
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Making organoruthenium complexes of 8-hydroxyquinolines more hydrophilic: impact of a novel L-phenylalanine-derived arene ligand on the biological activity†
Sanam
Movassaghi
a,
Muhammad
Hanif
 a,
Hannah U.
Holtkamp
a,
Tilo
Söhnel
a,
Stephen M. F.
Jamieson
b and
Christian G.
Hartinger
*a
a,
Hannah U.
Holtkamp
a,
Tilo
Söhnel
a,
Stephen M. F.
Jamieson
b and
Christian G.
Hartinger
*a
aSchool of Chemical Sciences, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand. E-mail: c.hartinger@auckland.ac.nz; Web: http://hartinger.auckland.ac.nz
bAuckland Cancer Society Research Centre, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
First published on 12th January 2018
Abstract
Ru(arene) compounds have many desirable features making them promising candidates for further development in anticancer drug research. While a lot of emphasis has been placed on the modification of the ancillary ligands, there are not many examples of arene ligands bearing functional groups. Herein, we report the preparation of [Ru(arene)(8-oxyquinolinato)Cl] complexes with the arene being a protected form of the amino acid L-phenylalanine and 8-oxyquinolinato ligand substituted with halogens. With this approach we aimed to alter the pharmacological properties of the complexes and address issues with the aqueous solubility of the analogous p-cymene complexes. The complexes were shown to be stable in DMSO and water and reacted readily with L-histidine and 9-ethylguanine as protein and DNA models, respectively. Assaying the antiproliferative activity in cancer cells gave IC50 values in the low μM range. While the lipophilicity of the p-cymene analogues correlated well with their in vitro cytotoxicity, the potency of the complexes with the L-phenylalanine-derived arene was independent of lipophilicity.
Introduction
Metallopharmaceuticals have flourished since the discovery of the antineoplastic properties of cisplatin. However, despite their major impact as anticancer agents, the observed side effects as well as intrinsic or acquired resistance call for the development of novel platinum and non-platinum compounds.1–3 This has led to the development of complexes featuring a range of metal ions,4–7 and Ru compounds are at the forefront with different drug candidates studied in clinical trials.8–12RuII(arene) complexes based on the half-sandwich “piano-stool” scaffold may be equipped with unique structural features to form specific interactions with biomolecules and have revealed promising anticancer activity.13–20 RAPTA-type complexes are one of the most widely investigated classes of this compound type and have the general formula [Ru(arene)(PTA)X2] (PTA = 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decane; X = halido or biscarboxylato ligands). RAPTA-C (arene = η6-p-cymene [cym]) displayed antimetastatic activity both in vitro and in vivo.21 While RAPTA complexes were initially considered as cisplatin analogues to modify DNA through bifunctional modification, recent investigations showed that they tend to preferentially interact with proteins over DNA.15,22 In contrast to the RAPTA derivatives, the RAED compound class with the general formula [Ru(arene)Cl(en)]+ (en = 1,2-ethylenediamine) showed inhibition of primary tumour growth, and were effective in cisplatin-resistant tumour models.20,23 Unlike RAPTA-C, this compound class preferentially binds to DNA and the co-ligands impact both the cytotoxicity and reactivity toward biological targets.24,25
The arenes most commonly found in Ru(arene) complexes are cym and other related structures. Usually they are simple hydrocarbons and do not feature functional groups. Sadler and co-workers have shown that an expanded π-system leads to higher anticancer activity of RAED complexes, probably through intercalation between the DNA bases. In the case of the RAPTA compounds, the arene had only a minor impact on the biological activity.19,26 More recently a few reports have been published where the arene was further functionalised providing the compounds with additional features.14,16,19,27–29
Both RAED and RAPTA organometallics are simple structures and do not have a means incorporated that leads necessarily to selectivity and therefore lower side effects. The use of bioactive ligands in complexes is a promising approach towards multifunctional compounds, ideally resulting in synergistic effects between the metal centre and the ligands. Examples of biomolecules that were coordinated to metal centres include flavonols,30 8-oxyquinoline,31 quinolones,32 curcumin derivatives,33 chlorambucil,14 non-steroidal antiinflammatory drugs,13,34 and ethacrynic acid.29,35 This concept has gained considerable attention in the design of new organoruthenium antitumour drugs and is currently widely applied.
One of the structures often observed in natural products is the quinoline framework. Quinolines (and also quinolones) have been widely investigated in recent years and are known for their broad biological activities that include anticancer, anti-HIV, antifungal, antileishmanial, antischistosomal, antioxidant, antibacterial, and neuroprotective properties.36,37 The 8-oxyquinoline clioquinol (5-chloro-7-iodoquinolin-8-ol) is effective against Parkinson's and Alzheimer's diseases.38,39
In addition to their biological properties, such structures have been shown to be good ligands to many different metal centres.40 One important example is the GaIII anticancer complex KP46 [tris(8-oxyquinolinato)gallium(III)] which has been investigated in clinical trials.41,42 These observations have sparked interest in the design of anticancer agents based on bioactive quinolines and their metal complexes. We and others have developed ruthenium compounds of 8-oxyquinoline-derived ligands.31,43–47 Organoruthenium compounds of 8-oxyquinoline derived from clioquinol have shown promising antiproliferative activities, but they have poor aqueous solubility.
Herein we report the replacement of the cym ligand in [Ru(cym)(8-oxyquinolinato)Cl]Cl complexes with a natural compound, i.e., L-phenylalanine, to alter its pharmacological properties and aqueous solubility. We discuss the synthesis of the new organoruthenium dimeric precursor and its use to prepare a range of 8-oxyquinolinato complexes. The synthesis was complemented with studies on the physicochemical and biological properties to elucidate the potential of the compounds as anticancer agents.
Results and discussion
The design of anticancer drugs requires a good balance between hydrophilicity and lipophilicity to make them sufficiently soluble in aqueous media but still allow for efficient cell membrane penetration in order to exert their tumour-inhibiting potential. Several studies have shown that [Ru(cym)(8-oxyquinolinato)Cl] complexes are highly potent in cancer cells with IC50 values in the low μM range.31,43 However, their aqueous solubility is poor which has also been found for other promising anticancer organoruthenium complexes of bioactive ligands like flavones.48,49 Therefore, we introduced here L-phenylalanine (Phe) as a biologically relevant arene ligand to replace cym. We expected that this approach would increase the solubility of the compound and in addition it would provide functional groups for further modification in future.To prepare the target compounds, the acetyl-protected Phe was converted to its 1,4-cyclohexadiene analogue A by Birch reduction. Diene A was refluxed with RuCl3·xH2O in ethanol over night to give [(η6-N-acetyl-L-phenylalanine ethyl ester)RuIICl2]2B as a red solid (Scheme 1). To the best of our knowledge, B is the first organoruthenium dichlorido bridged dimeric compound bearing phenylalanine as the arene co-ligand, although Phe was introduced as an arene co-ligand to ruthenocenes such as [(η5-Cp)Ru(η6-N-acetyl-L-phenylalanine ethyl ester)]PF6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 50 and [(η5-C6H3Me4)Ru(η6-N-acetyl-L-phenylalanine ethyl ester)]PF6.51 In the latter cases, the compounds were studied for applications in the labelling of amino acids and peptides. Both A and B were characterised by 1H and 13C{1H} NMR spectroscopy and the 1H NMR spectrum of B showed the typical signals for the CH3 protons of ethyl ester and the acetyl group at 1.14 and 1.80 ppm, respectively.
