
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Steric control in the metal–ligand electron transfer of iminopyridine–ytterbocene complexes†
Alexander A.
Trifonov
 *ab,
Tatyana V.
Mahrova
a,
Lapo
Luconi
c,
Giuliano
Giambastiani
*cd,
Dmitry M.
Lyubov
a,
Anton V.
Cherkasov
a,
Lorenzo
Sorace
*e,
Elisa
Louyriac
f,
Laurent
Maron
*f and
Konstantin A.
Lyssenko
b
*ab,
Tatyana V.
Mahrova
a,
Lapo
Luconi
c,
Giuliano
Giambastiani
*cd,
Dmitry M.
Lyubov
a,
Anton V.
Cherkasov
a,
Lorenzo
Sorace
*e,
Elisa
Louyriac
f,
Laurent
Maron
*f and
Konstantin A.
Lyssenko
b
aG. A. Razuvaev Institute of Organometallic Chemistry of Russian Academy of Sciences, Tropinina str. 49, 603950 Nizhny Novgorod, GSP-445, Russia. E-mail: trif@iomc.ras.ru
bA. N. Nesmeyanov Institute of Organoelement Compounds of Russian Academy of Sciences, 28 Vavilova str., 119991, Moscow, GSP-1, Russia
cIstituto di ChimicadeiCompostiOrganometallici (ICCOM - CNR), Via Madonna del Piano 10, 50019, Sesto Fiorentino, Italy
dKazan Federal University, 420008 Kazan, Russian Federation
eDipartimento di Chimica “U. Schiff” and UdR INSTM, Università di Firenze, 50019 Sesto Fiorentino, Italy
fUniversité de Toulouse, INSA, UPS, CNRS-UMR5215, LPCNO Avenue de Rangueil 135, 31077 Toulouse, France
First published on 21st December 2017
Abstract
A systematic study of reactions between Cp*2Yb(THF) (Cp* = η5-C5Me5, 1) and iminopyridine ligands (IPy = 2,6-iPr2C6H3N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(C5H3N-R), R = H (2a), 6-C4H3O (2b), 6-C4H3S (2c), 6-C6H5 (2d)) featuring similar electron accepting properties but variable denticity and steric demand, has provided a new example of steric control on the redox chemistry of ytterbocenes. The reaction of the unsubstituted IPy 2a with 1, either in THF or toluene, gives rise to the paramagnetic species Cp*2YbIII(IPy)˙− (3a) as a result of a formal one-electron oxidation of the YbII ion along with IPy reduction to a radical-anionic state. The reactions of 1 with substituted iminopyridines 2b–d, bearing aryl or hetero-aryl dangling arms on the 6 position of the pyridine ring occur in a non-coordinating solvent (toluene) only and afford coordination compounds of a formally divalent ytterbium ion, coordinated by neutral IPy ligands Cp*2YbII(IPy)0 (3b–d). The X-ray diffraction studies revealed that 2a–c act as bidentate ligands; while the radical-anionic IPy in 3a chelates the YbIII ion with both nitrogens, neutral IPy ligands in 3b and 3c participate in the metal coordination sphere through the pyridine nitrogen and O or S atoms from the furan or thiophene moieties, respectively. Finally, in complex 3d the neutral IPy ligand formally adopts a monodentate coordination mode. However, an agostic interaction between the YbII ion and an ortho C–H bond of the phenyl ring has been detected. Imino-nitrogens in 3b–d are not involved in the metal coordination. Variable temperature magnetic measurements on 3a are consistent with a multiconfigurational ground state of the Yb ion and suggest that the largest contribution arises from the 4f13-radical configuration. For complexes 3b and 3c the data of magnetic measurements are indicative of a YbII-closed shell ligand electronic distribution. Complex 3d is characterized by a complex magnetic behavior which does not allow for an unambiguous estimation of its electronic structure. The results are rationalized using DFT and CSSCF calculations. Unlike diazabutadiene analogues, 3a does not undergo a solvent mediated metal–ligand electron transfer and remains paramagnetic in THF solution. On the other hand, complexes 3b–d readily react with THF to afford 1 and free IPy 2b–d.
CH(C5H3N-R), R = H (2a), 6-C4H3O (2b), 6-C4H3S (2c), 6-C6H5 (2d)) featuring similar electron accepting properties but variable denticity and steric demand, has provided a new example of steric control on the redox chemistry of ytterbocenes. The reaction of the unsubstituted IPy 2a with 1, either in THF or toluene, gives rise to the paramagnetic species Cp*2YbIII(IPy)˙− (3a) as a result of a formal one-electron oxidation of the YbII ion along with IPy reduction to a radical-anionic state. The reactions of 1 with substituted iminopyridines 2b–d, bearing aryl or hetero-aryl dangling arms on the 6 position of the pyridine ring occur in a non-coordinating solvent (toluene) only and afford coordination compounds of a formally divalent ytterbium ion, coordinated by neutral IPy ligands Cp*2YbII(IPy)0 (3b–d). The X-ray diffraction studies revealed that 2a–c act as bidentate ligands; while the radical-anionic IPy in 3a chelates the YbIII ion with both nitrogens, neutral IPy ligands in 3b and 3c participate in the metal coordination sphere through the pyridine nitrogen and O or S atoms from the furan or thiophene moieties, respectively. Finally, in complex 3d the neutral IPy ligand formally adopts a monodentate coordination mode. However, an agostic interaction between the YbII ion and an ortho C–H bond of the phenyl ring has been detected. Imino-nitrogens in 3b–d are not involved in the metal coordination. Variable temperature magnetic measurements on 3a are consistent with a multiconfigurational ground state of the Yb ion and suggest that the largest contribution arises from the 4f13-radical configuration. For complexes 3b and 3c the data of magnetic measurements are indicative of a YbII-closed shell ligand electronic distribution. Complex 3d is characterized by a complex magnetic behavior which does not allow for an unambiguous estimation of its electronic structure. The results are rationalized using DFT and CSSCF calculations. Unlike diazabutadiene analogues, 3a does not undergo a solvent mediated metal–ligand electron transfer and remains paramagnetic in THF solution. On the other hand, complexes 3b–d readily react with THF to afford 1 and free IPy 2b–d.
Introduction
Since the pioneering studies of Cloke and Edelmann1 in the early 1980s who introduced diimines in organolanthanide chemistry, this field of coordination chemistry has received a large boost due to the diversity of coordination modes offered by this class of ligands along with their intriguing redox properties.2 The idea to combine redox active diimine ligands3 and ytterbium ions possessing two stable oxidation states4 has proven to be particularly fruitful5 thus leading to the discovery of a series of challenging phenomena such as solvent mediated redox transformations,5c,6 sterically induced reduction7 and redox isomerism.7,8 The course of the reactions between ytterbocenes and diimine ligands has been found to be affected by a number of factors. Depending on the nature of cyclopentadienyl-type ligands (Cp = cyclopentadienyl, indenyl, fluorenyl), their binding mode to the YbII ion (propensity to Cp-haptotropic rearrangements),9 and the steric demand of a diimine ligand, the reactions of ytterbocenes with diimines can result in either YbII/YbIII oxidation,6,10 formation of new C–C bonds11 or C–H bond activation at the diamine skeleton.12 The one-electron oxidation of ytterbocenes by diazabutadienes proved to be a sterically governed redox process.13 Depending on the steric hindrance of the ytterbium coordination sphere, YbIII complexes containing either radical-anionic diazabutadiene or covalently bonded imino-amido ligands can be formed.Iminopyridine (IPy) ligands belong to the same family of redox active diimine systems but their coordination chemistry with lanthanide ions still remains only moderately investigated14 and certainly much less studied than that of their saturated aminopyridine (APy) counterparts.14
In recent years our group has shown how the reaction of variably hindered ytterbocenes [L]2Yb(THF)n (L = η5-C13H9 (fluorenyl); η5-C9H7 (indenyl); η5-C5Me5; η5-C5H4Me) with a bidentate IPy ligand [2,6-iPr2C6H3NCH(C5H4N)] can lead to a dramatic rearrangement of the metal coordination sphere through unusual reactivity paths together with a permanent change of the ion oxidation state (Fig. 1).
 | ||
| Fig. 1 Representative examples of recently prepared Yb-complexes with IPy ligands. | ||
The YbII bis(fluorenyl) complex (η5-C13H9)2Yb(THF)2 in the presence of an excess of IPy, proceeds through the complete oxidative cleavage of the two η5-coordinated fragments, affording the paramagnetic YbIII species coordinated by three iminopyridyl radical-anions (I).11b Similarly, the YbII bis(indenyl) complex (η5-C9H7)2Yb(THF)2 undergoes an unusual NC bond insertion into a formal η1-Yb–C9H7 bond thus leading to the half-sandwich and oxidized compound (II).11b When bis(cyclopentadienyl) ytterbium complexes Cp2Yb(THF)n (Cp = η5-C5Me5, η5-C5H4Me) are reacted with two equivalents of the same IPy a similar oxidative cleavage at one Yb–Cp bond takes place and YbIII species (III) coordinated by two iminopyridyl radical-anions are formed.11a,15 For all these reactions, steric factors and the inherent tendency of the η5-coordinated ligands to undergo haptotropic rearrangement are claimed to play a fundamental role in the whole transformation.
