
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe atomic level mechanism of white phosphorous demolition by di-iodine†
Carlo
Mealli
 ,
Andrea
Ienco
*,
Maurizio
Peruzzini
and
Gabriele
Manca
*
,
Andrea
Ienco
*,
Maurizio
Peruzzini
and
Gabriele
Manca
*
Istituto di Chimica dei Composti Organometallici – Consiglio Nazionale delle Ricerche (CNR-ICCOM), Via Madonna del Piano 10, 50019, Sesto Fiorentino, FI, Italy. E-mail: gabriele.manca@iccom.cnr.it
First published on 8th December 2017
Abstract
A detailed mechanism of the I2-induced transformation of white phosphorus into PI3 emerges from a DFT analysis. This multi-step process implies that at any stage one P–P and two I–I bonds cleavages, associated with the formation of two P–I bonds plus an in situ generated brand new I2 molecule. Significant electron transfer between the atoms is observed at any step, but the reactions are better defined as concerted rather than redox. Along the steepest descent to the product, no significant barrier is encountered except for the very first P4 activation, which costs +14.6 kcal mol−1. At the atomic level, one first I2 molecule, a typical mild oxidant, is first involved in a linear halogen bonding interaction (XB) with one P donor, while its terminal I atom is engaged in an additional XB adduct with a second I2. Significant electron transfer through the combined diatomics allows the external I atom of the dangling I3 grouping to convey electrons into the σ* level of one P–P bond with its consequent cleavage. This implies at some point the appearance of a six-membered ring, which alternatively switches its bonding and no-bonding interactions. The final transformation of the P2I4 diphosphine into two PI3 phosphines is enlightening also for the specific role of the I substituents. In fact, it is proved that an organo-diphosphine analogue hardly undergoes the separation of two phosphines, as reported in the literature. This is attributable to the particularly high donor power of the carbo-substituted P atoms, which prevents the concertedness of the reaction but favors charge separation in an unreactive ion pair.
Introduction
The interconversion between various allotropes of elemental phosphorus depends on several factors, in particular temperature and pressure but also on the involvement of other chemical species.1 Our group has devoted much attention towards the molecular white phosphorus, P4, and its behavior toward transition-metal fragments from both the experimental and theoretical viewpoints.2 Such a know-how is also deemed important for interpreting the behavior of other P allotropes such as that of the polymeric red3 and black4 phosphorus. In particular, our interest is being presently attracted by the latter, as a precursor of the 2D material, phosphorene, which is obtainable upon exfoliation of the black phosphorus.5 As a matter of fact, we are currently involved in a project6 aimed at exploring the functionalization of the rugged phosphorene sheets with organic and inorganic species. As in P4, all the phosphorene sp3 P-atoms are pyramidally connected to the other three equivalent ones, hence a good understanding of the white phosphorus reactivity may offer an interpretational key for studying the behavior of the other allotropes. For instance, it has been reported that phosphorophilic di-iodine molecules contribute to the transformation of red phosphorus into the more ordered black allotrope,7 thus suggesting that in this case also some P–P bonds cleave and recombine into a different structure with also containing pyramidal atoms.In solution, the P4 molecule is known to be transformed by I2 into four PI3 molecules, as summarized by eqn (1):8,9
| P4 + 6I2 → 4PI3 | (1) |
In spite of relatively small number of atoms involved, no detailed atomic level mechanism of the process has been reported yet. Only a limited number of reaction intermediates have emerged from NMR spectroscopic studies,10 while more species have remained undisclosed, likely due to their short lifetime. Since modern computational chemistry allows for the detection of even feeble stationary points (including transition states), we present here a reconstruction of the multi-step process of eqn (1) based on a series of sequential energy profiles. In particular, since the diphosphine P2I4 is perhaps the best experimentally characterized intermediate in the process,9 and possibly the most immediate precursor of PI3, eqn (1) was split into two sequential parts, such as eqn (2) and (3), respectively:
| P4 + 4I2 → 2P2I4 | (2) |
| P2I4 + I2 → 2PI3 | (3) |
As an emblematic step, eqn (3) suggests the inner P–P σ bond cleavage, accompanied by the I–I bond to give two new P–I bonds, thus suggesting a formal 2 + 2 symmetry forbidden reaction. From the orbital viewpoint, this implies the interconversion of σ bonding into σ* MOs or the other way around, with an invariably difficult electron transfer between the levels.11 Another doubt is with respect to the reported method of synthesis for P2I4, based on the disproportionation of PI3.12 By assuming that eqn (3), in the given form is reverse, there would be again a problem of forbidden symmetry, to the point that one starts doubting on the correct proposition of eqn (3). Also the occurrence of similar problems for some of the steps summarized in eqn (1) cannot be a priori excluded.
With the previous points in mind, an in silico DFT analysis of the whole process was attempted to monitor the mode of electron redistribution and its implications for redox behavior. The latter is more probable for the parallel P4 + O2 stepwise process, given the larger electronegativity difference between the atoms. In fact, in the derivatives P4O6 and P4O10 the electrons are preferentially assigned to the oxygen atoms implying that the phosphorus oxidation state evolves from the zero value in P4 to +3 and +5, respectively.13 Such a conclusion appears more questionable for the reactivity of P4 with I2, given that the larger electropositivity of the element is closer to that of phosphorus. On the other hand, the participation of I2 in redox processes is rather general, a previous example studied by us being the oxidation of classic 44e− phosphide bridged Pt3 clusters to 42e− derivatives with the reduced iodides as terminal ligands.14
In our computational approach to the P4 activation, the DFT-D functional was used,15 since the dispersion forces help detecting particularly feeble adducts, some of which are endergonic and clearly disfavored by entropy. In particular, the latter highlights incipient Halogen Bonding (XB), still far from its canonic formulation emerging in frozen crystal structures,16 which clearly indicate the occurrence of a major electron density redistribution. In any case, the feeble adducts are fundamental to monitor the energy profile of any given step, characterized by the dynamic evolution of the XB species, which encompasses at some point the features found in the frozen crystal structures and beyond. Accordingly, the calculations were based on the dichloromethane PCM model,17 in search of consecutive minima and transition states, in order to obtain a complete picture of the progressive electron redistribution at the linear P–I–I assembly and the eventual P–P bond cleavage.
All the steps contributing to eqn (1) seem to imply a concerted process, mainly because of the relatively low basicity of the various P atoms interacting with di-iodine. This point also holds for the allegedly final activation of P2I4 to give PI3 (eqn (3)), but we became somewhat dubious about the result, after reading a recent paper by Cummins and coworkers,18 who showed how some related organo-substituted diphosphines does not afford any phosphine product with I2. Eventually, a reasonable explanation of the difference has been found and will be presented toward the end of this paper, while here we anticipate that it most likely depends on the donor power locally available at the atoms of the P–P bond on whether the latter may be cleaved or not.
Other studies in the literature concern the P4 + I2 reactivity in the presence of additional co-reactants. For instance, the role of the Ag+ cation in the homoleptic silver complex [(P4)2Ag]+ and others19 has been summarized in a review article.20 Otherwise, the P4 activation has been performed in the presence of carbene moieties, which afford partial but incomplete P–P demolition and formation of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P double bond.21 The latter feature has never been observed by us, excluding such a possibility in our mechanism (vide infra). In other cases, the reaction is promoted by organic radicals22 including a transition metal fragment such as Cp(CO)2Fe, originating from the homolysis of its dimeric precursor and carrying an unpaired spin at the d7 metal.23 In this regard, other authors have remarked how an unassisted P4 + I2 reaction excludes the access to any radical species.24 Also, the calculations corroborated such a result, since our attempts of isolating reaction radical intermediates invariably failed.
P double bond.21 The latter feature has never been observed by us, excluding such a possibility in our mechanism (vide infra). In other cases, the reaction is promoted by organic radicals22 including a transition metal fragment such as Cp(CO)2Fe, originating from the homolysis of its dimeric precursor and carrying an unpaired spin at the d7 metal.23 In this regard, other authors have remarked how an unassisted P4 + I2 reaction excludes the access to any radical species.24 Also, the calculations corroborated such a result, since our attempts of isolating reaction radical intermediates invariably failed.
A final point concerns the various energy profiles, which emerged from tested analyses of the possible, alternative steps. Most of them occur with evident energy gain and the lack of substantial barriers, with the most important exception of the very first P4 activation, estimated to be as large as +14.6 kcal mol−1, with an approximate half one later in the process. This implies that the P4 demolition requires in any case an initial but not excessive activation energy, after which it proceeds rather smoothly to the end.
