
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling with light the interaction between trans-tetrapyridyl ruthenium complexes and an oligonucleotide†
Vincent H. S.
van Rixel
a,
Geri F.
Moolenaar
a,
Maxime A.
Siegler
 b,
Luigi
Messori
c and
Sylvestre
Bonnet
*a
b,
Luigi
Messori
c and
Sylvestre
Bonnet
*a
aLeiden University, Leiden Institute of Chemistry, Gorlaeus Laboratories, P.O. Box 9502, 2300 RA Leiden, The Netherlands. E-mail: bonnet@chem.leidenuniv.nl
bSmall Molecule X-ray Crystallography Facility, Johns Hopkins University 3400N, Charles St. Baltimore, MD 21218, USA
cUniversity of Florence, Department of Chemistry, Via della Lastruccicia 3, 50019 Sesto Fiorentino, FI, Italia
First published on 23rd November 2017
Abstract
Three new trans-ruthenium(II) complexes coordinated to tetrapyridyl ligands, namely [Ru(bapbpy)(dmso)Cl]Cl ([2]Cl), [Ru(bapbpy)(Hmte)2](PF6)2 ([3](PF6)2), and [Ru(biqbpy)(Hmte)2](PF6)2 ([4](PF6)2), were prepared as analogues of [Ru(biqbpy)(dmso)Cl]Cl ([1]Cl), a recently described photoactivated chemotherapy agent. The new complexes were characterized, and their crystal structures showed the distorted coordination octahedron typical of this family of complexes. Their photoreactivity in solution was analyzed by spectrophotometry and mass spectrometry, which showed that the sulfur ligand was substituted upon blue light irradiation. The binding of the ruthenium complexes to a reference single-stranded oligonucleotide (s(5′CTACGGTTTCAC3′)) was explored both in the dark and under light irradiation by gel electrophoresis and high-resolution mass spectrometry. While adduct formation in the dark was negligible for the four complexes, light irradiation led to the formation of adducts with one or two ruthenium centers per oligonucleotide. The absence of interactions in the dark and the presence of complex–oligonucleotide adducts demonstrate that visible light controls the interaction of these ruthenium complexes with nucleic acids.
Introduction
The leading anti-cancer drug cisplatin is one of the landmarks in modern inorganic chemistry.1 In the body cisplatin enters cells, hydrolyzes, and forms irreversible adducts with DNA, ultimately triggering cell death via apoptosis.2–5 However, because of its lack of selectivity, severe side-effects are commonly associated with the use of cisplatin, including nephrotoxicity, neurotoxicity, ototoxicity, and nausea.6 Furthermore, cancer cells can acquire increasing levels of cisplatin-resistance.7 Alternatively, as transplatin is not active in vivo and less cytotoxic than cisplatin in vitro, anticancer metallodrugs with a trans geometry have not been seriously considered until recently.8–12Photodynamic therapy (PDT) is used in the clinic to locally activate anticancer pro-drugs, improve their selectivity, and reduce their side effects. PDT relies on the local activation of ground state triplet oxygen to highly reactive excited state singlet oxygen, thus critically depending on the presence of dioxygen. In anticancer therapy, photo-activated chemotherapy (PACT) is an emerging alternative strategy based on compounds that are chemically modified upon light irradiation without depending on the presence of oxygen.13–21 Notably, platinum-based PACT compounds have been described and often rely on the photoreduction of an octahedral platinum(IV) center to a square planar platinum(II) complex.14,22–25 Transplatin is also much more cytotoxic after UVA light irradiation than in the dark.25 However, light-activated platinum complexes often lack strong absorption in the visible region, which is a problem for photochemotherapy in vivo. Ruthenium(II) polypyridyl compounds have emerged as a promising alternative due to their excellent visible-light absorption properties.26,27 Most of the ruthenium-based PACT agents known to date contain two bidentate ligands based on the 2,2-bipyridine scaffold.13,28–31 After light activation, a cis-bisaqua complex is obtained that can bind to DNA in a fashion similar to cisplatin.32
By contrast, the trans compound [Ru(biqbpy)(dmso)Cl]Cl ([1]Cl) (biqbpy = 6,6′-bis[N-(isoquinolyl)-1-amino]-2,2′-bipyridine) has recently been reported as a phototoxic PACT agent that induces apoptosis upon green light activation.33 This new type of octahedral ruthenium complex consists of a tetrapyridyl biqbpy ligand that coordinates in the basal plane of the octahedron, allowing the coordination of two trans axial monodentate ligands that can be photochemically or thermally substituted. In our initial study we reported that a significant amount of ruthenium ends up in the nucleus of the cell, which justified DNA-binding studies using gel electrophoresis and circular pUC19 DNA plasmid. These studies clearly showed that the interaction with DNA was switched on by light irradiation. However, the nature of the metal–DNA adducts was unclear. In this work, we present three new derivatives of [1]Cl: [Ru(biqbpy)(Hmte)2](PF6)2 ([2](PF6)2, Hmte = 2-methylthioethanol), and two bapbpy analogues [Ru(bapbpy)(dmso)Cl]Cl ([3]Cl) and [Ru(bapbpy)(Hmte)2](PF6)2 ([4](PF6)2, (bapbpy = 6,6′-bis[N-(pyridyl)-1-amino]-2,2′-bipyridine)), and studied their interaction, in the dark and under light irradiation, with the oligodeoxyribonucleotide (ODN) s(5′CTACGGTTTCAC3′) (ODN1). Oligonucleotides have been widely used as models for DNA, to study its interaction with potential anti-cancer compounds.34–38 For the first time in PACT we introduce here a combined gel electrophoresis and high resolution electrospray injection mass spectrometry approach to study the binding of a light-activated drug to oligonucleotides.