Despite the intriguing reactivity issues observed in the reaction of (η5-C9H7)2Yb(THF)2 with the bidentate IPy ligand, 6-(hetero)aryl substituted analogues [2,6-iPr2(C6H3)NCH(C5H3N)-6-R′ (R′ = 2-furyl, 2-thiophenyl, phenyl)] behave unexpectedly as neutral (κ2-or κ3-coordinated) systems by replacing THF molecules from the metal coordination sphere, without affecting the metal oxidation state of the targeted coordinative compounds (IV).16 The reason for the inhibition of metal-to-ligand electron transfer in the latter compounds is likely due to the lack of convergence between the ytterbium center and the bulkier substituted IPy ligands as a consequence of a metal ion size decrease in the case of a YbII/YbIII oxidation.17 To gain additional insight into the steric regulation of these redox processes and to clarify the role of the steric and electronic properties of ligands bound to the ytterbium ion, hereafter we performed the study of the model ytterbocene complexes Cp*2Yb(THF) (Cp* = η5-C5Me5) with unsubstituted and 6-(hetero)aryl-substituted iminopyridine ligands. A series of di- and trivalent ytterbium complexes coordinated by neutral or radical anionic ligands have been synthesized and fully characterized.
Experimental section
General considerations and material characterization
All experiments were performed under an inert atmosphere, using standard Schlenk-tube and glove-box techniques. After drying over KOH, THF was purified by distillation from sodium/benzophenone ketyl and hexane and toluene were dried by distillation from sodium/triglyme benzophenone ketyl prior to use. (η5-C5Me5)2Yb(THF),18 IPy 2a19 and IPy 2b–d13,16,20 were prepared according to literature procedures. Unless otherwise stated, all commercially available chemicals were used as provided from commercial supplies. 1H and 13C{1H} NMR spectra were obtained on either a Bruker Avance-II 400 MHz NMR spectrometer or a Bruker DPX 200 MHz NMR spectrometer. Chemical shifts (δ) are reported in ppm relative to tetramethylsilane (TMS), referenced to the chemical shifts of residual solvent resonances (1H and 13C). IR spectra were recorded as Nujol mulls on a Bruker-Vertex 70 spectrophotometer. The N, C, and H elemental analyses were carried out in the microanalytical laboratory of the IOMC by means of a Carlo Erba Model 1106 elemental analyzer with an accepted tolerance of 0.4 unit on carbon (C), hydrogen (H), and nitrogen (N). Lanthanide metal analysis was carried out by complexometric titration.21General procedure for the synthesis of 3a–d
In a typical procedure, a toluene solution (20 mL) of (η5-C5Me5)2Yb(THF) (1) (0.50 g, 0.97 mmol) was treated dropwise with a toluene solution (10 mL) of the appropriate ligand (2a–d) (0.97 mmol) and the reaction mixtures were maintained under stirring at 50 °C for the expected reaction times (24 h for 3a and 3c; 12 h for 3b; 28 h for 3d). For 3a, after removing volatiles under vacuum, the brownish-black solid was re-dissolved in 20 mL of nonane and the solution was cooled down to −20 °C to give brownish-black crystals of the complex in 76% yield. For 3b–d, brownish-black crystals were obtained directly from slow concentration of the toluene mixtures at room temperature in 70, 48 and 32% yield, respectively. Characterization data for (C5Me5)2Yb[κ2–2,6-iPr2(C6H3)NCH(C5H4N)]˙− (3a): elemental analysis calcd (%) for C38H48N2Yb: C, 64.29; H, 7.38; N, 3.95; Yb, 24.38; found (%): C, 63.88; H, 7.01; N, 4.17; Yb, 24.56. IR (Nujol, KBr) ν/cm−1: 1575 (s), 1380 (s), 1265 (s), 1235 (s), 1155 (s), 1100 (m), 995 (s), 895 (s), 850 (m), 820 (s), 725 (s). 1H NMR (200 MHz, C6D6, 293 K, sw = 200 ppm): −70.3 (s), −32.4 (s), −16.3 (br s), −12.1 (br s), −7.9 (br s), −7.4 (s), −6.11 (s), 3.41 (s, CH3 Cp*), 11.8 (s), 18.7 (s), 23.2 (s), 26.4 (s), 33.2 (s), 43.2 (s), 80.8 (s) ppm. Characterization data for (C5Me5)2Yb[κ2-2,6-iPr2C6H3NCH(C5H4N)-6-(C4H3O)] (3b): elemental analysis calcd (%) for C42H54N2OYb: C, 65.01; H, 7.01; N, 3.61; Yb, 22.30; found (%): C, 65.36; H, 7.12; N, 3.31; Yb, 22.04. IR (Nujol, KBr), ν/cm−1:1635 (s), 1560 (s), 1315 (m), 1270 (s), 1160 (m), 1020, 810 (s), 755 (m). 1H NMR (200 MHz, C6D6, 293 K): 1.33 (d, 3JHH = 6.5 Hz, 12H, CH3iPr), 1.92 (s, 30H, CH3 Cp*), 3.41 (sept, 3JHH = 6.5 Hz, 2H, CH iPr), 6.01 (br s, 1H, NCH), 6.47 (br s, 1H, CH Ar), 6.69 (br d, 3JHH = 7.5 Hz, 1H, CH Ar), 7.20 (m, 3H, CH Ar), 7.83 (m, 2H, CH Ar), 8.14 (br d, 3JHH = 7.5 Hz, 1H, CH Ar), 8.59 (br s, 1H, CH Ar) ppm. 13C{1H} NMR (50 MHz, C6D6, 293 K): 10.7 (s, CH3, C5Me5), 22.2 (s, CH3iPr), 28.4 (s, CH iPr), 110.6 (s, CH Ar), 111.3 (s, C C5Me5), 113.3 (s, CH Ar), 122.7 (s, CH Ar), 123.2 (s, CH Ar), 123.7 (s, CH Ar), 125.4 (s, C Ar), 134.9 (s, CH Ar), 136.1 (s, CH Ar), 137.2 (s, C Ar), 137.6 (s, CH Ar), 143.8 (s, C Ar), 148.5 (s, C Ar), 152.3 (s, C Ar), 158.4 (s, CHN) ppm. Characterization data for (C5Me5)2Yb[κ2-2,6-iPr2C6H3NCH(C5H4N)-6-(C4H3S)] (3c): elemental analysis calcd (%) for C42H54N2SYb: C, 63.69; H, 6.87; N, 3.54; Yb, 21.85; found (%): C, 63.36; H, 7.12; N, 3.31; Yb, 22.14. IR (Nujol, KBr), ν/cm−1: 1685 (m), 1645 (m), 1570 (s), 1380 (s), 1320 (w), 1255 (w), 1180 (m), 1080 (m), 930 (w), 850 (m), 715 (m). 1H NMR (200 MHz, C6D6, 293 K): 1H NMR (200 MHz, C6D6, 293 K): 1.77 (d, 3JHH = 7.0 Hz, 12H, CH3iPr), 2.11 (br s, 30H, CH3 Cp*), 3.42 (br sept, 3JHH = 7.0 Hz, 2H, CH iPr), 6.51 (d, 3JHH = 7.6 Hz, 1H, CH Ar), 6.73 (m, 2H, NCH and CH Ar), 6.97 (m, 4H, CH Ar), 7.33 (br s, 1H, CH Ar), 7.55 (br s, 1H, CH Ar), 7.79 (d, 3JHH = 7.6 Hz, 1H, CH Ar) ppm. Characterization data for (C5Me5)2Yb[κ1-2,6-iPr2C6H3NCH(C5H4N)-6-Ph] (3d): elemental analysis calcd (%) for C44H56N2Yb: C, 67.24; H, 7.18; N, 3.56; Yb, 22.02; found (%): C, 66.98; H, 7.12; N, 3.31; Yb, 22.14. IR (Nujol, KBr), ν/cm−1: 1645 (s), 1585 (s), 1320 (s), 1180 (m), 1029 (s), 820 (s), 740 (s), 725 (m). 1H NMR (200 MHz, C6D6, 293 K): 1.31 (d, 3JHH = 7.0 Hz, 6H, CH3iPr), 1.77 (d, 3JHH = 7.0 Hz, 6H, CH3iPr), 2.24 (br s, 30H, CH3 Cp*), 3.39 (m, 1H, CH iPr), 3.65 (m, 1H, CH iPr), 6.07 (s, 1H, NCH), 6.48–7.03 (complex m, 7H, CH Ar), 7.42 (m, 2H, CH Ar), 7.97 (m, 1H, CH Ar), 8.29 (m, 1H, CH Ar) ppm.
