
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rise of A-site columnar-ordered A2A′A′′B4O12 quadruple perovskites with intrinsic triple order
Alexei A.
Belik

Research Center for Functional Materials, National Institute for Materials Science (NIMS), Namiki 1-1, Tsukuba, Ibaraki 305-0044, Japan. E-mail: Alexei.Belik@nims.go.jp
First published on 15th January 2018
Abstract
A-site-ordered AA′3B4O12 quadruple perovskites (with twelve-fold coordinated A and square-planar coordinated A′ sites) were discovered in 1967. Since then, there have been considerable research efforts to synthesize and characterize new members of such perovskites. These efforts have led to the discoveries of many interesting physical and chemical properties, such as inter-site charge transfer and disproportionation, giant dielectric constant, multiferroic properties, reentrant structural transitions and high catalytic activity. The first member of A-site columnar-ordered A2A′A′′B4O12 quadruple perovskites (with ten-fold coordinated A, square-planar coordinated A′ and tetrahedrally coordinated A′′ sites), CaFeTi2O6, was discovered in 1995, and for 19 years it was the only representative of this family. In the last few years, A2A′A′′B4O12 perovskites have experienced rapid growth. Herein, we present a brief overview of the recent developments in this field and highlight an under-investigated status and great potential of A2A′A′′B4O12, which can be prepared mainly at high pressure and high temperature. The presence of the A′′ site gives an additional degree of freedom in designing such perovskites. The A2A′A′′B4O12 perovskites are discussed in comparison with well-known AA′3B4O12 perovskites.
 Alexei A. Belik | Alexei Belik graduated from the Department of Materials Science of Moscow State University (MSU), Russia, in 1997 and received his PhD in inorganic chemistry at MSU in 1999. He did his first postdoc (2000–2002) at the National Institute for Materials Science (NIMS), Japan, under the supervision of Prof. Fujio Izumi and his second postdoc (2002–2004) at the Institute for Chemical Research, Kyoto University, under the supervision of Profs. Mikio Takano and Masaki Azuma. In 2004, he joined the NIMS again as an independent scientist under the ICYS program. In 2006, he took a permanent position at the NIMS. He was an MANA independent scientist for 9 years. |
Introduction
ABO3 compounds crystallize in a number of structure types [e.g., perovskites, LiNbO3, corundum, ilmenite, hexagonal BaMnO3-type structures (2H, 4H, 6H, 15R and so on), hexagonal LuMnO3-type, pyroxenes, rare earth sesquioxide structures (A, B and C (bixbyite)), AlFeO3, KSbO3, PbReO3, CaIrO3 and others] depending on the relative size of the A and B cations and on the nature of cations.1 The perovskite-type structure is one of the most important and adaptive structures in inorganic chemistry, and it has the largest number of representatives.2–4 It is built from a framework of corner-shared BO6 octahedra that form cavities, which are filled with A cations. The ideal perovskite structure is cubic (space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) with ap ≈ 3.8 Å. However, the cubic structure is rarely realized at room temperature because of the mismatch among the sizes of A, B and O. The mismatch, which is usually described by the Goldschmidt tolerance factor,5 leads to different distortions. Distortions can be considered as tilts of rigid BO6 octahedra in a first approximation. And tilts, in turn, are described by using the Glazer tilt system and B–O–B bond angles.6 There are basically 15 tilt systems in ABO3 perovskites.4 In simple ABO3 perovskites, the B–O–B tilt angles are within the range of 140–180°.
m) with ap ≈ 3.8 Å. However, the cubic structure is rarely realized at room temperature because of the mismatch among the sizes of A, B and O. The mismatch, which is usually described by the Goldschmidt tolerance factor,5 leads to different distortions. Distortions can be considered as tilts of rigid BO6 octahedra in a first approximation. And tilts, in turn, are described by using the Glazer tilt system and B–O–B bond angles.6 There are basically 15 tilt systems in ABO3 perovskites.4 In simple ABO3 perovskites, the B–O–B tilt angles are within the range of 140–180°.
A special class of the perovskite family is formed for the a+a+a+ tilt system with very large B–O–B tilt angles of about 140–145° (space group of the parent structure is Im).7,8 It is the so-called A-site-ordered quadruple perovskites, AA′3B4O12, which have a twelve-fold coordinated A site and a square-planar coordinated A′ site. The A′ site is typically occupied by Jahn–Teller cations [such as, Cu2+ and Mn3+] or other cations which allow a square-planar coordination: Cu3+,8 Fe2+,9,10 Co2+,11 Pd2+,12 Pb4+ (ref. 13) and Mn2+ (ref. 14), and most of the AA′3B4O12 compounds need high pressure (HP) and high temperature (HT) for their preparation. The AA′3B4O12 quadruple perovskite subfamily of the perovskite family has also numerous representatives with A = Na+, Mn2+, Cd2+, Ca2+, Sr2+, Pb2+, R3+ (R = rare earths), Ce4+, Bi3+ and Th4+, and B = Mn3+, Mn4+, Fe3+, Cr3+, Al3+, Ti4+, V4+, Ge4+, Sn4+, Ru4+, Ir4+, Ta5+, Nb5+, Sb5+ and others.7 This subfamily was discovered in 1967 in CaCu3Ti4O12 during Cu2+ doping of CaTiO3.15 Since then, there have been considerable research efforts to synthesize and characterize new members of such quadruple perovskites. These efforts have led to the discoveries of many interesting physical and chemical properties, for example, giant dielectric constant,7 inter-site charge transfer and disproportionation,8 multiferroic properties, reentrant structural transitions,16 and high catalytic activity.17
Another special class of the perovskite family can be formed with the a+a+c− tilt system and with very large B–O–B tilt angles of about 140–150° (space group of the parent structure is P42/nmc).3,4 In such a case, compounds with the A2A′A′′B4O12 stoichiometry are formed. Therefore, they can also be called A-site-ordered quadruple perovskites, where A′ is the site with a square-planar coordination and A′′ is the site with a tetrahedral coordination. To distinguish them from AA′3B4O12, we suggest calling them A-site columnar-ordered18 quadruple perovskites. The first representative of this subfamily, CaFeTi2O6, was discovered just in 1995.9,19,20 CaFeTi2O6 was the only representative of such quadruple perovskites for 19 years, and this distortion of perovskites was considered to be highly exotic and rare.3,4 Only in 2014 was another representative reported, CaMnTi2O6.21 In the last few years, A2A′A′′B4O12 quadruple perovskites have experienced rapid growth.18,22–24 The purpose of this frontier article is to highlight their under-investigated status and great potential and to attract attention of the perovskite and high-pressure communities. The A2A′A′′B4O12 quadruple perovskites are compared with the AA′3B4O12 ones.
Crystal structure descriptions
Simple ABO3 perovskites can have, and AA′3B4O12 and A2A′A′′B4O12 quadruple perovskites have very strong tilts of the BO6 octahedral sublattice with the B–O–B bond angles reaching 140–145°. In simple ABO3 perovskites, such strong tilts are realized in compounds with quite small A cations, for example, A = Sc and In.25,26 The ABO3 perovskites with such strong tilts usually have GdFeO3-related distortions26 with the (ideal) space group Pnma (√2ap × 2ap × √2ap) and the tilt system of a−b+a−, which means that BO6 octahedra are rotated in one direction (in-phase) along the b axis and in opposite directions (out-of-phase) along the a and c axes (in cubic axes of the original Pmm cell) (Fig. 1a). The AA′3B4O12 quadruple perovskites have a 2ap × 2ap × 2ap cell and the a+a+a+ tilt system, which means that BO6 octahedra are rotated in one direction along all three crystallographic directions (Fig. 1b). The A2A′A′′B4O12 quadruple perovskites have a 2ap × 2ap × 2ap cell and the a+a+c− tilt system, which means that BO6 octahedra are rotated in one direction along the a and b axes and in opposite directions along the c axis (Fig. 1c). Therefore, on considering the B sublattice, all three perovskites are very similar to each other and differ from each other by a number of the in-phase (very strong) tilts of the BO6 octahedra.
