
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anti-tumour active gold(I), palladium(II) and ruthenium(II) complexes with thio- and selenoureato ligands: a comparative study†
Anja
Molter
a,
Stefanie
Kathrein
b,
Brigitte
Kircher‡
bc and
Fabian
Mohr‡
 *a
*a
aFakultät für Mathematik und Naturwissenschaften, Anorganische Chemie, Bergische Universität Wuppertal, 42119 Wuppertal, Germany. E-mail: fmohr@uni-wuppertal.de
bInnsbruck Medical University, Department of Internal Medicine V (Hematology & Oncology), Immunobiology and Stem Cell Laboratory, 6020 Innsbruck, Austria
cTyrolean Cancer Research Institute, 6020 Innsbruck, Austria
First published on 12th March 2018
Abstract
We report the results of a comparative study of the biological activity of a series of gold(I), palladium(II) and ruthenium(II) complexes containing deprotonated thio- and selenoureato ligands. A library of compounds was prepared and characterised by spectroscopic methods and the solid-state structures of several derivatives were determined by single crystal X-ray diffraction. The in vitro activity of these compounds was evaluated in mammary and ovarian carcinoma, acute lymphatic and acute and chronic myeloid leukemia cell lines. At lower concentrations Ru- and Pd-containing compounds displayed stronger anti-cancer effects than the gold compounds. In all cases, the selenium derivatives proved to be more active than the corresponding sulfur compounds.
Introduction
Acylselenoureas and acylthioureas of the type ArC(O)NHC(E)NR2 (E = S, Se) have been known since the early 19th century1,2 and their metal complexes, particularly those with acylthiourea ligands, have been thoroughly investigated for a number of applications.3–6 For example, some acylthio- and acylselenoureas have been used for the extraction of gold from aqueous solutions.7 Gold(I) complexes, especially those with phosphine ligands, are known to display biological activity.8–14 A cytotoxicity study of a series of gold(I) triethylphosphine complexes [Au(X)PEt3] (X = Cl, Br, CN, SCN, or thiolato ligand) revealed that the sulfur containing complexes exhibited a higher activity compared to those containing halide ligands.10 We have observed similar results in several gold(I) phosphine or N-heterocyclic carbene complexes containing heterocyclic sulfur ligands.15,16 There are several reports describing studies of acylthiourea palladium complexes focusing on their anti-microbial,17 fungicidal,18 thermal19 or mesomorphic20 properties. Palladium(II) complexes with phenanthroline or its derivatives can intercalate with DNA thus exhibiting significant in vitro anti-tumour activity.8,14,21–23 The anticancer activity of palladium complexes has recently been subject of a detailed review.24 Apart from some homoleptic bis(selenoureato) palladium(II) complexes, there are only very few examples of heteroleptic Pd(II) derivatives containing monoanionic acylselenoureas.6 Ruthenium compounds, in particular organometallic arene ruthenium(II) species, are emerging as a new class of promising anti-tumour active substances.8,14,25–32 Ruthenium complexes with selenoureato ligands are unknown and only a few complexes with monoanionic thioureato ligands have been reported, although none have been examined for their biological properties.33–38 Given our interest in the study of biologically active metal complexes with chalcogen ligands,15,16,39–44 we wished to carry out a comparative study of a family of compounds containing metal centres with known anticancer activity [Au(PTA), Pd(1,10-Phen) and Ru(p-cymol)] in combination with both acylthio- and acylselenoureato ligands.Results and discussion
Synthesis and crystal structures
The new selenourea N,N-diethyl-N′-4-nitrobenzoylselenourea (denoted hereafter as HLSe) was prepared from KSeCN, 4-nitrobenzoyl chloride and Et2NH in CH2Cl2 in the presence of PEG-400 as phase-transfer catalyst following a literature procedure.45 Orange crystals of the pure material were obtained in about 35% yield after recrystallisation from EtOH. Alternatively, the compound can also be obtained in similar yield using the original method of Douglass.1 The known thiourea N,N-diethyl-N′-4-nitrobenzoylthiourea (denoted hereafter as HLS) was prepared analogously from KSCN, 4-nitrobenzoyl chloride and Et2NH in acetone. The identity and purity of HLSe was confirmed by 1H, 13C and 77Se NMR spectroscopy as well as by single crystal X-ray diffraction. The molecular structure of the compound is shown in Fig. 1. Overall, the bond distances and angles of HLSe (Table S1†) are similar to those reported for PhC(O)NHC(Se)NEt2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 46 with some minor differences, which are detailed in the following section. The C
46 with some minor differences, which are detailed in the following section. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CSe units in HLSe lie almost perpendicular to each other (dihedral angle Se–C–N–C = 90.2°), whilst in the Ph-derivative this angle is approximately 108°.46 Furthermore, the Ph-derivative forms dimers through intermolecular H-bonds between the NH group and the selenium atom in the solid-state, whereas HLSe forms an infinite chain through N–H⋯O hydrogen bonds [N(H)⋯O = 2.905(3) Å]. In HLS similar features can be observed: infinite chain formation through N–H⋯O hydrogen bonds [N(H)⋯O = 2.915(2) Å] and perpendicular CO and CS units (dihedral angle S–C–N–C = 91.1°).47
O and CSe units in HLSe lie almost perpendicular to each other (dihedral angle Se–C–N–C = 90.2°), whilst in the Ph-derivative this angle is approximately 108°.46 Furthermore, the Ph-derivative forms dimers through intermolecular H-bonds between the NH group and the selenium atom in the solid-state, whereas HLSe forms an infinite chain through N–H⋯O hydrogen bonds [N(H)⋯O = 2.905(3) Å]. In HLS similar features can be observed: infinite chain formation through N–H⋯O hydrogen bonds [N(H)⋯O = 2.915(2) Å] and perpendicular CO and CS units (dihedral angle S–C–N–C = 91.1°).47
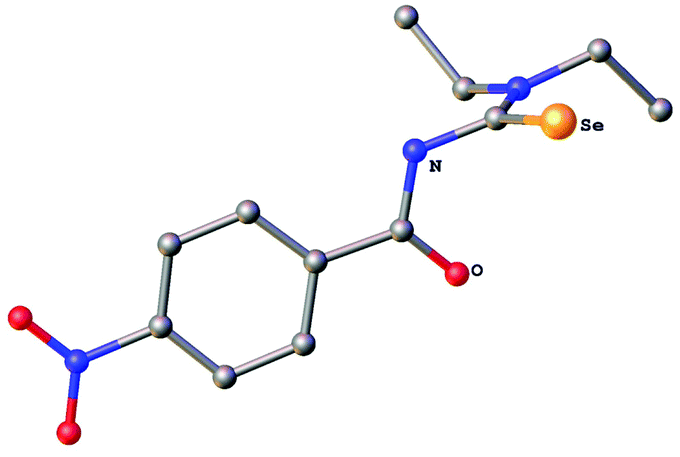 | ||
| Fig. 1 Molecular structure of N,N-diethyl-N′-4-nitrobenzoylselenourea HLSe. Ellipsoids show 30% probability levels. Hydrogen atoms have been omitted for clarity. | ||
The seleno- or thioureas HLSe and HLS react with [AuCl(PTA)] (PTA: 1,3,5-triaza-7-phosphaadamantane) in the presence of base to give neutral gold(I) PTA complexes containing the deprotonated seleno- or thioureato ligands (Scheme 1). Similarly, the reaction of HLSe and HLS with cis-[PdCl2(1,10-Phen)] in the presence of base affords the cationic Pd(II) species cis-[Pd{4-O2NC6H4C(O)NC(E)NEt2}(1,10-Phen)]+ (E = S, Se), which were isolated as their PF6 salts as shown in Scheme 1. The organoruthenium(II) complexes [Ru{4-O2NC6H4C(O)NC(E)NEt2)(p-cymol)(PPh3)]PF6 (E = S, Se) were prepared from the reaction of [RuCl2(p-cymol)]2 with the corresponding ligand in the presence of Ph3P, base and NH4PF6 (Scheme 1).