Magnetic characterization
Magnetic measurements of crystalline samples of 3a–d were carried out by using an MPMS-XL SQUID magnetometer in the temperature range of 1.8–300 K with an applied magnetic field of 1000 Oe (up to 40 K) and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Oe (from 40 K to 350 K), to avoid saturation effects. Samples were prepared in a glove box by wrapping them in Teflon and then loaded in the SQUID magnetometer in less than 30′′. The susceptibility is evaluated in the low field limit as χm = Mm/H. The raw data have been corrected for diamagnetic contribution of the sample holder, and the intrinsic diamagnetism of the sample, estimated by using Pascal's constant to obtain the paramagnetic susceptibility.
000 Oe (from 40 K to 350 K), to avoid saturation effects. Samples were prepared in a glove box by wrapping them in Teflon and then loaded in the SQUID magnetometer in less than 30′′. The susceptibility is evaluated in the low field limit as χm = Mm/H. The raw data have been corrected for diamagnetic contribution of the sample holder, and the intrinsic diamagnetism of the sample, estimated by using Pascal's constant to obtain the paramagnetic susceptibility.
X-ray crystallography
The X-ray data for 3a–d were collected on Bruker Smart Apex (3b,d), Bruker D8 Quest (3a) and Agilent Xcalibur E (3c) diffractometers (MoKα-radiation, ω-scans technique, λ = 0.71073 Å, T = 100(2) K) using SMART,22 APEX223 and CrysAlis PRO24 software packages. The structures were solved by direct and dual-space25 methods and were refined by full-matrix least squares on F2 for all data using SHELX.25a SADABS26 and CrysAlis PRO were used to perform area-detector scaling and absorption corrections. All non-hydrogen atoms were found from Fourier syntheses of electron density and were refined anisotropically. Hydrogen atoms H(10A) in 3b,c and H(3A) in 3d also were found from Fourier syntheses of electron density and were refined isotropically. Other hydrogen atoms in 3a–d were placed in calculated positions and were refined in the “riding” model with U(H)iso = 1.2 Ueq of their parent atoms (U(H)iso = 1.5 Ueq for methyl groups). 1552992 (3a), 1552993 (3b), 1552994 (3c), 1552995 (3d)† contain the supplementary crystallographic data for this paper. The crystallographic data and structure refinement details are given in Table S1.†Computational details
The calculations have been carried out with the Gaussian09 software27 at the DFT level using the hybrid functional B3PW91.28 Ytterbium atoms were treated with the small-core pseudopotential from the Stuttgart group,29 that explicitly account for the 4f electrons. Oxygen, carbon and hydrogen atoms have been treated with the all electron 6-31G** Pople basis set.30 The geometry optimization has been performed without any symmetry constraints taking the geometry obtained from the X-ray diffraction measurement of the product. Analytical calculations of the vibrational frequencies of the optimized geometry were done to confirm that it's a minimum. CASSCF calculations were also carried out based on the SCF orbitals obtained for the triplet state and the active space choice is detailed in the manuscript.Results and discussion
When a dark-red THF solution of Cp*2Yb(THF) (1) is treated at room temperature with an equimolar amount of IPy ligand 2a, the colour of the reaction mixture changed rapidly to deep brown thus indicating that a reaction takes place. Following the process by 1H NMR (THF-d8, 293 K) spectroscopy proved the rapid oxidation of the metal ion to YbIII and the subsequent formation of a paramagnetic species. This result contrasts with earlier observations dealing with the reactivity of ytterbocenes in the presence of N,N-disubstituted diazabutadienes7 where only non-coordinating aromatic and aliphatic hydrocarbons5,6,16 allowed the reaction to take place, whereas no interaction occurred in the presence of coordinating solvents (i.e. THF, py). Hereby, 2a reacts with ytterbocene 1 in THF to give the paramagnetic complex (C5Me5)2Yb[κ2-2,6-iPr2(C6H3)NCH(C5H4N)]˙− (3a) as a result of a formal one-electron oxidation of the metal ion and IPy ligand reduction to the radical-anionic state (Scheme 1). In the present case, THF-to-IPy ligand displacement at the ytterbium center is likely facilitated by the reduced steric hindrance of IPy16 compared to the more commonly used and sterically demanding N,N-disubstituted diazabutadiene systems.7 | ||
| Scheme 1 Reaction scheme for the generation of the paramagnetic species 3a. | ||
3a was isolated in 76% yield as dark brown, air- and moisture-sensitive, microcrystals. Single crystals suitable for X-ray study were obtained from a concentrated toluene solution of the compound, cooled down to −20 °C for days. A perspective view of the 3a structure is given in Fig. 2 along with a selection of the main structural details [bond lengths (Å) and angles (°)]. The structure refinement data are given in Table S1 (see the ESI†).