Results
Synthesis
The complex [Ru(bapbpy)(dmso)Cl]Cl ([2]Cl) was synthesized by reacting bapbpy with 1.1 equivalents of [Ru(dmso)4Cl2] in ethanol overnight at 80 °C (Scheme 1) following the synthetic approach to [1]Cl.33 Reacting [1]Cl or [2]Cl with an excess of Hmte overnight at 80 °C in water, followed by anion exchange using KPF6, and size-exclusion chromatography, afforded [3](PF6)2 and [4](PF6)2, respectively (Scheme 1). In this reaction the chloride ligand instantly hydrolyzes, but an excess of thioether and heat were required to also substitute thermally the dmso ligand. In [3](PF6)2 and [4](PF6)2 an upfield shift of the 1H NMR signals corresponding to the methyl group of Hmte was observed (1.53 and 1.56 ppm in acetone-d6, respectively), compared to free Hmte (2.11 ppm). This upfield shift is characteristic of the shielding cone of polypyridyl ligands coordinated to the metal center, and indicated that Hmte was coordinated to ruthenium.33,39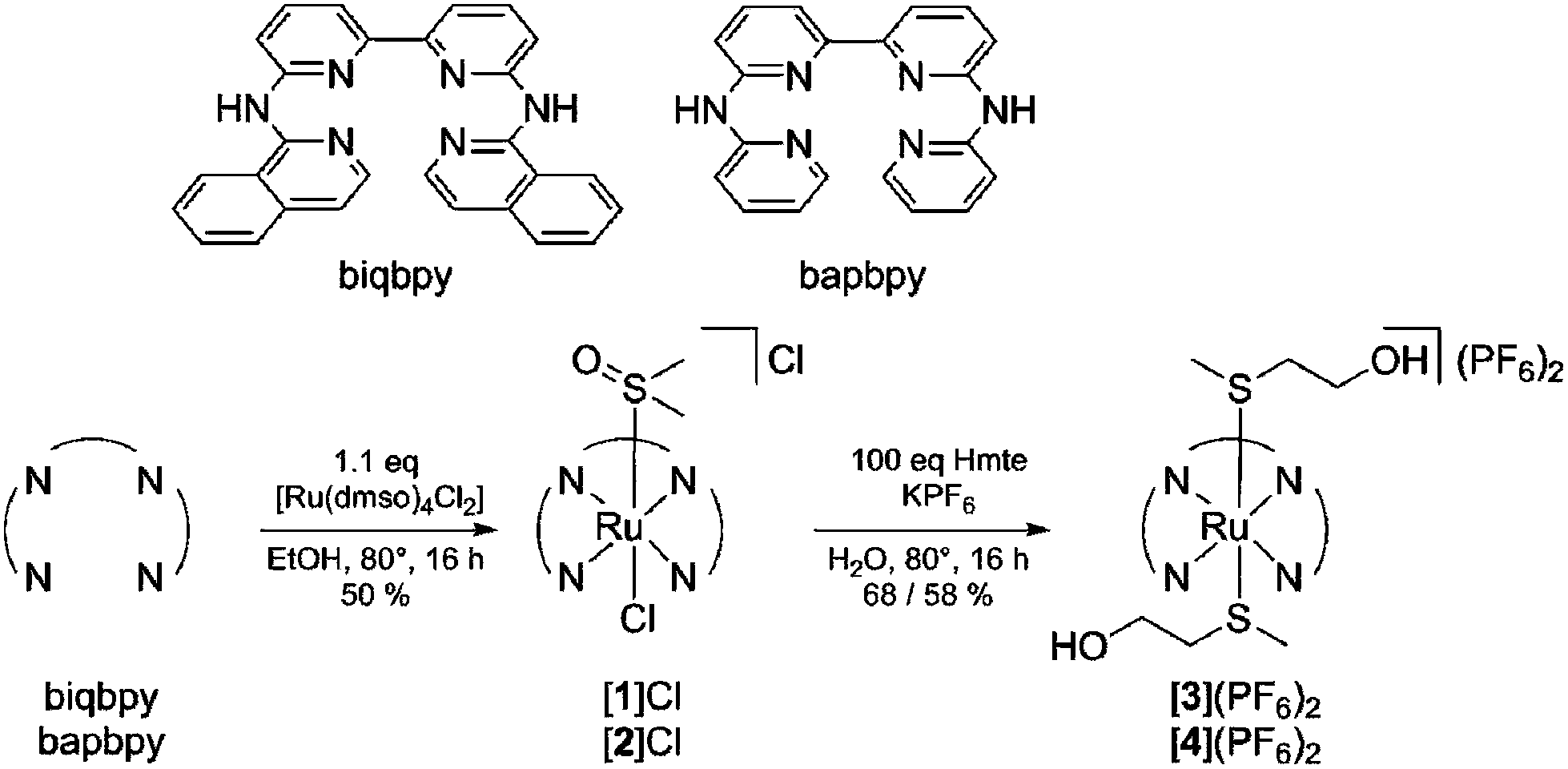 | ||
| Scheme 1 Synthesis of tetrapyridyl ruthenium complexes [1]Cl and [2]Cl, and their trans Hmte analogues [3](PF6)2 and [4](PF6)2. The conditions were adapted from Van Rixel et al.33 | ||
Crystal structures of the ruthenium compounds
Slow vapor diffusion of ethyl acetate into a methanol solution containing [2]Cl, and vapor diffusion of diethyl ether into an acetone solution of [3](PF6)2 or [4](PF6)2 yielded ruby-colored crystals suitable for X-ray structure determination. In the structure of [2]Cl, [3](PF6)2 and [4](PF6)2, the dmso or Hmte ligands were found to bind via their sulfur atom to ruthenium, as expected from the softness of the sulfur and ruthenium(II) atoms (Fig. 1). In the structure of [2]Cl, two slightly different geometries were found in the asymmetric unit. Selected bond distances, angles, torsion angles, and dihedral angles are summarized in Tables 1 and 2. The bond distances Ru–N1 and Ru–N6 fall between 2.09 and 2.11 Å, compared to 2.02–2.04 Å for Ru–N3 and Ru–N4. For [2a]Cl like for [1]Cl (Van Rixel et al.33) the Ru–S1 bond was 2.2266(13) Å, whereas the Ru–S1 bonds for [3](PF6)2 and [4](PF6)2 were 2.3661(7) and 2.3822(8) Å, respectively. This difference in bond length can be explained by the more electron-accepting character of the sulfoxide ligand and the better π-donating character of the chloride ligand trans to dmso, compared to Hmte. In [1]Cl and [2]Cl back-bonding into the dmso π* orbital is increased thereby resulting in a shorter Ru–S bond length. | ||
| Fig. 1 Displacement ellipsoid plots (50% probability level) of the cationic part, as observed in the asymmetric unit of (2 × [2]Cl) (A), ([3](PF6)2) (B), and ([4](PF6)2) (C) at 110(2) K. Counter anions and H atoms have been omitted for clarity. | ||
| Bond | [1]Cla | [2a]Cl | [2b]Cl | [3](PF6)2 | [4](PF6)2 |
|---|---|---|---|---|---|
| a Values are taken from a study by Van Rixel et al.33 | |||||
| Ru–N1 | 2.1018(19) | 2.102(4) | 2.098(4) | 2.109(3) | 2.101(3) |
| Ru–N3 | 2.020(2) | 2.027(4) | 2.023(4) | 2.036(2) | 2.029(3) |
| Ru–N4 | 2.0220(19) | 2.031(4) | 2.028(4) | 2.025(3) | 2.029(3) |
| Ru–N6 | 2.087(2) | 2.098(4) | 2.089(4) | 2.098(2) | 2.101(3) |
| Ru–Cl | 2.4363(5) | 2.4513(13) | 2.4360(11) | — | — |
| Ru–S1 | 2.2262(6) | 2.2266(13) | 2.2260(11) | 2.3661(7) | 2.3822(8) |
| Ru–S2 | — | — | — | 2.3847(7) | 2.3847(7) |
| Angle | [1]Cla | [2a]Cl | [2b]Cl | [3](PF6)2 | [4](PF6)2 |
|---|---|---|---|---|---|
| a Values are taken from a study by Van Rixel et al.33 b The dihedral angle α–β is formed by the planes between the terminal pyridyls (N1–C1–C2–C3–C4–C5 and N6–C16–C17–C18–C19–C20) or quinolyls (N1–C1–C2–C3–C4–C5–C6–C7–C8–C9 and N6–C20–C21–C22–C23–C24–C25–C26–C27–C28). | |||||
| N1–Ru–N3 | 91.77(8) | 89.89(14) | 91.63(14) | 90.13(9) | 91.56(11) |
| N1–Ru–N4 | 170.80(8) | 165.10(15) | 164.72(14) | 166.35(10) | 169.80(10) |
| N1–Ru–N6 | 97.90(8) | 97.98(15) | 96.03(14) | 99.08(9) | 96.68(14) |
| N3–Ru–N4 | 80.78(8) | 80.63(14) | 80.22(14) | 80.77(10) | 80.94(15) |
| N3–Ru–N6 | 164.83(8) | 170.92(14) | 171.25(14) | 168.14(9) | 169.80(10) |
| N4–Ru–N6 | 90.51(8) | 92.51(14) | 93.22(14) | 91.38(10) | 91.56(11) |
| N1–N3–N4–N6 | 12.78(9) | 12.5(2) | 13.3(2) | 13.7(1) | 9.9(1) |
| α–βb | 45.38 | 42.9(2) | 47.0(2) | 44.50(9) | 30.43(15) |
The torsion angle N1–N3–N4–N6 in all four complexes varied between 9.88 and 13.72°. The dihedral angle between the average planes of the terminal pyridyl or quinolyl moiety of the ligand, here labelled as the N1 and N6 plane, is also a measure of the flatness of the tetrapyridyl ligand. For [1]Cl, [2]Cl, [3](PF6)2 and [4](PF6)2 these angles are 45.38, 30.36 or 42.89, 44.50 and 30.40°, respectively. These values for [2]Cl and [4](PF6)2 strongly deviate due to the crystal lattice effects (polar π and hydrogen bonding interactions). Nonetheless, all the data demonstrate that the ruthenium complexes are distorted, due to the steric hindrance between the two hydrogen atoms borne by the C1 and C20 or C28 carbon atoms that constrains the ligand conformation to wrap in a helical fashion around the metal center.40,41 Neither the nature of the trans ligands (Hmte vs. dmso) nor the replacement of the terminal pyridyl moiety (in bapbpy) by an isoquinolyl moiety (in biqbpy) has a strong effect on the bond angles and coordination bond distances of the tetrapyridyl ligand.