50 and [(η5-C6H3Me4)Ru(η6-N-acetyl-L-phenylalanine ethyl ester)]PF6.51 In the latter cases, the compounds were studied for applications in the labelling of amino acids and peptides. Both A and B were characterised by 1H and 13C{1H} NMR spectroscopy and the 1H NMR spectrum of B showed the typical signals for the CH3 protons of ethyl ester and the acetyl group at 1.14 and 1.80 ppm, respectively.
 | ||
| Scheme 1 Preparation of the Ru complexes 1a–5a from 8-oxyquinoline 1 or its derivatives 2–5 and Ru precursor B. The numbering scheme was used to assign the peaks observed in the NMR spectra. | ||
Single crystals of B were obtained by slow diffusion of ethyl ether into a methanol solution and they were analysed by X-ray diffraction (Fig. 1). The compound crystallised in the monoclinic space group P21. The molecular structure of the dimer shows ‘piano-stool’ geometry around the Ru atom, where three chlorido ligands act as the legs of the chair and the seat is formed from the η6-coordinated arene ring of Phe to the RuII centre. The individual molecules form hydrogen bonds with each other through the amide NH and the acetyl O atoms with bond lengths for N2H⋯O3 of 2.027(7) and N1H⋯O6 of 2.153(5) Å.
 | ||
| Fig. 1 Molecular structure of B given at 50% probability level. | ||
The dimeric Ru structure was found to be not symmetrical with regard to the substituents on the η6-coordinated phenyl moiety. The [Ru(μ-Cl2)Ru] moiety is virtually planar and comparison of the bond lengths around the Ru centre with those observed for [Ru(cym)Cl2]2 revealed that the bond lengths were slightly shorter compared to the same bonds in [Ru(cym)Cl2]2 (Table 1).
The structure of B was also confirmed by electrospray ionisation mass spectrometry in the positive ion mode. The base peak at m/z 371.9944 was assigned to the singly-charged cation [½B − Cl]+ (mtheor = 371.9935). In addition peaks of lesser intensity were assigned to the ions [½B − 2Cl − H]+ (m/z 336.0180, mtheor = 336.0172) and [B − Cl]+ (m/z 780.9566, mtheor = 780.9559).
Ru dimer B was then used to prepare a series of organometallic compounds of the general formula of [Ru(arene)(L)Cl] (arene = η6-N-acetyl-L-phenylalanine ethyl ester) with L = 8-oxyquinoline 1 and its derivatives 2–5 with different halogen substitution pattern on the quinoline ring. The 8-oxyquinoline derivatives were deprotonated using NaOMe and then dimer B was added in MeOH. The reaction mixture was stirred at room temperature for about 5–6 h and 1a–5a were then precipitated by addition of diethyl ether and n-hexane after transferring the compounds into dichloromethane.
The complexes were characterised by elemental analysis, NMR spectroscopy and electrospray ionisation mass spectrometry (ESI-MS). The presence of the Phe chiral centre in combination with the chiral RuII centre resulted in the formation of diastereomers, as shown by NMR spectroscopy. Both 1H and 13C{1H} NMR spectra featured two sets of signals (Fig. 2 for the 1H NMR spectra of 2a and 4a). We used the singlets assigned to the amide proton and H19, neighbouring the halogen substituents, to determine the ratios between the two diastereomers. The diastereomers were found to be present at different ratios depending on the size of the halogen substituents at the 8-oxyquinolinato ligand. While for the 8-oxyquinolinato a ratio of 1:1 was determined, the diiodo derivative 4a featured in a 3.2:0.8 ratio. The clioquinol derivative 5a with a iodo and a chloro substituent was found in the middle between the two extremes at 2.2:1.8 (Table 2).
 | ||
| Fig. 2 Aromatic region of the 1H NMR spectra of 2a and 4a showing two sets of signals due to presence of the chiral centres at the arene ligand and the ruthenium centre. The two sets of diastereomers are colour-labelled. The peaks of H15 and H16 for the two diastereomers overlap in the NMR spectra. | ||
| Compound | R1, R2 | Ratio |
|---|---|---|
| 1a | H, H | 1.0:1.0 |
| 2a | Cl, Cl | 2.1:1.9 |
| 3a | Br, Br | 2.4:1.6 |
| 4a | I, I | 3.2:0.8 |
| 5a | Cl, I | 2.2:1.8 |
Comparison of the chemical shifts observed in the 1H NMR spectra reveals a dependence of the peaks assigned to H19 on the halogen substitution pattern. The peaks shift gradually from 1a–4a towards lower field while for the mixed halogen compound 5a the resonance was detected close to the one for the dibromo derivative (at around 7.8 ppm for both diastereomers). All the other protons resonate at approximately the same frequencies and are substitution independent.
The nature of the compounds was confirmed by ESI-MS studies in positive mode. All mass spectra for 1a–5a featured the [M − Cl]+ base peak.
Stability in DMSO and aqueous solution
The stability in DMSO and water was determined for 1a as a representative example for the presented compound class. Stability under these conditions is a prerequisite for biological studies and it was determined with 1H NMR spectroscopy over a time span of 3 days. The complexes were very stable in water (Fig. 3) and only after 24 h minor peaks around 6.2 ppm were observed in the 1H NMR spectra. Addition of 1 eq. of AgNO3 induced abstraction of the chlorido ligand and formation of the aqua complex which resulted in a clear change in the NMR spectrum, especially in the region were the Ru(arene) protons would be expected (Fig. 3). Complex 1a was also found to be stable in D6-DMSO for at least 24 h (Fig. S1†), after which another set of signals appeared in the 1H NMR spectra, possibly due to DMSO coordination.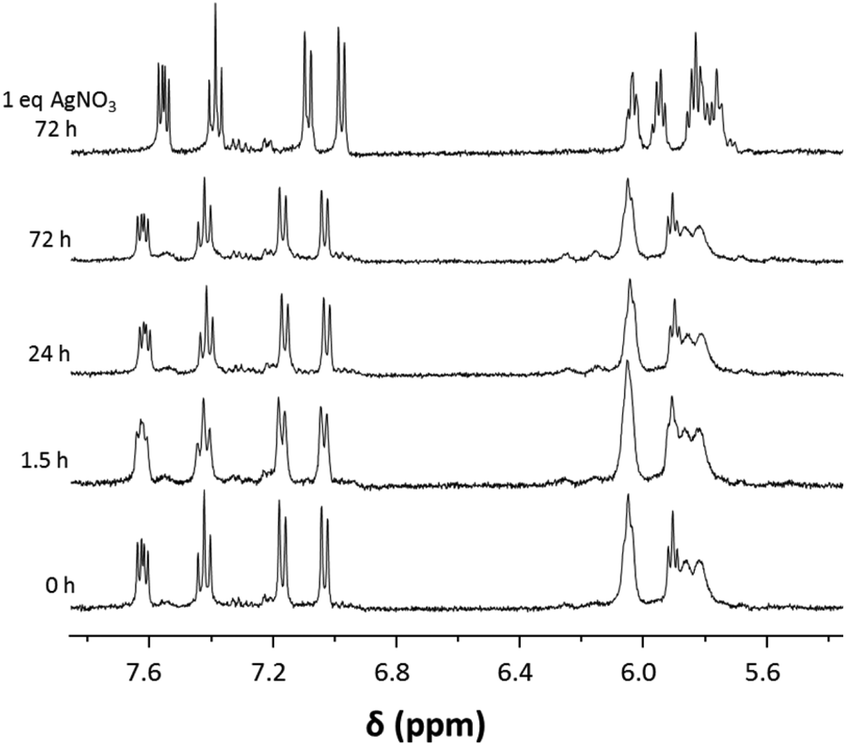 | ||