 | ||
| Fig. 2 ORTEP diagram (30% probability thermal ellipsoids) of complex (C5Me5)2Yb[κ2-2,6-iPr2(C6H3)NCH(C5H4N)]˙− (3a). Hydrogen atoms and Me-substituents at the iPr-groups are omitted for clarity. Selected bond distances (Å) and bond angles (°): Yb(1)–C(29) 2.601(2), Yb(1)–C(30) 2.618(2), Yb(1)–C(33) 2.624(2), Yb(1)–C(22) 2.660(2), Yb(1)–C(20) 2.662(2), Yb(1)–C(32) 2.665(2), Yb(1)–C(31) 2.672(2), Yb(1)–C(19) 2.670(2), Yb(1)–C(23) 2.668(2), Yb(1)–C(21) 2.671(2), Yb(1)–N(1) 2.325(2), Yb(1)–N(2) 2.385(2), N(2)–C(6) 1.348(3), C(5)–C(6) 1.408(3), N(1)–Yb(1)–N(2) 72.84(7), CpCentr–Yb–CpCentr 132.9(2). | ||
The ytterbium ion is η5-coordinated by two Cp* ligands together with the two nitrogen atoms from the IPy framework that formally increase the final complex coordination number to eight (assuming CN 3 assigned for the Cp ligand). The Yb–C bond lengths in 3a [Yb–Cmean: 2.651(6) Å; Yb–Cpcentre: 2.345(2) Å] are shorter than those measured on both the IPy-free ytterbocene 1 (Yb–Cmean 2.69 Å; Yb–Cpcentre: 2.41 Å)31 and the diamagnetic eight-coordinated pyridine adduct (Cp*)2Yb(Py)2 (Yb–Cmean 2.74 Å).32 On the other hand, they are in good accord with Yb–C bond distances measured in related eight-coordinated YbIII complexes of the state-of-the-art {i.e. (η5-C5Me4H)2YbI(THF) [Yb–Cmean 2.60(4) Å],33 (η5-C5Me4H)2Yb(μ-Cl)2Li(THF)2 [Yb–Cmean 2.604(6) Å],34 (C5Me5)2YbF(THF) [Yb–Cmean 2.628(5) Å]35 and (C5Me5)2YbF(Et2O) [Yb–Cmean 2.631(8) Å]}.35 The 5-membered YbNCCN metallacycle in 3a is not planar and it forms a dihedral angle of 27.0(2)° between the planes defined by YbNN and NCCN atoms. Yb–N bond lengths [2.325(2), 2.385(2) Å] fall in a close range of those formerly reported for the related paramagnetic ytterbocene complexes coordinated by radical-anionic diazabutadiene ligands {i.e. (η5-C5Me5)2YbIII(RNCHCHNR)˙− [R = tBu, Yb–N 2.358(3), 2.394(3) Å;6a R = p-tolyl, Yb–N 2.340(5), 2.368(5) Å;5b R = p-anisyl, Yb–N 2.339(7), 2.337(6) Å]}.5b Finally, bond distances within the NCCN fragment are markedly different from those typically measured for transition metal-coordinated36 neutral IPy ligands [for 3a: d(CN) = 1.348(3) Å; d(C–C) = 1.408(3) Å]. For model transition metal compounds containing neutral IPy ligands: IPyMIICl2 (MII = Pd,36 Ni;36b IPy = 2a): d(CN) = 1.279(4)–1.278(3) Å; d(C–C) = 1.457(5)–1.463(4) Å]. All these data taken together largely support the hypothesis of a trivalent oxidation state for the ytterbium ion in 3a as a result of one-electron metal oxidation along with a reduction of the coordinated IPy ligand to the state of a radical-anionic species.11b,15,37 However, all our attempts to detect the paramagnetic radical-anionic iminopyridine ligand in 3a (both in the solid state and in solution – hexane, toluene; 203–293 K) by the EPR technique failed. This outcome is in line with our previous studies where we demonstrated that YbIII complexes coordinated by paramagnetic radical-anionic diimino ligands are EPR-silent.6,10 In addition, this behavior was not surprising at all given the fast electronic relaxation characterizing YbIII complexes and the even number of unpaired electrons of the resulting species.38
The temperature dependence of the magnetic moment and of the molar susceptibility of 3a is reported in Fig. 3. A maximum in χMvs. T data is observed, together with the presence of an unavoidable paramagnetic impurity of YbIII coordinated to diamagnetic ligands, which is responsible for the paramagnetic tail observed at T < 50 K (Fig. 3A). By a fit of the low temperature region using a Curie law this is estimated to be 8.1%. This allowed correction of the data of Fig. 3A by following the procedure reported by Booth and co-workers.39 The corrected curves shown in Fig. 3B present a maximum in χM at 170 K along with a temperature independent paramagnetism contribution of 5.4 × 10−3 emu mol−1. Both these values are consistent with a multiconfigurational ground state39 and the observed μeff value at the highest measured temperature (4.19 at 350 K) suggests that the largest contribution is from the 4f13-radical configuration (expected μeff = 4.8 for uncoupled spins), in agreement with the results from the X-ray diffraction data.
 | ||
| Fig. 3 (A) μeffvs. T and χMvs. T curves of 3a before correction for the paramagnetic impurity. The dotted line represents the fit to estimate the amount of paramagnetic YbIII impurity assuming a Curie–Weiss behavior; (B) μeffvs. T and χMvs. T curves of 3a after correction for the paramagnetic impurity. The horizontal dashed line represents the μeff value expected at high temperature for a system formed by uncoupled YbIII and S = 1/2. | ||
The 1H NMR spectroscopy is a sensitive tool that allows for determining the magnetism of complexes in solution. The proton NMR spectrum can qualitatively indicate whether the complex is diamagnetic or paramagnetic. The 1H NMR spectrum of 3a (benzene-d6, 293 K) shows a set of broadened signals that are substantially shifted with respect to those of related diamagnetic species (Fig. S1, ESI†) thus giving an additional proof of its paramagnetic nature. The methyl protons of the C5Me5 ring appear as a slightly broadened singlet at 3.41 ppm, in good agreement with the chemical shifts measured for related paramagnetic species containing YbIII ions coordinated by radical-anionic bipyridine, phenanthroline or related ligands. The protons of the radical-anionic iminopyridine ligand give rise to a set of signals in a broad interval from −70 to 80 ppm.
Finally, the room-temperature electronic absorption spectra of 3a (recorded in nonane), the radical-anionic potassium derivative [2,6-iPr2(C6H3)NCH(C5H4N)]˙−K+ (recorded in THF) and of the neutral Ipy 2a (recorded in hexane) were put at comparison for the sake of completeness. The spectra are shown in Fig. S3 (see the ESI†). The UV-Vis spectrum of 3a shows two absorption bands at 355 and 455 nm, respectively. While the neutral IPy ligand 2a displays a single absorbance at 340 nm, its potassium radical-anionic derivative [2,6-iPr2(C6H3)NCH(C5H4N)]˙−K+ also displays two main absorbances at 355 and 525 nm, respectively. This comparative analysis is in line with the presence of a radical-anionic iminopyridine ligand in solution and corroborates with the NMR data on the paramagnetic nature of 3a.
With the aim of gaining additional insight into the role of steric factors driving the metal-to-ligand electron transfer in the above-mentioned coordinative compounds, we studied the reaction of 1 in the presence of bulkier IPy ligands (2b–d), bearing coordinating or not-coordinating (hetero)aryl fragments attached to the six-position of the pyridine core (Scheme 2).
 | ||
| Scheme 2 Reaction scheme for the generation of the coordination compounds 3b–d. | ||
This systematic investigation takes advantage of the previous outcomes of our study of the coordination modes and electronic effects resulting from the reaction of the same class of IPy ligands with the bulkier bis(indenyl) ytterbium complex (η5-C9H7)2YbII(THF)2.16 In that case, the coordination of 6-(hetero)aryl-substituted IPy ligands occurred with the generation of diamagnetic species as a result of the simple replacement of the two coordinated THF molecules.
Unlike the above-mentioned reaction with the bidentate IPy 2a, Cp*2Yb(THF) (1) does not react in THF with the more sterically demanding ligands 2b–d. This was preliminary confirmed by the absence of any appreciable color change in the solutions of 1 upon treatment with an equimolecular amount of 2b–d. In addition, monitoring the reaction course over several hours by 1H NMR (THF-d8, 293 K) spectroscopy has showed only signals from unreacted starting materials. On the contrary, the addition of 2a–d to a toluene solution of 1 at room temperature (Scheme 2) resulted in the immediate change of the solution colour from dark-red to brownish-black. After slow concentration of each solution, brownish-black microcrystals of complexes (C5Me5)2Yb[κ2-2,6-iPr2C6H3NCH(C5H4N)-6-(C4H3O)] (3b), (C5Me5)2Yb[κ2-2,6-iPr2C6H3NCH(C5H4N)-6-(C4H3S)] (3c), (C5Me5)2Yb[κ1-2,6-iPr2C6H3NCH(C5H4N)-6-Ph] (3d) were obtained in 70, 48 and 32% isolated yields, respectively. Complexes 3b–d were isolated as highly air-and moisture-sensitive solids showing moderate solubility in aromatic and aliphatic hydrocarbons thus hampering their full NMR characterization (13C{1H} NMR spectra).
