
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembly of singlet-emitting double-helical silver dimers: the curious coordination chemistry and fluorescence of bisquinolylpyridone†
Charlotte M. A.
Farrow
a,
Geoffrey R.
Akien
 a,
Nathan R.
Halcovitch
a,
James A.
Platts
*b and
M. P.
Coogan
*a
a,
Nathan R.
Halcovitch
a,
James A.
Platts
*b and
M. P.
Coogan
*a
aDepartment of Chemistry, Lancaster University, Bailrigg, Lancaster, LA1 4YB, UK. E-mail: m.coogan@lancaster.ac.uk
bSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: platts@cardiff.ac.uk
First published on 9th February 2018
Abstract
2,6-Bis(2-quinolyl)-4(1H)-pyridone 1, a novel quinoline analogue of the well-known ligand 2-terpyridone, shows unusual fluorescence with a large Stokes shift and low energy emission. Pyridine-pyridone tautomerism is investigated by NMR and theoretical methods and indicates that the low energy emission is from the pyridine form. 1 reacts with Ag(I) salts to give a double helical Ag2N6 core showing a BLUE shift in fluorescence with respect to the free ligand, which has been characterised experimentally and theoretically as involving an unusual mixed MLCT/ILCT excited state and emission from a singlet state.
Introduction
Terpyridines have a rich and varied coordination chemistry1 and amongst these a hydroxylated example 2,6-bis(2-pyridyl)-4(1H)-pyridone2 (terpyridone hereafter) is widely applied in coordination chemistry. This ligand forms a variety of types of complex in which it bonds in a tridentate N^N^N, bidentate N^N and bridging N^N:N and N^N:O modes forming monomeric and supramolecular species including some with interesting photophysical properties.1–3 It is known that substitution of pyridines for quinolines red-shifts absorption and emission properties of derived metal complexes4 and many oligopyridine ligands have been investigated with one or more pyridines replaced with quinolines for this reason. However, the bisquinoline analogue of terpyridone, 2,6-bis(2-quinolyl)-4(1H)-pyridone 1, is unknown. In light of the rich coordination chemistry and luminescence potentially available an investigation of this ligand was undertaken.Results and discussion
Synthesis of ligand 1
The synthesis of 1 was achieved by a variation on the Constable method for terpyridone,2 reacting methyl quinaldate with acetone in the presence of sodium t-butoxide to give 1,5-bis(2-quinolyl)-pentane-1,3,5-trione following a double Claisen condensation. We find sodium tbutoxide a convenient and less hazardous replacement for sodium hydride in the synthesis of both 1 and 2,6-bis(2-pyridyl)-4(1H)-pyridone and it does not appear to lead to significant loss of yield in either case. 1,5-Bis(2-quinolyl)-pentane-1,3,5-trione is recovered following aqueous work up as a fibrous yellow solid and is a difficult material to characterise as it is only sparingly soluble and appears to exists in solution as a gross mix of keto–enol tautomers and/or enol double bond isomers. However, condensation of the material recovered from the double Claisen condensation with ammonium acetate in methanol, following aqueous work up gave 1 in good yield (Scheme 1).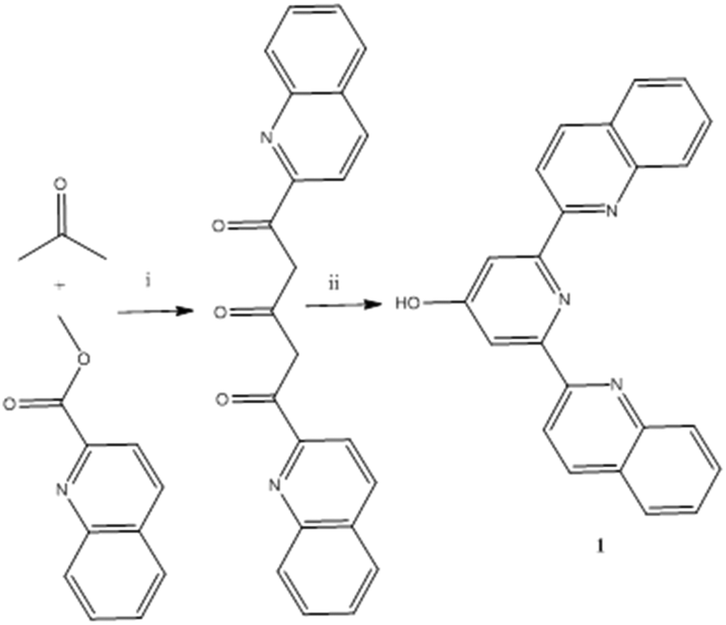 | ||
| Scheme 1 Synthesis of 2,6-bis(2-quinolyl)-4(1H)-pyridone 1. (i) NaOBut, THF 60 °C 16h; (ii) NH4OAc, MeOH, 60 °C, 16h. | ||
NMR Studies of bis-quinolylpyridone 1