Experimental section
All the compounds were optimized, within the Gaussian09 program,25 by using the hybrid density functional B97-D.15 The dispersion forces in the latter are fundamental for allowing some aggregation of the species with consequent definition of the step profiles. All the free energies, derived after the calculations of the vibrational frequencies, refer to a temperature of 298 K. All the calculations were based on the CPCM model17 for the dichloromethane solvent, most typically used in the various experiments. The basis set 6-31G inclusive of polarization functions was basically used, although as a simplification the Stuttgart/Dresden (SDD) pseudo-potential26 was used for iodine. This appears to be at the origin of some overestimation of the calculated distances vs. the experimentally available ones. The coordinates of the optimized structures and their energetic parameters are reported in the ESI.†Results and discussion
General aspects of the reactivity between white phosphorus and di-iodine
Eqn (1) is exergonic at −76.4 kcal mol−1, of which −47.4 kcal mol−1 is for the double diphosphine production in eqn (2). The final derivation of the PI3 product (eqn (3)) affords a further free energy gain of −14.5 kcal mol−1 per P2I4 molecule. The computational analysis started with the initial formation of the di-iodine adduct with P4 and continued with other subsequent derivatives, whose evolution was monitored by a series of relaxed scans of reasonable reaction coordinates. All the encountered key points were then fully optimized, as minima or transition states, thus allowing precise stepwise profiles, based on structural motifs most of which are present in the literature.To facilitate the reading of the paper, all the steps, detected through the strategy, are anticipated in Fig. 1 and refer to the evolution of the P4 skeleton, without addressing yet the role of di-iodine molecules to be illustrated case by case. Also, Fig. 1 is subdivided into two main streams, with the left side essentially representing the succession of eqn (2) and (3), while the right side is indicative of possible alternative steps. Cut down to the bone, each step mainly corresponds to a new P–P cleavage, hence it is identified by a progressive Roman number, which is accompanied by an alphabetic character in the case of alternative routes.
 | ||
| Fig. 1 Flowchart showing the computed phosphorus intermediates in the process of eqn (1). The mode of action of the I2 reactants is similar in the various steps and will be separately illustrated. | ||
The diphosphine cleavage in eqn (3) occurs twice in the left side cascade, but only once in the right side, where some PI3 product forms at earlier stages, suggesting eqn (4) in place of eqn (2):
| P4 + 5I2 → P2I4 + 2PI3 | (4) |
The energy evaluation of the overall eqn (4) shows that the P2I4 diphosphine formation is more exergonic than that through eqn (2) (−61.9 vs. −47.4 kcal mol−1), but only for a half amount of the product, since, as expected, the energy balance of the whole process must be the same. The following detailed analyses show that basically any step implies a nucleophilic interaction of an electron pair at P4 (or one of the fragments derived from its degradation) with I2 molecule(s). Some behavior difference arises because the P donor power is not constant but varies with local geometry and interconnections. For instance, the original P4 basicity is the smallest detected, likely due to its strained tetrahedral structure. In any case, however, any incipient P⋯I–I adduct is linear, even suggesting a native Halogen Bonding (XB), which can still be far from its classic equilibrium features, emerging from frozen crystal structures. The latter are generalized with a generic D base (the P lone pair in our case) and a X–Y halogen donor or a X2 dihalogen, one of which is I2 itself.16 The stronger the base, the larger the P–I bonding interaction, while the I⋯I elongation is enhanced with accumulation of the electron density at the most external I atom, closer to an iodide. According to the XB definition,16 the residual I⋯I interaction is mainly of electrostatic forces, which somehow hide the electron transfer and/or polarization effects occurring to reach the point. On the other hand, the monitoring of the evolution from the initial adduct as well as the underpinning of the ultimate XB scission have been scarcely documented to date. Moreover, the steps for the P4 reactivity are peculiar not because of the actual heterotypic scission of the terminal iodide at the end of each step, but for the subsequent P–P cleavage associated with the formation of two new P–I bonds. Previously, we have already addressed some other peculiar behavior of XB adducts formed at a metal center upon the I2 addition to a chloride ligand, which is intriguingly substituted by an in situ formed less electronegative iodide.27,28 Also in this case, the key information was extracted by the computational monitoring of the system's evolution involving a XB dynamism. The general electronic underpinnings of the latter are briefly summarized in the next section.
Dynamic aspects of the halogen bonding (XB)
In a typical Dδ+–X⋯Xδ− XB interaction (where D is a generic base), the scission of the terminal halide is controlled by the D's donor power. It has been found that the latter is also related to the surrounding crystalline environment, as it has emerged from a number of X-ray structures, with an asymmetric I3− anion in place of the symmetric expected one.29 This may be seen as a peculiar XB species, where the more distal lateral iodide has for some reason accumulated a larger electron density, hence has a somewhat lower donor power than its trans-axial analogue. Our proposed explanation27 was based on the different interactions of the lateral I3− atoms with the surrounding cations in the cell. Thus, the more affected I atom is forced a higher accumulation of electron density and behaves as a weaker donor toward the associated I2 moiety. The extent of the effect varies crystal to crystal, as shown by the various degrees of I3− asymmetry in different structures. Consequently, the asymmetric I3− species are like snapshots along the dynamic evolution path of XB. To further clarify the point, we refer to some of our experimental and theoretical analyses27,28 of generic D+–I⋯I− adducts. The larger the D donor power, more electron density is drifted into the I2 σ* level, which behaves as a unique acceptor because of its relatively low energy. The consequence is that the abstraction of the terminal iodide is progressively easier, especially if facilitated by the presence of a polar solvent. For instance, this occurs in the preparation of organo iodo-compounds from a strong donor such as a carbo-anion and I2, where the dynamism of the XB intermediate is particularly relevant.30 As mentioned, even a XI2− mixed trihalide (formally similar to an asymmetric I3−) may undergo a dynamic electron redistribution to the point that a coordinated Cl–I–I− trihalide separates one iodide from the hetero-diatomic I–Cl, which was experimentally detected by Raman spectra.28To generalize the expected trends of electronic redistribution in a linear XB system, we emphasize the qualitative implications of the generic HOMO in Scheme 1.27 In the latter, the sp hybridization of the central atom depends on the lateral donor power. Only for a fully symmetric triiodide, the central p orbital is uninvolved, with the s orbital alone being equally antibonding, hence repulsive at both sides because of its formal population.31 This is consistent with the intrinsic hypervalent character of the symmetric trihalides and more in general XB, because the feature persists upon asymmetrization.
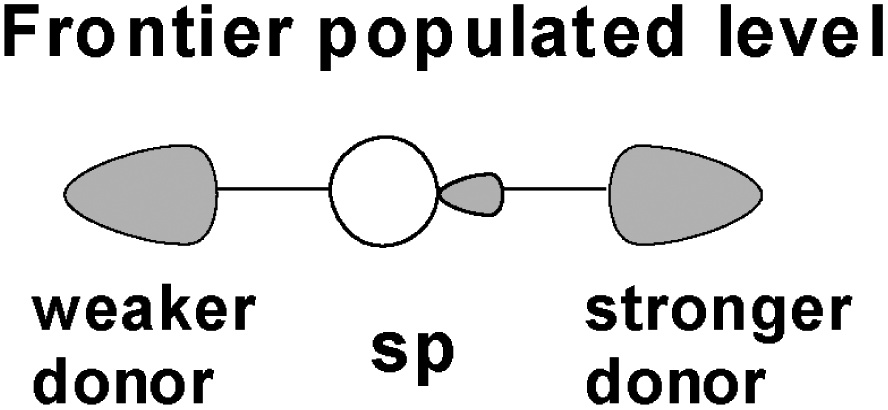 | ||
| Scheme 1 HOMO of a generic halogen bonded Y–X–D species suggesting the stretching trends. | ||
The control of the relative power of the lateral donors is responsible for the p mixing, which reinforces the bonding of the stronger donor to the central atom. Conversely, a large sp lobe develops toward the opposite weaker donor, which is most likely the terminal halide of the XB system. The latter is affected by bond stretching, if not an actual scission. The fact that the HOMO has larger contributions to the antibonding area, implies a corresponding electron density accumulation, with increasing possibility of the terminal halide scission, especially with the support of an abstractor.
The limiting adducts of an organo-phosphine with di-iodine
The previously indicated trends of XB interactions are experimentally corroborated by the available structures of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 adducts of the organo-phosphine PiPr3 with di-iodine.32,33 Already in PiPr3·I2 shown in Fig. 2a,32 the I–I distance of 3.38 Å is definitely weakened with respect to the free diatomic of 2.73 Å, while the P–I bond could be considered as single (2.41 Å).
:1 and 1:2 adducts of the organo-phosphine PiPr3 with di-iodine.32,33 Already in PiPr3·I2 shown in Fig. 2a,32 the I–I distance of 3.38 Å is definitely weakened with respect to the free diatomic of 2.73 Å, while the P–I bond could be considered as single (2.41 Å).
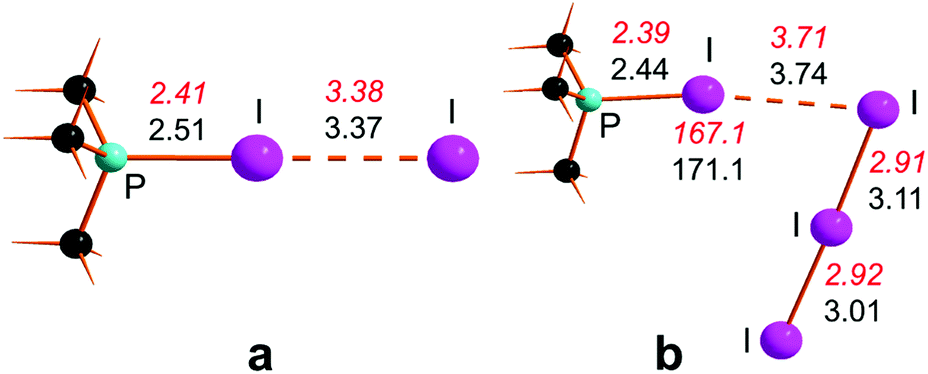 | ||
| Fig. 2 Experimental structures of PiPr3·I2 (a) and PiPr3·2I2 (b) with geometric parameters shown in red italics, while the black ones are those of the corresponding optimized P(CH3)3 models. | ||
The mentioned experimental values (in red italics) are satisfactorily reproduced in the DFT optimized model P(CH3)3·I2, shown in black in Fig. 2a.34 Moreover, the distal I atom is found to have a −0.7 charge, consistent with the realistic XB character of the adduct. The structure of the 1:2 PiPr3·2I2 adduct in Fig. 2b33 indicates that a second and residually acidic I2 molecule has the possibility of acting as a real extractor of the first formed iodide, as indicated by the large and non-bonding I⋯I separation of 3.71 Å, attained by the first molecule and the ±0.92 charge separation of the phosphonium and tri-iodide counterions. As a matter of fact, the optimized model best formulates as the ion pair {[P(CH3)3I]+[I3]−}, although the computed I⋯I and P–I distances and the I3− asymmetry are somewhat more pronounced than in the experimental data. A better consistency instead is observed for the P–I⋯I angles of 167.1° and 171.1° in the experimental and in silico 1:2 species, respectively. The latter deviation from linearity confirms some lost XB character of the first adduct, after the formation of the classic I3− species. In any case, the computed free energies of the P(CH3)3 adducts are consistent with a very high donor power of the organo substituted phosphines. In fact, P(CH3)3·I2 is already exergonic at −23.5 kcal mol−1, while the 1:2 adduct gains an additional −7.8 kcal mol−1. In this respect the aggregation of up to three molecules is entropically penalized up to the estimated value of +16.7 kcal mol−1, which is nonetheless overwhelmed by the highly exothermic electron transfer in these species. The entropy seems instead to favor the physical separation of the counterions [PR3I]+ and I3− by the endergonic amount of −5.6 kcal mol−1. An important conclusion about the phosphine adducts is the occurrence of an actual 1e− redox process independently from the actual separations of the counterions [P(CH3)3I]+ and I3− or their combination as an ion pair. Such a picture greatly differs from any of those encountered in the overall P4 demolition by I2, because of the limited possibilities of electron transfer with scarcely dative P atoms, such as the I-substituted ones. This point will reappear frequently in the subsequent discussion.
Domino evolution of the P4 reactivity
Independent of the routes in Fig. 1, the large exergonic balance of the overall process is consistent with its easy experimental occurrence. The possible barriers in various steps have not yet been remarked upon, but they are either small or absent in most cases, except in the very early stages (e.g., steps I or IIa) because of reasons to be clarified. Importantly, the general mode of attack of a P lone pair into I2 compares with that already illustrated in Fig. 2 relative to the collinear interaction of an organo-phosphine lone pair with a diatomic, with further enhancement of electron transfer upon the addition of a second one. This occurs also in the various steps of the P4 activation with the basic difference that the less dative P atom transfers a reduced amount of electrons, without preventing the further evolution of the systems in the form of a new P–P bond cleavage. In contrast, the latter event will be shown to be definitely more difficult upon a large shift of the electron density, which leads to an apparently inert ion pair. These aspects, as well as other problems, encountered step by step, will be addressed in the following discussion.First P–P bond cleavage in P4
Since no adduct between I2 and P4, 1, has ever been experimentally reported or detected, its identification was attempted in silico. To have more chances of identifying such a feeble adduct, the DFT-D functional15 was adopted, also because the dispersion forces have already enabled us to detect some otherwise metastable metallo-organic initiators of catalytic processes for the transformation of azides.35 Also in this case, an initial minimum of formula P4·I2, 1·I2 (on the left side of Fig. 3) was optimized, although endergonic by as much as +6.2 kcal mol−1. Any lone pair of a P4 tetrahedron is a potential donor, as shown for instance by its behavior as a η1 ligand toward a vacant σ metal acceptor. An example is the complex [(np3)Ni(η1-P4)] (where np3 = N(CH2CH2PPh2)3),36 first reported by our institute, which was followed by other η1-P4 complexes.2 Conversely, no structure is available to support the dative interaction of P4 toward I2, whose residual acceptor capabilities are given by its σ* level. As a matter of fact, the latter must be higher in energy than any reasonable metal σ hybrid with d character. | ||
| Fig. 3 Optimized adducts between P4 and one or two I2 molecule(s). | ||
It is evident that in 1·I2 the dative interaction is still very poor, although its incipient XB character is indicated by the collinearity of the P⋯I and I–I vectors. The lengths of the latter however (3.23 and 2.79 Å, respectively) prove a minimum electron redistribution, which suggested a search for possible alternatives. Thus, by assuming residual basicity for any bent and strained P–P linkage, the species 1·(I2)br, in the middle of Fig. 3, was optimized with I2 in the perpendicular orientation. Also, in this case the diatomic seems minimally perturbed in view of the two large P–Ibridge distances of 3.64 Å. On the other hand, in view of the 0.05 Å P–P elongation and the smaller endergonic balance of +3.9 kcal mol−1, a second diatomic was added to favor the electron withdrawing from P4, similarly to what happens for the phosphine's bis-adduct of Fig. 2b. As a matter of fact, the new species 1·2I2, on the right side of Fig. 3, was equally optimized starting from either one of the mono-adducts 1·I2 or 1·(I2)br. An enhanced electron redistribution is indicated by the 0.04 Å stretching of the first added I2, while its terminal I1* atom is already forming a quasi-orthogonal I3 grouping. Also, the 0.03 Å elongation of the P1–P2 bridged bond and the average ∼0.15 Å shortening of the P–Ibridge distances are indicative of some activation as well as the almost null free energy cost of the adduct (only +0.7 kcal mol−1). In trying to guess how the species 1·2I2 may further evolve toward the expected P–P cleavage, we noticed the large spatial freedom of the terminal I3 grouping. Hence we tested, as a possible reaction coordinate, the shortening of the I2⋯P2 distance down to a bonding value. The corresponding relaxed scan from 8.47 to 2.55 Å highlighted a promising profile, since the initial energy loss of only 10 kcal mol−1 later converted to a double size energy gain, all through reasonable chemical species. In fact, the encountered key points could then be fully optimized, affording for step I the precise free energy profile as shown in Fig. 4. Indeed, such a strategy based on relaxed scans and full optimizations allowed us to construct the flowchart of Fig. 1. The process shown in Fig. 4 starts from the left with the separated P4 and I2 reactants, which assemble into the adducts 1·(I2)br and 1·2I2 shown in Fig. 3 with a total energy cost of +4.6 kcal mol−1. Another +10.0 kcal mol−1 must be added to reach the transition state (1–2)TS, whose overall barrier of +14.6 kcal mol−1 is the highest in the overall P4 demolition process, which hence requires an activation energy to be triggered. Remarkably at (1–2)TS, the first added I2 molecule is stretched by 3.45 Å, suggesting that an important electron delocalization has already occurred, as also underlined by the 0.6 Å shortening of the P–I bridging linkages, still almost symmetric. On the other hand, the I1–I1* vector is no more perpendicular to P1–P2, but reoriented to align with the terminal I2 atom of the I3 grouping with the P1–P2 linkage (angle of 171°).
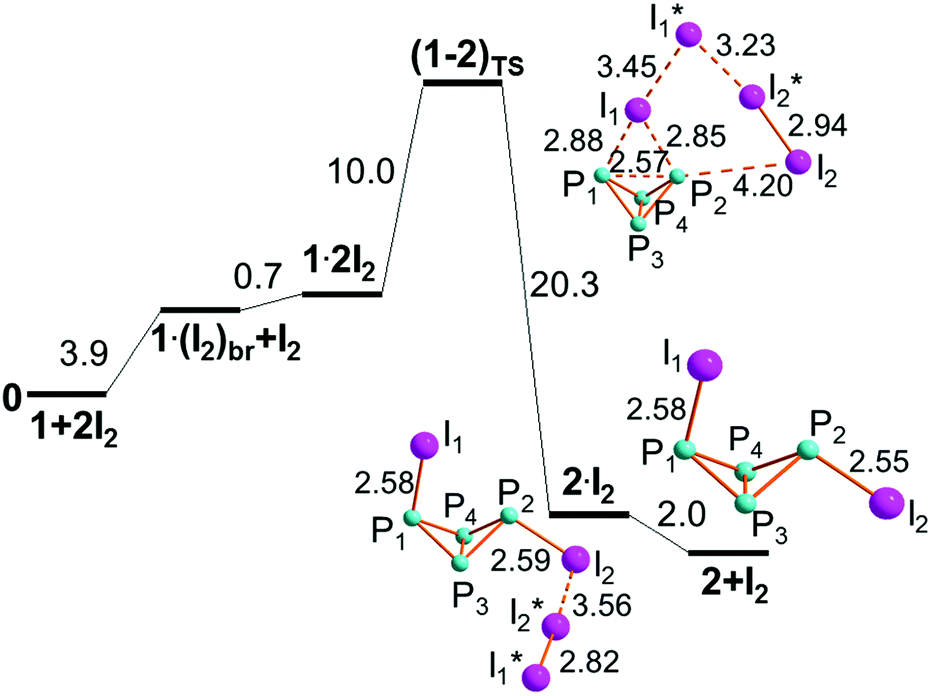 | ||
| Fig. 4 Profile of step I for the first activation of P4 by two I2 molecules (free energies in kcal mol−1). | ||
Although at (1–2)TS the I2⋯P2 separation is still as large as 4.20 Å, it seems that some iodine electron density may start transferring to the P1–P2 σ* level, as also indicated by the 2.57 Å P1–P2 elongation. An effect of this sort for the P–P bond weakening was previously remarked by other authors.37 Also, the vibrations of the unique imaginary frequency for (1–2)TS is indicative of the intended I2⋯P2 bond shortening. Another point worth noting is that the mentioned 3.45 Å elongation of the first added diatomic is still ∼0.3 Å shorter than that of the organo-phosphine aggregate PiPr3·2I2 shown in Fig. 2b, hence one cannot yet propose an ion pair formulation for it, a point, whose importance will become more evident later.
After (1–2)TS, the P1–P2 bond definitely cleaves, as it emerges from the two subsequent minima, namely the metastable adduct IP4I·I2, 2·I2, and the isolated butterfly 2. Obtaining the former species is exergonic by −20.3 kcal mol−1, while another −2.0 kcal mol−1 is gained on dismantling the residual XB interaction which holds together 2 with the I2 molecule. The nature of the latter must be emphasized, because it is generated in situ from the two original I2 molecules, cooperating in the step. Such an event will be repeatedly observed throughout the P4 demolition, and emphasizes the importance of having I2 and the associated phosphorous reactant in the 2:1 ratio.38
In closing the description of step I, we address the chemical reliability of the butterfly 2 based on the data in the literature and CCDC database.29 From previous NMR investigation of the P4 + X2 reaction in CS2 (X = Cl, Br),10b the formation of distinct butterfly isomers emerged. In the latter, the two X substituents may point in either opposite direction (as in 2) or both hang over the butterfly's cavity. The former isomer was 4:1 more abundant, although information for X = I is definitely less clear in this respect. Also, various structures of the carbo-substituted RP4R analogues are known in isomeric forms.39 To find possible implications for our system, optimization of the isomer alternative to 2 was found to be +12.2 kcal mol−1 less stable (see Fig. S1†) and moreover the interconversion energy is as high as +35 kcal mol−1. These results suggested continuing the analysis of any possible steps II, exclusively starting from the isomer 2.