Photoreactivity of [2]Cl, [3](PF6)2, and [4](PF6)2
The photoreactivity of [2]Cl, [3](PF6)2, and [4](PF6)2 was tested under blue light irradiation (λexc = 445 nm) in aqueous solution and monitored by mass spectrometry and UV-vis spectroscopy. For [2]Cl, initially dissolved in CD3OD, mass spectrometry before irradiation and for the dark control samples (Fig. S2A†) showed peaks at m/z = 280.3 corresponding to [Ru(bapbpy)(dmso)(CD3OD)]2+ (calc. m/z = 280.1), indicating that the chloride ligand hydrolyzed quickly and spontaneously upon dissolution in water, but that dmso was thermally stable. After light irradiation, however, these peaks were replaced by a new set of peaks at m/z = 249.7 corresponding to [Ru(bapbpy)(H2O)(CD3OD)]2+ (calc. m/z = 250.0) (Fig. S2B†). This indicates that the dmso ligand in [2]Cl (similar to [1]Cl) is quantitatively photosubstituted by a solvent molecule. When the photoreaction was followed by UV-vis spectroscopy, irradiation of [2]Cl was characterized by isosbestic points at 281 and 302 nm, and resulted, in the steady-state, in a new spectrum characterized by absorption maxima at 306, 335, 404 and 495 nm, similar to what was previously reported for [1]Cl (Fig. 2A). The quantum yield for the photosubstitution of dmso by water was 0.004 and comparable to that found for [1]Cl.33 | ||
| Fig. 2 Evolution of the electronic absorption spectra of a solution of [2]Cl (A) in demineralized water (5 v/v% CD3OD) of [3](PF6)2 (B) in demineralized water (5 v/v% acetone-d6) upon blue light irradiation (λirr = 445 nm (Δλ1/2 = 22 nm), photon flux Φ = 1.81 × 10−7 mol s−1, tirr = 15 min). Time: 0 min (red curve) to 15 min (black curve). Conditions [Ru]0 = 0.05 mM and the irradiated volume was 3.0 mL at 298 K. Inset: a plot of the absorbance at 350 nm (A) or 480 nm (B) as a function of irradiation time. | ||
For [3](PF6)2 and [4](PF6)2 blue light irradiation in aqueous solution analyzed afterwards by mass spectrometry showed peaks at m/z = 317.2 and m/z = 266.9 corresponding to [3 – Hmte]2+ and [4 – Hmte]2+, respectively (calc. m/z = 317.1 and 267.0, see Fig. S3†). The dark control samples also showed these peaks, but in addition peaks at m/z = 363.0 and m/z = 313.1 corresponding to [3]2+ and [4]2+ (calc. m/z = 363.1 and 313.1, Fig. S4†) were detected. These data indicate that blue light irradiation of complexes [3](PF6)2 and [4](PF6)2 results in the photosubstitution of one Hmte ligand for a weakly bound solvent molecule that is removed in the mass spectrometer. When the irradiation reaction of [3](PF6)2 was followed by UV-vis spectroscopy the spectrum evolved over time showing an overall increase in intensity and a slight red-shift of the metal-to-ligand-charge transfer (MLCT) from 451 nm to 465 nm (Fig. 2B). Upon irradiation of a solution of [4](PF6)2, a general increase in the visible domain through an isosbestic point at 398 nm was observed (Fig. S5†). Overall, the three new complexes show and confirm that the coordination of bapbpy or biqbpy to ruthenium enables photosubstitution of the axial trans ligands.
Gel electrophoresis with ODN1
ODNs have proven to be valuable model targets for anti-cancer compounds because they can be specifically designed, and easily studied by, for instance, mass spectrometry.42,43 As [1]Cl is phototoxic, cytotoxic, and enters the nucleus, we aimed to study our small library of tetrapyridyl ruthenium complexes in their reactions with a standard ODN. Thus, the ODN s(5′CTACGGTTTCAC3′) – hereafter denoted as ODN1 – was reacted with [1]Cl, [2]Cl, [3](PF6)2 and [4](PF6)2 to start building a structure–activity relationship. Like in previous studies ODN1 contained a GG-box, an important binding motif for cisplatin.43First, the outcomes of the dark and photochemical reactions were analyzed using gel electrophoresis to study the influence of light on the interaction of the Ru complexes with ODN1. The binding of a ruthenium complex to ODN1 significantly adds to the molecular weight of the oligonucleotide, and can add up to two positive charges per ruthenium complex to an otherwise negatively charged oligonucleotide. Both factors contribute to retardation during the migration of ODN1 on the gel. In these experiments, 3 equivalents of [1]Cl, [2]Cl, [3](PF6)2, or [4](PF6)2 were added to ODN1, light-activated, and incubated for 6 hours with 1 eq. of the ODN1 ([ODN] = 0.25 mM and [Ru] = 0.75 mM]). The mixtures were then pipetted on a gel, the gel was run, and the adducts were visualized using toluidine blue (Fig. 3).44 Without light-activation, all complexes (Fig. 3, lanes 1, 3, 5, and 7) showed identical band migration as the untreated ODN1 control (lane 9), indicating that under such conditions no ruthenium–ODN1 adducts formed in the dark, irrespective of the structure of the complex. When the compounds were light-activated in the presence of ODN1, all free ODN1 bands decreased in intensity, and two extra bands appeared above the original ODN1 band. This strongly indicates that each light-activated ruthenium complex forms two distinct adducts with ODN1.