As 2-terpyridone has been reported to exist in equilibrium between hydroxypyridine and pyridone forms5 an investigation of the tautomerism of 1 was undertaken by NMR. As with terpyridone, 1 predominantly exists in the keto (pyridone) form (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O δC 182 ppm) in the less polar CDCl3, but primarily in the enol (pyridinol) form in the more polar DMSO-d6 (C–O δC 166.5 ppm).6
O δC 182 ppm) in the less polar CDCl3, but primarily in the enol (pyridinol) form in the more polar DMSO-d6 (C–O δC 166.5 ppm).6
A closer examination showed some of the peaks to be unexpectedly broad, which made determining the quaternary carbon chemical shifts challenging, and completely prevented the observation of the potentially useful 15N chemical shifts via1H,15N-HMBC.7 Gradual dilution of a 176 mM DMSO-d6 solution of 1 to 5 mM improved the linewidth (FWHM) of the OH from 68.0 to 17.5 Hz, but slightly increased that of the CH from 2.3 to 5.3 Hz, indicating that intramolecular processes are not the main cause of the line-broadening. Instead, titration of a 20 mM solution with small amounts of a 100 mM NEt3/DMSO-d6 stock solution was much more successful, with a final 1 mol% (with respect to the ligand) NEt3 reducing the linewidths to 0.73 and 0.49 Hz for the OH and CH, respectively. We attribute this improvement to the neutralisation of acidic impurities still remaining after the recrystallization, as evidenced by the appearance of the NEt3 CH2 as a quartet of doublets (J 7.3, 4.3 Hz) due to coupling with the methyl and NH+. Approximately 75% of the impurities were due to residual acetic acid, but 25% could not be directly accounted for. The presence of acid is likely to increase the rate of proton exchange between OH, NH and water and increase the rate of tautomerisation, both of which would serve to increase the linewidths. Recent work has shown that acetic acid solvates are readily accessible,10 so it is quite possible that small amounts of this crystalline form were simultaneously formed during the synthesis procedure.
4-Pyridones are known to self-associate in solution,8 but the molecular weight calculated from diffusion experiments on 5–100 mM solutions in DMSO-d6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 compared favourably with its true molecular weight, so it seems likely that this is not a significant factor. This is in keeping with observations that increasing steric bulk decreases the tendency towards self-association.8,9
9 compared favourably with its true molecular weight, so it seems likely that this is not a significant factor. This is in keeping with observations that increasing steric bulk decreases the tendency towards self-association.8,9
The greatly improved linewidth in DMSO-d6 allowed for the identification of the minor keto tautomer by 2D NOESY and its quantification at 1.5 mol%. The 1H linewidths of the keto form were notably broader than that of the enol tautomer, but the S/N was too low to establish whether this was due to rotational isomerism or simply a relatively higher acidity of the NH over the OH. It was also possible to use 15N-HMBC to measure 15N chemical shifts of 279.4 ppm for the central pyridine ring, and 302.0 ppm for the quinoline nitrogen.
The solubility of 1 in CDCl3 was lower at 10 mM and gave similarly broad peaks, but again, the addition of NEt3 greatly improved the linewidth, allowing quantification of the minor enol tautomer at 4.5 mol%, in comparison to 0.9 mol% for terpyridone at the same concentration. As expected, 15N-HSQC and 15N-HMBC experiments yielded substantially different chemical shifts for the major keto tautomer: 127.3 ppm for the pyridone NH, and 297.2 ppm for the quinolyl N.