Crystals suitable for single-crystal X-ray diffraction studies were grown by slow concentration of the respective toluene solutions at ambient temperature under a gentle stream of nitrogen. Complex 3b crystallizes as a solvate with one molecule of toluene while crystals of 3c and 3d do not contain any crystallization solvent. ORTEP representations of the three crystal structures are given in Fig. 4–6 along with a selection of the main structural details [bond lengths (Å) and angles (°)]; Table S1† lists their main crystal data and structural refinement details.
 | ||
| Fig. 4 ORTEP diagram (30% probability thermal ellipsoids) of complex (C5Me5)2Yb[κ2-2,6-iPr2C6H3NCH(C5H4N)-6-(C4H3O)] (3b). Hydrogen atoms, Me-substituents of iPr-groups and one toluene molecule of crystallization are omitted for clarity. Selected bond distances (Å) and bond angles (°): Yb(1)–O(1) 2.468(2), Yb(1)–N(1) 2.535(2), Yb(1)–C(33) 2.676(2), Yb(1)–C(34) 2.678(2), Yb(1)–C(23) 2.679(3), Yb(1)–C(35) 2.687(2), Yb(1)–C(24) 2.690(3), Yb(1)–C(27) 2.692(3), Yb(1)–C(37) 2.694(2), Yb(1)–C(25) 2.695(3), Yb(1)–C(36) 2.697(2), Yb(1)–C(26) 2.709(2), N(2)–C(10) 1.264(3), C(5)–C(10) 1.475(4), O(1)–Yb(1)–N(1) 66.8(6). | ||
 | ||
| Fig. 5 ORTEP diagram (30% probability thermal ellipsoids) of complex (C5Me5)2Yb[κ2-2,6-iPr2C6H3NCH(C5H4N)-6-(C4H3S)] (3c). Hydrogen atoms and Me-substituents of iPr-groups are omitted for clarity. Selected bond distances (Å) and bond angles (°): Yb(1)–N(1) 2.623(2), Yb(1)–S(1) 3.0103(7), Yb(1)–C(26) 2.645(3), Yb(1)–C(27) 2.669(3), Yb(1)–C(35) 2.696(3), Yb(1)–C(25) 2.699(3), Yb(1)–C(36) 2.712(2), Yb(1)–C(37) 2.723(3), Yb(1)–C(34) 2.730(3), Yb(1)–C(23) 2.732(3), Yb(1)–C(24) 2.741(3), Yb(1)–C(33) 2.764(3), N(2)–C(10) 1.258(4), C(5)–C(10) 1.468(4), N(1)–Yb(1)–S(1) 65.15(4). | ||
 | ||
| Fig. 6 ORTEP diagram (30% probability thermal ellipsoids) of complex (C5Me5)2Yb[κ1-2,6-iPr2C6H3NCH(C5H4N)-6-Ph] (3d). Hydrogen atoms [except H(7)] and Me-substituents of iPr-groups are omitted for clarity. Selected bond distances (Å) and bond angles (°): Yb(1)–N(1) 2.602(2), Yb(1)–C(7) 2.969(3), Yb(1)–C(39) 2.683(3), Yb(1)–C(28) 2.683(2), Yb(1)–C(29) 2.692(2), Yb(1)–C(35) 2.695(3), Yb(1)–C(38) 2.702(3), Yb(1)–C(36) 2.712(2), Yb(1)–C(37) 2.718(2), Yb(1)–C(25) 2.736(2), Yb(1)–C(27) 2.758(2), Yb(1)–C(26) 2.776(2), C(5)–C(12) 1.488(4), N(1)–Yb(1)–C(7) 62.48(7). | ||
At odds with previously reported bis(indenyl)ytterbium/IPy coordination derivatives16 where ligands 2b and 2c–d behaved as tridentate and bidentate systems, respectively, the reaction of 1 with 2b–d leads to complexes 3b–d featuring different denticity of the coordinated IPys. As shown in Fig. 4 and 5, the potentially tridentate ligands 2b–c coordinate the metal ion as bidentate (κ2) systems involving donor atoms of both heterocycles thus resulting in a formal coordination number of eight for the ytterbium ion, whereas imino nitrogens point away from the metal coordination sphere. In the case of the bidentate ligand 2d, only the pyridine core coordinates the ytterbium ion (κ1) along with an agostic interaction at a C–H bond of the phenyl substituent [Yb(1)–C(3) = 2.969(3) Å, Yb(1)–H(3A) = 2.49(3) Å, Yb(1)–H(3A)–C(3) = 162.2(7)°]. Therefore, 2d formally behaves as a monodentate (κ1) system, again keeping away the imino nitrogen from the metal center as for previously described complexes 3b–c. The increased steric demand of η5-Cp* with respect to indenyl in the ytterbocene complex is largely responsible of the different IPy coordination mode around the metal ion. Although Yb–C bond distances in 3b–d are reasonably longer than those measured in precursor 1 [d(Yb–Cmean) for 1: 2.651(6) Å; for 3b: 2.689(8) Å; for 3c: 2.711(9) Å, for 3d: 2.715(7) Å] as well as in the (Cp*)2Yb(bipy) adduct from the literature40 (Yb–Cmean: 2.62 Å), an appreciable shortening is measured with respect to another eight-coordinate ytterbocene/pyridine adduct of the state-of-the-art [d(Yb–Cmean) for (Cp*)2Yb(Py)2: 2.74 Å].32 In spite of close stereo-electronic and redox properties of IPy ligands of this series (2b–d)16 such a shortening effect is more evident in 3b compared to the other related complexes 3c,d. This trend is also observed for the Yb–N bond distances. Indeed, complex 3b presents a Yb–N bond length [2.535(2) Å] that is noticeably shorter than that measured on the related compounds 3c,d [3c: 2.623(2) Å; 3d: 2.602(2) Å].
The magnetic properties of 3b–d are outlined in Fig. 7 as μeffvs. T. For compounds 3b and 3c only a weak paramagnetic contribution is observed, likely due to the presence of an YbIII containing impurity. This behavior is in agreement with structural features observed for these systems, which point to an YbII-closed shell ligand electronic distribution. On the other hand, 3d is characterized by a much more complex magnetic behavior. It presents a μeff value of 2.4 up to 280 K that is much lower than that expected for a single YbIII-species and a radical anionic ligand, even assuming them to be antiferromagnetically coupled. Such a μeff value is also completely different from that presumed for a diamagnetic YbII-closed shell system. On the other hand, no maximum in χMvs. T, which would be indicative of a multiconfigurational ground state, is observed (see Fig. S17 of the ESI†). Notably, heating 3d from 270 K to 350 K gives rise to a clear-cut increase of the μeff value to 3.55. Such a variation is irreversible as witnessed by repeating the measurement after cooling down the sample. The value observed at high temperature is consistent with that expected for a strong antiferromagnetically coupled YbIII-radical system, resulting in a 1F3 state due to the combination of the 2F7/2 term of YbIII and the 2S1/2 term of a radical-anionic ligand.40
 | ||
| Fig. 7 μ eff vs. T curve for 3b (blue triangles), 3c (green rhombus) and 3d. For the latter, after the first heating cycle (red and white circles) the sample has been cooled down to 2 K and heated again up to 350 K (red squares). | ||
It is noteworthy that the μeffvs. T profile obtained after the first heating/cooling cycle can be perfectly rescaled, up to 270 K, on the pristine curve (see Fig. S18 of the ESI†). This result suggests that the species formed after heating above 270 K is also responsible for the intermediate μeff value measured before heating. Such a behavior can be reasonably explained by considering that the process leading to the increased μeff is already operative at room temperature (since the increase is visible above 270 K). Therefore, transformation already partially occurred when the sample is initially cooled down for measuring its magnetic properties. In other terms, the results of the magnetic investigation coupled with the outcomes of X-ray diffraction suggest that what we are actually measuring is not the structurally characterized form of 3d, but rather a product which evolves from 3d at T > 270 K. In this framework, it is tempting to attribute to 3d a diamagnetic character and to the evolution product a YbIII-radical one. However, in the absence of definitive clues or clear-cut experimental evidence, this is a purely speculative hypothesis. Finally, we wish to stress here that a simple redox isomeric transition involving 3d cannot be invoked to explain the observed behaviour, since this should favour a closed shell configuration at high temperature.41 However, if a structural rearrangement takes place on heating, this might favour the YbIII-radical charge distribution; such a coupled structural/electronic transition would be in analogy to what is often observed in the case of CoII complexes presenting similar temperature dependence of magnetic moment.19,42
In contrast to 3a, the 1H NMR spectra of 3b–d (benzene-d6, 293 K) prove their diamagnetic nature in solution. The methyl protons of C5Me5 rings in 3b, 3c and 3d appear as singlets at 1.92, 2.11 and 2.24 ppm respectively, and they are in line with the chemical shifts earlier reported for diamagnetic complexes (C5Me5)2Yb(L)n with coordinated O-, N-, and P-containing Lewis bases.43 At the same time, the signals of IPy ligands (2b–d) coordinated to the Yb ions in the 1H NMR spectra give rise to the expected sets of signals with typical chemical shifts (see the ESI†). Finally, the UV-Vis spectra of 3b–d are consistent with the diamagnetic nature of these complexes in solution.
