
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biologically inspired oxidation catalysis using metallopeptides
Laia
Vicens
and
Miquel
Costas
 *
*
Institut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, Campus Montilivi, Girona E-17071, Catalonia, Spain. E-mail: miquel.costas@udg.edu
First published on 2nd January 2018
Abstract
The stereoselective oxidation of hydrocarbons is one of the most challenging reactions for synthetic chemists. However, this transformation is one of the most common reactions in nature. Metalloenzymes that catalyze this transformation are taken as inspiration for the development of new catalysts. There are several examples in the literature where either peptides or metal catalysts are used in the stereoselective oxidation reaction, but the synergistic combination of both systems is still a non-explored field. The use of metallopeptides in biologically inspired oxidation reactions is discussed in this perspective.
Introduction
Reactions of selective oxidation are important in organic synthesis because oxygenated hydrocarbon chains are ubiquitous in organic molecules of relevance in pharmaceutical chemistry and biochemistry. Efficient preparation of these molecules requires the development of novel oxidation methodologies operating with high levels of chemo, regio and stereoselectivity. These methods should exhibit high atom economy and need to be based in earth abundant elements. These aspects make these systems more sustainable than current methodologies. Catalytic methods operating under mild conditions and using first row transition metals and benign oxidants such as O2 and H2O2 are particularly appealing.1Selective oxidation is common in nature. Metalloenzymes catalyze a large variety of reactions with high efficiency using mild conditions,2 and one of the most distinctive and valuable aspects of enzymatic reactions is their high selectivity in terms of chemo, regio and stereoselectivity. Key to this selectivity is the highly elaborated structure of the enzyme active site.3 In the enzyme active site the metal is ligated by amino acid residues, which are key to define its reactivity. Moreover, large polypeptide chains define elaborated second sphere structures, creating a cavity that embeds the metal and defines a three-dimensional network of chemical functionalities that can engage in non-covalent interactions with the substrate. These interactions determine the proper orientation of the substrate in the enzyme active site, and may also activate or deactivate specific substrate sites. These factors translate into highly selective oxidative transformations.4 Furthermore, due to the inherent chirality of amino acids, proteins contain chiral active centers, susceptible to highly stereoselective oxidations.
Enzymes thus constitute paradigmatic evidence that peptides can endow catalytic metal centers with extraordinary selectivity properties and constitute an inspiration motif to develop synthetic metal based oxidation catalysts. Along these lines, the combination of peptides and metal ions can be viewed as a powerful strategy to create artificial catalysts. In this approach, it is envisioned that the high reactivity of metals can be combined with the high structural and functional group versatility of peptides, ideally translating into reactive and selective catalysts.
Despite the combination of peptides and metal catalysts is a promising platform to be used in oxidative transformations, examples in the literature still remain scarce, presumably because peptides and metal catalysts may be expected to face compatibility problems. These examples are discussed in the following sections. In contrast, peptides have been actively explored as oxidation organocatalysts, and highly stereoselective transformations have been described. These studies are particularly interesting because they have delineated mechanisms to govern the regio and stereoselectivity in oxidation reactions, which ideally will be very interesting to implement in metal catalyzed reactions. These reactions are therefore of fundamental interest in the design of metal based catalysts, and have also been briefly discussed.
Organocatalysis
Organic chemists have used small peptides (<50 amino acids) as catalysts for a number of asymmetric transformations, including oxidations.5 Despite a limited number of commonly used amino acid residues, they can be combined in a large number of different sequences to produce a variety of secondary structures. This offers a powerful tool to rapidly create catalyst diversity, a key factor to pursue tunable selectivity (specially stereoselectivity).4b,5,6 Moreover, the reactivity of peptides can be easily tuned by modifying a single amino acid residue of the chain.6bIn the specific field of oxidation reactions, most of the studies described so far have focused on asymmetric epoxidation reactions.7 Relevant examples are described in the following lines, and the mechanisms that govern selectivity are briefly discussed (Scheme 1).
 | ||
| Scheme 1 Representative examples of peptide-based organocatalytic systems applied in oxidation reactions. | ||