Structural studies of bis-quinolylpyridone 1
Crystals of 1 demonstrate polymorphism, those grown from cooling a mixture of EtOH and H2O give the monoclinic form in space group I2/a. In this structure the molecules are all in the pyridone form, with a C–O distance of 1.265(2) Å. This structure also contains continuous voids running along the b-axis occupied by solvent molecules. The other two polymorphs were formed by slow diffusion of Et2O into concentrated solutions of 1 in either CDCl3 or CD3CN. From CD3CN another monoclinic form in P21/n is obtained (1-a), this time with one molecule in pyridone form and another in the pyridinol form in the asymmetric unit. The C–O bond lengths are 1.264(2) and 1.343(3) Å and the two oxygen atoms are hydrogen-bound as a pair along the c-axis. Finally, from CDCl3, we have obtained the triclinic polymorph (1-b) that has an asymmetric unit containing 4 different molecules, in similar hydrogen-bound pairs (Fig. 1 and 2). | ||
| Fig. 1 1 in I2/a showing void channels running along b-axis. | ||
 | ||
| Fig. 2 Solid-state molecular structure 1-a demonstrating the pyridine-pyridyl tautomers. | ||
It is notable that in 1-b one of molecules in the pyridinol form deviates significantly from planarity. Indeed, in both structures examination of the quinoline arms in relation to the central pyridine in these structures, one can observe (Table 1) that in general the pyridone molecules remain very close to planar, whereas the pyridinol forms demonstrate larger deviations from planarity.
| Structure | Torsion angles in pyridone-form | Torsion angles in pyridinol-form |
|---|---|---|
| 1 (I2/a) | 2.6(2) and 2.6(2) | |
| 1-a (P21/n) | 2.3(3) and 8.8(3) | 21.29(17) and 20.84(17) |
1-b (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) ) |
1.58(16) and 2.74(16) | 5.30(13) and 1.49(13) |
| 1.82(18) and 5.12(18) | 23.35(15) and 13.8(3) (major) and 1.8(4) (minor) |
Silver complexation of bis-quinolylpyridone 1
As terpyridone is known to form a trimeric cage11 upon reaction with rhenium pentacarbonyl halides followed by activation with silver salts, a similar approach was taken, treating 1 with equimolar Re(CO)5Br followed by activation of the solid product with AgBF4. However, upon slow diffusion of diethyl ether into an acetonitrile solution of the product of this reaction, instead of a rhenium-based trimer, a dimeric silver species was isolated in which each Ag2 unit is bound by two molecules of 1. Closer examination revealed that the Ag2L2 unit showed an unusual connectivity and geometry with each of the terminal quinoline nitrogens of each ligand binding a different silver ion, while the central pyridone nitrogen of each ligand bridges between both silvers. The two molecules of 1 are themselves mutually entwined in a double helical arrangement around the central Ag–Ag axis (Fig. 3). Although coordination to rhenium had been intended it is known that terpyridines only coordinate to the rhenium fac-tricarbonyl core in a bidentate mode and the derived complexes are labile4 whereas the isolated product involves coordination all of the nitrogen donors and thus appears to be the thermodynamically preferred product of the reaction. | ||
| Fig. 3 Solid-state molecular structures of 2 and 2-a (respectively) demonstrating the bridging-helical arrangement of the ligand system. | ||
In order to demonstrate that the formation of the dimeric helical structure was not a feature of the unusual reaction conditions, ligand 1 was treated with AgBF4 and AgOTf, and in each case crystals were obtained in which the double helical Ag2L2 structure was repeated (Fig. 3). A number of other examples of Ag dimers featuring a single nitrogen bridging between the two centres are known12 including a similar helical example derived from a chiral terpyridine in which the handedness of the helix is controlled by the ligand in solution, but not in the solid state.13
Photophysical studies of bis-quinolylpyridone 1 and silver helicate 2
Formation of helicates from coordination of oligopyridines with a variety of metals is well precedented,1,13 but unexpectedly 2 was found to show intense luminescence, prompting an investigation of the photophysical properties of both 1 and 2. Ligand 1 shows a remarkably low energy emission for a simple organic molecule, with λmax emission 485 nm, with broad structured emission bands extending beyond 600 nm (Fig. 4) giving visually yellow emission. In light of the unusually large Stokes shift the emission lifetime of 1 was investigated by Time Correlated Single Photon Counting (TCSPC) methods and found to have an intensity-weighted average lifetime of 1.4 ns, strongly indicating fluorescence from a singlet excited state. This suggests that the large Stokes shift derives from a geometrical arrangement in the singlet excited state, rather than a change of electronic configuration. | ||
| Fig. 4 Excitation and emission spectra of ligand 1. | ||
The UV-vis absorption spectrum (see ESI†) of complex 2 showed an unexpectedly low energy band appearing as a shoulder on the more intense higher energy absorptions with an apparent maximum around 350 nm. Upon excitation at 350 nm, 2 unexpectedly showed a broad emission band centred around 440 nm and extending to much lower energy (Fig. 5). Further investigation revealed an excitation maximum at 355 nm and a series of emission bands of varying intensity contributing to the low energy emission profile.