Calculations were carried out in order to determine the electronic nature of complexes 3a,b,d. The computational strategy is the same as that was developed by one of us (LM) to study multireference ground state complexes in bipyridine/phenanthroline Yb complexes.39,40,44 In summary, first DFT(B3PW91) geometry optimization using small core Relativistic Effective Core Potentials (RECP) was carried out and in a second step CASSCF calculations were performed. For complex 3a, within the precision of the DFT calculations, the displacement of a THF molecule from Cp*2Yb(THF) by the ligand 2a is expected to generate a YbIII complex through an exothermic process (3.8 kcal mol−1) whereas the formation of a YbII species is virtually thermoneutral (0.7 kcal mol−1).45 Due to this very small energy difference between the YbII and YbIII complexes, CASSCF calculations were carried out. As in previous CASSCF calculations on ytterbium, different active spaces were used. A first one distributing 14 electrons into the 8 orbitals (the 7 4f of the Yb and the π* one of the ligand) was used and leads to similar results to the 8 electrons/5 orbitals (4 4f + π*) one. Finally, a reduced active space of 4 electrons/3 orbitals (2 4f + π*) was tested and gave the same qualitative results as the two others. Therefore, only the results obtained with the last one are discussed here for the sake of simplicity. Two singlet states, a closed shell one (f14) and an open-shell one (multiconfigurational state f14 + f13), and a triplet state were computed. The open-shell singlet state was found to be the ground state with the triplet state only 4.1 kcal mol−1 higher in energy and the closed-shell singlet is 5.3 kcal mol−1 higher in energy than the ground-state. The open-shell singlet state is formed by 78% of YbIII and 22% of YbII. This is in line with the magnetic measurement that indicates that complex 3a is exhibiting a multiconfigurational character in the ground state with a major contribution from YbIII.
In the same way, calculations were carried out on complex 3b. Unlike complex 3a, the coordination of the iminopyridine is not exothermic but rather slightly endothermic (5.8 kcal mol−1). The formation of the YbIII complex from the YbII one is also endothermic by up to 8.9 kcal mol−1, making this highly unfavorable. To ensure this value, similar CASSCF calculations were carried out and the closed-shell singlet [YbII] turned out to be the lowest by up to 10.6 kcal mol−1 with respect to the triplet state (3b); the open-shell singlet being even higher in energy at 12.3 kcal mol−1. This is again in line with the magnetic measurement that indicates that the ground state of 3b is diamagnetic.
Finally, the calculations were conducted on the interesting 3d complex. At the DFT level, the coordination of the iminopyridine is found to be even more endothermic (14.6 kcal mol−1) and the formation of the YbIII complex is endothermic by 3.3 kcal mol−1 from the YbII one. Unlike the other cases, it is the coordination that appears to be complicated, it might not be the CASSCF calculations that would yield the answer. Indeed, at this level, the open-shell singlet is also found to be the ground state but with the triplet state almost degenerate (1.2 kcal mol−1) and the closed-shell singlet only 5.3 kcal mol−1 higher in energy. With such low energy differences between the states, it is hard to conclude on the nature of the ground state.
For a family of metallocene-type YbIII complexes coordinated by radical-anionic diazabutadiene ligands, the existence of a reversible solvent mediated ligand-to-metal electron transfer has been formerly discovered.5c,6 This phenomenon consists of the displacement of the radical-anionic diazabutadiene ligand by molecules of a coordinating solvent (THF, DME, Py) followed by electron transfer from the diazabutadiene radical anion to the ytterbium ion: as a whole, the process results in the oxidation of the ligand to the neutral diazabutadiene and the reduction of YbIII to YbII (Scheme 3).
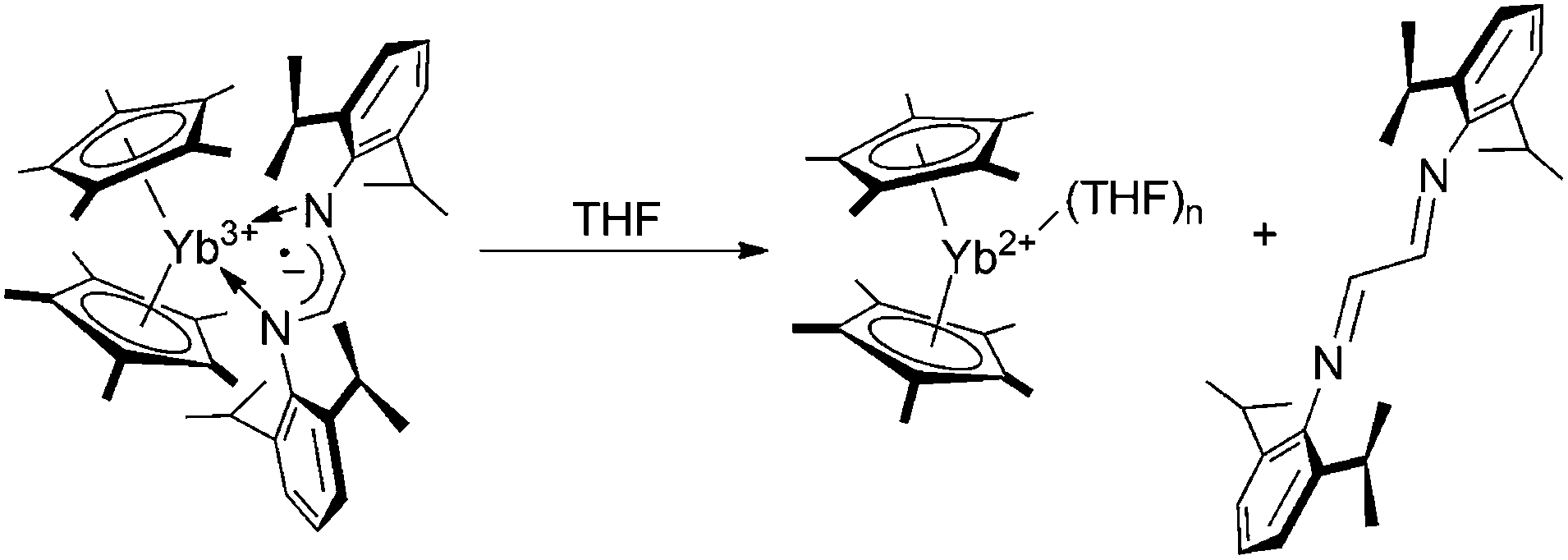 | ||
| Scheme 3 Solvent mediated ligand-to-metal electron transfer on a metallocene YbIII complex coordinated by a radical-anionic diazabutadiene ligand. | ||
In order to check whether this behavior applies to related coordination compounds featuring IPy ligands, the reaction of 3a with THF was carried out. Unlike diazabutadiene congeners, 3a remains paramagnetic in THF-d8 solution and no redox replacement of the iminopyridine ligand by THF occurs (Scheme 4). In contrast to 3a, the addition of a stoichiometric amount of THF-d8 to benzene-d6 solutions of 3b–d resulted in the immediate replacement of the IPy ligands in the Yb coordination sphere by THF. The 1H NMR spectra of 3b–d recorded in THF-d8/benzene-d6 mixtures have unambiguously demonstrated the generation of (C5Me5)2Yb(THF)n and free IPy ligands 2b–d (Scheme 4). This result highlights the different reactivity of complexes 3b–d from 3a with respect to the replacement of IPy ligands by THF most likely caused by the different nature of metal–ligand interaction in these complexes. In addition, it demonstrates that metallocene type YbIII complexes coordinated by chelating radical-anionic diazabutadiene and IPy ligands behave differently in the presence of a coordinating solvent, despite their similar nature.
 | ||
| Scheme 4 Solvent mediated ligand-to-metal electron transfer on coordination compounds 3a–d. | ||
It can be inferred that such a different behavior between diazabutadienes and IPys is reasonably ascribed to the higher steric demand of the former class of ligands that is claimed to weaken the metal–ligand interaction. In 3a the energy of formation for Cp*2YbII-THF is not sufficient to compensate for the energy loss for the cleavage of Coulombic interaction between the YbIII ion and the radical-anionic iminopyridine ligand. At the same time, the quick IPy displacement by stoichiometric amounts of THF in complexes 3b–d coordinated by a neutral IPy ligand is in line with the energies of coordination bonds in YbII complexes with N- and O-containing ligands.