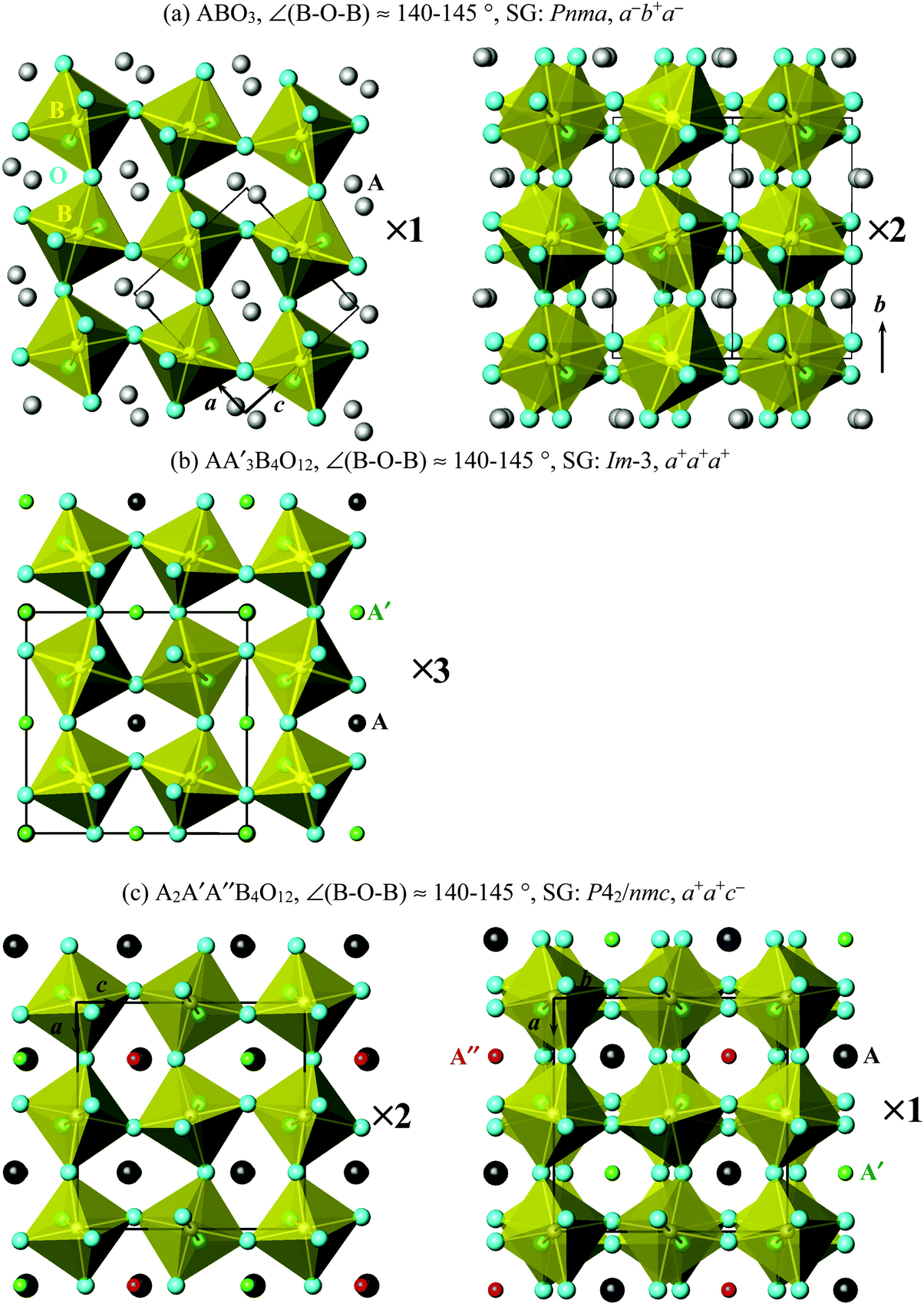 | ||
| Fig. 1 Connections of BO6 octahedra along different directions in (a) ABO3 simple perovskites, (b) AA′3B4O12 quadruple perovskites and (c) A2A′A′′B4O12 quadruple perovskites, emphasizing different tilt systems. A, A′ and A′′ sites are shown by spheres. SG: space group. | ||
The different numbers of the in-phase BO6 tilts (determined, of course, primarily by the stoichiometry) create different numbers and types of cavities for A cations (Fig. 2). There is one type of cavity in the case of ABO3, and the coordination number is reduced from twelve (in the ideal cubic ABO3) to eight. The AO8 polyhedra are connected by edges to form a three-dimensional (3D) network (Fig. 2a).
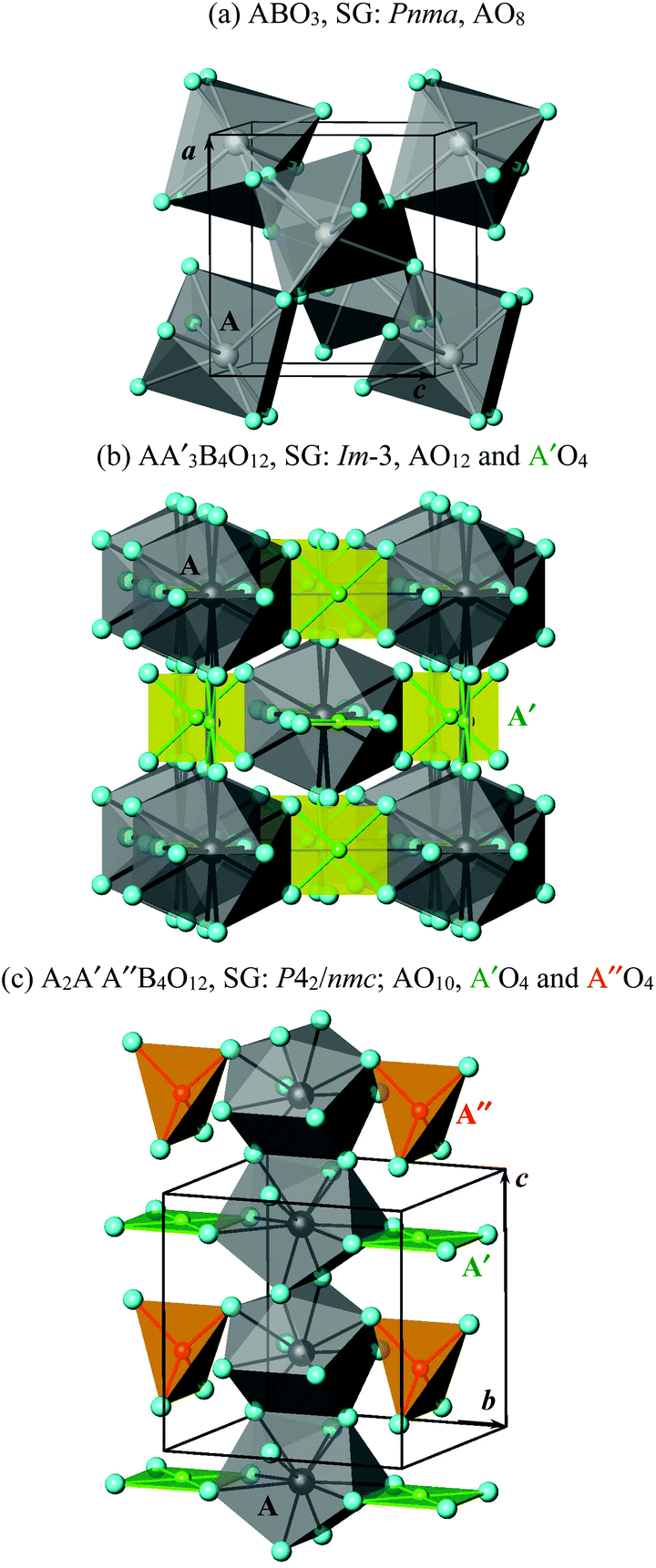 | ||
| Fig. 2 Fragments of structures with polyhedra for the A, A′ and A′′ sites in (a) ABO3 simple perovskites, (b) AA′3B4O12 quadruple perovskites and (c) A2A′A′′B4O12 quadruple perovskites. B atoms are omitted for clarity. SG: space group. | ||
There are two types of cavities in the case of AA′3B4O12 with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. The A site keeps a twelve-fold coordination as in the ideal cubic ABO3, and the A′ site has four very short A′–O bond lengths, four much longer A′–O bond lengths, and four very long A′–O bond lengths (in total, still twelve). The four very short A′–O bond lengths form a square-planar coordination. The AO12 polyhedra are isolated from each other. The A′O4 squares are also isolated from each other (Fig. 2b), and they are oriented in such a way that they are orthogonal to each other, but they are 3D-connected through the longer A′–O bond lengths. The AO12 and A′O4 polyhedra are 3D-connected through edges.