 | ||
| Fig. 5 Excitation and emission spectra of complex 2. | ||
Unusually, it appears that upon formation of complex 2 the emission profile of ligand 1 suffers a blue-shift, presumably as a result of the loss of planarity leading to reduced conjugation and thus higher energy photophysics (average C–N–N–C torsion angle quinoline – pyridine = 33°, overall planar offset quinoline – quinoline = 66°, see Fig. 6, Table 2). However, the emission profile observed is highly unusual and in no way characteristic of a single isolated quinoline ring coordinated to a metal which is not photophysically active, with a Stokes shift of around 100 nm and structured emission tailing to >550.
 | ||
| Fig. 6 Simplified solid-state structure of 2 with anions and hydrogen atoms omitted, highlighting the torsion angles between pyridine and quinoline moieties. | ||
| Structure | Torsion angle (degrees) for Npyr–C–C–Nquin |
|---|---|
| 2 | 31.5(7); 30.9(7); 37.8(6); 25.9(8) and 30.2(7); 30.8(7); 30.8(7); 31.2(8) |
| 2-a | Chelate: 22.1(3); 27.5(3); Bridge: 34.5(3); 42.6(3) |
Theoretical studies of bis-quinolylpyridone 1 and silver helicate 2
In light of the unexpectedly large Stokes shift of ligand 1 and the blue-shifted photophysics of its complex, 2, theoretical calculations were used to probe the structure and photophysics of 1 and 2 in more detail. DFT in simulated CH3CN indicates that pyridone tautomer is 6 kJ mol−1 lower in energy than the pyridine form. Both forms are predicted to have similar absorption (strong bands at 296 nm for pyridone, 283 nm for pyridine) and singlet emission (376 nm for pyridone, 388 nm for pyridine) spectra, where all bands are π–π* in nature. Given the poor agreement between experiment and theory for emission, we also tested the performance of the semi-empirical ZINDO approach: this predicts singlet emission at 364 nm for pyridone and 496 nm for pyridine. The latter is in striking agreement with the main peak in Fig. 3. ZINDO emission for the 1 pyridinol form at 496 nm is dominated by transition HOMO−4 ← LUMO, at the excited state geometry: as shown by orbital plots this is essentially π–π* in nature (Fig. 7, predicted spectra and orbital and compositions are reported in ESI†).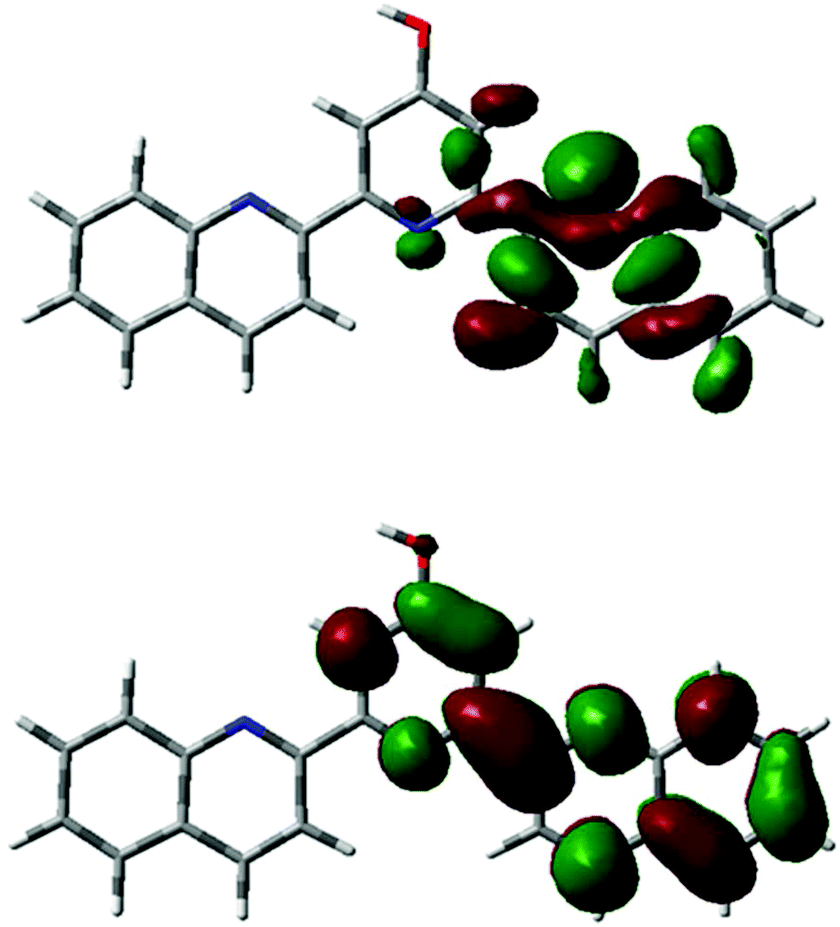 | ||
| Fig. 7 HOMO-4 (top) and LUMO (bottom) of 1 (pyridinol form, ZINDO). | ||
Large Stokes shift and tailing emission bands are often indicative of d-metal involvement in photophysics, so the photophysics of 2 were also examined with theoretical methods. TD-DFT prediction of the absorption spectrum is in broad agreement with experiment, with lowest energy band predicted to lie at 330 nm, a transition dominated by HOMO–LUMO excitation. As shown in Fig. 8, the former is largely metal d in character, with