
Concentration dependent supramolecular interconversions of triptycene-based cubic, prismatic, and tetrahedral structures†
Sourav
Chakraborty,  a
Kevin J.
Endres,
a
Ranajit
Bera,
b
Lukasz
Wojtas,
c
Charles N.
Moorefield,
d
Mary Jane
Saunders,
e
Neeladri
Das,
b
Chrys
Wesdemiotis
*af
and
George R.
Newkome
*afg
a
Kevin J.
Endres,
a
Ranajit
Bera,
b
Lukasz
Wojtas,
c
Charles N.
Moorefield,
d
Mary Jane
Saunders,
e
Neeladri
Das,
b
Chrys
Wesdemiotis
*af
and
George R.
Newkome
*afg
a
Kevin J.
Endres,
a
Ranajit
Bera,
b
Lukasz
Wojtas,
c
Charles N.
Moorefield,
d
Mary Jane
Saunders,
e
Neeladri
Das,
b
Chrys
Wesdemiotis
*af
and
George R.
Newkome
*afg
Abstract
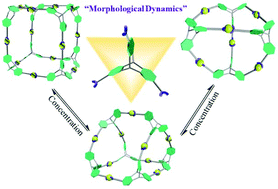
The quantitative, single step, self-assembly of a shape-persistent, three-dimensional C3v-symmetric, triptycene-based tris-terpyridinyl ligand initially gives a platonic-based cubic architecture, which was unequivocally characterized by 1D and 2D NMR spectroscopy, mass spectrometry, and single crystal X-ray structural analysis. The unique metal–ligand binding properties of the Cd2+ analogue of this construct give rise to a concentration-dependent dynamic equilibrium between cube, prism, and tetrahedron-shaped architectures. Dilution transforms this cube into two identical tetrahedra through a stable prism-shaped intermediate; increasing the concentration reverses the process.
- This article is part of the themed collection: Molecular metal-containing soft materials
 Please wait while we load your content...
Please wait while we load your content...

