
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
[Ru3(6-NHC)(CO)10]: synthesis, characterisation and reactivity of rare 46-electron tri-ruthenium clusters†
Charles E.
Ellul
,
John P.
Lowe
,
Mary F.
Mahon
,
Paul R.
Raithby
 * and
Michael K.
Whittlesey
*
* and
Michael K.
Whittlesey
*
Department of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: m.k.whittlesey@bath.ac.uk
First published on 21st February 2018
Abstract
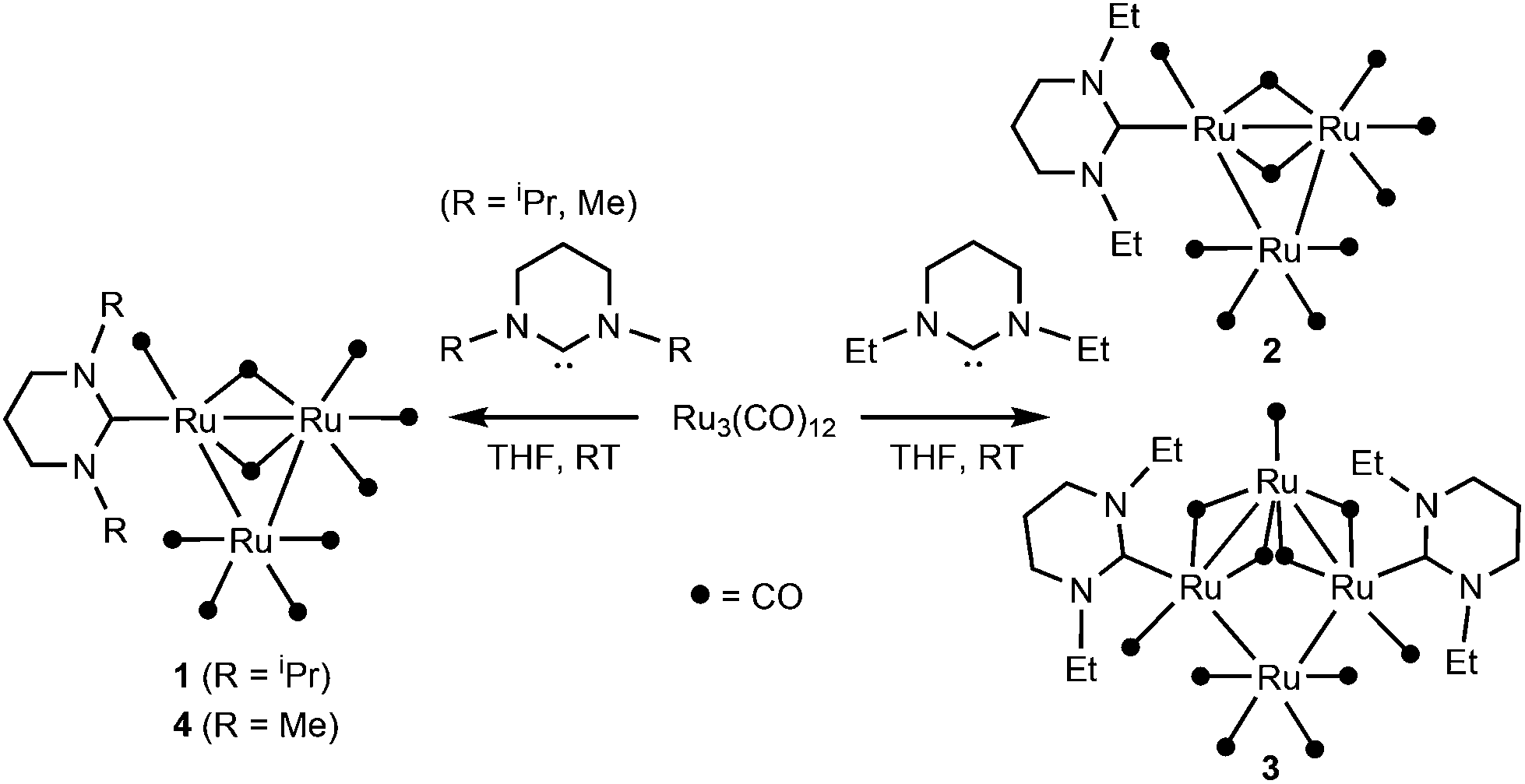
[Ru3(CO)12] reacts at room temperature with N-alkyl substituted 6-membered ring N-heterocyclic carbenes (6-NHC) to form [Ru3(6-NHC)(CO)10] (6-NHC = 6-iPr 1, 6-Et 2 and 6-Me 4), rare examples of coordinatively unsaturated (46-electron) ruthenium clusters. Complexes 1, 2 and 4 have been structurally characterised, along with the tetranuclear ruthenium cluster [Ru4(6-Et)2(CO)11] 3 that is formed along with 2. The degradation of the 6-iPr derivative 1 by pyrimidinium salt elimination helped to explain the poor activity of the complex in the catalytic acylation of pyridine.