some contribution from N lone pairs, while the latter (see Fig. 9) is a π* ligand-based orbital, such that excitation is assigned to be MLCT. Singlet emission, following relaxation of the first excited state, is predicted to come at 497 nm (TD-DFT) or 443 nm (ZINDO), the latter again in better agreement with experiment. Once again, this transition involves HOMO–LUMO combination, but at the optimal geometry of the excited state, the character of these orbitals changes. Now, both are ligand based, HOMO localised mainly on quinolines and LUMO on pyridines, with small contributions from Ag, such that the photophysics of 2 seems to be best characterised as ILCT emission resulting from MLCT/ILCT absorption.
 | ||
| Fig. 8 Ground state HOMO of 2 (ZINDO). | ||
 | ||
| Fig. 9 Ground state LUMO of 2 (ZINDO). | ||
In light of the theoretical finding that a singlet excited state gave the best fit for the observed emission bands, regardless of the large Stokes shift we re-examined the photophysical properties of complex 2 using time resolved methods. TCSPC measurements in both solution and solid state gave identical results indicating an intensity-weighted average lifetime of 2.5 ns, strongly suggestive of singlet emission, regardless of the involvement of a second row transition metal. The close match between lifetime in the solid and solution states suggest that the emissive species is the same in solution and solid states, i.e. that the structure of the helicate observed crystallographically is maintained in solution. As it is important to establish that the complex maintains the dimeric structure in solution, and mass spectrometry did not identify an intact [Ag2L2] ion (showing only a variety of fragments the largest of which matches an [AgL2] ion) 1H NMR DOSY experiments were undertaken to assess the solution speciation. As the complex is insoluble in solvents for which reliable DOSY molecular mass determination is established internal standards were used (see ESI†) and indicate that the molecular mass of the solution species closely matches that of a mass standard chosen to be close to that of the helicate and far greater than a standard approximately the mass of a monomeric complex (see ESI†).
Further evidence is provided by the considerable 15N chemical shift changes on coordination of nitrogen to silver. The helicate 2 gave 15N shifts of 247.8 for the pyridone and 268.0 for the quinolyl group, or −31.6 and −34.0 ppm compared to the free ligand in DMSO-d6. These coordination shifts are entirely typical for Ag–N complexes.7 While the solubility of 1 in MeCN-d3 was too low to measure 15N shifts in the same solvent, the coordination shift is more than an order of magnitude larger than any known solvent-induced shift changes.