| Fig. 3 1H NMR spectroscopic study on stability of 1a in D2O monitored over 3 d. After a 3 d-incubation period 1 eq. of AgNO3 was added to induce abstraction of the chlorido ligand and formation of the aqua complex. | ||
Biomolecule interaction
The formation of covalent bonds with DNA is the foundation of the anticancer activity of cisplatin. Likewise, in the blood stream metal complexes are prone to undergo ligand exchange and coordinate to donor atoms of proteins or other blood components. In order to understand the nature of interactions between the organoruthenium compounds and biomolecules, 1a was studied for its reactions with small biomolecules such as L-cysteine (Cys), L-methionine (Met), L-histidine, and 9-ethylguanine (EtG), which were monitored by 1H NMR spectroscopy in 10% D6-DMSO/D2O.The reaction of 1a with His at a molar ratio of 1:1 was monitored over a period of 3 days and resulted in the formation of a dative bond between the imidazole-N and the ruthenium centre by exchange of a chlorido ligand. Within the first hours of reaction, significant changes occurred in the 1H NMR spectra (Fig. 4). The peaks assigned to His and some of those attributed to 1a disappeared over time, while new species were forming as indicated by the appearance of additional signals in the 1H NMR spectra. One example of a pair of disappearing peaks was found at about 9.6 ppm (Fig. 4 and S2†). While the original peak assigned to H14 of the complex, which neighbours the Ru-coordinating quinoline nitrogen atom, decreases over time, a new signal forms at about 9.5 ppm. Similar observations were made for a series of other peaks (compare Fig. 4). Notably, while initially the signals in the 1H NMR spectrum were quite broad, after 72 h of reaction the peaks of the His adduct were sharp and well defined.
 | ||
| Fig. 4 1H NMR spectroscopic study of the reaction between 1a and His in 10% D6-DMSO/D2O, monitored for a period of 3 d. The disappearing peaks assigned to imidazole-CH of His are highlighted in grey. | ||
The adduct formation of His with 1a was also confirmed by ESI-MS which helped to identify a His adduct in the samples used for the NMR studies and after dilution with acetonitrile. All samples featured as the base peak [1a − Cl]+, in which proton(s) had partly been exchanged with deuterium (Fig. S3†). In addition, two ions were detected in which His was coordinated to the Ru centre. The signal at m/z 637.1445 was assigned to [1a + His − Cl]+ (mtheor = 637.1461; partly H/D exchanged), while the peak at m/z 441.0718 was identified as [1a + His + 2 D2O − Cl − arene]+ (mtheor = 441.0649). The latter two species increased in relative intensity over time and after 48 h of incubation, they were detected at about 1/3 and 2/3 relative intensity to the [1a–Cl]+ base peak, respectively.
The reaction of 1a with Cys (1:1) resulted in the quick decomposition of the complex (Fig. S4†), while it did not react with Met under the same conditions. The decomposition of Ru(arene) complexes in the presence of Cys has been observed before and is also indicated by a release of the arene ligand from the metal centre. This results in the disappearance of the typical Ru(arene) signals in the range of 5.5–6.5 ppm and the appearance of additional peaks in the aromatic region of the 1H NMR spectrum. The low reactivity with Met was confirmed by ESI-MS which indicated that [1a − Cl]+ was by far the major species in solution, followed by a transesterification product with MeOH, which was used in the MS experiment for dilution, and an unidentified dimeric compound.
DNA was identified as the cellular target for anticancer platinum drugs and also the ruthenium anticancer agent RAED with its bidentate en ligand. Therefore, we assayed the reaction between 1a and 9-ethylguanine (EtG) as a DNA model base to estimate the ability of the complex to form DNA adducts. Incubation mixtures of molar ratios of 1:1 and 1:2 (1a:EtG) were analysed immediately after mixing by 1H NMR spectroscopy (Fig. 5). The reaction occurred very quickly, as shown by following the H8 signal, and the NMR spectra contained an additional peak with only a minor highfield shift compared to that of H8 in 9-EtG. Addition of a second equivalent of EtG, which cannot coordinate to the Ru centre as there is only a single labile ligand that can be substituted, resulted in an increase of the minor peak of unreacted EtG in the reaction mixture. The formation of the adduct was confirmed by the presence of a peak at m/z 663.1691 in the ESI-mass spectrum which was assigned to [1a + EtG − Cl]+ (mtheor = 663.1715; partly H/D exchanged, compare Fig. S5†). The base peak in the spectrum was however again assigned to the [1a − Cl]+ ion.
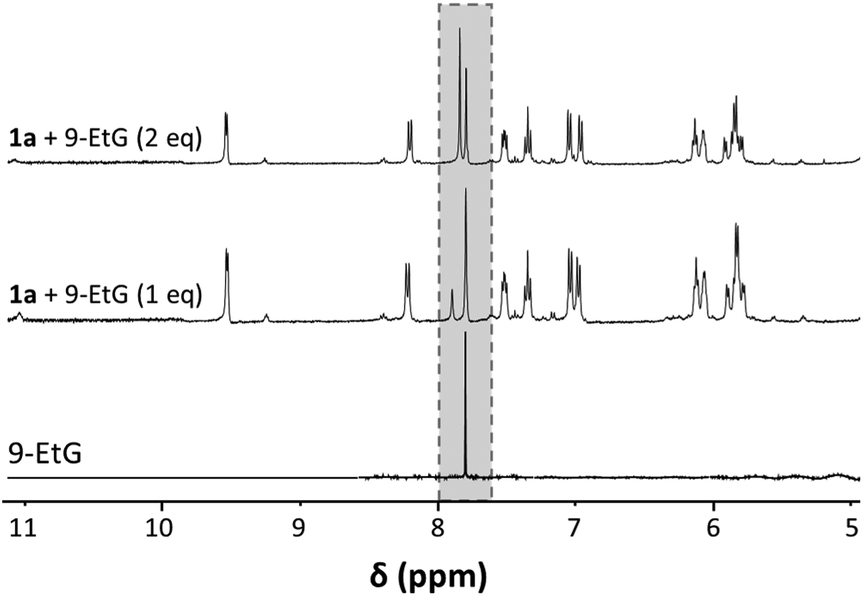 | ||
| Fig. 5 Aromatic region of the 1H NMR spectra recorded for the reaction of 1a with 1 and 2 eq. 9-EtG, as well as 9-EtG for comparison. The grey box indicates the region that features the H8 signal for coordinated and free 9-EtG. | ||
These studies show that the compounds have a clear preference for the reaction with nitrogen donors, while both DNA and proteins could be targets for coordinative bond formation.
In vitro anticancer activity and lipophilicity
The in vitro antiproliferative activity of complexes 1a–5a was studied in HCT116 human colorectal, NCI-H460 non-small cell lung, SiHa cervical carcinoma, and SW480 colon adenocarcinoma cells (Table 3) and compared to the cytotoxic activity of the analogous cym complexes 1a′–5a′ as well as dimeric precursor B. While B was non-cytotoxic, all the 8-oxyquinolinato complexes were potent antiproliferative agents with IC50 values in the low micromolar range. The cytotoxicity was virtually independent of the cell line used in these studies. The IC50 values increased gradually in the order 1a < 2a < 3a < 4a, much in line with the addition of larger halogens. The mixed Cl,I-substituted 5a showed IC50 values between those of the Br,Br and I,I derivatives 3a and 4a. This follows the trend of the calculated logP (clogP) values for the ligands 1–5 (Table S1†). It is, however, in contrast to the p-cymene analogues for which the non-halogenated compound was less active than the other derivatives which showed very similar IC50 values considering the standard deviations.
| Compound | IC50 values (μM) | |||
|---|---|---|---|---|
| HCT116 | NCI-H460 | SiHa | SW480 | |
| n.d., not determined. | ||||
| B | 152 ± 24 | 317 ± 63 | 132 ± 17 | 196 ± 40 |
| 1a | 2.5 ± 0.3 | 3.6 ± 0.5 | 5.7 ± 0.5 | 3.8 ± 0.2 |
| 2a | 4.7 ± 0.8 | 4.2 ± 0.1 | 14 ± 1 | 9.0 ± 1.0 |
| 3a | 7.5 ± 1.1 | 7.1 ± 1.0 | 20 ± 0.1 | 14 ± 1 |
| 4a | 16 ± 2 | 14 ± 1 | 27 ± 0.2 | 26 ± 1 |
| 5a | 12 ± 3 | 11 ± 0.3 | 25 ± 4 | 21 ± 0.4 |
| 1a′ | 12 ± 1 | 11 ± 2 | 19 ± 2 | n.d. |
| 2a′ | 5.0 ± 0.7 | 4.0 ± 0.7 | 7.6 ± 1.3 | n.d. |
| 3a′ | 6.3 ± 0.9 | 5.8 ± 0.6 | 8.2 ± 0.9 | n.d. |
| 4a′ | 5.2 ± 1.9 | 4.6 ± 1.3 | 7.3 ± 0.9 | n.d. |
| 5a′ | 7.7 ± 0.6 | 5.6 ± 0.3 | 8.5 ± 0.4 | n.d. |
Comparison with the cym analogues 1a′–5a′ shows that introduction of the protected Phe group as the arene has not much impact on the cytotoxicity and there is no clear trend between the two series of compounds. This is an interesting observation given the fact that the novel arene ligand impacted the solubility of some of the compounds, especially those which were found to be very cytotoxic in our previous studies and had the lowest solubility (Table 4).31 This suggests that in some cases the introduction of a more hydrophilic arene ligand could make the compounds more accessible for studies in aqueous environment, while only a minor impact on the biological activity is achieved.
P) of 1a–5a with the analogous cym complexes 1a′–5a′, the latter were taken from ref. 31
| Complex | Solubility (mM) | LogP |
|---|---|---|
| 1a | 5.625 | −1.33 ± 0.131 |
| 2a | 3.501 | −0.43 ± 0.06 |
| 3a | 2.778 | −0.20 ± 0.02 |
| 4a | 0.135 | −0.15 ± 0.01 |
| 5a | 0.125 | −0.47 ± 0.01 |
| 1a′ | 0.458 | 0.46 ± 0.01 |
| 2a′ | 0.450 | 0.43 ± 0.07 |
| 3a′ | 0.222 | 0.61 ± 0.02 |
| 4a′ | 0.026 | 0.85 ± 0.09 |
| 5a′ | 0.028 | 0.24 ± 0.02 |
To validate the observations, we also determined the n-octanol–water partition, expressed as the octanol–water partition coefficient (logP), using the shake flask method. The logP value is used to give an estimation of the lipophilicity of a compound, which has a major impact on cell membrane penetration and therefore cellular accumulation, as well as oral bioactivity of drugs. The logP values of 1a–5a were found to be in the range of −1.33 to −0.15 which is significantly lower than those found for 1a′–5a′ (Table 4).31 This would be expected based on the introduction of a more hydrophilic π-bound arene ligand. The trend found for the logP values also matches the clogP values for the ligands 1–5 well (Table S1†), taking into consideration the standard deviations, and being found in the negative logP range. Compound 1a with the unsubstituted 8-oxyquinolinato ligand had the lowest logP value. In contrast the diiodo derivative 4a has the highest logP values while the mixed halogen compound 5a gave a logP similar to that of 2a. This again confirms the impact of the size of the halogen substituent on the properties of the compounds. Interestingly, the correlation is inverse to the cytotoxic activity, which is a noteworthy observation as most commonly cytotoxic activity goes hand in hand with lipophilicity.53 However, we speculated for anthracene-derivatised compounds that the lipophilicity may shadow effects such as cellular accumulation and target interaction.27
Conclusions
In recent years we and others have reported the high cytotoxic activity of [Ru(cym)(8-oxyquinolinato)Cl] complexes.31,43 In order to alter their pharmacological properties and improve their limited aqueous solubility, we report here the introduction of an L-phenylalanine-derived arene ligand to replace the commonly used cym and vary the halogen substituents on the 8-oxyquinolinato ligand. The complexes were thoroughly characterised and the molecular structure of the dimeric precursor was determined by X-ray diffraction analysis.The organometallic compounds were found to be less lipophilic than their cym counterparts. The novel arene ligand had only a significant impact on the solubility of the compounds which were found to be very cytotoxic in our previous study.31 The complexes were demonstrated to have sufficient stability in aqueous solution and in DMSO. While they did not react with Met and decomposed in the presence of Cys, His and 9-EtG reacted readily with 1a after a ligand exchange reaction with the chlorido ligand.
In general, the replacement of cym with the protected Phe ligand did not alter the in vitro anticancer activity of the complexes significantly, which was found in the low μM range. Notably, the most active compounds featuring cym as the arene were not the most antiproliferative agents with the Phe-derived ligand. The most potent derivative was complex 1a with the unsubstituted 8-oxyquinolinato ligand, as also found in the clinically tested Ga complex KP46.
Experimental
All reactions were performed in Schlenk flasks with dry solvents under nitrogen atmosphere. Chemicals acquired from commercial supplier were used without any prior purification. Dry solvents were prepared according to literature procedures.54 Ruthenium(III) chloride hydrate (99%) was obtained from Precious Metals Online. N-Acetyl-L-phenylalanine (99%), 5,7-dibromo-8-hydroxyquinoline (98%) and 8-hydroxyquinoline (99%) were purchased from AK Scientific. N-Octanol, 5,7-diiodo-8-hydroxyquinoline (97%), and (1S)-(+)-10-camphorsulfonic acid were obtained from Sigma-Aldrich. 5-Chloro-7-iodo-8-hydroxyquinoline (ultrapure) was bought from OFC Inc., 5,7-dichloro-8-hydroxyquinoline (99%) from Acros, and sodium methoxide from Fluka.Elemental analyses were conducted on a vario EL cube (Elementar Analysensysteme GmbH, Hanau, Germany). 1D and multinuclear 2D (1H–13C HSQC, and HMBC) NMR spectra were recorded on Bruker Avance AVIII 400 MHz NMR spectrometer at ambient temperature at 400.13 MHz (1H) or 100.57 MHz (13C{1H}).
High resolution mass spectra were recorded on a Bruker microOTOF-Q II ESI-MS in positive ion mode.
X-ray diffraction measurements of single crystals of [(η6-ethyl 2-acetamido-3-phenylpropanoate)RuIICl2]2B were performed on a Siemens/Bruker SMART APEX II single-crystal diffractometer with a CCD area detector using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å; Table S2†). The data were processed with the SHELX2016 software packages.55 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were inserted at calculated positions and refined with a riding model or without restrictions. Mercury 3.9. was used to visualise the molecular structure.
Syntheses
General procedures for the synthesis of (8-oxyquinolinato)Ru complexes
Two different methods were used to prepare the complexes:Method I. Compound B (0.45 equiv.) was added to a stirred solution of sodium methoxide (1.1 equiv.) and 8-oxyquinoline derivative (1.0 equiv.) in methanol. The reaction mixture was refluxed for 1.5–4 h under a nitrogen atmosphere. The solvent was evaporated, the residue was dissolved in dichloromethane, the solution was filtered, and the complex was precipitated with n-hexane.
Method II. An 8-oxyquinoline derivative (1.0 equiv.) and sodium methoxide (1.1 equiv.) were added to a mixture of chloroform (8 mL) and methanol (15 mL). Compound B (0.45 equiv.) was added to the reaction mixture. The reaction mixture was stirred for 1 h at room temperature under a nitrogen atmosphere. The formed precipitate was collected by filtration, washed with n-hexane, and dried under vacuum.
Chlorido(8-quinolinolato-κ2N,O)(η6-ethyl-2-acetamido-3-phenylpropanoate)ruthenium(II) (1a)
The reaction was performed following method I and using 8-oxyquinoline 1 (50 mg, 0.34 mmol) and B (130 mg, 0.16 mmol) to afford a dark green solid (122 mg, 69%). Anal. calcd for C22H23Cl N2O4Ru·H2O·0.2CH2Cl2: C, 48.27; H, 4.34; N, 5.39%. Found: C, 48.40; H, 4.65; N, 5.08. ESI+: m/z 481.0714 [M − Cl]+ (mtheor = 481.0780). The reaction gave a mixture of diastereomers in a ratio of 1.0:1.0, indicated as d1 and d2 in the NMR data. 1H NMR (400.13 MHz, D6-DMSO): δ 1.15 (t, 3JH9,H8 = 7, 6H, H11), 1.80 (s, 3H, H13d1), 1.81 (s, 3H, H13d2), 2.69–3.97 (m, 4H, H7), 4.03–4.14 (m, 4H, H10), 4.49–4.58 (m, 2H, H8), 5.58–5.72 (m, 6H, Harom), 5.87–5.97 (m, 4H, Harene), 6.64–6.71 (m, 2H, H18), 6.80 (d, 3JH20,H19 = 5 Hz, 1H, H20d1), 6.82 (d, 3JH20,H19 = 5 Hz, 1H, H20d2), 7.20–7.26 (m, 3JH19,H18 = 8 Hz, 3JH19,H20 = 8 Hz, 2H, H19), 7.46 (d, 3JH16,H15 = 9 Hz, 3JH14,H15 = 9 Hz, 1H, H15d1), 7.48 (d, 3JH16,H15 = 9 Hz, 3JH14,H15 = 9 Hz, 1H, H15d2), 8.20–8.26 (d, 3JH16,H15 = 9 Hz, 2H, H16), 8.42 (d, 3JH17,NH = 8 Hz, 1H, NHd1), 8.53 (d, 3JH17,NH = 8 Hz, 1H, NHd2), 9.21–9.28 (m, 2H, H14) ppm. 13C{1H} NMR (100.57 MHz, D6-DMSO): δ 14.5 (C11), 22.7 (C13), 34.9 (C7d1), 35.1 (C7d2), 52.5 (C8d1), 52.7 (C8d2), 61.2 (C10), 79.4 (C4d1), 79.5 (C4d2), 80.7 (C6), 81.1 (C3d1), 81.2 (C3d2), 84.7 (C2), 86.3 (C5), 97.9 (C1), 109.8 (C18d1), 109.9 (C18d2), 113.6 (C20), 122.8 (C15), 130.2 (C19), 123.8 (C17), 137.6 (C16), 150.7 (C14), 169.6 (C21), 169.8 (C12), 170.3 (C9) ppm.
Chlorido(5,7-dichloro-8-quinolinolato-κ2N,O)(η6-ethyl-2-acetamido-3-phenylpropanoate)ruthenium(II) (2a)
The reaction was performed following method I and using 5,7-dichloro-8-oxyquinoline 2 (133 mg, 0.62 mmol) and B (114 mg, 0.32 mmol) to afford an ochre solid (135 mg, 74%). Anal. calcd for C22H21Cl3N2O4Ru·H2O: C, 43.59; H, 3.50 N, 4.76%. Found: C, 43.83; H, 3.85; N, 4.65%. ESI+: m/z 548.9915 [M − Cl]+ (mtheor = 548.9994). The reaction gave a mixture of diastereomers in a ratio of 2.1:1.9, indicated as d1 and d2 in the NMR data. 1H NMR (400.13 MHz, D6-DMSO): δ 1.15 (t, 3JH9,H8 = 7 Hz, 6H, H11), 1.80 (s, 3H, H13d1), 1.81 (s, 3H, H13d2), 2.71–3.01 (m, 4H, H7), 4.04–4.14 (m, 4H, H10), 4.52–4.61 (m, 2H, H8), 5.68–5.81 (m, 6H, Harom), 5.99–6.08 (m, 4H, Harom), 7.58 (s, 1H, H19d1), 7.59 (s, 1H, H19d2), 7.66–7.72 (m, 1H, H15), 8.36–8.40 (m, 2H, H16), 8.44 (d, 3JH17,NH = 8 Hz, 1H, NHd1), 8.49 (d, 3JH17,NH = 8 Hz, 1H, NHd2), 9.43 (d, 3JH14,H15 = 5 Hz, 2H, H14) ppm. 13C{1H} NMR (100.57 MHz, D6-DMSO): δ 14.1 (C11), 22.3 (C13), 34.3 (C7d1), 34.7 (C7d2), 51.8 (C8d1) 52.1 (C8d2), 60.8 (C10d1), 60.8 (C10d2), 78.5 (C4d1), 78.9 (C4d2), 80.0 (C6d1), 79.8 (C6d2), 81.0 (C3d1), 80.1 (C3d2), 85.2 (C2d2), 85.3 (C2d1), 86.9 (C5d1), 86.1 (C5d2), 98.3 (C1d2), 98.7 (C1d1), 110.3 (C18d1), 110.1 (C18d2), 116.1 (C20d1), 116.1 (C20d2), 123.7 (C15), 125.6 (C17), 128.8 (C19), 134.1 (C16), 144.3 (C22), 152.3 (C14d1), 152.2 (C14d2), 163.1 (C21d1), 162.9 (C21d2), 169.5 (C12), 171.0 (C9) ppm.
Chlorido(5,7-dibromo-8-quinolinolato-κ2N,O)(η6-ethyl-2-acetamido-3-phenylpropanoate)ruthenium(II) (3a)
The reaction was performed according to method II using 5,7-dibromo-8-oxyquinoline 3 (220 mg, 0.73 mmol) and B (200 mg, 0.33 mmol) to afford an ochre solid (357 mg, 73%). Anal. calcd for C22H21Br2ClN2O4Ru·1.3H2O·0.2CH2Cl2: C, 37.51; H, 3.01; N, 3.96%. Found: C, 37.34; H, 3.39; N, 3.92. ESI+: m/z 638.8883 [M − Cl]+ (mtheor = 638.8973). The reaction gave a mixture of diastereomers in a ratio of 2.4:1.6, indicated as d1 and d2 in the NMR data. 1H NMR (400.13 MHz, D6-DMSO): 1.15 (t, 3JH9,H8 = 7 Hz, 6H, H11), 1.80 (s, 3H, H13d1), 1.81 (s, 3H, H13d2), 2.71–3.03 (m, 4H, H7), 4.04–4.14 (m, 4H, H10), 4.53–4.67 (m, 2H, H8), 5.65–5.81 (m, 6H, Harom), 5.98–6.08 (m, 4H, Harom), 7.67–7.73 (m, 1H, H15), 7.79 (s, 1H, H19d1), 7.81 (s, 1H, H19d2), 8.28–8.33 (m, 1H, H16), 8.43 (d, 3JH17,NH = 8 Hz, 1H, NHd1), 8.49 (d, 3JH17,NH = 8 Hz, 1H, NHd2), 9.40 (d, 3JH14,H15 = 5 Hz, 2H, H14) ppm. 13C{1H} NMR (100.57 MHz, D6-DMSO): δ 14.1 (C11), 22.3 (C13d1), 22.4 (C13d2), 34.2 (C7d1), 34.7 (C7d2), 51.7 (C8d1), 52.1 (C8d2), 60.9 (C10), 78.3 (C4d1), 78.7 (C4d2), 79.6 (C6d1), 79.7 (C6d2), 80.0 (C3d1), 80.7 (C3d2), 85.3 (C2d1), 85.5 (C2d2), 86.5 (C5d1), 87.4 (C5d2), 98.5 (C1d1), 98.9 (C1d2), 99.0 (C18d1), 99.2 (C18d2), 106.2 (C20d1), 106.3 (C20d2), 124.1 (C15), 127.4 (C17), 134.1 (C19), 136.5 (C16), 144.1 (C22), 152.3 (C14), 164.9 (C21d1), 164.8 (C21d2), 169.5 (C12d1), 169.5 (C12d2), 171.0 (C9) ppm.
Chlorido(5,7-diiodo-8-quinolinolato-κ2N,O)(η6-ethyl-2-acetamido-3-phenylpropanoate)ruthenium(II) (4a)
The synthesis was performed following method II and using 5,7-diiodo-8-oxyquinoline 4 (300 mg, 0.76 mmol) and B (208 mg, 0.34 mmol) to afford an ochre product (408 mg, 70%). Anal. calcd for C22H21ClI2N2O4Ru·H2O: C, 33.77; H, 2.69; N, 3.57. Found: C, 33.63; H, 2.95; N, 3.57. ESI+: m/z 732.8652 [M − Cl]+ (mtheor = 732.8713). The reaction gave a mixture of diastereomers in a ratio of 3.2:0.8, indicated as d1 and d2 in the NMR data. 1H NMR (400.13 MHz, D6-DMSO): δ 1.16 (t, 3JH9,H8 = 7 Hz, 6H, H11), 1.80 (s, 3H, H13d1), 1.82 (s, 3H, H13d2), 2.69–3.06 (m, 4H, H7), 4.05–4.15 (m, 4H, H10), 4.54–4.63 (m, 2H, H8), 5.58–5.81 (m, 6H, Harom), 5.97–6.09 (m, 4H, Harom), 7.63–7.70 (m, 2H, H15), 8.05 (s, 1H, H19d1), 8.06 (s, 1H, H19d2), 8.13–8.20 (m, 2H, H16), 8.41 (d, 3JH17,NH = 8 Hz, 1H, NHd1), 8.46 (d, 3JH17,NH = 8 Hz, 1H, NHd2), 9.34 (d, 3JH14,H15 = 5 Hz, 2H, H14) ppm. 13C{1H} NMR (100.57 MHz, D6-DMSO): δ 14.5 (C11), 22.8 (C13), 34.6 (C7d1), 35.0 (C7d2), 52.1 (C8d1), 52.5 (C8d2), 61.3 (C10), 74.0 (C18), 78.3 (C4), 79.6 (C6), 79.7 (C3), 83.3 (C20), 85.9 (C2d1), 86.1 (C2d2), 88.6 (C5), 99.8 (C1), 124.8 (C15), 131.1 (C17), 141.4 (C16), 142.9 (C22), 145.6 (C19), 152.5 (C14), 168.7 (C21), 169.9 (C12d1), 170.0 (C12d2), 171.5 (C9) ppm.
Chlorido(5-chloro-7-iodo-8-quinolinolato-κ2N,O)(η6-ethyl-2-acetamido-3-phenylpropanoate)ruthenium(II) (5a)
The synthesis was performed following method I and using 5-chloro-7-iodo-8-oxyquinoline 5 (300 mg, 0.98 mmol) and B (270 mg, 0.44 mmol) to afford an olive-green product (504 mg, 76%). Anal. calcd for C22H21Cl2IN2O4Ru·0.2H2O·0.5CH2Cl2: C, 37.69; H, 2.92; N, 3.99. Found: C, 37.41; H, 3.13; N, 3.88. ESI+: m/z 640.9332 [M − Cl]+ (mtheor = 640.9353). The reaction gave a mixture of diastereomers in a ratio of 2.2:1.8, indicated as d1 and d2 in the NMR data. 1H NMR (400.13 MHz, D6-DMSO): δ 1.16 (t, 6H, H11), 1.79 (s, 3H, H13d1), 1.82 (s, 3H, H13d2), 2.68–3.05 (m, 4H, H7), 4.03–4.15 (m, 4H, H10), 4.54–4.63 (m, 2H, H8), 5.60–5.81 (m, 6H, Harom), 5.97–6.07 (m, 4H, Harom), 7.67–7.72 (m, 2H, H15), 7.77 (s, 1H, H19d1), 7.78 (s, 1H, H19d2), 8.33–8.38 (m, 2H, H16), 8.42 (d, 3JH17,NH = 8 Hz, 1H, NHd1), 8.47 (d, 3JH17,NH = 8 Hz, 1H, NHd2), 9.39 (d, 3JH14,H15 = 5 Hz, 2H, H14) ppm. 13C{1H} NMR (100.57 MHz, D6-DMSO): δ 14.1 (C11), 22.3 (C13d1), 22.4 (C13d2), 34.1 (C7d1), 34.6 (C7d2), 51.6 (C8d1), 52.1 (C8d2), 60.8 (C10), 77.8 (4d1), 78.3 (4d2), 79.2 (C6), 80.0 (C20d1), 80.1 (C20d2), 79.7 (C3d1), 80.4 (C3d2), 85.5 (C2d1), 85.7 (C2d2), 87.2 (C4 d1), 88.2 (C4d2), 98.8 (C1d1), 99.3 (C1d2), 111.0 (C18), 123.9 (C15), 126.6 (C17), 134.2 (C16), 135.8 (C19), 141.6 (C22), 152.0 (C14d1), 152.1 (C14d2), 167.0 (C21), 169.5 (C12d1), 169.6 (C12d1), 171.0 (C9) ppm.
:2 ratios were recorded immediately after mixing. The mass spectra were recorded on a Bruker microOTOF-Q II ESI-MS in positive ion mode.
The cells were seeded at 750 (HCT116, NCI-H460), 4000 (SiHa) or 5000 (SW480) cells per well in 96-well plates and left to settle for 24 h. The compounds were added to the plates in a series of 3-fold dilutions, containing a maximum of 0.5% DMSO at the highest concentration. The assay was terminated after 72 h by addition of 10% trichloroacetic acid (Merck Millipore) at 4 °C for 1 h. The cells were stained with 0.4% sulforhodamine B (Sigma-Aldrich) in 1% acetic acid for 30 min in the dark at room temperature and then washed with 1% acetic acid to remove unbound dye. The stain was dissolved in unbuffered Tris base (10 mM; Serva) for 30 min on a plate shaker in the dark and quantified on a BioTek EL808 microplate reader at an absorbance wavelength of 490 nm with 450 nm as the reference wavelength to determine the percentage of cell growth inhibition by determining the absorbance of each sample relative to a negative (no inhibitor) and a no-growth control (day 0). The IC50 values were calculated with SigmaPlot 12.5 using a three-parameter logistic sigmoidal dose–response curve between the calculated growth inhibition and the compound concentration. The presented IC50 values are the mean of at least 3 independent experiments, where 10 concentrations were tested in duplicate for each compound.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) P).
A previously published procedure was followed,31 where the OECD guidelines56 for the logP determination via the shake flask method were slightly modified. A known amount of each complex (1a–5a) was suspended in water (pre-saturated with n-octanol) and shaken for four days on an orbital shaker. Afterwards, the solution was centrifuged for 5 min at 2000 rpm to allow phase separation and the ruthenium content of the saturated aqueous solution was measured by ICP-MS to give the solubility of the compounds in H2O. To obtain the partition coefficient, different ratios (0.5:1, 1:1, and 2:1) of the saturated solutions were shaken with pre-saturated n-octanol for 30 min on an orbital shaker. After shaking for an additional 5 min by hand and centrifugation for 5 min at 10000 rpm, the aqueous phase was collected with a syringe according to OECD guidelines. For the analysis, the samples were diluted 1:100 with 5% HNO3. The Ru content was determined on an Agilent 7700 ICP-MS equipped with a MicroMist nebuliser, a Scott double pass spray chamber, and an ASX-500 autosampler (CETAC Technologies) in a Serie SuSi laminar flow hood (SPECTEC).
P).
A previously published procedure was followed,31 where the OECD guidelines56 for the logP determination via the shake flask method were slightly modified. A known amount of each complex (1a–5a) was suspended in water (pre-saturated with n-octanol) and shaken for four days on an orbital shaker. Afterwards, the solution was centrifuged for 5 min at 2000 rpm to allow phase separation and the ruthenium content of the saturated aqueous solution was measured by ICP-MS to give the solubility of the compounds in H2O. To obtain the partition coefficient, different ratios (0.5:1, 1:1, and 2:1) of the saturated solutions were shaken with pre-saturated n-octanol for 30 min on an orbital shaker. After shaking for an additional 5 min by hand and centrifugation for 5 min at 10000 rpm, the aqueous phase was collected with a syringe according to OECD guidelines. For the analysis, the samples were diluted 1:100 with 5% HNO3. The Ru content was determined on an Agilent 7700 ICP-MS equipped with a MicroMist nebuliser, a Scott double pass spray chamber, and an ASX-500 autosampler (CETAC Technologies) in a Serie SuSi laminar flow hood (SPECTEC).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Auckland (Doctoral Scholarship to H. H.) for financial support. The authors are grateful to Tanya Groutso for collecting the single crystal X-ray diffraction data, and Tony Chen for ESI-MS analyses.References
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- A. L. Noffke, A. Habtemariam, A. M. Pizarro and P. J. Sadler, Chem. Commun., 2012, 48, 5219–5246 RSC.
- C. G. Hartinger, A. D. Phillips and A. A. Nazarov, Curr. Top. Med. Chem., 2011, 11, 2688–2702 CrossRef CAS PubMed.
- M. Hanif, M. V. Babak and C. G. Hartinger, Drug Discovery Today, 2014, 19, 1640–1648 CrossRef CAS PubMed.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS.
- N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
- E. Alessio, Eur. J. Inorg. Chem., 2017, 1549–1560 CrossRef CAS.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- A. Notaro and G. Gasser, Chem. Soc. Rev., 2017, 46, 7317–7337 RSC.
- E. Păunescu, S. McArthur, M. Soudani, R. Scopelliti and P. J. Dyson, Inorg. Chem., 2016, 55, 1788–1808 CrossRef PubMed.
- A. A. Nazarov, S. M. Meier, O. Zava, Y. N. Nosova, E. R. Milaeva, C. G. Hartinger and P. J. Dyson, Dalton Trans., 2015, 44, 3614–3623 RSC.
- M. V. Babak, S. M. Meier, K. V. M. Huber, J. Reynisson, A. A. Legin, M. A. Jakupec, A. Roller, A. Stukalov, M. Gridling, K. L. Bennett, J. Colinge, W. Berger, P. J. Dyson, G. Superti-Furga, B. K. Keppler and C. G. Hartinger, Chem. Sci., 2015, 6, 2449–2456 RSC.
- K. J. Kilpin, C. M. Clavel, F. Edafe and P. J. Dyson, Organometallics, 2012, 31, 7031–7039 CrossRef CAS.
- Y. Q. Tan, P. J. Dyson and W. H. Ang, Organometallics, 2011, 30, 5965–5971 CrossRef CAS.
- P. Nowak-Sliwinska, J. R. van Beijnum, A. Casini, A. A. Nazarov, G. Wagnieres, H. van den Bergh, P. J. Dyson and A. W. Griffioen, J. Med. Chem., 2011, 54, 3895–3902 CrossRef CAS PubMed.
- M. Hanif, A. A. Nazarov, A. Legin, M. Groessl, V. B. Arion, M. A. Jakupec, Y. O. Tsybin, P. J. Dyson, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2012, 48, 1475–1477 RSC.
- A. Habtemariam, M. Melchart, R. Fernandez, S. Parsons, I. D. H. Oswald, A. Parkin, F. P. A. Fabbiani, J. E. Davidson, A. Dawson, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2006, 49, 6858–6868 CrossRef CAS PubMed.
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306, 86–114 CrossRef CAS.
- B. Wu, M. S. Ong, M. Groessl, Z. Adhireksan, C. G. Hartinger, P. J. Dyson and C. A. Davey, Chem. – Eur. J., 2011, 17, 3562–3566 CrossRef CAS PubMed.
- R. E. Morris, R. E. Aird, S. Murdoch Pdel, H. Chen, J. Cummings, N. D. Hughes, S. Parsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS PubMed.
- Z. Adhireksan, G. E. Davey, P. Campomanes, M. Groessl, C. M. Clavel, H. J. Yu, A. A. Nazarov, C. H. F. Yeo, W. H. Ang, P. Droge, U. Rothlisberger, P. J. Dyson and C. A. Davey, Nat. Commun., 2014, 5, 1–13 Search PubMed.
- Z. J. Ma, G. Palermo, Z. Adhireksan, B. S. Murray, T. von Erlach, P. J. Dyson, U. Rothlisberger and C. A. Davey, Angew. Chem., Int. Ed., 2016, 55, 7441–7444 CrossRef CAS PubMed.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed.
- A. A. Nazarov, J. Risse, W. H. Ang, F. Schmitt, O. Zava, A. Ruggi, M. Groessl, R. Scopelitti, L. Juillerat-Jeanneret, C. G. Hartinger and P. J. Dyson, Inorg. Chem., 2012, 51, 3633–3639 CrossRef CAS PubMed.
- E. Paunescu, M. Soudani, C. M. Clavel and P. J. Dyson, J. Inorg. Biochem., 2017, 175, 198–207 CrossRef CAS PubMed.
- W. H. Ang, L. J. Parker, A. De Luca, L. Juillerat-Jeanneret, C. J. Morton, M. Lo Bello, M. W. Parker and P. J. Dyson, Angew. Chem., Int. Ed., 2009, 48, 3854–3857 CrossRef CAS PubMed.
- A. Kurzwernhart, W. Kandioller, C. Bartel, S. Bächler, R. Trondl, G. Mühlgassner, M. A. Jakupec, V. B. Arion, D. Marko, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2012, 48, 4839–4841 RSC.
- M. Kubanik, H. Holtkamp, T. Söhnel, S. M. F. Jamieson and C. G. Hartinger, Organometallics, 2015, 34, 5658–5668 CrossRef CAS.
- J. Kljun, A. K. Bytzek, W. Kandioller, C. Bartel, M. A. Jakupec, C. G. Hartinger, B. K. Keppler and I. Turel, Organometallics, 2011, 30, 2506–2512 CrossRef CAS PubMed.
- R. Pettinari, F. Marchetti, F. Condello, C. Pettinari, G. Lupidi, R. Scopelliti, S. Mukhopadhyay, T. Riedel and P. J. Dyson, Organometallics, 2014, 33, 3709–3715 CrossRef CAS.
- F. Aman, M. Hanif, M. Kubanik, A. Ashraf, T. Söhnel, S. M. F. Jamieson, W. A. Siddiqui and C. G. Hartinger, Chem. – Eur. J., 2017, 23, 4893–4902 CrossRef CAS PubMed.
- G. Agonigi, T. Riedel, S. Zacchini, E. Păunescu, G. Pampaloni, N. Bartalucci, P. J. Dyson and F. Marchetti, Inorg. Chem., 2015, 54, 6504–6512 CrossRef CAS PubMed.
- S. N. Al-Busafi, F. E. O. Suliman and Z. R. Al-Alawi, Res. Rev.: J. Chem., 2014, 3, 1–10 Search PubMed.
- Y. Song, H. Xu, W. Chen, P. Zhan and X. Liu, MedChemComm, 2015, 6, 61–74 RSC.
- D. Kaur, F. Yantiri, S. Rajagopalan, J. Kumar, J. Q. Mo, R. Boonplueang, V. Viswanath, R. Jacobs, L. Yang, M. F. Beal, D. DiMonte, I. Volitaskis, L. Ellerby, R. A. Cherny, A. I. Bush and J. K. Andersen, Neuron, 2003, 37, 899–909 CrossRef CAS PubMed.
- P. A. Adlard, R. A. Cherny, D. I. Finkelstein, E. Gautier, E. Robb, M. Cortes, I. Volitakis, X. Liu, J. P. Smith, K. Perez, K. Laughton, Q.-X. Li, S. A. Charman, J. A. Nicolazzo, S. Wilkins, K. Deleva, T. Lynch, G. Kok, C. W. Ritchie, R. E. Tanzi, R. Cappai, C. L. Masters, K. J. Barnham and A. I. Bush, Neuron, 2008, 59, 43–55 CrossRef CAS PubMed.
- V. Oliveri, V. Lanza, D. Milardi, M. Viale, I. Maric, C. Sgarlata and G. Vecchio, Metallomics, 2017, 9, 1439–1446 RSC.
- S. M. Valiahdi, P. Heffeter, M. A. Jakupec, R. Marculescu, W. Berger, K. Rappersberger and B. K. Keppler, Melanoma Res., 2009, 19, 283–293 CrossRef CAS PubMed.
- É. Enyedy, O. Dömötör, K. Bali, A. Hetényi, T. Tuccinardi and B. Keppler, J. Biol. Inorg. Chem., 2015, 20, 77–88 CrossRef PubMed.
- R. Schuecker, R. O. John, M. A. Jakupec, V. B. Arion and B. K. Keppler, Organometallics, 2008, 27, 6587–6595 CrossRef CAS.
- C. Gemel, R. John, C. Slugovc, K. Mereiter, R. Schmid and K. Kirchner, J. Chem. Soc., Dalton Trans., 2000, 2607–2612 RSC.
- T.-T. Thai, B. Therrien and G. Süss-Fink, J. Organomet. Chem., 2009, 694, 3973–3981 CrossRef CAS.
- S. L. Nongbri, B. Therrien and K. M. Rao, Inorg. Chim. Acta, 2011, 376, 428–436 CrossRef CAS.
- M. Gobec, J. Kljun, I. Sosic, I. Mlinaric-Rascan, M. Ursic, S. Gobec and I. Turel, Dalton Trans., 2014, 43, 9045–9051 RSC.
- A. Kurzwernhart, W. Kandioller, E. A. Enyedy, M. Novak, M. A. Jakupec, B. K. Keppler and C. G. Hartinger, Dalton Trans., 2013, 42, 6193–6202 RSC.
- A. Kurzwernhart, W. Kandioller, S. Bachler, C. Bartel, S. Martic, M. Buczkowska, G. Muhlgassner, M. A. Jakupec, H. B. Kraatz, P. J. Bednarski, V. B. Arion, D. Marko, B. K. Keppler and C. G. Hartinger, J. Med. Chem., 2012, 55, 10512–10522 CrossRef CAS PubMed.
- R. M. Moriarty, Y. Y. Ku and U. S. Gill, J. Chem. Soc., Chem. Commun., 1987, 1837–1838 RSC.
- E. A. Trifonova, D. S. Perekalin, N. L. Loskutova, Y. V. Nelyubina and A. R. Kudinov, J. Organomet. Chem., 2014, 770, 1–5 CrossRef CAS.
- C. S. Allardyce, P. J. Dyson, D. J. Ellis, P. A. Salter and R. Scopelliti, J. Organomet. Chem., 2003, 668, 35–42 CrossRef CAS.
- F. Caruso, R. Pettinari, M. Rossi, E. Monti, M. B. Gariboldi, F. Marchetti, C. Pettinari, A. Caruso, M. V. Ramani and G. V. Subbaraju, J. Inorg. Biochem., 2016, 162, 44–51 CrossRef CAS PubMed.
- D. B. G. Williams and M. Lawton, J. Org. Chem., 2010, 75, 8351–8354 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- OECD, Test No. 107: Partition Coefficient (n-octanol/water): Shake Flask Method, OECD Publishing, 1995 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: clogP data, additional 1H NMR spectroscopy and mass spectrometry data, X-ray crystallography measurement data. CCDC 1585951. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt04451h |
| This journal is © The Royal Society of Chemistry 2018 |