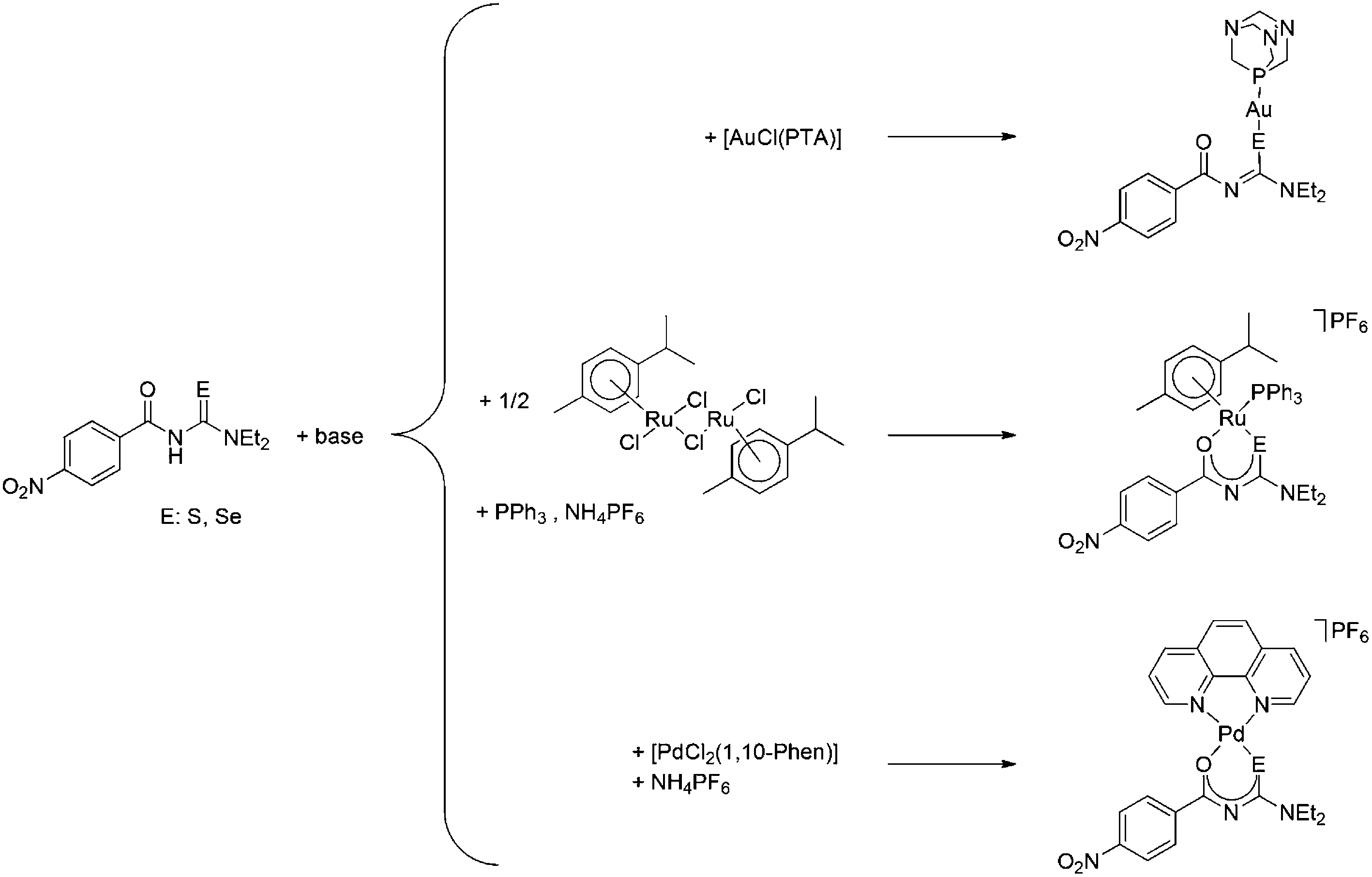 | ||
| Scheme 1 | ||
The compounds were characterised by 1H, 13C, 77Se and 31P NMR spectroscopy, electrospray mass spectrometry and several derivatives also by X-ray crystallography. In the gold(I) complexes the monoanionic thio- or selenoureato ligands coordinate only via the sulfur or selenium atom to the gold(I) centre. In case of the ruthenium and palladium species, bidentate coordination of both chalcogen atoms of the thio- and selenourea ligands is observed. In all the compounds reported here, the thio- or selenourea ligands are deprotonated, which is evident from the disappearance of the signal for the N–H proton in the 1H NMR spectra of the compounds. For the gold derivatives, a shift of the position of the singlet due to the PTA ligand in the 31P NMR spectra is observed upon coordination of the seleno- or thioureato ligands. This resonance is observed at −52.0 ppm in [AuCl(PTA)] and at −51.7 and −49.8 ppm in the thio- and selenoureato derivatives, respectively. In addition, the 13C NMR signals of the quaternary carbon atom bound to the sulfur or selenium atom are shifted to 176.8 (S) or 167.1 (Se) ppm to higher field upon coordination. Whilst the 77Se NMR signal for the unbound selenourea is found at 502 ppm, that of the gold selenoureato species is observed at 244 ppm and that of the ruthenium derivative is shifted upfield to −26 ppm. Unfortunately, the palladium species were not soluble enough to obtain any 77Se NMR spectra. The metal compounds were stable in solution for at least 48 h, as observed by NMR spectroscopy. Furthermore, we did not observe any ligand exchange reactions in dmso solutions. X-ray quality crystals of two ruthenium and palladium complexes were obtained and studied by X-ray diffraction. In the organoruthenium complexes (Fig. 2), the typical piano stool conformation about the metal centre is observed. The metal is thus bound to the arene ring (in η6 fashion), the chelating thio- or selenoureato ligand as well as to the phosphorus atom of the PPh3 ligand. The presence of four different atoms around the metal centre results in metal-centred chirality. The O–Ru–E angles (E = S, Se) are comparable to those reported for homoleptic, octahedral ruthenium(III) tris(chelate) complexes.34 The distances between the ruthenium atom and the centre of the arene rings (S 1.745 Å, Se 1.746 Å) are typical for such η6-arene ruthenium compounds.48 The palladium complexes (Fig. 3) show square-planar coordination with one bidentate chalcogenoureato and a 1,10-phenanthroline ligand bound to the metal. In both the ruthenium and the palladium compounds, the chalcogenoureato ligands are nearly planar due to electron delocalisation in the ligand backbone. A comparison of the two C–N distances as well as the C–O and C–Se bond lengths of the free and coordinated ligand, shows that upon coordination to the metal atom the NH–CSe unit is transformed into an NC–Se− moiety (the selenolato tautomer). The C–Se bond distance increases from 1.82 Å to 1.88 Å (Pd) or 1.90 Å (Ru) which is in agreement with literature data for ruthenium-μ-phenylselenido compounds.49 Due to the slightly smaller radius of the sulfur atom, the analogous bond distances in the sulfur complexes are correspondingly shorter. The C–S bond distance increases from 1.66 Å (HLS) to 1.73 Å in the complex. In contrast, the CO and C(O)N bond distances remain virtually unchanged upon coordination when compared with those of the free seleno- or thiourea, respectively. A table listing important bond distances and angles of the metal complexes and the thio- and selenourea ligands is included in the ESI.†
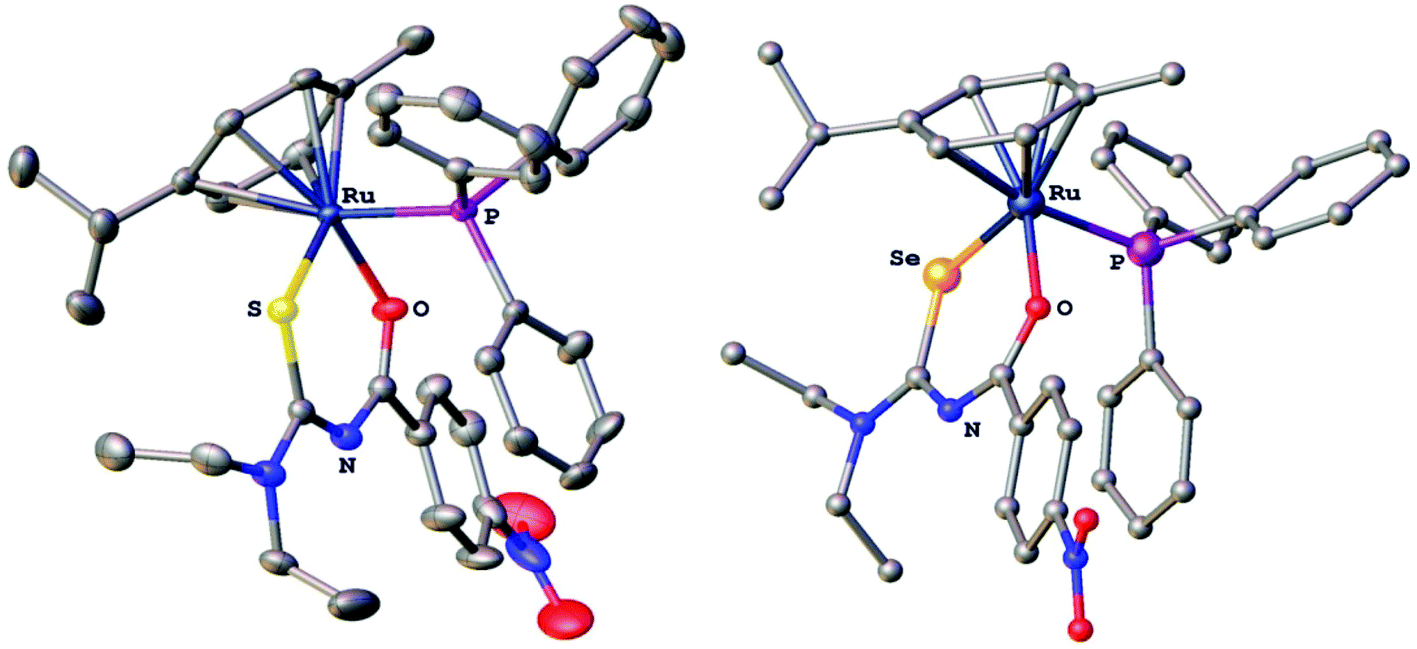 | ||
| Fig. 2 Molecular structures of [Ru{4-O2NC6H4C(O)NC(S)NEt2}(p-cym)(PPh3)]PF6 and [Ru{4-O2NC6H4C(O)NC(Se)NEt2}(p-cym)(PPh3)]PF6. Ellipsoids show 50% probability levels. Hydrogen atoms and the PF6− anions have been omitted for clarity. | ||
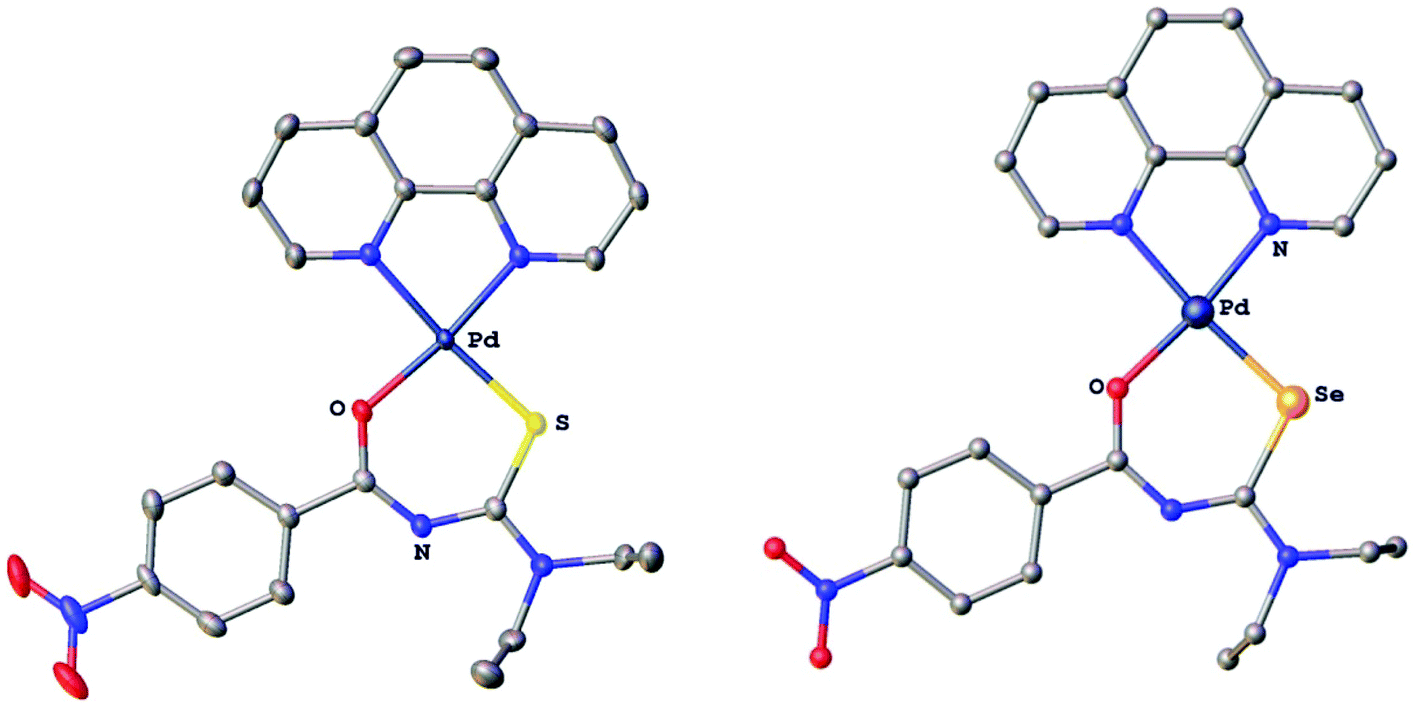 | ||
| Fig. 3 Molecular structures of cis-[Pd{4-O2NC6H4C(O)NC(S)NEt2}(1,10-phen)]PF6 and cis-[Pd{4-O2NC6H4C(O)NC(Se)NEt2}(1,10-phen)]PF6. Ellipsoids show 50% probability levels. Hydrogen atoms, the PF6− anions as well as the dmso of solvation have been omitted for clarity. | ||
Biological studies
The biological activity of HLS, HLSe as well as selected Au(I), Pd(II) and Ru(II) complexes including their chlorido-precursors (Chart 1) and cisplatin as further control was determined by their effect on proliferation, metabolic activity, programmed cell death and induction of oxidative stress of various tumor and leukemia cell lines.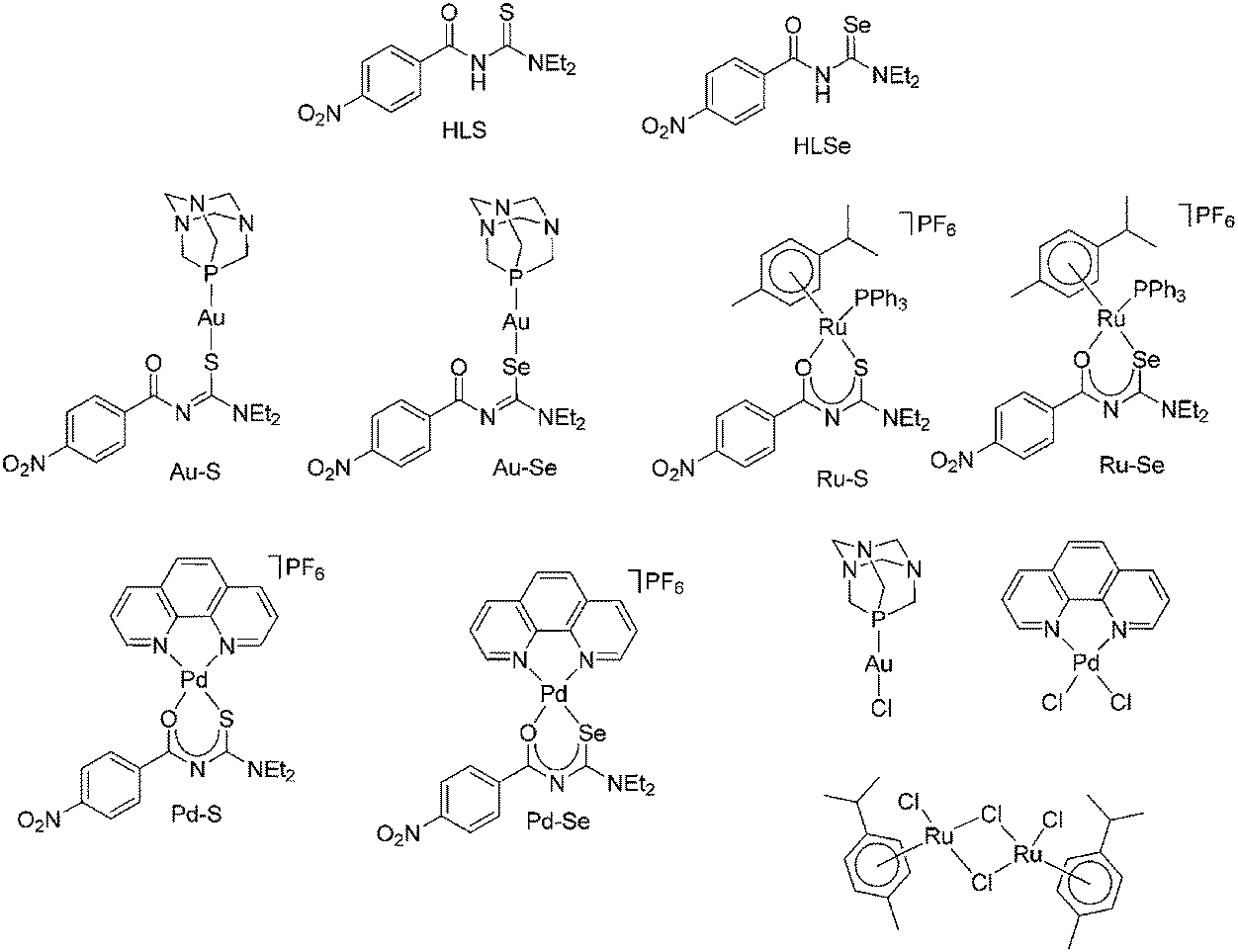 | ||
| Chart 1 Compounds selected for biological studies. | ||
All Au(I), Pd(II) and Ru(II) compounds dose-dependently reduced the proliferation of mammary and ovarian carcinoma, acute lymphatic as well as acute and chronic myeloid leukemia cell lines (ESI Fig. S1A–S1F†), whereby, in general, the selenium derivatives induced stronger effects than the sulfur compounds. For example, a concentration of 5 μM Pd–S significantly reduced the proliferation of SUP-B15 cells from 100% (without compound; data not shown) to 44.5 ± 8.4% and a concentration of 10 μM to 18.7 ± 3.1% (p < 0,05; Fig. 4) so that the IC50 was calculated as 4.9 ± 1.2 μM (Table 1). Interestingly, in the mammary carcinoma as well as the myeloid leukemia cell lines Pd–S was almost ineffective. In contrast, Pd–Se induced strong anti-proliferative effects and both Ru(II) compounds completely abolished the proliferation in all cell lines tested. Weaker anti-proliferative activity (again in all cell lines) was observed for the two Au(I) complexes. Cisplatin dose-dependently reduced the proliferation of the tumor cell lines, but did not show any activity on the leukemia cell lines. Whereas [RuCl2(p-cymol)]2 was completely ineffective in all cell lines, [AuCl(PTA)] and the thio- and selenoureas HLSe and HLS induced anti-proliferative effects at higher concentrations (25 μM and 50 μM, ESI Fig. S1A–S1F†). cis-[PdCl2(1,10-Phen)] dose-dependently reduced the proliferation of all cell lines almost as effective as Pd–Se which may be explained by the ability of cis-[PdCl2(1,10-Phen)] to intercalate with DNA.
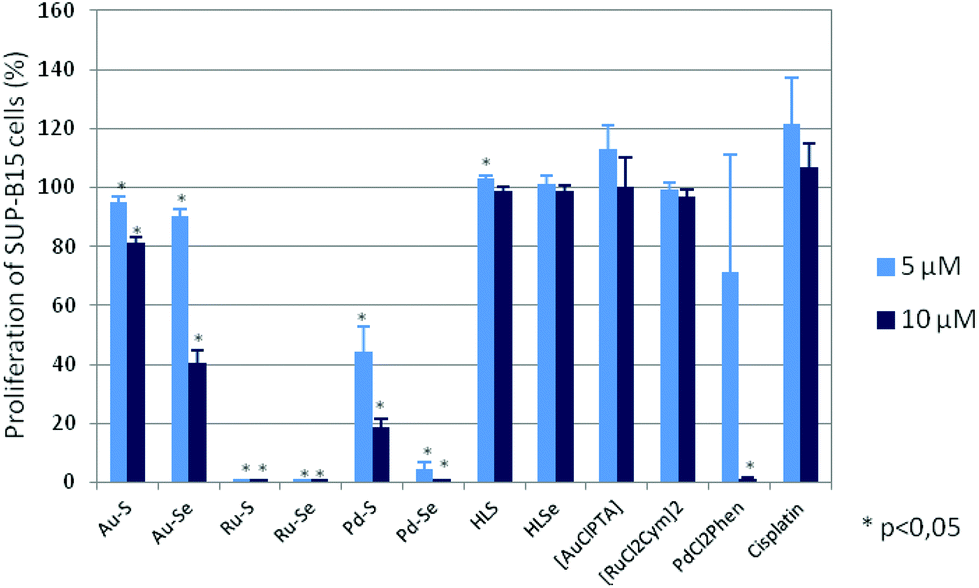 | ||
| Fig. 4 Proliferation of SUP-B15 cells after a three-day incubation with the Au(I), Ru(II), Pd(II) complexes, HLS, HLSe, the chlorido complexes [AuCl(PTA)], [RuCl2(p-cymol)]2, cis-[PdCl2(1,10-Phen)] as well as cisplatin. Proliferation in the absence of the compounds was set to 100%. Mean proliferation and standard error of four experiments is shown. Statistical significance was determined with the Wilcoxon Rank Sum Test (*p < 0.05 against the untreated control). | ||
| Compound | IC50 (μM) | |
|---|---|---|
| Proliferation | Metabolic activity | |
| IC50 values are calculated as the concentration at which the compounds induce 50% inhibition of proliferation or metabolic activity of SUP-B15 cells in comparison to untreated cells. The IC50 values are presented as means ± standard error of four experiments. | ||
| Au–S | 17.9 ± 3.2 | 18.6 ± 3.2 |
| Au–Se | 9.1 ± 0.5 | 10.4 ± 1.0 |
| Ru–S | 3.5 ± 0.2 | 3.7 ± 0.7 |
| Ru–Se | 2.6 ± 0.5 | 4.1 ± 0.2 |
| Pd–S | 4.9 ± 1.2 | 13.9 ± 1.9 |
| Pd–Se | 3.7 ± 0.4 | 4.4 ± 0.0 |
The anti-proliferative activity of the substances was also examined on A-2780 cells resistant to cisplatin and revealed that the Au(I), Pd(II) and Ru(II) compounds have the potential to overcome cisplatin resistance (Table 2).
| Compound | A-2780 | Compound | A-2780 | ||
|---|---|---|---|---|---|
| 5 μM | Cisplatin-sensitive | Cisplatin-resistant | 10 μM | Cisplatin-sensitive | Cisplatin-resistant |
| Au–S | 46.8 | 79.8 | Au–S | 37.0 | 28.1 |
| Au–Se | 11.7 | 12.2 | Au–Se | 0.1 | 3.2 |
| Ru–S | 0.1 | 0.6 | Ru–S | 0.0 | 0.2 |
| Ru–Se | 0.1 | 0.2 | Ru–Se | 0.0 | 0.1 |
| Pd–S | 1.0 | 1.5 | Pd–S | 0.1 | 0.2 |
| Pd–Se | 0.2 | 0.3 | Pd–Se | 0.0 | 0.1 |
| HLS | 80.2 | 92.4 | HLS | 74.4 | 111.6 |
| HLSe | 76.2 | 91.7 | HLSe | 66.5 | 108.7 |
| AuClPTA | 107.2 | 76.8 | AuClPTA | 80.9 | 88.6 |
| [Cl2Cym]2 | 84.7 | 97.1 | [Cl2Cym]2 | 97.1 | 83.8 |
| PdCl2Phen | 24.1 | 66.4 | PdCl2Phen | 14.3 | 54.1 |
| Cisplatin | 15.0 | 67.2 | Cisplatin | 9.1 | 64.9 |
To evaluate if the reduced cell growth was also accompanied by cell death, the metabolic activity of the cell lines after compound incubation was analysed. Metabolic activity is determined by the reduction of tetrazolium salts into formazan derivatives. Since this process requires functional mitochondria, the assay discriminates living and dead cells whose mitochondria are deactivated within a few minutes after cell death. As shown in Fig. 5 and ESI Fig. S2A–S2F,† the metabolic activity of the cell lines was also dose-dependently diminished after 72 hours of incubation with the test compounds and cis-[PdCl2(1,10-Phen)]. Again, the Ru(II) compounds and Pd–Se were the most active compounds with an IC50 of ∼4 μM (Table 1), whereas the chlorido precursors [AuCl(PTA)] and [RuCl2(p-cymol)]2, HLS, HLSe as well as cisplatin were almost ineffective.
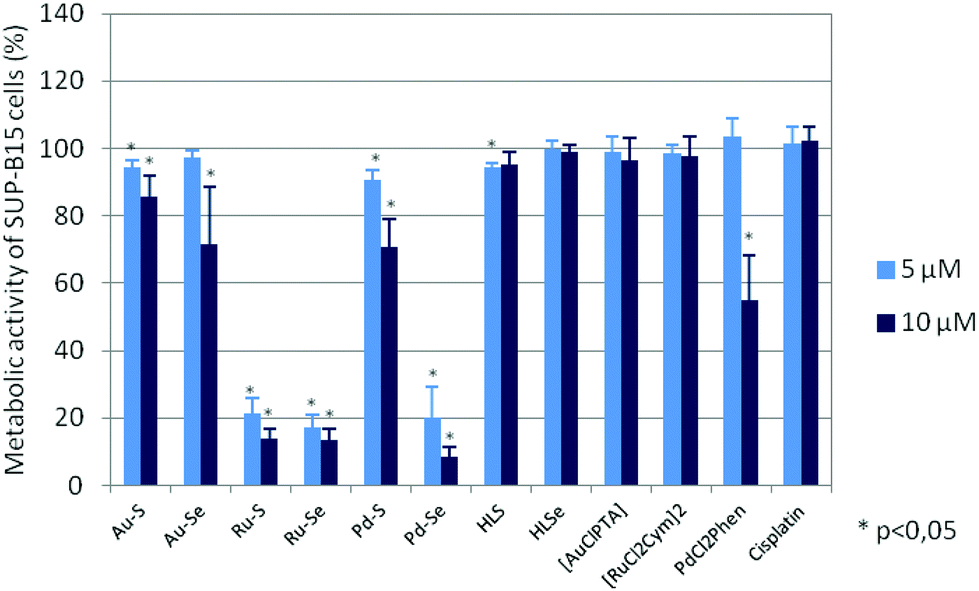 | ||
| Fig. 5 Metabolic activity of SUP-B15 cells after a three-day incubation with the Au(I), Ru(II), Pd(II) complexes, HLS, HLSe, [AuCl(PTA)], [RuCl2(p-cymol)]2, cis-[PdCl2(1,10-Phen)] as well as cisplatin. Metabolic activity in the absence of the compounds was set to 100%. Mean metabolic activity and standard error of four experiments is shown. Statistical significance was determined with the Wilcoxon Rank Sum Test (*p < 0.05 against the untreated control). | ||
To confirm the cytotoxicity of the compounds, their capacity to induce programmed cell-death was determined by ssDNA apoptosis ELISA, which is based on the selective denaturation of DNA in apoptotic cells by formamide and further detection of denatured DNA with a monoclonal antibody to single-stranded DNA (ssDNA). Compounds which reduced proliferation and metabolic activity also induced apoptosis of the leukemia and tumor cells, whereby the sensitivity of the cell lines to programmed cell-death induction varied (ESI Fig. S3A–S3E†). For example, apoptosis of the cell line SUP-B15 was 3.7 ± 0.8 times higher after addition of 5 μM Pd–Se and 5.2 ± 0.6 times higher when 10 μM Pd–Se was added to the cultures in comparison to cells without compound incubation (Fig. 6).
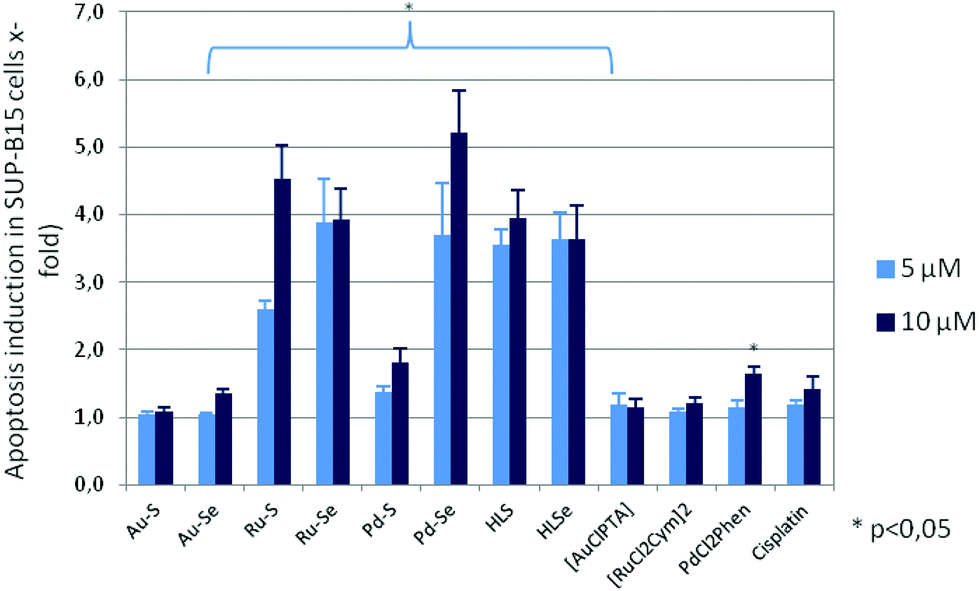 | ||
| Fig. 6 Induction of apoptosis in SUP-B15 cells after a three-day incubation with the Au(I), Ru(II), Pd(II) complexes, HLS, HLSe, [AuCl(PTA)], [RuCl2(p-cymol)]2, cis-[PdCl2(1,10-Phen)] as well as cisplatin. Mean apoptosis induction (x-fold) and standard error of four experiments is shown. Apoptosis rate in the absence of the respective compound was set to 1. Statistical significance was determined with the Wilcoxon Rank Sum Test (*p < 0.05 against the untreated control). | ||
A similar pro-apoptotic effect was observed for both Ru(II) compounds, whereas 10 μM Au–Se and 5 μM as well as 10 μM Pd–S caused a lower, but significant apoptosis rate. Consistent with the weak anti-proliferative and anti-metabolic activity, incubation with Au–S did not lead to programmed cell death in any of the cell lines. [AuCl(PTA)], [RuCl2(p-cymol)]2 and cisplatin were also ineffective in inducing apoptosis. Interestingly, despite no activity on proliferation and metabolism, HLS and HLSe significantly induced apoptosis in all leukemic cell lines, but not in the carcinoma cell lines (Fig. 6 and ESI Fig. S3A–S3E†). A similar effect was observed for cis-[PdCl2(1,10-Phen)], which decreased proliferation and metabolic activity of the cell lines, however, without inducing their cell death. These rather contradictory results are currently under investigation.
The pro-apoptotic effect of Ru–Se (Fig. 7A) and the lacking effect of [RuCl2(p-cymol)]2 (Fig. 7B) can be readily detected by morphological changes of the cisplatin-resistant A-2780 cells.
 | ||
| Fig. 7 Morphologic changes after a one-hour and 24-hours incubation, respectively, with 10 μM Ru–Se (A) and 10 μM [RuCl2(p-cymol)]2 (B). One representative experiment is shown. | ||
One mechanism involved in the induction of apoptosis is oxidative stress triggered by the formation of reactive oxygen species (ROS) due to deregulated mitochondrial activity. It has been shown for a variety of gold,50–52 ruthenium31,32,53–55 and palladium56 complexes that their anti-tumour activity is mediated by an enhanced ROS production. Therefore, ROS levels were measured after a two-hour incubation of SUP-B15 cells with 10 μM of the respective compound. As shown in Fig. 8, both Ru(II) compounds and Pd–Se significantly induced oxidative stress in this cell line, in agreement with an increase of programmed cell death. Interestingly, the pro-apoptotic effect of HLS and HLSe cannot be explained by an increase in ROS formation. Neither the chlorido compounds nor cisplatin did induce oxidative stress of SUP-B15 cells.
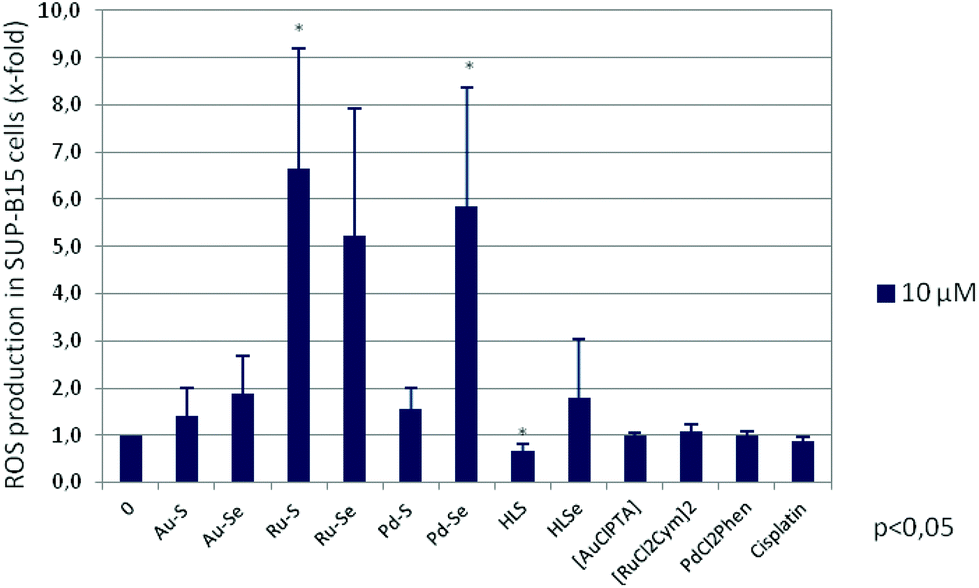 | ||
| Fig. 8 Induction of oxidative stress in SUP-B15 cells after a two-hours incubation with the Au(I), Ru(II), Pd(II) complexes, HLS, HLSe, [AuCl(PTA)], [RuCl2(p-cymol)]2, cis-[PdCl2(1,10-Phen)] as well as cisplatin. Mean ROS production (x-fold) with standard error of six (for the chloro precursors and cisplatin four) experiments is shown. Statistical significance was determined with the Wilcoxon Rank Sum Test (*p < 0.05 against the untreated control). | ||
Conclusions
In summary, gold(I), palladium(II) and ruthenium(II) compounds containing anionic nitro-substituted acylseleno- or acylthioureas display strong anti-tumor and anti-leukemia effects. The ligands HLS, HLSe as well as the chlorido complexes [AuCl(PTA)] and [RuCl2(p-cymol)]2 induced only weak effects or only at the highest concentrations tested, whereas cis-[PdCl2(1,10-Phen)] reduced the proliferation and the metabolic activity almost as effective as the test compounds. The mechanisms of action include inhibition of proliferation and metabolic activity as well as induction of apoptosis and oxidative stress by ROS formation. The strongest effects were observed for the Ru(II) compounds followed by the Pd(II) complexes whereby the selenium-containing compounds displayed stronger biological activity than the sulfur derivatives. The Au(I) complexes were only effective at higher concentrations.Materials and methods
Chemistry
Unless otherwise stated, all manipulations were carried out without taking precautions to exclude air and moisture. All chemicals and solvents (HPLC quality) were sourced commercially and used as received. [AuCl(PTA)] was prepared from the reaction of [AuCl(tht)]57 with the appropriate amount of the phosphine. The other metal precursors cis-[PdCl2(1,10-phen)],58 [Ru(p-cymol)Cl2]259 as well as N,N-diethyl-N′-4-nitrobenzoylthiourea (HLS)60 were prepared by standard procedures described in the literature. The purity of all compounds was confirmed to be >95% by elemental analysis or high-resolution mass spectrometry.
Instrumentation
Syntheses
1H NMR (400 MHz, CDCl3): δ = 1.33 (t, J = 7.1 Hz, 3H, NCH2CH3), 1.41 (t, J = 6.3 Hz, 3H, NCH2CH3), 3.60 (br. s, 2H, NCH2CH3), 4.14 (br. s, 2H, NCH2CH3), 8.03 (d, J = 8.2 Hz, 2H, H-2), 8.32 (d, J = 8.2 Hz, 2H, H-3), 8.65 (br. s, 1H, NH) ppm. 13C{1H} NMR (101 MHz, CDCl3): δ = 11.6 (NCH2CH3), 12.9 (NCH2CH3), 48.6 (NCH2CH3), 51.4 (NCH2CH3), 124.1 (C-3), 129.1 (C-2), 137.9 (C-1), 150.4 (C-4), 160.4 (CO), 179.8 (CSe) ppm. 77Se NMR (76 MHz, CDCl3): δ = 502 ppm. IR (KBr disk) ν: 1347 (s, NO2), 1526 (s, NO2), 1649 (s, CO amido urea), 2933 (w, Csp3–H), 2975 (w, Csp3–H), 3055 (w, Csp2–H), 3289 (m, N–H) cm−1. Elemental analysis calcd (%) for C12H15N3O3Se (328.23 g mol−1): C 43.91, H 4.61, N 12.80; found C 43.22, H 4.49, N 13.02.
Preparation of the gold(I) complexes
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) NC(Se)NEt2}(PTA)].
HLSe (92.7 mg, 0.28 mmol) and NaOMe (16.8 mg, 0.31 mmol) were dissolved in methanol (10 mL) under dinitrogen. The clear orange solution was stirred for 20 min at room temperature, then [AuCl(PTA)] (100 mg, 0.26 mmol) was added to give a yellow suspension. After 18 h at room temperature, the yellow solution with little black precipitate was evaporated to dryness. The residue was taken up in dichloromethane, passed through Celite and concentrated in vacuum. Addition of diethyl ether afforded a yellow solid, which was isolated by filtration and dried in air. Yield: 0.150 g (86%). 1H NMR (400 MHz, CDCl3): δ = 1.28 (br. s, 6H, NCH2CH3), 3.66 (br. s, 2H, NCH2CH3), 3.81 (br. s, 2H, NCH2CH3), 4.02 (s, 6H, NCH2N), 4.42 (q, 2JH–H = 13.2 Hz, 6H, PCH2N), 8.21 (d, J = 8.9 Hz, 2H, H-3), 8.28 (d, J = 8.9 Hz, 2H, H-2) ppm. 31P{H} NMR (162 MHz, CDCl3): δ = −49.8 ppm. 13C{H} NMR (101 MHz, CDCl3): δ = 13.0 (NCH2CH3), 13.4 (NCH2CH3), 46.0 (NCH2CH3), 48.4 (NCH2CH3), 52.3 (d, 1JP–C = 18.7 Hz, PCH2N), 72.9 (d, 3JP–C = 7.7 Hz, NCH2N), 123.1 (C-2), 130.2 (C-3), 143.68 (C-4), 149.2 (C-1), 163.1 (CO), 167.1 (C–Se) ppm. 77Se NMR (76 MHz, CDCl3, Me2Se): 244 ppm. IR (KBr disk) ν: 1342 (s, NO2), 1520 (s, NO2), 2932 (m, Csp3–H), 2972 (m, Csp3–H), 3058 (w, Csp2–H) cm−1. Elemental analysis calcd (%) for C18H26AuN6O3PSe (681.34 g mol−1): C 31.73, H 3.85, N 12.33; found: C 31.64, H 4.16, N 11.83.
NC(S)NEt2}(PTA)].
This was prepared as above using HLS (79.4 mg, 0.28 mmol), NaOMe (16.8 mg, 0.31 mmol) and [AuCl(PTA)] (100 mg, 0.26 mmol). The product was obtained as yellow solid in 83% yield (0.135 g). 1H NMR (400 MHz, CDCl3): δ = 1.26 (br. s, 6H, NCH2CH3), 3.63 (br. s, 2H, NCH2CH3), 3.80 (br. s, 2H, NCH2CH3), 4.02 (s, 6H, NCH2N), 4.40 (AB-q, 2JH–H = 13.2 Hz, 6H, PCH2N), 8.24 (4H, H-2, H-3) ppm. 31P{1H} NMR (162 MHz, CDCl3): δ = −51.7 ppm. 13C{1H} NMR (101 MHz, CDCl3): δ = 12.9 (NCH2CH3), 46.1 (NCH2CH3), 46.5 (NCH2CH3), 51.9 (d, 1JP–C = 20.0 Hz, PCH2N), 72.8 (d, 3JP–C = 7.9 Hz, NCH2N), 123.0 (C-2), 129.9 (C-3), 143.8 (C-1), 149.0 (C-4), 166.0 (CO), 176.8 (C–S) ppm. IR (KBr disk) ν: 1343 (s, NO2), 1518 (s, NO2), 1567 (s, CO), 2933 (w, Csp3–H), 2970 (w, Csp3–H), 3067 (w, Csp2–H) cm−1. Elemental analysis calcd (%) for C18H26AuN6O3PS (634.44 g mol−1): C 34.08, H 4.13, N 13.25, S 4.86; found: C 33.75, H 4.44, N 12.77, S 4.86.
NC(Se)NEt2}(PTA)].