 | ||
| Fig. 3 Gel electrophoresis of mixtures of ODN1 (5′CTACGGTTTCAC3′) and [2]Cl, [1]Cl, [4](PF6)2, or [3](PF6)2, on polyacrylamide gel. Lanes 1, 3, 5, and 7 correspond to the dark reaction between ODN1 and [2]Cl, [1]Cl, [4](PF6)2, or [3](PF6)2 respectively. Lanes 2, 4, 6, and 8 have the same composition but were irradiated. Lanes 9 and 10 correspond to ODN1 and 19–25 mer controls, respectively (in the dark). Conditions: T = 25 °C, [Ru] = 0.75 mM, [ODN] = 0.25 mM, and staining agent: toluidine blue. Conditions: irradiation λexc = 445 nm, Δλ1/2 = 22 nm, photon flux Φ = 2.2 × 10−7 mol s−1, tirr = 10 min, and tinc = 6 h. | ||
A second gel electrophoresis measurement was performed, including 19-, 23, 24, and 31-mer oligonucleotides, and an untreated ODN1 control sample that was irradiated (Fig. S10†). The first ruthenium–ODN1 adduct band ended up between the bands of the 24-mer and 31-mer control ODNs. The second ruthenium–ODN1 adduct band ended up between the 19-mer control and the untreated ODN1 irradiated control. As the coordination of one of the ruthenium complexes to ODN1 would increase the molecular weight equivalent of 1–2 base pairs, the band of such an adduct would be at the level of a 13- or 14-mer in the case of a monoruthenium-adduct, and at the level of a 15- or 16-mer for a bisruthenium-adduct. As migration of the adduct bands did not correspond to these levels, the (di)cationic charge of the ruthenium must have an extra lagging effect on the adduct band in gel migration. Lastly, the irradiated ODN1 control band runs at the same level as the dark ODN1 control band, indicating that irradiation did not have any effect on the ODN1 itself. Overall, whereas no reaction occurred in the dark, light-activation of all four ruthenium complexes induced strong interactions with ODN1. However, it was impossible at this stage to characterize these adducts.
High-resolution mass spectrometry
In order to identify these ruthenium–ODN1 adducts similar experiments were conducted but the samples were investigated using high-resolution mass spectrometry. For the dark reaction, ODN1 was incubated in LC-MS grade water at 25 °C for 6 hours – while shielded from light – in the presence of 3 equivalents of one of the four ruthenium complexes. Then, mass spectra in negative mode were recorded, and deconvoluted for clarity.45–47 Incubation of ODN1 with [1]Cl or [2]Cl without light-activation resulted in major signals at m = 3595.7 or 3617.6 corresponding to free ODN or ODN + Na+ (calc. m = 3595.7 or 3617.6), respectively. Minor signals at m = 4213.6 corresponding to [ODN1 + Ru(biqbpy)(dmso) − 2H] (calc. m = 4212.7, see Fig. 4A and S13A†) and minor signals at m = 4113.1 corresponding to [ODN1 + Ru(bapbpy)(dmso) − 2H] (calc. m = 4112.7, see Fig. S14A†), respectively, indicated that limited adduct formation in the dark was possible, but that the dmso remained bound to ruthenium. When comparing [1]Cl and [2]Cl the ratio between the adduct signals and that of the free ODN1 was found to be lower for [1]Cl than for [2]Cl, indicating that under such conditions [2]Cl was more susceptible to adduct formation with ODN1 than [1]Cl. When ODN1 was incubated with [3](PF6)2 or [4](PF6)2 the major peaks were found at m = 3595.7 corresponding to the free ODN1 (calc. m = 3594.6, Fig. S15A and S16A,† respectively). Whereas incubation with [3](PF6)2 resulted in virtually no ruthenium adduct signals, with [4](PF6)2 minor signals at m = 4034.7 corresponding to ruthenium adducts [ODN1 + Ru(bapbpy) − 2H] (calc. m = 4034.7) were found. On the one hand, no Hmte ligand stayed coordinated to the ruthenium center in such adducts; on the other hand, these signals were of lower intensity compared to that of the ruthenium adducts measured with [2]Cl (Fig. S14A†). Overall, for both the bapbpy and biqbpy complexes coordination of two trans thioethers inhibited the formation of ODN1–ruthenium adducts, compared to the coordination of dmso and a trans chloride ligand that is hydrolyzed thermally in water.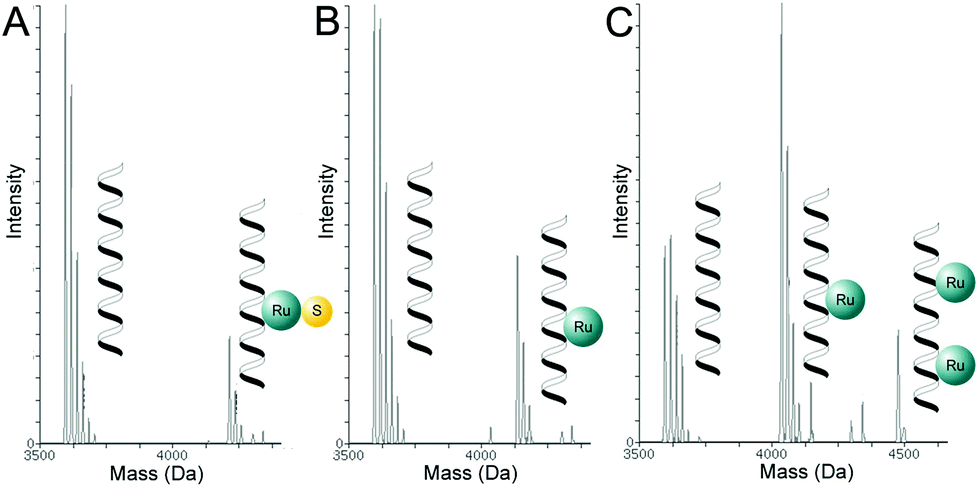 | ||
| Fig. 4 Representative scheme of the deconvoluted mass spectrometry data of ODN1 mixed with [1]Cl incubated (A) without light-activation, (B) with light-actviation, (C) and [2]Cl with light activation. The full dataset and interpretation can be found in the ESI.† Conditions: [ODN] = 0.03 mM, [Ru] = 0.09 mM, tirr = 10 min, tinc = 6 h, T = 298 K, λirr = 445 nm (Δλ1/2 = 22 nm), and the photon flux Φ = 2.17 × 10−7 mol s−1. Each helix represents a DNA single strand. | ||
Adduct-formation was also measured by ESI-MS after mixing ODN1 and the ruthenium complex, shining visible light (λirr = 445 nm, 10 min), followed by 6 h incubation. With [1]Cl the signals for free ODN1 were still dominant but a new set of peaks observed at m = 4136.3 corresponded to the adduct [ODN1 + Ru(biqbpy) − 2H] (calc. m = 4136.7, see Fig. 4B and Fig. S13B†). This peak was different from the peak observed in the dark, since the dmso ligand had been removed. With [2]Cl the strongest signals were observed at m = 4035.6 and corresponded to a similar adduct [ODN1 + Ru(bapbpy) − 2H] (calc. m = 4034.7), while a minor set of peaks for m = 4475.0 corresponded to the dinuclear adduct [ODN1 + (Ru(bapbpy))2 − 4H] (calc. m = 4474.7, Fig. 4C and Fig. S14B†). It is noteworthy that the signals at m = 3617.0 corresponding to [ODN1 + Na] (calc. m = 3617.6) remained, but they were significantly less intense compared to those of the dark control. These results confirmed, on the one hand, that when bound to ruthenium the dmso ligand has an inhibiting effect on the adduct formation with ODN1; on the other hand, the dmso ligand is cleaved off by light irradiation.