First considerations on the plausible concerted mechanism of the P4 demolition
From the results outlined to date, the following indications emerge:(i) The P–P cleavage does not exclusively produce only an I-substituted derivative of P4 but also a brand new I2 molecule. The latter is indicated as I*–I* to imply its generation from two distinct I–I* diatomics, whose I atoms form the new P–I linkages. Implicitly, step I corresponds to the ter-molecular reaction, shown in eqn (5), which affords two products. By the same token, the ultimate and emblematic diphosphine cleavage in eqn (3), better formulates as eqn (3′), as well as many other intermediate steps:
| P4 + 2I–I* → IP4I + I*–I* | (5) |
| P2I4 + 2I–I* → 2PI3 + I*–I*. | (3′) |
Certainly, the continuously reformed I2 molecules are reutilized in the process, so that overall eqn (1) does not necessarily imply a double number of diatomics (e.g., 12 rather than 6), while it is fundamental that the amount of di-iodine is constantly doubled compared to any P-based reactant.
(ii) The mechanistic study implies that step I and the following ones involve the combination of 3 + 3 bond breakings/makings, in place of the assumed 2 + 2 one.
(iii) The species (1–2)TS shown in Fig. 4 adumbrates a distorted six-membered P2I4 ring (this is the only case also featuring an endocyclic connection, such as the I1–P2 one), while somewhat more regular rings will appear in the subsequent steps. Importantly, the sides of the ring convert into two distinct sets 3 + 3 bonds/no-bonds when moving in alternative directions with respect to TS or a point in its proximity (see below). This apparently corroborates the idea of concertedness, implying a significant electron density redistribution between any pair of adjacent connections. With regard to this, it has been already mentioned that the P–P cleavage seems promoted by the σ* population, with the 4e−/2c configuration implying initial repulsion, which is eventually mitigated since the bonding and antibonding electron pairs become local and reoriented P lone pairs.
(iv) The concerted mechanism is in particular attributable to the relatively low donor power of the P atoms, such as those of P4 or its iodine-derivatives. In contrast, the larger donor power of organo-substituted analogues induces, as in the case of the phosphines shown in Fig. 2, a significant charge separation at the I2 reactant(s), with the resulting ion pair having no further possibility of concerted evolution. The most evident difference between the two situations will emerge, toward the end of this paper, from the behavior of the two differently substituted diphosphines (vide infra).
Second P–P bond cleavage
From the butterfly 2, the P4 demolition may continue in two possible ways, given that the P–P bond to be cleaved may be either the hinge (step IIa)40 or a peripheral one (step IIb). The former is closer to any one of the P4 precursor, given that its atoms all form P–P bonds, hence one may predict a difficult aggregation with the I2 molecule(s). As a matter of fact, step IIa encounters some, but not insurmountable, difficulty, which seem to favor the alternative step IIb.Cleavage of the IP4I butterfly's hinge (step IIa)
Although the initial aggregate (2·2I2)hinge could be reasonably expected to have the P–P edge bridged by I2, as in 1·2I2 shown in Fig. 4, the feature is not found in the profile given in Fig. 5, given that only one P atom (P3) is associated with two I2 molecules. | ||
| Fig. 5 Step IIa for the di-iodine attack on the hinge of the butterfly 2 to give the final puckered four-membered ring 3 (free energies in kcal mol−1). | ||
Apparently, the P basicity must be somewhat larger, possibly due to the less strained character of the P–P bond. A more effective interaction is confirmed by the P3–I3 and I3⋯I3* distances of 2.67 and 3.05 Å, respectively, which are indicative of somewhat more pronounced effects than in any adduct of 1. Moreover, (2·2I2)hinge has about half energy cost of +2.2 kcal mol−1. Also, the externally dangling I3 grouping is again suited for the relaxed scan analysis based on the I4⋯P4 shortening. This allowed the optimization of the key points down to the cleavage of the P–P hinge and beyond it. The precise profile features at (2–3)TS the second significant barrier detected in the whole process. Its +8.4 kcal mol−1 height is about halved with respect to (1–2)TS, while a significant difference is of the lack of bridge-bonding at the P–P linkage to be cleaved. The first diatomic is instead practically cleaved at 3.90 Å, with the quasi-symmetric terminal I3 grouping already significantly charged (−0.7). The P4–P3 bond is not however significantly stretched (2.37 Å), apparently because the I4⋯P4–P3 angle of 153° is still somewhat bent to limit the electron transfer to the P4–P3 σ* level. Most likely, the situations change on the following descent from TS, given that the distance becomes as large as 3.10 Å in the product (PI)4, 3, with the shape of a puckered four-membered ring. As usual, the product 3 is preceded by an adduct (3·I2 in Fig. S2†), where the in situ formed I3*–I4* diatomic is still slightly interacting. The estimated free energy gains are −17.6 kcal mol−1 at 3·I2 and −19.6 kcal mol−1 upon separation of the components.
The chemical reliability of the product 3 is corroborated by other analogues in the literature, although without the prove of ad hoc NMR studies.10 In any case, the stereochemistry of 3 is peculiar for the two pairs of consecutive P–I linkages (not even parallel to each other), which simultaneously point to opposite sides of the ring. No other example of this is instead present in the CCDC,29 which in no case has a P4 ring with only halogen substituents, but at most two of them in the species P4I2(SiR3)2.41 Since other structures feature pairs of trans-diagonal organo-substituents on the same side of the P4 ring, we computationally tested such a stereochemistry also for (PI)4. Hence, the corresponding species 3′ in Fig. S3† was compared with that of 3, in order to find out an insignificant energy difference of only <1 kcal mol−1 with a negligible interconversion barrier, as shown by the flat PES near TS. On the other hand, the unique relevance of 3vs. 3′ clearly emerged on analyzing the subsequent step IIIa (see below), which is unique, given that the terminal I3 rearrangement is highly hindered by the substituents, when starting from 3′.
Cleavage of one peripheral P–P linkage of the IP4I butterfly (step IIb)
The aggregation of two I2 molecules is less unhindered at one I-substituted P atom of 2, possibly because the flexibility of the terminal P–I vector confers a larger degree of basicity to the P lone pair. Indeed, the aggregate (2·2I2)periph is less endergonic than the (2·2I2)hinge one, as it emerges from the comparison of Fig. 5 and 6. Also, the analogue of (2·2I2)periph involving the P2 rather than the P1 atom has been discarded because the terminal I3 rearrangement would be hindered by the P1–I1 linkage already oriented over the cavity of the butterfly. Hence, step IIb uniquely proceeds as shown in Fig. 6, almost being also barrierless. In fact, a destabilization of only +0.6 kcal mol−1 was estimated by our initial scan, which showed a plateau around (2–4)TS‡, which could not be fully optimized. After the latter, our strategy afforded the detailed energy profile given in Fig. 6, still apparently consistent with a concerted mechanism. | ||
| Fig. 6 Step IIb for the I2 attack on one external I-substituted atom of the butterfly 2. The species (2–4)TS‡, resulting from a relaxed scan, could not be fully optimized for lying on a flat plateau, indicated by the helical connection (free energies in kcal mol−1). | ||
Although the evolution of the butterfly 2 seems easier through step IIb, step IIa cannot be automatically excluded under the conditions for triggering the P4 activation, since the barrier, is only half of that already passed in step I. On the other hand, the missing barrier for step IIb seems to imply a subsequent energy gain of only −10.6 kcal mol−1 at the species 4·I2 (see Fig. S4†), with an additional −1.5 kcal mol−1 gained for the ultimate separation of 4. The latter can be synthetically formulated as I2P-P3I2, to indicate in the right a P3 ring with three exocyclic bonds, i.e., two equally oriented P–I ones and another P–PI2 one in opposite directions with respect to the triangle. Again, experimental analogues are known with organo- in place of halogen substituents. Perhaps, the most related species is RClP-P3ClR with R being a substituted phenyl ring.42 In some cases, the two substituents on the same side of the P3 ring are interconnected by a chain,43 while in 4 two adjacent P–I linkages are not exactly parallel to avoid a close contact.
Third P–P bond cleavage
The cascade of steps through the general mechanism continues from both products of steps IIa and IIb, provided that 3 can be effectively formed in spite of the barriers to be passed. In this case, the left side cascade of Fig. 1, may continue with the opening of the (PI)4 ring to give the chain P4I6, 5, with two terminal PI2 groupings (step IIIa). Conversely on the right side, the three-membered cycle 4 may follow alternative paths, such as steps IIIb and IIIc, depending on the phosphorus/di-iodine docking mode. In the former case, the three-membered ring (PI)3, 6, may form together with a dissociating (PI)3 phosphine 7 or, in the latter, the tripod-like dendrimer P(PI2)3, 8, is consequent to the opening of the P3 cycle at the bond involving the I-substituted P atoms.Opening of the four-membered ring (PI)4 (step IIIa)
The stereochemistry of the intermediate 3, based on the precursor 2, can in principle afford different adducts of the type 3·2I2. The one shown on the left side of Fig. 7 does not encounter hindrance problems in the important I3 grouping rearrangement. | ||
| Fig. 7 Step IIIa for transforming 3 into the open chain product 5 (free energies in kcal mol−1). | ||
Remarkably, 3·2I2 is the first exergonic aggregate encountered in the present P4 chemistry (−3.8 kcal mol−1), implying that the donor power of the P1 atom has become larger. This is also corroborated by the more evidently perturbed P1–I5 and I5–I5* distances of 2.56 and 3.10 Å, respectively. Also in this case, our strategy afforded a reliable energy profile, which includes the low lying transition state (3–5)TS (+2.5 kcal mol−1). The latter is −1.3 kcal mol−1 more stable than the separated reactants, suggesting an essentially barrierless evolution. The −0.6 charge of the terminal I3 unit at TS is again consistent with a concerted electron transfer, which determines the cleavage of the P1–P4 bond to give the open chain P4I6, 5, stabilized by −11.8 kcal mol−1.
As usual, the intermediate 5·I2 precedes the final mentioned product upon release of the in situ generated I2 molecule with the exergonic balance of −3.7 kcal mol−1. The still rather closed I6–P4–P1 angle of 135° at (3–5)TS indicates poor activation of the P4–P1 bond, still as short as 2.36 Å. On the other hand, the electron flow to its σ* level becomes possibly on the descent to 5·I2, once again consistent with the concerted mechanism.
Alternative steps IIIb and IIIc from 4
As the product of step IIb, I2P-P3I2 may aggregate two I2 molecules in different ways to afford the species (4·2I2)a, (4·2I2)b and (4·2I2)c shown in Fig. 8, whose energy balances are similarly exergonic, namely −2.7, −3.9 and −0.3 kcal mol−1, respectively.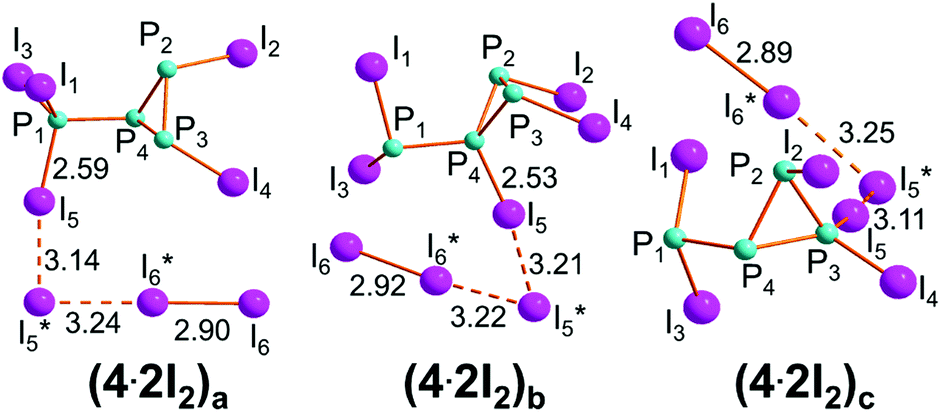 | ||
| Fig. 8 Optimized structures of the isomeric adducts between I2P-P3I2, 4 and two I2 molecules. | ||
Both (4·2I2)a and (4·2I2)b may be almost equivalent precursors of the P1–P4 cleavage with separation of the phosphine PI3, 7, from the cycle (PI)3, 6. Fig. 9 shows the profile of step IIIb only departing from (4·2I2)a. On the other hand, the aggregate (4·2I2)b could in principle have the chance of undergoing the triangular P3 ring opening at either the P4–P2 or P4–P3 linkage to give the four-membered open chain I2P-PI-PI-PI2, 5. The latter process is inhibited by the two P–I vectors departing from the ring, which hinders the I3 rearrangement. Conversely, the P2–P3 side may open up starting from (4·2I2)c to afford the dendrimer P(PI2)3, 8, as shown by the profile of step IIIc shown in Fig. 10. The mentioned possibilities are here described in some detail.
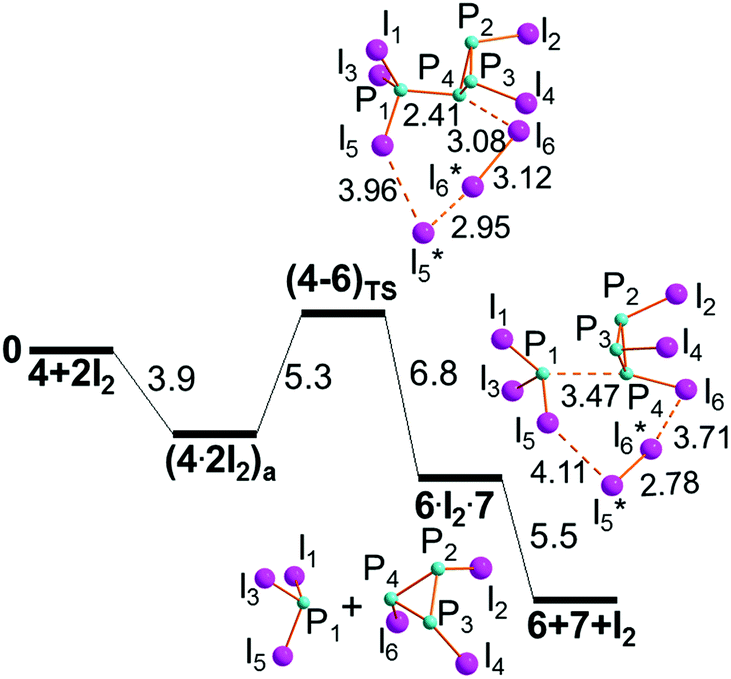 | ||
| Fig. 9 Step IIIb for the first cracking of the P4 skeleton into a PI3, 7 and the (PI)3 ring 6 (free energies in kcal mol−1). | ||
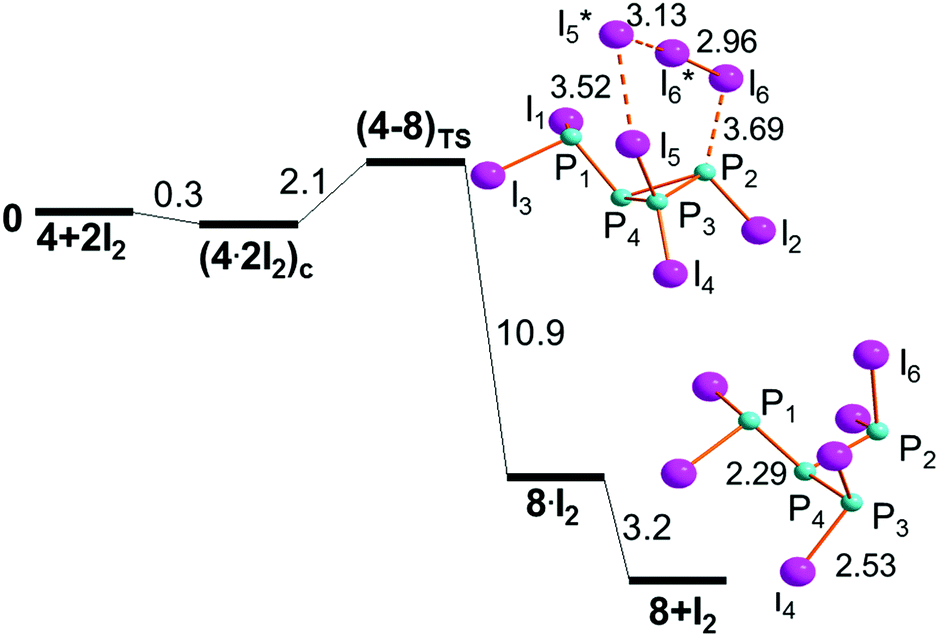 | ||
| Fig. 10 Step IIIc for the opening of the cycle I2P-P3I2, 4, to give the dendrimer P(PI2)3, 8 (free energies in kcal mol−1). | ||
Fragmentation of the P4 skeleton in 4 to give the PI3 and the ring (PI)3 (step IIIb)
The cleavage of the P1–P3 exocyclic bond in (4·2I2)a is monitored as shown in Fig. 9. The encountered +5.3 kcal mol−1 barrier at (4–6)TS is in actuality as small as +1.4 kcal mol−1 given that the aggregation of the components in (4·2I2)a is somewhat exergonic (−3.9 kcal mol−1). This is because of the relatively good basicity of the exocyclic atom P1, which determines the actual cleavage of the first I5–I5* diatomic at TS (separation of 3.96 Å), hence a significant charge delocalization at the terminal I3 grouping, which is not too asymmetric given the 2.95 and 3.12 Å distances.The situation is not so dissimilar from that of R3P·2I2 shown in Fig. 2b, although it is improper to discuss an ion pair formulation, since the terminal I6 atom is already at 3.08 Å from the P4 atom, consistent with the idea of a concerted mechanism at work.