Introduction
Despite the unquestionable impact that N-heterocyclic carbene (NHC) ligands have had on organometallic chemistry over the last 25 years, their application to low-valent metal cluster chemistry has received only limited attention.1 However, on the basis of what has been observed, it is clear that combining NHCs with metal clusters frequently leads to very interesting observations. Thus, there are examples in which cluster structure is retained upon reaction with one type of NHC substitution pattern,2 but cleaved upon only relatively small changes to either the substituents or reaction stoichiometry.3,4 In very recent cases, clusters with unusually high NHC content have been identified as catalyst deactivation products.5 Arguably of most interest have been the observations of atypical NHC binding modes,6 the detection of unprecedented reaction intermediates7 and the formation of multiply activated carbene ligands.2b,8The group 8 tricarbonyl precursors [M3(CO)12] (M = Fe,9 Ru,1b,2,3,6,7 Os)1b,2c,7,10,11 have proven to be the most fertile area of cluster chemistry for reaction with NHCs, largely due to their ease of accessibility and the well-known differences in properties that are seen upon descending the group.12 For example, osmium exhibits a willingness to adopt 46-electron counts in some Os3 clusters (e.g. [Os3(CO)10(μ-H)2]), whereas ruthenium shows a greater tendency to maintain coordinative saturation (i.e. 48 electrons) meaning that electron-deficient Ru3 systems are not very common.13 Indeed, [Ru3(CO)10(μ-H)2] has only been generated photchemically14 and its chemistry explored only to a very limited extent.15
We16 (and others)17 have shown that NHCs with a ring size of >5 can be used to stabilise low coordination numbers in a wide range of mononuclear transition metal complexes, but as far as we are aware, reactions between this class of so-called ‘ring-expanded carbenes’18 and transition metal clusters have not been described.19 Herein, we report that N-alkylated, 6-membered ring NHCs (denoted as 6-NHC) react with [Ru3(CO)12] at room temperature to afford novel 46-electron Ru3 clusters of general formula [Ru3(6-NHC)(CO)10]. Their structures, together with studies of reactivity alongside other Ru3 clusters in catalytic C–H bond functionalisation, are described.
Results and discussion
Synthesis and characterisation of [Ru3(6-NHC)(CO)10] (6-NHC = 6iPr, 6-Et, 6-Me)
Infra-red analysis of a reaction of 6-iPr and Ru3(CO)12 (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, Scheme 1) in THF showed replacement of the ν(CO) bands, characteristic of the starting material, by a new set of terminal carbonyl stretches between 2084–1963 cm−1, as well as a lower frequency feature at 1802 cm−1, over a period of ca. 2.5 days at room temperature. Removal of the solvent, extraction of the red-brown residue into hexane and slow evaporation confirmed that all of the IR bands arose from the formation of a single product, the 46-electron cluster, [Ru3(6-iPr)(CO)10] (1, Scheme 1), which could be isolated as a red crystalline product in 65% yield.20
:1 ratio, Scheme 1) in THF showed replacement of the ν(CO) bands, characteristic of the starting material, by a new set of terminal carbonyl stretches between 2084–1963 cm−1, as well as a lower frequency feature at 1802 cm−1, over a period of ca. 2.5 days at room temperature. Removal of the solvent, extraction of the red-brown residue into hexane and slow evaporation confirmed that all of the IR bands arose from the formation of a single product, the 46-electron cluster, [Ru3(6-iPr)(CO)10] (1, Scheme 1), which could be isolated as a red crystalline product in 65% yield.20
 | ||
| Scheme 1 Summary of the reactivity of [Ru3(CO)12] with 6-iPr, 6-Me and 6-Et. | ||
The X-ray structure of 1 (Fig. 1) revealed an asymmetric arrangement of the three Ru atoms (Ru(1)–Ru(2) 2.7287(2) Å, Ru(1)–Ru(3) 2.8090(2) Å, Ru(2)–Ru(3) 2.8815(2) Å), a single 6-iPr ligand bound to Ru(1) (Ru(1)–C(11) 2.1178(17) Å) and two asymmetrically bridging CO ligands (Ru(1)–C(2) 1.9791(19) Å, Ru(2)–C(2) 2.125(2) Å, Ru(1)–C(2)–Ru(2) 83.26(7)°; Ru(1)–C(3) 2.0128(19) Å, Ru(2)–C(3) 2.118(2) Å, Ru(1)–C(3)–Ru(2) 82.65(7)°) on the Ru(1)–Ru(2) edge. The two shorter Ru–C(bridging carbonyl) distances are associated with Ru(1), which is also bound to the 6-iPr ligand; this may result from the carbene exerting less steric pressure than the two carbonyl ligands associated with Ru(2), or may alternatively reflect a greater electron density on Ru(1) due to the strongly σ-donating NHC. As anticipated, the carbene ligand lay in the equatorial plane of the Ru3 core, as is common for the vast majority of nitrogen and phosphorus donor ligands coordinated to Ru3 triangular clusters, although the dihedral twist between the pyrimidine ring and the Ru3 core is significantly greater (87.3°) than in either of the 5-membered ring NHC derivatives [Ru3(IMes)(CO)11] (37.7°; IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene)2b or [Ru3(ab-ItBu)(CO)11]6a (9.8°; ab denotes an ‘abnormal’ (or mesionic) ligated NHC,21 ItBu = 1,3-di-tert-butylimidazol-2-ylidene). As a consequence, the N-iPr groups lie in the same plane as the axial carbonyl ligands. The inherent strain of this conformation may be alleviated somewhat by the presence of the μ-CO ligands. The Ru–Ru bond lengths in 1 are reminiscent of the Os–Os bond lengths in [Os3(CO)10(μ-H)2] where the ‘unsaturated’ dihydrido-bridged bond is substantially shorter, at 2.680(2) Å, than the mean of the other two bonds, at 2.814 Å.22 However, in a series of 48-electron tri-ruthenium clusters containing a ‘Ru(μ-CO)2Ru’ unit, the dicarbonyl-bridged Ru–Ru bond is also shorter than the two unbridged Ru–Ru bonds.23
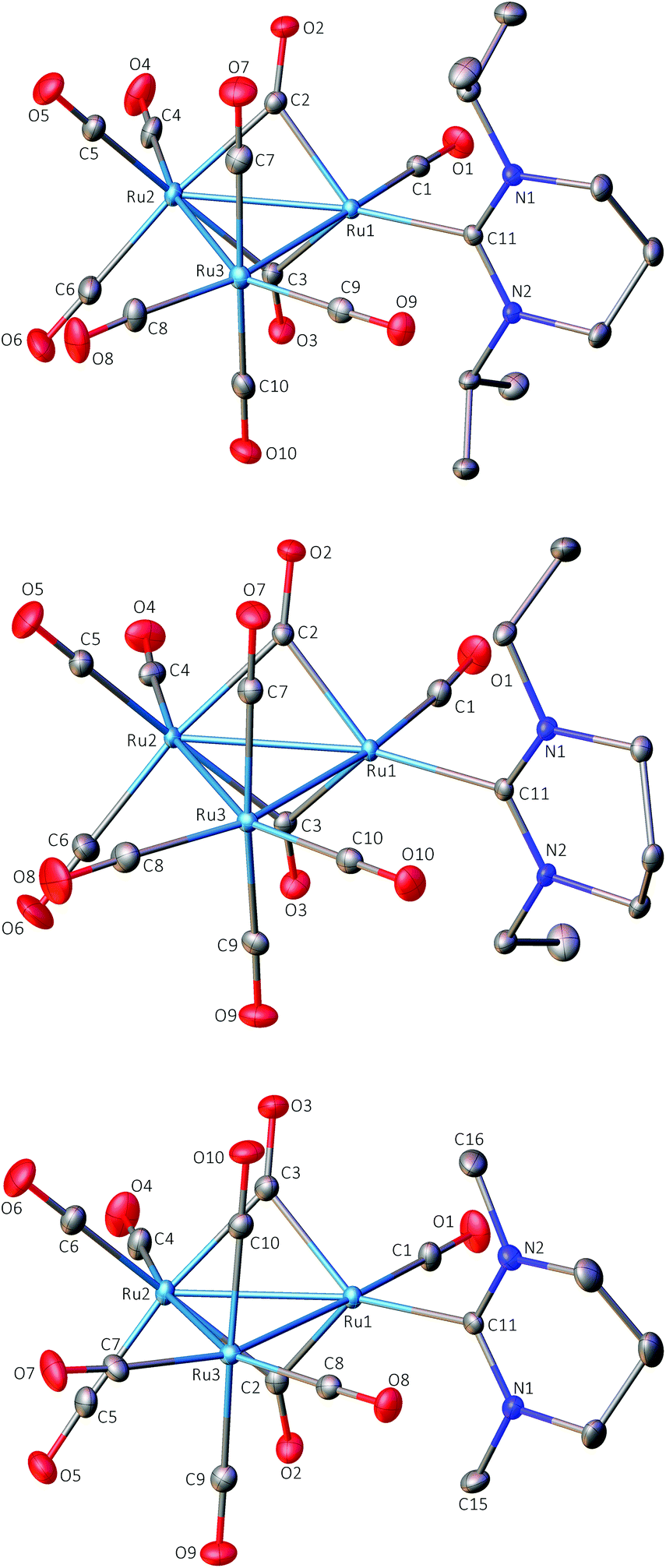 | ||
| Fig. 1 Molecular structures of 1 (top), 2 (centre) and 4 (bottom). Ellipsoids are shown at 30% probability. The minor disordered component in 3 has been omitted for clarity, as have hydrogen atoms in all cases. | ||
Changing to the less bulky 6-Et ligand led to the formation of an analogous product, [Ru3(6-Et)(CO)10] (2, Scheme 1), albeit in much lower yield. Moreover, the crystallisation of deep red 2 also afforded very small amounts of a second, less intensely coloured product, which upon manual separation, was structurally characterised as the unusual Ru4 cluster, [Ru4(6-iPr)2(CO)11] 3 (Fig. S7†).
The solution IR spectrum of 2 was essentially identical to that of 1. However, in contrast to the sharp, well-resolved room temperature 1H NMR spectrum of 1, the spectrum of 2 comprised of three broad resonances at δ 3.43, 3.27 and 2.07, together with a sharp triplet at δ 1.31. The two higher frequency broad signals resolved into three sharper multiplets (relative integrals of 2:2:4) for the eight NCH2 protons upon cooling to 235 K.
The molecular geometry of 2 (Fig. 1) is similar to that of 1 with the dicarbonyl-bridged Ru(1)–Ru(2) bond (2.7277(2) Å) being the shortest, while the Ru(1)–Ru(3) and Ru(2)–Ru(3) distances are 2.8018(2) and 2.8644(3) Å. The Ru(1)–C(11) metal–carbene distance is 2.121(2) Å and, as for 1, the ligand lies close to the equatorial plane of the Ru3 core. The bridging carbonyl ligands display asymmetry (Ru(1)–C(2) 1.993(3) Å, Ru(2)–C(2) 2.121(2) Å, Ru(1)–C(2)–Ru(2) 83.02(9)°; Ru(1)–C(3) 1.985(2) Å, Ru(2)–C(3) 2.115(3) Å, Ru(1)–C(3)–Ru(2) 83.36(9)°), again with the shortest Ru–C(bridging carbonyl) distances associated with Ru(1), which is also bound to the carbene ligand.
The N-Me substituted ligand 6-Me behaved similarly to 6-iPr in yielding only the [Ru3(6-NHC)(CO)10] product, [Ru3(6-Me)(CO)10] 4 (Scheme 1). The carbonyl absorption bands in the IR spectrum of 4 partially merged to give a total of nine bands compared to the eleven bands seen for both 1 and 2. In the proton NMR spectrum, both the N-Me singlet and NCH2CH2 quintet were sharp, while the NCH2 triplet was noticeably broader, suggestive of fluxionality (cf.2).
The molecular structure of 4 (Fig. 1) also closely resembles that of 1 and 2 with the three Ru–Ru distances following the same trend (Ru(1)–Ru(2) 2.7320(3) Å, Ru(1)–Ru(3) 2.8104(3) Å, Ru(2)–Ru(3) 2.8688(3) Å), and the same distribution of the carbonyls and the carbene ligand. The asymmetry in the bonding of the two bridging carbonyls is again apparent with the shortest Ru–C(carbonyl) distance being associated with Ru(1) which also has the carbene ligand bonded to it (Ru(1)–C(11) (carbene) 2.107(3) Å; Ru(1)–C(2) 1.980(3) Å and Ru(1)–C(3) 1.996(3) Å for the two carbonyl bonds). By comparison the bridging-carbonyl Ru(2)–C bond lengths are 2.128(3) Å (Ru(2)–C(2)) and 2.113(3) Å (Ru(2)–C(3)).
Stoichiometric and catalytic reactions of 1 involving CO
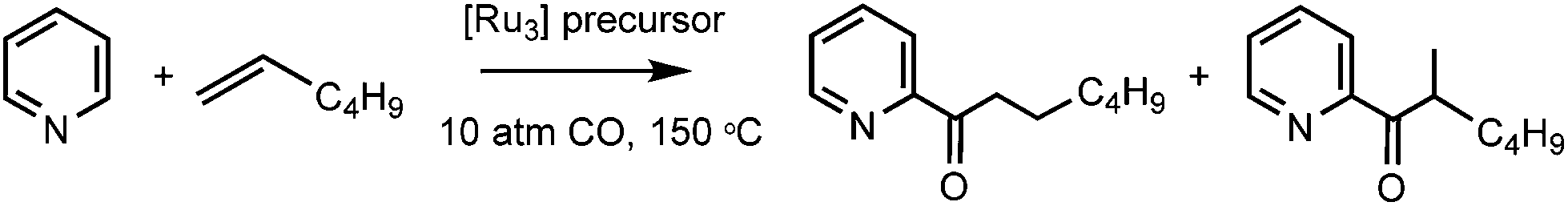
The comparatively poor yields of 2 and 4, as well as the need to manually separate 2 from 3, led us to use 1 for investigations into the reactivity of the [Ru3(6-NHC)(CO)10] complexes. Given the coordinative unsaturation, we were surprised to find that there was no reaction of [Ru3(6-iPr)(CO)10] with CO (1 atm in THF-d8), even upon heating to 80 °C. However, exposure of 1 to 1 atm 13CO led to the appearance of a 13C enhanced carbonyl signal at δ 200 in the 13C{1H} NMR spectrum at room temperature (Fig. S11†), implying that although [Ru3(6-iPr)(CO)10] will not add CO, it can undergo facile CO exchange.24The stability of 1 to CO led us to test it as a precursor in the catalytic acylation of pyridine (Table 1). Moore and co-workers reported in 199225 that the insertion of CO and a terminal alkene into the ortho C–H position of pyridine was catalysed by [Ru3(CO)12] at high pressure (10 atm CO) under forcing conditions (150 °C, 16 h) to give predominantly linear acylation products. In our hands, we were unable to achieve the 65% yield with 1-hexene described by Moore using [Ru3(CO)12], achieving instead a more modest 31% average yield.26 Disappointingly, 1 exhibited lower activity than [Ru3(CO)12], as did the coordinatively saturated, abnormally bound 5-membered ring NHC clusters, [Ru3(ab-ItBu)(CO)11]6a and [Ru3(ab-IAd)(CO)11] (IAd = 1,3-bis(adamantyl)imidazol-2ylidene).6b
|
|
|
|---|---|
| Ru precursor | Product yieldb (%) |
| a Reaction conditions: Ru3 precursor (0.026 mmol), 1-hexene (0.25 mL), pyridine (7.5 mL), CO (10 atm), 150 °C, 16 h. b Yields (average of at least two runs) determined by integration of 1H NMR product resonances at δ 3.22 and/or δ 1.74 (linear product)25 and δ 4.1 (branched product)25versus the OMe resonance of 2,4,6-(MeO)3C6H3 as an internal standard. | |
| [Ru3(CO)12] | 31 |
| 1 | 12 |
| [Ru3(ab-ItBu)(CO)11] | 23 |
| [Ru3(ab-IAd)(CO)11] | 6 |
Degradation of 1 through loss of [6-iPrH]+ and 6-iPr
In an effort to rationalise the poor catalytic activity, 1 was heated in the presence of 4 equiv. pyridine in an NMR tube scale reaction. Warming to 85 °C brought about loss of the 1H NMR resonances of 1 and appearance of signals due to the pyridinium cation [6-iPrH]+.27 The presence of low frequency proton signals at δ −15.5 and δ −19.2 suggested that this was partnered with anionic ruthenium carbonyl hydride species and, indeed, an X-ray study of a small number of orange-yellow crystals isolated from the reaction yielded a structure of [6-iPrH]2[Ru4(CO)12H2] (Fig. S14†).28 The di-potassium salt of [Ru4(CO)12H2]2− has a hydride chemical shift of δ −19.3.29A likely pathway to [6-iPrH]+ formation involves C–H activation of pyridine by 1,23a,30 followed by reductive elimination from a resulting {(6-iPr)RuH} moiety. Support for reductive elimination from such a species came upon reacting 1 with H2 at 60 °C (THF-d8), which again generated pyrimidinium proton NMR signals, together with hydride signals at δ −12.1 and −12.5, in <1 h.
Further evidence for the low stability of the [Ru3(6-NHC)(CO)10] complexes comes from the reaction of 1 with phosphines, where we found that 1 reacted with 1–3 equiv. PPh3 at 60 °C with complete loss of the starting material and the formation of multiple phosphorus containing species. Efforts to characterise the product mixture led to isolation of just the known phosphine carbonyl cluster, [Ru3(PPh3)3(CO)9] (Fig. S18†),31 indicating that 1 also appears to be susceptible to loss of free carbene under quite mild conditions.
Conclusions
The synthesis and structural characterisation of rare examples of 46-electron tri-ruthenium clusters has been achieved upon reacting [Ru3(CO)12] with N-alkyl substituted, 6-membered ring N-heterocyclic carbenes under very mild conditions. Reactivity studies of the [Ru3(6-NHC)(CO)10] complexes carried out using the 6-iPr derivative 1 indicated that loss of the carbene ligand took place upon addition of PPh3, mild heating under H2 or in the presence of pyridine, thereby limiting the potential of these complexes in catalytic applications.The formation of very different products in the reaction of [Ru3(CO)12] with 6-NHCs to those formed with 5-membered ring analogues is notable,1b suggesting that (i) investigations with 6- and/or 7-membered ring NHCs bearing, for example, N-aryl substituents, and/ or (ii) the use of other group 8 carbonyl clusters as precursors, is worthy of investigation.
Experimental
All manipulations were carried out using standard Schlenk, high vacuum and glovebox techniques using dried and degassed solvents. NMR spectra were recorded on a Bruker Avance 500 and 400 MHz NMR spectrometers and run in THF-d8 (referenced to δ 3.58 (1H) and δ 67.6 (13C)). IR spectra were recorded in hexane solution on a Nicolet Nexus spectrometer. Elemental analyses were performed by Elemental Microanalysis Ltd, Okehampton, Devon, UK. [Ru3(CO)12] (Sigma Aldrich) was used as received, while the [PF6]− salts of [6-iPrH]+, [6-EtH]+ and [6-MeH]+ were prepared according to the literature.32[Ru3(6-iPr)(CO)10] 1
[6-iPrH][PF6] (108 mg, 0.34 mmol) and KHMDS (66 mg, 0.33 mmol) were stirred in THF for 1 h in a J. Young's resealable ampoule. The suspension was then reduced to dryness, [Ru3(CO)12] (110 mg, 0.17 mmol) added and the mixture redissolved in THF (6 mL). After stirring at room temperature for 3–4 days, the solvent was removed and the residue extracted exhaustively with warm hexane (4 × 25 mL). The hexane was removed under vacuum and the residue allowed to slowly evaporate in an argon-filled glovebox from ca. 2 mL hexane/THF (90:10) to afford deep-red crystalline 1. Yield 84 mg, 65%. 1H NMR (500 MHz, 298 K): δ = 3.59 (sept, 2H, 3JHH = 6.50 Hz, CHMe2), 3.40 (m, 2H, NCH2), 3.18 (m, 2H, NCH2), 2.11 (m, NCH2CHH, 1H), 1.88 (m, 1H, NCH2CHH), 1.36 (d, 6H, 3JHH = 6.50 Hz, CHMe2), 1.21 (d, 6H, 3JHH = 6.50 Hz, CHMe2). 13C{1H} NMR (101 MHz, 298 K): δ = 198.7 (s, Ru–CNHC), 57.5 (s, NCHMe2), 40.6 (s, NCH2), 21.6 (s, NCH2CH2), 20.6 (s, CHMe2), 20.0 (s, CHMe2). IR (cm−1): 2084, 2061, 2041, 2031, 2022, 2009, 2001, 1984, 1976, 1963, 1802 (all νCO). Analysis found: C, 31.96; H, 2.68; N, 3.73. C20H20N2O10Ru3 requires: C, 31.92; H, 2.77; N, 3.60.
[Ru3(6-Et)(CO)10] 2 and [Ru4(6-Et)2(CO)11] 3
As for 1 but with [6-EtH][PF6] (100 mg, 0.35 mmol), KHMDS (69 mg, 0.35 mmol) and [Ru3(CO)12] (111 mg, 0.17 mmol). Combined yield of 2 and 3: 24 mg. Selected spectroscopic data for 2. 1H NMR (500 MHz, 235 K): δ = 3.47 (m, 2H, NCH2CH3), 3.39 (m, 2H, NCH2CH3), 3.26 (m, 4H, NCH2CH3), 2.07 (m, 2H, NCH2CH2), 1.31 (t, 6H, 3JHH = 6.99 Hz, NCH2CH3). 13C{1H} NMR (126 MHz, 235 K): δ 206.1 (s), 201.0 (s), 199.1 (s), 198.2 (s), 189.5 (s), 186.8 (s), 52.5 (s, NCH2), 45.1 (s, NCH2), 20.9 (s, NCH2CH2), 13.9 (s, CH2CH3). IR (cm−1): 2085, 2061, 2042, 2031, 2022, 2009, 2002, 1984, 1977, 1964, 1803 (all νCO). Analysis found: C, 30.01; H, 2.20; N, 3.93. C18H16N2O10Ru3 requires: C, 29.87; H, 2.23; N, 3.87. Only a few single crystals of 3 could be isolated, restricting characterisation to just an X-ray crystal structure (Fig. S7†).[Ru3(6-Me)(CO)10] 4
As for 1 but with [6-MeH][PF6] (60 mg, 0.23 mmol), KHMDS (46 mg, 0.23 mmol) and [Ru3(CO)12] (75 mg, 0.12 mmol) to afford deep-red crystals of 4. Yield 16 mg, 20%. 1H NMR (500 MHz, 298 K): δ = 3.32 (br t, 4H, 3JHH = 4.8 Hz, NCH2), 3.14 (s, 6H, NMe), 2.09 (quint, 2H, 3JHH = 6.0 Hz, NCH2CH2). 13C{1H} NMR (101 MHz, 298 K): δ = 202.7 (s, Ru–CNHC), 47.5 (s, NCH2), 43.9 (s, NMe), 21.1 (s, NCH2CH2). IR (cm−1): 2086, 2061, 2043, 2023, 2010, 2003, 1981, 1962, 1806 (all νCO). Analysis found: C, 28.02; H, 1.66; N, 3.82. C16H12N2O10Ru3 requires: C, 27.63; H, 1.74; N, 4.03.Reactivity studies of 1
(a) with C5H5N: 1 (17.1 mg, 0.023 mmol) was combined with pyridine (7.5 μL, 0.093 mmol) in THF-d8 in a J. Young's resealable NMR tube and the reaction followed by 1H NMR spectroscopy. (b) with H2: 1 (11.1 mg, 0.015 mmol) was dissolved in THF-d8 in a J. Young's resealable NMR tube, the solution freeze–pump–thaw degassed (×3), placed under 1 atm H2 and the reaction followed by 1H NMR spectroscopy. (c) with PPh3: 1 (6.5 mg, 0.009 mmol) was combined with 3 equiv. PPh3 (6.8 mg, 0.026 mmol) in THF-d8 in a J. Young's resealable NMR tube and the reaction followed by 1H and 31P NMR spectroscopy.Catalysis
Solid samples of Ru3 precursors (0.0265 mmol), together with 2,4,6-(MeO)3C6H3 (0.0265 mmol) as an internal standard, were weighed into a Parr autoclave inside a glovebox. A solution of 1-hexene (2 mmol) in pyridine (7.5 mL, dried over activated 3 Å molecular sieves) was added by cannula, and the autoclave assembly put together under a flow of argon. After purging twice with CO, the autoclave was pressurised to 10 atm and heated at 150 °C for 16 h. After cooling and depressurising, a small amount of the red-orange solution was diluted with CDCl3 and analysed by 1H NMR spectroscopy. Product resonances were assigned by comparison to the literature.25X-ray crystallography
Data for 1 were collected on a Nonius kappaCCD diffractometer using Mo-Kα radiation, while those for 2, 3 (ESI†) and 4 were obtained using a Cu-Kα source and an Agilent SuperNova instrument. Refinements, achieved using SHELXL33via Olex2,34 were relatively straightforward and only points of note are mentioned hereafter. In 3 (ESI†), C22 was refined subject to taking 75:25 disorder with C22A into account. Meanwhile, the asymmetric unit in 4 was seen to comprise 2 molecules, which differ in the relative orientation of the apical carbene–carbon atoms within the respective molecules to which they belong.
Crystallographic data for all compounds have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications CCDC 1559943 (1), 1559944 (2), 1559945 (3, ESI), 1559946 (4), 1816567 ([6-iPrH]2[Ru4(CO)12H2], ESI) and 1816568 ([Ru3(PPh3)3(CO)9], ESI) respectively.†
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank EPSRC/University of Bath for support (DTA to CEE). PRR is grateful to the EPSRC for continued support through a Programme Grant (EP/K004956/1). MKW would like to thank Dr Matthew Jones for assistance with the catalysis.References
- (a) G. Hogarth, S. E. Kabir and E. Nordlander, Dalton Trans., 2010, 39, 6153 RSC; (b) J. A. Cabeza and P. García-Álvarez, Chem. Soc. Rev., 2011, 40, 5389 RSC.
- (a) M. F. Lappert and P. L. Pye, J. Chem. Soc., Dalton Trans., 1977, 2172 RSC; (b) J. A. Cabeza, I. del Río, D. Miguel and M. G. Sánchez-Vega, Chem. Commun., 2005, 3956 RSC; (c) J. A. Cabeza, I. del Río, D. Miguel, E. Pérez-Carreño and M. G. Sánchez-Vega, Organometallics, 2008, 27, 211 CrossRef CAS; (d) J. A. Cabeza, I. del Río, M. Fernández-Colinas, E. Pérez-Carreño, M. G. Sánchez-Vega and D. Vázquez-García, Organometallics, 2009, 28, 1832 CrossRef CAS.
- (a) M. I. Bruce, M. L. Cole, R. S. C. Fung, C. M. Forsyth, M. Hilder, P. C. Junk and K. Konstas, Dalton Trans., 2008, 4118 RSC; (b) C. E. Ellul, O. Saker, M. F. Mahon, D. C. Apperley and M. K. Whittlesey, Organometallics, 2008, 27, 100 CrossRef CAS; (c) J. A. Cabeza, M. Damonte, P. García-Álvarez, A. R. Kennedy and E. Pérez-Carreño, Organometallics, 2011, 30, 826 CrossRef CAS.
- L. Álvarez-Rodríguez, J. A. Cabeza, P. García-Álvarez, E. Pérez-Carreño and D. Polo, Inorg. Chem., 2015, 54, 2983 CrossRef PubMed.
- (a) J. Campos, L. S. Sharninghausen, R. H. Crabtree and D. Balcells, Angew. Chem., Int. Ed., 2014, 53, 12808 CrossRef CAS PubMed; (b) L. S. Sharninghausen, B. Q. Mercado, R. H. Crabtree, D. Balcells and J. Campos, Dalton Trans., 2015, 44, 18403 RSC; (c) L. S. Sharninghausen, B. Q. Mercado, C. Hoffmann, X. Wang, J. Campos, R. H. Crabtree and D. Balcells, J. Organomet. Chem., 2017, 849–850, 17 CrossRef CAS.
- (a) C. E. Ellul, M. F. Mahon, O. Saker and M. K. Whittlesey, Angew. Chem., Int. Ed., 2007, 46, 6343 CrossRef CAS PubMed; (b) M. R. Crittall, C. E. Ellul, M. F. Mahon, O. Saker and M. K. Whittlesey, Dalton Trans., 2008, 4209 RSC.
- Y. Liu, R. Ganguly, H. V. Huynh and W. K. Leong, Angew. Chem., Int. Ed., 2013, 52, 12110 CrossRef CAS PubMed.