Probably the best well-known enantioselective catalyzed oxidation reaction is the Juliá–Colonna epoxidation, discovered and developed in the early eighties.8 The reaction relies on the use of poly-(amino acids) as catalysts, and provides excellent enantioselectivities (up to 97% in 24–48 h) in the epoxidation of chalcones (Scheme 1A). Several advances have been subsequently made, including the modification of the original reaction conditions and of the structure of the catalysts, which have broadened the substrate scope.9
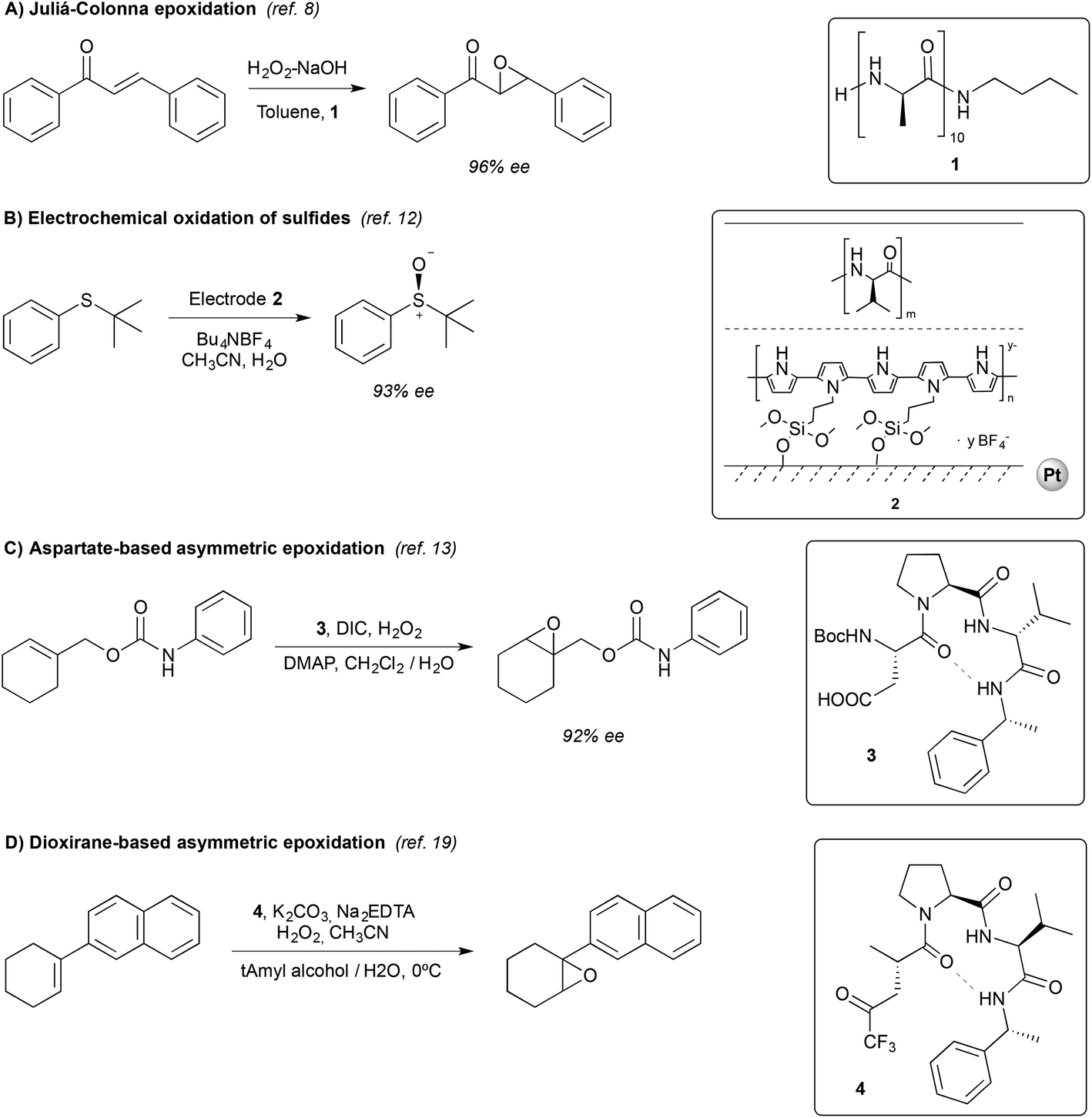
This reaction proceeds via the addition of hydroperoxide over the β carbon in the substrate, generating an enolate that subsequently displaces a hydroxide through an intramolecular nucleophilic attack.10 Stereoselectivity in the attack relies on the relative positioning of the substrate and the peroxide resulting from a network of hydrogen bond interactions between the oxygen atoms of the peroxide, and of the substrate with the N–H groups of the peptide (Fig. 1).11
 | ||
| Fig. 1 Schematic representation of the interaction between poly-leucine 1 and chalcone. The methyl groups of the side chain of the amino acids have been omitted for clarity. Adapted from ref. 11b. | ||
Peptides have also been used in the electrochemical oxidation of sulfides. The most successful strategy is the use of peptide-coated electrodes. Komori and Nonaka showed that platinum electrodes double-coated with polypyrrole and poly-L-valine provided the best results, allowing the preparation of sulfoxides in moderate to good yields and good to excellent enantioselectivities (Scheme 1B).12
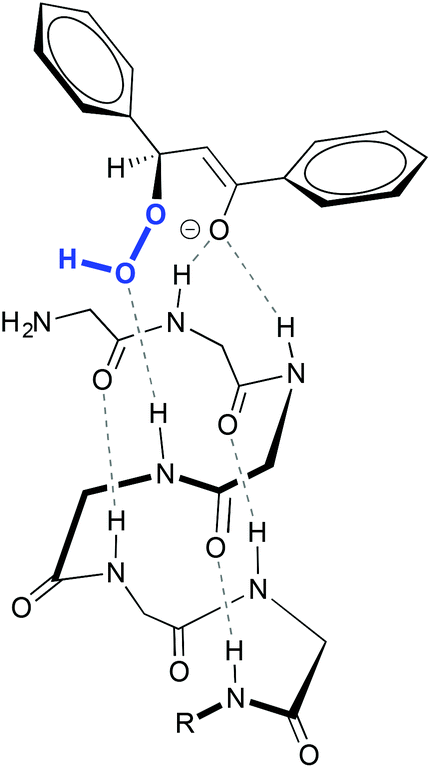
More elaborated peptides have been explored by Miller and co-workers, who developed an aspartate-derived peptide catalyst that epoxidizes 1-substituted carbamates with enantioselectivities up to 92% (Scheme 1C).13 Using aqueous H2O2 or urea hydrogen peroxide (UHP) as an oxidant, a peracid intermediate is generated in situ (Fig. 2, 3a), which is responsible for the oxidation of olefins. Of note, this represents the first example of a highly enantioselective electrophilic epoxidation using a peptide catalyst.
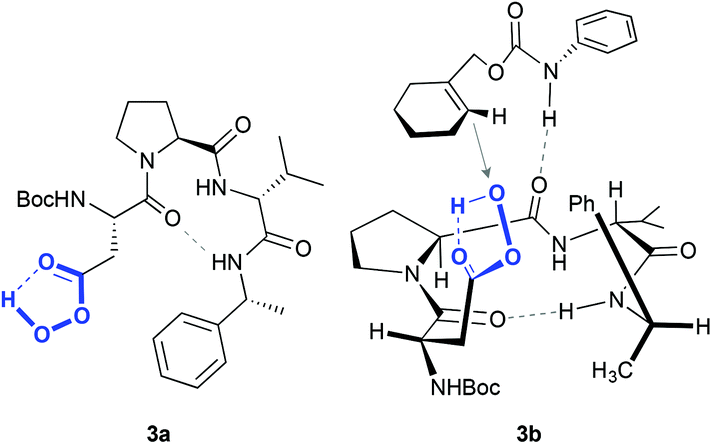 | ||
| Fig. 2 Peracid intermediate generated in the peptide (3a) and hypothetical transition state for the asymmetric epoxidation of cyclohex-1-en-1-ylmethylphenylcarbamate (see Scheme 1C), consistent with the absolute configuration of the major epoxide (3b). Adapted from ref. 13. | ||
The authors proposed that the epoxidation is directed by H-bonding interactions that occur between this intermediate and the carbamate moiety of the substrate (Fig. 2, 3b). In accordance with this proposal, when this functionality is not present in the molecule, the enantioselectivity decreases substantially (10% ee in the epoxidation of 1-phenyl-1-cyclohexene). These data, together with other mechanistic studies,14 reveal that these non-covalent interactions between the peptide and the olefin are key to define the stereoselectivity of the reactions.
Other aspartate-derived peptide catalysts have also been applied in the selective asymmetric epoxidation of polyenes,15 the enantioselective epoxidation of allylic alcohols16 and the enantioselective oxidation of indoles.17 In these examples, the structure of the peptide plays a crucial role in dictating the selectivity of the reactions due to the different non-covalent interactions that occur between the catalyst and the substrate. This fact is clearly demonstrated in the case of the epoxidation of polyenes.15 Through a combinatorial approach, the authors reported a peptide (Fig. 3A, 5) that epoxidizes 1-farnesol and other polyenes with high regioselectivity towards the 2,3-position and also with high enantioselectivity (86% ee). These results are comparable with those obtained with the Sharpless catalyst.18 Since no reaction is observed when the alcohol moiety of the olefin is substituted by a methyl ether, it is proposed that the hydroxyl group of the substrate interacts with the peptide, directing the selectivity of the reactions. The authors also found a peptide (Fig. 3A, 6) that shows regioselectivity towards the 6,7-position, but only low enantioselectivities were observed (10% ee). This loss of enantioselectivity is attributed to a high number of rotational bonds between the hydroxyl group and the double bond, since the olefin is located in a more remote position (Fig. 3B).
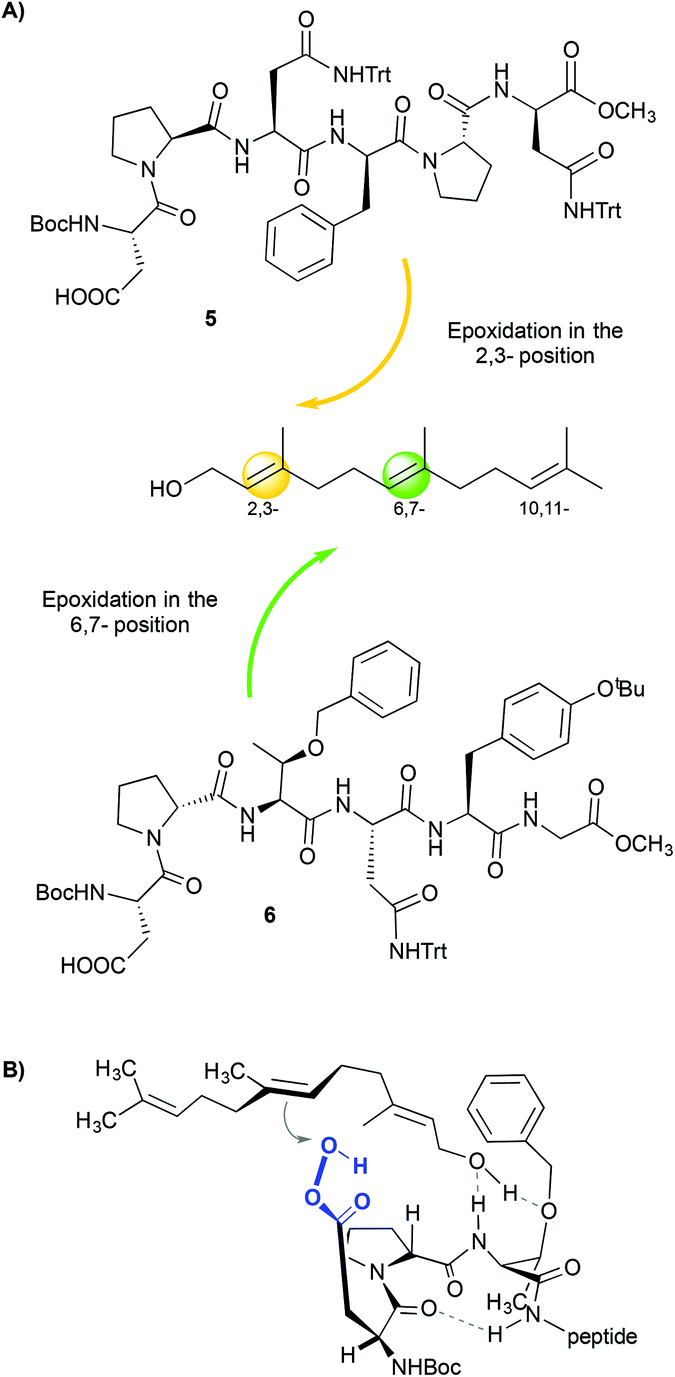 | ||
| Fig. 3 (A) Regioselective epoxidation of farnesol using different peptides. (B) Possible model that justifies the selectivity towards the 6,7-position. Adapted from ref. 15b. | ||
The same authors reported, in 2012, a new type of peptide for asymmetric epoxidation.19 In this case, a dioxirane was transiently generated (Fig. 4, 4a). This intermediate was catalytically active and yields high enantioselectivities in the epoxidation of aryl-substituted alkenes (Scheme 1D). It is proposed that in the transition state (Fig. 4, 4b) the peptide adopts a conformation that minimizes the allylic strain between the amide and the trifluoroketone center and the substrate may interact with the peptide by locating the smallest substituent of the alkene (H) near the largest dioxirane substituent (CF3). The oxygen of the oxirane can then interact with the double bond due to the overlap between the lone pairs on the oxygen atom with the π* orbitals of the double bond.