With [3](PF6)2 upon light irradiation new signals were observed at m = 4135.3 corresponding to the adduct [ODN1 + Ru(biqbpy) − 2H] (calc. m = 4134.7, see Fig. S15B†). However, the signals at m = 3617.4 corresponding to [ODN1 + Na] were still dominant. This behavior was similar to that found for [1]Cl in the presence of light, which confirmed the observations using gel electrophoresis that [1]Cl and [3](PF6)2 are, after light irradiation, similar. With [4](PF6)2 like with [2]Cl dominant signals were observed, after light activation, at m = 4034.8, which corresponded to the adduct [ODN1 + Ru(bapbpy) − 2H] (calc. m = 4034.7, see Fig. S16†). Here as well, both thioether ligands had been photosubstituted. This result confirmed that [4](PF6)2 and [2]Cl interact in a similar fashion with ODN1 after light irradiation, and that the bapbpy-based complexes interact better with ODN1 compared to their biqbpy-based analogues.
Overall, one should note that the ESI-MS method used in these studies was not calibrated for quantitative measurements. However, considering that all investigated reaction mixtures have a limited degree of molecular variation, a qualitative conclusion can be drawn based on these data. Three well-defined trends can be delineated. First, the light-activation of the complex strongly enhances the adduct formation between trans tetrapyridyl ruthenium complexes and ODN1, which depends on whether none, one, or two axial ligands are coordinated. Second, the nature of the tetrapyridyl ligand, bapbpy vs. biqbpy, influences significantly the degree of adduct formation after light activation: the bapbpy complexes are more prone to interact with ODN1 than the biqbpy complexes. Third, light activation of [2]Cl and [4](PF6)2 leads to the formation of adducts with ODN1 that can have either one or two bound ruthenium complexes.
Discussion & conclusion
In this work we have demonstrated that light can control the coordination interaction between trans ruthenium complexes with the bapbpy or biqbpy ligand and the oligonucleotide ODN1. No traces of adducts containing one ruthenium center and two oligonucleotides were found, indicating that under such conditions no inter-strand crosslinking occurs. The almost complete absence of adduct formation in the dark demonstrates that a single dmso ligand or two trans thioethers strongly inhibit the interaction of the metal complex with nucleotides. Meanwhile, by combining the gel electrophoresis and mass spectrometry results, we observed that the adducts formed upon light activation consist of one strand bound to one or two ruthenium complexes.trans Ruthenium complexes cannot mimic the cisplatin binding mode, suggesting that a different mechanism of binding takes place producing a different type of DNA distortion. While the ESI-MS data in our study suggest that after light-activation the trans complexes have two coordination sites available for nucleotide binding, it is hard to imagine both being employed by coordination to adjacent DNA bases like cisplatin. Transplatin, on the other hand, is known to form crosslinks between different strands. We found no indication here suggesting that the trans ruthenium complexes [1]+–[4]2+ would form adducts with two or more oligonucleotides. Other interactions responsible for the binding of metallodrugs to DNA involve hydrogen bonding with especially the phoshphate backbone,3,48,49 or pi–pi stacking interactions with the base pairs.50–52 However, these interactions dramatically depend on the supramolecular assembly of nucleic acids (double-strands vs. single-strands, G-quadruplexes vs. 3-way junctions, etc.), and short single-stranded oligonucleotides such as ODN1 cannot model such interactions. All four compounds in this study are similar in terms of hydrogen-bonding properties as they all have two non-coordinated NH bridges, and [1]+ and [2]+ on the one hand and [3]2+ and [4]2+ on the other hand share very similar coordination properties. However, the complexes based on biqbpy have a stronger aromatic and lipophilic character compared to those based on bapbpy, and [2]+ and [4]2+ indeed were found to bind slightly less to ODN1 compared to their bapbpy analogues [1]+ and [3]2+ (all other conditions being identical). Overall, non-coordinating interactions may play a role in the interaction of biqbpy- and bapbpy-based trans ruthenium complexes with DNA, but better DNA models, e.g. double-strands, would be necessary for such studies, as well as more quantitative analytical techniques.53 Still, oligonucleotides appear as an excellent tool to characterize the coordination properties of (light-activated) metal-based drugs to nucleic acids, with minimal interference from secondary interactions such as hydrogen bonding and pi–pi stacking.
Experimental part
Materials and methods
All 1H NMR spectra were recorded on a Bruker DPX-300, DMX-400, or AV-500 spectrometer. Chemical shifts are indicated in ppm relative to tetramethylsilane. Mass spectra were recorded by using a Thermoquest Finnagen AQA Spectrometer and an MSQ Plus Spectrometer positive ionization mode. For a schematic representation of [Ru(bapbpy)L2] and [Ru(biqbpy)L2] with atom numbering used in NMR attribution, see Fig. S1.† UV-vis experiments were performed on a Cary 50 Varian spectrometer equipped with a Cary Single Cell Peltier for temperature control.The ligands 6,6′-bis[N-(pyridyl)-1-amino]-2,2′-bipyridine (bapbpy), 6,6′-bis[N-(isoquinolyl)-1-amino]-2,2′-bipyridine (biqbpy), and [Ru(dmso)4Cl2] were synthesized according to literature procedures.33,40,41,54 [2-(Methylthio)ethanol (Hmte) and ODN1 (bought HPLC-purified) were purchased from Sigma-Aldrich.
Synthesis and crystallography
Single crystals of [2]Cl were obtained by crystallization via liquid–vapor diffusion using MeOH as the solvent and EtOAc as the counter-solvent. In short, 1.0 mg of [2]Cl was dissolved in 1 mL of MeOH and placed in a small GC vial. This vial was placed in a larger vial containing 2.8 mL of EtOAc. The large vial was closed and vapor diffusion occurred within a few days to afford X-ray quality crystals.
The structure is ordered. The crystal was found to be twinned non-merohedrally, and the twin relationship is a twofold axis found along the direct-space vector 0.0044 0.0086 1.0000. The batch scale factor of the minor component refines to 0.3188(11). The final structure refinement was performed using the HKL5 instruction (true hkl files including reflection sets for the twinned component 1 and the associated overlapped reflections from component 2).