- (a) J. A. Cabeza, I. del Río, D. Miguel, E. Pérez-Carreño and M. G. Sánchez-Vega, Dalton Trans., 2008, 1937 RSC; (b) J. A. Cabeza, I. del Río, D. Miguel and M. G. Sánchez-Vega, Angew. Chem., Int. Ed., 2008, 47, 1920 CrossRef CAS PubMed; (c) J. A. Cabeza and E. Pérez-Carreño, Organometallics, 2008, 27, 4697 CrossRef CAS; (d) C. Y. Zhang, B. Li, H. B. Song, S. S. Xu and B. Q. Wang, Organometallics, 2011, 30, 3029 CrossRef CAS; (e) J. A. Cabeza, M. Damonte and P. García-Álvarez, Organometallics, 2011, 30, 2371 CrossRef CAS; (f) J. A. Cabeza, M. Damonte and E. Pérez-Carreño, Organometallics, 2012, 31, 8355 CrossRef CAS; (g) J. A. Cabeza, M. Damonte, P. García-Álvarez and E. Pérez-Carreño, Chem. Commun., 2013, 49, 2813 RSC; (h) J. Yan, Z. Han, D. J. Zhang and C. B. Liu, RSC Adv., 2016, 6, 99625 RSC.
- S. Warratz, L. Postigo and B. Royo, Organometallics, 2013, 32, 893 CrossRef CAS.
- (a) C. E. Cooke, M. C. Jennings, M. J. Katz, R. K. Pomeroy and J. A. C. Clyburne, Organometallics, 2008, 27, 5777 CrossRef CAS; (b) Y. Liu, R. Ganguly, H. V. Huynh and W. K. Leong, Organometallics, 2013, 32, 7559 CrossRef CAS.
- For reactions of other Os–CO precursors, see; (a) J. A. Cabeza, I. da silva, I. del Río and M. G. Sánchez-Vega, Dalton Trans., 2006, 3966 RSC; (b) J. A. Cabeza, I. del Río, J. M. Fernández-Colinas, E. Pérez-Carreño, M. G. Sánchez-Vega and D. Vázquez-Garciá, Organometallics, 2010, 29, 3828 CrossRef CAS.
- (a) M. I. Bruce, in Comprehensive Organometallic Chemistry I, ed. G. Wilkinson, F. G. A. Stone and E. W. Abel, Pergamon, Oxford, 1982, vol. 4 Search PubMed; (b) R. D. Adams and J. P. Selegue, in Comprehensive Organometallic Chemistry I, ed. G. Wilkinson, F. G. A. Stone and E. W. Abel, Pergamon, Oxford, 1982, vol. 4 Search PubMed; (c) A. J. Deeming, in Comprehensive Organometallic Chemistry II, ed. E. W. Abel, F. G. A. Stone and G. Wilkinson, Elsevier, Oxford, 1995, vol. 7 Search PubMed.
- (a) S. Rivomanana, G. Lavigne, N. Lugan and J.-J. Bonnet, Inorg. Chem., 1991, 30, 4110 CrossRef CAS; (b) G. Süss-Fink, G. Rheinwald, H. Stoeckli-Evans, C. Bolm and D. Kaufmann, Inorg. Chem., 1996, 35, 3081 CrossRef; (c) H.-C. Böttcher, H. Thönnessen, P. G. Jones and R. Schmutzler, J. Organomet. Chem., 1996, 520, 15 CrossRef; (d) M. Graf, K. Merzweiler, C. Bruhn and H.-C. Böttcher, J. Organomet. Chem., 1998, 553, 371 CrossRef CAS; (e) H.-C. Böttcher, M. Graf, K. Merzweiler, T. Rösel, H. Schmidt and C. Wagner, Polyhedron, 2001, 20, 2011 CrossRef; (f) R. D. Adams, Y. Kan, Q. Zhang, M. B. Hall and E. Trufan, Organometallics, 2012, 31, 50 CrossRef CAS.
- N. E. Leadbeater, J. Lewis and P. R. Raithby, J. Organomet. Chem., 1997, 543, 251 CrossRef CAS.
- N. E. Leadbeater, J. Lewis, P. R. Raithby and M.-A. Rennie, Polyhedron, 1998, 17, 1755 CrossRef CAS.
- (a) C. J. E. Davies, M. J. Page, C. E. Ellul, M. F. Mahon and M. K. Whittlesey, Chem. Commun., 2010, 46, 5151 RSC; (b) M. J. Page, W. Y. Lu, R. C. Poulten, E. Carter, A. G. Algarra, B. M. Kariuki, S. A. Macgregor, M. F. Mahon, K. J. Cavell, D. M. Murphy and M. K. Whittlesey, Chem. – Eur. J., 2013, 19, 2158 CrossRef CAS PubMed; (c) R. C. Poulten, M. J. Page, A. G. Algarra, J. J. Le Roy, I. López, E. Carter, A. Llobet, S. A. Macgregor, M. F. Mahon, D. M. Murphy, M. Murugesu and M. K. Whittlesey, J. Am. Chem. Soc., 2013, 135, 13640 CrossRef CAS PubMed; (d) L. R. Collins, N. A. Rajabi, S. A. Macgregor, M. F. Mahon and M. K. Whittlesey, Angew. Chem., Int. Ed., 2016, 55, 15539 CrossRef CAS PubMed; (e) S. Pelties, E. Carter, A. Folli, M. F. Mahon, D. M. Murphy, M. K. Whittlesey and R. Wolf, Inorg. Chem., 2016, 55, 11006 CrossRef CAS PubMed; (f) S. Sabater, M. J. Page, M. F. Mahon and M. K. Whittlesey, Organometallics, 2017, 36, 1776 CrossRef CAS; (g) W. J. M. Blackaby, S. Sabater, R. C. Poulten, M. J. Page, A. Folli, V. Krewald, M. F. Mahon, D. M. Murphy, E. Richards and M. K. Whittlesey, Dalton Trans., 2018, 47, 769 RSC.
- (a) N. Phillips, J. Rowles, M. J. Kelly, I. Riddlestone, N. H. Rees, A. Dervisi, I. A. Fallis and S. Aldridge, Organometallics, 2012, 31, 8075 CrossRef CAS; (b) P. Hauwert, J. J. Dunsford, D. S. Tromp, J. J. Weigand, M. Lutz, K. J. Cavell and C. J. Elsevier, Organometallics, 2013, 32, 131 CrossRef CAS; (c) N. Phillips, T. Dodson, R. Tirfoin, J. I. Bates and S. Aldridge, Chem. – Eur. J., 2014, 20, 16721 CrossRef CAS PubMed; (d) O. S. Morozov, A. V. Lunchev, A. A. Bush, A. A. Tukov, A. F. Asachenko, V. N. Khrustalev, S. S. Zalesskiy, V. P. Ananikov and M. S. Nechaev, Chem. – Eur. J., 2014, 20, 6162 CrossRef CAS PubMed; (e) T. Wurm, F. Mulks, C. R. N. Bohling, D. Riedel, P. Zargaran, M. Rudolph, F. Rominger and A. S. K. Hashmi, Organometallics, 2016, 35, 1070 CrossRef CAS; (f) J. J. Dunsford, D. J. Evans, T. Pugh, S. N. Shah, N. F. Chilton and M. J. Ingleson, Organometallics, 2016, 35, 1098 CrossRef CAS; (g) Ł. Banach, P. A. Guńka and W. Buchowicz, Dalton Trans., 2016, 45, 8688 RSC; (h) A. J. Jordan, C. M. Wyss, J. Bacsa and J. P. Sadighi, Organometallics, 2016, 35, 613 CrossRef CAS.
- Cabeza and co-workers have reported that pyrimidine and 2-methylpyrimidine add and cyclometallate upon reaction with [Ru3(CO)12]: (a) J. A. Cabeza, I. del Río, E. Pérez-Carreño and V. Pruneda, Organometallics, 2011, 30, 1148 CrossRef CAS; (b) J. A. Cabeza, P. García-Álvarez, E. Pérez-Carreño and V. Pruneda, Chem. – Eur. J., 2013, 19, 3426 CrossRef CAS PubMed.
- J. Li, W. X. Shen and X. R. Li, Curr. Org. Chem., 2012, 16, 2879 CrossRef CAS.
-
1 is also formed at a 1:1 ratio of [Ru3(CO)12]: 6-iPr with no IR evidence for any other carbonyl products.
- M. Albrecht, Adv. Organomet. Chem., 2014, 62, 111 CrossRef CAS.
- A. G. Orpen, A. V. Rivera, E. G. Bryan, D. Pippard, G. M. Sheldrick and K. D. Rouse, J. Chem. Soc., Chem. Commun., 1978, 723 RSC.
- (a) M. I. Bruce, M. G. Humphrey, M. R. Snow, E. R. T. Tiekink and R. C. Wallis, J. Organomet. Chem., 1986, 314, 311 CrossRef CAS; (b) R. D. Adams and J. H. Yamamoto, Organometallics, 1995, 14, 3704 CrossRef CAS; (c) B. K. Maiti, H. Gorls, O. Klobes and W. Imhof, Eur. J. Inorg. Chem., 2011, 1545 CrossRef CAS.
- A number of groups have reported that [Ru3(CO)12] does not undergo direct exchange with 13CO, although the reaction can be promoted by the addition of some salts or metal additives: (a) KH: J. C. Bricker, C. C. Nagel and S. G. Shore, J. Am. Chem. Soc., 1982, 104, 1444 CrossRef CAS; (b) KOMe: D. J. Darensbourg, R. L. Gray and M. Pala, Organometallics, 1984, 3, 1928 CrossRef CAS; (c) Cp/Cp*Rh(CO)2: W. E. Lindsell, C. B. Knobler and H. D. Kaesz, J. Organomet. Chem., 1985, 296, 209 CrossRef CAS; (d) KH/NaBH4: J. C. Bricker, M. W. Payne and S. G. Shore, Organometallics, 1987, 6, 2545 CrossRef CAS.
- E. J. Moore, W. R. Pretzer, T. J. O‘Connell, J. Harris, L. Labounty, L. Chou and S. S. Grimmer, J. Am. Chem. Soc., 1992, 114, 5888 CrossRef CAS.
- This represents the average value from six catalytic runs, including ones in which (i) solutions were prepared both in and out of a glovebox and (ii) different batches of pyridine were used.
- There was no change by NMR spectroscopy when 1 was heated in just THF-d8 at 70 °C for 4 h.
- R. Suter, A. A. Bhattacharyya, L.-Y. Hsu, J. A. Krause Bauer and S. G. Shore, Polyhedron, 1998, 17, 2889 CrossRef CAS.
- K. E. Inkrott and S. G. Shore, Inorg. Chem., 1979, 18, 2817 CrossRef CAS.
- B. R. Cockerton and A. J. Deeming, J. Organomet. Chem., 1992, 426, C36 CrossRef CAS.
- N. E. Leadbeater and P. R. Raithby, J. Coord. Chem., 2001, 53, 311 CrossRef CAS.
- A. Aidouni, S. Bendahou, A. Demonceau and L. Delaude, J. Comb. Chem., 2008, 10, 886 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, A46, 467–473 CrossRef CAS; G. M. Sheldrick, SHELXL-97, a computer program for crystal structure refinement, University of Göttingen, 1997 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1559943, 1559944, 1559945, 1559946, 1816567 and 1816568. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt00189h |
| This journal is © The Royal Society of Chemistry 2018 |