Given the unusual structure of the Ag2N6 core in 2, we also examined the bonding and stabilisation of this complex using theoretical methods. Atoms-in-Molecules (AIM) analysis located a bond critical point (bcp) corresponding to an Ag–Ag bond (ρpcb = 0.024 au). Each Ag also has two strong Ag–N (ρpcb = 0.079 au) bonds to quinoline N and two weaker Ag–N (ρpcb = 0.036 au) to pyridine N, supporting the assignment of the latter as bridging interactions. In addition, stacking interactions between quinolines are found (8 bcp's with ρpcb between 0.005 and 0.007 au). Decomposing 2 into constituent parts sheds more light on the stability of this complex: breaking 2 into two ‘AgN3’ species in simulated CH3CN has ΔG = +123 kJ mol−1, i.e.2 is strongly stabilised due to Ag-Ag and Ag-N bonding. In contrast, stacking does not contribute significantly to overall stabilisation: removing Ag+ species from 2 and to estimate ΔG for L2 → 2L results in −23 kJ mol−1.
Conclusions
2,6-Bis(2-quinolyl)-4(1H)-pyridone 1, exists in solution as a mixture of tautomers with the pyridone tautomer favoured by less polar solvents and exists in the solid state in a variety of polymorphs including one which shows solvent-filled channels. Solutions of 1 show intense room temperature luminescence with an unusually large Stokes shift and low energy emission which is assigned to a π–π* transition of the hydroxypyridine tautomer. Coordination of 1 to Ag(I) gives binuclear double helicates which themselves show luminescence, which is, unusually, blue-shifted compared to the ligand. The emission from the binuclear helicates originates from singlet ILCT emission resulting from MLCT/ILCT absorption. Future studies will explore the coordination chemistry of this new ligand with other metal ions. In light of the fluorescence of the compounds reported herein it is possible that more soluble analogues may have applications in, inter alia, fluorescent cell imaging.Experimental
All starting materials, reagents and solvents were purchased from commercial suppliers and used as supplied unless otherwise stated. 1H, 13C, and 15N NMR were recorded at 400, 101 and 41 MHz respectively on a Bruker Avance III 400 equipped with a broadband observe probe (BBO) and referenced to the deuterium lock shift unless otherwise reported. Chemical shifts are reported in ppm, and coupling constants in Hz. IR spectra were recorded on an Agilent Technologies Cary 630 FTIR Spectrometer as thin films or solids and are reported in wavenumbers (cm−1). UV vis spectra were recorded on an Agilent Technologies Cary 60. Steady state emission and excitation spectra were recorded on an Agilent Technologies Cary Eclipse. Time-resolved spectra were recorded on a PicoQuant FluoTime 300 exciting with an LDH-P-C-375 and decays analysed with the program FluoFit.1,5-Bis(2-quinolyl)-pentane-1,3,5-trione
To a flame dried round bottom flask equipped with a stirrer bar and nitrogen bubbler was added methyl quinaldate (2.00 g, 10.7 mmol) and sodium t-butoxide (2.50 g, 26.0 mmol) and THF (30 ml). To the resulting slurry was added acetone (375 μL, 295 mg, 5.10 mmol) and the resulting thick red suspension was heated at 60 °C for 16 h. To the cooled mixture was added NH4Cl (sat. aq. 100 ml), the mixture stirred vigorously for 10 minutes and the allowed to stand for 1 h. The solid orange product was collected by filtration, dried under vacuum. The crude product was recrystallised from toluene, separated by vacuum filtration, washed with diethyl ether and dried in air to give the title compound as an orange fibrous solid (888 mg, 2.41 mmol, 47%). A second crop was obtained upon evaporation of the mother liquor over 72 h in air, identical to the material above (372 mg, 1.01 mmol, 19%). Total yield of recrystallised material, 66%. 1H NMR (400 MHz, DMSO-d6): δ 8.32 (1H, d, J = 8.3Hz) (H1,1′), 8.21 (1H, d, J = 5.0Hz) (H5,5′), 8.19 (1H, d, J = 5.0Hz) (H4,4′), 7.89 (1H, d, J = 8.0Hz) (H6,6′), 7.79 (1H, t, J = 13.8Hz) (H2,2′), 7.64 (1H, t, J = 13.7Hz) (H3,3′), 7.08 (0.7H, s) (H7′), 2.31 (2H, s) (H7). IR ν max: 1723 (CO), 1604, 1498, 1451, 1243 cm−1. Due to low solubility this material was used without further characterisation.