Crystal data: Formula: C22H22Cl2N6O1Ru1S1. Fw = 590.48, a small orange block, 0.22 × 0.17 × 0.14 mm3, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 12.6164(2), b = 13.6053(3), c = 14.3404(3) Å, α = 100.1729(17), β = 92.9120(16), γ = 106.0391(17)°, V = 2315.57(8) Å3, Z = 4, Dx = 1.694 g cm−3, μ = 1.027 mm−1, and Tmin–Tmax: 0.831–0.900. 36
(no. 2), a = 12.6164(2), b = 13.6053(3), c = 14.3404(3) Å, α = 100.1729(17), β = 92.9120(16), γ = 106.0391(17)°, V = 2315.57(8) Å3, Z = 4, Dx = 1.694 g cm−3, μ = 1.027 mm−1, and Tmin–Tmax: 0.831–0.900. 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 503 reflections were measured up to a resolution of (sinθ/λ)max = 0.59 Å−1. 9478 reflections were unique (Rint = 0.031, including the overlapped reflections from the twin component 2), of which 7962 were observed [I > 2σ(I)]. 616 parameters were refined using 4 restraints. R1/wR2 [I > 2σ(I)]: 0.0410/0.1012. R1/wR2 [all refl.]: 0.0536/0.1091. S = 1.096. The residual electron density was found between −0.75 and 0.87 e Å−3. Deposited as CCDC 1535298.†
503 reflections were measured up to a resolution of (sinθ/λ)max = 0.59 Å−1. 9478 reflections were unique (Rint = 0.031, including the overlapped reflections from the twin component 2), of which 7962 were observed [I > 2σ(I)]. 616 parameters were refined using 4 restraints. R1/wR2 [I > 2σ(I)]: 0.0410/0.1012. R1/wR2 [all refl.]: 0.0536/0.1091. S = 1.096. The residual electron density was found between −0.75 and 0.87 e Å−3. Deposited as CCDC 1535298.†
:2:2), the reaction was cooled down to room temperature and KPF6 (0.4 g, 2.2 mmol) was added. Then, the aqueous layer was extracted with dichloromethane (3 × 15 mL), and the organic layer was evaporated in vacuo. The compound was purified using size-exclusion chromatography (Sephadex LH-20, acetone). After evaporation, the product was reprecipitated from acetone (0.5 mL) by the addition of diethyl ether (10 mL). Yield: 19 mg (68%). 1H NMR (400 MHz, 300 K, acetone-d6): δ = 10.42 (s, 2H, NH), 8.87 (d, 2H, J = 8.8 Hz, Hqi-4), 8.68 (d, 2H, J = 7.6 Hz, Hqi-7), 8.64 (d, 2H, J = 6.8 Hz, Hqi-9), 8.40 (t, 2H, J = 8.4 Hz, Hqi-6), 8.16 (d, 4H, J = 7.6 Hz, H3 and H5), 8.02 (t, 2H, J = 7.2 Hz, Hqi-5), 7.94 (t, 2H, J = 8.4 Hz, Hqi-10), 3.39–3.32 (m, 4H, Hβ), 2.10–2.00 (m, 4H, Hγ), 2.10–2.00 (m, 4H, Hγ), 1.53 (s, 6H, Hα). High resolution ES MS m/z (calc.): 725.1301 (725.1309, [M − H]+). 13C NMR (125 MHz, 300 K, acetone-d6): δ = 156.70 (C2), 152.45 (C6), 151.62 (Cqi-2), 146.11 (Cqi-9), 139.01 (Cqi-6), 137.23 (Cqi-8), 133.42 (C*), 130.09 (Cqi-5), 128.77 (C5), 123.75 (C4), 120.90 (Cqi-3), 120.84 (Cqi-7), 120.35 (Cqi-10), 118.26 (C3), 59.28 (Cβ), 38.78 (Cγ), 16.85 (Cα). Elem. anal. calcd for C34H36F12N6O2P2RuS2 + ½acetone: C, 40.81; H, 3.76; N, 8.04. Found: C, 40.53; H, 4.11; N, 8.37.
Single crystals of [3](PF6)2 were obtained by crystallization via liquid–vapor diffusion using acetone as the solvent and diethylether as the counter-solvent. In short, 1.0 mg of [3](PF6)2 was dissolved in 0.4 mL of acetone and placed in a small GC vial. This vial was placed in a larger vial containing 2.8 mL of diethyl ether. The large vial was closed and vapor diffusion occurred within a few days to afford X-ray quality crystals.
The structure is partly disordered. One of the two counter-ions and O2/O2′ (OH group) are disordered over two orientations. The occupancy factors of the major components of the disorder refine to 0.624(13) and 0.77(5), respectively. The crystal that was mounted on the diffractometer was non-merohedrally twinned, and the twin relationship corresponds to a twofold axis along the 0.0009a* + 0.9998b* + 0.0201c* vector. The BASF scale factor refines to 0.2115(9).
Formula: C34H36F12N6O2P2Ru1S2. Fw = 1015.82, a red block, 0.21 × 0.19 × 0.14 mm3, monoclinic, P21/n (no. 14), a = 9.54745(18), b = 21.8432(4), c = 18.4565(3) Å, β = 98.2773(17)°, V = 3808.95(12) Å3, Z = 4, μ = 6.08 mm−1, and Tmin–Tmax: 0.401–0.563. 27773 reflections were measured up to a resolution of (sinθ/λ)max = 0.616 Å−1. 10943 reflections were unique (Rint = 0.022), of which 9630 were observed [I > 2σ(I)]. 620 parameters were refined using 256 restraints. R1/wR2 [I > 2σ(I)]: 0.029/0.0943. R1/wR2 [all refl.]: 0.0333/0.0963. S = 0.99. The residual electron density was found between −0.66 and 0.64 e Å−3. Deposited as CCDC 1535299.†
:2:2), the reaction mixture was cooled down to room temperature and KPF6 (0.5 g, 2.7 mmol) was added. Then, the aqueous layer was extracted with ethyl acetate (3 × 15 mL), and the organic layer was evaporated in vacuo. The compound was purified using size-exclusion chromatography (Sephadex LH-20, acetone). After evaporation, the product was precipitated from acetone (0.5 mL) by the addition of diethyl ether (10 mL). Yield: 45 mg (58%). 1H NMR (400 MHz, 300 K, acetone-d6): δ = 10.56 (s, 2H, N–H), 8.80 (d, 2H, J = 6.0 Hz py-6), 8.51 (d, 2H, J = 7.6 Hz, H5), 8.27 (d, 2H, J = 8.4 Hz, H4), 8.07 (dd, 2H, J = 8.8 Hz, py-4), 7.62 (d, 2H, J = 8.4 Hz, H5), 7.50 (d, 2H, J = 8.4 Hz, py-3), 7.36 (dd, 2H, J = 7.2 Hz, py-5), 3.42 (t, 4H, J = 5.6 Hz, Hγ), 2.01 (t, 4H, J = 5.6 Hz, Hβ), 1.56 (s, 6H, Hα). 13C NMR (100 MHz, 300 K, acetone-d6): δ = 162.1 (C2), 154.3 (Cpy-6), 139.9 (Cpy-4), 139.2 (C4), 133.9 (C6), 130.5 (Cpy-2), 120.3 (Cpy-5), 119.2 (C5), 117.7 (Cpy-3), 116.6 (Cpy-3), 116.1(C3), 59.5 (Cγ), 38.9 (Cβ), 17.0 (Cα). High resolution ES MS m/z (calc.): 574.0961 (574.0963, [M − H − Hmte + ACN]+). Elem. anal. calcd for C26H32F12N6O2P2RuS2 + ½acetone + ½H2O: C, 34.63; H, 3.80; N, 8.81. Found: C, 34.44; H, 4.33; N, 9.01.
Single crystals of [4](PF6)2 were obtained by crystallization through liquid–vapor diffusion using acetone as the solvent and diethyl ether as the counter-solvent. In short, 1.0 mg of [4](PF6)2 was dissolved in 0.4 mL of acetone and placed in a small vial. This vial was placed in a larger vial containing 2.8 mL of diethyl ether. The large vial was closed and vapor diffusion occurred within a few days to afford X-ray quality crystals.
The structure is ordered. The ruthenium complex is found at sites of twofold axial symmetry, and only one half of the molecule is found to be crystallographically independent.