Conclusions
In this paper we have described a new example of sterically controlled metal–ligand electron transfer by reacting (C5Me5)2Yb(THF) with IPy ligands featuring very similar electron accepting properties but variable denticity and steric demand. The reactions of (C5Me5)2Yb(THF) with ligands 2a–d proceed with the displacement of the coordinated THF molecule by means of an IPy framework. The observed outcomes highlight an important effect related to the steric demand of the iminopyridine systems on both their coordination mode and on their ability to foster metal-to-ligand electron transfer phenomena. Thus, the less sterically crowded 2a coordinates the metal ion as a bidentate system and a metal-to-ligand electron transfer takes place with the formation of a trivalent ytterbium species coordinated by a radical anionic IPy ligand. With the bulkier 2b and 2c, potentially featuring as tridentate systems, the coordination to the metal ions takes place through the pyridine nitrogen and either O or S atoms of the heteroaryl substituents, only. For these neutral coordinative YbII-complexes (3b and 3c) the imine nitrogen points away from the metal coordination sphere. For the bidentate 2d ligand, featuring steric hindrance similar to 2b–c, a monodentate coordination to the YbII ion is accomplished through the pyridine nitrogen only. The reason which blocks metal-to-ligand electron transfer in compounds 2b–d is likely the impossibility of a close approach between the ytterbium center and the bulkier substituted IPy ligands as a consequence of a metal ion size decrease in the case of a YbII/YbIII oxidation. We note that while this trend is clearly defined in solution by NMR and in the solid state by X-ray data, magnetic analysis in the solid state confirms it only for 3a–c. The former complex provides a magnetic response that is as expected for a multiconfigurational ground state with the largest contribution from the 4f13-radical configuration, while 3b and 3c are diamagnetic and thus consistent with the closed-shell singlet ground state configuration. On the contrary, the magnetic behavior of 3d turned out to be extremely complex, suggesting the instability of its structure on heating above 270 K.Unlike indenyl ligands cyclopentadienyl analogues are not prone to haptotropic rearrangements and the use of Cp*2Yb(THF) provided us with an opportunity to demonstrate clearly and unambiguously that bulkiness and the electronic properties of the aromatic carbocyclic ligands coordinated to the YbII ion are also decisive for the occurrence of intramolecular metal-to-ligand electron transfer and the type of coordination adopted by IPy ligands. The bidentate coordination of less sterically demanding Ipy 2a allows for a metal-to-ligand electron transfer resulting in a shortening of the Yb–Cp* bonds and the formation of rather short Yb–N bonds (Yb(1)–N(1) 2.325(2) Å). The introduction of bulky substituents into the Py ring excludes the possibility of such structural changes associated with oxidation and blocks the metal-to-ligand electron transfer. Indeed the resulting IPy complexes 2b–d feature Yb–Cp* and Yb–N distances characteristic for YbII species.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Russian Science Foundation (Project 17-73-30036).References
- (a) F. G. N. Cloke, H. C. de Lemos and A. A. Sameh, J. Chem. Soc., Chem. Commun., 1986, 1344–1345 RSC; (b) F. G. N. Cloke, Chem. Soc. Rev., 1993, 22, 17–21 RSC; (c) A. Recknagel, M. Noltemeyer and F. T. Edelmann, J. Organomet. Chem., 1991, 410, 53–61 CrossRef CAS; (d) P. Poremba and F. T. Edelmann, J. Organomet. Chem., 1997, 549, 101–104 CrossRef CAS.
- (a) T. V. Mahrova, G. K. Fukin, A. V. Cherkasov, A. A. Trifonov, N. Ajellal and J.-F. Carpentier, Inorg. Chem., 2009, 48, 4258–4266 CrossRef CAS PubMed; (b) H. Kaneko, H. Nagae, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2011, 133, 19626–19629 CrossRef CAS PubMed; (c) A. A. Kissel, D. M. Lyubov, T. V. Mahrova, G. K. Fukin, A. V. Cherkasov, T. A. Glukhova, D. Cui and A. A. Trifonov, Dalton Trans., 2013, 42, 9211–9225 RSC; (d) H. Kaneko, H. Tsurugi, T. K. Panda and K. Mashima, Organometallics, 2010, 29, 3463–3466 CrossRef CAS; (e) K.-H. Thiele, V. Lorenz, G. Thiele, P. Zoennchen and J. Scholz, Angew. Chem., Int. Ed. Engl., 1994, 33, 1372–1373 CrossRef; (f) H. Görls, B. Neumüller, A. Scholz and J. Scholz, Angew. Chem., Int. Ed. Engl., 1995, 34, 673–676 CrossRef; (g) A. A. Kissel, T. V. Mahrova, D. M. Lyubov, A. V. Cherkasov, G. K. Fukin, A. A. Trifonov, I. Del Rosal and L. Maron, Dalton Trans., 2015, 44, 12137–12148 RSC.
- (a) H. tom Dieck and I. W. Renk, Chem. Ber., 1971, 104, 110–130 CrossRef CAS; (b) H. tom Dieck, K.-D. Franz and F. Hoffmann, Chem. Ber., 1975, 108, 163–173 CrossRef CAS; (c) J. Reinhold, R. Benedix, P. Birner and H. Hennig, Inorg. Chem., 1979, 33, 209–213 CAS.
- (a) L. R. Morss, Chem. Rev., 1976, 76, 827–841 CrossRef CAS; (b) R. G. Finke, S. R. Keenan, D. A. Shirardi and P. L. Watson, Organometallics, 1986, 5, 598–601 CrossRef CAS; (c) A. M. Bond, G. B. Deacon and R. H. Newnham, Organometallics, 1986, 5, 2312–2316 CrossRef CAS.
- (a) A. A. Trifonov, Eur. J. Inorg. Chem., 2007, 3151–3167 CrossRef CAS; (b) M. D. Walter, D. J. Berg and R. A. Andersen, Organometallics, 2007, 26, 2296–2307 CrossRef CAS; (c) P. Cui, Y. Chen, G. Wang, G. Li and W. Xia, Organometallics, 2008, 27, 4013–4016 CrossRef CAS; (d) K. Vasudevan and A. H. Cowley, Chem. Commun., 2007, 3464–3466 RSC.
- (a) A. A. Trifonov, Yu. A. Kurskii, M. N. Bochkarev, S. Muehle, S. Dechert and H. Schumann, Russ. Chem. Bull., 2003, 52, 601–606 CrossRef CAS; (b) A. A. Trifonov, E. A. Fedorova, G. K. Fukin, V. N. Ikorskii, Yu. A. Kurskii, S. Dechert, H. Schumann and M. N. Bochkarev, Russ. Chem. Bull., 2004, 53, 2736–2743 CrossRef CAS; (c) A. A. Trifonov, E. A. Fedorova, V. N. Ikorskii, S. Dechert, H. Schumann and M. N. Bochkarev, Eur. J. Inorg. Chem., 2005, 2812–2818 CrossRef CAS.
- A. A. Trifonov, I. A. Borovkov, E. A. Fedorova, G. K. Fukin, J. Larionova, N. O. Druzhkov and V. K. Cherkasov, Chem. – Eur. J., 2007, 13, 4981–4987 CrossRef CAS PubMed.
- I. L. Fedushkin, O. V. Maslova, A. G. Morozov, S. Dechert, S. Demeshko and F. Meyer, Angew. Chem., Int. Ed., 2012, 51, 10584–10587 CrossRef CAS PubMed.
- I. D. Gridnev, Coord. Chem. Rev., 2008, 252, 1798–1818 CrossRef CAS.
- A. A. Trifonov, E. N. Kirillov, M. N. Bochkarev, H. Schumann and S. Muehle, Russ. Chem. Bull., 1999, 48, 382–384 CrossRef CAS.
- (a) A. A. Trifonov, E. A. Fedorova, G. K. Fukin, N. O. Druzhkov and M. N. Bochkarev, Angew. Chem., Int. Ed., 2004, 43, 5045–5048 CrossRef CAS PubMed; (b) A. A. Trifonov, E. A. Fedorova, I. A. Borovkov, G. K. Fukin, E. V. Baranov, J. Larionova and N. O. Druzhkov, Organometallics, 2007, 26, 2488–2491 CrossRef CAS.