:3. The A site keeps a twelve-fold coordination as in the ideal cubic ABO3, and the A′ site has four very short A′–O bond lengths, four much longer A′–O bond lengths, and four very long A′–O bond lengths (in total, still twelve). The four very short A′–O bond lengths form a square-planar coordination. The AO12 polyhedra are isolated from each other. The A′O4 squares are also isolated from each other (Fig. 2b), and they are oriented in such a way that they are orthogonal to each other, but they are 3D-connected through the longer A′–O bond lengths. The AO12 and A′O4 polyhedra are 3D-connected through edges.
There are three types of cavities in the case of A2A′A′′B4O12 with a ratio of 2:1:1. Therefore, such perovskites have intrinsic triple A-site ordering. The A site has a ten-fold coordination (plus two very long bond lengths), and both A′ and A′′ sites have four very short A′–O and A′′–O bond lengths with similar values of 1.9–2.1 Å, four much longer bond lengths, and four very long bond lengths (in total, still twelve). The four very short A′–O bond lengths form a square-planar coordination. However, the four very short A′′–O bond lengths form a tetrahedral coordination (Fig. 2c). Therefore, the A′ and A′′ sites have fundamentally different coordination environments, and they should be distinguished even though they have similar bond lengths. The AO10 polyhedra are connected through edges and form chains (columns) along the c axis. The A′O4 squares and A′′O4 tetrahedra are separated from each other, but they are connected through the longer A′–O bonds to form chains (columns) along the c axis. Because of the existence of the –AO10–AO10– columns and the –A′O4+4–A′′O4– columns (or columns consisting of isolated A′O4 and A′′O4 units), A2A′A′′B4O12 perovskites can be called A-site columnar-ordered perovskites. Connections of the AO10 and A′O4 polyhedra through edges, the AO10 and A′′O4 polyhedra through corners and the –AO10–AO10– columns through corners create a 3D network. Tetrahedral coordination is usually not realized in oxygen-stoichiometric ABO3 perovskites; only the removal of oxygen in ordered manners creates tetrahedral sites.4 The A2A′A′′B4O12 perovskites are oxygen-stoichiometric perovskites with tetrahedral sites.
Known examples and expected compositions of A2A′A′′B4O12
The number of reported A2A′A′′B4O12 perovskites is so limited at the moment that they all can be simply listed here (for example, in the chronological order of discovery and in the short formulae): CaFeTi2O6 (space group P42/nmc),9,10,19,20,27 CaMnTi2O6 (space group P42mc),21,28,29 RMnMnSbO6 (R = La, Pr, Nd and Sm) (space group P42/n),22 RMnGaTiO6 (R = Sm and Gd) (space group P42/nmc),18 RMnMn2O6![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) RMn3O6 (R = Gd–Tm and Y) (space group Pmmn),23 CaMnFeReO6,24 CaMnMnReO624 and Ca2CuMnFe2Re2O12 (space group P42/n).24 We recently prepared R3+2Mn3+Mn2+[Mn3+3Ti4+]BO12 (space group P42/nmc), R3+2Mn2+Mn2+[Mn3+2Ti4+2]BO12 (space group P42/nmc), R3+2Mn3+Ga3+[Mn3+4]BO12 (space group P42/nmc) and R3+2Cu2+Mn2+[Mn3+2]B1[Mn4+2]B2O12 (space group Pmmn), where []B means the presence of one B site and []B1[]B2 means a B-site order.30 (We note that it was not possible to precisely determine the degree of Cu2+ and Mn2+ ordering and Mn3+ and Ga3+ ordering in the last two compounds with X-ray powder diffraction).
RMn3O6 (R = Gd–Tm and Y) (space group Pmmn),23 CaMnFeReO6,24 CaMnMnReO624 and Ca2CuMnFe2Re2O12 (space group P42/n).24 We recently prepared R3+2Mn3+Mn2+[Mn3+3Ti4+]BO12 (space group P42/nmc), R3+2Mn2+Mn2+[Mn3+2Ti4+2]BO12 (space group P42/nmc), R3+2Mn3+Ga3+[Mn3+4]BO12 (space group P42/nmc) and R3+2Cu2+Mn2+[Mn3+2]B1[Mn4+2]B2O12 (space group Pmmn), where []B means the presence of one B site and []B1[]B2 means a B-site order.30 (We note that it was not possible to precisely determine the degree of Cu2+ and Mn2+ ordering and Mn3+ and Ga3+ ordering in the last two compounds with X-ray powder diffraction).
Because of the similarities in the B sublattice (Fig. 1), it is expected that the B site of A2A′A′′B4O12 can be occupied by most of the B-type cations for perovskites and their different combinations (a list of examples is given in the second paragraph of Introduction).