2,6-Bis(2-quinolyl)-4(1H)-pyridone 1
To a stirred suspension of 1,5-bis(2-quinolyl)-pentane-1,3,5-trione (888 mg, 24.1 mmol) in methanol (25 ml) at 60° C was added ammonium acetate (1.50 g, 19.4 mmol) and the stirred mixture heated at 60 °C for 66 h. After cooling to room temperature, the reaction mixture was treated with water (100 ml) and a yellow gel-like substance separated by prolonged vacuum filtration. The product was recrystallised from an ethanol acetone mixture to give the title compound as a white solid (595 mg, 1.77 mmol, 74%). 1H NMR (400 MHz, CD3OD): δ 8.51 (1H d, J = 8.1 Hz, H8,8′) 8.28 (1H, d, J = 8.2 Hz, H4,4′) 8.20 (1H, d, J = 8.2 Hz, H3,3′) 8.03 (1H, d, J = 8.2 Hz, H5,5′) 7.91 (1H, m, H6,6′) 7.72 (1H, m, H7,7′) 7.33 (1H, s, pyridone H3,3′) 13C{1H} NMR (400 MHz, DMSO-d6): δ 166.6, 156.7, 155.6, 147.6, 137.5, 130.5, 129.7, 128.6, 128.4, 127.6, 119.3, 109.8. IR ν max: 3271, 3055, 1634 (CO), 1564 1502, 1429, 1258 cm−1. m/z (ESI, HRMS 350.1277, C23H15N3O predicts 350.1288). C23H15N3O·2H2O requires C 71.7 H 4.97 N 10.9 found C 71.7 H 4.67 N 11.1%.
Disilver(I)-bis(2,6-bis(2-quinolyl)-4(1H)-pyridonyl) tetrafluoroborate 2
To a round bottom flask was added ligand 1 (67.0 mg, 0.191 mmol) and silver(I) tetrafluoroborate (40 mg, 0.206 mg) and acetonitrile (20 ml) and the mixture heated at reflux for 5 minutes during which time the reactants passed into solution. The mixture was then concentrated under vacuum to 2 ml and treated with diethyl ether (2 ml) giving a white precipitate of the title compound (68 mg, 0.128 mmol, 67%). 1H NMR (400 MHz, CD3CN): δ 8.07 (1H, d, J = 8.1 Hz, H8,8′) 7.80 (1H, d, J = 7.7 Hz, H5,5′) 7.63 (1H, s, pyridine H3,3′) 7.58 (3H, m, H3,3′,4,4′,7,7′) 7.49 (1H, m, H6,6′). 13C{1H} NMR (400 MHz, CD3CN, saturated solution, some peaks not observed): δ 206.6, 138.8, 131.2, 129.4, 128.4, 128.2, 128.1, 112.6. m/z (ESI, HRMS) observed 805.1483 C46H30N6O2Ag (L2Ag) requires 805.1476. C46H30N6O2Ag2B2F8·H2O requires C 50.0 H 2.92 N 7.60 found C 49.8 H 2.71 N 7.99%.Theoretical calculations
Geometry of 1 and 2 were fully optimised without any symmetry constraint using the M06-2X functional14 and a basis set consisting of 6-31G(d) on light atoms15 and Stuttgart-Dresden basis/ECP on Ag.16 TD-DFT calculations were performed using the range-separated CAM-B3LYP method,17 with the same basis set. All such calculations used Gaussian09.18 ZINDO/S19 calculations on DFT optimised geometries were carried out using the ORCA package.20Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr David Rochester, Lancaster University, for Mass Spectrometry.Notes and references
- See e.g. A. M. W. Cargill Thompson, Coord. Chem. Rev., 1997, 160, 1 CrossRef CAS; E. C. Constable, Chem. Soc. Rev., 2007, 36, 246–253 RSC; H. Hofmeier and U. S. Schubert, Chem. Soc. Rev., 2004, 33, 373–399 RSC; U. S. Schubert and C. Eschbaumer, Angew. Chem., Int. Ed., 2002, 41, 2892 CrossRef PubMed; A. Wild, A. Winter, F. Schlutter and U. S. Schubert, Chem. Soc. Rev., 2011, 40, 1459–1511 Search PubMed.
- E. C. Constable and M. D. Ward, J. Chem. Soc., Dalton Trans., 1990, 1405 RSC.
- M. A. Hayes, C. Meckel, E. Schatz and M. D. Ward, J. Chem. Soc., Dalton Trans., 1992, 703 RSC.
- S. Hostachy, C. Policar and N. Delsuc, Coord. Chem. Rev., 2017, 351, 172 CrossRef CAS; L. Wei, J. W. Babich, W. Ouellette and J. Zubieta, Inorg. Chem., 2006, 45, 3057 CrossRef PubMed; R. H. Platel, M. P. Coogan and J. A. Platts, Dalton Trans., 2015, 44, 1170 RSC.
- E. Murguly, T. B. Norsten and N. Branda, J. Chem. Soc., Perkin Trans. 2, 1999, 2789–2794 RSC.
- S. Wang, J. Chen and L. Li, Int. Res. J. Pure Appl. Chem., 2016, 11, 1–6 CrossRef.
- L. Pazderski, Annu. Rep. NMR Spectrosc., 2013, 80, 33–179 CrossRef CAS.
- P. Beak, J. B. Covington, S. G. Smith, J. M. White and J. M. Zeigler, J. Org. Chem., 1980, 45, 1354–1362 CrossRef CAS.
- R. Evans, Z. Deng, A. K. Rogerson, A. S. McLachlan, J. J. Richards, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2013, 52, 3199–3202 CrossRef CAS PubMed.
- P. Florio, C. J. Coghlan, C.-P. Lin, K. Saito, E. M. Campo, W. R. Jackson and M. T. W. Hearn, Aust. J. Chem., 2014, 67, 651–656 CrossRef CAS.
- M. P. Coogan, V. Fernandez-Moreira, B. M. Kariuki, S. J. Pope and F. L. Thorp-Greenwood, Angew. Chem., Int. Ed., 2009, 48, 4965–4968 CrossRef CAS PubMed.
- I. Bassanetti, M. Mattarozzi, M. Delferro, T. J. Marks and L. Marchio, Eur. J. Inorg. Chem., 2016, 2626, DOI:10.1002/ejic.201501236; M. P. Carranza, B. R. Manzano, F. A. Jalon, A. M. Rodriguez, L. Santos and M. Moreno, Inorg. Chem., 2010, 49, 3828, DOI:10.1021/ic902563d; A. Cingolani, F. Effendy, C. Marchetti, R. Pettinari, B. W. Skelton and A. H. White, Inorg. Chem., 2004, 43, 4387, DOI:10.1021/ic0497376; F. Hintermaier, S. Mihan, M. Gerdan, V. Schunemann, A. Trautwein and W. Beck, Chem. Ber., 1996, 129, 571, DOI:10.1002/cber.19961290515.
- G. Baum, E. C. Constable, D. Fenske, C. E. Housecroft and T. Kulke, Chem. Commun., 1998, 2659 Search PubMed; F.-L. Hu, H.-F. Wang, D. Guo, H. Zhang, J.-P. Lang and J. E. Beve, Chem. Commun., 2016, 52, 7990–7993 RSC; Z.-H. Wei, H.-X. Li, W.-H. Zhang, Z.-G. Ren, Y. Zhang, J.-P. Lang and B. F. Abrahams, Inorg. Chem., 2008, 47, 10461–10468 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS; P. C. Hariharan and J. A. Pople, Theor. Chem. Acc., 1973, 28, 213 CrossRef.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chem. Acc., 1990, 77, 123 CrossRef CAS.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CrossRef CAS.
- M. C. Zerner, G. H. Lowe, R. F. Kirchner and U. T. Mueller-Westerhoff, J. Am. Chem. Soc., 1980, 102, 589 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1572595–1572599. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt04744d |
| This journal is © The Royal Society of Chemistry 2018 |