- A. A. Trifonov, E. A. Fedorova, G. K. Fukin, E. V. Baranov, N. O. Druzhkov and M. N. Bochkarev, Chem. – Eur. J., 2006, 12, 2752–2757 CrossRef CAS PubMed.
- A. A. Trifonov, B. G. Shestakov, K. A. Lyssenko, J. Larionova, G. K. Fukin and A. V. Cherkasov, Organometallics, 2011, 30, 4882–4889 CrossRef CAS.
- G. Giambastiani and J. Campora, in Olefin Upgrading Catalysis by Nitrogen-based Metal Complexes, I and II, Springer, London, 2011, p. 287 and 265 Search PubMed.
- A. A. Trifonov, I. D. Gudilenkov, J. Larionova, C. Luna, G. K. Fukin, A. V. Cherkasov, A. I. Poddel'sky and N. O. Druzhkov, Organometallics, 2009, 28, 6707–6713 CrossRef CAS.
- A. A. Trifonov, B. G. Shestakov, I. D. Gudilenkov, G. K. Fukin, G. Giambastiani, C. Bianchini, A. Rossin, L. Luconi, J. Filippi and L. Sorace, Dalton Trans., 2011, 40, 10568–10575 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- T. Don Tilley, R. A. Andersen, B. Spencer, H. Ruben, A. Zalkin and D. H. Templeton, Inorg. Chem., 1980, 19, 2999–3003 CrossRef.
- Z. Benko, S. Burck, D. Gudat, M. Nieger, L. Nyulaszi and N. Shore, Dalton Trans., 2008, 4937–4945 RSC.
- (a) C. Bianchini, G. Giambastiani, G. Mantovani, A. Meli and D. Mimeau, J. Organomet. Chem., 2004, 689, 1356–1361 CrossRef CAS; (b) C. Bianchini, D. Gatteschi, G. Giambastiani, I. Guerrero Rios, A. Ienco, F. Laschi, C. Mealli, A. Meli, L. Sorace, A. Toti and F. Vizza, Organometallics, 2007, 26, 726–739 CrossRef CAS; (c) C. Bianchini, G. Mantovani, A. Meli, F. Migliacci and F. Laschi, Organometallics, 2003, 22, 2545–2547 CrossRef CAS.
- S. J. Lyle and M. M. Rahman, Talanta, 1953, 10, 1177–1182 CrossRef.
- M. Bruker, SMART, Bruker AXS Inc., Wisconsin, USA, 2012 Search PubMed.
- M. Bruker, APEX2, Bruker AXS Inc., Wisconsin, USA, 2012 Search PubMed.
- Y. Agilent, CrysAlis PRO, Agilent Technologies Ltd, Oxfordshire, England, 2014 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- M. Bruker, SADABS, Bruker AXS Inc., Wisconsin, USA, 2001 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef; (c) J. P. Perdew, Y. Wand and Y. K. Burke, in Electronic Density Functional Theory: Recent Progress and New Directions, ed. J. F. Dobson, G. Vignale and M. P. Das, Plenum, New York, 1998 Search PubMed.
- (a) M. Dolg, A. Stoll, A. Savin and H. Preuss, Theor. Chim. Acta, 1989, 75, 173–194 CrossRef CAS; (b) M. Dolg, H. Stoll and H. Rreuss, Theor. Chim. Acta, 1993, 85, 441–450 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. Schultz, C. J. Burns, D. J. Schwartz and R. A. Andersen, Organometallics, 2000, 19, 781–789 CrossRef CAS.
- T. Don Tilley, R. A. Andersen, B. Spencer and A. Zalkin, Inorg. Chem., 1982, 21, 2647–2649 CrossRef.
- I. A. Borovkov, G. K. Fukin and A. A. Trifonov, Russ. Chem. Bull., 2008, 57, 541–545 CrossRef CAS.
- G. Zi, Q. Yang, T. C. W. Mak and Z. Xie, Organometallics, 2001, 20, 2359–2366 CrossRef CAS.
- P. L. Watson, T. H. Tulip and I. Williams, Organometallics, 1990, 9, 1999–2009 CrossRef CAS.
- (a) V. C. Gibson, R. K. O'Reily, D. F. Wass, A. J. P. White and D. J. Williams, Dalton Trans., 2003, 2824–2830 RSC; (b) T. V. Laine, M. Klinga and M. Leskelä, Eur. J. Inorg. Chem., 1999, 959–964 CrossRef CAS; (c) T. V. Laine, U. Piironen, K. Lappalainen, M. Klinga, E. Aitolla and M. Leskelä, J. Organomet. Chem., 2000, 606, 112–124 CrossRef CAS.
- (a) C. C. Lu, E. Bill, T. Weyhermüller, E. Bothe and K. Wieghardt, J. Am. Chem. Soc., 2008, 130, 3181–3197 CrossRef CAS PubMed; (b) C. C. Lu, G. S. De Beer, T. Weyhermüller, E. Bill, E. Bothe and K. Wieghardt, Angew. Chem., Int. Ed., 2008, 47, 6384–6387 CrossRef CAS PubMed; (c) O. T. Summerscales, T. W. Myers and L. A. Berben, Organometallics, 2012, 31, 3463–3465 CrossRef CAS.
- A. Abragam and B. Bleaney, Electron paramagnetic resonance of transitionions, Oxford University Press, Oxford, England, 1970 Search PubMed.
- C. H. Booth, M. D. Walter, D. Kazhdan, Y.-J. Hu, W. W. Lukens, E. D. Bauer, L. Maron, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2009, 131, 6480–6491 CrossRef CAS PubMed.
- M. Schultz, J. M. Boncella, D. J. Berg, T. Don Tilley and R. A. Andersen, Organometallics, 2002, 21, 460–472 CrossRef CAS.
- I. L. Fedushkin, O. V. Maslova, E. V. Baranov and A. S. Shavyrin, Inorg. Chem., 2009, 48, 2355–2357 CrossRef CAS PubMed.
- (a) K. G. Alley, G. Poneti, P. S. D. Robinson, A. Nafady, B. Moubaraki, J. B. Aitken, S. C. Drew, C. Ritchie, B. F. Abrahams, R. K. Hocking, K. S. Murray, A. M. Bond, H. H. Harris, L. Sorace and C. Boskovic, J. Am. Chem. Soc., 2013, 135, 8304–8323 CrossRef CAS PubMed; (b) O. Drath, R. W. Gable, B. Moubaraki, K. S. Murray, G. Poneti, L. Sorace and C. Boskovic, Inorg. Chem., 2016, 55, 4141–4151 CrossRef CAS PubMed.
- (a) A. G. Avent, M. A. Edelman, M. F. Lappert and G. A. Lawless, J. Am. Chem. Soc., 1989, 111, 3423–3425 CrossRef CAS; (b) D. J. Schwartz and R. A. Andersen, Organometallics, 1995, 14, 4308–4318 CrossRef CAS.
- (a) G. Nocton, C. H. Booth, L. Maron and R. A. Andersen, Organometallics, 2013, 32, 1150–1158 CrossRef CAS; (b) G. Nocton, C. H. Booth, L. Maron and R. A. Andersen, Organometallics, 2013, 32, 5305–5312 CrossRef CAS; (c) C. H. Booth, D. Kazhdan, E. L. Werkema, M. D. Walter, W. W. Lukens, E. D. Bauer, Y.-J. Hu, L. Maron, O. Eisenstein, M. Head-Gordon and R. A. Andersen, J. Am. Chem. Soc., 2010, 132, 17537–17549 CrossRef CAS PubMed; (d) G. Nocton, C. H. Booth, L. Maron, L. Ricard and R. A. Andersen, Organometallics, 2014, 33, 6819–6829 CrossRef CAS; (e) G. Nocton, W. W. Lukens, C. H. Booth, S. S. Rozenel, S. A. Medling, L. Maron and R. A. Andersen, J. Am. Chem. Soc., 2014, 136, 8626–8641 CrossRef CAS PubMed; (f) M. D. Walter, D. J. Berg and R. A. Andersen, Organometallics, 2006, 25, 3228–3237 CrossRef CAS.
- (a) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed; (b) N. E. Schultz, Y. Zhao and D. G. Truhlar, J. Comput. Chem., 2008, 29, 185–189 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1552992–1552995. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt04299j |
| This journal is © The Royal Society of Chemistry 2018 |