The A sites in all three families retain large environments with some variations in the coordination numbers. Therefore, it is expected that the A site of A2A′A′′B4O12 can be occupied by many typical A cations for perovskites (a list of examples is given in the second paragraph of Introduction). Despite the twelve-fold coordination of the A site in AA′3B4O12, we are not aware of any such perovskites with A = Ba2+. This fact shows that strong and almost fixed tilts of the BO6 octahedra put limits on the possible A–O bond lengths. It is well illustrated by the crystal structures of AMn7O12 with A = Cd, Ca, Sr and Pb,31,32 where the A–O bond lengths are almost the same in all these compounds (l(A–O) ∼ 2.6–2.7 Å) independent of the size of A cations. With high enough pressure, small cations can be forced into the A site resulting in rattling (e.g., in CuCu3V4O12),33 while too large cations (e.g., A = Ba) cannot. In A2A′A′′B4O12, the A-site coordination number and A–O bond lengths (l(A–O) ∼ 2.3–2.5 Å (×6) and l(A–O) ∼ 2.7 Å (×4)) are further reduced in comparison with AA′3B4O12. This fact can put additional limits on the size of A-type cations that can be forced into the A site. All rare-earth elements (except Ce and Pm) and Ca2+ were found so far at the A site of A2A′A′′B4O12. The size of Na+ is close to those of Ca2+ and La3+;34 therefore, we can also expect such compounds with A = Na+. We also emphasize that depending on compositions at the A′, A′′ and B sites, ranges of stability of A2A′A′′B4O12 (A = R = rare-earths) vary from R = La–Sm22 for RMnMnSbO6 to R = Gd–Tm and Y23 for RMnMn2O6 (in all cases, we always mean the synthesis conditions used; stability ranges can change if the synthesis pressure is increased or other conditions are altered). It means that a number of different rare-earth elements should always be checked when preparing such compounds with different B-site contents. A similar tendency was also observed in AA′3B4O12: for example, RMn3(AlTi3)O12 is formed for R = La–Sm, but not for R = Gd–Lu;14 RMn7O12 is formed for R = La–Er and Y, but not for R = Tm–Lu.23 On the other hand, RCu3Mn4O12 can be prepared for all R at a relatively low pressure of 2 GPa.35
The A′O4 sites of AA′3B4O12 and A2A′A′′B4O12 are identical. Therefore, it is expected that they can be occupied by the same cations. They are listed and highlighted by bold letters in Table 1, which also gives some other cations with a possible square-planar coordination. We note that there are reports about the presence of Li+ (e.g., in (Li1.333Cu1.333)Ta4O12)36 and very small amounts of Fe3+ and Ti4+ at the A′ site of AA′3B4O12.37,38 Mn3+, Cu2+, Fe2+ and Mn2+ were found so far in the A′ site of A2A′A′′B4O12 quadruple perovskites.
| Cation | Radius (Å) | Cation | Radius (Å) |
|---|---|---|---|
| Radii are taken from ref. 34 (for the square-planar (SQ) coordination if available) except for Mn3+ and Cu3+ (rSQ(Mn3+/Cu3+) = l(Mn/Cu–O) − r(O2−), where l(Mn–O) = 1.91 Å and l(Cu–O) = 1.88 Å are the average Mn–O and Cu–O bond distances in AMn3+3B4O12 and ACu3+3B4O12 quadruple perovskites (taking the four shortest Mn/Cu–O bonds) and r(O2−) = 1.4 Å). Cations marked by bold letters (Cu3+, Mn3+, Cu2+, Co2+, Fe2+, Pd2+, Pb4+ and Mn2+) were found in the A′ site of AA′3B4O12 quadruple perovskites; note that Li+ (together with Cu2+)36 and very small amounts of Ti4+ and Fe3+ were also reported.37,38 Mn3+, Cu2+, Fe2+ and Mn2+ were found so far in the A′ site of A2A′A′′B4O12 quadruple perovskites. LS: low-spin. | |||
| Cu 3+ | ≈0.48 | Fe 2+ | 0.64 |
| Ni2+(LS) | 0.49 | Pd 2+ | 0.64 |
| Mn 3+ | ≈0.51 | Pb 4+ | 0.65 |
| Cu 2+ | 0.57 | Mn 2+ | 0.66 |
| Co 2+ | 0.58 | Ag3+ | 0.67 |
| Pt2+ | 0.60 | Au3+ | 0.68 |
| Cr2+ | Not available | ||
The A′′O4 tetrahedral site is unique in A2A′A′′B4O12. Table 2 gives a list of some cations that can have a tetrahedral coordination environment and their ionic radii.34 Cu2+, Fe2+ and Mn2+ were found so far in the A′′ site of A2A′A′′B4O12 and they probably accidentally overlap with cations found at the A′ site (Fig. 3). The charge and size of Co2+, Mg2+ and Zn2+, for example, are close to those of Cu2+, Fe2+ and Mn2+, but they should preferably occupy the tetrahedral A′′ site instead of the square-planar A′ site.39 On the other hand, however, all cations shown in Table 2 can have an octahedral coordination, and some of them (e.g., Ni2+, Sn4+ and Ti4+) show a very strong preference for such an environment.39 Therefore, strong competitions with the octahedral B sites or the formation of other phases where such cations have an octahedral coordination are possible. One strategy to force them into the A′′ site is to use cations at the B sites with an even stronger preference for octahedral environments (similar to NiCr2O4 and CuCr2O4 spinels). We can expect that with high enough pressure during the synthesis some elements from Table 2 (in addition to Cu2+, Fe2+ and Mn2+) can be stabilized at the A′′ site. For example, 5 GPa was enough to stabilize Fe2+ in the square-planar A′ site of CaFe3Ti4O12, while more than 12 GPa was required to prepare CaFeTi2O6 with Fe2+ in both the square-planar A′ and tetrahedral A′′ sites.9
 | ||
| Fig. 3 A schematic figure with some cations that can have a tetrahedral coordination and a square-planar coordination in oxides and their overlap.39 Bold letters show cations found at the A′ site of AA′3B4O12. Cu2+, Fe2+ and Mn2+ (underlined) were found so far in the A′′ site of A2A′A′′B4O12 quadruple perovskites. | ||
| Cation | Radius (Å) | Cation | Radius (Å) |
|---|---|---|---|
| Radii are taken from ref. 34. Cations marked by bold letters (Cu2+, Fe2+ and Mn2+) were found so far in the A′′ site of A2A′A′′B4O12 quadruple perovskites. HS: high-spin. | |||
| Ge4+ | 0.39 | Cu 2+ | 0.57 |
| Al3+ | 0.39 | Co2+(HS) | 0.58 |
| Ti4+ | 0.42 | Li+ | 0.59 |
| Ga3+ | 0.47 | Zn2+ | 0.60 |
| Fe3+ | 0.49 | Fe 2+ (HS) | 0.63 |
| Ni2+ | 0.55 | Pb4+ | 0.65 |
| Sn4+ | 0.55 | Mn 2+ (HS) | 0.66 |
| Mg2+ | 0.57 | Cd2+ | 0.78 |
| Co3+ | Not available | ||
An attempt to prove the above concept (about the location of different elements at the A′ and A′′ sites) was made as shown in ref. 24 by the synthesis of Ca2CuMnFe2Re2O12 aiming at Cu2+ at the A′ site and Mn2+ at the A′′ site. We synthesized R2CuMnMn4O12. We also tried to insert Ga3+ and prepared R2MnGaMn4O12 (space group P42/nmc).30 We emphasize that because Mn3+ can disproportionate during the synthesis as found in RMnMn2O6 = [R3+2]A[Mn3+]A′[Mn2+]A′′[Mn3+2]B1[Mn3+Mn4+]B2O12,23 any cation/charge distributions are possible between [R3+2]A[Mn3+]A′[Ga3+]A′′[Mn3+4]BO12 with Ga3+ at the A′′ site and [R3+2]A[Mn3+]A′[Mn2+]A′′[Mn3+2Mn4+Ga3+]BO12 with Ga3+ at the B site (assuming39 that the A′′ site cannot be occupied by Mn3+ and Mn4+). Our structural analysis with X-ray powder diffraction data gave evidence that the cation distribution is more close to the former case (A′′–O bond lengths of 1.89 Å were more consistent with Ga–O bond lengths). However, a precise model can only be determined with neutron diffraction, and this method is an essential tool to study cation distributions in A2A′A′′B4O12.