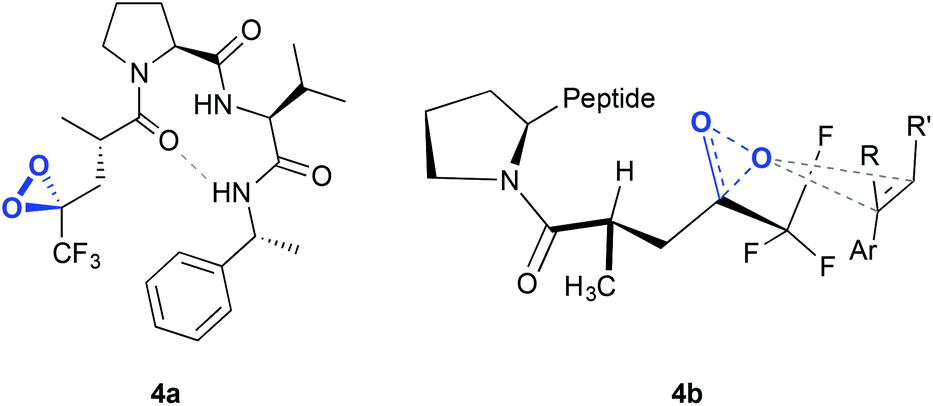 | ||
| Fig. 4 Dioxirane intermediate generated in the peptide (4a) and hypothetical transition state for the asymmetric epoxidation of an olefin, consistent with the absolute configuration of the major epoxide (4b). Adapted from ref. 19. | ||
Overall, these studies demonstrate the ability of peptides to govern stereoselectivity in oxidation reactions on the basis of a combination of a series of weak interactions between the peptide with the oxidant and the substrate. Particular limitations of these reactions are the need for relatively large catalyst loadings and long reaction times, aspects that a priori may be addressed by introducing highly reactive metal ions.
Metallopeptides
The combination of peptides and metal catalysts has been used in several asymmetric transformations such as hydrosilylation,20 cyclopropanation,21 allylic alkylation,22 conjugate additions to enones23 and addition of cyanides to aldehydes (Strecker reaction),24 among others.25 However, a few examples of the use of metallopeptides as catalysts in oxidation reactions have been reported, and moreover, all of them are applied in the asymmetric epoxidation of olefins.In 1992, Inoue and co-workers reported the catalytic asymmetric epoxidation of allylic alcohols using titanium metallopeptides.26 In a similar way as in the Katsuki–Sharpless epoxidation,27 titanium alkoxides are used in this system. However, the tartrates that are used as partners were substituted by dipeptides whose amino terminal site is modified to a phenolic Schiff base (Fig. 5). Although this system does not improve the results obtained by Sharpless, it represents a pioneering example in the field of metallopeptides.
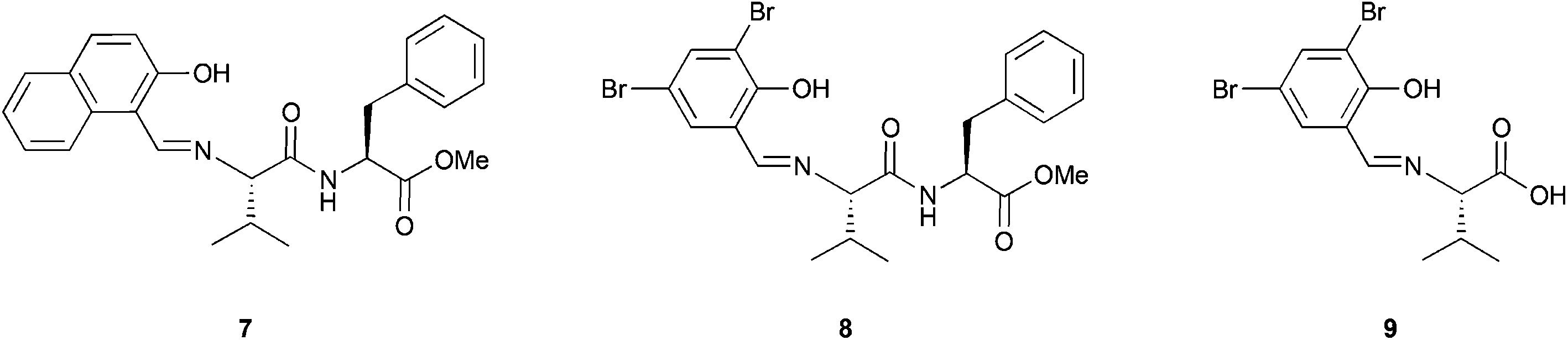 | ||
| Fig. 5 Representative ligands used in the work of Inoue and co-workers. | ||
This system, previously tested in the asymmetric synthesis of cyanohydrins,6a was used as a catalyst in the epoxidation of nerol using TBHP as an oxidant at −20 °C in the presence of 10 mol% of a mixture formed by equimolar quantities of Ti(OiPr)4 and Nap-S-Val-S-Phe-OMe (7), which was the most efficient peptide in the synthesis of cyanohydrins. The corresponding epoxide was obtained, even though only low enantioselectivities (13% ee) were achieved. However, modifying the structure of the peptide or even using only one amino acid and using 1,1-diphenylethyl hydroperoxide (DPEHP) as an oxidant instead of TBHP, the enantioselectivity increases up to 66% ee (Scheme 2A). This methodology allows the asymmetric epoxidation of several allylic alcohols in good yields and enantioselectivities (Scheme 2B).