Formula: C26H32F12N6O2P2Ru1S2. Fw = 915.70, orange-red needles, 0.25 × 0.05 × 0.02 mm3, monoclinic, I2/a, a = 18.9321(3), b = 11.23471(18), c = 15.6066(2) Å, β = 96.7047(15)°, V = 3296.77(9) Å3, Z = 4, μ = 6.94 mm−1, and Tmin–Tmax: 0.391–0.881. 10972 reflections were measured up to a resolution of (sinθ/λ)max = 0.616 Å−1. 3243 reflections were unique (Rint = 0.024), of which 3045 were observed [I > 2σ(I)]. 239 parameters were refined using 2 restraints. R1/wR2 [I > 2σ(I)]: 0.0378/0.1016. R1/wR2 [all refl.]: 0.0399/0.1034. S = 1.06. The residual electron density was found between −0.74 and 2.11 e Å−3. Deposited as CCDC 1535297.†
Additional notes:
(i) The residual electron density peak of 2.11 e− Å−3 found at 0.75 Å from Ru1 is chemically meaningless. All remaining residual electron density peaks are lower than 1 e− Å−3.
(ii) The occupancy factor of Ru1 was refined freely (its value was 1.034(3) × 0.5 = 0.517(2)) before being constrained to 0.5 in the final refinement.
UV-vis spectroscopy absorbance studies
Photoreactivity and stability studies were monitored using a UV-vis spectrometer equipped with the temperature set at 298 K and a magnetic stirrer. The measurements were performed in a quartz cuvette, containing 3 mL of solution (0.010–0.050 mM). The stirred sample was irradiated perpendicularl to the axis of the spectrometer with a blue LED (λ = 445 nm, Δλ1/2 = 22 nm, photon flux Φ = 2.1 × 10−8 mol s−1, tirr = 10 minutes) fitted to the top of the cuvette, and an absorption spectrum was obtained at regular time intervals and analyzed using Microsoft Excel. Mass spectrometry was performed at the beginning and at the end of the irradiation to confirm the nature of the reagent and products.Gel studies
HPLC purified ODN1 was ordered from Sigma-Aldrich and received as a solid (fixed amount), and LC-MS water was added in such amounts that a 1 mM solution was obtained. [1]Cl and [2]Cl were dissolved in methanol and [3](PF6)2 and [4](PF6)2 were dissolved in acetone to obtain 1 mM solutions. Then, 5 μL of ODN solution, 15 μL of Ru solution, and 5 μL of MilliQ water were transferred to a 1 mL quartz cuvette, and irradiated for 10 minutes with blue LED (λirr = 445 nm, Δλ1/2 = 22 nm, photon flux Φ = 2.2 × 10−7 mol s−1, irradiation time 10 minutes). Following irradiation, the sample was transferred to a 1.5 mL Eppendorf microtube and incubated in the dark for 6 hours at 298 K. Then, 1–2 μl of the mixture (containing 400 pmol of ODN1 per lane) was added to 3 μl formamide/dye solution (formamide 0.01% (w/v), bromophenol blue 0.01% (w/v), xylenecyanol 0.01% (w/v)) and loaded (without heating) on a 20% polyacrylamide gel containing 8 M urea in 1× Tris/borate/EDTA buffer. After electrophoresis (22 mA per gel) the gel was stained for 5 min in 0.01% w/v Toluidine Blue O ((7-amino-8-methyl-phenothiazin-3-ylidene)-dimethyl-ammonium) in water on a rotary shaker. The background staining was removed by washing the gel multiple times with tap water. The procedure was adapted from Popa and Bosch.44ESI-MS ODN experiments
HPLC purified ODN1 (s(5′CTACGGTTTCAC3′) or C116H149N40O72P11) was ordered from Sigma-Aldrich and received as a solid (fixed amount). LC-MS water was added in such amounts that a 1 mM solution was obtained. [1]Cl and [2]Cl were dissolved in methanol and [3](PF6)2 and [4](PF6)2 were dissolved in acetone to obtain 1 mM solutions. Then, 15 μL of ODN1 aqueous solution, 45 μL of Ru solution, and 450 μL of LC-MS grade water were transferred to a 1 mL quartz cuvette, and irradiated for 10 minutes with a blue LED (λirr = 445 nm, Δλ1/2 = 22 nm, photon flux Φ = 2.2 × 10−7 mol s−1, irradiation time 10 minutes). Following irradiation, the sample was transferred to a 1.5 mL Eppendorf microtube and incubated in the dark for 6 hours at 298 K. For every sample a control sample shielded from light in a parallel experiment was performed. Then, the solutions were directly injected (flow rate = 5 μl min−1, 1:1 water/methanol) in an Orbitrap high-resolution mass spectrometer (Thermo, San Jose, CA, USA), equipped with a conventional ESI source. The mass spectra have been recorded in the range of 300–2000 m/z values. The following standardized working conditions were applied: spray voltage 2.7 kV, tube lens voltage −113 V, capillary voltage −20 V and capillary temperature 280 °C. Sheath and auxiliary gases were set at 23 a.u. and 4 a.u., respectively. The m/z spectra were deconvoluted using ProMass for Xcalibur (ThermoQuest, Finnigan). The chemical structure of ODN1 is shown in Scheme S12.†
Conflicts of interest
There is no conflict of interest to report.Acknowledgements
This work was supported by the Dutch Organisation for Scientific Research (NWO-CW) via a VIDI grant to S. B. The European Research Council is also kindly acknowledged for a Starting Grant to S. B. Prof. E. Bouwman is kindly acknowledged for the support and input. The COST action CM1105 “Functional metal complexes that bind to biomolecules” is gratefully acknowledged for stimulating scientific discussion and for a Short Term Scientific Mission to VvR to work in the laboratory of LM.References
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- V. Brabec, in Progress in Nucleic Acid Research and Molecular Biology, Academic Press, 2002, vol. 71, pp. 1–68 Search PubMed.
- J. Reedijk, Proc. Nat. Acad. Sci. U. S. A., 2003, 100, 3611–3616 CrossRef CAS PubMed.
- S. E. Sherman and S. J. Lippard, Chem. Rev., 1987, 87, 1153–1181 CrossRef CAS.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- A.-M. Florea and D. Büsselberg, Cancers, 2011, 3, 1351 CrossRef CAS PubMed.
- Z. H. Siddik, Oncogene, 2003, 22, 7265–7279 CrossRef CAS PubMed.
- U. Kalinowska-Lis, J. Ochocki and K. Matlawska-Wasowska, Coord. Chem. Rev., 2008, 252, 1328–1345 CrossRef CAS.
- E. Wachter, A. Zamora, D. K. Heidary, J. Ruiz and E. C. Glazer, Chem. Commun., 2016, 52, 10121–10124 RSC.
- A. G. Quiroga, J. Inorg. Biochem., 2012, 114, 106–112 CrossRef CAS PubMed.
- L. Messori, L. Cubo, C. Gabbiani, A. Álvarez-Valdés, E. Michelucci, G. Pieraccini, C. Ríos-Luci, L. G. León, J. M. Padrón, C. Navarro-Ranninger, A. Casini and A. G. Quiroga, Inorg. Chem., 2012, 51, 1717–1726 CrossRef CAS PubMed.
- F. J. Ramos-Lima, A. G. Quiroga, B. García-Serrelde, F. Blanco, A. Carnero and C. Navarro-Ranninger, J. Med. Chem., 2007, 50, 2194–2199 CrossRef CAS PubMed.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed.
- N. J. Farrer, J. A. Woods, L. Salassa, Y. Zhao, K. S. Robinson, G. Clarkson, F. S. Mackay and P. J. Sadler, Angew. Chem., Int. Ed., 2010, 49, 8905–8908 CrossRef CAS PubMed.