The presence of the A′′ site gives an additional degree of freedom in A2A′A′′B4O12, and the number of elements that can have a tetrahedral environment is larger than that with a square-planar environment (Fig. 3). Therefore, it is expected that A2A′A′′B4O12 perovskites should be numerous and versatile.
Known distortions
The parent structure of AA′3B4O12 has the space group Im (no. 204). All distortions from the direct group–subgroup relations are experimentally realized (except for the same Im symmetry with 6ap × 6ap × 6ap) (Fig. 4). These distortions are exemplified in CaCu3Ti4O12 (the parent structure),15,40 CaCu3Ga2Sb2O12,41 Ce0.5Cu3Ti4O12,42 (Li1.333Cu1.333)Nb4O12,36 CaCu3Fe4O12,8,38 A2+Mn7O12 (A = Cd, Ca, Sr and Pb)31,32 and BiCu0.2Mn6.8O12.16 Most of these distortions are temperature-driven, that is, they are observed on cooling for a particular composition, and there exists a high-temperature parent modification. However, there are a few composition-driven distortions, that is, they are determined by a particular composition and take place during the synthesis.41,42 The I23 distortion in (Li1.333Cu1.333)Nb4O12 could be temperature-driven (caused by Nb displacements), but no evidence (no phase transitions to the parent structure) was reported;36 this is why we call it composition-driven. There are a number of temperature-driven distortions that cannot be described by the direct group–subgroup paths, for example, in LaMn7O12 (Im ⇒ (Immm) ⇒ I2/m),43 NaMn7O12 (Im ⇒ (Immm) ⇒ (I2/m) ⇒ C2/m)44,45 and BiMn7O12 (Im ⇒ (Immm) ⇒ I2/m ⇒ Im ⇒ I1),46 and there is one such composition-driven distortion in Cu2+xTa4O12 (Im ⇒ (Pm) ⇒ Pmmm),47 where space groups in parentheses give group–subgroup links. Despite a large number of AA′3B4O12-type compounds, proper polar distortions are realized only in one case of BiMn7O12 due to the effects of the lone electron pair of Bi3+.46 But the ferroelectricity of BiMn7O12 has not been demonstrated yet.
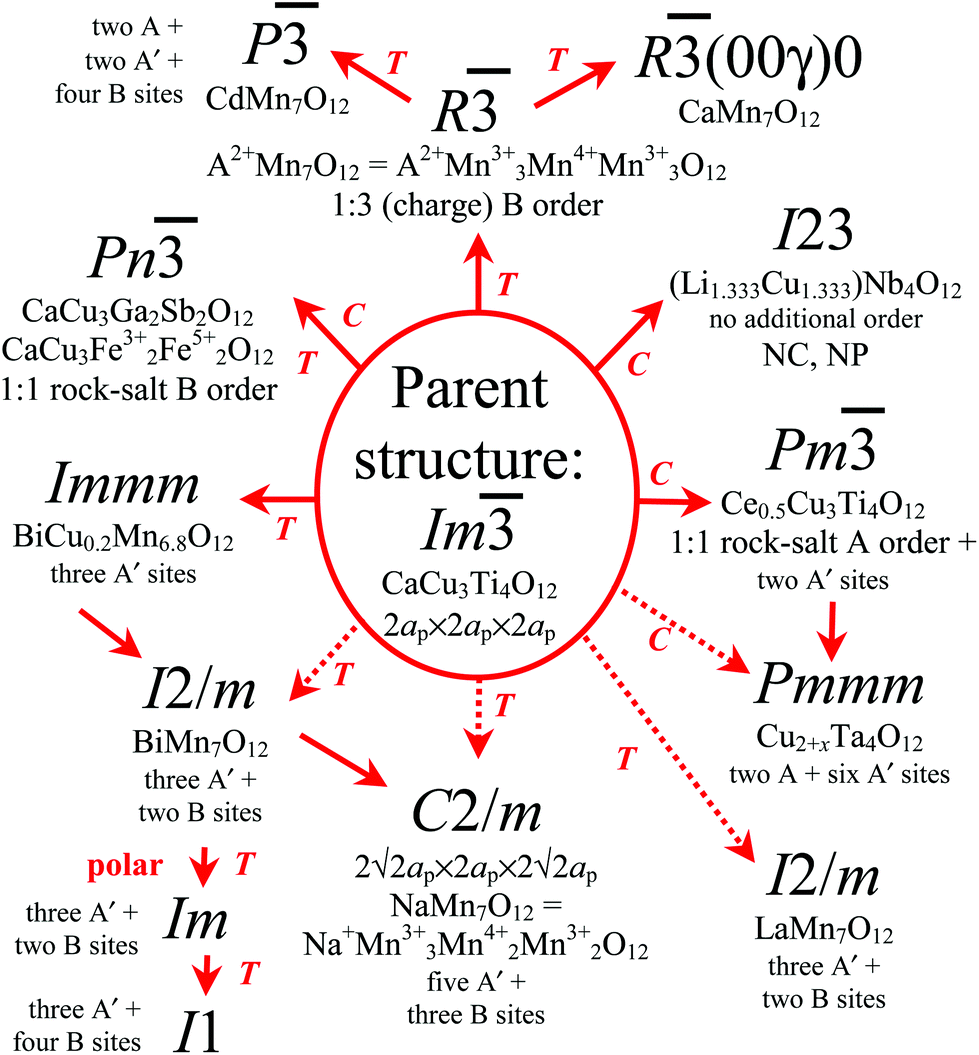 | ||
| Fig. 4 A schematic diagram of all direct group–subgroup distortions and other known distortions of the parent structure of AA′3B4O12 quadruple perovskites (space group Im). Direct group–subgroup distortions are shown by solid arrows; other distortions are shown by dashed arrows. Examples of known distortions are given. Types of A- and B-site orders or number of distinct sites are highlighted. All distortions keep the 2ap × 2ap × 2ap cell except R, P, R(00γ)0 and C2/m. T: a temperature-driven distortion, C: a composition-driven distortion, NC: non-centrosymmetric, NP: non-polar. | ||
The parent structure of A2A′A′′B4O12 has the space group P42/nmc (no. 137). There are seven distortions from the direct group–subgroup relations (except for the same P42/nmc symmetry with 2ap × 2ap × 6ap and 6ap × 6ap × 2ap) (Fig. 5). Three of them have already been found experimentally. Surprisingly, a temperature-driven proper polar distortion takes place in CaMnTi2O6 without any effects of lone electron pairs.21 This is a quite promising fact in comparison with the AA′3B4O12 family. Rock-salt B-site (space group P42/n)22,24 and layered B-site (space group Pmmn)23 orders have already been realized in A2A′A′′B4O12. The reported P42/n distortion is composition-driven. The Pmmn distortion in RMnMn2O6 could be temperature-driven, but no evidence (no phase transitions to the parent structure) was found so far; this is why we call it composition-driven. It is interesting that the Pmmn model has potential for an additional 1:1 rock-salt A-site order (that is, it has two independent A sites: A1 and A2), which might be realized, for example, in a hypothetical compound [Ca2+]A1[Dy3+]A2[Mn3+]A′[Mn2+]A′′[Mn3+2]B1[Mn4+2]B2O12 (it is just given to illustrate compositional design principles). The polar P42mc distortion has potential for an additional 1:1 columnar A-site order: [Na+]A1[Nd3+]A2[Mn2+]A′[Mn2+]A′′[Ti4+4]BO12 (a hypothetical compound to illustrate compositional design principles). A1–O and A2–O distances in the Pmmn (e.g., RMnMn2O6) and P42mc (e.g., CaMnTi2O6) models are different; therefore, there might be driving forces for the ordering of cations with different oxidation states and sizes. The rock-salt A-site ordering is rare in ABO3;3,4 however, the ordering would not be direct in the Pmmn model because the A sublattice is ‘diluted’ by A′ and A′′.