HLSe (92.7 mg, 0.28 mmol) and NaOMe (16.8 mg, 0.31 mmol) were dissolved in methanol (10 mL) under dinitrogen. The clear orange solution was stirred for 20 min at room temperature, then [AuCl(PTA)] (100 mg, 0.26 mmol) was added to give a yellow suspension. After 18 h at room temperature, the yellow solution with little black precipitate was evaporated to dryness. The residue was taken up in dichloromethane, passed through Celite and concentrated in vacuum. Addition of diethyl ether afforded a yellow solid, which was isolated by filtration and dried in air. Yield: 0.150 g (86%). 1H NMR (400 MHz, CDCl3): δ = 1.28 (br. s, 6H, NCH2CH3), 3.66 (br. s, 2H, NCH2CH3), 3.81 (br. s, 2H, NCH2CH3), 4.02 (s, 6H, NCH2N), 4.42 (q, 2JH–H = 13.2 Hz, 6H, PCH2N), 8.21 (d, J = 8.9 Hz, 2H, H-3), 8.28 (d, J = 8.9 Hz, 2H, H-2) ppm. 31P{H} NMR (162 MHz, CDCl3): δ = −49.8 ppm. 13C{H} NMR (101 MHz, CDCl3): δ = 13.0 (NCH2CH3), 13.4 (NCH2CH3), 46.0 (NCH2CH3), 48.4 (NCH2CH3), 52.3 (d, 1JP–C = 18.7 Hz, PCH2N), 72.9 (d, 3JP–C = 7.7 Hz, NCH2N), 123.1 (C-2), 130.2 (C-3), 143.68 (C-4), 149.2 (C-1), 163.1 (CO), 167.1 (C–Se) ppm. 77Se NMR (76 MHz, CDCl3, Me2Se): 244 ppm. IR (KBr disk) ν: 1342 (s, NO2), 1520 (s, NO2), 2932 (m, Csp3–H), 2972 (m, Csp3–H), 3058 (w, Csp2–H) cm−1. Elemental analysis calcd (%) for C18H26AuN6O3PSe (681.34 g mol−1): C 31.73, H 3.85, N 12.33; found: C 31.64, H 4.16, N 11.83.
NC(S)NEt2}(PTA)].
This was prepared as above using HLS (79.4 mg, 0.28 mmol), NaOMe (16.8 mg, 0.31 mmol) and [AuCl(PTA)] (100 mg, 0.26 mmol). The product was obtained as yellow solid in 83% yield (0.135 g). 1H NMR (400 MHz, CDCl3): δ = 1.26 (br. s, 6H, NCH2CH3), 3.63 (br. s, 2H, NCH2CH3), 3.80 (br. s, 2H, NCH2CH3), 4.02 (s, 6H, NCH2N), 4.40 (AB-q, 2JH–H = 13.2 Hz, 6H, PCH2N), 8.24 (4H, H-2, H-3) ppm. 31P{1H} NMR (162 MHz, CDCl3): δ = −51.7 ppm. 13C{1H} NMR (101 MHz, CDCl3): δ = 12.9 (NCH2CH3), 46.1 (NCH2CH3), 46.5 (NCH2CH3), 51.9 (d, 1JP–C = 20.0 Hz, PCH2N), 72.8 (d, 3JP–C = 7.9 Hz, NCH2N), 123.0 (C-2), 129.9 (C-3), 143.8 (C-1), 149.0 (C-4), 166.0 (CO), 176.8 (C–S) ppm. IR (KBr disk) ν: 1343 (s, NO2), 1518 (s, NO2), 1567 (s, CO), 2933 (w, Csp3–H), 2970 (w, Csp3–H), 3067 (w, Csp2–H) cm−1. Elemental analysis calcd (%) for C18H26AuN6O3PS (634.44 g mol−1): C 34.08, H 4.13, N 13.25, S 4.86; found: C 33.75, H 4.44, N 12.77, S 4.86.
Preparation of the palladium(II) complexes
1H NMR (400 MHz, DMSO-d6, 32 °C): δ = 1.28 (t, J = 7.1 Hz, 3H, NCH2CH3), 1.42 (t, J = 7.2 Hz, 3H, NCH2CH3), 3.94 (q, J = 7.1 Hz, 2H, NCH2CH3), 3.97 (q, J = 7.2 Hz, 2H, NCH2CH3), 7.98 (dd, J = 8.1, 5.3 Hz, 1H, H3- or H8-phen), 8.24 (d, J = 7.9 Hz, 1H, H3- or H8-phen), 8.30 (m, 4H, H-2, H-3), 8.62 (dd, J = 5.3, 0.9 Hz, 1H, H2- or H9-phen), 8.91 (dd, J = 8.1, 0.6 Hz, 1H, H4- or H7-phen), 8.97 (m, 2H, H4- or H7-phen) ppm. 31P{1H} NMR (243 MHz, DMSO-d6, 25 °C): δ = −144.2 (sept, J = 711.2 Hz, PF6) ppm. HR-ESI-MS: m/z (obs., calc.) = 613.9920, 613.9923 [M]+. X-ray quality crystals were obtained by slow diffusion of diethyl ether into a saturated DMSO solution of the complex.
Preparation of the ruthenium complexes
Biological activity
:1000 with cell culture medium. Dmso in the appropriate concentration did not induce any effect on the cell lines.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
FM gratefully acknowledges a visiting scientist award from the Spanish CEI TRIANGULAR-E3los horizontes del hombre program. The authors wish to thank Petra Schumacher and Brigitte Floegel for excellent technical assistance.References
- I. B. Douglass, J. Am. Chem. Soc., 1937, 59, 740–742 CrossRef CAS.
- I. B. Douglass and F. B. Dains, J. Am. Chem. Soc., 1934, 56, 719–721 CrossRef CAS.
- J. Stach, R. Kirmse, A. Heinrich, W. Dietzsch, J. Hartung and L. Beyer, Z. Chem., 1983, 23, 453–454 CrossRef CAS.
- M. Schuster and K. H. König, Fresenius’ Z. Anal. Chem., 1988, 331, 383–386 CrossRef CAS.
- J. Akhtar, J. C. Bruce, M. A. Malik, K. R. Koch, M. Afzaal and P. O'Brien, Mater. Res. Soc. Symp. Proc., 2009, 1148E, 1148 Search PubMed.
- F. Fuge, C. Lehmann and F. Mohr, J. Organomet. Chem., 2009, 694, 2395–2401 CrossRef CAS.
- M. Petrich, J. L. Cortina, J. Hartung, M. Aguilar, A. Sastre, L. Beyer and K. Gloe, Solvent Extr. Ion Exch., 1993, 11, 51–66 CrossRef CAS.
- M. Gielen and E. R. T. Tiekink, Metallotherapeutic Drugs & Metal-based Diagnostic Agents, John Wiley & Sons, Chichester, 2005 Search PubMed.
- S. J. Berners Price, C. K. Mirabelli, R. K. Johnson, M. R. Mattern, F. L. McCabe, L. F. Faucette, C. Sung, P. J. Sadler and S. T. Crooke, Cancer Res., 1986, 46, 5486–5493 CAS.
- V. Gandin, A. P. Fernandes, M. P. Rigobello, B. Dani, F. Sorrentino, F. Tisato, M. Bjornstedt, A. Bindoli, A. Sturaro, R. Rella and C. Marzano, Biochem. Pharmacol., 2010, 79, 90–101 CrossRef CAS PubMed.
- E. R. T. Tiekink, Crit. Rev. Oncol. Hemat., 2002, 42, 225–248 CrossRef PubMed.
- E. R. T. Tiekink, Gold Bull., 2003, 36, 117–124 CrossRef CAS.
- Gold chemistry. Applications and future directions in the life sciences, ed. F. Mohr, Wiley-VCH, Weinheim, 2009 Search PubMed.
- S. Medici, M. Peana, V. M. Nurchi, J. I. Lachowicz, G. Crisponi and M. A. Zoroddu, Coord. Chem. Rev., 2015, 284, 329–350 CrossRef CAS.
- S. Miranda, E. Vergara, F. Mohr, D. de Vos, E. Cerrada, A. Mendía and M. Laguna, Inorg. Chem., 2008, 47, 5641–5648 CrossRef CAS PubMed.
- E. Schuh, C. Pflüger, A. Citta, A. Folda, M. P. Rigobello, A. Bindoli, A. Casini and F. Mohr, J. Med. Chem., 2012, 55, 5518–5528 CrossRef CAS PubMed.
- G. Binzet, H. Arslan, U. Flörke, N. Külcü and N. Duran, J. Coord. Chem., 2006, 59, 1395–1406 CrossRef CAS.
- Y. Nural, R. Kilincarslan, H. A. Dondas, B. Cetinkaya, M. S. Serin, R. Grigg, T. Ince and C. Kilner, Polyhedron, 2009, 28, 2847–2854 CrossRef CAS.
- A. C. Tenchiu, M. Iliş, F. Dumitraşcu, A. C. Whitwood and V. Cîrcu, Polyhedron, 2008, 27, 3537–3544 CrossRef CAS.
- V. Circu, D. Manaila-Maximean, C. Rosu, M. Ilis, Y. Molard and F. Dumitrascu, Liq. Cryst., 2009, 36, 123–132 CrossRef CAS.
- M. Cusumano, M. L. Di Pietro and A. Giannetto, Inorg. Chem., 2006, 45, 230–235 CrossRef CAS PubMed.
- M. Cusumano, M. L. Di Pietro, A. Giannetto and P. A. Vainiglia, J. Inorg. Biochem., 2005, 99, 560–565 CrossRef CAS PubMed.
- M. Cusumano, M. L. Di Pietro, A. Giannetto and P. A. Vainiglia, Eur. J. Inorg. Chem., 2005, 278–284 CrossRef CAS.
- M. N. Alam and F. Huq, Coord. Chem. Rev., 2016, 316, 36–67 CrossRef CAS.
- C. S. Allardyce, P. J. Dyson, D. J. Ellis and S. L. Heath, Chem. Commun., 2001, 1396–1397 RSC.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed.
- C. A. Vock, W. H. Ang, C. Scolaro, A. D. Phillips, L. Lagopoulos, L. Juillerat-Jeanneret, G. Sava, R. Scopelliti and P. J. Dyson, J. Med. Chem., 2007, 50, 2166–2175 CrossRef CAS PubMed.
- A. Dorcier, W. H. Ang, S. Bolaño, L. Gonsalvi, L. Juillerat-Jeannerat, G. Laurenczy, M. Peruzzini, A. D. Phillips, F. Zanobini and P. J. Dyson, Organometallics, 2006, 25, 4090–4096 CrossRef CAS.
- A. K. Renfrew, A. D. Phillips, A. E. Egger, C. G. Hartinger, S. S. Bosquain, A. A. Nazarov, B. K. Keppler, L. Gonsalvi, M. Peruzzini and P. J. Dyson, Organometallics, 2009, 28, 1165–1172 CrossRef CAS.
- R. S. Correa, K. M. de Oliveira, F. G. Delolo, A. Alvarez, R. Mocelo, A. M. Plutin, M. R. Cominetti, E. E. Castellano and A. A. Batista, J. Inorg. Biochem., 2015, 150, 63–71 CrossRef CAS PubMed.
- B. Tang, D. Wan, S. H. Lai, H. H. Yang, C. Zhang, X. Z. Wang, C. C. Zeng and Y. J. Liu, J. Inorg. Biochem., 2017, 173, 93–104 CrossRef CAS PubMed.
- L. Chen, G. Li, F. Peng, X. Jie, G. Dongye, K. Cai, R. Feng, B. Li, Q. Zeng, K. Lun, J. Chen and B. Xu, Oncotarget, 2016, 7, 80716–80734 Search PubMed.
- G. Roebisch, E. Ludwig and W. Bansse, Z. Anorg. Allg. Chem., 1982, 493, 26–32 CrossRef CAS.
- J. Sieler, R. Richter, E. Hoyer, L. Beyer, O. Lindqvist and L. Andersen, Z. Anorg. Allg. Chem., 1990, 580, 167–174 CrossRef CAS.
- K. H. König, M. Schuster, G. Schneeweis and B. Steinbrech, Fresenius’ Z. Anal. Chem., 1984, 319, 66–69 CrossRef.
- S. Pisiewicz, J. Rust, C. W. Lehmann and F. Mohr, Polyhedron, 2010, 29, 1968–1972 CrossRef CAS.
- M. Schuster and K. H. Koenig, Fresenius’ Z. Anal. Chem., 1988, 331, 383–386 CrossRef CAS.
- N. Gunasekaran and R. Karvembu, Inorg. Chem. Commun., 2010, 13, 952–955 CrossRef CAS.
- E. Guerrero, S. Miranda, S. Luttenberg, N. Frohlich, J. M. Koenen, F. Mohr, E. Cerrada, M. Laguna and A. Mendia, Inorg. Chem., 2013, 52, 6635–6647 CrossRef CAS PubMed.
- D. Dolfen, K. Schottler, V. Seied-Mojtaba, M. A. Jakupec, B. K. Keppler, E. R. T. Tiekink and F. Mohr, J. Inorg. Biochem., 2008, 102, 2067–2071 CrossRef CAS PubMed.
- P. Bippus, M. Skocic, M. A. Jakupec, B. K. Keppler and F. Mohr, J. Inorg. Biochem., 2011, 105, 462–466 CrossRef CAS PubMed.
- A. Molter, J. Rust, C. W. Lehmann, G. Deepa, P. Chiba and F. Mohr, Dalton Trans., 2011, 40, 9810–9820 RSC.
- E. Schuh, S. M. Valiahdi, M. A. Jakupec, B. K. Keppler, P. Chiba and F. Mohr, Dalton Trans., 2009, 10841–10845 RSC.
- R. Rubbiani, E. Schuh, A. Meyer, J. Lemke, J. Wimberg, N. Metzler-Nolte, F. Meyer, F. Mohr and I. Ott, MedChemComm, 2013, 4, 942–948 RSC.
- T. B. Wei, H. Wang, Q. Lin and Y. M. Zhang, Chin. J. Org. Chem., 2005, 25, 1565–1569 CAS.
- J. C. Bruce, N. Revaprasadu and K. R. Koch, New J. Chem., 2007, 31, 1647–1653 RSC.
- S. Saeed, N. Rashid, J. P. Jasinski, R. J. Butcher and H. Rizwan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, O2589–U2710 CAS.
- M. R. J. Elsegood, M. B. Smith and N. M. Sanchez-Ballester, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, M2838–M2840 CAS.
- P. L. Andreu, J. A. Cabeza, D. Miguel, V. Riera, M. A. Villa and S. Garciagranda, J. Chem. Soc., Dalton Trans., 1991, 533–536 RSC.
- J. F. Arambula, R. McCall, K. J. Sidoran, D. Magda, N. A. Mitchell, C. W. Bielawski, V. M. Lynch, J. L. Sessler and K. Arumugam, Chem. Sci., 2016, 7, 1245–1256 RSC.
- C. Sánchez-de-Diego, I. Mármol, R. Pérez, S. Gascón, M. J. Rodriguez-Yoldi and E. Cerrada, J. Inorg. Biochem., 2017, 166, 108–121 CrossRef PubMed.
- M. Altaf, M. Monim-Ul-Mehboob, A. N. Kawde, G. Corona, R. Larcher, M. Ogasawara, N. Casagrande, M. Celegato, C. Borghese, Z. H. Siddik, D. Aldinucci and A. A. Isab, Oncotarget, 2017, 8, 490–505 CrossRef PubMed.
- J. Yellol, S. A. Pérez, A. Buceta, G. Yellol, A. Donaire, P. Szumlas, P. J. Bednarski, G. Makhloufi, C. Janiak, A. Espinosa and J. Ruiz, J. Med. Chem., 2015, 58, 7310–7327 CrossRef CAS PubMed.
- L. S. Flocke, R. Trondl, M. A. Jakupec and B. K. Keppler, Invest. New Drugs, 2016, 34, 261–268 CrossRef CAS PubMed.
- C. C. Zeng, S. H. Lai, J. H. Yao, C. Zhang, H. Yin, W. Li, B. J. Han and Y. J. Liu, Eur. J. Med. Chem., 2016, 122, 118–126 CrossRef CAS PubMed.
- P. Banerjee, P. Majumder, S. Halder, M. G. Drew, S. Bhattacharya and S. Mazumder, Free Radical Res., 2015, 49, 253–268 CrossRef CAS PubMed.
- R. Usón, A. Laguna and M. Laguna, Inorg. Synth., 1989, 26, 85–91 Search PubMed.
- F. R. Hartley, Organomet. Chem. Rev., Sect. A, 1970, 6, 119–137 CAS.
- M. A. Bennett, T. N. Huang, T. W. Matheson and A. K. Smith, Inorg. Synth., 1982, 21, 74–78 CAS.
- W. Hernandez, E. Spodine, A. Vega, R. Richter, J. Griebel, R. Kirmse, U. Schroder and L. Beyer, Z. Anorg. Allg. Chem., 2004, 630, 1381–1386 CrossRef.
- CrysalisPro, Rigaku Oxford Diffraction, Oxford, UK, 2016 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables with crystallographic and refinement details, bond distances and angles as well as further information on the biological studies. CCDC 1584125–1584129. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt04180b |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |