δ = 583.0 ppm (4), 449.0 ppm (5)).
Since Cp rings are not directly bonded to selenium atoms, the substitution of hydrogen atoms creates a much smaller change in the 77Se NMR chemical shift. Therefore, the substitution of these Cp rings can be designed to increase the solubility of complexes in non-polar solvents without severe effects in the analytical process. Furthermore, substitution of Cp rings with specific substituents can be used to tune the reactivity of the resulting complex towards protic reagents,22 and thus increase their scope for use as precursors for electronic materials.
The use of ferrocene instead of bulky organic linkers to stabilize metallocene chalcogenide moieties both reduces carbon contamination upon decomposition and facilitates delivery of early transition metal, chalcogen and iron elements in one step for potential iron-doped metal diselenide synthesis. The design of suitable precursors that can undergo clean decomposition processes is essential in the development of selenides as functional materials.9,38–41
A thermogravimetric study of complexes 1–3 was carried out to further probe the suitability of complexes of this type as CVD precursors. Although complexes 4 and 5 can be synthesized through the same tandem synthetic route, their isolation requires a more complicated process, making them less adequate precursors for CVD of iron-intercalated TMDs and thus were not further investigated. TGA was performed up to 1000 °C to maximize intercalation of iron in TMDs, for which temperatures over 900 °C are required.10–12 The decomposition profiles of 1–3 exhibit a two-step decomposition route (see Fig. 2). The first mass loss appears in the temperature range 225–310 °C consistent with the loss of the two (C5H5) fragments bonded to the group 4 metallocene of the respective complexes 1, 2 and 3. Further decomposition of ferrocene begins at ∼350 °C, with a mass loss expected for the loss of two (C5H4) rings.
 | ||
| Fig. 2 Thermogravimetric analysis (TGA) of [Fe(η5-C5H4Se)2Ti(η5-C5H5)2]2 (1) in green, [Fe(η5-C5H4Se)2Zr(η5-C5H5)2]2 (2) in red and [Fe(η5-C5H4Se)2Hf(η5-C5H5)2]2 (3) in blue. Grey shaded region 1 (∼225–310 °C) encompasses the mass loss of cyclopentadienyl ring bonded group 4 metals; grey shaded region 2 (∼350–500 °C) refers to the mass loss due to decomposition of ferrocene moieties. | ||
In all three cases the mass percentage that remains after thermal decomposition corresponds to a ratio for Fe: M:Se of 1:1:2: ∼50%, ∼55% and ∼60% respectively for complexes 1, 2 and 3.
The polycrystalline product of the thermolysis of complexes 1–3 during 1 h were characterized by X-ray diffraction (see Fig. 3), showing the formation of iron-intercalated transition metal diselenides. The peak at 2θ ∼ 20° in all three diffractograms is likely due to the excess of α-iron which is not intercalated into the TMD structures. These results act as a proof of concept that these complexes can thus act as precursors to the described ternary mixed metal selenides.
 | ||
| Fig. 3 (Left) X-Ray diffraction patterns of standards for Fe0.48TiSe242 (solid black line) and α-iron43 (dashed grey line), and the product of thermolysis at 1000 °C of complex 1 (solid green line); (right) X-ray diffraction patterns of standards for Fe0.16ZrSe244 (solid black line) and α-iron43 (dashed grey line), and products of thermolysis at 1000 °C of complex 2 (solid red line) and complex 3 (solid blue line). For clarity, the displayed data are background-subtracted to remove the large contribution from Fe fluorescence. | ||
Conclusions
Complexes 1–3 and 5 containing 1,1′-ferrocenyldiselenolato ligands on group 4 metallocenes have been crystallographically characterized and compared to 4.2577Se NMR studies have confirmed a decreasing trend down the group. We have reported a synthetic route involving a high-yield tandem reaction for 1–3 with potential for large-scale application as a new class of single-source precursors for use in chemical vapour deposition (CVD). Complexes 1–3 crystallize as non-polymeric small macrocyles and have been studied as potentially suitable single source precursors for FexMSe2 materials (0 < x < 1; M = Ti, Zr, Hf) upon thermal decomposition, showing full decomposition at temperatures below 600 °C. Treatment of these complexes for 1 h at 1000 °C produced iron-intercalated group 4 transition metal diselenide materials, which were characterized by Powder X-Ray Diffraction (PXRD). Therefore, we present a potential new type of selenium-containing complex of the type [Fe(η5-C5H4Se)2M(η5-C5H5)2]2 as organometallic single-source precursors for an alternative synthesis of iron-intercalated TMDs by thermal decomposition, involving a notably faster process than that previously reported.Further study will continue to expand the research involving these single-source precursors and their application in CVD, with the scope of minimization of the iron contamination in the final product, and production of these materials as thin films.
Experimental
General
Reactions and manipulation of samples were carried out under an argon atmosphere using standard Schlenk techniques. Air- and moisture-sensitive reagents and products were stored in a glovebox (argon).Li2[Fe(η5-C5H4)2]45 and Li2[Fe(η5-SeC5H4)2]29 were prepared according to literature procedures. N,N,N′,N′-Tetramethylethylenediamine (Aldrich) was distilled over sodium and stored over molecular sieves. Ferrocene (Merck) was purified by Soxhlet extraction from hexane. Selenium (granules, Aldrich), nBuLi (2.5 M in hexane, Aldrich), tBuLi (1.7 M in hexane, Aldrich), (MCl2(η5-C5H4tBu)2) (M = Zr, Hf) (Alpha Aesar), and (MCl2(η5-C5H5)2) (M = Ti, Zr, Hf) (Aldrich), were used as supplied. Solvents were dried and distilled under argon prior to use. Toluene, tetrahydrofuran and n-hexane were dried over Na/benzophenone and stored over a Na mirror. CH2Cl2 was dried over P2O5 and stored over 3 Å molecular sieves (20% m/v).
Single crystal X-ray diffraction
Diffraction data for complexes 1·3CH3C6H5, 2·(CH2)4O, and 5 were collected on a Nonius Kappa CCD diffractometer at 120–155 K using graphite monochromated MoKα radiation (λ = 0.71073 Å; 55 kV, 25 mA). Those of 3·1.90CH2Cl2 were collected on a SuperNova diffractometer using graphite monochromated CuKα radiation (λ = 1.54184 Å; 50 kV, 0.8 mA). Crystal data and details of structure determination are shown in Table 2.Structures were solved by direct methods using SHELXS-2016 and refined using SHELXL-2016.46,47 After the full-matrix least-squares refinement of non-hydrogen atoms with anisotropic thermal parameters the hydrogen atoms were placed in calculated positions in the cyclopentadienyl rings (C–H = 0.95 Å) and in the methyl and methylene groups (C–H = 0.98 and 0.99 Å, respectively). The isotropic thermal parameters of the hydrogen atoms were fixed at 1.2 times that of the corresponding carbon atom. The scattering factors for the neutral atoms were those incorporated into the programs.
Solvent molecules in 1·3CH3C6H5 and 3·1.90CH2Cl2 are disordered. In the former complex, one of the toluene molecules assumed two orientations of an equal site occupancy of 0.5 around the inversion center. The anisotropic displacement parameters of all carbon atoms were constrained to be equal during the refinement. In 3·1.90CH2Cl2, there are two crystallographically independent solvent molecules. One of them is disordered in three different orientations. The disorder involving the three most abundant orientations was resolved by constraining the anisotropic displacement parameters of all atoms to be equal and refining the site occupancy factors of the three orientations. The sum of the site occupancy factors of the disordered solvent molecule was refined to the value of 0.905(9).
Powder X-ray diffraction
Diffraction data were collected using a STOE Stadi P diffractometer (Mo Kα1 radiation, 0.70903 Å, 50 kV, 30 mA). Data of the thermolysis products were collected over the 2θ ranges of 10–31° (complex 1) and 2–31° (complexes 2 and 3), with a step size of 0.5° and a count time of 5 s per step.Thermogravimetric analysis
The instrument used for simultaneous thermal analysis was a Netzsch STA 449C. All measurements were carried out with the precursor sample in an aluminium oxide crucible packed under an argon atmosphere. Data were recorded from room temperature (20 °C) to 1000 °C using a protective gas (helium).NMR spectroscopy
77Se NMR spectra were recorded on a Bruker Avance III 400 spectrometer operating at 76.31 MHz, with a pulse width of 16.75 μs and a pulse delay of 1.0 s. The measurements were performed using 10 mm NMR tubes, at room temperature. A saturated solution of SeO2 (aq) was used as an external standard and the 77Se chemical shifts were reported relative to neat Me2Se [δ (Me2Se) = δ (SeO2) + 1302.6].48 NMR data acquisition was performed unlocked in THF unless otherwise stated. 1H and 13C NMR spectra were recorded in benzene-d6 using a Bruker Avance III 600 Cryo spectrometer operating at 600.130 and 150.903 MHz, respectively. Typical respective spectral widths for proton and carbon were 8.22 and 24.04 kHz, pulse widths were 26.50 and 14.30 μs, and pulse delays were 1.0 and 2.0 s, respectively. The measurements were performed using 5 mm NMR tubes, at room temperature. The 1H and 13C NMR spectra were referenced to the solvent resonances and are reported relative to Me4Si.Synthesis of complexes
Selenium powder (0.474 g, 6.00 mmol) was added to a solution of Li2[Fe(η5-C5H4)2] (0.940 g, 3.00 mmol) in THF (30 mL) at −78 °C. After 40 min stirring at −78 °C and 45 min at RT, the mixture was transferred dropwise to a THF solution of [Ti(η5-C5H5)2Cl2] (0.747 g, 3.00 mmol), [Zr(η5-C5H5)2Cl2] (0.877 g, 3.00 mmol), [Hf(η5-C5H5)2Cl2] (1.139 g, 3.00 mmol), [Zr(η5-C5H4tBu)2Cl2] (1.214 g, 3.00 mmol) or [Hf(η5-C5H4tBu)2Cl2] (1.475 g, 3.00 mmol) at −78 °C and stirred for 5 minutes to form complexes 1, 2, 3, 4 and 5, respectively. The solvent was fully removed in vacuo, the solid was quickly filtered in dry CH2Cl2, and complexes 1–3 were subsequently washed with dry n-hexane (40 mL). Compounds 4 and 5 were purified by column chromatography performed under air-excluded conditions using a stationary phase of aluminum oxide and a mixture of n-hexane:dichloromethane (1:2) as the eluent, with a very significant decrease in yield. Only small amounts of complexes 4 and 5 were used to record their 77Se NMR chemical shifts and the crystal structure of 5.
A table of bond parameters and the packing diagrams of 1–3 and 5, as well as crystallographic/refinement data for compounds 1–3 and 5, respectively, can be found in the ESI.† CCDC 1810808, 1810810, 1810811 and 1810809.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Fortum Foundation, UCL and the Ramsay Memorial Trust.Notes and references
- G. W. Shim, W. Hong, S. Y. Yang and S.-Y. Choi, J. Mater. Chem. A, 2017, 5, 14950–14968 CAS.
- J. Li, M. Hong, L. Sun, W. Zhang, H. Shu and H. Chang, ACS Appl. Mater. Interfaces, 2018, 10, 458–467 CAS.
- B. Gautheron, G. Tainturier, S. Pouly, F. Theobald, H. Vivier and A. Laarif, Organometallics, 1984, 3, 1495–1499 CrossRef CAS.
- Y.-H. Wang, K.-J. Huang and X. Wu, Biosens. Bioelectron., 2017, 97, 305–316 CrossRef CAS PubMed.
- A. Ramasubramaniam, R. Selhorst, H. Alon, M. D. Barnes, T. Emrick and D. Naveh, J. Mater. Chem. C, 2017, 5, 11158–11164 RSC.
- G. Zhang and Y.-W. Zhang, J. Mater. Chem. C, 2017, 5, 7684–7698 RSC.
- H. Li, Y. Li, A. Aljarb, Y. Shi and L.-J. Li, Chem. Rev. DOI:10.1021/acs.chemrev.7b00212.
- J. Yang, Y. Zhang, Y. Zhang, J. Shao, H. Geng, Y. Zhang, Y. Zheng, M. Ulaganathan, Z. Dai, B. Li, Y. Zong, X. Dong, Q. Yan and W. Huang, Small, 2017, 13, 1702181 CrossRef PubMed.
- M. Abdulsalam and D. Joubert, Comput. Mater. Sci., 2016, 115, 177–183 CrossRef CAS.
- R. H. Plovnick, D. S. Perloff, M. Vlasse and A. Wold, J. Phys. Chem. Solids, 1968, 29, 1935–1940 CrossRef CAS.
- Y. Arnaud, M. Chevreton, A. Ahouandjinou, M. Danot and J. Rouxel, J. Solid State Chem., 1976, 18, 9–15 CrossRef CAS.
- E. G. Shkvarina, A. N. Titov, S. G. Titova and O. M. Fedorova, J. Struct. Chem., 2016, 57, 710–716 CrossRef CAS.
- J. Arnold, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2007, pp. 353–417 Search PubMed.
- J. S. Ritch, T. Chivers, M. Afzaal and P. O'Brien, Chem. Soc. Rev., 2007, 36, 1622 RSC.
- P. Meunier, B. Gautheron and A. Mazouz, J. Chem. Soc., Chem. Commun., 1986, 424–425 RSC.
- B. Gautheron, G. Tainturier and P. Meunier, J. Organomet. Chem., 1981, 209, C49–C51 CrossRef CAS.
- C. Legrand, P. Meunier, J. L. Petersen, P. Tavares, J. Bodiguel, B. Gautheron and G. Dousse, Organometallics, 1995, 14, 162–169 CrossRef CAS.
- P. Meunier, B. Gautheron and A. Mazouz, J. Organomet. Chem., 1987, 320, C39–C43 CrossRef CAS.
- H. Köpf and T. Klapötke, J. Organomet. Chem., 1986, 310, 303–309 CrossRef.
- W. A. Howard, T. M. Trnka and G. Parkin, Inorg. Chem., 1995, 34, 5900–5909 CrossRef CAS.
- D. E. Gindelberger and J. Arnold, Organometallics, 1994, 13, 4462–4468 CrossRef CAS.
- R. Broussier, A. Da Rold and B. Gautheron, J. Organomet. Chem., 1992, 427, 231–244 CrossRef CAS.
- C. Guimon, G. Pfister-Guillouzo, P. Meunier, B. Gautheron, G. Tainturier and S. Pouly, J. Organomet. Chem., 1985, 284, 299–312 CrossRef CAS.
- J. Conradie, J. Mol. Struct. (THEOCHEM), 2009, 915, 51–57 CrossRef CAS.
- T. Yamasaki, N. Suzuki and K. Motizuki, J. Phys. C: Solid State Phys., 1987, 20, 395–404 CrossRef CAS.
- R. Steudel, K. Hassenberg, J. Pickardt, E. Grigiotti and P. Zanello, Organometallics, 2002, 21, 2604–2608 CrossRef CAS.
- B. Gautheron and G. Tainturier, J. Organomet. Chem., 1984, 262, c30–c34 CrossRef CAS.
- R. Broussier, Y. Gobet, R. Amardeil, A. Da Rold, M. M. Kubicki and B. Gautheron, J. Organomet. Chem., 1993, 445, C4–C5 CrossRef CAS.
- R. Broussier, A. Abdulla and B. Gautheron, J. Organomet. Chem., 1987, 332, 165–173 CrossRef CAS.
- S. Zeltner, R.-M. Olk, M. Pink, S. Jelonek, P. Jörchel, T. Gelbrich, J. Sieler and R. Kirmse, Z. Anorg. Allg. Chem., 1996, 622, 1979–1986 CrossRef CAS.
- H. Sugiyama, Y. Hayashi, H. Kawaguchi and K. Tatsumi, Inorg. Chem., 1998, 37, 6773–6779 CrossRef CAS PubMed.
- S. Vincendeau, V. Collière and C. Faulmann, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, m268–m270 CAS.
- O. Jeannin, M. Nomura and M. Fourmigué, J. Organomet. Chem., 2007, 692, 4113–4118 CrossRef CAS.
- A. L. Hector, W. Levason, G. Reid, S. D. Reid and M. Webster, Chem. Mater., 2008, 20, 5100–5106 CrossRef CAS.
- W. A. Howard, G. Parkin and A. L. Rheingold, Polyhedron, 1995, 14, 25–44 CrossRef CAS.
- P. Granger, B. Gautheron, G. Tainturier and S. Pouly, Org. Magn. Reson., 1984, 22, 701–704 CrossRef CAS.
- L.-C. Song, C. Han, Q.-M. Hu and Z.-P. Zhang, Inorg. Chim. Acta, 2004, 357, 2199–2204 CrossRef CAS.
- N. D. Boscher, C. J. Carmalt and I. P. Parkin, Chem. Vap. Depos, 2006, 12, 54–58 CrossRef CAS.
- C. H. (Kees) de Groot, C. Gurnani, A. L. Hector, R. Huang, M. Jura, W. Levason and G. Reid, Chem. Mater., 2012, 24, 4442–4449 CrossRef.
- N. D. Boscher, C. S. Blackman, C. J. Carmalt, I. P. Parkin and A. G. Prieto, Appl. Surf. Sci., 2007, 253, 6041–6046 CrossRef CAS.
- K. George, C. H. (Kees) de Groot, C. Gurnani, A. L. Hector, R. Huang, M. Jura, W. Levason and G. Reid, Phys. Procedia, 2013, 46, 142–148 CrossRef CAS.
- G. Calvarin, J. R. Gavarri, M. A. Buhannic, P. Colombet and M. Danot, Rev. Phys. Appl., 1987, 22, 1131–1138 CrossRef CAS.
- M. Braun and R. Kohlhaas, Phys. Status Solidi B, 1965, 12, 429–444 CrossRef CAS.
- A. Gleizes, J. Revelli and J. A. Ibers, J. Solid State Chem., 1976, 17, 363–372 CrossRef CAS.
- J. J. Bishop, A. Davison, M. L. Katcher, D. W. Lichtenberg, R. E. Merrill and J. C. Smart, J. Organomet. Chem., 1971, 27, 241–249 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- R. C. Burns, M. J. Collins, R. J. Gillespie and G. J. Schrobilgen, Inorg. Chem., 1986, 25, 4465–4469 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1810808–1810811. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt00300a |
| This journal is © The Royal Society of Chemistry 2018 |