- Z. Li, A. David, B. A. Albani, J.-P. Pellois, C. Turro and K. R. Dunbar, J. Am. Chem. Soc., 2014, 136, 17058–17070 CrossRef CAS PubMed.
- D. A. Lutterman, P. K. L. Fu and C. Turro, J. Am. Chem. Soc., 2006, 128, 738–739 CrossRef CAS PubMed.
- R. E. Mahnken, M. A. Billadeau, E. P. Nikonowicz and H. Morrison, J. Am. Chem. Soc., 1992, 114, 9253–9265 CrossRef CAS.
- T. Joshi, V. Pierroz, C. Mari, L. Gemperle, S. Ferrari and G. Gasser, Angew. Chem., Int. Ed., 2014, 53, 2960–2963 CrossRef CAS PubMed.
- M. A. Sgambellone, A. David, R. N. Garner, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2013, 135, 11274–11282 CrossRef CAS PubMed.
- R. Deibel, K. Luebke and H. Morrison, Photochem. Photobiol., 1989, 49, 519–522 CrossRef.
- A. M. Angeles-Boza, P. M. Bradley, P. K. L. Fu, S. E. Wicke, J. Bacsa, K. R. Dunbar and C. Turro, Philos. Trans. R. Soc., A, 2013, 371, 20120128 CrossRef PubMed.
- F. S. Mackay, J. A. Woods, P. Heringová, J. Kašpárková, A. M. Pizarro, S. A. Moggach, S. Parsons, V. Brabec and P. J. Sadler, Proc. Nat. Acad. Sci. U. S. A., 2007, 104, 20743–20748 CrossRef CAS PubMed.
- F. Navas, S. Perfahl, C. Garino, L. Salassa, O. Novakova, C. Navarro-Ranninger, P. J. Bednarski, J. Malina and A. G. Quiroga, J. Inorg. Biochem., 2015, 153, 211–218 CrossRef CAS PubMed.
- N. J. Farrer, J. A. Woods, V. P. Munk, F. S. Mackay and P. J. Sadler, Chem. Res. Toxicol., 2010, 23, 413–421 CrossRef CAS PubMed.
- P. Heringova, J. Woods, F. S. Mackay, J. Kasparkova, P. J. Sadler and V. Brabec, J. Med. Chem., 2006, 49, 7792–7798 CrossRef CAS PubMed.
- C. Moucheron, New J. Chem., 2009, 33, 235–245 RSC.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC.
- A. N. Hidayatullah, E. Wachter, D. K. Heidary, S. Parkin and E. C. Glazer, Inorg. Chem., 2014, 53, 10030–10032 CrossRef CAS PubMed.
- S. J. Burya, A. M. Palmer, J. C. Gallucci and C. Turro, Inorg. Chem., 2012, 51, 11882–11890 CrossRef CAS PubMed.
- O. Filevich and R. Etchenique, Photochem. Photobiol. Sci., 2013, 12, 1565–1570 CAS.
- L. Salassa, E. Borfecchia, T. Ruiu, C. Garino, D. Gianolio, R. Gobetto, P. J. Sadler, M. Cammarata, M. Wulff and C. Lamberti, Inorg. Chem., 2010, 49, 11240–11248 CrossRef CAS PubMed.
- P. M. Takahara, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Nature, 1995, 377, 649–652 CrossRef CAS PubMed.
- V. H. S. van Rixel, B. Siewert, S. L. Hopkins, S. H. C. Askes, A. Busemann, M. A. Siegler and S. Bonnet, Chem. Sci., 2016, 7, 4922–4929 RSC.
- J. L. Beck, M. L. Colgrave, S. F. Ralph and M. M. Sheil, Mass Spectrom. Rev., 2001, 20, 61–87 CrossRef CAS PubMed.
- M. Groessl and P. J. Dyson, Curr. Top. Med. Chem., 2011, 11, 2632–2646 CrossRef CAS PubMed.
- E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS PubMed.
- J.-M. Teuben and J. Reedijk, J. Biol. Inorg. Chem., 2000, 5, 463–468 CAS.
- J. Rodríguez, J. Mosquera, J. R. Couceiro, M. E. Vázquez and J. L. Mascareñas, Angew. Chem., Int. Ed., 2016, 55, 15615–15618 CrossRef PubMed.
- R. E. Goldbach, I. Rodriguez-Garcia, J. H. van Lenthe, M. A. Siegler and S. Bonnet, Chem. – Eur. J., 2011, 17, 9924–9929 CrossRef CAS PubMed.
- S. Bonnet, G. Molnár, J. Sanchez Costa, M. A. Siegler, A. L. Spek, A. Bousseksou, W.-T. Fu, P. Gamez and J. Reedijk, Chem. Mater., 2009, 21, 1123–1136 CrossRef CAS.
- Z. Arcis-Castíllo, S. Zheng, M. A. Siegler, O. Roubeau, S. Bedoui and S. Bonnet, Chem. – Eur. J., 2011, 17, 14826–14836 CrossRef PubMed.
- D. Musumeci, L. Rozza, A. Merlino, L. Paduano, T. Marzo, L. Massai, L. Messori and D. Montesarchio, Dalton Trans., 2015, 44, 13914–13925 RSC.
- D. Musumeci, C. Platella, C. Riccardi, A. Merlino, T. Marzo, L. Massai, L. Messori and D. Montesarchio, Dalton Trans., 2016, 45, 8587–8600 RSC.
- L. M. Popa and L. Bosch, FEBS Lett., 1969, 4, 143–146 CrossRef CAS PubMed.
- A. Casini, C. Gabbiani, E. Michelucci, G. Pieraccini, G. Moneti, P. J. Dyson and L. Messori, J. Biol. Inorg. Chem., 2009, 14, 761–770 CrossRef CAS PubMed.
- A. Casini, A. Guerri, C. Gabbiani and L. Messori, J. Inorg. Biochem., 2008, 102, 995–1006 CrossRef CAS PubMed.
- A. Casini, G. Mastrobuoni, C. Temperini, C. Gabbiani, S. Francese, G. Moneti, C. T. Supuran, A. Scozzafava and L. Messori, Chem. Commun., 2007, 156–158, 10.1039/B611122J.
- Y. Mantri, S. J. Lippard and M.-H. Baik, J. Am. Chem. Soc., 2007, 129, 5023–5030 CrossRef CAS PubMed.
- J. Grau, R. F. Brissos, J. Salinas-Uber, A. B. Caballero, A. Caubet, O. Roubeau, L. Korrodi-Gregório, R. Pérez-Tomás and P. Gamez, Dalton Trans., 2015, 44, 16061–16072 RSC.
- H. Song, J. T. Kaiser and J. K. Barton, Nat. Chem., 2012, 4, 615–620 CrossRef CAS PubMed.
- Y.-H. Hsiang, L. F. Liu, M. E. Wall, M. C. Wani, A. W. Nicholas, G. Manikumar, S. Kirschenbaum, R. Silber and M. Potmesil, Cancer Res., 1989, 49, 4385–4389 CAS.
- M. R. Gill, S. N. Harun, S. Halder, R. A. Boghozian, K. Ramadan, H. Ahmad and K. A. Vallis, Sci. Rep., 2016, 6, 31973 CrossRef CAS PubMed.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- E. Dulière, M. Devillers and J. Marchand-Brynaert, Organometallics, 2003, 22, 804–811 CrossRef.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1535297–1535299. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt03613b |
| This journal is © The Royal Society of Chemistry 2018 |