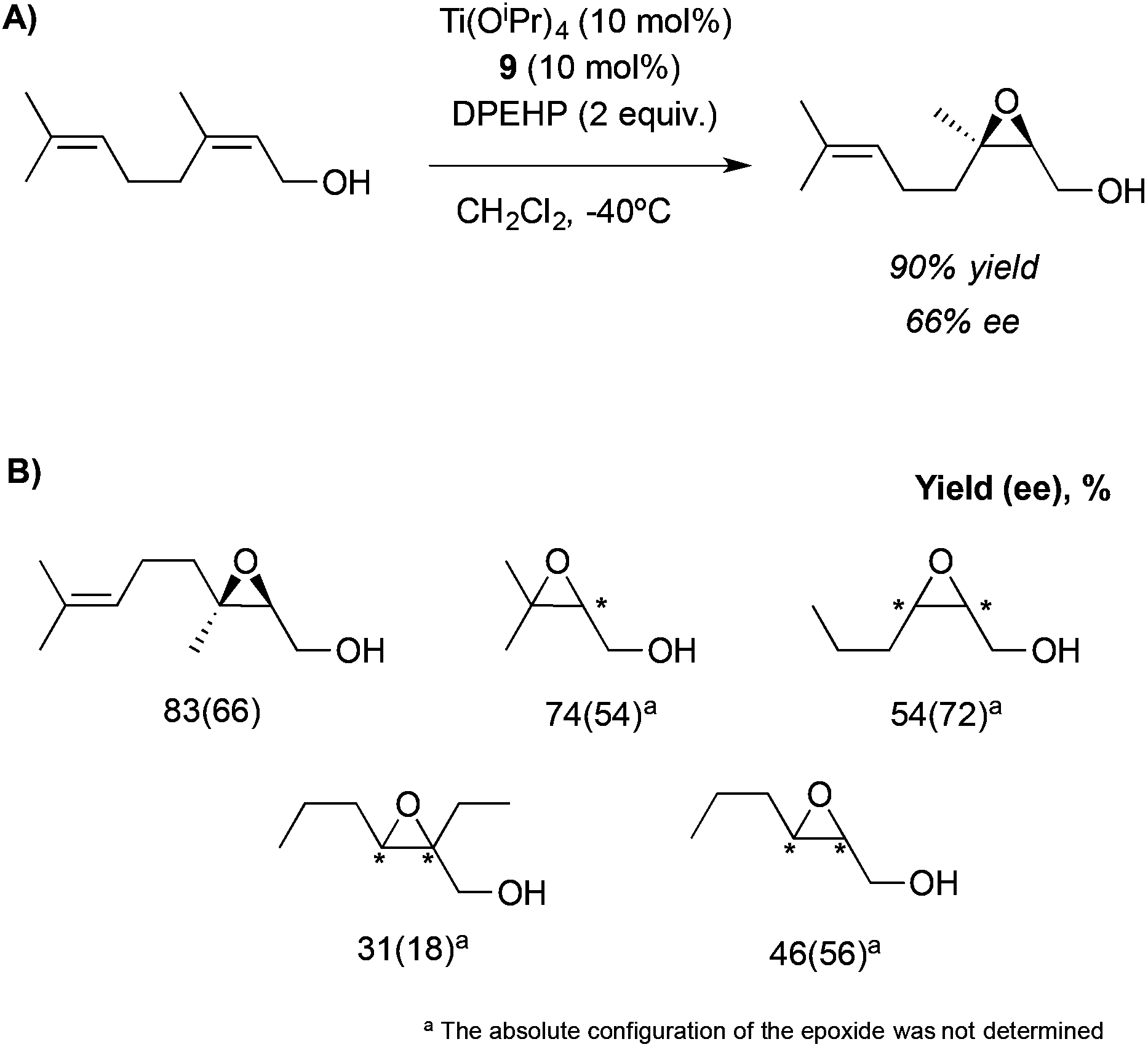 | ||
| Scheme 2 (A) Asymmetric epoxidation of nerol catalysed by the Ti-9 complex. (B) Substrate scope for this methodology. | ||
In 1999, Jacobsen and co-workers reported that the combination of metal ions and peptides can be effective in the asymmetric epoxidation of trans-β-methylstyrene.28 The structure of the ligands was based on a chiral peptide chain that contains some moieties that can interact with the metal center. It was envisioned that this structure would create a binding pocket accessible for the metal ions.
To design selective epoxidation catalysts, the authors used an elegant combinatorial chemistry approach (Fig. 6). Five different amino acids that present side chains with electron-donor character (A, B, C, D and E in Fig. 6) were attached to a polystyrene-based support. To these blocks, three linking elements were attached. A salicylimine ligand was also prepared, resembling the well-known salen-type ligands,29 highly effective for asymmetric epoxidation. Finally, 12 different end capping agents incorporating different heterocycles were attached to the previous 16 structures, generating a total of 192 ligands, which were combined with a series of 30 metal ion sources.
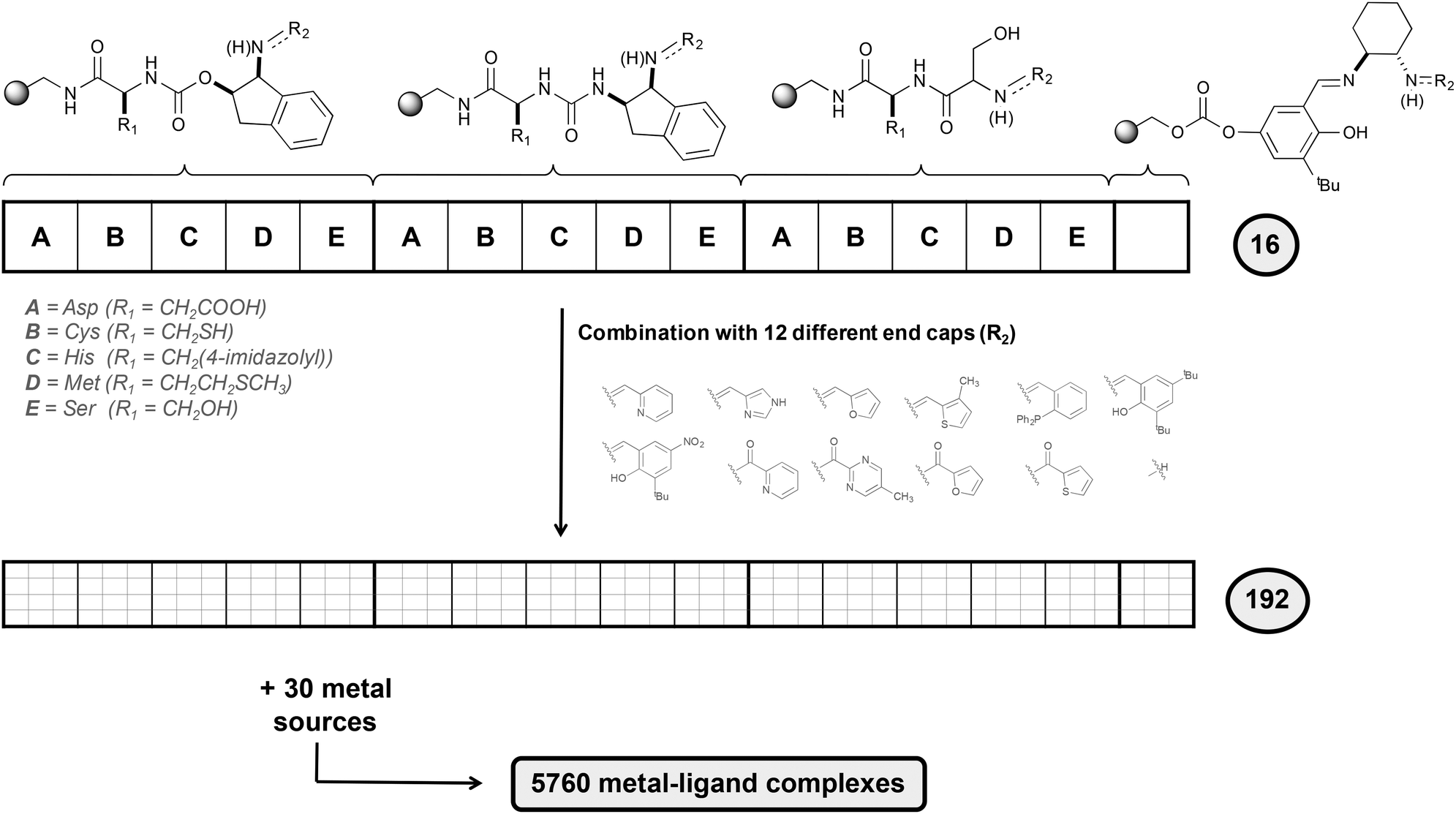 | ||
| Fig. 6 Schematic representation of the synthesis of the library following a combinatorial approach. | ||
A three-step procedure was then applied to identify efficient epoxidation catalysts from all the 5760 metal–ligand complexes synthesized. In the first step, the full library was tested in the epoxidation of trans-β-methylstyrene and the conditions of the reaction were optimized. The use of H2O2 as oxidant in combination with a mixture of dichloromethane and tert-butanol as solvent was found to be the best conditions. In the second step, a total of 30 sub-libraries were prepared, each containing a mixture of all 192 ligands and a metal source. It was observed that libraries containing FeII centers were the most active ones, in particular the library that contains FeCl2. Finally, in the third step the ligand components needed for the catalytic activity were determined by preparing 12 different sub-libraries, each containing the 16 basic structures with a unique ending cap. The results obtained showed that the most active libraries were those containing a pyridine moiety.
The enantioselectivity of the reaction was determined in the asymmetric epoxidation of trans-β-methylstyrene using the three most active complexes (Scheme 3A) however only low ee values were obtained (up to 7% ee). To improve the enantioselectivity of the catalysts, a parallel library was synthesized and three new structures were identified as efficient catalysts (Scheme 3B), obtaining the epoxide in up to 78% yield and 20% ee when 13 was used. This system reported by Jacobsen and co-workers constitutes also a pioneering work of the use of metallopeptides in oxidation reactions. Surprisingly, although the potential of peptides to be combined with metal centers is demonstrated, this field has not been further developed.
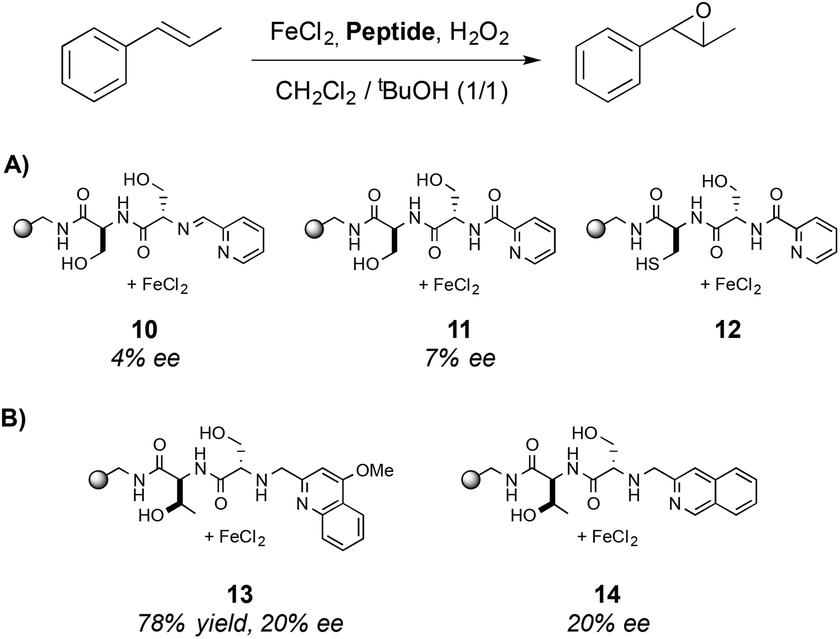 | ||
| Scheme 3 Metal–ligand complexes that present the highest activity in the asymmetric epoxidation of trans-β-methylstyrene. | ||
This work is one of the first examples of non-heme iron catalyzed asymmetric epoxidation using H2O2 as the oxidant. However, the levels of stereoselection and the activity of the catalysts were modest. It can be reasoned that the peptides employed are quite flexible, and contain several potential binding sites. Therefore, the peptides employed in this work do not provide a unique, rigid, and well-defined coordination sphere around the metal. Instead, binding of the peptide to the metal may result in multiple and distinct catalytically active species. In the following years there has been an increasing understanding of the key elements in the coordination chemistry architecture of iron complexes that are necessary to create efficient oxidation catalysts.30 Novel generations of iron and manganese coordination complexes, which contain rigid and oxidatively robust tetradentate ligands, have been described to activate H2O2via paths that resemble those operating in O2 activating enzymes, furnishing high product yields and enantioselectivities in C–H oxidation,31 epoxidation32 and syn-dihydroxylation.33
A synergistic combination of one of these catalysts with peptides has been recently described to create a highly enantioselective epoxidation catalytic system.34 In this work, peptides were employed with the aim to shape the second coordination sphere of the metal and also to establish non-covalent interactions with the substrates.4b,6b These features were envisioned to play an important role in defining the reactivity of the metal center and the chemo- and regioselectivity of the reactions.
In this work, the iron complex (S,S)-[Fe(OTf)2(Me2Npdp)] (15), was employed as a catalyst. It was previously shown that this catalyst activates hydrogen peroxide efficiently employing catalytic amounts of a carboxylic acid,32e and even an amino acid, as co-catalysts.32i Taking advantage of this feature, peptides with a terminal carboxylic acid were used as co-catalysts.
Different peptides were tested in combination with 15 in the asymmetric epoxidation of α-methylstyrene, which was taken as a model substrate (Scheme 4). It was found that these systems were able to activate hydrogen peroxide and epoxidize the olefin with excellent chemoselectivity. Interestingly, peptides did not alter the reactivity of the catalyst and were not oxidized under the experimental conditions, proving a remarkable compatibility. From the screening of peptides, there were different features that were found to be crucial to obtain high enantioselectivities.
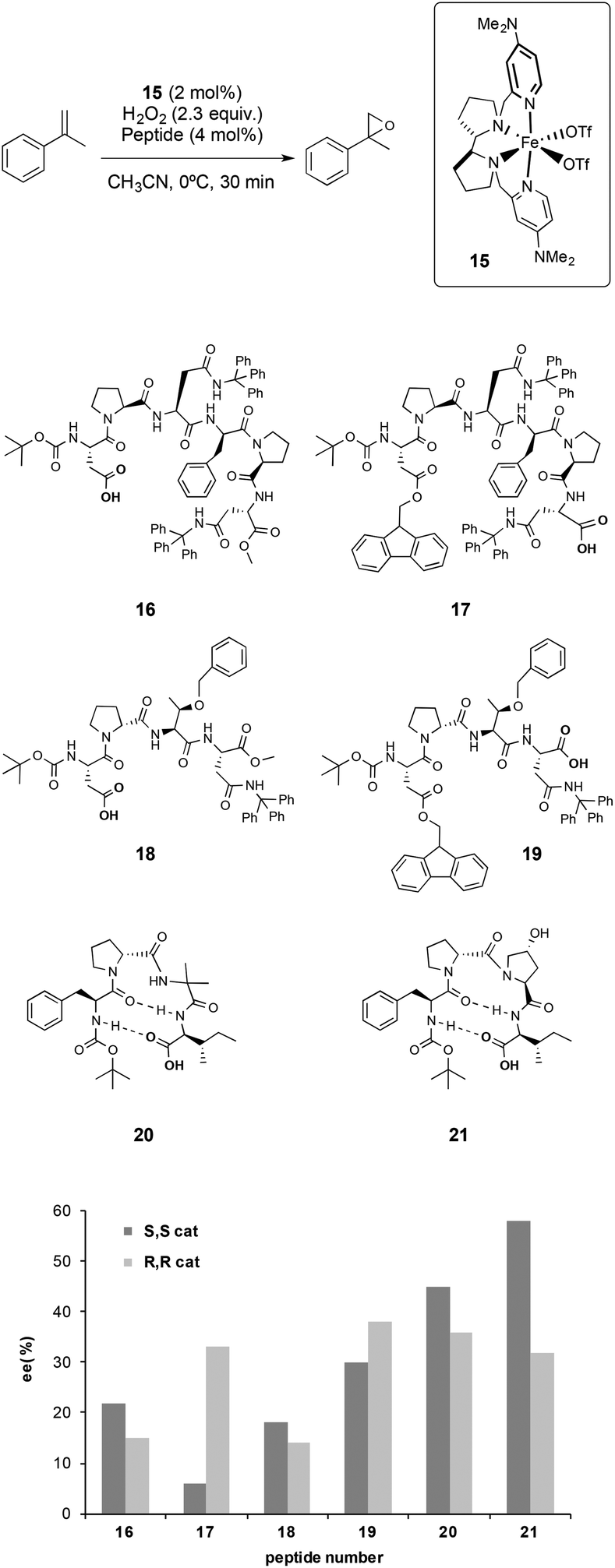 | ||
| Scheme 4 Diagrams of representative peptides studied in the asymmetric epoxidation of α-methylstyrene catalysed by 13 and enantioselectivities obtained. For each peptide, the dark grey bar corresponds to the ee obtained with (S,S)-15 while the light grey bar corresponds to the ee obtained with (R,R)-15. | ||
(i) The enantiomeric ratio of the product is sensitively dependent on the relative chirality of the metal catalyst and the peptides. Matching–mismatching effects resulting from the combination of the two chiralities were observed for a number of peptides, indicating that the peptide can play a crucial role in dictating the enantioselectivity of the oxidations, and that synergistic effects between the metal catalyst and peptide can be induced.
(ii) The position of the carboxylic acid in the structure of the peptide also plays an important role in determining the selectivity of the reactions, as different enantioselectivities were observed when peptides with identical structures except for the position of the carboxylic acid moiety were used (16vs.17 and 18vs.19 in Scheme 4). These observations, in combination with the results obtained in previous studies on the activity of non-heme iron catalysts with simple carboxylic acids;32c,e,35 suggest that the peptide acts as a ligand, likely via the carboxylic acid moiety.
(iii) Peptides that present a flexible backbone provide worse enantioselectivities than those obtained with more rigid peptides. The β-turn structure that the peptide adopts seems to be important to improve the stereoselectivity of the reactions, however the identification of structure–activity correlations is difficult due to the high complexity of the peptides tested by the authors, and because the number of peptides examined is relatively small.
All the set of reactions tested allows the identification of compound 21 as the best peptide within the series tested, which was subsequently studied in the asymmetric epoxidation of different α-methylstyrene derivatives (Scheme 5), substrates that are particularly challenging for current epoxidation technologies.32i,36 Moderate enantioselectivities were obtained with meta-, para- and non-substituted derivatives, but the values increase when ortho-substituted α-methylstyrene derivatives were tested, obtaining up to 92% ee.
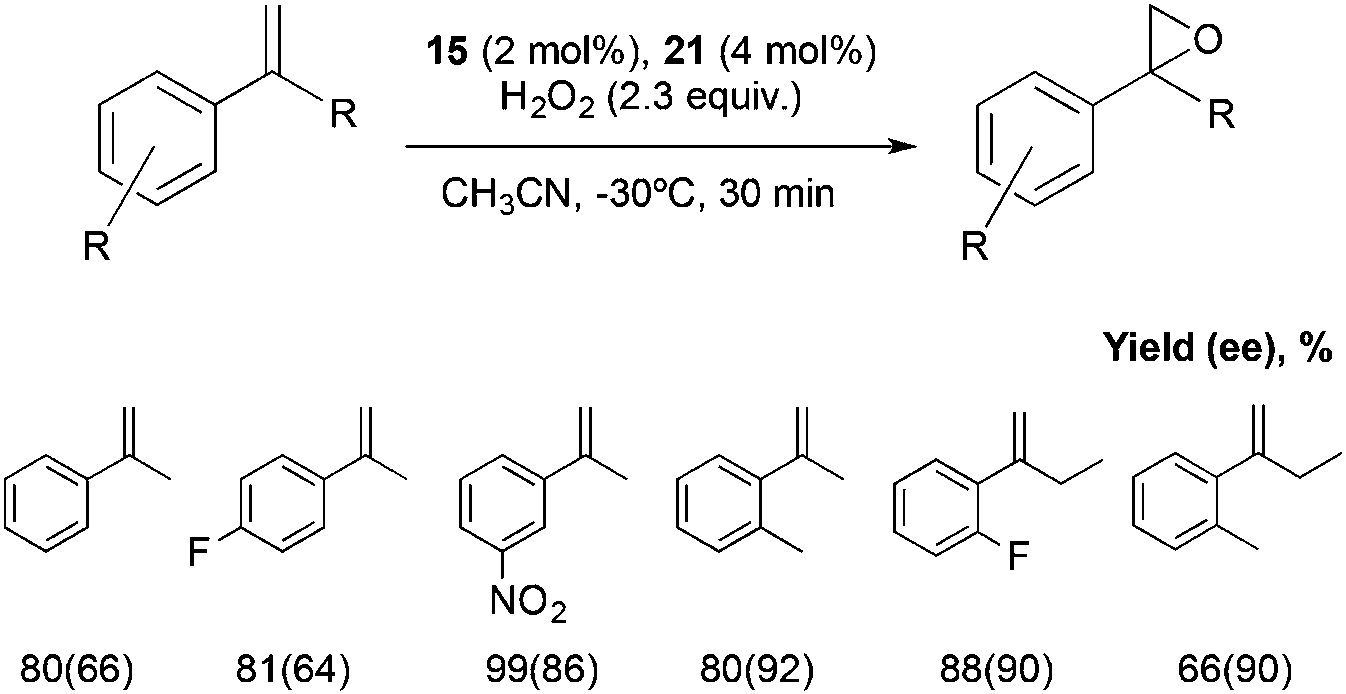 | ||
| Scheme 5 Representative examples of the substrate scope for this system. | ||
Different control experiments demonstrated the synergistic cooperation of the peptide and the metal catalyst in the activation of hydrogen peroxide and also in the highly chemo- and enantioselective oxygen atom transfer to the olefin.
In summary, it was also concluded that the carboxylic acid group of the peptide connects the metal center to the amino acid chain of the peptide, which is then defining the second coordination sphere around the metal (Fig. 7). Therefore, this system can be considered as an approach towards the design of artificial oxygenases.
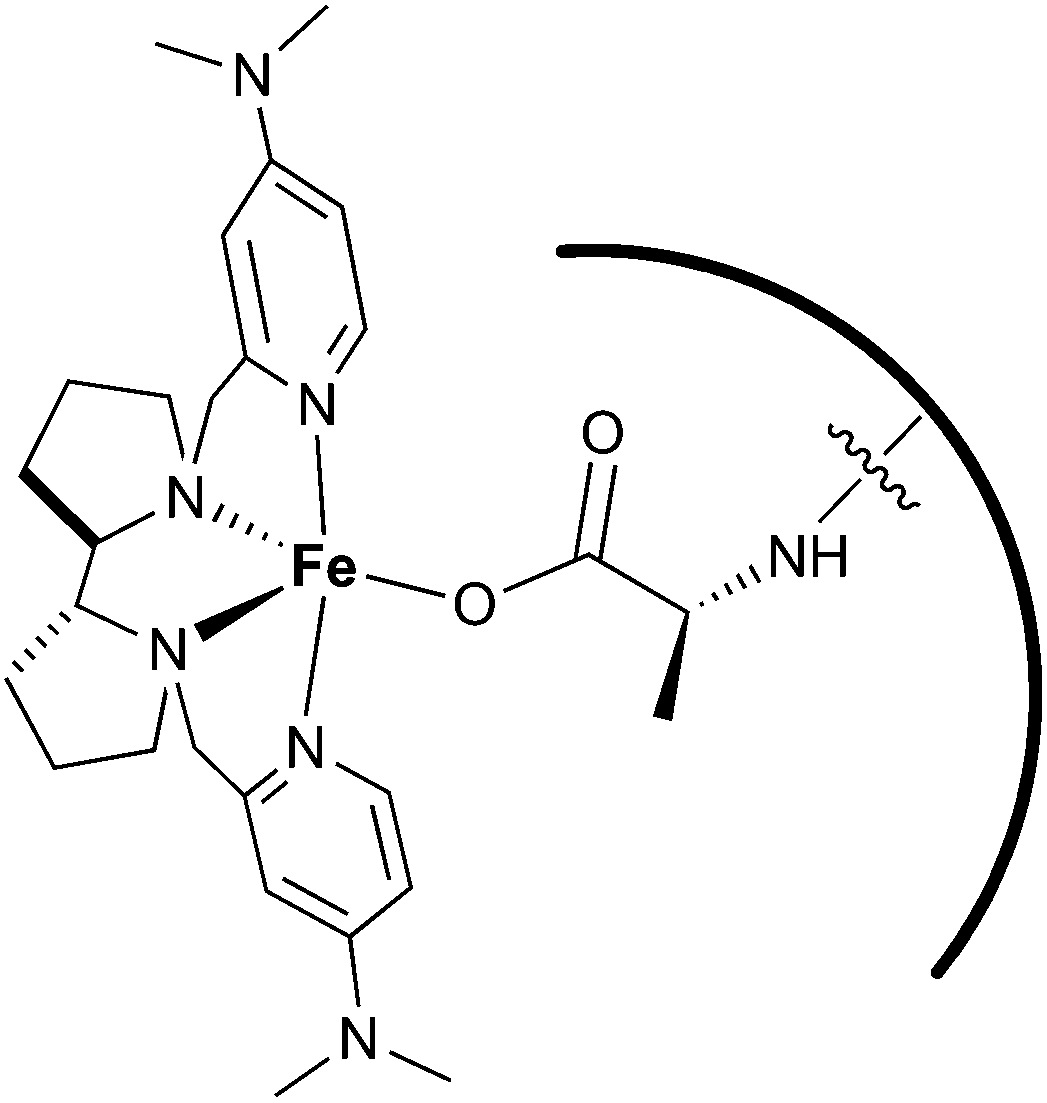 | ||
| Fig. 7 Schematic representation of the interaction between the peptide and the metal catalyst. | ||
The strategy described in this work relies on the accurate control of the first coordination sphere around the metal. At the same time, it also represents a promising platform for the rapid identification of peptides that will improve the enantioselectivity of previously described iron catalysts. However, this approach presents some weak points. Most importantly, the metal catalysts and the peptide are exclusively connected via the carboxylic acid moiety, so their interaction is non-directional. Therefore, this catalyst–peptide system is structurally very flexible, which can be considered as a drawback for favoring the selectivity of the system on the basis of specific interactions between the peptide and the substrate, which in turn must result in dominant catalyst–substrate relative orientations. Furthermore, the current study is based on unfunctionalized substrates, which are very unlikely to engage in weak interactions with the peptide, and therefore the selectivity role of the peptide may be almost exclusively based on steric interactions. Studies with substrates containing polar groups susceptible to engage in weak interactions such as H-bonding or electrostatic and hydrophobic interactions may represent an attractive future avenue.
Conclusions and outlook
In summary, metallopeptides have been used as catalysts in several asymmetric reactions, but only very few examples are applied so far to oxidation reactions. The current studies show that peptides appear to be compatible with oxidation catalysis and thus they can represent a valuable choice for ligand design. It is envisioned that the rich architecture of peptides, readily accessible via standard peptide synthesis methodologies, may allow introducing high structural versatility in metal oxidation catalysts, endowing the metals with high selectivity properties. The use of weak interactions between the substrate and peptides is expected to enable selectivities that are still inaccessible with the current synthetic methods used in organic synthesis, thus expanding the substrate scope of the reactions.Overall, metallopeptides may become a powerful tool in the development of new catalytic systems for bioinspired oxidation reactions, a field in which metallopeptides have rarely been used. The combination of metal catalysts and peptides would combine the potential of both systems, which can suppose an important step towards the development of novel oxidation technologies with utility in organic synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the MINECO of Spain (CTQ2015-70795-P) and the Catalan DIUE of the Generalitat de Catalunya (2009SGR637). M. C. is thankful for an ICREA-Academia award. We thank the MECD of Spain for an FPU grant to L. V. (FPU16/04231).References
- (a) M. Beller, Adv. Synth. Catal., 2004, 346, 107 CrossRef CAS; (b) R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2007 Search PubMed; (c) V. K. Ahluwalia, Oxidation in Organic Synthesis, CRC Press, Boca Raton, 2012 Search PubMed.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939 CrossRef CAS PubMed.
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239 CrossRef CAS PubMed.
- (a) W. J. Shaw, Catal. Rev., 2012, 54, 489 CrossRef CAS; (b) J. C. Lewis, ACS Catal., 2013, 3, 2954 CrossRef CAS.
- (a) E. R. Jarvo and S. J. Miller, Tetrahedron, 2002, 58, 2481 CrossRef CAS; (b) E. A. C. Davie, S. M. Mennen, Y. Xu and S. J. Miller, Chem. Rev., 2007, 107, 5759 CrossRef PubMed.
- (a) A. Mori, H. Nitta, M. Kudo and S. Inoue, Tetrahedron Lett., 1991, 32, 4333 CrossRef CAS; (b) A. Mori, H. Abet and S. Inoue, Appl. Organomet. Chem., 1995, 9, 189 CrossRef CAS.
- R. L. Davis, J. Stiller, T. Naicker, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2014, 53, 7406 CrossRef CAS PubMed.
- (a) S. Juliá, J. Masana and J. C. Vega, Angew. Chem., Int. Ed. Engl., 1980, 19, 929 CrossRef; (b) S. Julia, J. Guixer, J. Masana, J. Rocas, S. Colonna, R. Annuziata and H. Molinari, J. Chem. Soc., Perkin Trans. 1, 1982, 1317 RSC.
- (a) S. Colonna, H. Molinari, S. Banfi, S. Juliá, J. Masana and A. Alvarez, Tetrahedron, 1983, 39, 1635 CrossRef CAS; (b) S. Itsuno, M. Sakakura and K. Ito, J. Org. Chem., 1990, 55, 6047 CrossRef CAS; (c) P. A. Bentley, S. Bergeron, M. W. Cappi, D. E. Hibbs, M. B. Hursthouse, T. C. Nugent, R. Pulido, S. M. Roberts and L. Eduardo Wu, Chem. Commun., 1997, 739 RSC; (d) R. W. Flood, T. P. Geller, S. A. Petty, S. M. Roberts, J. Skidmore and M. Volk, Org. Lett., 2001, 3, 683 CrossRef CAS PubMed; (e) K. Kudo, K. Akagawa and T. Hirata, Synlett, 2015, 27, 1217 CrossRef.
- G. Carrea, S. Colonna, A. D. Meek, G. Ottolina and S. M. Roberts, Chem. Commun., 2004, 1412 RSC.
- (a) A. Berkessel, N. Gasch, K. Glaubitz and C. Koch, Org. Lett., 2001, 3, 3839 CrossRef CAS PubMed; (b) D. R. Kelly and S. M. Roberts, Chem. Commun., 2004, 2018 RSC; (c) S. P. Mathew, S. Gunathilagan, S. M. Roberts and D. G. Blackmond, Org. Lett., 2005, 7, 4847 CrossRef CAS PubMed.
- T. Komori and T. Nonaka, J. Am. Chem. Soc., 1983, 105, 5690 CrossRef CAS.
- G. Peris, C. E. Jakobsche and S. J. Miller, J. Am. Chem. Soc., 2007, 129, 8710 CrossRef CAS PubMed.
- C. E. Jakobsche, G. Peris and S. J. Miller, Angew. Chem., Int. Ed., 2008, 47, 6707 CrossRef CAS PubMed.
- (a) P. A. Lichtor and S. J. Miller, Nat. Chem., 2012, 4, 990 CrossRef CAS PubMed; (b) P. A. Lichtor and S. J. Miller, J. Am. Chem. Soc., 2014, 136, 5301 CrossRef CAS PubMed.
- N. C. Abascal, P. A. Lichtor, M. W. Giuliano and S. J. Miller, Chem. Sci., 2014, 5, 4504 RSC.
- F. Kolundzic, M. N. Noshi, M. Tjandra, M. Movassaghi and S. J. Miller, J. Am. Chem. Soc., 2011, 133, 9104 CrossRef CAS PubMed.
- (a) D. C. Dittmer, R. P. Discordia, Y. Zhang, C. K. Murphy, A. Kumar, A. S. Pepito and Y. Wang, J. Org. Chem., 1993, 58, 718 CrossRef CAS; (b) M. Uyanik, H. Ishibashi, K. Ishihara and H. Yamamoto, Org. Lett., 2005, 7, 1601 CrossRef CAS PubMed; (c) J. A. Marshall and R. K. Hann, J. Org. Chem., 2008, 73, 6753 CrossRef CAS PubMed; (d) T. Kotaki, T. Shinada, K. Kaihara, Y. Ohfune and H. Numata, Org. Lett., 2009, 11, 5234 CrossRef CAS PubMed; (e) J. Tanuwidjaja, S.-S. Ng and T. F. Jamison, J. Am. Chem. Soc., 2009, 131, 12084 CrossRef CAS PubMed; (f) A. Koohang, J. L. Bailey, R. M. Coates, H. K. Erickson, D. Owen and C. D. Poulter, J. Org. Chem., 2010, 75, 4769 CrossRef CAS PubMed.
- D. K. Romney and S. J. Miller, Org. Lett., 2012, 14, 1138 CrossRef CAS PubMed.
- (a) R. Sambasivan and Z. T. Ball, J. Am. Chem. Soc., 2010, 132, 9289 CrossRef CAS PubMed; (b) A. Monney and M. Albrecht, Chem. Commun., 2012, 48, 10960 RSC; (c) A. Monney, F. Nastri and M. Albrecht, Dalton Trans., 2013, 42, 5655 RSC; (d) Z. T. Ball, Acc. Chem. Res., 2013, 46, 560 CrossRef CAS PubMed.
- (a) R. Sambasivan and Z. T. Ball, Angew. Chem., Int. Ed., 2012, 51, 8568 CrossRef PubMed; (b) R. Sambasivan and Z. T. Ball, Chirality, 2013, 25, 493 CrossRef CAS PubMed; (c) R. Sambasivan, W. Zheng, S. J. Burya, B. V. Popp, C. Turro, C. Clementi and Z. T. Ball, Chem. Sci., 2014, 5, 1401 RSC.