The next intermediate is still an aggregate, namely 6·I2·7, where the newly formed I5*–I6* diatomic holds this time together two distinct molecules. Clearly, there is no direct interaction between the latter as shown by the large P4⋯P1 separation of 3.47 Å, but in any case this allows an energy gain of −6.8 kcal mol−1, which almost doubles (another −5.5 kcal mol−1) with the final scission of the three molecular components. In this case, the process seems particularly favored by entropy, whose contribution is evaluated to be about −17 kcal mol−1. The formation of the first of the four expected PI3 molecules in the overall process is remarkable for preceding that of the diphosphine P2I4, until now considered its natural precursor. An early presence of PI3 could be possibly presumed on the basis of the early NMR data, although not explicitly proved.10
Opening of the three-membered ring in 4 to give the dendrimer 8 (step IIIc)
The P3–P2 cyclic bond in (4·2I2)c does not encounter hindrance problems on its cleavage, because the terminal I3 grouping occupies the space opposite to the two P–I linkages, as shown in Fig. 10. Being the least stable of the isomers in Fig. 8, (4·2I2)c reaches the transition state (4–8)TS with a cost of only +2.1 kcal mol−1, which is reduced to +1.8 kcal mol−1 with respect to its separated components.The (4–8)TS stereochemistry is particularly remarkable for allowing an easy identification of the P2I4 six membered ring, which switches the 3 + 3 formed/broken linkages in the ideal concerted nature of the process. In this case, not only the turning TS point is easily reached but also its subsequent descent to the dendrimer 8 seems smooth because of the total energy gain of −14.1 kcal mol−1, of which −3.2 kcal mol−1 is associated with the scission of the intermediate 8·I2. Again, halogenated species such as 8 have not been structurally characterized, but again the molecular type is supported by related tripodal triphosphanes with organic substituents.44
Fourth P–P bond cleavage
In both cascades of Fig. 1, there remain three of the six original P4 bonds to be cleaved. The precursor on the left side is the open chain P4I6, 5, which in principle may be cracked in the middle to give two P2I4 diphosphines or allow the scission of a PI3 phosphine from the chain P3I5, 10. On the right side, the fourth P–P cleavage may occur at either the ring (PI)3, 6, or the dendrimer P(PI2)3, 8, which equally afford the chain 10.Chain-breaking at P4I6 (step IVa)
Alternative conformations of 5 are possible as shown by the structure of some organo-substituted P4 chains in the CCDC.29 In some cases, the chain is overall planar with zig-zag conformation,45 while in others it is puckered and similar to a trapezoid, in which one of the cyclic sides has opened up without a major conformational rearrangement.46 The latter seems to be the case of compound 5, which derives from the closed cycle 3 without any significant conformational change (see step IIIa in Fig. 7). In other words, the product 5 maintains a chelate shape. This in principle causes some repulsion between the terminal P lone pairs, which is somewhat mitigated by a torsion around the central P2–P3 linkage (the corresponding PPPP dihedral angle is 125°). In this manner, the P4 chain stabilizes, whereas its potential adduct with two I2 molecules at one lateral P atom encounters major stereochemical problems, which emerge from its attempted optimization. This is clearly indicated by the corresponding species 5·2I2‡ (Fig. S5†) with illogical chemical features. For instance, one I atom is found bridging between the two lateral P atoms with an energy cost of about +10 kcal mol−1, hence the species was abandoned as a precursor of the chain breaking. In contrast, the adduct 5·2I2 involving an inner P atom of 5 has a stabilization energy of −4.4 kcal mol−1 and was used as the starting point of step IVa. For the sake of brevity, the corresponding standard profile is presented in the ESI (Fig. S6†) and highlights the central split of 5 into the two P2I4 molecules, 9, through a practically barrierless process and an overall energy gain of −15.3 kcal mol−1. As expected, an intermediate with the in situ formed I2 molecule, namely 9·I2·9, precedes the final dissociation of the molecular components, which is exergonic by −4.4 kcal mol−1, likely because of the favourable entropy.It is worth mentioning at this point an additional feature of 5·2I2, which has been overlooked until the submission of this paper. Namely, the species is not only the precursor of the diphosphine 9, but it alternatively affords the separation of the phosphine 7 from the shorter chain P3I5, 10, whose behavior will be illustrated below. Importantly, this shows an interlink missing until now between the two cascades shown in Fig. 1. Very briefly, the alternative step starting from 5·2I2, whose detailed profile IVa-bis is shown in Fig. S7,† is also smooth, for having the (5–10)TS −0.9 kcal mol−1 lower than the reactants and a subsequent free energy gain of −14.1 kcal mol−1. The new results lead to the conclusion that the final product PI3 is in any case partially attainable even before the achievement of diphosphine 9.
Opening of the three-membered ring 6 in step IVb
On the right side of Fig. 1, the 4th P–P cleavage may start from the (PI)3 ring. The observed conformation of the latter with all the I substituents on the same side is somewhat unusual and it is most likely imposed by that of precursor 4 (see step IIIb in Fig. 9). As a matter of fact, the known organo-substituted P3 analogues have the 2 + 1 distribution of the substituents47 and also a calculation for such a (PI)3 isomer shows a larger stability of −3.9 kcal mol−1. Also, no interconversion of the isomer seems possible in view of a free energy cost >25 kcal mol−1. The computed profile for step IVb shown in Fig. S8† shows that the initial adduct 6·2I2 leaves large space for the rearrangement of the terminal I3 grouping and the eventual formation of the open chain P3I5, 10. Also, the mechanism is usual with a small barrier of +2.5 kcal mol−1 to be passed at the transition state (6–10)TS, followed by an overall energy gain of −18.4 kcal mol−1. NMR evidence of P3I5 has been provided,48 while it is worth mentioning that other similar halogenated chains have been recognized as relevant in the chemistry of small Pn units.33,44Alternative generation of the phosphine 7 and the chain 10 from the dendrimer 8 (step IVc)
The pyramidal dendrimer P(PI2)3, obtained through step IIIc, can add two di-iodine molecules at either the central P atom, (8·2I2)centr, or one of the terminal ones, (8·2I2)lat. From both precursors one may expect the analogous Pcentr–Plat bond cleavage but, since (8·2I2)centr is more stable by −5.0 kcal mol−1, it was the isomer of choice for step IVc. A new cracking of the P4 skeleton generates phosphine 7 and the open chain 10, which at this point appears to have various modes of formation. The profile of the step IVc shown in Fig. S9† confirms its unquestionable feasibility, given that (8–10)TS lies −3.2 kcal mol−1 lower than the separated reactants and the overall exergonic balance is −17.2 kcal mol−1, consistent with the concerted mechanism.Final P–P cleavages (steps V and VI)
The two last P–P cleavages are equivalent on the left side cascade of Fig. 1 with the generation of four phosphines, 7, from two diphosphines, 9. On the right side, no P2I4 molecule has yet formed at the 4th P–P cleavage, while one-fourth of the PI3 product is already present (it has alternatively formed through step IIIb or IVc). The amount of phosphine doubles in step V through the cracking of the chain 10, still excluding the mandatory involvement of the diphosphine for its formation. The latter is instead produced with the mentioned 5th P–P cleavage on the right side, which is in any case based on the cracking of 10, as shown by the subsequent profile.Cracking in step V of the open chain 10
Irrespective of its formation, the three-membered chain 10 easily undergoes a P–P cleavage under the general mechanism. The profile shown in Fig. S10† assumes that the di-iodine molecule is anchored at the central P atom, although a lateral aggregation should not be dramatically different. The (10–9/7)TS is again more stable than the separated reactants and from it the splitting of 9 and 7 occurs with an overall energy gain of −14.3 kcal mol−1, of which −4.0 kcal mol−1 is for the final scission of the ter-molecular intermediate 9·I2·7 shown in Fig. 11, for the particularly favourable entropic contribution.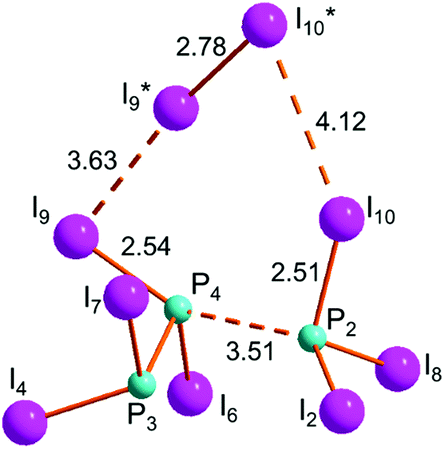 | ||
| Fig. 11 Optimized structure of ter-molecular intermediate 9·I2·7. | ||
The just outlined process prompts an interesting comparison with the inverse, Ag+ promoted, combination of PI3 and P2I4 to give the cationic open chain P3I6+.19 With the lack of a detailed reaction profile, Scheme 2a suggests how the AgI precipitation may favour the extraction of one iodide from PI3,35 with the consequent orbital vacancy at the PI2+ grouping being saturated by a P2I4 lone pair and the formation of the new P–P bond.
 | ||
| Scheme 2 Comparison of the acidic action of Ag+vs. I2 in the potential formation of the cationic chain P3I6+. | ||
A similar role could be exerted by the residual I2 acidity (see Scheme 2b), which could in principle extract an iodide from PI3 and give I3−, hence the ion pair I3−/P3I6+. The intermediate 9·I2·7 shown in Fig. 11 would be the turning point of the reaction, which is energetically penalized in forming the P–P bond, hence excluding the cationic chain P3I6+ (Fig. S10†).
The ultimate I2-promoted transformation of the diphosphine P2I4 into PI3 and the inhibition for organo-substituted analogues
The diphosphine's dissociation step to give PI3 is a ter-molecular process, as it already emerged by the proposal of eqn (3′)vs. eqn (3). The latter implies a 2 + 2 addition, which is a symmetry forbidden reaction,11 because of the inter-crossing of the σ and σ* levels along the pathway and the high energy costs to invert the population. On the other hand, the concept of σ bond metathesis49 has been raised for some organometallic compounds of d0 elements, with interchanging some coordination bonds associated with the alternative formation/cleavage of metal–element bonds, while the inverse behaviour applies to the associated element–element bonds. Possibly, some relation with eqn (3) may be established because of the electronic equivalence between the P atoms and d0 metals, but the point has not been further pursued. Conversely, we continued with the general picture emerging from the various P4 demolition steps, which is based on the 3:3 and not the 2:2 ratio of broken/formed bonds. As a matter of fact, P2I4 behaves in the known manner toward di-iodine, while other organo-diphosphine analogues do not undergo the P–P cleavage under the action of I2. We refer in particular to the species P2(dmb)2, 11, recently reported by Cummins et al.,18 where dmb are chains of four carbon atoms, which close two cycles by sharing the same P–P linkage. For the comparative calculations shown in Fig. 12, the realistic model of 11 was preferred to the simpler diphosphine P2(CH3)4 one. A first important difference concerns the initial 1:1 adduct 9·I2vs. 11·I2. As a matter of fact, an organo-substituted P atom must be a stronger donor, more effectively perturbing the added I2 molecule, as already noticed for the phosphine adduct PR3·I2 in Fig. 2a.50 Thus, the I11–I11* elongation in 11·I2 is definitely larger than in 9·I2 (3.31 vs. 2.97 Å), while P1–I11 distance is closer to a single bond (2.53 vs. 2.73 Å). Experimentally, in 11, the two distances are 2.411 and 3.417 Å, respectively. Since no experimental information is available on the aggregate of the organo-diphosphine with two diatomics, we also optimized the species 11·2I2 and compared it with 9·2I2. What emerges is the definite cleavage of the first I2 molecule (I11–I11* = 3.97 Å) and the lost collinearity with the P linearity with the P1–I11 vector. More remarkable are the free energy changes. While in Fig. 12a, the maximum free energy gain for 9·2I2 is as usual not large (−2.25 kcal mol−1), the addition of two diatomics to 11 is −28.2 kcal mol−1 (i.e., −19.6 kcal mol−1 and −8.6 kcal mol−1, in the order).
 | ||
| Fig. 12 (a) Final I2-promoted dissociation of P2I4 into two PI3; and (b) different behavior of the organo-diphosphine 10 with disfavoured P–P cleavage (free energies in kcal mol−1). | ||
Also the terminal and asymmetric I3− in 11·2I2 is almost monoanionic (charge = −0.93), the adduct is best formulated as ion pair [11I]+[I3]−, whose cation closely resembles the well-known and stable [P2I5]+ one, which is rather unreactive.51 It may be deduced at this point that the maximized electron transfer at the species in question is inconsistent with the until now proposed concerted mechanism in these processes. Nonetheless, we continued with our strategy of relaxed scans and subsequent optimizations to evaluate the possible difficulties of the Cummins's system in forming the new I12–P2 bond and the eventual P1–P2 cleavage. As a matter of fact, the profile b in Fig. 12 follows the trends already highlighted in a, although the energies are much different.
Thus, to reach (11–12)TS the energy cost is +21.0 kcal mol−1vs. the +3.0 kcal mol−1 of (9–7)TS. While from the latter the generation of distinct PI3 molecules is as smooth as in all of the previous cases (the energy gains are −9.5 and −5.7 kcal mol−1 at 7·I2·7 and 2PI3, respectively), the structure of (11–12)TS is highly questionable. In fact, this already features broken P1–P2 bonds with a separation as large as 3.53 Å, while after the high free energy cost to reach the point, the subsequent energy gains to obtain 12 or its immediate precursor 12·I2 are as small as −4.2 and −0.5 kcal mol−1, respectively. Likely, our computational strategy has forced the attainment of these minima, which are spontaneously attainable at least through the mechanism proposed in this paper. Incidentally, it is worth mentioning that a compound of type 12 with halogen atoms other than iodine was spectroscopically detected after treating 11 with halogenating reactants other than diatomics, e.g., C2Cl618 or the Grignard species.18,52 Due to the lack of an X-ray structure, we did not perform any computational analysis.
In closing this section, we mention a final intriguing problem about the reported method of preparation of the diphosphine P2I4 upon the disproportionation of PI3.12,53 Without deepening any mechanistic aspect of this process, a reductive elimination of I2 seems unlikely for the same reasons illustrated for the reverse 2 + 2 direct addition implied by eqn (3). Alternatively, the reverse process of Fig. 12a (from right to left) cannot be excluded a priori in spite of the +15 kcal mol−1 barrier to be passed. On the other hand, such an event would not correspond to the actual PI3 disproportionation but its reaction with an I2 co-reactant, which is excluded from the environment if not specifically added. Perhaps, other factors may become important as for instance the role played by some specific solvents,53 which we have not explored at this time.
Conclusions and extensions
The computational analysis of the P4 activation by I2 to generate the phosphine PI3 confirms the value of the in silico approach in identifying unclear mechanisms with otherwise unknown intermediates. The overall process, which is exergonic by ∼−76 kcal mol−1, occurs through a sequence of energetically favourable steps, except for an initial barrier of about +15 kcal mol−1. The latter seems due to the still low donor power of the unperturbed white phosphorus, which hardly transfers electron density from its compact and strained structure. After the barrier, a multi-step process is triggered with a repeating concerted action, given the occurrence of 3 + 3 bond breakings/makings in each case. The common feature is a dynamic electron transfer throughout six atoms, cyclically arranged. The process starts with a delocalization of some P electron density through the first formed P–I–I linear unit and continues at the subsequent tri-iodide XB adduct involving a second diatomic. Some electron density ends up to a P–P σ* level with the cleavage of one of the original P4 sides. The mechanism is evidently ter-molecular at any step with the peculiar reformation of a new di-iodine molecule.Fig. 1 has already been a useful reference throughout the paper, but provides no energy indication relative to the occurrence of alternative steps. For this, we present the conclusive Fig. 13, outlining the most probable cascade of steps, with the significant barrier, (1–2)TS, appearing only at the very beginning.
 | ||
| Fig. 13 Favoured cascade of events in the P4 demolition by I2, except for the unique initial barrier (1–2)TS. Half amount of the PI3 product forms in the earlier stages rather than at the very end. | ||
The analysis indicates a fundamental role played in the attack of I2 by the P atom donor power. The latter must not be large to trigger a concerted electron transfer without altering the oxidation states of the involved atoms. In fact, an excessive donor power, as in the case of organo-substituted P atoms, determines a major electron transfer and charge separation (ion pair formation), which does not allow concertedness. This point clearly emerged by comparing the P2I4 diphosphine with an organo-substituted analogue of the type P2R4, which was reported by Cummins and co-workers as being unable to induce P–P cleavage.18
The present elucidation of the basic intermediates in the P4 + I2 process may be relevant for the widely investigated chemistry of phosphorus-based molecules. Moreover, it seems to have a direct impact on our ongoing studies on the functionalization of phosphorene 2D material.6 As in P4, any P atom is pyramidal and directly linked to three equal ones, hence its coordination to an acidic metal center seems plausible, as found for some known P4 complexes.2 As a matter of fact, we have gained some computational evidence, by using the solid state package CRYSTAL,54 that the P donor power is rather similar in the two cases, hence a number of metal fragments suitable for phosphorene's coordination in the η1, η2 and η3 modes have been already individuated.55 By the same token, the reactivity of phosphorene toward di-iodine molecules could also follow a concerted mechanism in place of a prompt charge separation. On the other hand, a number of disfavouring factors have emerged from our preliminary analyses, such as the reduced freedom of the initial P–I–I and I–I–I pendants of the XB-type to span over the 2D surface and attack a specific σ* level of a P–P to be broken. Possibly, some well tailored modelling is necessary on which we are working now.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant Agreement No. 670173) for funding the project PHOSFUN “Phosphorene functionalization: a new platform for advanced multifunctional materials” through an ERC Advanced Grant. ISCRA-CINECA HP Grants HP10C2QI78 and HP10CFMSCC for the computational resources are also acknowledged.References
- (a) D. E. C. Corbridge, The structural Chemistry of Phosphorus, Elsevier, Amsterdam, NL, 1974 Search PubMed; (b) F. Bachuber, J. von Appen, R. Dronskowski, P. Schmidt, T. Nilges, A. Pfitzner and R. Weihrich, Angew. Chem., Int. Ed., 2014, 53, 11629–11633 CrossRef PubMed.
- (a) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed; (b) I. de los Rios, J.-R. Hamon, P. Hamon, C. Lapinte, L. Toupet, A. Romerosa and M. Peruzzini, Angew. Chem., Int. Ed., 2001, 40, 3910–3912 CrossRef CAS; (c) M. Peruzzini, L. Gonsalvi and A. Romerosa, Chem. Soc. Rev., 2005, 34, 1038–1047 RSC; (d) P. Barbaro, M. Di Vaira, M. Peruzzini, S. Costantini and P. Stoppioni, Chem. – Eur. J., 2007, 13, 6682–6690 CrossRef CAS PubMed; (e) C. Mealli, F. Costanzo, A. Ienco, M. Peruzzini and E. Perez-Carreño, Inorg. Chim. Acta, 1998, 275–276, 366–372 CrossRef CAS.
- (a) M. E. Schlesinger, Chem. Rev., 2002, 102, 4267–4301 CrossRef CAS PubMed; (b) C. C. Stephenson, R. L. Potter, T. G. Maple and J. C. Morrow, J. Chem. Thermodyn., 1969, 1, 59–76 CrossRef CAS.
- (a) D. Warschauer, J. Appl. Phys., 1963, 34, 1853–1860 CrossRef CAS; (b) A. Brown and S. Rundquist, Acta Crystallogr., 1965, 19, 684–685 CrossRef CAS.
- (a) H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tomanek and P. D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS PubMed; (b) L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372–377 CrossRef CAS PubMed; (c) D. Hanlon, C. Backes, E. Doherty, C. S. Cucinotta, N. C. Berner, C. Boland, K. Lee, A. Harvey, P. Lynch, Z. Gholamvand, S. Zhang, K. Wang, G. Moynihan, A. Pokle, Q. M. Ramasse, N. McEvoy, W. J. Blau, J. Wang, G. Abellan, F. Hauke, A. Hirsch, S. Sanvito, D. D. O'Reagan, G. S. Duesberg, V. Nicolosi and J. N. Coleman, Nat. Commun., 2015, 6(1–11), 8563 CrossRef CAS PubMed; (d) M. Serrano-Ruiz, M. Caporali, A. Ienco, V. Piazza, S. Heun and M. Peruzzini, Adv. Mater. Interfaces, 2016, 3(1–8), 1500441 CrossRef PubMed.
- The project PHOSFUN aimed at understanding the basic properties of the phosphorene layers and their applications in the fields of catalysis and nanoelectronics has been financed with an Advanced ERC Grant of the European Commission (see the Acknowledgements).
- (a) S. Lange, P. Schmidt and T. Nigels, Inorg. Chem., 2007, 46, 4028–4035 CrossRef CAS PubMed; (b) Z. Zhang, X. Xin, G. Yan, Q. Li, Y. Yang and T.-L. Ren, Sci. China Mater., 2016, 59, 122–134 CrossRef CAS.
- F. E. E. Germann and R. N. Traxler, J. Am. Chem. Soc., 1927, 49, 307–312 CrossRef CAS.
- D. Wyllie, M. Ritchie and E. B. Ludlam, J. Chem. Soc., 1940, 583–587 RSC.
- (a) R. L. Carroll and R. P. Carter, Inorg. Chem., 1967, 6, 401–403 CrossRef CAS; (b) B. W. Tattershall and N. L. Kendall, Polyhedron, 1994, 13, 1517–1521 CrossRef CAS.
- R. B. Woodward and R. Hoffmann, J. Am. Chem. Soc., 1965, 87, 395–397 CrossRef CAS.
- I. P. Beletskay, E. I. Tomilenko, N. I. Kamenichnaya, V. I. Staninets, Zh. K. Gorbatenko and N. G. Feshchenko, Khim. Elementoorg. Soedin., 1976, 170 Search PubMed.
- (a) D. J. Heinze, Pure Appl. Chem., 1975, 141–172 Search PubMed; (b) N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, 2nd edn, 1997 Search PubMed.
- T. Funaioli, P. Leoni, L. Marchetti, A. Albinati, S. Rizzato, F. Fabrizi de Biani, A. Ienco, G. Manca and C. Mealli, Inorg. Chem., 2013, 52, 4635–4647 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- (a) G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS; (b) P. Metrangolo and G. Resnati, Science, 2008, 321, 918–919 CrossRef CAS PubMed and references therein; (c) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Lang, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed; (d) L. P. Wolters, P. Schyman, M. J. Pavan, W. L. Jorgensen, F. M. Bickelhaupt and S. Kozuch, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 523–546 CrossRef CAS; (e) S. V. Rosokha, C. L. Stern and J. T. Ritzert, Chem. – Eur. J., 2013, 19, 8774–8788 CrossRef CAS PubMed.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- I. Knopf, D. Tofan, D. Beetstra, A. Al-Nezari, K. Al-Bahily and C. C. Cummins, Chem. Sci., 2017, 8, 1463–1468 RSC.
- (a) I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2001, 40, 4406–4408 CrossRef CAS; (b) M. Gonsior, I. Krossing, L. Muller, I. Raabe, M. Jansen and L. van Wullen, Chem. – Eur. J., 2002, 8, 4475–4492 CrossRef CAS PubMed.
- M. H. Holthause and J. J. Weigand, Chem. Soc. Rev., 2014, 43, 6639–6657 RSC.
- (a) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 7052–7055 CrossRef CAS PubMed; (b) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2007, 129, 14180–14181 CrossRef CAS PubMed; (c) O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 5530–5533 CrossRef CAS PubMed.
- B. M. Cossairt and C. Cummins, New J. Chem., 2010, 34, 1533–1536 RSC.
- S. Heinl and M. Scheer, Chem. Sci., 2014, 5, 3221–3225 RSC.
- T. Kennedy and R. S. Sinclair, J. Inorg. Nucl. Chem., 1970, 32, 1125–1133 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- M. Dolg, H. Stoll, H. Preuss and R. M. Pitzer, J. Phys. Chem., 1993, 97, 5852–5859 CrossRef CAS.
- (a) F. Bigoli, P. Deplano, A. Ienco, C. Mealli, M. L. Mercuri, M. A. Pellinghelli, G. Pintus, G. Saba and E. F. Trogu, Inorg. Chem., 1999, 38, 4626–4636 CrossRef CAS PubMed; (b) G. Manca, A. Ienco and C. Mealli, Cryst. Growth Des., 2012, 12, 1762–1771 CrossRef CAS.
- M. E. G. Mosquera, P. Gomez-Sal, L. Diaz, L. M. Aguirre, A. Ienco, G. Manca and C. Mealli, Inorg. Chem., 2016, 55, 283–291 CrossRef CAS PubMed.
- Cambridge Structural Database System, version 5.38, Cambridge Crystallographic Data Centre, Cambridge, UK Search PubMed.
- M. Schlosser, Organoalkali Chemistry, in Organometallics in Synthesis: Third Manual, John Wiley & Sons, Inc., Hoboken, NJ, 2013, p. 61 Search PubMed.
- The important involvement of the s filled central orbital is not addressed in the widely used Pimentel and Rundle models. (a) C. G. Pimentel, J. Chem. Phys., 1951, 19, 446–448 CrossRef; (b) R. J. Hach and R. E. Rundle, J. Am. Chem. Soc., 1951, 73, 4321–4324 CrossRef CAS.
- F. Ruthe, P. G. Jones, W.-W. du Mont, P. Deplano and M. L. Mercuri, Z. Anorg. Allg. Chem., 2000, 626, 1105–1111 CrossRef CAS.
- W. I. Cross, S. M. Godfrey, C. A. McAuliffe, R. G. Pritchard, J. M. Sheffield and G. M. Thompson, J. Chem. Soc., Dalton Trans., 1999, 2795–2798 RSC.
- Calculation performed with the CPCM modeling of the CH2Cl2 solvent.
- G. Manca, E. Gallo, D. Intrieri and C. Mealli, ACS Catal., 2014, 4, 823–832 CrossRef CAS.
- P. Dapporto, S. Midollini and L. Sacconi, Angew. Chem., Int. Ed., 1979, 18, 469 CrossRef.
- I. Krossing, J. Chem. Soc., Dalton Trans., 2002, 500–512 RSC.
- I 2 was instead reported to be often in defect in some of the previous spectroscopic studies of this reactivity, such as the one of ref. 10.
- (a) R. Fox, R. J. Wright, E. Rivard and P. P. Power, Angew. Chem., Int. Ed., 2005, 44, 7729–7733 CrossRef PubMed; (b) J. Bresien, K. Faust, C. Hering-Junghans, J. Rothe, A. Schulz and A. Villinger, Dalton Trans., 2016, 45, 1998–2007 RSC.
- Pairs of the P4I2 molecules are not fully equivalent because of the orientation of the two I substituents.
- A. Lorbach, A. Nadj, S. Tullmann, F. Dornhaus, F. Schödel, I. Sänger, G. Magraf, J. W. Bats, M. Bolte, M. C. Holthausen, M. Wagner and H.-W. Lerner, Inorg. Chem., 2009, 48, 1005–1017 CrossRef CAS PubMed.
- J. Bresien, C. Hering, A. Schulz and A. Villinger, Chem. – Eur. J., 2014, 20, 12607–12615 CrossRef CAS PubMed.
- K.-O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 7545–7549 CrossRef CAS PubMed.
- (a) G. Fritz, E. Matern, H. Krautscheid, R. Ahlrichs, J. W. Olkowska and J. Pikies, Z. Anorg. Allg. Chem., 1999, 625, 1604–1607 CrossRef CAS; (b) G. Fritz, K. Stoll, W. Hönle and H. G. V. Schnering, Z. Anorg. Allg. Chem., 1986, 544, 127–136 CrossRef; (c) C. Jones, P.-C. Junk and T. C. Williams, J. Chem. Soc., Dalton Trans., 2002, 2417–2418 RSC; (d) A. Wisniewska, K. Baranowska, E. Matern and J. Pikies, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, 01364 Search PubMed.
- R. Wolf, S. Gomez-Ruiz, J. Reinhold, W. Nohlmann and E. Hey-Hawkins, Inorg. Chem., 2006, 45, 9107–9113 CrossRef CAS PubMed.
- K.-F. Tebbe and R. Frohlich, Z. Naturforsch., B: Chem. Sci., 1982, 37, 534 Search PubMed.
- (a) J. Borm, G. Huttner and O. Orama, J. Organomet. Chem., 1986, 306, 29–38 CrossRef CAS; (b) R. A. Barlett, H. V. Rasika Dias, K. M. Flynn, H. Hope, B. D. Murray, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 1987, 109, 5693–5698 CrossRef; (c) N. Tokitoh, A. Tsurusaki and T. Sasamori, Phosphorus, Sulfur Silicon Relat. Elem., 2009, 184, 979–986 CrossRef CAS; (d) A. Baldy and J. Estienne, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 747–749 CrossRef; (e) N. Burford, C. A. Dyker, M. Lumsden and A. Decken, Angew. Chem., Int. Ed., 2005, 44, 6196–6199 CrossRef CAS PubMed.
- K. B. Dillon and B. Y. Xue, Inorg. Chim. Acta, 2003, 320, 172–173 CrossRef.
- R. Waterman, Organometallics, 2013, 32, 7249–7263 CrossRef CAS.
- In the paper of Cummins,18 it is not specified whether a different ratio between the reactants can alter the system, as it emerged in the case of Fig. 2.
- M. Gonsior, I. Krossing, L. Muller, I. Raabe, M. Jansen and L. von Wullen, Chem. – Eur. J., 2002, 8, 4475–4492 CrossRef CAS.
- R. W. Alder, D. D. Ellis, J. K. Hogg, A. Martin, A. G. Orpen and P. N. Taylor, Chem. Commun., 1996, 537–538 RSC.
- A. V. Kirsanov, Zh. K. Gorbatenko and N. G. Feshchenko, Chemistry of Phosphorus Iodides in IUPAC Inorganic Phosphorus Compounds, Prague, 1974, 125–138 Search PubMed.
- R. Dovesi, R. Orlando, A. Erba, C. M. Zicovich-Wilson, B. Civalleri, S. Casassa, L. Maschio, M. Ferrabone, M. De La Pierre, P. D'Arco, Y. Noel, M. Causa, M. Rerat and B. Kirtman, Int. J. Quantum Chem., 2014, 114, 1287–1317 CrossRef CAS.
- C. Mealli, A. Ienco, M. Peruzzini and G. Manca, manuscript in preparation.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7dt04034b |
| This journal is © The Royal Society of Chemistry 2018 |