 | ||
| Fig. 5 A schematic diagram of all direct group–subgroup distortions of the parent structure of A2A′A′′B4O12 quadruple perovskites (space group P42/nmc). Examples of known distortions are given. Types of A- and B-site orders or number of distinct sites are highlighted. All distortions keep the 2ap × 2ap × 2ap cell except Ccca. T: a temperature-driven distortion, C: a composition-driven distortion, NC: non-centrosymmetric, NP: non-polar. | ||
Physical and chemical properties of A2A′A′′B4O12
As already mentioned, CaMnTi2O6 crystallizes in a polar space group. This compound shows a structural phase transition from P42mc to the parent P42/nmc structure at 630 K. The phase transition is accompanied by sharp dielectric constant anomalies and the loss of second-harmonic generation signals. Moreover, a room-temperature ferroelectric P–E hysteresis loop was demonstrated.21 These results proved that CaMnTi2O6 is ferroelectric at a ferroelectric Curie temperature of 630 K. The shifts of Mn2+ cations at the A′ site from the square-planar plane and Ti4+ cations from the center of the TiO6 octahedra are responsible for the polar distortion.21,29 Polarization was calculated to be about 0.3 C m−2, and a highly tunable semiconducting energy band gap was predicted in CaMnTi2O6.29 The P42mc-to-P42/nmc ferroelectric transition was also found at room temperature at about 7 GPa.28Without the assistance from the B sublattice, the interaction among magnetic cations at the A′ and A′′ sites is usually very weak (at least, among Mn2+): CaMnTi2O6 has the Néel temperature (TA,N) of about 10 K,21 while SmMnGaTiO6 was reported to order at TA,N = 3 K, and no ordering was found in GdMnGaTiO6.18 In the case of SmMnGaTiO6 and GdMnGaTiO6, even small disordering of Ga3+ and Mn2+ between the A′′ and B sites could suppress a long-range magnetic ordering. On the other hand, magnetic interactions through Ti4+ could be enhanced in CaMnTi2O6 resulting in higher TN similar to a higher magnetic transition temperature in CaCu3Ti4O12 in comparison with CaCu3Ge4O12 and CaCu3Sn4O12.3 No cooperative long-range magnetic order of the parent compound, CaFeTi2O6, was found down to 4.2 K.27
The presence of magnetic cations at the B sites significantly enriched magnetic behaviours. First, the ordering temperature of Mn2+ at the A′ and A′′ sites can reach TA,N = 70 K in CaMnFeReO624 and TA,C = 100 K in CaMnMnReO6.24 Second, the magnetic ordering of Mn2+ at the A′ and A′′ sites can be either antiferromagnetic (TA,N in CaMnFeReO6)24 or ferromagnetic (TA,C in CaMnMnReO624 and TC = 76 K in NdMnMnSbO6).22 Magnetic cations at the B sublattice can order first as in CaMnMnReO6 at TB,C = 120 K (a ferrimagnetic structure),24 can order first with much higher temperatures as in CaMnFeReO6 at TB,C = 500 K and in Ca2CuMnFe2Re2O12 at TB,C = 560 K (a ferrimagnetic structure of Fe3+ and Re5+)24 or can order at the same temperature with the A′ and A′′ sites as in NdMnMnSbO6.22 There is a spin reorientation transition in NdMnMnSbO6 at 42 K driven by order of the Nd moments.22 RMnMn2O6 compounds also show complex magnetic behaviours at several transition temperatures (two or three magnetic transitions depending of R and stoichiometry);23 but magnetic structures have not been investigated yet. The maximum magnetic transition temperature is about 80 K in RMnMn2O6,23 but it jumps to about 180 K in R2CuMnMn4O12 after Cu2+ doping.30 This jump could be caused by the increase of the Mn4+ concentration at one of the B sites from [Mn3+2]B1[Mn3+Mn4+]B2 to [Mn3+2]B1[Mn4+2]B2. Most of the R2CuMnMn4O12 compounds exhibit three magnetic transitions (e.g., at 15, 145 and 180 K for R = Tm).30 Significant magnetoresistance was found in CaMnFeReO6 and magnetoresistance switching was observed in Ca2CuMnFe2Re2O12.24 Therefore, A2A′A′′B4O12 perovskites show complex magnetic properties.
It was found that RMnMn2O6 (RMn3O6) compounds are prone to non-stoichiometry, R1−δMn3O6−1.5δ, with δ = −(0.059–0.071) for R = Gd, δ = 0 for R = Dy, δ = 0.05–0.1 for R = Ho and Y, and δ = 0.12 for R = Er and Tm, and their magnetic properties are highly sensitive to the δ values.23 The real rare-earth cation deficiency was formed in the case of Er0.88Mn3O5.82 and Tm0.88Mn3O5.82; and the cation distribution can be written as [Tm3+1.76]A[Mn3+]A′[Mn2+]A′′[Mn3+2]B1[Mn3+Mn4+]B2O12−δ.30 However, the manganese deficiency in GdMn3−yO6−1.5y is realized through the doping of a small amount of Gd into the tetrahedral A′′ site: [Gd3+2]A[Mn3+]A′[Mn2+0.86Gd3+0.14]A′′[Mn3+2]B1[Mn3.43+2]B2O12.30 The location of rare-earth elements at tetrahedral sites is very unusual.39 RMnMn2O6 compounds were prepared from R2O3 and Mn3+2O3.23 However, during the synthesis at HP and HT, charge disproportionation takes place because Mn can enter the A′′ site only in the oxidation state of +2.
Neutron diffraction showed that Fe3+ cations occupy the A′ site (8%) and the A′′ site (16%) in CaMnFeReO6.24 This fact shows that Fe3+ can in principle be at the tetrahedral A′′ site (Fig. 3). The location of a very small amount of Fe3+ cations at the square-planar A′ site was also observed in AA′3B4O12 by Mössbauer spectroscopy, but the concentration never reaches a few percent.38 A very small antisite disorder at the B sites was found in CaMnFeReO6 (3% Fe/Re) and CaMnMnReO6 (4% Mn/Re); CaMnMnReO6 also had about 5% of Mn at the Ca site,24 and small amounts of Mn were found at the Nd and Sb sites of NdMnMnSbO6.22
Different atoms (e.g., Cu2+ and Mn2+) that can occupy both tetrahedral and square-planar sites (Fig. 3) order poorly between the A′ and A′′ sites as found by neutron diffraction in the case of Ca2CuMnFe2Re2O12 (60% Cu and 40% Mn at A′ and 23–36% Cu and 64–77% Mn at A′′);24 and the degree of ordering can depend on synthesis conditions.24 On the other hand, different charge orderings take place (at the A′ and A′′ sites and/or at the B site) for the same atom; examples with Mn known so far are: RMnMn2O6, R2MnMn[Mn3Ti]BO12 and R2CuMnMn4O12. Mn2+ and Mn3+ at the A′ site can be distinguished by Mn–O bond lengths: l(Mn2+–O) ∼ 2.1 Å and l(Mn3+–O) ∼ 1.91 Å. Other good candidates for such same-atom charge-ordered scenarios are Cu, Co and Fe (Fig. 3). Another interesting observation is that when the B site is solely occupied by Mn with the average oxidation state higher than +3, there seems to be a tendency for a layered B-site ordering with the appearance of one layer consisting of highly distorted (by the Jahn–Teller effect) Mn3+O6 octahedra.
Very few compositions of AA′3B4O12 can be prepared at ambient pressure (for example, (Li1.333Cu1.333)Nb4O12,36 Cu2+xTa4O12,47 CaMn7O12 and CaCu3Ti4O12).3 The majority of AA′3B4O12 requires HP and HT synthesis conditions. All known A2A′A′′B4O12 require HP and HT conditions. They were prepared at 12 GPa–1670 K for CaFeTi2O6,20 7 GPa–1470 K for CaMnTi2O6,21 10 GPa–1470 K for RMnMnSbO6,22 6 GPa–1370 K for RMnGaTiO6,18 6 GPa–1670 K for RMnMn2O6,23 and 10 GPa–1670 K for CaMnFeReO6 (and others).24
The symmetry of A2A′A′′B4O12 under synthesis conditions is crucial for the realization of ordering of different cations between the A′ and A′′ sites. In general, three situations are possible for compounds with the A2A′A′′B4O12 stoichiometry. First, the structure can have one crystallographic A site (e.g., with the maximum symmetry of Pmm and ap × ap × ap). In this case, we will probably have GdFeO3-type structures (simple ABO3 perovskites) with random distribution of all A cations at one site after quenching and pressure release.22 Second, the structure can have two crystallographic A sites with the 1:1 ratio (e.g., with P4/mmm symmetry and √2ap × √2ap × ap considered in ref. 29). In this case, it is difficult to expect noticeable ordering of different cations between the A′ and A′′ sites. Third, the structure can have three A sites with the 2:1:1 ratio (e.g., with I4/mmm symmetry and 2ap × 2ap × 2ap). In this case, the ordering of different cations between the A′ and A′′ sites can be realized. If the second case takes place (for some compositions), a modification of synthesis conditions could help to increase the ordering. In most cases, samples are quenched after the synthesis, but slow cooling could drive (partial) ordering. We are not aware of any in situ HP and HT studies of either AA′3B4O12 or A2A′A′′B4O12.
Some arguments in favor of the terminology
The AA′3B4O12 and A2A′A′′B4O12 quadruple perovskites have many common structural features (Fig. 1 and 2). The parent structures of both families have the doubled lattice parameter along all three crystallographic directions, 2ap × 2ap × 2ap, in comparison with a simple ABO3 perovskite with ap ≈ 3.8 Å resulting in eight ABO3 formula units in their unit cell. The simplest composition with integer numbers can be written as AA′3B4O12 (Z = 2), corresponding to four ABO3 units. This is why such perovskites are called quadruple. This formula reflects the presence of the A and A′ cations with quite different coordination environments with the 1:3 ratio. Because 1 and 3 are odd numbers (and A and A′ are quite different cations), this formula is retained in the majority of cases. However, there are very few exceptions when A′ = A (for example, CuCu3V4O12 and MnMn3Mn4O12).33,48 For such cases, the formula can be reduced to simple ABO3 (Z = 8). However, this formal formula reduction does not eliminate the structural features of such perovskites – they still belong to the A-site-ordered quadruple perovskite family. Note that perovskites and perovskite-related compounds with the 1:3 B-site ordering can also be referred to as (B-site-ordered) quadruple perovskites.49,50 By analogy, the simplest composition with integer numbers can be written as A2A′A′′B4O12 (Z = 2), which reflects quite different coordination environments for the A, A′ and A′′ sites with the 2:1:1 ratio. The A2A′A′′B4O12 formula corresponds to quadruple perovskites. It turns out that A′′ = A′ in many cases for perovskites reported so far, and the formula can be reduced to the double perovskite one, AA′B2O6. However, this formal formula reduction does not reflect the structural features of such perovskites, which still belong to the A-site columnar-ordered quadruple perovskite family, A2A′A′′B4O12. Ca2CuMnFe2Re2O12,24 R2CuMnMn2Mn2O12 (or R2CuMn5O12)30 and R2MnGaMn4O12 (or R2GaMn5O12)30 already require the quadruple formula to write them in integer numbers.
Conclusion
A-site columnar-ordered A2A′A′′B4O12 quadruple perovskites are in the incipient stage of research. They can be considered as an extended and more complex version of the A-site-ordered AA′3B4O12 quadruple perovskites (with twelve-fold coordinated A and square-planar coordinated A′ sites) because A2A′A′′B4O12 has ten-fold coordinated A and square-planar coordinated A′ sites plus an additional tetrahedrally coordinated A′′ site. This A′′ site gives an additional degree of freedom in A2A′A′′B4O12 because the number of elements that can have a tetrahedral environment is larger than that with a square-planar environment. The A2A′A′′B4O12 perovskites are the only oxygen-stoichiometric perovskites with tetrahedral sites. This article highlights great potential of A2A′A′′B4O12 and gives some design principles.Note added in proof
After this paper was accepted, the synthesis of ferroelectric Ca2−xMnxTi2O6 (x = 0.6) by a spark plasma sintering method was reported.51Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by JSPS KAKENHI Grant Numbers JP15K14133 and JP16H04501 and JSPS Bilateral Open Partnership Joint Research Projects. We thank Mr L. Zhang (NIMS), Dr Y. Matsushita (NIMS) and Dr K. Yamaura (NIMS) for their help with unpublished experimental data mentioned in this article and Profs. E. V. Antipov (Moscow State University) and I. A. Presniakov (Moscow State University) for discussion.References
- D. M. Giaquinta and H.-C. zur Loye, Chem. Mater., 1994, 6, 365–372 CrossRef CAS.
- R. H. Mitchell, Perovskites: Modern and Ancient, Almaz Press, Thunder Bay, Ontario, Canada, 2002 Search PubMed.
- G. King and P. M. Woodward, J. Mater. Chem., 2010, 20, 5785–5796 RSC.
- A. M. Abakumov, A. A. Tsirlin and E. V. Antipov, Transition-Metal Perovskites, in Comprehensive Inorganic Chemistry II (Second Edition): From Elements to Applications, ed. J. Reedijk and K. R. Poeppelmeier, Elsevier, Amsterdam, 2013, vol. 2, pp. 1–40 Search PubMed.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- A. M. Glazer, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1975, 31, 756–762 CrossRef.
- A. N. Vasil'ev and O. S. Volkova, Low Temp. Phys., 2007, 33, 895–914 CrossRef.
- I. Yamada, J. Ceram. Soc. Jpn., 2014, 122, 846–851 CrossRef.
- K. Leinenweber, J. Linton, A. Navrotsky, Y. Fei and J. B. Parise, Phys. Chem. Miner., 1995, 22, 251–258 CrossRef CAS.
- J. Linton, A. Navrotsky and Y. Fei, Phys. Chem. Miner., 1998, 25, 591–596 CrossRef CAS.
- S. V. Ovsyannikov, Y. G. Zainulin, N. I. Kadyrova, A. P. Tyutyunnik, A. S. Semenova, D. Kasinathan, A. A. Tsirlin, N. Miyajima and A. E. Karkin, Inorg. Chem., 2013, 52, 11703–11710 CrossRef CAS PubMed.
- K. Shiro, I. Yamada, N. Ikeda, K. Ohgushi, M. Mizumaki, R. Takahashi, N. Nishiyama, T. Inoue and T. Irifune, Inorg. Chem., 2013, 52, 1604–1609 CrossRef CAS PubMed.
- Y. Sakai, et al. , J. Am. Chem. Soc., 2017, 139, 4574–4581 CrossRef CAS PubMed.
- G. Shimura, Y. Shirako, K. Niwa and M. Hasegawa, J. Solid State Chem., 2016, 242, 55–62 CrossRef CAS.
- A. Deschanvres, B. Raveau and F. Tollemer, Bull. Soc. Chim. Fr., 1967, 4077–4078 CAS.
- A. A. Belik, Y. Matsushita and D. D. Khalyavin, Angew. Chem., Int. Ed., 2017, 56, 10423–10427 CrossRef CAS PubMed.
- I. Yamada, Sci. Technol. Adv. Mater., 2017, 18, 541–548 CrossRef PubMed.
- G. Shimura, K. Niwa, Y. Shirako and M. Hasegawa, Eur. J. Inorg. Chem., 2017, 835–839 CrossRef CAS.
- K. Leinenweber and J. Parise, J. Solid State Chem., 1995, 114, 277–281 CrossRef CAS.
- N. Yao, A. Navrotsky and K. Leinenweber, J. Solid State Chem., 1996, 123, 73–82 CrossRef CAS.
- A. Aimi, D. Mori, K. Hiraki, T. Takahashi, Y. J. Shan, Y. Shirako, J. S. Zhou and Y. Inaguma, Chem. Mater., 2014, 26, 2601–2608 CrossRef CAS.
- E. Solana-Madruga, A. M. Arevalo-Lopez, A. J. Dos Santos-Garcia, E. Urones-Garrote, D. Avila-Brande, R. Saez-Puche and J. P. Attfield, Angew. Chem., Int. Ed., 2016, 55, 9340–9344 CrossRef CAS PubMed.
- L. Zhang, Y. Matsushita, K. Yamaura and A. A. Belik, Inorg. Chem., 2017, 56, 5210–5218 CrossRef CAS PubMed.
- G. M. McNally, A. M. Arévalo-López, P. Kearins, F. Orlandi, P. Manuel and J. P. Attfield, Chem. Mater., 2017, 29, 8870–8874 CrossRef CAS.
- A. A. Belik, Y. Matsushita, M. Tanaka and E. Takayama-Muromachi, Chem. Mater., 2012, 24, 2197–2203 CrossRef CAS.
- A. A. Belik and W. Yi, J. Phys.: Condens. Matter, 2014, 26, 163201 CrossRef PubMed.
- (a) W. M. Reiff, K. Leinenweber and J. Parise, Mater. Res. Soc. Symp. Proc., 1997, 453, 387–392 CrossRef CAS; (b) H. Li, S. Liu, L. Chen, C. Li and Z. Wang, Physica E, 2015, 69, 133–137 CrossRef CAS.
- J. Ruiz-Fuertes, T. Bernert, D. Zimmer, N. Schrodt, M. Koch-Muller, B. Winkler, L. Bayarjargal, C. Popescu, S. MacLeod and K. Glazyrin, Phys. Rev. B, 2017, 96, 094101 CrossRef.
- G. Y. Gou, N. Charles, J. Shi and J. M. Rondinelli, Inorg. Chem., 2017, 56, 11854–11861 CrossRef CAS PubMed.
- L. Zhang, Y. Matsushita, K. Yamaura and A. A. Belik, unpublished results.
- Y. S. Glazkova, N. Terada, Y. Matsushita, Y. Katsuya, M. Tanaka, A. V. Sobolev, I. A. Presniakov and A. A. Belik, Inorg. Chem., 2015, 54, 9081–9091 CrossRef CAS PubMed.
- A. A. Belik, Y. S. Glazkova, Y. Katsuya, M. Tanaka, A. V. Sobolev and I. A. Presniakov, J. Phys. Chem. C, 2016, 120, 8278–8288 CAS.
- Y. Akizuki, I. Yamada, K. Fujita, K. Taga, T. Kawakami, M. Mizumaki and K. Tanaka, Angew. Chem., Int. Ed., 2015, 54, 10870–10874 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- J. Sanchez-Benitez, J. A. Alonso, M. J. Martinez-Lope, A. de Andres and M. T. Fernandez-Diaz, Inorg. Chem., 2010, 49, 5679–5685 CrossRef CAS PubMed.
- M. Sato and Y. Hama, J. Mater. Chem., 1993, 3, 233–236 RSC.
- J. Li, M. A. Subramanian, H. D. Rosenfeld, C. Y. Jones, B. H. Toby and A. W. Sleight, Chem. Mater., 2004, 16, 5223–5225 CrossRef CAS.
- I. Yamada, M. Murakami, N. Hayashi and S. Mori, Inorg. Chem., 2016, 55, 1715–1719 CrossRef CAS PubMed.
- D. Waroquiers, X. Gonze, G.-M. Rignanese, C. Welker-Nieuwoudt, F. Rosowski, M. Göbel, S. Schenk, P. Degelmann, R. André, R. Glaum and G. Hautier, Chem. Mater., 2017, 29, 8346–8360 CrossRef CAS.
- M. A. Subramanian, D. Li, N. Duan, B. A. Reisner and A. W. Sleight, J. Solid State Chem., 2000, 151, 323–325 CrossRef CAS.
- S.-H. Byeon, M. W. Lufaso, J. B. Parise, P. M. Woodward and T. Hansen, Chem. Mater., 2003, 15, 3798–3804 CrossRef CAS.
- T. Saito, R. Yamada, C. Ritter, M. S. Senn, J. P. Attfield and Y. Shimakawa, Inorg. Chem., 2014, 53, 1578–1584 CrossRef CAS PubMed.
- H. Okamoto, M. Karppinen, H. Yamauchi and H. Fjellvag, Solid State Sci., 2009, 11, 1211–1215 CrossRef CAS.
- S. V. Streltsov and D. I. Khomskii, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 201115 CrossRef.
- A. Prodi, A. Daoud-Aladine, F. Gozzo, B. Schmitt, O. Lebedev, G. van Tendeloo, E. Gilioli, F. Bolzoni, H. Aruga-Katori, H. Takagi, M. Marezio and A. Gauzzi, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 180101 CrossRef.
- A. A. Belik, Y. Matsushita, Y. Kumagai, Y. Katsuya, M. Tanaka, S. Yu. Stefanovich, B. I. Lazoryak, F. Oba and K. Yamaura, Inorg. Chem., 2017, 56, 12272–12281 CrossRef CAS PubMed.
- S. G. Ebbinghaus, Prog. Solid State Chem., 2007, 35, 421–431 CrossRef CAS.
- S. V. Ovsyannikov, A. M. Abakumov, A. A. Tsirlin, W. Schnelle, R. Egoavil, J. Verbeeck, G. Van Tendeloo, K. V. Glazyrin, M. Hanfland and L. Dubrovinsky, Angew. Chem., Int. Ed., 2013, 52, 1494–1498 CrossRef CAS PubMed.
- M. A. Subramanian, J. Solid State Chem., 1994, 111, 134–140 CrossRef CAS.
- Y. Shimoda, Y. Doi, M. Wakeshima and Y. Hinatsu, J. Solid State Chem., 2010, 183, 1962–1969 CrossRef CAS.
- Z. Y. Li, et al. , J. Am. Chem. Soc., 2018 DOI:10.1021/jacs.7b11219.
| This journal is © The Royal Society of Chemistry 2018 |