- (a) S. J. Greenfield, A. Agarkov and S. R. Gilbertson, Org. Lett., 2003, 5, 3069 CrossRef CAS PubMed; (b) A. Agarkov, S. J. Greenfield, T. Ohishi, S. E. Collibee and S. R. Gilbertson, J. Org. Chem., 2004, 69, 8077 CrossRef CAS PubMed; (c) C. R. Landis and T. P. Clark, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5428 CrossRef CAS PubMed; (d) C. A. Christensen and M. Meldal, J. Comb. Chem., 2007, 9, 79 CrossRef CAS PubMed.
- (a) S. J. Degrado, H. Mizutani and A. H. Hoveyda, J. Am. Chem. Soc., 2001, 123, 755 CrossRef CAS PubMed; (b) B. Breit and A. C. Laungani, Tetrahedron: Asymmetry, 2003, 14, 3823 CrossRef CAS.
- (a) H. Nitta, D. Yu, M. Kudo, A. Mori and S. Inoue, J. Am. Chem. Soc., 1992, 114, 7969 CrossRef CAS; (b) N. S. Josephsohn, K. W. Kuntz, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2001, 123, 11594 CrossRef CAS PubMed.
- (a) J. F. Jensen, K. Worm-Leonhard and M. Meldal, Eur. J. Org. Chem., 2008, 3785 CAS; (b) G. Guisado-Barrios, B. K. Munoz, P. C. J. Kamer, B. Lastdrager, G. van der Marel, M. Overhand, M. Vega-Vazquez and M. Martin-Pastor, Dalton Trans., 2013, 42, 1973 RSC; (c) B. Kandemir, L. Kubie, Y. Guo, B. Sheldon and K. L. Bren, Inorg. Chem., 2016, 55, 1355 CrossRef CAS PubMed; (d) B. Kim, A. J. Chinn, D. R. Fandrick, C. H. Senanayake, R. A. Singer and S. J. Miller, J. Am. Chem. Soc., 2016, 138, 7939 CrossRef CAS PubMed.
- Y. Iseki, M. Kudo, A. Mori and S. Inoue, J. Org. Chem., 1992, 57, 6329 CrossRef CAS.
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974 CrossRef CAS.
- M. B. Francis and E. N. Jacobsen, Angew. Chem., Int. Ed., 1999, 38, 937 CrossRef CAS.
- (a) K. Srinivasan, P. Michaud and J. K. Kochi, J. Am. Chem. Soc., 1986, 108, 2309 CrossRef CAS PubMed; (b) W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobsen, J. Am. Chem. Soc., 1990, 112, 2801 CrossRef CAS; (c) E. N. Jacobsen, W. Zhang, A. R. Muci, J. R. Ecker and L. Deng, J. Am. Chem. Soc., 1991, 113, 7063 CrossRef CAS.
- (a) G. Olivo, O. Cussó and M. Costas, Chem. - Asian J., 2016, 11, 3148 CrossRef CAS PubMed; (b) G. Olivo, O. Cussó, M. Borrell and M. Costas, J. Biol. Inorg. Chem., 2017, 22, 425 CrossRef CAS PubMed.
- (a) K. Hamachi, R. Irie and T. Katsuki, Tetrahedron Lett., 1996, 37, 4979 CrossRef CAS; (b) A. Miyafuji and T. Katsuki, Tetrahedron, 1998, 54, 10339 CrossRef CAS; (c) T. Hamada, R. Irie, J. Mihara, K. Hamachi and T. Katsuki, Tetrahedron, 1998, 54, 10017 CrossRef CAS; (d) S.-I. Murahashi, S. Noji and N. Komiya, Adv. Synth. Catal., 2004, 346, 195 CrossRef CAS; (e) S.-I. Murahashi, S. Noji, T. Hirabayashi and N. Komiya, Tetrahedron: Asymmetry, 2005, 16, 3527 CrossRef CAS; (f) M. Milan, M. Bietti and M. Costas, ACS Cent. Sci., 2017, 3, 196 CrossRef CAS PubMed; (g) C. Miao, X.-X. Li, Y.-M. Lee, C. Xia, Y. Wang, W. Nam and W. Sun, Chem. Sci., 2017, 8, 7476 RSC; (h) E. P. Talsi, D. G. Samsonenko, R. V. Ottenbacher and K. P. Bryliakov, ChemCatChem, 2017, 9, 4580 CrossRef CAS.
- (a) Y. Nishikawa and H. Yamamoto, J. Am. Chem. Soc., 2011, 133, 8432 CrossRef CAS PubMed; (b) B. Wang, S. Wang, C. Xia and W. Sun, Chem. – Eur. J., 2012, 18, 7332 CrossRef CAS PubMed; (c) O. Y. Lyakin, R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2012, 2, 1196 CrossRef CAS; (d) X. Wang, C. Miao, S. Wang, C. Xia and W. Sun, ChemCatChem, 2013, 5, 2489 CrossRef CAS; (e) O. Cussó, I. Garcia-Bosch, X. Ribas, J. Lloret-Fillol and M. Costas, J. Am. Chem. Soc., 2013, 135, 14871 CrossRef PubMed; (f) O. Cussó, I. Garcia-Bosch, D. Font, X. Ribas, J. Lloret-Fillol and M. Costas, Org. Lett., 2013, 15, 6158 CrossRef PubMed; (g) L. Luo and H. Yamamoto, Eur. J. Org. Chem., 2014, 7803 CrossRef CAS PubMed; (h) R. V. Ottenbacher, D. G. Samsonenko, E. P. Talsi and K. P. Bryliakov, ACS Catal., 2014, 4, 1599 CrossRef CAS; (i) O. Cussó, X. Ribas, J. Lloret-Fillol and M. Costas, Angew. Chem., Int. Ed., 2015, 54, 2729 CrossRef PubMed; (j) O. Cusso, X. Ribas and M. Costas, Chem. Commun., 2015, 51, 14285 RSC; (k) O. Cussó, M. Cianfanelli, X. Ribas, R. J. M. Klein Gebbink and M. Costas, J. Am. Chem. Soc., 2016, 138, 2732 CrossRef PubMed; (l) D. Shen, B. Qiu, D. Xu, C. Miao, C. Xia and W. Sun, Org. Lett., 2016, 18, 372 CrossRef CAS PubMed; (m) W. Wang, Q. Sun, D. Xu, C. Xia and W. Sun, ChemCatChem, 2017, 9, 420 CrossRef CAS; (n) X. Chen, B. Gao, Y. Su and H. Huang, Adv. Synth. Catal., 2017, 359, 2535 CrossRef CAS.
- (a) M. Costas, A. K. Tipton, K. Chen, D.-H. Jo and L. Que, J. Am. Chem. Soc., 2001, 123, 6722 CrossRef CAS PubMed; (b) J. W. de Boer, W. R. Browne, S. R. Harutyunyan, L. Bini, T. D. Tiemersma-Wegman, P. L. Alsters, R. Hage and B. L. Feringa, Chem. Commun., 2008, 3747 RSC; (c) T. W.-S. Chow, Y. Liu and C.-M. Che, Chem. Commun., 2011, 47, 11204 RSC; (d) C. Zang, Y. Liu, Z.-J. Xu, C.-W. Tse, X. Guan, J. Wei, J.-S. Huang and C.-M. Che, Angew. Chem., Int. Ed., 2016, 55, 10253 CrossRef CAS PubMed.
- O. Cusso, M. W. Giuliano, X. Ribas, S. J. Miller and M. Costas, Chem. Sci., 2017, 8, 3660 RSC.
- (a) M. C. White, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 7194 CrossRef CAS PubMed; (b) R. Mas-Ballesté and L. Que, J. Am. Chem. Soc., 2007, 129, 15964 CrossRef PubMed; (c) S. R. Iyer, M. M. Javadi, Y. Feng, M. Y. Hyun, W. N. Oloo, C. Kim and L. Que, Chem. Commun., 2014, 50, 13777 RSC.
- (a) A. F. Dexter, F. J. Lakner, R. A. Campbell and L. P. Hager, J. Am. Chem. Soc., 1995, 117, 6412 CrossRef CAS; (b) B. Wang, O. A. Wong, M.-X. Zhao and Y. Shi, J. Org. Chem., 2008, 73, 9539 CrossRef CAS PubMed; (c) O. A. Wong, B. Wang, M.-X. Zhao and Y. Shi, J. Org. Chem., 2009, 74, 6335 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |