
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and applications to catalysis of novel cyclopentadienone iron tricarbonyl complexes†
Alessandro
Del Grosso
,
Alexander E.
Chamberlain
,
Guy J.
Clarkson
and
Martin
Wills
 *
*
Department of Chemistry, The University of Warwick, Coventry, CV4 7AL, UK. E-mail: m.wills@warwick.ac.uk
First published on 5th January 2018
Abstract
A series of cyclopentadienone iron tricarbonyl complexes with diverse structures were prepared, in each case using the intramolecular cyclisation of a diyne as a key step. The complexes were generated as enantiomerically enriched through (i) asymmetric synthesis of a C2-symmetric diol following a reported protocol, (ii) resolution of enantiomerically-enriched diastereoisomers formed from a chiral alcohol and (iii) kinetic resolution of a racemic ketone-containing iron tricarbonyl complex. The approaches underline the diversity of the synthetic routes which can be employed in the synthesis of homochiral cyclopentadienone iron tricarbonyl complexes. Although the complexes proved to be effective as catalysts for the reduction of ketones, the alcohol products were formed in low ees (not exceeding ca. 35%), highlighting the challenging nature of asymmetric catalysis using complexes of this type.
Introduction
Cyclopentadienone iron tricarbonyl complexes 1 have recently found a significant number of applications in the catalysis of a number of organic transformations,1–3 notably hydrogenation4 and transfer hydrogenation,5 and formation of C–N bonds via reductive amination6 and ‘hydrogen borrowing’ reactions.7 The hydrogen transfer is believed to proceed via formation of the hydride 2 and 16-electron species 3, with the hydride transfer itself through the transition state also illustrated in Fig. 1.8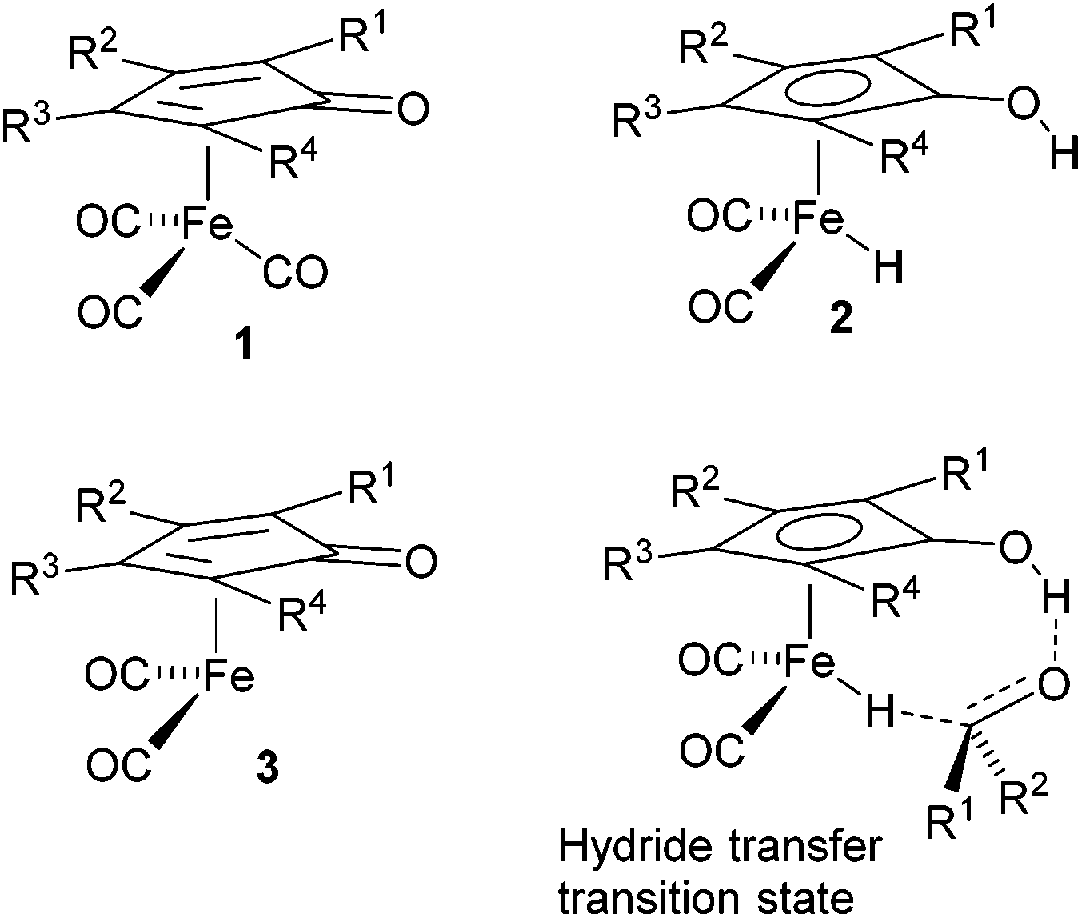 | ||
| Fig. 1 Iron tricarbonyl cyclopentadienone structures. | ||
A number of derivatives of this class of catalyst have been reported, for example 4–11, and some of these have been applied in enantioselective catalysis of ketone and imine reduction reactions.9 In other examples, non-chiral cyclopentadienone iron tricarbonyl complexes have been used in conjunction with an asymmetric reagent, such as a phosphonic acid, in the catalysis of asymmetric reduction reactions (Fig. 2).10
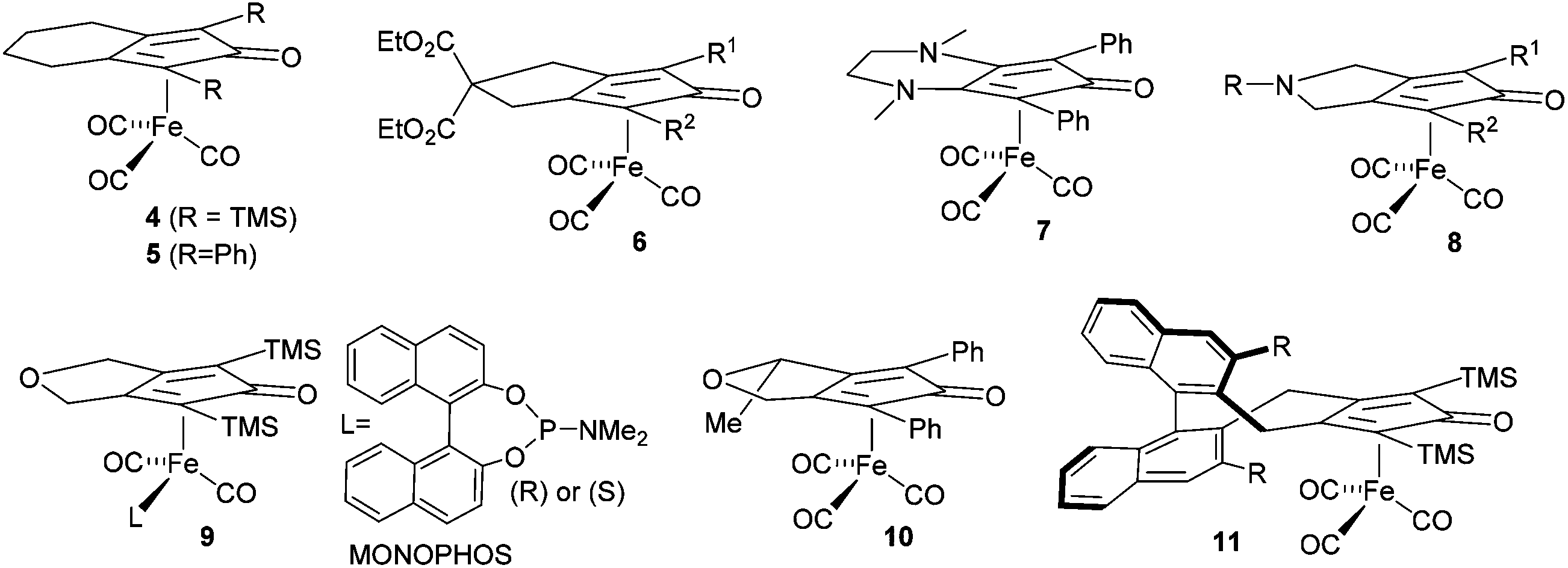 | ||
| Fig. 2 Reported asymmetric and non-asymmetric catalysts based on iron tricarbonyl cyclopentadienone structures. | ||
In a recent study, we described an efficient route to the synthesis of catalysts such as 12, through the reaction of iron pentacarbonyl with the derivatives of a C2-symmetric diol 13 (Scheme 1).11
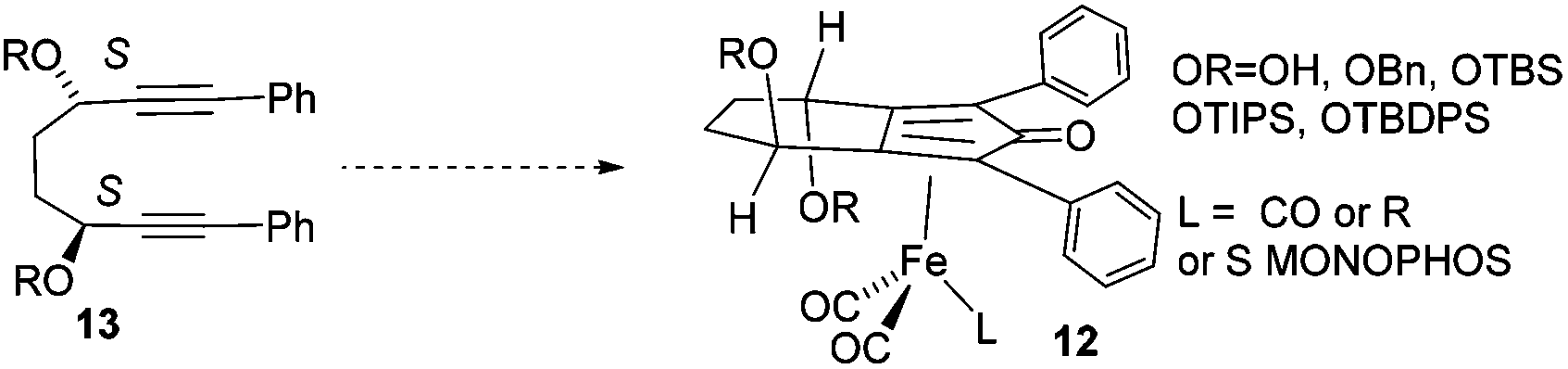 | ||
| Scheme 1 Previously-reported asymmetric iron complexes derived from C2-symmetric diols. | ||
Herein we describe the synthesis and applications of a series of cyclopentadienone iron tricarbonyl complexes with diverse structures, and efficient routes to their formation. Whilst the complexes are competent pre-catalysts for asymmetric reactions, the induced enantioselectivities remain low, reflecting the challenge of achieving high enantioselectivities with this class of complex.
Results and discussion
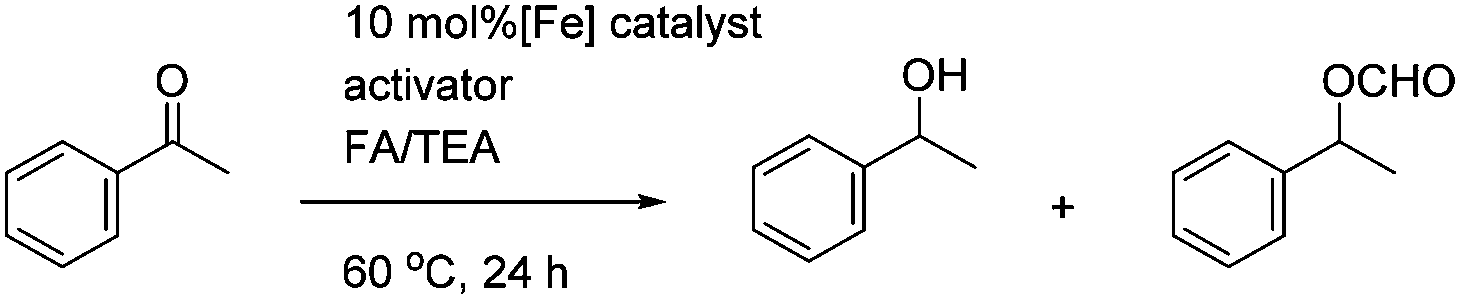
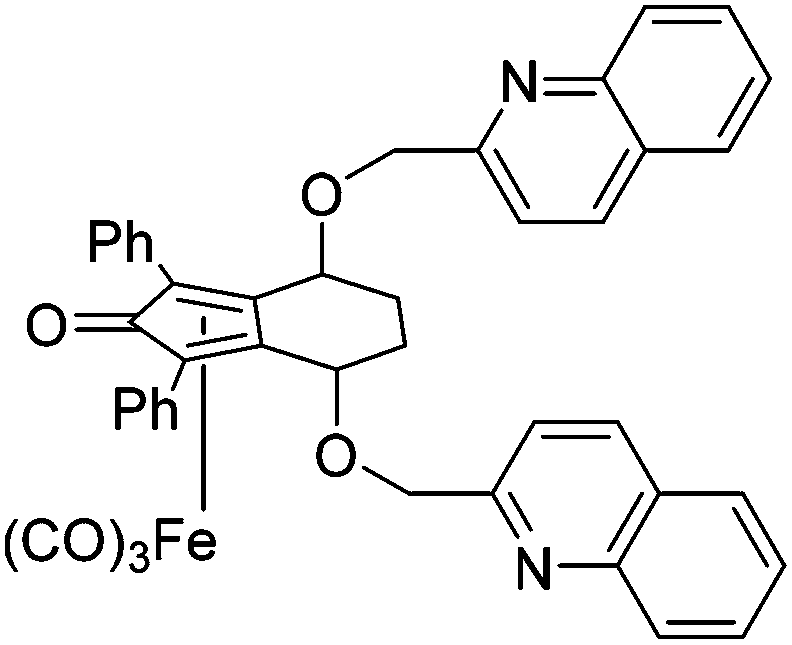
In the first extension of our studies, we sought to determine what effect an electron-donating group on the side chains of the catalyst might have. Towards this end we prepared the pyridine-containing complex 14, from diol 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 and via the dialkyne precursor 16, following the route illustrated in Scheme 2a. Treatment of 14 with slightly more than 1 equivalent of trimethylamine N-oxide (TMAO)13 resulted in formation of a new species which appears to match the structure 17 in which one CO group was replaced by the pyridine in an intramolecular reaction. The addition of triphenylphosphine to the cyclic complex 17 led to formation of a new product with a peak at δ 57.57 in the 31P NMR which is indicative of the formation of an iron triphenylphosphine complex. In addition, a further complex, 18, containing a quinoline, was prepared from the dialkyne 19 (Scheme 2b). Complexes 14, 17 and 18 proved to be effective catalysts for the reduction of ketones, with selected results shown in Table 1 for hydrogenation and Table 2 for transfer hydrogenation (full results are in the ESI†).
12 and via the dialkyne precursor 16, following the route illustrated in Scheme 2a. Treatment of 14 with slightly more than 1 equivalent of trimethylamine N-oxide (TMAO)13 resulted in formation of a new species which appears to match the structure 17 in which one CO group was replaced by the pyridine in an intramolecular reaction. The addition of triphenylphosphine to the cyclic complex 17 led to formation of a new product with a peak at δ 57.57 in the 31P NMR which is indicative of the formation of an iron triphenylphosphine complex. In addition, a further complex, 18, containing a quinoline, was prepared from the dialkyne 19 (Scheme 2b). Complexes 14, 17 and 18 proved to be effective catalysts for the reduction of ketones, with selected results shown in Table 1 for hydrogenation and Table 2 for transfer hydrogenation (full results are in the ESI†).
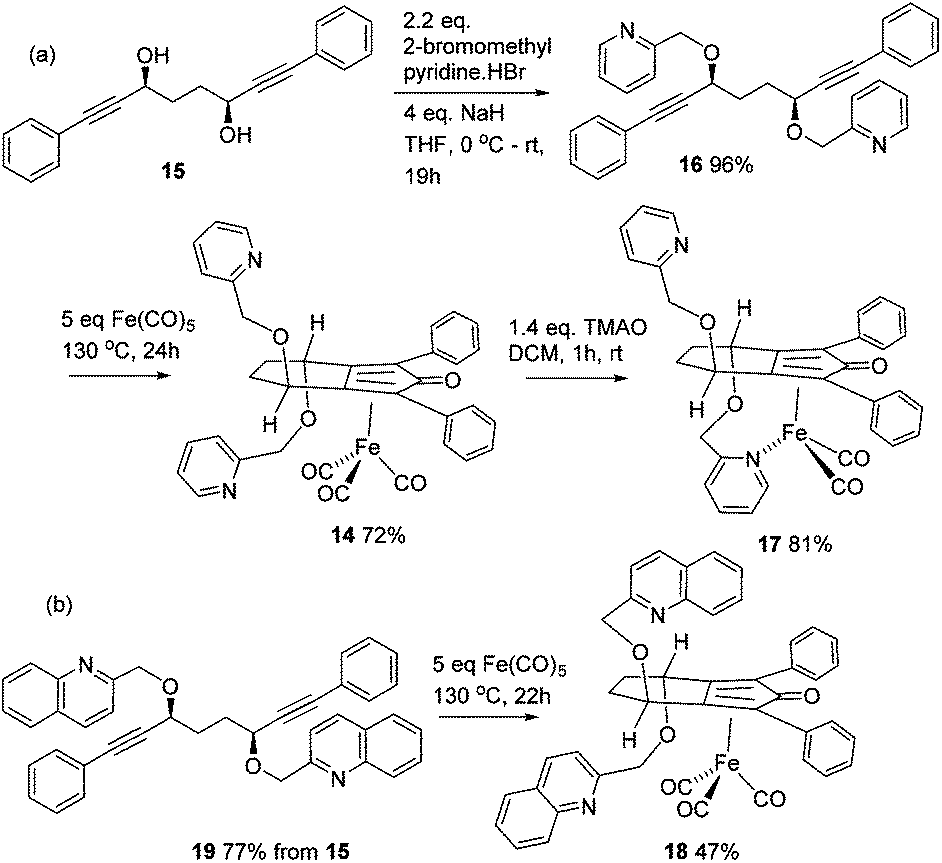 | ||
| Scheme 2 Synthesis of pyridine-containing complexes. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Activator/% | Solvent | Conv./% |
| 1 | 14 | K2CO3 (3%) | IPA/H2O | 17.3 |
| 2 | 14 | K2CO3 (2%) | IPA/H2O | 18.6 |
| 3 | 14 | TMAO (1%) | IPA/H2O | 37.1 |
| 4 | 17 | K2CO3 (5%) | IPA/H2O | 15.4 |
| 5 | 17 | TMAO (1%) | IPA/H2O | 36.3 |
| 6 | 17 | TMAO (2%) | IPA/H2O | 30.3 |
| 7 | 18 | — | IPA/H2O | 70.9 |
| 8 | 18 | K2CO3 (5%) | IPA/H2O | 51.2 |
| 9 | 18 | TMAO (1%) | IPA/H2O | 99.8 |
| 10 | 18 | TMAO (2%) | IPA/H2O | 99.7 |
| 11 | 18 | TMAO (3%) | IPA/H2O | 99.8 |
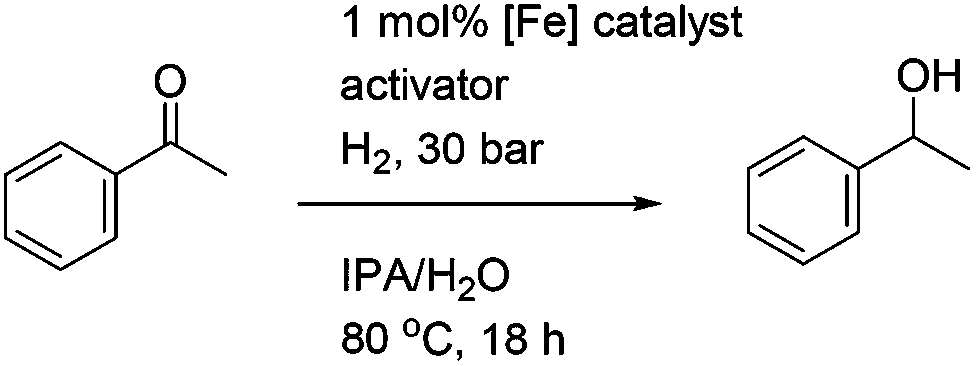
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Activator/% | Conv./% | Alcohol/% (ee/%) | Formate/% (ee/%) |
| a Hetereogenous reaction (catalyst is not soluble). | |||||
| 1 | 14 | — | 73.7 | 63.1 (2.4 R) | 10.6 (2.6 R) |
| 2 | 14 | TMAO (10%) | 99.7 | 90.8 (2 R) | 8.9 (2.8 R) |
| 3 | 17 | — | 99.9 | 85.5 (4.2 R) | 14.4 (3 R) |
| 4 | 17 | TMAO (10%) | 99.9 | 87.8 (3.4 R) | 11.8 (3 R) |
| 5a | 18 | TMAO (10%) | 58.1 | 45.4 (3.4 R) | 12.7 (5.2 R) |
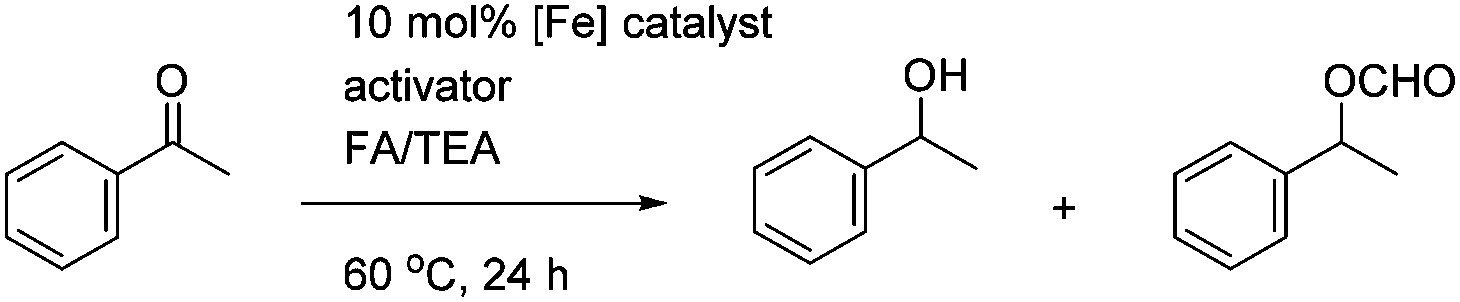
In the hydrogenation reactions, catalysts 14 and 17 gave products in low conversions, although the addition of K2CO3 or TMAO as an activating agent resulted in the highest conversions. In common with previous results, the addition of triphenylphosphine served to inhibit the reaction (see ESI†). The similarity between the results obtained with 14 and 17 suggest that they are reacting through a common intermediate, i.e. the formation (possibly reversible) of 17 from 14 upon treatment with TMAO. The quinoline-derived complex 18 gave improved conversions compared to the pyridine-containing complexes, although the addition of TMAO was again essential for optimal activation, and an excess of TMAO did not reduce the conversions. In all cases the ees were low however and no higher than ca. 7–8% (full results for these and other tests are given in the ESI†).
In the attempted asymmetric transfer hydrogenations (ATH), using a formic acid/triethylamine 5:2 azeotrope (FA/TEA) mixture as both reducing agent and solvent, the pyridine-containing complex 14 required the addition of TMAO to give optimal results however the cyclic complex 17 proved to be just as active with or without the addition of TMAO. This suggests that 17 may represent an ‘activated’ version of 14, with reversible coordination of the pyridine acting to free a coordination site for hydride delivery and subsequent hydrogen transfer. The quinoline complex 18 gave poor conversion even with the addition of TMAO, however it appeared to not be soluble in the reaction mixture. Again the ees were low in all cases (between 2–4% only).
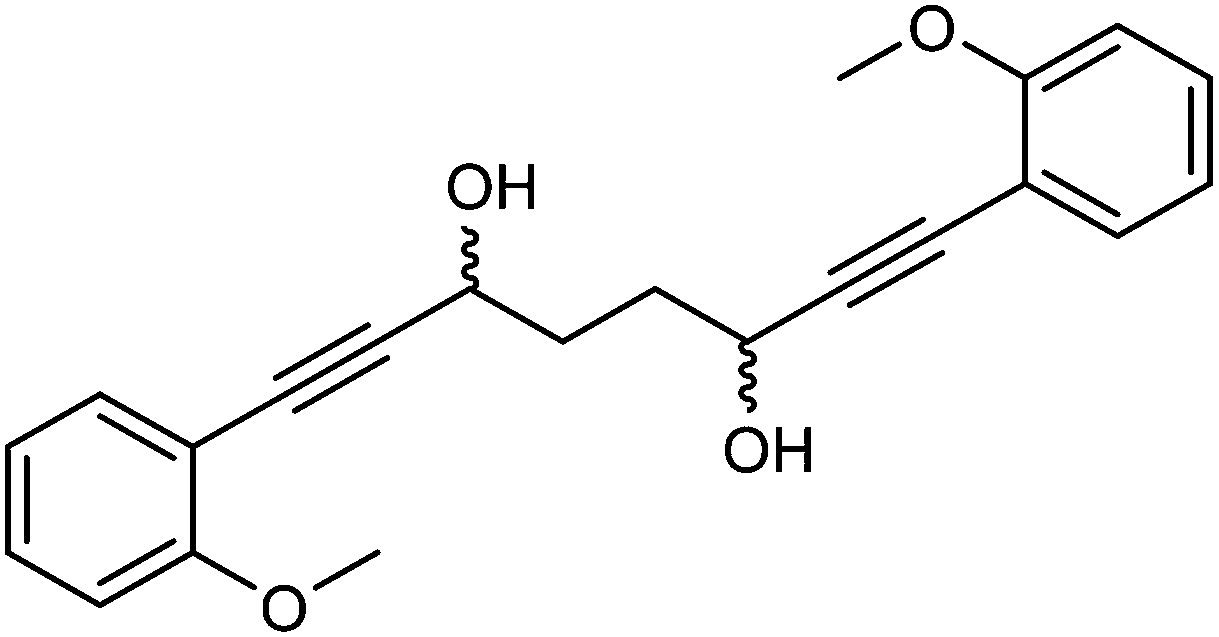
Previously reported catalysts 12, and those illustrated in Scheme 2, contain unsubstituted phenyl rings adjacent to the central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the cyclopentadienone, therefore we sought to see if an improvement could be made by the introduction of an ortho-substituent to these rings, since the extra steric hindrance has the potential to further influence the asymmetry of the reduction reactions. The synthesis of a series of compounds containing ortho-methoxy groups on the aromatic rings was completed (Scheme 3) following a route based on the method previously reported, using the reaction of the lithium salt of alkyne 20 with bis-Weinreb reagent 21, to give diketone 22, followed by ATH catalysed by tethered Ru(II) complex 23, to the corresponding diol 24 in high ee. Both enantiomers, and the racemic standard (for HPLC analysis) were prepared for comparison, although the (S,S)-configuration products 24 were converted to catalysts for evaluation. Conversion of diol 24 to the dibenzyl derivative 25 was achieved efficiently. Both the diol and its two derivatives were treated at 130 °C for 24 h with three equivalents of Fe(CO)5 to form the required complexes 26 and 27 respectively. In each case, a stable complex was formed and fully characterised, including by X-ray crystallography (Fig. 3).
O of the cyclopentadienone, therefore we sought to see if an improvement could be made by the introduction of an ortho-substituent to these rings, since the extra steric hindrance has the potential to further influence the asymmetry of the reduction reactions. The synthesis of a series of compounds containing ortho-methoxy groups on the aromatic rings was completed (Scheme 3) following a route based on the method previously reported, using the reaction of the lithium salt of alkyne 20 with bis-Weinreb reagent 21, to give diketone 22, followed by ATH catalysed by tethered Ru(II) complex 23, to the corresponding diol 24 in high ee. Both enantiomers, and the racemic standard (for HPLC analysis) were prepared for comparison, although the (S,S)-configuration products 24 were converted to catalysts for evaluation. Conversion of diol 24 to the dibenzyl derivative 25 was achieved efficiently. Both the diol and its two derivatives were treated at 130 °C for 24 h with three equivalents of Fe(CO)5 to form the required complexes 26 and 27 respectively. In each case, a stable complex was formed and fully characterised, including by X-ray crystallography (Fig. 3).
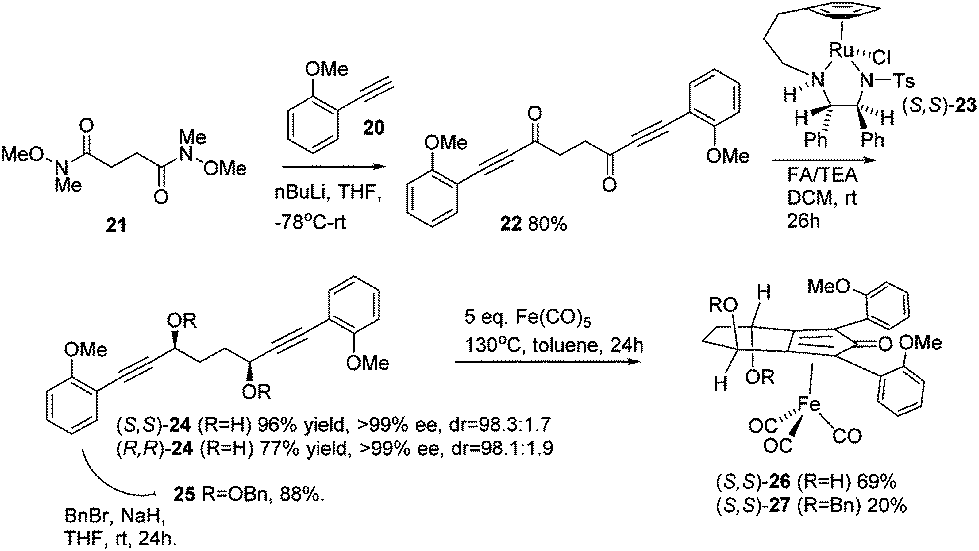 | ||
| Scheme 3 Synthesis of ortho-methoxy substituted complexes. | ||
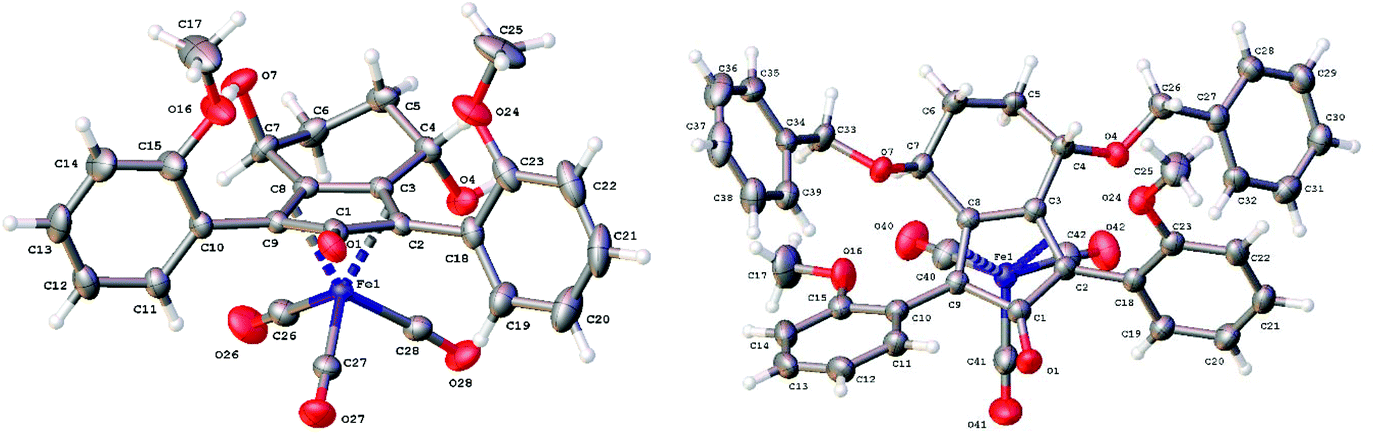 | ||
| Fig. 3 X-ray crystallographic structures of complexes 26 and 27. | ||
The X-ray crystallographic analysis of 26 and 27 (Fig. 3) confirmed the expected structures of the complexes, however there was no obvious transfer of chirality to the o-OMe aromatic rings. This flexibility may be responsible for the lack of selectivity in the subsequent reductions in which the catalysts were tested.
In the hydrogenation tests (Table 3; further results are listed in the ESI†), we focussed initially on the diol complex 26. Again it was found that activation was required for best results, with full conversions being generated using TMAO. Previously, and in initial tests, a combination of IPA and water was used as solvent, as this had been found to give good results. A series of alternative solvents were tested however these gave, with the exception of THF, inferior results in terms of conversion, and no significant change to the ee. Reactions at slightly lower temperatures (60 °C and 40 °C) resulted in lower conversions, and the reduction of a series of acetophenone derivatives were also tested. In these tests, ortho-chloroacetophenone gave a product in lower conversion than the less hindered or electron-rich ketones. The dibenzyloxy-substituted catalyst 27 was also effective although slightly more active in THF than isopropanol/water (IPA/H2O), and gave a product in up to 21% ee, which was higher than the average for the diol. This indicates that the extra steric hindrance created by the combined ring and ortho-aromatic substrates had a small positive effect on the selectivity (Table 4).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Substrate | Activator/% | Solvent/other changes | Conv./% | ee/% |
| 1 | 26 | PhCOMe | — | IPA/H2O | 54.4 | 10 (R) |
| 2 | 26 | PhCOMe | K2CO3 (5%) | IPA/H2O | 19.5 | 9.2 (R) |
| 3 | 26 | PhCOMe | TMAO (1%) | IPA/H2O | 99.8 | 9.4 (R) |
| 4 | 26 | PhCOMe | TMAO (1%) | IPA | 89.3 | 3.6 (R) |
| 5 | 26 | PhCOMe | TMAO (1%) | H2O | 57.2 | 7.4 (R) |
| 6 | 26 | PhCOMe | TMAO (1%) | Toluene | 98.1 | 2.2 (R) |
| 7 | 26 | PhCOMe | TMAO (1%) | Chlorobenzene | 85.9 | 0.2 (R) |
| 8 | 26 | PhCOMe | TMAO (1%) | THF | 98.6 | 9.4 (R) |
| 9 | 26 60 °C | PhCOMe | TMAO (1%) | IPA/H2O | 87.5 | 9.0 (R) |
| 10 | 26 40 °C | PhCOMe | TMAO (1%) | IPA/H2O | 21.8 | 12.0 (R) |
| 11 | 26 | 2-(OMe)C6H4COMe | TMAO (1%) | IPA/H2O | >99 | 8.6 (S) |
| 12 | 26 | 4-(OMe)C6H4COMe | TMAO (1%) | IPA/H2O | 91.6 | 6.0 (R) |
| 13 | 26 | 2-ClC6H4COMe | TMAO (1%) | IPA/H2O | 56.8 | 3.6 (R) |
| 14 | 26 | 4-ClC6H4COMe | TMAO (1%) | IPA/H2O | 96.6 | 10.2 (S) |
| 15 | 27 | PhCOMe | TMAO (1%) | IPA/H2O | 77.8 | 21.0 (R) |
| 16 | 27 | PhCOMe | TMAO (1%) | THF | 100 | 16.0 (R) |
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Activator/% | Conv./% | Alcohol/% (ee/%) | Formate/% (ee/%) |
| a Followed over time and at lower temp. of 30 °C. | |||||
| 1 | 26 | — | 54.9 | 46.8 (28.4 R) | 8.2 (25 R) |
| 2 | 26 | TMAO (10%) | 99.6 | 87.2 (30.2 R) | 12.3 (28 R) |
| 3a | 26 30 °C 24 h | TMAO (10%) | 25.7 | 25.2 (35.2 R) | 0.5 (n/a) |
| 4a | 26 30 °C 48 h | TMAO (10%) | 51.5 | 49.9 (34.4 R) | 1.6 (n/a) |
| 5a | 26 30 °C 72 h | TMAO (10%) | 87.8 | 81.9 (34.6 R) | 5.9 (n/a) |
| 6 | 27 | TMAO (10%) | 93.3 | 89.4 (28.0 R) | 10.6 (29.3 R) |
In the case of the ATH reactions, again the conversions were improved by the use of an activator at 80 °C, the ees of acetophenone reduction, however, were improved to 25–30%. A reaction was followed over time at a lower temperature of ca. 30 °C, and in this case the ee improved to ca. 35%, although the reaction required 3 days to reach ca. 82%. In this case, as was the case for all reductions using FA/TEA, some formate was also formed, presumably through formylation of the initial alcohol product, although the ee of the formate was similar to that of the alcohol. In previous work, we have established that the alcohol and formate products in these reactions are of the same configuration.11 The result with the dibenzyl-substituted complex 27 was comparable to that for the diol catalyst 26, indicating that the increase of steric bulk had little effect on the reaction enantioselectivity.
In order to improve the enantioselectivity, we sought to place a larger group on the ortho position of the aromatic rings adjacent to the CO of the cyclopentadienone and towards this end we prepared the acetylene reagent precursor o(OBn)C6H4CCH (OBn = OCH2Ph), however the subsequent reaction was very problematic with deprotonation at the benzylic CH2 resulting in the formation of side products. In an alternative, successful, approach, we prepared the tetrahedropyran (THP)-protected diketone 28 using alkyne 29, and subsequently the diol 30 through the sequence shown in Scheme 4. Unfortunately the cyclisation of this diol failed to give a clean product. However the hydrolysis of the THP groups gave the tetrol 31 which was either di- or tetrabenzylated to give 32 and 33 respectively. Both dialkynes were cyclised with Fe(CO)5 in good yield to form complexes 34 and 35. Reaction of the dibenzyl intermediate 32 with t-butyldimethylsilyl chloride (TBSCl) resulted in formation of 36 which was cyclised successfully but in low isolated yield to 37. Peaks corresponding to the product in the 1H NMR spectrum of 37 were very broad, however the mass spectrum indicated formation of the desired complex.
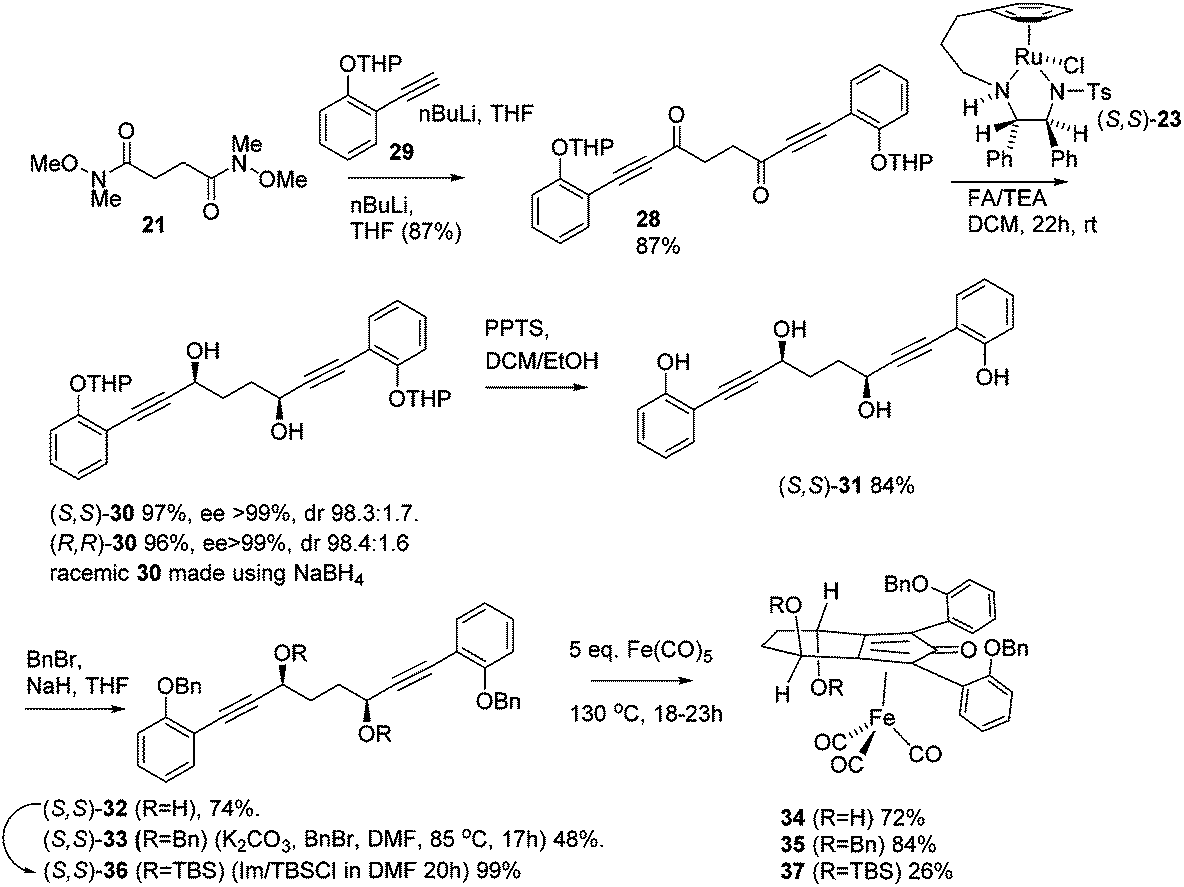 | ||
| Scheme 4 Synthesis of benzyloxy-substituted complexes via OTHP intermediate 30. | ||
The application of complexes 34, 35 and 37 to the reduction of acetophenone was tested, through both hydrogenation with hydrogen gas and ATH. Further results are given in the ESI.† In the pressure hydrogenation of acetophenone (Table 5) the tetrabenzyl complex 35 required activation but gave a product of ca. 23% ee consistently. The dibenzyl 34 (i.e. containing hydroxyl groups in the ring fused to the cyclopentadienone) worked without activation and indeed were less active in the presence of K2CO3; a trend we have noticed before.11 Further solvents were tested (Table 5 and ESI†) and of these, EtOAc, THF, IPA and tBuOH gave good but not improved results. A reaction at 60 °C gave a lower conversion with no improvement to the ee. A similar result was obtained using complex 37. In ATH reactions (Table 6) the ee (26%) generated by the dibenzyloxy complex 34 was not as high as the 36% ee achieved using the tetra OBn complex 35 however in both cases the reactions in FA/TEA gave a higher ee than the hydrogenation reaction with the same substrate. Although not fully purified, the bis-OTBS-substituted complex 37 gave a product of ca. 34% ee in a preliminary ATH test. Once again, the formate side-product was of similar ee to the alcohol in each case.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Activator/% | Solvent | Conv./% | ee/% |
| 1 | 35 | — | IPA/H2O | 4.8 | 25.2 (R) |
| 2 | 35 | K2CO3 (5%) | IPA/H2O | >99 | 23.2 (R) |
| 3 | 35 | TMAO (1%) | IPA/H2O | >99 | 23 (R) |
| 4 | 34 | — | IPA/H2O | >99 | 3.4 (R) |
| 5 | 34 | K2CO3 (5%) | IPA/H2O | 12 | 5.2 (R) |
| 6 | 34 | TMAO (1%) | IPA/H2O | >99 | 3.2 (R) |
| 7 | 35 | 1% TMAO | EtOAc | >99 | 24.5 (R) |
| 8 | 35 | 1% TMAO | Toluene | 54.2 | 16.2 (R) |
| 9 | 35 | 1% TMAO | THF | 91.6 | 25.2 (R) |
| 10 | 35 | 1% TMAO | IPA | >99 | 19.2 (R) |
| 11 | 35 | 1% TMAO | Neat | 87.6 | 18.8 (R) |
| 12 | 35 | 1% TMAO | tBuOH | 98.8 | 19.6 (R) |
| 13 | 37 | TMAO 1% | IPA/H2O | 85.2% | 12 (R) |
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Activator/% | Conv./% | Alcohol/% (ee/%) | Formate/% (ee/%) |
| 1 | 35 | TMAO (10%) | 55.7 | 47.9 (35.6 R) | 7.8 (35.8 R) |
| 2 | 34 | TMAO (10%) | 96.2 | 88.3 (25.8 R) | 7.9 (26.4 R) |
| 3 | 37 | TMAO (10%) | 98.6 | 91.1 (34.2 R) | 8.5 (32.6 R) |
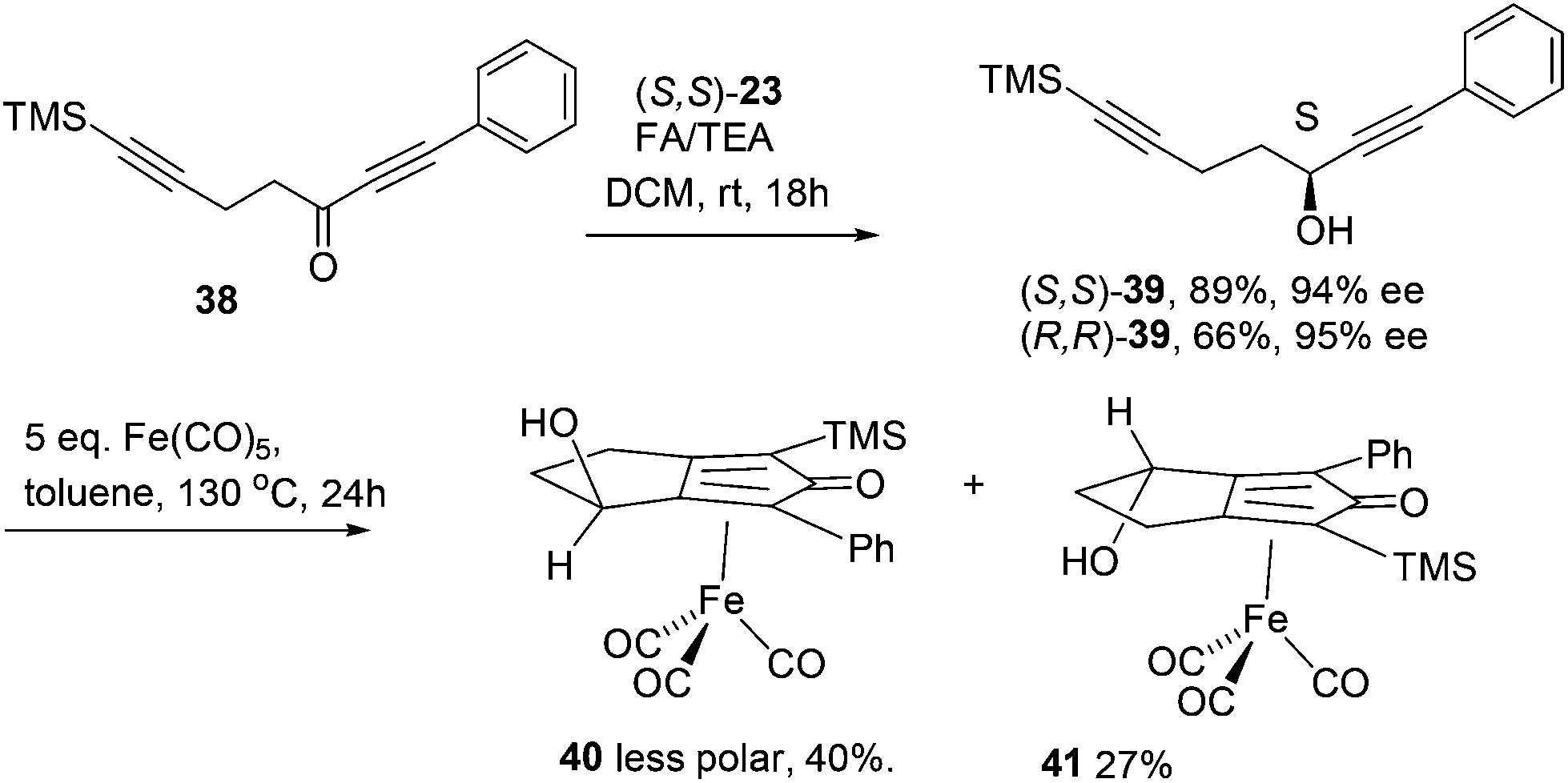
Further extension of the range of complexes for investigation was achieved through the asymmetric synthesis of complexes containing a single chiral centre and non-identical groups flanking the central CO of the cyclone. This concept of planar chirality has been applied before in similar reductions9c,14 however the opportunity was taken to explore new routes to the synthesis of Fe cyclone catalysts (Scheme 5). Addition of phenylacetylide anion to TMSCCCH2CH2CHO followed by oxidation gave ketone 38 which was subsequently reduced to 39 in 94–95% ee using ATH with both enantiomers of tethered catalyst 23. Cyclisation of 39 gave a mixture of two diastereomeric complexes 40 and 41 which were readily resolved by flash chromatography on silica gel and characterised by X-ray crystallography to confirm the relative stereochemistry in each isomer (Fig. 4). These create compounds with planar chirality about the centre CO of the cyclone group.
 | ||
| Scheme 5 Synthesis of diastereoisomeric complexes 40 and 41. | ||
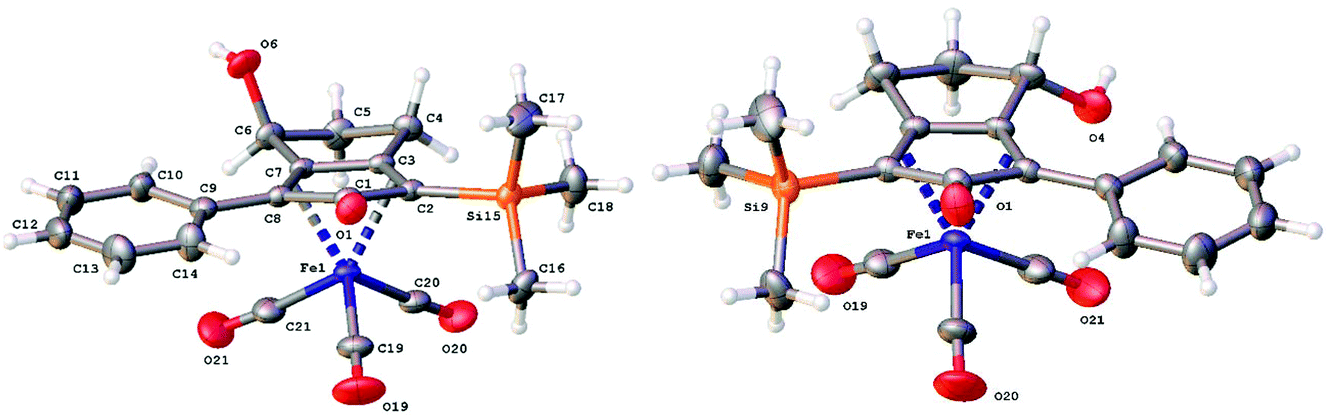 | ||
| Fig. 4 X-ray crystallographic structures of complexes 40 and 41 respectively. | ||
In the reduction tests on acetophenone, both hydrogenation and transfer hydrogenation (Table 7), the diastereoisomeric complexes 40 and 41 gave alcohols of opposite configuration, indicating that the planar symmetry of the molecule created the primary directing effect. However the enantioselectivity was very low in all cases. Once again, for the hydrogenation reaction, activation was required, with TMAO giving the best results.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Activator/% | Solvent | Conv./% | ee/% |
| 1 | 40 | — | IPA/H2O | 4 | 5 (R) |
| 2 | 40 | K2CO3 (5%) | IPA/H2O | 68 | 7.4 (R) |
| 3 | 40 | TMAO (1%) | IPA/H2O | 99.7 | 6.2 (R) |
| 4 | 41 | — | IPA/H2O | 11.9 | 11.4 (S) |
| 5 | 41 | K2CO3 (5%) | IPA/H2O | 82.3 | 11.6 (S) |
| 6 | 41 | TMAO (1%) | IPA/H2O | 99.8 | 12.2 (S) |
| Entry | Catalyst | Activator/% | Solvent | Alcohol/% (ee/%) | Formate/% (ee/%) |
|---|---|---|---|---|---|
| 7 | 40 | TMAO (10%) | FA/TEA | 91.1 (3.8 R) | 8.1 (4 R) |
| 8 | 41 | TMAO (10%) | FA/TEA | 88.4 (5.8 S) | 9.9 (5.6 S) |
A kinetic resolution approach was also used to prepare a final class of asymmetric catalyst (Scheme 6); we prepared the bistrimethylsilyl (bisTMS) complex 42 (previously prepared by Pearson et al.), in racemic form2c from aldehyde 43via ketone 44. Asymmetric reduction, following a precedent set by previous work in our group,15 with tethered catalyst 23, led to preferential reduction of one enantiomer of racemic 42 to leave enriched ketone 42 and alcohol 45, both in high ee, which could be separated by chromatography on silica gel (Scheme 6). The X-ray crystallographic structure of alcohol 45 was also obtained (Fig. 5), in order to confirm the endo relationship of the alcohol to the iron centre, as would be anticipated from the expected addition of hydride to the face of the ketone away from the Fe(CO)3 unit due to steric hindrance.15
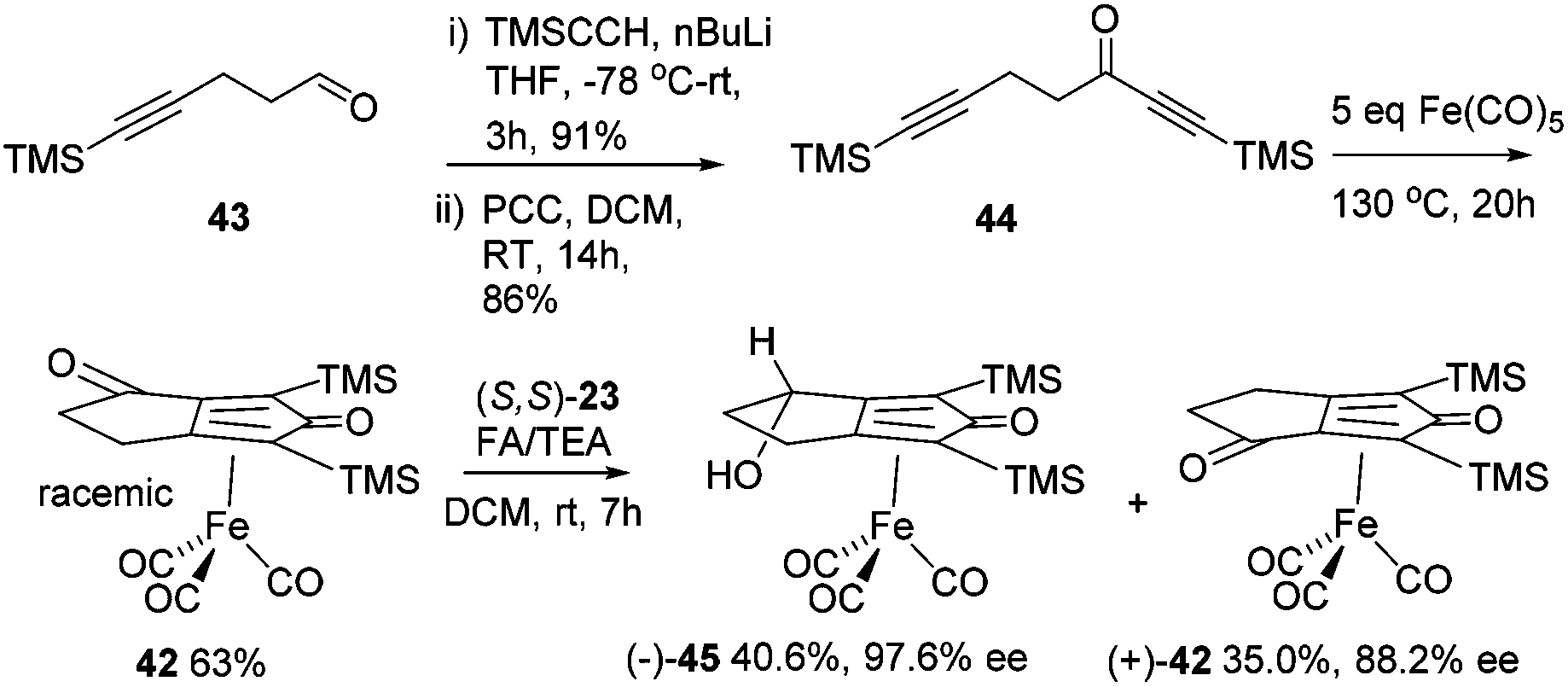 | ||
| Scheme 6 Synthesis of enantiomerically-enriched complexes via a dynamic kinetic resolution. | ||
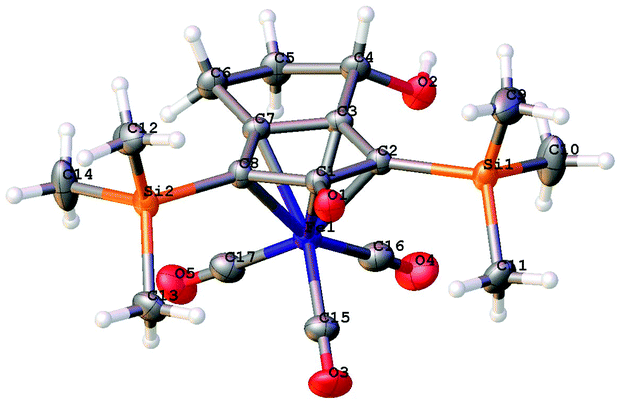 | ||
| Fig. 5 Solid state structure of (−)-45 with atom labels and thermal ellipsoids at 50% probability level. | ||
Although TMS groups flank both sides of the central ketone in this case, the asymmetric complexes provide the opportunity to gauge the effect of distant chiral centres on any asymmetric induction. Both new catalysts were also tested in asymmetric reductions of acetophenone (Table 8) but gave only moderate results, although it was reassuring to see the opposite sense of induction between 42 and 45 in the asymmetric hydrogenation reactions, as would be predicted on the basis of planar chirality control. Although in reality they are too low to indicate any significant directing effect.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Activator/% | Solvent | Conv./% | ee/% |
| 1 | 45 | — | IPA/H2O | 13.7 | 6.8 (S) |
| 2 | 45 | K2CO3 (5%) | IPA/H2O | 99.4 | 9.4 (S) |
| 3 | 45 | TMAO (10%) | IPA/H2O | 99.6 | 4.2 (S) |
| 4 | 42 | — | IPA/H2O | >99 | 3.8 (R) |
| 5 | 42 | K2CO3 (5%) | IPA/H2O | 9.6 | 2.6 (R) |
| 6 | 42 | TMAO (1%) | IPA/H2O | >99 | 0.4 (R) |
| Entry | Catalyst | Activator (%) | Solvent | Alcohol/% (ee/%) | Formate/% (ee/%) |
|---|---|---|---|---|---|
| 7 | 42 | TMAO (10%) | FA/TEA | 3.9 (3.8 ee R) | 24.2 (4 ee R) |
Although the measured enantiomeric excesses are low, some trends emerge from the data which suggest that the steric bulk and substitution on the groups flanking the central CO bond influence the process of asymmetric induction. Hydrides 46–50 (Fig. 6a), formed through activation of 34, 35, 37, 40 and 41 respectively, are representative of those that would be formed from the iron tricarbonyl precursors. Assuming that the proposed concerted mechanism for hydride transfer (illustrated in Fig. 6b for each complex) is operating, then our working model for 46–48 is that the tilt of the aromatic rings is influenced by the alkoxy groups on the 4-carbon backbone and this creates a chiral environment in the region where hydride transfer takes place (Fig. 6c). In turn this is likely to create a difference in the steric hindrance of one face of the ketone to the complex relative to the other face, and has in turn the potential to differentiate between addition of the hydride to the Re or Si face of the ketone substrate. The ees of the reduction products are generally higher using catalysts with larger alkoxy groups on the 4-C backbone, which may suggest that the ‘tilt’ of the aromatic rings is slightly better-defined and this leads in turn to improved differentiation of the faces of the ketone in the reduction step.
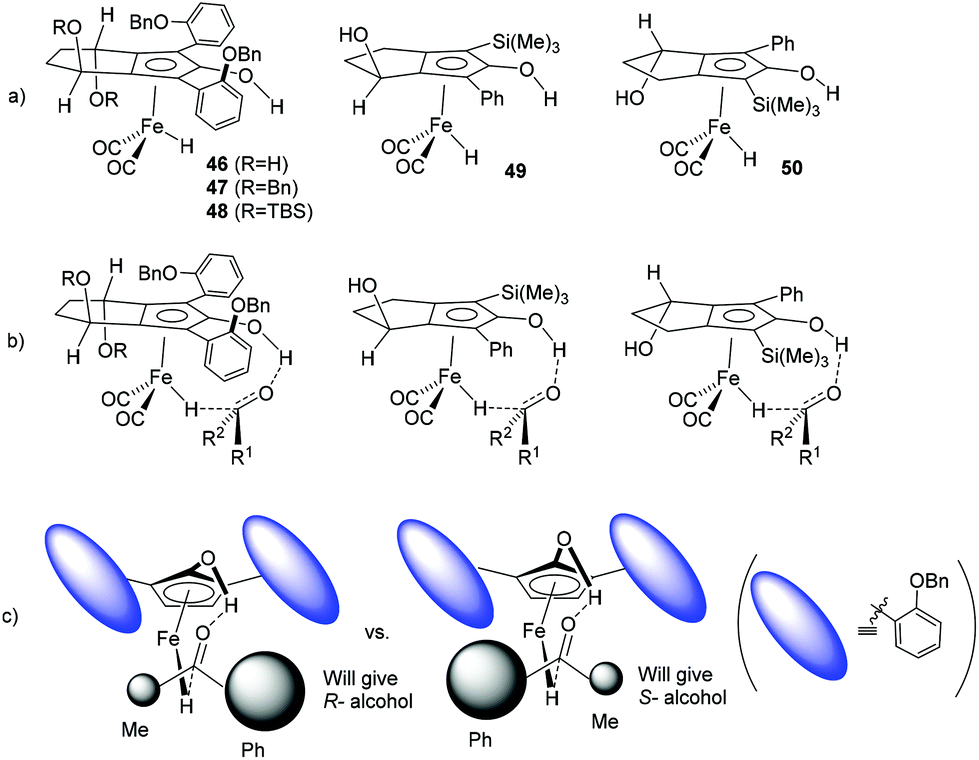 | ||
| Fig. 6 (a) Hydrides formed from complexes 34, 35, 37, 40 and 41 respectively. (b) Likely modes of hydride transfer from complexes 46–50 based on literature precedents. (c) Transition state control model for asymmetric hydride transfer from 46–48 to each face of the ketone substrate (the ovals representing positions of the tilted aromatic rings) indicating their diastereomeric nature and potential for enantiodifferentiation. | ||
In the case of 49 and 50, the complexes have a pseudo-planar chirality and in this case a similar differentiation of the faces of the ketone substrate can operate (not illustrated) due to the difference in the steric bulk of the substituents either side of the central CO bond. This would be likely to disfavour approach from one face over the other sufficiently to generate an enantioselectivity, and would account for the reversal in enantioselectivity from 49 to 50.
However the limited enantioselectivity may be the result of the significant distance from the groups flanking the central CO of the cyclopentadienone ring from the ketone substituents in the proposed reduction model. Until this distance can be closed, or the mechanism better understood, progress towards high enantioselectivities with this class of catalyst will remain challenging. The improved ees obtained using the ATH process also cannot be directly explained with the model in Fig. 6c, and suggest the operation of an additional directing effect by the solvent.
In conclusion, we have completed the synthesis of a range of enantiomerically-enriched cyclopentadienone iron tricarbonyl complexes through a range of diverse approaches, thus opening up routes to a range of valuable derivatives for testing as reduction and oxidation catalysts. Larger functional groups flanking the central CO bond in the catalyst appear to give improved enantioselectivities in reductions, however even the best enantiomeric excesses were relatively modest. Whilst the synthesis of the iron-based catalysts was successful, the results indicate that significant work remains to be carried out in order to translate our understanding of the mechanism to the synthesis of a highly enantioselective catalyst for the target applications. This remains the subject of ongoing studies in our group.
Experimental section
General experimental methods
All solvents and reagents were degassed before use and all reactions were carried out under a nitrogen atmosphere. Reactions were monitored by TLC using aluminium backed silica gel plates, visualized using UV 254 nm and phosphomolybdic acid or potassium permanganate. Flash column chromatography was carried out routinely on silica gel. Reagents and solvents were used as received from commercial sources unless otherwise stated. The synthesis of iron complexes was carried out in ACE 15 Ml 150 psi pressure tested pressure tubes and heated in aluminium heating blocks. Pressure hydrogenation reactions were carried out in a Parr pressure vessel. 1H NMR spectra were recorded on a Bruker DPX (400 or 500 MHz) spectrometer. Chemical shifts are reported in δ units, parts per million relative to 7.26 ppm for chloroform and 0.00 ppm for TMS. Mass spectra for analysis of synthetic products were recorded on a Bruker Esquire 2000 or a Bruker MicroTOF mass spectrometer. IR spectra were recorded on a PerkinElmer Spectrum One FT-IR Golden Gate instrument. Melting points were recorded on a Stuart Scientific SMP 1 instrument and are uncorrected.This compound is novel. In an round bottom flask under a nitrogen atmosphere (3S,6S)-1,8-diphenylocta-1,7-diyne-3,6-diol 15 (812 mg, 2.80 mmol) and 2-(bromomethyl)pyridine hydrobromide (1.56 g, 6.17 mmol) was dissolved/suspended in anhydrous THF (80 cm3). The mixture was cooled at 0 °C and NaH (60% in mineral oil, 447 mg, 11.2 mmol) was added in small portions. After 19 hours, H2O (100 cm3) was added dropwise and the mixture was stirred 10 minutes. Then THF was removed and the product was extracted with DCM (3 × 100 cm3). The reunited organic layers were washed with brine (50 cm3) and dried over MgSO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether/EtOAc = 1
 :1 to petroleum ether/EtOAc = 1:4) to give 2,2′-((((3S,6S)-1,8-diphenylocta-1,7-diyne-3,6-diyl)bis(oxy))bis(methylene))dipyridine 16 (1.27 g, 2.69 mmol, yield: 96%) as colourless solid. M.p. 64.2–66.4 °C. [α]25D −88.6 (c 0.50, CHCl3). IR(neat) 3058, 2966, 2931, 2893, 2868, 2836, 2220, 1591, 1571, 1489, 1476, 1457, 1441, 1388, 1343, 1329, 1299, 1242. δH (500 MHz, CDCl3), 8.56 (2H, d, J = 4.9 Hz, PyH), 7.68 (2H, td, J = 7.7, 1.7 Hz, PyH), 7.51 (2H, d, J = 7.8 Hz, PyH), 7.40–7.46 (4H, m, ArH), 7.23–7.34 (6H, m, ArH), 7.18 (2H, dd, J = 6.9, 5.3 Hz, PyH), 5.02 (2H, d, J = 13.3 Hz, PyCHH), 4.74 (2H, d, J = 13.3 Hz, PyCHH), 4.49–4.61 (2H, m, CHOR), 2.11–2.32 (4H, m, CH2). δC (125 MHz, CDCl3), 158.4 (C), 148.9 (CH), 136.6 (CH), 131.8 (CH), 128.4 (CH), 128.2 (CH), 122.5 (C), 122.3 (CH), 121.5 (CH), 87.4 (C), 86.5 (C), 71.5 (CH2), 69.8 (CH), 31.6 (CH2). m/z (ESI) [M + H]+, 473.2; [M + Na]+, 495.2. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C32H28N2O2Na 495.2043; found 495.204.
:1 to petroleum ether/EtOAc = 1:4) to give 2,2′-((((3S,6S)-1,8-diphenylocta-1,7-diyne-3,6-diyl)bis(oxy))bis(methylene))dipyridine 16 (1.27 g, 2.69 mmol, yield: 96%) as colourless solid. M.p. 64.2–66.4 °C. [α]25D −88.6 (c 0.50, CHCl3). IR(neat) 3058, 2966, 2931, 2893, 2868, 2836, 2220, 1591, 1571, 1489, 1476, 1457, 1441, 1388, 1343, 1329, 1299, 1242. δH (500 MHz, CDCl3), 8.56 (2H, d, J = 4.9 Hz, PyH), 7.68 (2H, td, J = 7.7, 1.7 Hz, PyH), 7.51 (2H, d, J = 7.8 Hz, PyH), 7.40–7.46 (4H, m, ArH), 7.23–7.34 (6H, m, ArH), 7.18 (2H, dd, J = 6.9, 5.3 Hz, PyH), 5.02 (2H, d, J = 13.3 Hz, PyCHH), 4.74 (2H, d, J = 13.3 Hz, PyCHH), 4.49–4.61 (2H, m, CHOR), 2.11–2.32 (4H, m, CH2). δC (125 MHz, CDCl3), 158.4 (C), 148.9 (CH), 136.6 (CH), 131.8 (CH), 128.4 (CH), 128.2 (CH), 122.5 (C), 122.3 (CH), 121.5 (CH), 87.4 (C), 86.5 (C), 71.5 (CH2), 69.8 (CH), 31.6 (CH2). m/z (ESI) [M + H]+, 473.2; [M + Na]+, 495.2. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C32H28N2O2Na 495.2043; found 495.204.
This compound is novel. In an ACE pressure tube under a nitrogen atmosphere 2,2′-((((3S,6S)-1,8-diphenylocta-1,7-diyne-3,6-diyl)bis(oxy))bis(methylene))dipyridine 16 (500 mg, 1.06 mmol) was dissolved in anhydrous toluene (5 cm3) previously degassed by freeze–pump–thaw cycles. Fe(CO)5 (0.79 cm3, 5.86 mmol) was added, the pressure tube was sealed and the mixture was heated at 130 °C. After 24 hours the mixture was cooled to room temperature and the pressure tube was carefully opened into the fumehood to release CO pressure. Then the mixture was diluted with EtOAc (15 cm3) and filtered through a Celite plug washing through with EtOAc (100 cm3). The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: EtOAc to EtOAc/MeOH = 9/1) to give tricarbonyl-((4S,7S)-1,3-diphenyl-4,7-bis(pyridin-2-ylmethoxy)-4,5,6,7-tetrahydro-2H-inden-2-one) iron 14 (485 mg, 0.76 mmol, yield: 72%) as an orange solid.
M.p. 55.6 °C dec. [α]25D +82.3 (c 0.52, CHCl3). IR(neat) 3056, 3013, 2931, 2864, 2062, 1983, 1633, 1590, 1571, 1499, 1475, 1435, 1388, 1362, 1329 cm−1. δH (500 MHz, CDCl3), 8.53 (1 H, dq, J = 4.9, 0.8 Hz, PyH), 8.47 (1 H, dq, J = 4.9, 0.6 Hz, PyH), 7.78–7.83 (2 H, m, ArH), 7.71–7.77 (2 H, m, ArH), 7.58 (1 H, td, J = 7.7, 1.7 Hz, PyH), 7.51 (1 H, td, J = 7.7, 1.7 Hz, PyH), 7.27–7.42 (6 H, m, ArH), 7.18 (1 H, ddd, J = 7.3, 5.0, 0.8 Hz, PyH), 7.14 (1 H, ddd, J = 7.3, 5.0, 0.8 Hz, PyH), 7.03 (1 H, d, J = 7.8 Hz, PyH), 6.71 (1 H, d, J = 7.8 Hz, PyH), 4.82 (1 H, t, J = 3.2 Hz, CHOR), 4.73 (1 H, d, J = 12.1 Hz, PyCHH), 4.60 (1 H, d, J = 12.1 Hz, PyCHH), 4.57 (1 H, d, J = 12.1 Hz, PyCHH), 4.52 (1 H, t, J = 3.3 Hz, CHOR), 4.28 (1 H, d, J = 12.1 Hz, PyCHH), 2.14–2.35 (3 H, m, CH2), 2.03–2.12 (1 H, m, CH2). δC (125 MHz, CDCl3), 208.16 (C), 170.08 (C), 157.18 (C), 156.91 (C), 148.93 (CH), 148.80 (CH), 136.60 (CH), 136.50 (CH), 131.23 (C), 131.06 (C), 130.08 (CH), 129.71 (CH), 128.38 (CH), 128.34 (CH), 128.02 (CH), 122.62 (CH), 122.52 (CH), 122.25 (CH), 121.69 (CH), 100.15 (C), 100.08 (C), 83.28 (C), 81.69 (C), 72.82 (CH2), 72.62 (CH2), 70.80 (CH), 68.74 (CH), 22.09 (CH2), 21.98 (CH2). m/z (ESI) [M + H]+, 641.1; [M + Na]+, 663.1. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C36H28FeN2O6Na 663.1190; found 663.1191.
This compound is novel. In an round bottom flask under a nitrogen atmosphere, tricarbonyl-((4S,7S)-1,3-diphenyl-4,7-bis(pyridin-2-ylmethoxy)-4,5,6,7-tetrahydro-2H-inden-2-one) Iron 14 (500 mg, 0.78 mmol) was dissolved in anhydrous DCM (20 cm3). Then trimethylamine N-oxide (77 mg, 1.10 mmol) was added and the reaction mixture was stirred for 1 hour and the volatiles were removed. The product was purified by flash chromatography on silica gel (eluent: hexane/EtOAc = 3
 :7 to 1:4) to give dicarbonyl-((4S,7S)-1,3-diphenyl-4,7-bis(pyridin-2-ylmethoxy)-4,5,6,7-tetrahydro-2H-inden-2-one) iron 17 (386 mg, 0.63 mmol, yield: 81%) as orange solid. M.p. 150 °C dec. [α]25D +985.3 (c 6.8 × 10−3, CHCl3). IR(neat) 3095, 3077, 3052, 2988, 2952, 2910, 1992, 1934, 1617, 1597, 1571, 1497, 1477, 1447, 1437, 1393, 1367, 1314, 1242 cm−1.
:7 to 1:4) to give dicarbonyl-((4S,7S)-1,3-diphenyl-4,7-bis(pyridin-2-ylmethoxy)-4,5,6,7-tetrahydro-2H-inden-2-one) iron 17 (386 mg, 0.63 mmol, yield: 81%) as orange solid. M.p. 150 °C dec. [α]25D +985.3 (c 6.8 × 10−3, CHCl3). IR(neat) 3095, 3077, 3052, 2988, 2952, 2910, 1992, 1934, 1617, 1597, 1571, 1497, 1477, 1447, 1437, 1393, 1367, 1314, 1242 cm−1.
δ H (500 MHz, CDCl3), 8.62 (1H, d, J = 5.0 Hz, PyH), 8.40 (1H, d, J = 4.3 Hz, PyH), 7.82–7.88 (2H, m, ArH), 7.48–7.58 (3H, m, 2× ArH and 1× PyH), 7.45 (1H, dd, J = 7.7, 1.7 Hz, PyH), 7.20–7.32 (3H, m, ArH), 7.18 (1H, d, J = 6.9 Hz, PyH), 7.07 (1H, dd, J = 6.7, 5.2 Hz, PyH), 6.95–7.02 (3H, m, ArH), 6.76 (1H, ddd, J = 7.3, 6.0, 1.4 Hz, PyH), 6.55 (1H, d, J = 7.8 Hz, PyH), 5.65 (1H, d, J = 13.0 Hz, PyCHH), 5.32 (1H, d, J = 4.4 Hz, CHOR), 4.71–4.79 (2H, m, 1× PyCHH and 1× CHOR), 4.42 (1H, d, J = 12.0 Hz, PyCHH), 3.93 (1H, d, J = 12.0 Hz, PyCHH), 2.65 (1H, tt, J = 14.4, 3.0 Hz CHH), 2.54 (1H, tdd, J = 14.4, 5.6, 3.5 Hz CHH), 2.43 (1H, dq, J = 14.4, 3.0 Hz, CHH), 2.22–2.31 (1H, m, CHH). δC (125 MHz, CDCl3), 217.0 (C), 211.6 (C), 168.7 (C), 162.3 (C), 159.7 (CH), 157.5 (C), 148.5 (CH), 138.1 (CH), 136.4 (CH), 133.5 (C), 132.0 (C), 129.4 (CH), 128.3 (CH), 128.0 (CH), 127.7 (CH), 127.2 (CH), 126.0 (CH), 125.5 (CH), 124.6 (CH), 122.3 (CH), 121.8 (CH), 99.4 (C), 82.7 (C), 81.7 (C), 73.1 (CH2), 71.5 (C), 70.7, 69.3 (CH2), 69.1 (CH), 27.5 (CH2), 23.9 (CH2). m/z (ESI) [M + H]+, 613.1; [M + Na]+, 635.1. HRMS (ESI-Q-TOF) m/z: [M + H]+ calcd for C35H29FeN2O5 613.1421; found 613.1427.
This compound is novel. In an round bottom flask under a nitrogen atmosphere (3S,6S)-1,8-diphenylocta-1,7-diyne-3,6-diol 15 (1.00 g, 3.44 mmol) was dissolved in anhydrous DMF (40 cm3). The mixture was cooled at 0 °C and NaH (60% in mineral oil, 826 mg, 20.65 mmol) was added in small portions. Then and 2-(chloromethyl)quinoline hydrochloride (1.56 g, 7.94 mmol) was added in small portions and the reaction mixture was warmed at room temperature. After 24 hours, NH4Cl aqueous (30 cm3) and H2O (50 cm3) were added. The product was extracted with 3 × 150 cm3 and the reunited organic layers were washed with 3 × 100 cm3 of H2O. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: pentane/EtOAc = 9
 :1 to 3:7) to give 2,2′-((((3S,6S)-1,8-diphenylocta-1,7-diyne-3,6-diyl)bis(oxy))bis(methylene))diquinoline 19 (1.52 g, 2.65 mmol, yield: 77%) as brown solid which was enough pure by NMR to be used without further purification. A sample for full characterisation was purified by double crystallisation from hot hexane giving a white solid. M.p. 98.4–99.6 °C. [α]25D −148.0 (c 0.66, CHCl3). IR(neat) 3056, 2961, 2937, 2868, 2228, 1735, 1709, 1616, 1596, 1561, 1503, 1490, 1444, 1425, 1384, 1335, 1313, 1254, 1228, 1206. δH (500 MHz, CDCl3), 8.13 (2H, d, J = 8.5 Hz, ArH), 8.05 (2H, d, J = 8.5 Hz, ArH), 7.78 (2H, d, J = 8.1 Hz, ArH), 7.64–7.73 (4H, m, ArH), 7.48–7.54 (2H, m, ArH), 7.38–7.44 (4H, m, ArH), 7.24–7.33 (6H, m, ArH), 5.17 (2H, d, J = 13.3 Hz, ArCHH), 4.92 (2H, d, J = 13.3 Hz, ArCHH), 4.57–4.68 (2H, m, CHOR), 2.18–2.36 (4H, m, CH2). δC (125 MHz, CDCl3), 159.1 (C), 147.4 (C), 136.6 (CH), 131.8 (CH), 129.5 (CH), 128.9 (CH), 128.4 (CH), 128.2 (CH), 127.6 (CH), 127.5 (C), 126.2 (CH), 122.5 (C), 119.6 (CH), 87.5 (C), 86.7 (C), 72.2 (CH2), 70.0 (CH), 31.7 (CH2). m/z (ESI) [M + H]+, 573.2; [M + Na]+, 595.2. HRMS (ESI-Q-TOF) m/z: [M + H]+ calcd for C40H33N2O2N 573.2537; found 573.2537.
:1 to 3:7) to give 2,2′-((((3S,6S)-1,8-diphenylocta-1,7-diyne-3,6-diyl)bis(oxy))bis(methylene))diquinoline 19 (1.52 g, 2.65 mmol, yield: 77%) as brown solid which was enough pure by NMR to be used without further purification. A sample for full characterisation was purified by double crystallisation from hot hexane giving a white solid. M.p. 98.4–99.6 °C. [α]25D −148.0 (c 0.66, CHCl3). IR(neat) 3056, 2961, 2937, 2868, 2228, 1735, 1709, 1616, 1596, 1561, 1503, 1490, 1444, 1425, 1384, 1335, 1313, 1254, 1228, 1206. δH (500 MHz, CDCl3), 8.13 (2H, d, J = 8.5 Hz, ArH), 8.05 (2H, d, J = 8.5 Hz, ArH), 7.78 (2H, d, J = 8.1 Hz, ArH), 7.64–7.73 (4H, m, ArH), 7.48–7.54 (2H, m, ArH), 7.38–7.44 (4H, m, ArH), 7.24–7.33 (6H, m, ArH), 5.17 (2H, d, J = 13.3 Hz, ArCHH), 4.92 (2H, d, J = 13.3 Hz, ArCHH), 4.57–4.68 (2H, m, CHOR), 2.18–2.36 (4H, m, CH2). δC (125 MHz, CDCl3), 159.1 (C), 147.4 (C), 136.6 (CH), 131.8 (CH), 129.5 (CH), 128.9 (CH), 128.4 (CH), 128.2 (CH), 127.6 (CH), 127.5 (C), 126.2 (CH), 122.5 (C), 119.6 (CH), 87.5 (C), 86.7 (C), 72.2 (CH2), 70.0 (CH), 31.7 (CH2). m/z (ESI) [M + H]+, 573.2; [M + Na]+, 595.2. HRMS (ESI-Q-TOF) m/z: [M + H]+ calcd for C40H33N2O2N 573.2537; found 573.2537.
This compound is novel. In an ACE pressure tube under a nitrogen atmosphere 2,2′-((((3S,6S)-1,8-diphenylocta-1,7-diyne-3,6-diyl)bis(oxy))bis(methylene))diquinoline 19 (400 mg, 0.70 mmol) was dissolved in anhydrous toluene (5 cm3) previously degassed by freeze–pump–thaw cycles. Fe(CO)5 (0.47 cm3, 3.49 mmol) was added, the pressure tube was sealed and the mixture was heated at 130 °C. After 22 hours the mixture was cooled to room temperature and the pressure tube was carefully opened into the fumehood to release CO pressure. Then the mixture was diluted with EtOAc (15 cm3) and filtered through a Celite plug washing through with EtOAc (100 cm3). The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: pentane/EtOAc = 1
 :1 to 3:7) to give tricarbonyl-(4S,7S)-1,3-diphenyl-4,7-bis(quinolin-2-ylmethoxy)-4,5,6,7-tetrahydro-2H-inden-2-one iron 18 (244 mg, 0.33 mmol, 47%) as yellow/orange solid. M.p. 74.7 °C dec. [α]25D +23.0 (c 0.10, CHCl3). IR(neat) 3056, 2930, 2862, 2064, 2011, 1988, 1733, 1633, 1599, 1565, 1501, 1442, 1427, 1364, 1311, 1218, 1070 cm−1. δH (500 MHz, CDCl3), 7.91–8.07 (4H, m, ArH), 7.75–7.82 (6H, m, ArH), 7.63–7.75 (2H, m, ArH), 7.49–7.58 (2H, m, ArH), 7.27–7.39 (6H, m, ArH), 7.08 (1H, d, J = 8.4 Hz, ArH), 6.75 (1H, d, J = 8.4 Hz, ArH), 4.85–4.95 (2H, m, 1× CHOR and 1× ArCHH), 4.76 (2H, dd, J = 15.2, 12.1 Hz, ArCHH), 4.60–4.65 (1H, m, CHOR), 4.47 (1H, d, J = 12.1 Hz, ArCHH), 2.18–2.40 (3H, m, CH2), 2.08–2.15 (1H, m, CH2); δC (125 MHz, CDCl3), 208.2 (C), 170.1 (C), 157.7 (C), 157.4 (C), 147.3 (C), 147.2 (C), 136.6 (CH), 136.5 (CH), 131.2 (C), 131.1 (C), 130.2 (CH), 129.64 (CH), 129.57 (CH), 129.0 (CH), 128.9 (CH), 128.44 (CH), 128.40 (CH), 128.1 (CH), 127.6 (CH), 127.53 (C), 127.50 (C), 126.5 (CH), 126.4 (CH), 120.1 (CH), 119.5 (CH), 100.2 (C), 100.0 (C), 83.5 (C), 81.6 (C), 73.4 (CH2), 73.3 (CH2), 71.0 (CH), 69.0 (CH), 22.2 (CH2), 22.1 (CH2). m/z (ESI) [M + H]+, 741.2; [M + Na]+, 763.2. HRMS (ESI-Q-TOF) m/z: [M + H]+ calcd for C44H33FeN2O6 741.1683; found 741.1670.
:1 to 3:7) to give tricarbonyl-(4S,7S)-1,3-diphenyl-4,7-bis(quinolin-2-ylmethoxy)-4,5,6,7-tetrahydro-2H-inden-2-one iron 18 (244 mg, 0.33 mmol, 47%) as yellow/orange solid. M.p. 74.7 °C dec. [α]25D +23.0 (c 0.10, CHCl3). IR(neat) 3056, 2930, 2862, 2064, 2011, 1988, 1733, 1633, 1599, 1565, 1501, 1442, 1427, 1364, 1311, 1218, 1070 cm−1. δH (500 MHz, CDCl3), 7.91–8.07 (4H, m, ArH), 7.75–7.82 (6H, m, ArH), 7.63–7.75 (2H, m, ArH), 7.49–7.58 (2H, m, ArH), 7.27–7.39 (6H, m, ArH), 7.08 (1H, d, J = 8.4 Hz, ArH), 6.75 (1H, d, J = 8.4 Hz, ArH), 4.85–4.95 (2H, m, 1× CHOR and 1× ArCHH), 4.76 (2H, dd, J = 15.2, 12.1 Hz, ArCHH), 4.60–4.65 (1H, m, CHOR), 4.47 (1H, d, J = 12.1 Hz, ArCHH), 2.18–2.40 (3H, m, CH2), 2.08–2.15 (1H, m, CH2); δC (125 MHz, CDCl3), 208.2 (C), 170.1 (C), 157.7 (C), 157.4 (C), 147.3 (C), 147.2 (C), 136.6 (CH), 136.5 (CH), 131.2 (C), 131.1 (C), 130.2 (CH), 129.64 (CH), 129.57 (CH), 129.0 (CH), 128.9 (CH), 128.44 (CH), 128.40 (CH), 128.1 (CH), 127.6 (CH), 127.53 (C), 127.50 (C), 126.5 (CH), 126.4 (CH), 120.1 (CH), 119.5 (CH), 100.2 (C), 100.0 (C), 83.5 (C), 81.6 (C), 73.4 (CH2), 73.3 (CH2), 71.0 (CH), 69.0 (CH), 22.2 (CH2), 22.1 (CH2). m/z (ESI) [M + H]+, 741.2; [M + Na]+, 763.2. HRMS (ESI-Q-TOF) m/z: [M + H]+ calcd for C44H33FeN2O6 741.1683; found 741.1670.
This compound is novel. In a round bottom flask under a nitrogen atmosphere 1-ethynyl-2-methoxybenzene 20 (0.5 cm3, 3.86 mmol) was dissolved in anhydrous THF (10 cm3). The mixture was cooled at −78 °C and n-butyllithium (2.5 M in hexanes, 1.55 cm3, 3.88 mmol) was added dropwise. The mixture was warmed to room temperature and stirred at this temperature for 1 hour. Then the mixture was cooled to 0 °C and transferred via cannula dropwise to a flask containing N1,N4-dimethoxy-N1,N4-dimethylsuccinamide 21 (359 mg, 1.76 mmol) dissolved/suspended in THF (15 cm3) at −78 °C. After 5 minutes the mixture was warmed at room temperature and stirred at this temperature for 90 minutes. Then the mixture was cooled to −40 °C and saturated NaHCO3 aqueous solution (10 cm3) was added. The mixture was warmed to room temperature and H2O (30 cm3) was added. The product was extracted with DCM (3 × 50 cm3) and the reunited organic layers were washed with brine (50 cm3) and dried over Na2SO4. The volatiles were removed and the product was purified by filtration through a silica plug washing with DCM to give 1,8-bis(2-methoxyphenyl)octa-1,7-diyne-3,6-dione 22 (458 mg, 1.32 mmol, yield: 80%) as colourless solid.
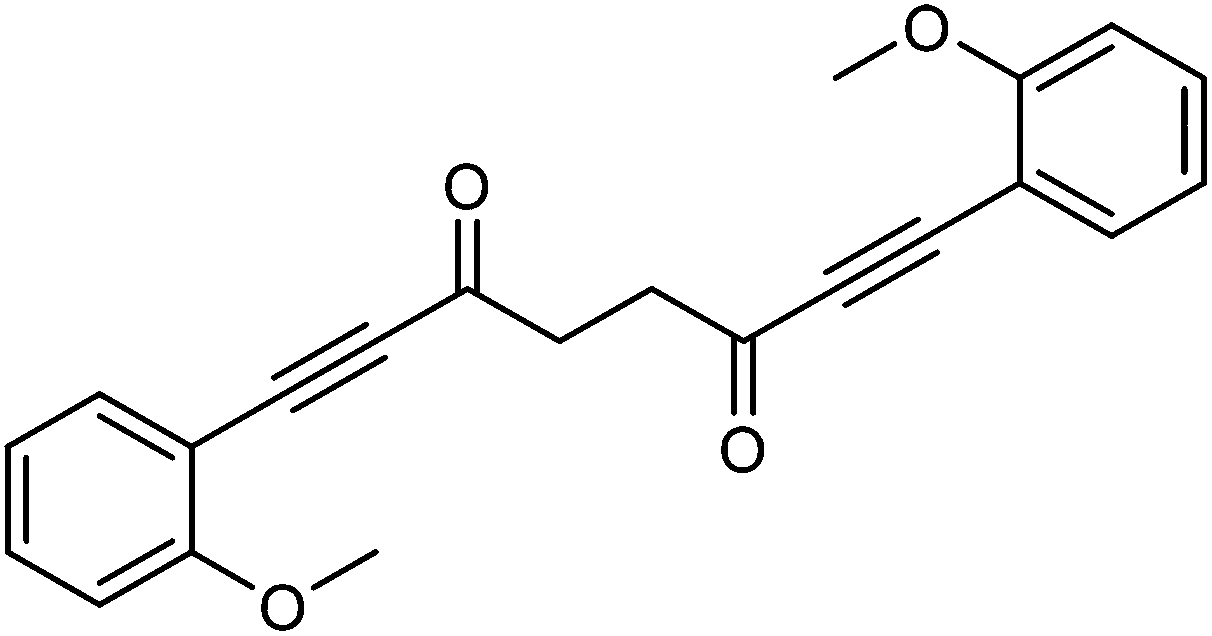
M.p. 128.8–130.1 °C. IR(neat) 2983, 2948, 2919, 2840, 2183, 1656, 1593, 1569, 1488, 1460, 1427, 1402, 1314, 1282, 1252, 1216 cm−1. δH (500 MHz, CDCl3), 7.52 (2H, dd, J = 7.5, 1.2 Hz, ArH), 7.39–7.46 (2H, m, ArH), 6.85–6.99 (4H, m, ArH), 3.90 (6H, s, CH3), 3.15 (4H, s, CH2). δC (125 MHz, CDCl3), 185.4 (C), 161.6 (C), 135.0 (CH), 132.6 (CH), 120.6 (CH), 110.8 (CH), 109.0 (C), 91.5 (C), 89.1 (C), 55.8 (CH3), 39.2 (CH2). m/z (ESI) [M + Na]+ 369.0. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C22H18O4Na 369.1097; found 369.1094.
This compound is novel. In an round bottom flask under a nitrogen atmosphere 1,8-bis(2-methoxyphenyl)octa-1,7-diyne-3,6-dione 22 (450 mg, 1.30 mmol) was dissolved in anhydrous DCM (1.5 cm3). [(S,S)-Teth-TsDpen-RuCl] 23 (8 mg, 0.013 mmol) and an azeotropic mixture of formic acid/triethylamine (5
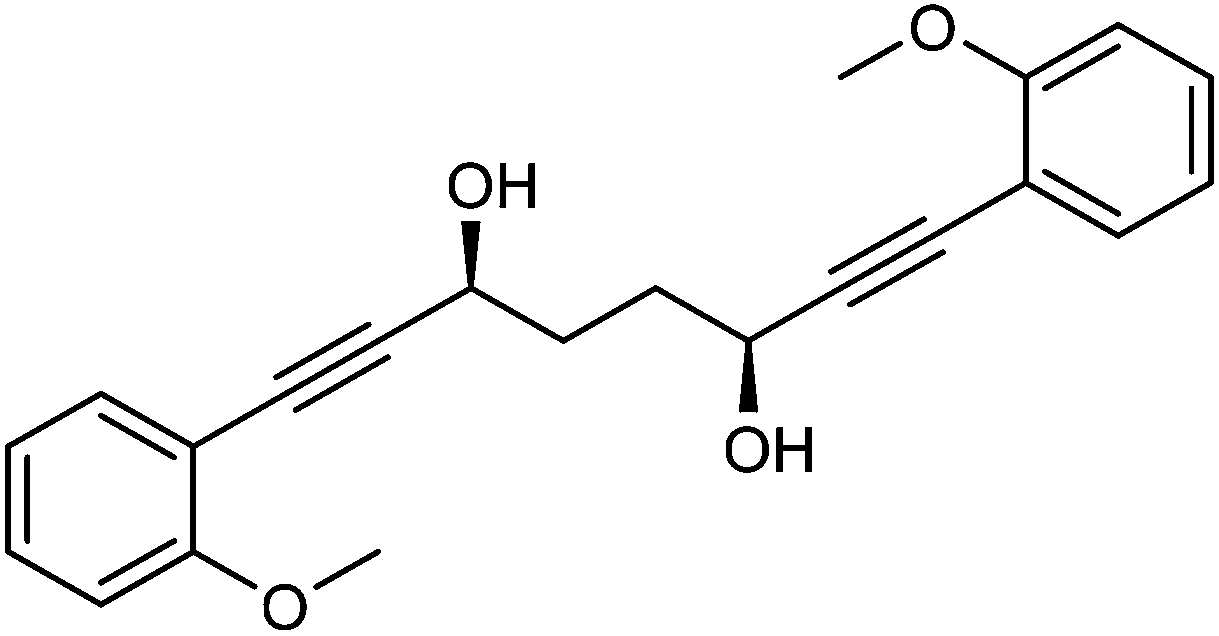 :2 mixture, 1.5 cm3) were added. After 26 hours, saturated NaHCO3 aqueous solution (5 cm3) and H2O (5 cm3) were added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 20 cm3). The reunited organic layers were washed with brine (15 cm3) and dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether/EtOAc = 7:3 to 3:7) to give (3S,6S)-1,8-bis(2-methoxyphenyl)octa-1,7-diyne-3,6-diol 24 (438 mg, 1.25 mmol, yield: 96%, dr = 98.3:1.7, >99% ee) as colourless solid. The enantiomeric and diastereomeric excess were determined by HPLC analysis with a ChiralPak IB column: 0.46 cm × 25 cm, mobile phase EtOAc:hexane 3:2, flow rate 1 mL min−1, temperature 30 °C, UV detection at λ = 254 nm; tR = 6.4 min (R,R), tR = 11.8 min (R,S), tR = 22.9 min (S,S). M.p. 113.3–114.8 °C. [α]25D −16.8 (c 0.62, CHCl3). IR(neat) 3489, 3070, 3021, 2968, 2948, 2892, 2867, 2838, 2226, 1596, 1574, 1488, 1428, 1406, 1346, 1316, 1257, 1232, 1205 cm−1. δH (500 MHz, CDCl3), 7.40 (2H, dd, J = 7.6, 1.6 Hz, ArH), 7.29 (2H, ddd, J = 8.4, 7.5, 1.7 Hz, ArH), 6.90 (2H, dd, J = 7.5, 0.8 Hz, ArH), 6.87 (2H, d, J = 8.4 Hz, ArH), 4.83 (2H, q, J = 4.7 Hz, CHOH), 3.88 (6H, s, CH3), 2.64 (2H, d, J = 5.6 Hz, OH), 2.06–2.24 (4H, m, CH2).
:2 mixture, 1.5 cm3) were added. After 26 hours, saturated NaHCO3 aqueous solution (5 cm3) and H2O (5 cm3) were added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 20 cm3). The reunited organic layers were washed with brine (15 cm3) and dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether/EtOAc = 7:3 to 3:7) to give (3S,6S)-1,8-bis(2-methoxyphenyl)octa-1,7-diyne-3,6-diol 24 (438 mg, 1.25 mmol, yield: 96%, dr = 98.3:1.7, >99% ee) as colourless solid. The enantiomeric and diastereomeric excess were determined by HPLC analysis with a ChiralPak IB column: 0.46 cm × 25 cm, mobile phase EtOAc:hexane 3:2, flow rate 1 mL min−1, temperature 30 °C, UV detection at λ = 254 nm; tR = 6.4 min (R,R), tR = 11.8 min (R,S), tR = 22.9 min (S,S). M.p. 113.3–114.8 °C. [α]25D −16.8 (c 0.62, CHCl3). IR(neat) 3489, 3070, 3021, 2968, 2948, 2892, 2867, 2838, 2226, 1596, 1574, 1488, 1428, 1406, 1346, 1316, 1257, 1232, 1205 cm−1. δH (500 MHz, CDCl3), 7.40 (2H, dd, J = 7.6, 1.6 Hz, ArH), 7.29 (2H, ddd, J = 8.4, 7.5, 1.7 Hz, ArH), 6.90 (2H, dd, J = 7.5, 0.8 Hz, ArH), 6.87 (2H, d, J = 8.4 Hz, ArH), 4.83 (2H, q, J = 4.7 Hz, CHOH), 3.88 (6H, s, CH3), 2.64 (2H, d, J = 5.6 Hz, OH), 2.06–2.24 (4H, m, CH2).
δ C (125 MHz, CDCl3), 160.0 (C), 133.6 (CH), 129.9 (CH), 120.4 (CH), 111.7 (C), 110.6 (CH), 94.0 (C), 81.5 (C), 62.6 (CH), 55.7 (CH3), 33.4 (CH2). m/z (ESI) [M + Na]+ 373.1. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C22H22O4Na 373.1410; found 373.1403.
This compound is novel. In an round bottom flask under a nitrogen atmosphere 1,8-bis(2-methoxyphenyl)octa-1,7-diyne-3,6-dione 22 (100 mg, 0.29 mmol) was dissolved in anhydrous DCM (1.0 cm3). [(R,R)-Teth-TsDpen-RuCl] 23 (1.8 mg, 0.0029 mmol) and an azeotropic mixture of formic acid/triethylamine (5
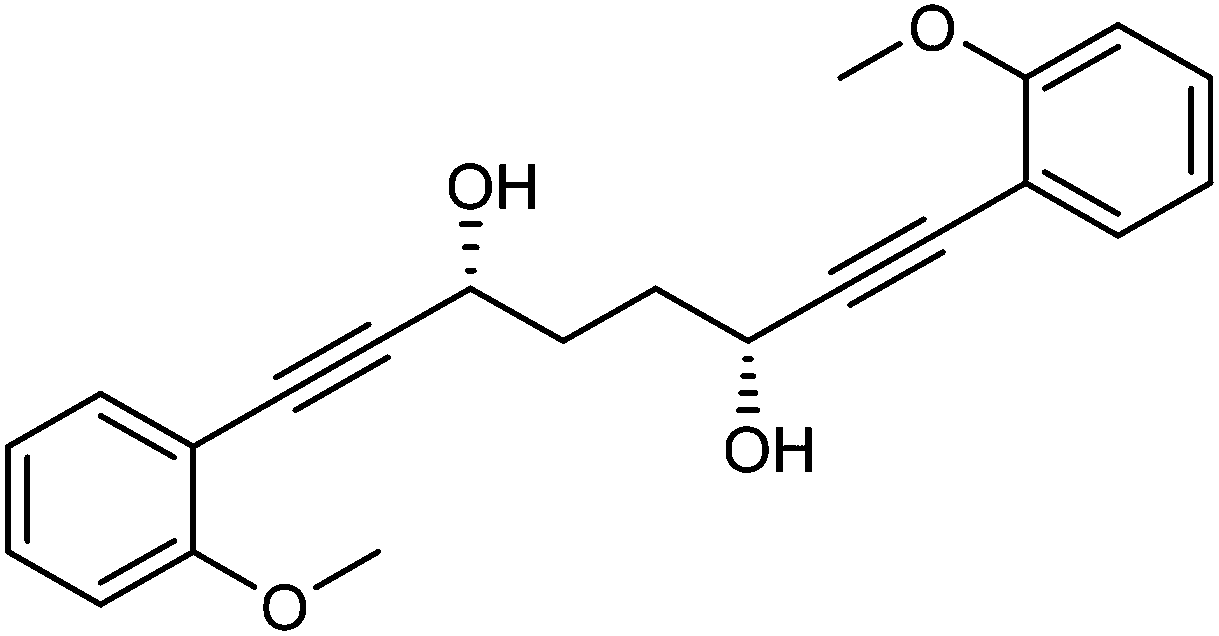 :2 mixture, 0.5 cm3) were added. After 26 hours saturated NaHCO3 aqueous solution (5 cm3) and H2O (5 cm3) were added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 20 cm3). The reunited organic layers were washed with brine (15 cm3) and dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether/EtOAc = 7:3 to 3:7) to give (3R,6R)-1,8-bis(2-methoxyphenyl)octa-1,7-diyne-3,6-diol 24 (78 mg, 0.22 mmol, yield: 77%, dr = 98.1:1.9, >99% ee) as colourless solid. The enantiomeric and diastereomeric excess were determined by HPLC analysis with a ChiralPak IB column: 0.46 cm × 25 cm, mobile phase EtOAc:hexane 3:2, flow rate 1 mL min−1, temperature 30 °C, UV detection at λ = 254 nm; tR = 6.4 min (R,R), tR = 11.8 min (R,S), tR = 22.9 min (S,S). [α]25D +16.6 (c 0.46, CHCl3).
:2 mixture, 0.5 cm3) were added. After 26 hours saturated NaHCO3 aqueous solution (5 cm3) and H2O (5 cm3) were added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 20 cm3). The reunited organic layers were washed with brine (15 cm3) and dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether/EtOAc = 7:3 to 3:7) to give (3R,6R)-1,8-bis(2-methoxyphenyl)octa-1,7-diyne-3,6-diol 24 (78 mg, 0.22 mmol, yield: 77%, dr = 98.1:1.9, >99% ee) as colourless solid. The enantiomeric and diastereomeric excess were determined by HPLC analysis with a ChiralPak IB column: 0.46 cm × 25 cm, mobile phase EtOAc:hexane 3:2, flow rate 1 mL min−1, temperature 30 °C, UV detection at λ = 254 nm; tR = 6.4 min (R,R), tR = 11.8 min (R,S), tR = 22.9 min (S,S). [α]25D +16.6 (c 0.46, CHCl3).
This compound is novel. In an round bottom flask under a nitrogen atmosphere 1,8-bis(2-methoxyphenyl)octa-1,7-diyne-3,6-dione 22 (50 mg, 0.14 mmol) was dissolved in DCM/MeOH 1
 :1 (2 cm3). NaBH4 (14 mg, 0.37 mmol) was added and the reaction was stirred for 2 hours. Then H2O (10 cm3) was added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 10 cm3). The reunited organic layers were washed with brine (10 cm3) and dried over Mg2SO4. The volatiles were removed and the product was purified by filtration through a silica plug washing with EtOAc to give 1,8-bis(2-methoxyphenyl)octa-1,7-diyne-3,6-diol 24 (46 mg, 0.13 mmol, yield: 90.9%) as light brown solid. δH (500 MHz, CDCl3), 7.37–7.42 (4H, m, ArH), 7.26–7.32 (4H, m, ArH), 6.84–6.92 (8H, m, ArH), 4.82 (1.7H, br. s., CHOH), 4.76 (2.3H, br. s., CHOH), 3.88 (12H, d, J = 5.2 Hz, CH3), 3.03 (2H, d, J = 5.5 Hz, OH), 2.69 (2H, d, J = 2.7 Hz, OH), 2.06–2.25 (8H, m, CH2). δC (125 MHz, CDCl3), 160.05 (C), 160.03 (C), 133.6 (CH), 133.5 (CH), 129.9 (CH), 120.45 (CH), 120.42 (CH), 111.7 (C), 110.6 (CH), 94.1 (C), 94.0 (C), 81.6 (C), 81.4 (C), 62.8 (CH), 62.6 (CH), 55.72 (CH3), 55.69 (CH3), 33.9 (CH2), 33.3 (CH2).
:1 (2 cm3). NaBH4 (14 mg, 0.37 mmol) was added and the reaction was stirred for 2 hours. Then H2O (10 cm3) was added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 10 cm3). The reunited organic layers were washed with brine (10 cm3) and dried over Mg2SO4. The volatiles were removed and the product was purified by filtration through a silica plug washing with EtOAc to give 1,8-bis(2-methoxyphenyl)octa-1,7-diyne-3,6-diol 24 (46 mg, 0.13 mmol, yield: 90.9%) as light brown solid. δH (500 MHz, CDCl3), 7.37–7.42 (4H, m, ArH), 7.26–7.32 (4H, m, ArH), 6.84–6.92 (8H, m, ArH), 4.82 (1.7H, br. s., CHOH), 4.76 (2.3H, br. s., CHOH), 3.88 (12H, d, J = 5.2 Hz, CH3), 3.03 (2H, d, J = 5.5 Hz, OH), 2.69 (2H, d, J = 2.7 Hz, OH), 2.06–2.25 (8H, m, CH2). δC (125 MHz, CDCl3), 160.05 (C), 160.03 (C), 133.6 (CH), 133.5 (CH), 129.9 (CH), 120.45 (CH), 120.42 (CH), 111.7 (C), 110.6 (CH), 94.1 (C), 94.0 (C), 81.6 (C), 81.4 (C), 62.8 (CH), 62.6 (CH), 55.72 (CH3), 55.69 (CH3), 33.9 (CH2), 33.3 (CH2).
This compound is novel. In an ACE pressure tube under a nitrogen atmosphere (3S,6S)-1,8-bis(2-methoxyphenyl)octa-1,7-diyne-3,6-diol 24 (190 mg, 0.54 mmol) was dissolved/suspended in anhydrous toluene (5 cm3) previously degassed by freeze–pump–thaw cycles. Fe(CO)5 (0.37 cm3, 2.74 mmol) was added, the pressure tube was sealed and the mixture was heated at 130 °C for 24 hours. After the mixture was cooled to room temperature and the pressure tube was carefully opened into the fumehood to release CO pressure. Then the mixture was diluted with EtOAc (15 cm3) and filtered through a Celite plug washing through with EtOAc (100 cm3). The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: pentane/EtOAc = 7
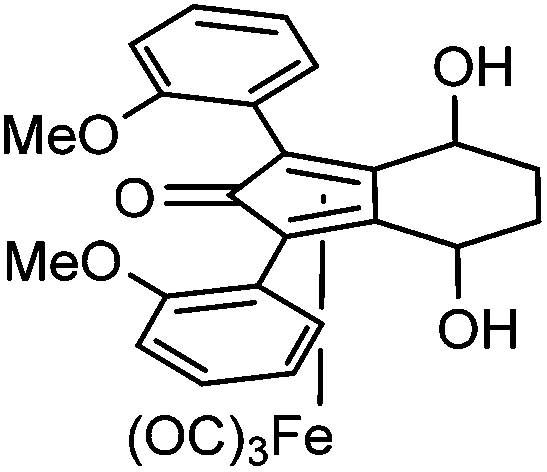 :3 to 1:4) to give tricarbonyl-(4S,7S)-4,7-dihydroxy-1,3-bis(2-methoxyphenyl)-4,5,6,7-tetrahydro-2H-inden-2-one iron 26 (193 mg, 0.37 mmol, yield: 69%) as yellow solid. M.p. 183.0–184.0 °C. [α]25D −88.8 (c 0.49, CHCl3). IR(neat) 3367 (broad), 3077, 3008, 2936, 2856, 2837, 2071, 2027, 1979, 1737, 1625, 1600, 1497, 1466, 1435, 1388, 1310, 1278, 1244 cm−1. δH (500 MHz, CDCl3), 7.69 (1H, dd, J = 7.6, 1.0 Hz, ArH), 7.43 (1H, dd, J = 7.5, 1.2 Hz, ArH), 7.33–7.41 (2H, m, ArH), 6.95–7.10 (4H, m, ArH), 4.91–5.00 (1H, m, CHOH), 4.76–4.83 (1H, m, CHOH), 3.904 (3H, s, CH3), 3.895 (3H, s, CH3), 2.80 (1H, d, J = 1.4 Hz, OH), 2.42 (1H, d, J = 3.2 Hz, OH), 2.22–2.35 (2H, m, CH2), 1.75–1.88 (1H, m, CHH), 1.56–1.71 (1H, m, CHH). δC (125 MHz, CDCl3), 208.5 (C), 169.1 (C), 157.0 (C), 156.5 (C), 134.4 (CH), 133.1 (CH), 130.3 (CH), 130.2 (CH), 121.7 (CH), 121.2 (CH), 119.98 (C), 118.96 (C), 111.8 (CH), 111.6 (CH), 105.1 (C), 104.5 (C), 80.1 (C), 79.1 (C), 63.7 (CH), 63.5 (CH), 55.7 (CH3), 55.5 (CH3), 28.5 (CH2), 28.3 (CH2). m/z (ESI) [M + H]+ 519.0; [M + Na]+ 540.9. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C26H22FeO8Na 541.0556; found 541.0558.
:3 to 1:4) to give tricarbonyl-(4S,7S)-4,7-dihydroxy-1,3-bis(2-methoxyphenyl)-4,5,6,7-tetrahydro-2H-inden-2-one iron 26 (193 mg, 0.37 mmol, yield: 69%) as yellow solid. M.p. 183.0–184.0 °C. [α]25D −88.8 (c 0.49, CHCl3). IR(neat) 3367 (broad), 3077, 3008, 2936, 2856, 2837, 2071, 2027, 1979, 1737, 1625, 1600, 1497, 1466, 1435, 1388, 1310, 1278, 1244 cm−1. δH (500 MHz, CDCl3), 7.69 (1H, dd, J = 7.6, 1.0 Hz, ArH), 7.43 (1H, dd, J = 7.5, 1.2 Hz, ArH), 7.33–7.41 (2H, m, ArH), 6.95–7.10 (4H, m, ArH), 4.91–5.00 (1H, m, CHOH), 4.76–4.83 (1H, m, CHOH), 3.904 (3H, s, CH3), 3.895 (3H, s, CH3), 2.80 (1H, d, J = 1.4 Hz, OH), 2.42 (1H, d, J = 3.2 Hz, OH), 2.22–2.35 (2H, m, CH2), 1.75–1.88 (1H, m, CHH), 1.56–1.71 (1H, m, CHH). δC (125 MHz, CDCl3), 208.5 (C), 169.1 (C), 157.0 (C), 156.5 (C), 134.4 (CH), 133.1 (CH), 130.3 (CH), 130.2 (CH), 121.7 (CH), 121.2 (CH), 119.98 (C), 118.96 (C), 111.8 (CH), 111.6 (CH), 105.1 (C), 104.5 (C), 80.1 (C), 79.1 (C), 63.7 (CH), 63.5 (CH), 55.7 (CH3), 55.5 (CH3), 28.5 (CH2), 28.3 (CH2). m/z (ESI) [M + H]+ 519.0; [M + Na]+ 540.9. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C26H22FeO8Na 541.0556; found 541.0558.
This compound is novel. In an round bottom flask under a nitrogen atmosphere (3S,6S)-1,8-bis(2-methoxyphenyl)octa-1,7-diyne-3,6-diol 24 (383 mg, 1.09 mmol) was dissolved in anhydrous THF (10 cm3) and NaH (60% in mineral oil, 109 mg, 2.72 mmol) was added in small portions. After 30 minutes, benzyl bromide (0.29 cm3, 2.44 mmol) and tetrabutylammonium iodide (158 mg, 0.43 mmol) were added. After 24 hours, saturated aqueous NH4Cl solution (50 cm3) was added and the product was extracted with DCM (3 × 50 cm3). The reunited organic layers were washed with brine (50 cm3) and dried over MgSO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether/EtOAc = 9
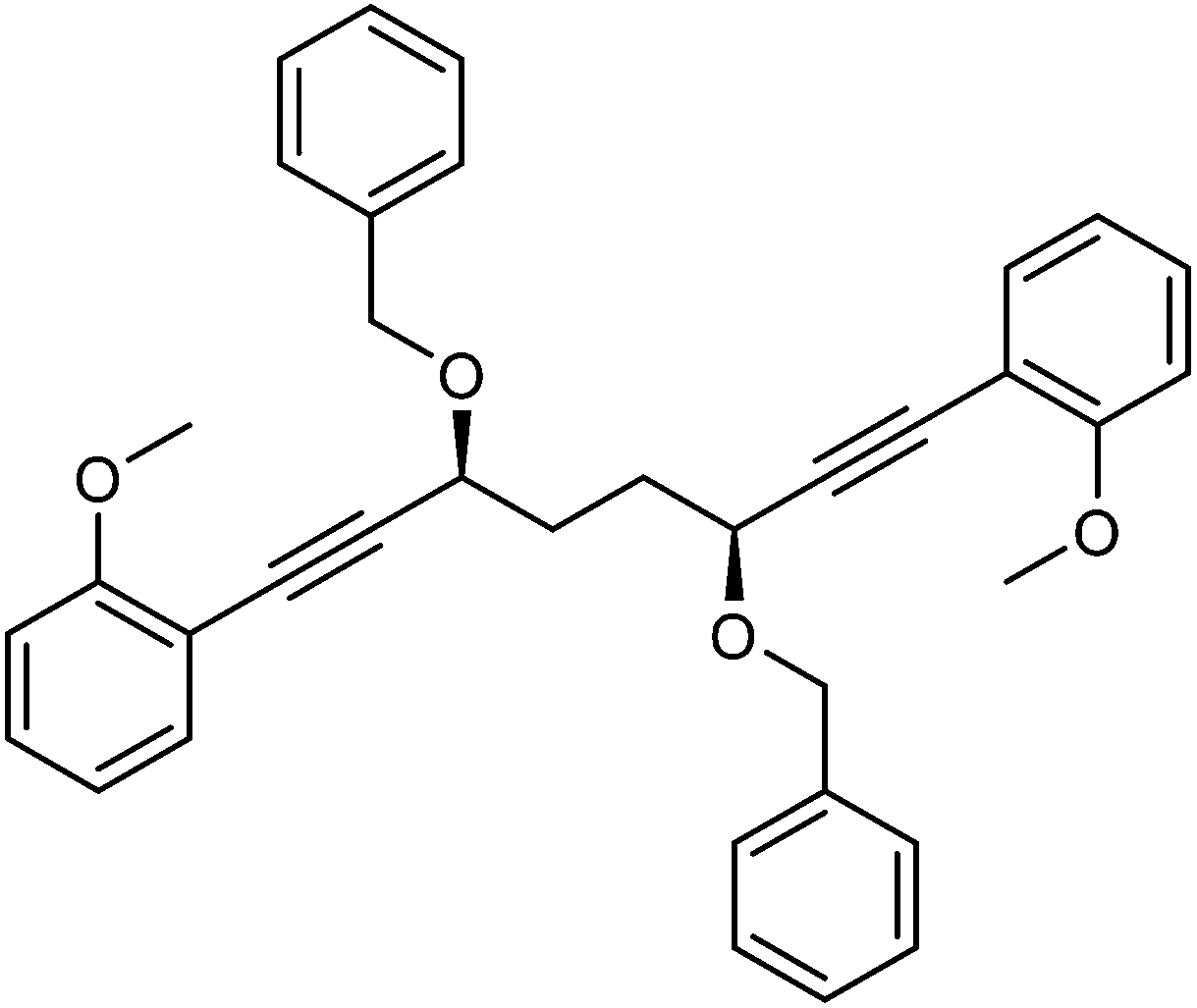 :1) to give 2,2′-((3S,6S)-3,6-bis(benzyloxy)octa-1,7-diyne-1,8-diyl)bis(methoxybenzene) 25 (511 mg, 0.96 mmol, 88%) as yellow oil. [α]30D −133.4 (c 0.515, CHCl3). IR(neat) 3062, 3029, 3004, 2961, 2934, 2836, 2224, 1596, 1575, 1547, 1492, 1454, 1434, 1333, 1291, 1261 cm−1. δH (500 MHz, CDCl3), 7.41 (6H, d, J = 7.5 Hz, ArH), 7.33 (4H, t, J = 7.4 Hz, ArH), 7.24–7.30 (4H, m, ArH), 6.83–6.92 (4H, m, ArH), 4.89 (2H, d, J = 11.7 Hz, PhCHH), 4.62 (2H, d, J = 11.7 Hz, PhCHH), 4.40–4.48 (2H, m, CHOR), 3.85 (6H, s, CH3), 2.06–2.24 (4H, m, CH2). δC (125 MHz, CDCl3), 160.2 (C), 138.2 (C), 133.5 (CH), 129.7 (CH), 128.3 (CH), 128.1 (CH), 127.5 (CH), 120.3 (CH), 112.0 (C), 110.6 (CH), 92.2 (C), 82.5 (C), 70.3 (CH2), 69.0 (CH), 55.6 (CH3), 31.7 (CH2). m/z (ESI) [M + Na]+, 553.1. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C36H34O4Na 553.2349; found 553.2344.
:1) to give 2,2′-((3S,6S)-3,6-bis(benzyloxy)octa-1,7-diyne-1,8-diyl)bis(methoxybenzene) 25 (511 mg, 0.96 mmol, 88%) as yellow oil. [α]30D −133.4 (c 0.515, CHCl3). IR(neat) 3062, 3029, 3004, 2961, 2934, 2836, 2224, 1596, 1575, 1547, 1492, 1454, 1434, 1333, 1291, 1261 cm−1. δH (500 MHz, CDCl3), 7.41 (6H, d, J = 7.5 Hz, ArH), 7.33 (4H, t, J = 7.4 Hz, ArH), 7.24–7.30 (4H, m, ArH), 6.83–6.92 (4H, m, ArH), 4.89 (2H, d, J = 11.7 Hz, PhCHH), 4.62 (2H, d, J = 11.7 Hz, PhCHH), 4.40–4.48 (2H, m, CHOR), 3.85 (6H, s, CH3), 2.06–2.24 (4H, m, CH2). δC (125 MHz, CDCl3), 160.2 (C), 138.2 (C), 133.5 (CH), 129.7 (CH), 128.3 (CH), 128.1 (CH), 127.5 (CH), 120.3 (CH), 112.0 (C), 110.6 (CH), 92.2 (C), 82.5 (C), 70.3 (CH2), 69.0 (CH), 55.6 (CH3), 31.7 (CH2). m/z (ESI) [M + Na]+, 553.1. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C36H34O4Na 553.2349; found 553.2344.
This compound is novel. In an ACE pressure tube under a nitrogen atmosphere 2,2′-((3S,6S)-3,6-bis(benzyloxy)octa-1,7-diyne-1,8-diyl)bis(methoxybenzene) 25 (323 mg, 0.61 mmol) was dissolved in anhydrous toluene (5 cm3) previously degassed by freeze–pump–thaw cycles. Fe(CO)5 (0.40 cm3, 3.04 mmol) was added, the pressure tube was sealed and the mixture was heated at 130 °C for 18 hours. After the mixture was cooled to room temperature and the pressure tube was carefully opened into the fumehood to release CO pressure. Then the mixture was diluted with EtOAc (15 cm3) and filtered through a Celite plug washing through with EtOAc (100 cm3). The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: hexane to hexane/EtOAc = 1
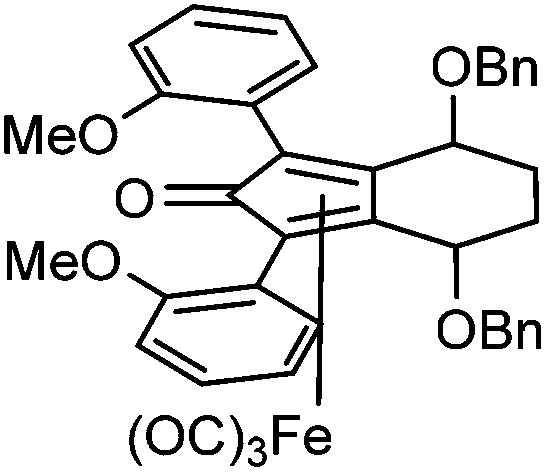 :1) to give tricarbonyl-(4S,7S)-4,7-bis(benzyloxy)-1,3-bis(2-methoxyphenyl)-4,5,6,7-tetrahydro-2-2H-inden-2-one iron 27 (85 mg, 0.12 mmol, yield: 20%) as yellow solid. M.p. 148.0–149.0 °C. [α]25D −123.4 (c 0.23, CHCl3). IR(neat) 3059, 3029, 2996, 2930, 2900, 2834, 2061, 2008, 1987, 1632, 1586, 1562, 1484, 1456, 1421, 1344, 1312, 1239 cm−1.
:1) to give tricarbonyl-(4S,7S)-4,7-bis(benzyloxy)-1,3-bis(2-methoxyphenyl)-4,5,6,7-tetrahydro-2-2H-inden-2-one iron 27 (85 mg, 0.12 mmol, yield: 20%) as yellow solid. M.p. 148.0–149.0 °C. [α]25D −123.4 (c 0.23, CHCl3). IR(neat) 3059, 3029, 2996, 2930, 2900, 2834, 2061, 2008, 1987, 1632, 1586, 1562, 1484, 1456, 1421, 1344, 1312, 1239 cm−1.
δ H (500 MHz, CDCl3), 7.93 (1H, d, J = 5.2 Hz, ArH), 7.27–7.39 (3H, m, ArH), 7.09–7.22 (6H, m, ArH), 6.95–7.07 (2H, m, ArH), 6.84–6.94 (2H, m, ArH), 6.69 (4H, d, J = 7.2 Hz, ArH), 4.70–4.77 (1H, m, CHOR), 4.62–4.69 (1H, m, CHOR), 4.43 (1H, d, J = 11.0 Hz, CHH), 4.34 (1H, d, J = 10.5 Hz, CHH), 4.22 (1H, d, J = 11.0 Hz, CHH), 4.07 (1H, d, J = 10.5 Hz, CHH), 3.84 (3H, s, CH3), 3.79 (3H, br. s., CH3), 2.27–2.45 (2H, m, CH2), 1.83–1.95 (1H, m, CHH), 1.65–1.77 (1H, m, CHH). δC (125 MHz, CDCl3), 209.0 (C), 157.4 (C), 156.5 (C), 137.3 (C), 133.1 (CH), 129.4 (CH), 129.3 (CH), 128.2 (CH), 127.9 (CH), 127.68 (CH), 127.67 (CH), 127.63 (CH), 127.3 (CH), 120.5 (CH), 119.88 (C), 119.83 (C), 110.9 (CH), 71.8 (CH2), 71.38 (CH2), 71.36 (CH), 70.3 (br., CH), 55.2 (CH3), 54.5 (CH3), 25.5 (CH2), 24.7 (CH2). m/z (ESI) [M + H]+ 699.0; [M + Na]+ 721.0.
HRMS (ESI-Q-TOF) m/z: [M + H]+ calcd for C40H35FeO8 699.1676; found 699.1685.
This compound is novel. In an round bottom flask under a nitrogen atmosphere 2-(2-ethynylphenoxy)tetrahydro-2H-pyran 29 (6.76 g, 33.42 mmol) was dissolved in anhydrous THF (100 cm3). The mixture was cooled at −78 °C and n-butyllithium (2.5 M in hexanes, 14 cm3, 35.00 mmol) was added dropwise over a period of 15 minutes. The mixture was warmed to −40 °C and stirred at this temperature for 30 minutes. Then the mixture was transferred via cannula dropwise to a flask containing N1,N4-dimethoxy-N1,N4-dimethylsuccinamide 21 (3.1 g, 15.18 mmol) dissolved/suspended in THF (80 cm3) at −78 °C. After 5 minutes the mixture was warmed at 0 °C and stirred at this temperature for 30 minutes. Then the mixture was cooled to −40 °C and saturated NaHCO3 aqueous solution (100 cm3) was added. The mixture was warmed to room temperature and H2O (200 cm3) was added. The product was extracted with DCM (3 × 500 cm3) and the reunited organic layers were dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: hexane/DCM = 1
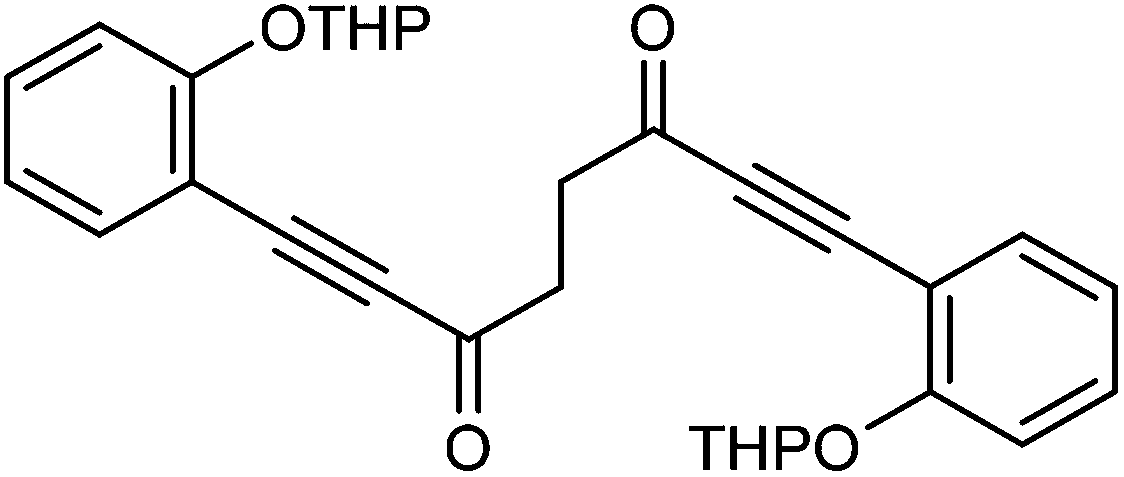 :1 to DCM/EtOAc = 4:1) to give 1,8-bis(2-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)octa-1,7-diyne-3,6-dione 28 (6.46 g, 13.27 mmol, yield: 87%) as a white solid. M.p. 127.6–129.4 °C. IR(neat) 2965, 2940, 2874, 2859, 2193, 1662, 1595, 1572, 1486, 1447, 1405, 1388, 1355, 1316, 1283, 1251, 1203 cm−1. δH (500 MHz, CDCl3), 7.52 (2H, dd, J = 7.6, 1.5 Hz), 7.35–7.43 (2H, m), 7.17 (2H, d, J = 8.4 Hz), 6.98 (2H, td, J = 7.6, 0.6 Hz), 5.56 (2H, t, J = 2.7 Hz), 3.90 (2H, td, J = 11.1, 2.7 Hz), 3.56–3.66 (1H, m), 3.13 (4H, s), 2.03–2.15 (2H, m), 1.95–2.03 (2H, m), 1.84–1.93 (2H, m), 1.59–1.76 (6H, m). δC (125 MHz, CDCl3), 185.1 (C), 159.1 (C), 134.7 (CH), 132.5 (CH), 121.5 (CH), 115.1 (CH), 110.1 (C), 96.4 (CH), 91.4 (C), 89.1 (C), 61.8 (CH2), 39.2 (CH2), 30.1 (CH2), 25.1 (CH2), 18.2 (CH2). m/z (ESI) [M + Na]+ 509.2. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C30H30O6Na 509.1935; found 509.1928.
:1 to DCM/EtOAc = 4:1) to give 1,8-bis(2-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)octa-1,7-diyne-3,6-dione 28 (6.46 g, 13.27 mmol, yield: 87%) as a white solid. M.p. 127.6–129.4 °C. IR(neat) 2965, 2940, 2874, 2859, 2193, 1662, 1595, 1572, 1486, 1447, 1405, 1388, 1355, 1316, 1283, 1251, 1203 cm−1. δH (500 MHz, CDCl3), 7.52 (2H, dd, J = 7.6, 1.5 Hz), 7.35–7.43 (2H, m), 7.17 (2H, d, J = 8.4 Hz), 6.98 (2H, td, J = 7.6, 0.6 Hz), 5.56 (2H, t, J = 2.7 Hz), 3.90 (2H, td, J = 11.1, 2.7 Hz), 3.56–3.66 (1H, m), 3.13 (4H, s), 2.03–2.15 (2H, m), 1.95–2.03 (2H, m), 1.84–1.93 (2H, m), 1.59–1.76 (6H, m). δC (125 MHz, CDCl3), 185.1 (C), 159.1 (C), 134.7 (CH), 132.5 (CH), 121.5 (CH), 115.1 (CH), 110.1 (C), 96.4 (CH), 91.4 (C), 89.1 (C), 61.8 (CH2), 39.2 (CH2), 30.1 (CH2), 25.1 (CH2), 18.2 (CH2). m/z (ESI) [M + Na]+ 509.2. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C30H30O6Na 509.1935; found 509.1928.
This compound is novel. In an round bottom flask under a nitrogen atmosphere 1,8-bis(2-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)octa-1,7-diyne-3,6-dione 28 (110 mg, 0.23 mmol) was dissolved/suspended in DCM/MeOH (1
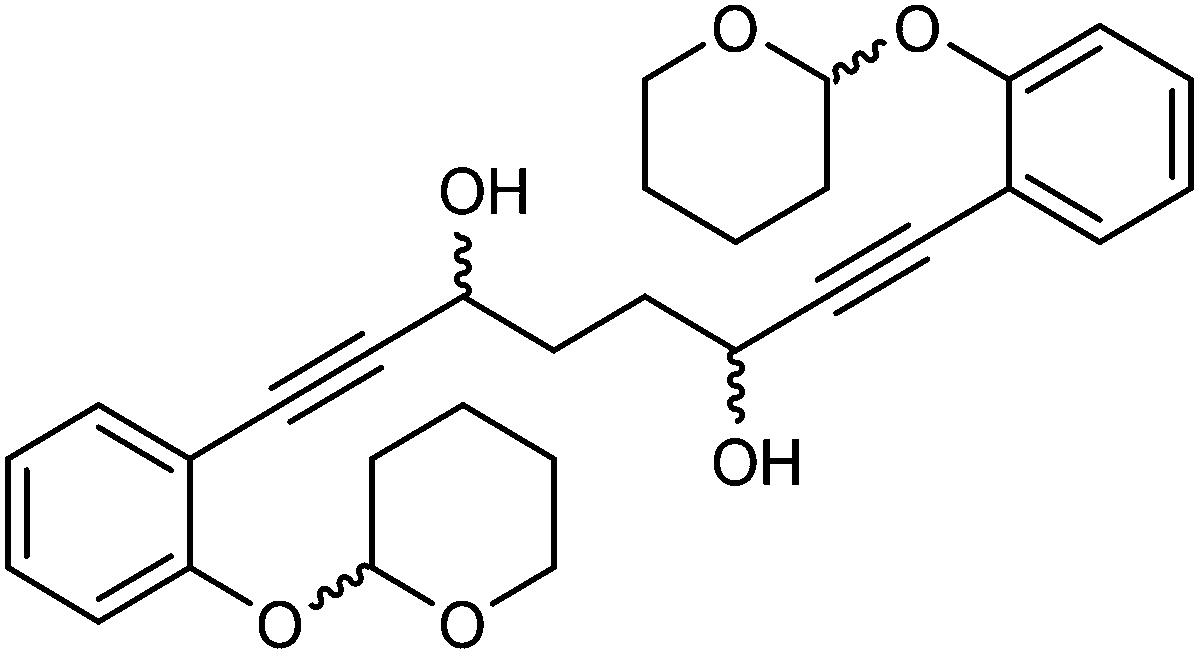 :1, 3 cm3). NaBH4 (26 mg, 0.69 mg) was added. After 4 hours H2O (10 cm3) was added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 15 cm3). The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: hexane/EtOAc = 1:4 to 3:2) to give 1,8-bis(2-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)octa-1,7-diyne-3,6-diol 30 (83 mg, 0.17 mmol, yield: 74.8%) as sticky colourless solid. δH (500 MHz, CDCl3), 7.37 (4H, d, J = 7.6 Hz, ArH), 7.20–7.28 (4H, m, ArH), 7.10 (4H, d, J = 8.2 Hz, ArH), 6.92 (4H, t, J = 7.6 Hz, ArH), 5.49 (4H, br. s., CH), 4.62–4.83 (4H, 2 overlapping br. s. at 4.78 and 4.74, CHOH), 3.82–4.04 (4H, m, CHH), 3.47–3.68 (4H, m, CHH), 2.62–2.85 (4H, m, OH), 1.52–2.24 (32H, m, CH2). δC (125 MHz, CDCl3), 157.6 (C), 133.24 (CH), 133.21 (CH), 129.7 (CH), 121.48 (CH), 121.45 (CH), 115.6 (CH), 115.52 (CH), 115.51 (CH), 113.10 (C), 113.07 (C), 96.72 (CH), 96.68 (CH), 96.66 (CH), 93.56 (C), 93.46 (C), 81.6 (C), 62.70 (CH2), 62.68 (CH2), 62.55 (CH2), 62.54 (CH2), 62.96 (CH2), 61.94 (CH2), 33.80 (CH2), 33.77 (CH2), 33.74 (CH2), 33.4 (CH2), 30.2 (CH2), 25.1 (CH2), 18.5 (CH2).
:1, 3 cm3). NaBH4 (26 mg, 0.69 mg) was added. After 4 hours H2O (10 cm3) was added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 15 cm3). The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: hexane/EtOAc = 1:4 to 3:2) to give 1,8-bis(2-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)octa-1,7-diyne-3,6-diol 30 (83 mg, 0.17 mmol, yield: 74.8%) as sticky colourless solid. δH (500 MHz, CDCl3), 7.37 (4H, d, J = 7.6 Hz, ArH), 7.20–7.28 (4H, m, ArH), 7.10 (4H, d, J = 8.2 Hz, ArH), 6.92 (4H, t, J = 7.6 Hz, ArH), 5.49 (4H, br. s., CH), 4.62–4.83 (4H, 2 overlapping br. s. at 4.78 and 4.74, CHOH), 3.82–4.04 (4H, m, CHH), 3.47–3.68 (4H, m, CHH), 2.62–2.85 (4H, m, OH), 1.52–2.24 (32H, m, CH2). δC (125 MHz, CDCl3), 157.6 (C), 133.24 (CH), 133.21 (CH), 129.7 (CH), 121.48 (CH), 121.45 (CH), 115.6 (CH), 115.52 (CH), 115.51 (CH), 113.10 (C), 113.07 (C), 96.72 (CH), 96.68 (CH), 96.66 (CH), 93.56 (C), 93.46 (C), 81.6 (C), 62.70 (CH2), 62.68 (CH2), 62.55 (CH2), 62.54 (CH2), 62.96 (CH2), 61.94 (CH2), 33.80 (CH2), 33.77 (CH2), 33.74 (CH2), 33.4 (CH2), 30.2 (CH2), 25.1 (CH2), 18.5 (CH2).
This compound is novel. In an round bottom flask under a nitrogen atmosphere 1,8-bis(2-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)octa-1,7-diyne-3,6-dione 28 (2.0 g, 4.11 mmol) was dissolved/suspended in anhydrous DCM (7 cm3). [(S,S)-Teth-TsDpen-RuCl] 23 (26 mg, 0.042 mmol) and an azeotropic mixture of formic acid/triethylamine (5
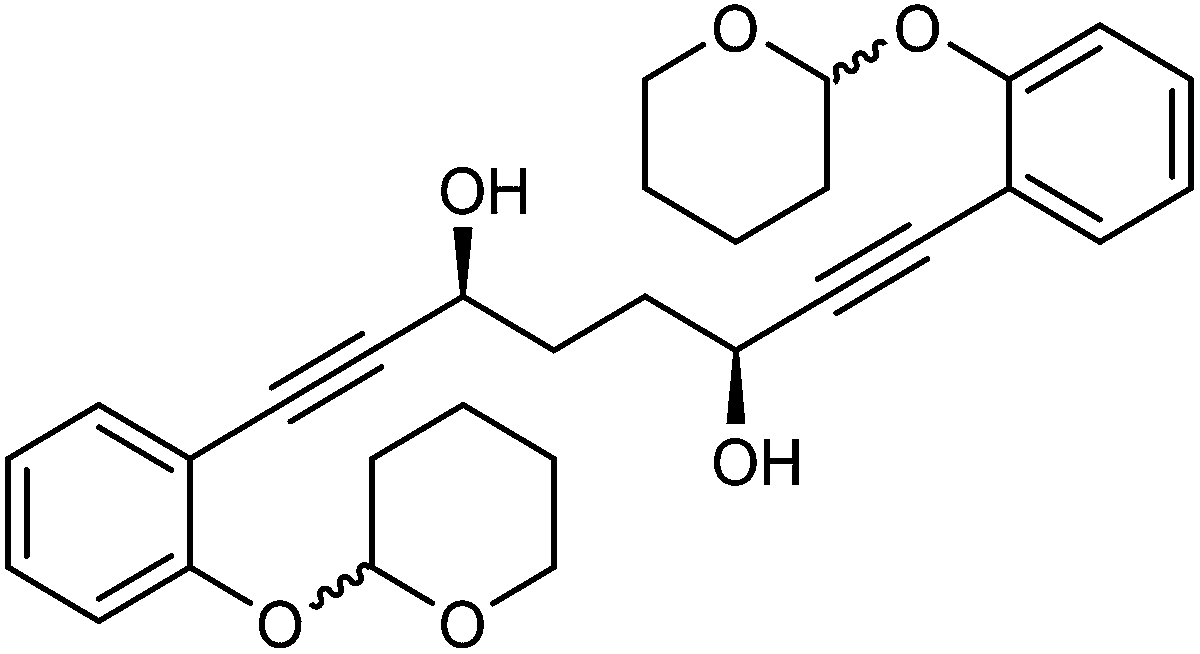 :2 mixture, 5.0 cm3) were added. After 22 hours saturated NaHCO3 aqueous solution (50 cm3) and H2O (50 cm3) were added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 150 cm3). The reunited organic layers were washed with brine (100 cm3) and dried over MgSO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether/EtOAc = 7:3 to 1:1) to give (3S,6S)-1,8-bis(2-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)octa-1,7-diyne-3,6-diol 30 (1.95 g, 3.99 mmol, yield: 96.7%, dr = 98.3:1.7, >99% ee) as a sticky colourless solid. The enantiomeric and diastereomeric excess were determined by HPLC analysis with a ChiralPak IB column: 0.46 cm × 25 cm, mobile phase EtOAc:hexane 7:3, flow rate 1 mL min−1, temperature 30 °C, UV detection at λ = 254 nm; tR = 13.4 and 13.9 min (R,R), tR = 16.2, 16.6 and 17.2 min (R,S), tR = 26.2, 28.3 and 30.2 min (S,S). [α]25D −0.58 (c 1.3, CHCl3). IR(neat) 3400 (br), 2942, 2869, 2853, 2228, 1596, 1574, 1488 1445, 1389, 1356, 1326, 1285, 1254, 1229, 1200, 1182 cm−1. δH (500 MHz, CDCl3), 7.37 (2H, d, J = 7.6 Hz, ArH), 7.24 (2H, t, J = 8.3 Hz, ArH), 7.10 (2H, d, J = 8.4 Hz, ArH), 6.92 (2H, t, J = 7.5 Hz, ArH), 5.50 (2H, br. s., CH), 4.78 (2H, br. s., CHOH), 3.85–4.02 (2H, m, CHH), 3.50–3.65 (2H, m, CHH), 2.74 (2H, br. d, OH), 1.99–2.23 (6H, m, CH2), 1.90–1.99 (2H, m, CH2), 1.81–1.90 (2H, m, CH2), 1.50–1.73 (6H, m, CH2) δC (125 MHz, CDCl3), 157.5 (C), 133.25 (CH), 133.23 (CH), 129.7 (CH), 121.4 (CH), 115.54 (CH), 115.50 (CH), 113.07 (C), 113.05 (C), 96.7 (CH), 93.5 (C), 81.61 (C), 81.59 (C), 62.54 (CH2), 62.52 (CH), 61.9 (CH2), 33.3 (CH2), 30.2 (CH2), 25.1 (CH2), 18.5 (CH2). m/z (ESI) [M + Na]+ 513.1. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C30H34O6Na 513.2248; found 513.2254.
:2 mixture, 5.0 cm3) were added. After 22 hours saturated NaHCO3 aqueous solution (50 cm3) and H2O (50 cm3) were added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 150 cm3). The reunited organic layers were washed with brine (100 cm3) and dried over MgSO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether/EtOAc = 7:3 to 1:1) to give (3S,6S)-1,8-bis(2-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)octa-1,7-diyne-3,6-diol 30 (1.95 g, 3.99 mmol, yield: 96.7%, dr = 98.3:1.7, >99% ee) as a sticky colourless solid. The enantiomeric and diastereomeric excess were determined by HPLC analysis with a ChiralPak IB column: 0.46 cm × 25 cm, mobile phase EtOAc:hexane 7:3, flow rate 1 mL min−1, temperature 30 °C, UV detection at λ = 254 nm; tR = 13.4 and 13.9 min (R,R), tR = 16.2, 16.6 and 17.2 min (R,S), tR = 26.2, 28.3 and 30.2 min (S,S). [α]25D −0.58 (c 1.3, CHCl3). IR(neat) 3400 (br), 2942, 2869, 2853, 2228, 1596, 1574, 1488 1445, 1389, 1356, 1326, 1285, 1254, 1229, 1200, 1182 cm−1. δH (500 MHz, CDCl3), 7.37 (2H, d, J = 7.6 Hz, ArH), 7.24 (2H, t, J = 8.3 Hz, ArH), 7.10 (2H, d, J = 8.4 Hz, ArH), 6.92 (2H, t, J = 7.5 Hz, ArH), 5.50 (2H, br. s., CH), 4.78 (2H, br. s., CHOH), 3.85–4.02 (2H, m, CHH), 3.50–3.65 (2H, m, CHH), 2.74 (2H, br. d, OH), 1.99–2.23 (6H, m, CH2), 1.90–1.99 (2H, m, CH2), 1.81–1.90 (2H, m, CH2), 1.50–1.73 (6H, m, CH2) δC (125 MHz, CDCl3), 157.5 (C), 133.25 (CH), 133.23 (CH), 129.7 (CH), 121.4 (CH), 115.54 (CH), 115.50 (CH), 113.07 (C), 113.05 (C), 96.7 (CH), 93.5 (C), 81.61 (C), 81.59 (C), 62.54 (CH2), 62.52 (CH), 61.9 (CH2), 33.3 (CH2), 30.2 (CH2), 25.1 (CH2), 18.5 (CH2). m/z (ESI) [M + Na]+ 513.1. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C30H34O6Na 513.2248; found 513.2254.
This compound is novel. In an round bottom flask under a nitrogen atmosphere 1,8-bis(2-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)octa-1,7-diyne-3,6-dione 28 (150 mg, 0.31 mmol) was dissolved/suspended in anhydrous DCM (1.0 cm3). [(R,R)-Teth-TsDpen-RuCl] 23 (1.9 mg, 0.003 mmol) and an azeotropic mixture of formic acid/triethylamine (5
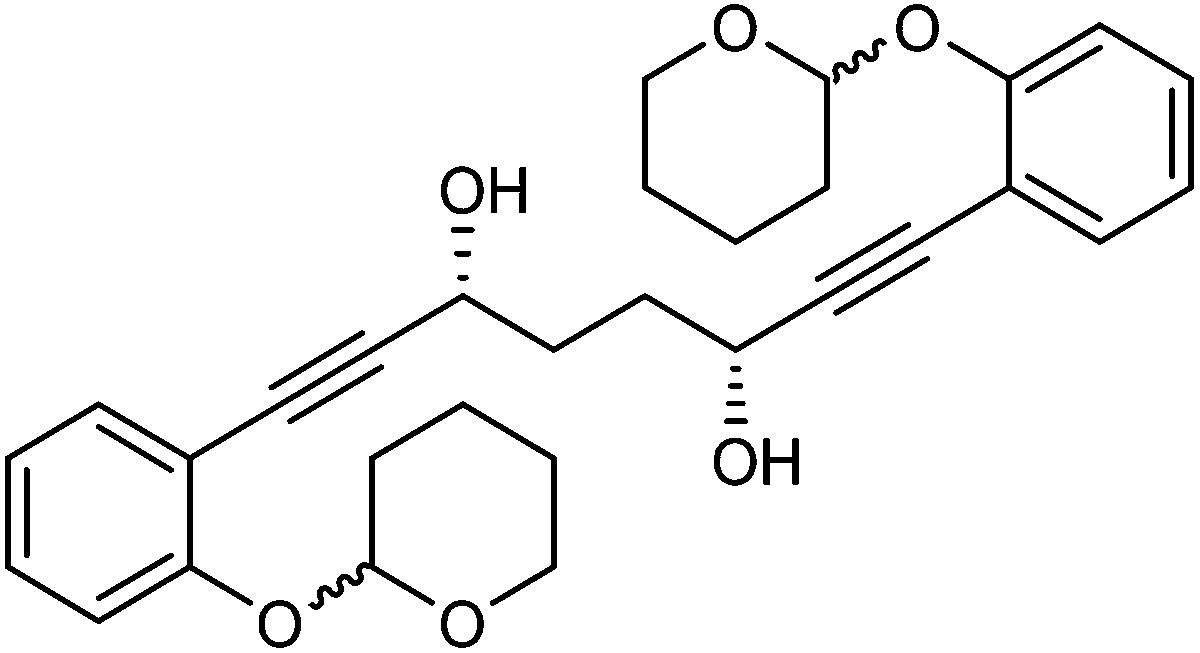 :2 mixture, 0.5 cm3) were added. After 23 hours saturated NaHCO3 aqueous solution (5 cm3) and H2O (10 cm3) were added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 20 cm3). The reunited organic layers were washed with brine (20 cm3) and dried over MgSO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether/EtOAc = 7:3 to 1:1) to give (3R,6R)-1,8-bis(2-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)octa-1,7-diyne-3,6-diol 31 (146 mg, 0.30 mmol, yield: 96%, dr = 98.4:1.6, >99% ee) as a sticky colourless solid. The enantiomeric and diastereomeric excess were determined by HPLC analysis with a ChiralPak IB column: 0.46 cm × 25 cm, mobile phase EtOAc:hexane 7:3, flow rate 1 mL min−1, temperature 30 °C, UV detection at λ = 254 nm; tR = 13.4 and 13.9 min (R,R), tR = 16.2, 16.6 and 17.2 min (R,S), tR = 26.2, 28.3 and 30.2 min (S,S). 1H and 13C NMR were identical to the reported data for (3S,6S)-1,8-bis(2-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)octa-1,7-diyne-3,6-diol. [α]27D +0.58 (c 1.8, CHCl3).
:2 mixture, 0.5 cm3) were added. After 23 hours saturated NaHCO3 aqueous solution (5 cm3) and H2O (10 cm3) were added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 20 cm3). The reunited organic layers were washed with brine (20 cm3) and dried over MgSO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether/EtOAc = 7:3 to 1:1) to give (3R,6R)-1,8-bis(2-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)octa-1,7-diyne-3,6-diol 31 (146 mg, 0.30 mmol, yield: 96%, dr = 98.4:1.6, >99% ee) as a sticky colourless solid. The enantiomeric and diastereomeric excess were determined by HPLC analysis with a ChiralPak IB column: 0.46 cm × 25 cm, mobile phase EtOAc:hexane 7:3, flow rate 1 mL min−1, temperature 30 °C, UV detection at λ = 254 nm; tR = 13.4 and 13.9 min (R,R), tR = 16.2, 16.6 and 17.2 min (R,S), tR = 26.2, 28.3 and 30.2 min (S,S). 1H and 13C NMR were identical to the reported data for (3S,6S)-1,8-bis(2-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)octa-1,7-diyne-3,6-diol. [α]27D +0.58 (c 1.8, CHCl3).
This compound is novel. In an round bottom flask (3S,6S)-1,8-bis(2-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)octa-1,7-diyne-3,6-diol 30 (1.55 g, 3.17 mmol) was dissolved in a mixture of DCM/EtOH (1
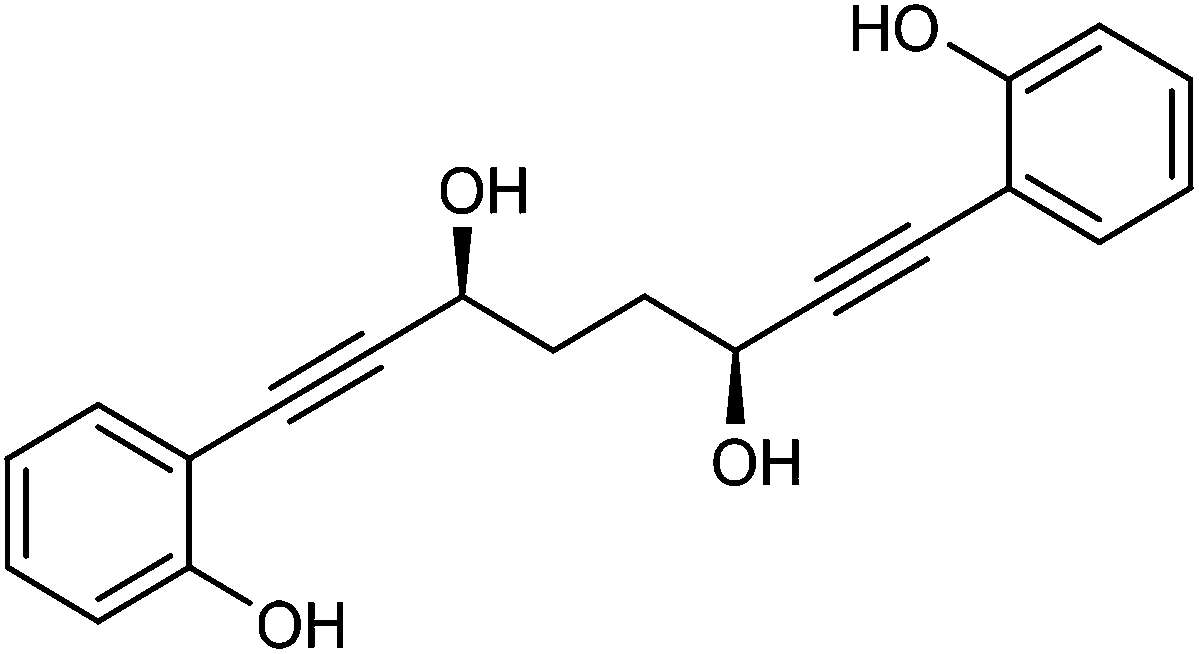 :3, 25 cm3) and pyridinium p-toluenesulfonate (107 mg, 0.43 mmol) was added. The mixture was stirred for 19 hours and the volatiles were removed. The product was purified by flash chromatography on silica gel (eluent: hexane/EtOAc = 3:2 to EtOAc) to give (3S,6S)-1,8-bis(2-hydroxyphenyl)octa-1,7-diyne-3,6-diol 31 (859 g, 2.66 mmol, yield: 84%) as colourless solid. [α]27D +15.2 (c 0.9, CH3OH). M.p. 144.0–145.6 °C. IR(neat) 3419 (br.), 3145 (br.), 2225, 1604, 1585, 1502, 1450, 1391, 1365, 1324, 1288, 1258 cm−1. δH (500 MHz, DMSO-d6), 9.79 (2H, s, OH), 7.24 (2H, dd, J = 7.6, 1.5 Hz, ArH), 7.12–7.18 (2H, m, ArH), 6.86 (2H, d, J = 8.2 Hz, ArH), 6.75 (2H, t, J = 7.5 Hz, ArH), 5.41 (2H, d, J = 5.5 Hz, OH), 4.50 (2H, d, J = 5.3 Hz, CHOH), 1.73–1.92 (4H, m, CH2). δC (125 MHz, DMSO-d6), 158.2 (C), 132.9 (CH), 129.6 (CH), 119.0 (CH), 115.4 (CH), 109.9 (C), 95.4 (C), 80.1 (C), 60.8 (CH), 33.8 (CH2). m/z (ESI) [M + Na]+ 345.1. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C20H18O4Na 345.1097; found 345.1101.
:3, 25 cm3) and pyridinium p-toluenesulfonate (107 mg, 0.43 mmol) was added. The mixture was stirred for 19 hours and the volatiles were removed. The product was purified by flash chromatography on silica gel (eluent: hexane/EtOAc = 3:2 to EtOAc) to give (3S,6S)-1,8-bis(2-hydroxyphenyl)octa-1,7-diyne-3,6-diol 31 (859 g, 2.66 mmol, yield: 84%) as colourless solid. [α]27D +15.2 (c 0.9, CH3OH). M.p. 144.0–145.6 °C. IR(neat) 3419 (br.), 3145 (br.), 2225, 1604, 1585, 1502, 1450, 1391, 1365, 1324, 1288, 1258 cm−1. δH (500 MHz, DMSO-d6), 9.79 (2H, s, OH), 7.24 (2H, dd, J = 7.6, 1.5 Hz, ArH), 7.12–7.18 (2H, m, ArH), 6.86 (2H, d, J = 8.2 Hz, ArH), 6.75 (2H, t, J = 7.5 Hz, ArH), 5.41 (2H, d, J = 5.5 Hz, OH), 4.50 (2H, d, J = 5.3 Hz, CHOH), 1.73–1.92 (4H, m, CH2). δC (125 MHz, DMSO-d6), 158.2 (C), 132.9 (CH), 129.6 (CH), 119.0 (CH), 115.4 (CH), 109.9 (C), 95.4 (C), 80.1 (C), 60.8 (CH), 33.8 (CH2). m/z (ESI) [M + Na]+ 345.1. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C20H18O4Na 345.1097; found 345.1101.
In a round bottom flask under a nitrogen atmosphere (3S,6S)-1,8-bis(2-hydroxyphenyl)octa-1,7-diyne-3,6-diol 31 (200 mg, 0.62 mmol) was dissolved in anhydrous DMF (2 cm3). Benzyl bromide (0.17 cm3, 1.43 mmol) and anhydrous potassium carbonate (214 mg, 1.55 mmol) were added and the mixture was heated to 85 °C. After 17 hours, the mixture was cooled to room temperature and H2O (15 cm3) was added. The product was extracted with DCM (20 cm3) and the organic layer was washed with H2O (15 cm3) and brine (15 cm3). The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: hexane/EtOAc = 3
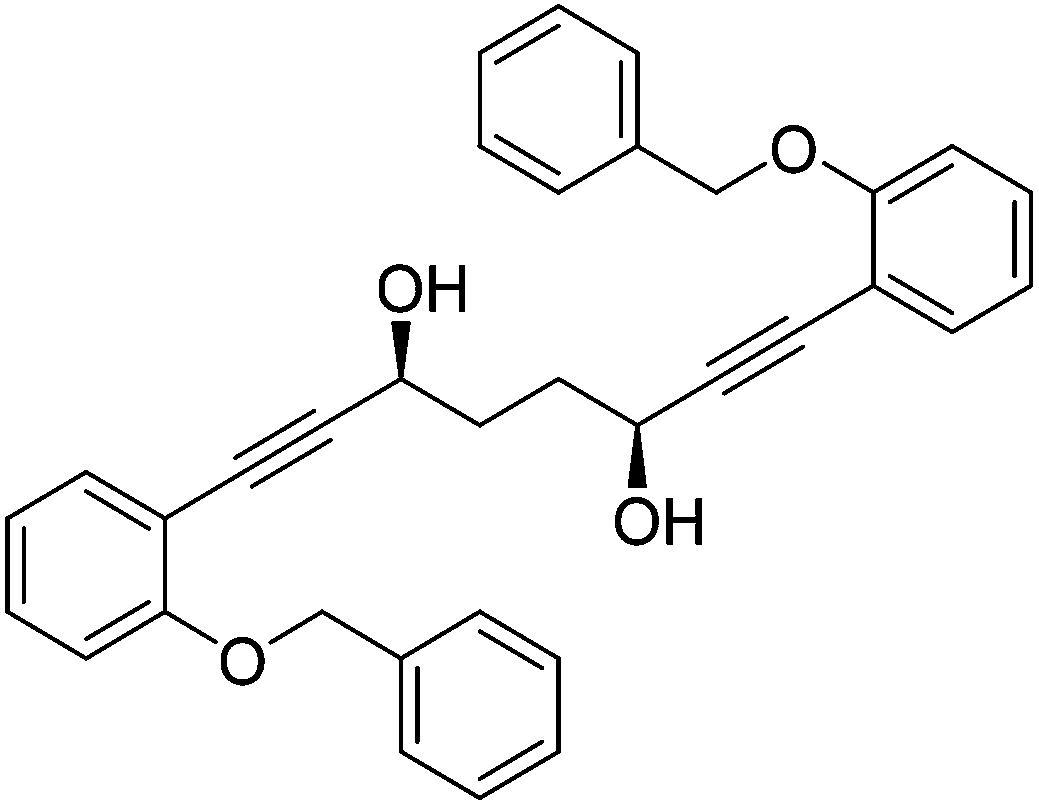 :7 to 1:4) to give (3S,6S)-1,8-bis(2-(benzyloxy)phenyl)octa-1,7-diyne-3,6-diol 32 (231 mg, 0.46 mmol, yield: 74%) as colourless solid. M.p. 122.3–123.5 °C. [α]27D −15.5 (c 0.10, CH3CN). IR(neat) 3257 (broad), 3078, 3033, 2951, 2900, 2859, 1599, 1575, 1494, 1444, 1414, 1377, 1312, 1290, 1281, 1264, 1241 cm−1. δH (500 MHz, CD3CN), 7.48 (4H, d, J = 7.6 Hz, ArH), 7.36 (6H, ddd, J = 7.6, 4.7, 3.1 Hz, ArH), 7.23–7.32 (4H, m, ArH), 7.01 (2H, d, J = 8.2 Hz, ArH), 6.91 (2H, td, J = 7.5, 0.9 Hz, ArH), 5.13 (4H, s, PhCH2), 4.56–4.64 (2H, m, CHOR), 3.49 (2H, d, J = 5.5 Hz, OH), 1.91–1.95 (4H, m, CH2). δC (125 MHz, CD3CN), 160.1 (C), 138.2 (C), 134.4 (CH), 130.9 (CH), 129.5 (CH), 128.8 (CH), 128.3 (CH), 121.9 (CH), 114.0 (CH), 113.6 (C), 96.0 (C), 81.3 (C), 71.0 (CH2), 62.7 (CH), 34.6 (CH2). m/z (ESI) [M + Na]+ 525.2; [M + K]+ 541.2. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C34H30O4Na 525.2036; found 525.2045.
:7 to 1:4) to give (3S,6S)-1,8-bis(2-(benzyloxy)phenyl)octa-1,7-diyne-3,6-diol 32 (231 mg, 0.46 mmol, yield: 74%) as colourless solid. M.p. 122.3–123.5 °C. [α]27D −15.5 (c 0.10, CH3CN). IR(neat) 3257 (broad), 3078, 3033, 2951, 2900, 2859, 1599, 1575, 1494, 1444, 1414, 1377, 1312, 1290, 1281, 1264, 1241 cm−1. δH (500 MHz, CD3CN), 7.48 (4H, d, J = 7.6 Hz, ArH), 7.36 (6H, ddd, J = 7.6, 4.7, 3.1 Hz, ArH), 7.23–7.32 (4H, m, ArH), 7.01 (2H, d, J = 8.2 Hz, ArH), 6.91 (2H, td, J = 7.5, 0.9 Hz, ArH), 5.13 (4H, s, PhCH2), 4.56–4.64 (2H, m, CHOR), 3.49 (2H, d, J = 5.5 Hz, OH), 1.91–1.95 (4H, m, CH2). δC (125 MHz, CD3CN), 160.1 (C), 138.2 (C), 134.4 (CH), 130.9 (CH), 129.5 (CH), 128.8 (CH), 128.3 (CH), 121.9 (CH), 114.0 (CH), 113.6 (C), 96.0 (C), 81.3 (C), 71.0 (CH2), 62.7 (CH), 34.6 (CH2). m/z (ESI) [M + Na]+ 525.2; [M + K]+ 541.2. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C34H30O4Na 525.2036; found 525.2045.
This compound is novel. In an ACE pressure tube under a nitrogen atmosphere (3S,6S)-1,8-bis(2-(benzyloxy)phenyl)-octa-1,7-diyne-3,6-diol 32 (200 mg, 0.40 mmol) was dissolved/suspended in anhydrous toluene (3 cm3) previously degassed by freeze–pump–thaw cycles. Fe(CO)5 (0.27 cm3, 2.00 mmol) was added, the pressure tube was sealed and the mixture was heated at 130 °C for 22 hours. After the mixture was cooled to room temperature and the pressure tube was carefully opened into the fumehood to release CO pressure. Then the mixture was diluted with EtOAc (15 cm3) and filtered through a Celite plug washing through with EtOAc (100 cm3). The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: hexane/EtOAc = 1
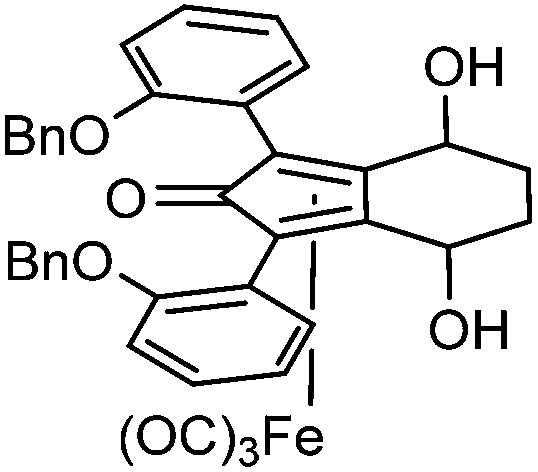 :4 to 1:1) to give tricarbonyl-(4S,7S)-1,3-bis(2-(benzyloxy)-phenyl)-4,7-dihydroxy-4,5,6,7-tetrahydro-2H-inden-2-one iron 34 (193 mg, 0.29 mmol, yield: 72%) as yellow solid. M.p. 155 °C dec. [α]26D −82.5 (c 0.19, CHCl3). IR(neat) 3370 (broad), 3062, 3034, 2958, 2924, 2856, 2063, 2011, 1988, 1737, 1717, 1616, 1597, 1579, 1494, 1450, 1373, 1297, 1278, 1226 cm−1. δH (500 MHz, CDCl3), 7.16–7.40 (14H, m, ArH), 7.07 (2H, t, J = 7.5 Hz, ArH), 6.99 (1H, d, J = 8.1 Hz, ArH), 6.90 (1H, d, J = 8.2 Hz, ArH), 5.00–5.15 (4H, m, CH2), 4.76–4.83 (1H, m, CHOH), 4.59–4.65 (1H, m, CHOH), 3.34 (1H, s, OH), 2.18–2.27 (1H, m, CHH), 2.09–2.18 (2H, m, CHH and OH), 1.66–1.79 (1H, m, CHH), 1.50–1.63 (1H, m, CHH). δC (125 MHz, CDCl3), 208.2 (C), 170.8 (C), 156.51 (C), 156.47 (C), 136.6 (C), 136.2 (C), 135.0 (CH), 134.4 (CH), 130.4 (CH), 130.2 (CH), 128.59 (CH), 128.52 (CH), 127.89 (CH), 127.80 (CH), 127.2 (CH), 126.9 (CH), 122.5 (CH), 121.8 (CH), 120.1 (C), 119.9 (C), 114.8 (CH), 113.3 (CH), 106.5 (C), 105.1 (C), 81.1 (C), 80.8 (C), 71.9 (CH2), 70.6 (CH2), 62.6 (CH), 62.4 (CH), 27.8 (CH2), 27.4 (CH2). m/z (ESI) [M + H]+ 671.1; [M + Na]+ 693.1.
:4 to 1:1) to give tricarbonyl-(4S,7S)-1,3-bis(2-(benzyloxy)-phenyl)-4,7-dihydroxy-4,5,6,7-tetrahydro-2H-inden-2-one iron 34 (193 mg, 0.29 mmol, yield: 72%) as yellow solid. M.p. 155 °C dec. [α]26D −82.5 (c 0.19, CHCl3). IR(neat) 3370 (broad), 3062, 3034, 2958, 2924, 2856, 2063, 2011, 1988, 1737, 1717, 1616, 1597, 1579, 1494, 1450, 1373, 1297, 1278, 1226 cm−1. δH (500 MHz, CDCl3), 7.16–7.40 (14H, m, ArH), 7.07 (2H, t, J = 7.5 Hz, ArH), 6.99 (1H, d, J = 8.1 Hz, ArH), 6.90 (1H, d, J = 8.2 Hz, ArH), 5.00–5.15 (4H, m, CH2), 4.76–4.83 (1H, m, CHOH), 4.59–4.65 (1H, m, CHOH), 3.34 (1H, s, OH), 2.18–2.27 (1H, m, CHH), 2.09–2.18 (2H, m, CHH and OH), 1.66–1.79 (1H, m, CHH), 1.50–1.63 (1H, m, CHH). δC (125 MHz, CDCl3), 208.2 (C), 170.8 (C), 156.51 (C), 156.47 (C), 136.6 (C), 136.2 (C), 135.0 (CH), 134.4 (CH), 130.4 (CH), 130.2 (CH), 128.59 (CH), 128.52 (CH), 127.89 (CH), 127.80 (CH), 127.2 (CH), 126.9 (CH), 122.5 (CH), 121.8 (CH), 120.1 (C), 119.9 (C), 114.8 (CH), 113.3 (CH), 106.5 (C), 105.1 (C), 81.1 (C), 80.8 (C), 71.9 (CH2), 70.6 (CH2), 62.6 (CH), 62.4 (CH), 27.8 (CH2), 27.4 (CH2). m/z (ESI) [M + H]+ 671.1; [M + Na]+ 693.1.
This compound is novel. In an round bottom flask under a nitrogen atmosphere (3S,6S)-1,8-bis(2-hydroxyphenyl)octa-1,7-diyne-3,6-diol 31 (250 mg, 0.78 mmol) was dissolved in anhydrous THF (2 cm3). NaH (60% in mineral oil, 155 mg, 3.88 mmol) and benzyl bromide (0.46 cm3, 3.87 mmol) were added and the mixture was heated at 50 °C. After 18 hours, the mixture was cooled to room temperature and saturated NH4Cl aqueous solution (5 cm3) and H2O (10 cm3) were added. The product was extracted with DCM (3 × 20 cm3) and the reunited organic layers were dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: hexane to hexane/EtOAc = 5
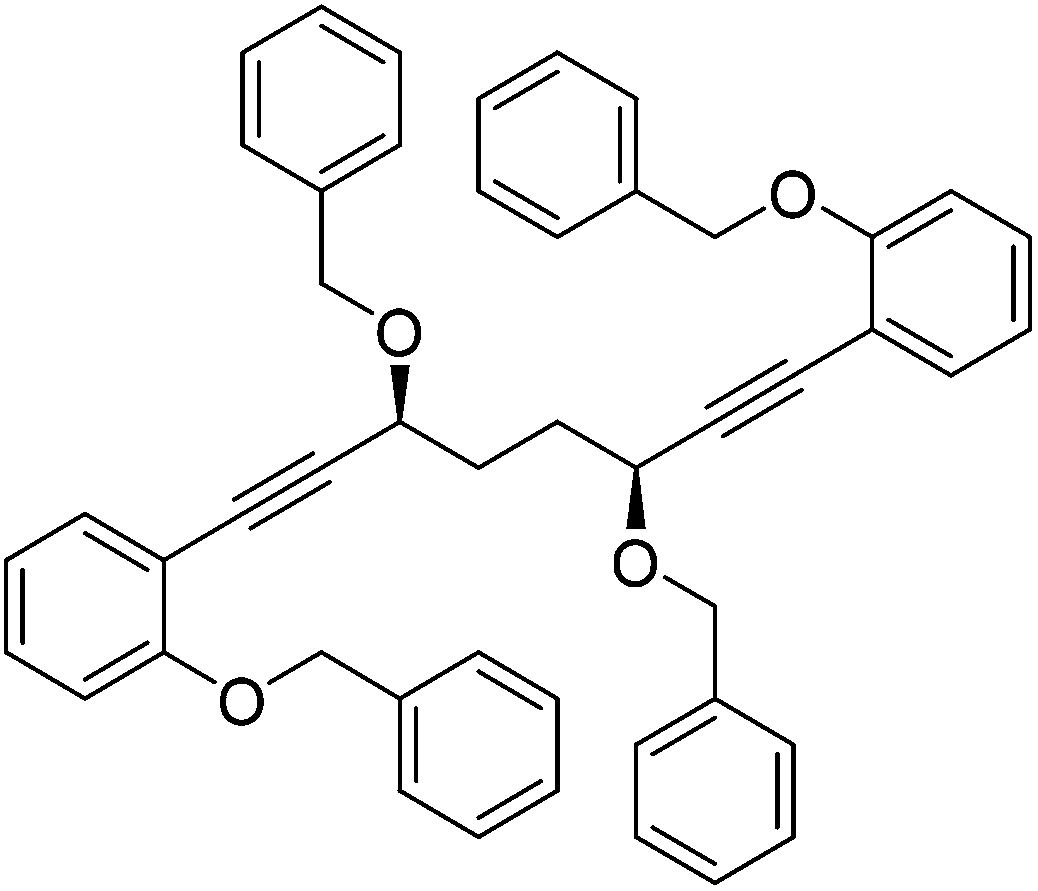 :1) to give 2,2′-((3S,6S)-3,6-bis(benzyloxy)octa-1,7-diyne-1,8-diyl)bis((benzyloxy)benzene) 33 (256 mg, 0.37 mmol, yield: 48%) as yellow oil. [α]28D −127.3 (c 0.41, CHCl3). IR(neat) 3086, 3063, 3030, 2928, 2860, 1595, 1574, 1489, 1444, 1380, 1332, 1289, 1261, 1227 cm−1. δH (500 MHz, CDCl3), 7.45 (4H, d, J = 7.6 Hz), 7.40 (2H, d, J = 7.5 Hz), 7.21–7.33 (16H, m), 7.16–7.21 (2H, m), 6.85–6.94 (4H, M), 5.12 (4H, s), 4.80 (2H, d, J = 11.9 Hz), 4.53 (2H, d, J = 11.9 Hz), 4.37 (2H, t, J = 5.5 Hz), 2.02–2.20 (4H, m). δC (125 MHz, CDCl3), 159.4 (C), 138.1 (C), 136.8 (C), 133.6 (CH), 129.6 (CH), 128.5 (CH), 128.2 (CH), 128.1 (CH), 127.8 (CH), 127.5 (CH), 127.1 (CH), 120.7 (CH), 112.8 (C), 112.4 (CH), 92.3 (C), 82.5 (C), 70.34 (CH2), 70.30 (CH2), 68.8 (CH), 31.7 (CH2). m/z (ESI) [M + Na]+ 705.3; [M + K]+ 721.3. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C48H42O4Na 705.2975; found 705.2976.
:1) to give 2,2′-((3S,6S)-3,6-bis(benzyloxy)octa-1,7-diyne-1,8-diyl)bis((benzyloxy)benzene) 33 (256 mg, 0.37 mmol, yield: 48%) as yellow oil. [α]28D −127.3 (c 0.41, CHCl3). IR(neat) 3086, 3063, 3030, 2928, 2860, 1595, 1574, 1489, 1444, 1380, 1332, 1289, 1261, 1227 cm−1. δH (500 MHz, CDCl3), 7.45 (4H, d, J = 7.6 Hz), 7.40 (2H, d, J = 7.5 Hz), 7.21–7.33 (16H, m), 7.16–7.21 (2H, m), 6.85–6.94 (4H, M), 5.12 (4H, s), 4.80 (2H, d, J = 11.9 Hz), 4.53 (2H, d, J = 11.9 Hz), 4.37 (2H, t, J = 5.5 Hz), 2.02–2.20 (4H, m). δC (125 MHz, CDCl3), 159.4 (C), 138.1 (C), 136.8 (C), 133.6 (CH), 129.6 (CH), 128.5 (CH), 128.2 (CH), 128.1 (CH), 127.8 (CH), 127.5 (CH), 127.1 (CH), 120.7 (CH), 112.8 (C), 112.4 (CH), 92.3 (C), 82.5 (C), 70.34 (CH2), 70.30 (CH2), 68.8 (CH), 31.7 (CH2). m/z (ESI) [M + Na]+ 705.3; [M + K]+ 721.3. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C48H42O4Na 705.2975; found 705.2976.
This compound is novel. In an ACE pressure tube under a nitrogen atmosphere (3S,6S)-1,8-bis(2-(benzyloxy)phenyl)-octa-1,7-diyne-3,6-diol 33 (1.00 g, 1.46 mmol) was dissolved/suspended in anhydrous toluene (7 cm3) previously degassed by freeze–pump–thaw cycles. Fe(CO)5 (1.0 cm3, 7.42 mmol) was added, the pressure tube was sealed and the mixture was heated at 130 °C for 23 hours. After the mixture was cooled to room temperature and the pressure tube was carefully opened into the fumehood to release CO pressure. Then the mixture was diluted with EtOAc (15 cm3) and filtered through a Celite plug washing through with EtOAc (100 cm3). The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: hexane/EtOAc = 9
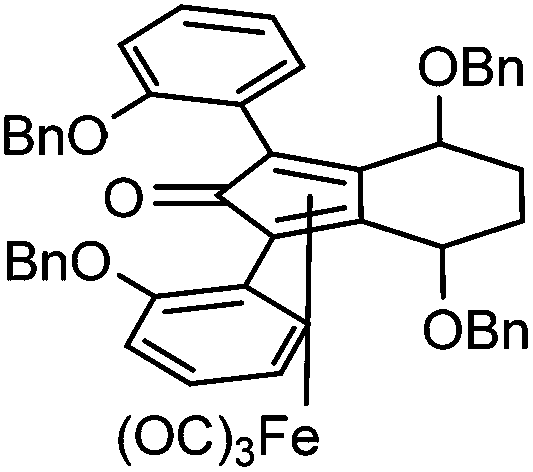 :1 to 7:3) to give tricarbonyl-(4S,7S)-4,7-bis(benzyloxy)-1,3-bis(2-(benzyloxy)phenyl)-4,5,6,7-tetrahydro-2H-inden-2-one iron 35 (1.04 g, 1.22 mmol, yield: 84%) as yellow solid. M.p. 65.4–67.0 °C. [α]21D −90.1 (c 0.25, CHCl3). IR(neat) 3060, 3030, 2926, 2860, 2060, 1981, 1644, 1599, 1579, 1495, 1450, 1364, 1338, 1277, 1224 cm−1. δH (500 MHz, CDCl3), 7.59 (1H, br. s., ArH), 6.84–7.42 (25H, m, ArH), 6.77 (1H, d, J = 8.2 Hz, ArH), 6.70 (2H, d, J = 7.2 Hz, ArH), 4.66–5.33 (4H, m, CH2 and CHOR), 4.17–4.64 (4H, m, CH2 and CHOR), 3.90–4.12 (2H, m, CH2), 2.14 (1H, br. s., CHH), 1.97 (1H, br. s., CHH), 1.83 (1H, br. s., CHH), 1.59 (2H, br. s., CHH). δC (125 MHz, CDCl3), 208.8 (C), 156.8 (C), 137.8 (C), 137.4 (C), 137.2 (C), 136.9 (C), 133.0 (CH), 129.6 (CH), 129.5 (CH), 128.4 (CH), 128.3 (CH), 128.1 (CH), 127.95 (CH), 127.92 (CH), 127.87 (CH), 127.6 (CH), 127.34 (CH), 127.29 (CH), 127.23 (CH), 126.68 (CH), 126.56 (CH), 121.7 (br., CH), 120.4 (CH), 119.9 (C), 113.2 (CH), 112.8 (CH), 71.7 (CH2), 71.4 (CH2), 70.5 (CH2), 70.4 (CH), 69.6 (br., CH2), 68.7 (CH), 24.0 (CH2), 23.5 (CH2). m/z (ESI) [M + H]+ 851.2; [M + Na]+ 873.1. HRMS (ESI-Q-TOF) m/z: [M + H]+ calcd for C52H43FeO8 851.2303; found 851.2302.
:1 to 7:3) to give tricarbonyl-(4S,7S)-4,7-bis(benzyloxy)-1,3-bis(2-(benzyloxy)phenyl)-4,5,6,7-tetrahydro-2H-inden-2-one iron 35 (1.04 g, 1.22 mmol, yield: 84%) as yellow solid. M.p. 65.4–67.0 °C. [α]21D −90.1 (c 0.25, CHCl3). IR(neat) 3060, 3030, 2926, 2860, 2060, 1981, 1644, 1599, 1579, 1495, 1450, 1364, 1338, 1277, 1224 cm−1. δH (500 MHz, CDCl3), 7.59 (1H, br. s., ArH), 6.84–7.42 (25H, m, ArH), 6.77 (1H, d, J = 8.2 Hz, ArH), 6.70 (2H, d, J = 7.2 Hz, ArH), 4.66–5.33 (4H, m, CH2 and CHOR), 4.17–4.64 (4H, m, CH2 and CHOR), 3.90–4.12 (2H, m, CH2), 2.14 (1H, br. s., CHH), 1.97 (1H, br. s., CHH), 1.83 (1H, br. s., CHH), 1.59 (2H, br. s., CHH). δC (125 MHz, CDCl3), 208.8 (C), 156.8 (C), 137.8 (C), 137.4 (C), 137.2 (C), 136.9 (C), 133.0 (CH), 129.6 (CH), 129.5 (CH), 128.4 (CH), 128.3 (CH), 128.1 (CH), 127.95 (CH), 127.92 (CH), 127.87 (CH), 127.6 (CH), 127.34 (CH), 127.29 (CH), 127.23 (CH), 126.68 (CH), 126.56 (CH), 121.7 (br., CH), 120.4 (CH), 119.9 (C), 113.2 (CH), 112.8 (CH), 71.7 (CH2), 71.4 (CH2), 70.5 (CH2), 70.4 (CH), 69.6 (br., CH2), 68.7 (CH), 24.0 (CH2), 23.5 (CH2). m/z (ESI) [M + H]+ 851.2; [M + Na]+ 873.1. HRMS (ESI-Q-TOF) m/z: [M + H]+ calcd for C52H43FeO8 851.2303; found 851.2302.
This compound is novel. In an round bottom flask under a nitrogen atmosphere (3S,6S)-1,8-bis(2-benzyloxyphenyl)octa-1,7-diyne-3,6-diol 32 (500 mg, 0.99 mmol) was dissolved in anhydrous DMF (10 cm3). Imidazole (339 mg, 4.98 mmol) and tert-butylchlorodimethylsilane (375 mg, 2.49 mmol) were added. After 20 hours, H2O (50 cm3) was added and the product was extracted with EtOAc (200 cm3). The organic layer was washed with brine (3 × 50 cm3) and dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: hexane to hexane/EtOAc = 19
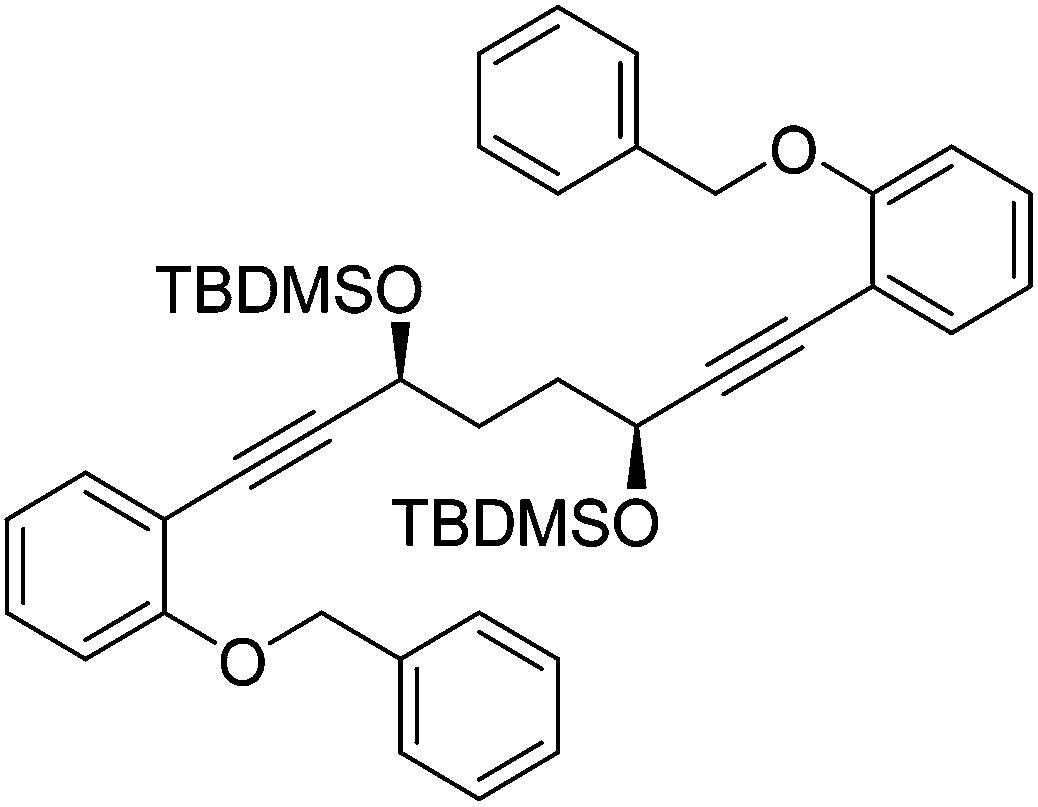 :1) to give (5S,8S)-5,8-bis((2-(benzyloxy)phenyl)ethynyl)-2,2,3,3,10,10,11,11-octamethyl-4,9-dioxa-3,10-disiladodecane: 36 (718 mg, 0.98 mmol, yield: 99%) as colourless oil. [α]27D −23.2 (c 1.6, CHCl3). IR(neat) 3067, 3033, 2953, 2928, 2884, 2855, 1596, 1574, 1490, 1471, 1444, 1407, 1380, 1360, 1338, 1289, 1257, 1229 cm−1. δH (500 MHz, CD3Cl), 7.43 (4H, d, J = 7.5 Hz, ArH), 7.30–7.40 (6H, m, ArH), 7.15–7.26 (4H, m, ArH), 6.82–6.90 (4H, m, ArH), 5.11 (4H, s, CH2OR), 4.59–4.72 (2H, m, CHOR), 1.90–2.14 (4H, m, CH2), 0.90 (18H, d, J = 2.6 Hz, CH3), 0.13 (12H, dd, J = 8.9, 2.5 Hz, CH3). δC (125 MHz, CDCl3), 159.2 (C), 136.9 (C), 133.6 (CH), 129.3 (CH), 128.4 (CH), 127.6 (CH), 127.0 (CH), 120.6 (CH), 113.2 (C), 112.6 (CH), 95.1 (C), 80.6 (C), 70.2 (CH2), 63.4 (CH), 34.5 (CH2), 25.8 (CH3), 18.3 (C), −4.4 (CH3), −5.0 (CH3). m/z (ESI) [M + Na]+ 753.4; [M + K]+ 769.3. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C46H58O4Si2Na 753.3766; found 753.3759.
:1) to give (5S,8S)-5,8-bis((2-(benzyloxy)phenyl)ethynyl)-2,2,3,3,10,10,11,11-octamethyl-4,9-dioxa-3,10-disiladodecane: 36 (718 mg, 0.98 mmol, yield: 99%) as colourless oil. [α]27D −23.2 (c 1.6, CHCl3). IR(neat) 3067, 3033, 2953, 2928, 2884, 2855, 1596, 1574, 1490, 1471, 1444, 1407, 1380, 1360, 1338, 1289, 1257, 1229 cm−1. δH (500 MHz, CD3Cl), 7.43 (4H, d, J = 7.5 Hz, ArH), 7.30–7.40 (6H, m, ArH), 7.15–7.26 (4H, m, ArH), 6.82–6.90 (4H, m, ArH), 5.11 (4H, s, CH2OR), 4.59–4.72 (2H, m, CHOR), 1.90–2.14 (4H, m, CH2), 0.90 (18H, d, J = 2.6 Hz, CH3), 0.13 (12H, dd, J = 8.9, 2.5 Hz, CH3). δC (125 MHz, CDCl3), 159.2 (C), 136.9 (C), 133.6 (CH), 129.3 (CH), 128.4 (CH), 127.6 (CH), 127.0 (CH), 120.6 (CH), 113.2 (C), 112.6 (CH), 95.1 (C), 80.6 (C), 70.2 (CH2), 63.4 (CH), 34.5 (CH2), 25.8 (CH3), 18.3 (C), −4.4 (CH3), −5.0 (CH3). m/z (ESI) [M + Na]+ 753.4; [M + K]+ 769.3. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C46H58O4Si2Na 753.3766; found 753.3759.
This compound is novel. In an ACE pressure tube under a nitrogen atmosphere (5S,8S)-5,8-bis((2-(benzyloxy)phenyl)ethynyl)-2,2,3,3,10,10,11,11-octamethyl-4,9-dioxa-3,10-disiladodecane 36 (500 mg, 0.68 mmol) was dissolved/suspended in anhydrous toluene (5 cm3) previously degassed by freeze–pump–thaw cycles. Fe(CO)5 (0.46 cm3, 3.41 mmol) was added, the pressure tube was sealed and the mixture was heated at 130 °C for 19 hours. After the mixture was cooled to room temperature and the pressure tube was carefully opened into the fumehood to release CO pressure. Then the mixture was diluted with EtOAc (15 cm3) and filtered through a Celite plug washing through with EtOAc (100 cm3). The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: hexane to hexane/EtOAc = 1
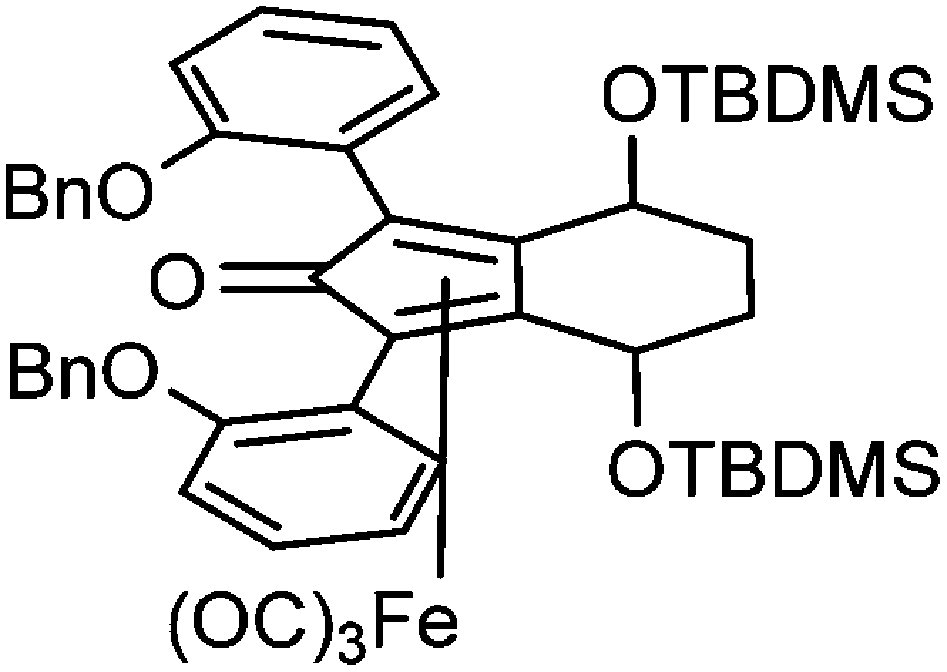 :1) to give tricarbonyl-(4S,7S)-1,3-bis(2-(benzyloxy)phenyl)-4,7-bis((tert-butyldimethylsilyl)oxy)-4,5,6,7-tetrahydro-2H-inden-2-one iron 37 (162 mg, 0.18 mmol, yield: 26%) as yellow solid. M.p. 92 °C dec. [α]21D −192.3 (c 0.1, CHCl3). IR(neat) 2950, 2927, 2882, 2854, 2061, 2008, 1980, 1648, 1599, 1579, 1497, 1450, 1363, 1277, 1248 cm−1. δH (500 MHz, CDCl3, −50 °C), 7.75 (0.5H, dd, J = 23.0, 7.5 Hz, ArH), 7.14–7.50 (10H, m, ArH), 6.72–7.13 (7H, m, ArH), 6.64 (1H, d, J = 8.6 Hz, ArH), 4.40–5.50 (6H, m, CH2OR and CHOR), 1.32–2.37 (4H, m, CH2), 0.48–0.78 (18H, m, CH3), −0.35–0.05 (6H, m, CH3), −0.98–−0.60 (6H, m, CH3). m/z (ESI) [M + H]+ 899.2; [M + Na]+ 921.2. HRMS (ESI-Q-TOF) m/z: [M + H]+ calcd for C50H59FeO8Si2 899.3094; found 899.3102.
:1) to give tricarbonyl-(4S,7S)-1,3-bis(2-(benzyloxy)phenyl)-4,7-bis((tert-butyldimethylsilyl)oxy)-4,5,6,7-tetrahydro-2H-inden-2-one iron 37 (162 mg, 0.18 mmol, yield: 26%) as yellow solid. M.p. 92 °C dec. [α]21D −192.3 (c 0.1, CHCl3). IR(neat) 2950, 2927, 2882, 2854, 2061, 2008, 1980, 1648, 1599, 1579, 1497, 1450, 1363, 1277, 1248 cm−1. δH (500 MHz, CDCl3, −50 °C), 7.75 (0.5H, dd, J = 23.0, 7.5 Hz, ArH), 7.14–7.50 (10H, m, ArH), 6.72–7.13 (7H, m, ArH), 6.64 (1H, d, J = 8.6 Hz, ArH), 4.40–5.50 (6H, m, CH2OR and CHOR), 1.32–2.37 (4H, m, CH2), 0.48–0.78 (18H, m, CH3), −0.35–0.05 (6H, m, CH3), −0.98–−0.60 (6H, m, CH3). m/z (ESI) [M + H]+ 899.2; [M + Na]+ 921.2. HRMS (ESI-Q-TOF) m/z: [M + H]+ calcd for C50H59FeO8Si2 899.3094; found 899.3102.
In a round bottom flask, under nitrogen, oxalyl chloride (2 M in DCM, 14.2 cm3, 28.40 mmol) was diluted with DCM (15 cm3). The solution was cooled to −78 °C and anhydrous DMSO (4 cm3, 56.31 mmol) was added dropwise. Then the mixture was stirred for 30 minutes and a solution of 5-(trimethylsilyl)pent-4-yn-1-ol (3.7 g, 23.7 mmol) in DCM (25 cm3) was added dropwise. After 30 minutes Et3N (16.5 cm3, 118.4 mmol) was added dropwise. The mixture was continued to stir at −78 °C for 1 hour then warmed to room temperature. After 16 hours a saturated aqueous solution of NaHCO3 (100 cm3) was added and the product was extracted with DCM (3 × 100 cm3). The reunited organic layers were washed with brine (50 cm3) and dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether to petroleum ether/EtOAc = 9/1) to give 5-(trimethylsilyl)pent-4-ynal (2.69 g, 17.43 mmol, 74%) as yellow oil. 1H NMR and 13C NMR were identical to the reported data;16δH (500 MHz, CDCl3), 9.79 (1H, s, O
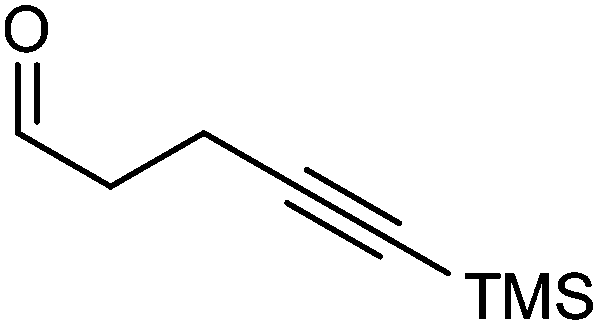 CH), 2.64–2.70 (2H, m, CH2), 2.52–2.58 (2H, m, CH2), 0.14 (9H, s, CH3). δC (125 MHz, CDCl3), 200.4 (CH), 104.7 (C), 85.8 (CH), 42.5 (CH2), 13.1 (CH2), 0.0 (CH3).
CH), 2.64–2.70 (2H, m, CH2), 2.52–2.58 (2H, m, CH2), 0.14 (9H, s, CH3). δC (125 MHz, CDCl3), 200.4 (CH), 104.7 (C), 85.8 (CH), 42.5 (CH2), 13.1 (CH2), 0.0 (CH3).
In a Schlenk tube under a nitrogen atmosphere phenylacetylene (0.85 cm3, 7.74 mmol) was dissolved in anhydrous THF (20 cm3). The mixture was cooled at −78 °C and n-butyllithium (2.5 M in hexanes, 3.1 cm3, 7.75 mmol) was added dropwise. The mixture was warmed to room temperature and stirred at this temperature for 1 hour. Then the mixture was cooled to −78 °C and transferred via cannula dropwise to a flask containing 5-(trimethylsilyl)pent-4-ynal (1.0 g, 6.48 mmol) dissolved in anhydrous THF (20 cm3) at −78 °C. After 5 minutes the mixture was warmed at room temperature and stirred at this temperature for 3 hours. Then H2O (50 cm3) was added dropwise and the product was extracted with DCM (3 × 50 cm3). The reunited organic layers were washed with brine (50 cm3) and dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether to petroleum ether/EtOAc = 13/1) to give racemic 1-phenyl-7-(trimethylsilyl)hepta-1,6-diyn-3-ol 39 (1.54 g, 6.00 mmol, 93%) as yellow oil.
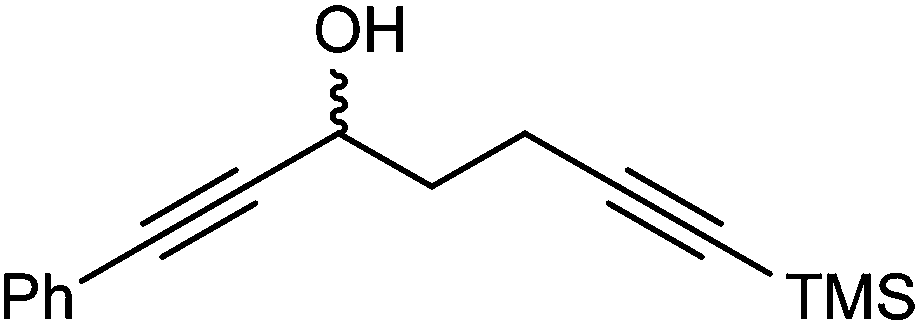
1H NMR and 13CNMR were identical to the reported data.17δH (500 MHz, CDCl3), 7.40–7.47 (2H, m, ArH), 7.28–7.34 (3H, m, ArH), 4.76 (1H, q, J = 5.8 Hz, CHOH), 2.41–2.59 (2H, m, CH2), 2.15 (1H, d, J = 5.3 Hz, CHOH), 2.02 (2H, q, J = 6.9 Hz, CH2), 0.16 (9H, s, CH3). δC (125 MHz, CDCl3), 131.7 (CH), 128.5 (CH), 128.3 (CH), 122.4 (C), 106.1 (C), 89.2 (C), 85.6 (C), 85.3 (C), 62.0 (CH), 36.4 (CH2), 16.0 (CH2), 0.1 (CH3).
This compound is novel. In an round bottom flask under a nitrogen atmosphere, racemic 1-phenyl-7-(trimethylsilyl)hepta-1,6-diyn-3-ol 39 (820 mg, 3.20 mmol) was dissolved in DCM (20 cm3). Then PCC (1.38 g, 6.40 mmol) was added. After 24 hours the mixture was filtered through a Celite plug washing through with DCM (30 cm3). The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether to petroleum ether/EtOAc = 49/1) to give 1-phenyl-7-(trimethylsilyl)hepta-1,6-diyn-3-one 38 (504 mg, 1.98 mmol, yield: 62%) as pale yellow oil. IR(neat) 2972, 2959, 2935, 2925, 2896, 2201, 2177, 1671, 1489, 1443, 1407, 1358, 1334, 1278, 1249, 1190 cm−1. δH (500 MHz, CDCl3), 7.56–7.61 (2H, m, ArH), 7.44–7.49 (1H, m, ArH), 7.36–7.42 (2H, m, ArH), 2.89–2.96 (2H, m, CH2), 2.59–2.67 (2H, m, CH2), 0.14 (9H, s, CH3). δC (125 MHz, CDCl3), 185.4 (C), 133.1 (CH), 130.9 (CH), 128.6 (CH), 119.8 (C), 104.7 (C), 91.6 (C), 87.3 (C), 85.6 (C), 44.2 (CH2), 14.7 (CH2), 0.0 (CH3). m/z (ESI) [M + H]+ 255.1; [M + Na]+ 277.1.
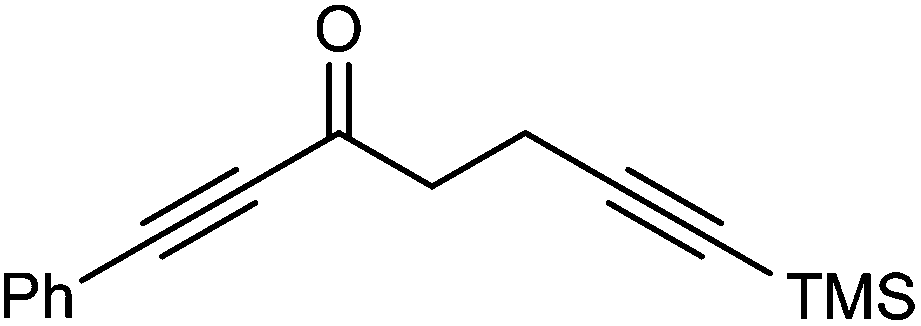
HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C16H18OSiNa 277.1019; found 277.1024.
In an round bottom flask under a nitrogen atmosphere 1-phenyl-7-(trimethylsilyl)hepta-1,6-diyn-3-one 38 (476 mg, 1.87 mmol) was dissolved in anhydrous DCM (1.3 cm3). [(S,S)-Teth-TsDpen-RuCl] 23 (11.6 mg, 0.019 mmol) and an azeotropic mixture of formic acid/triethylamine (5
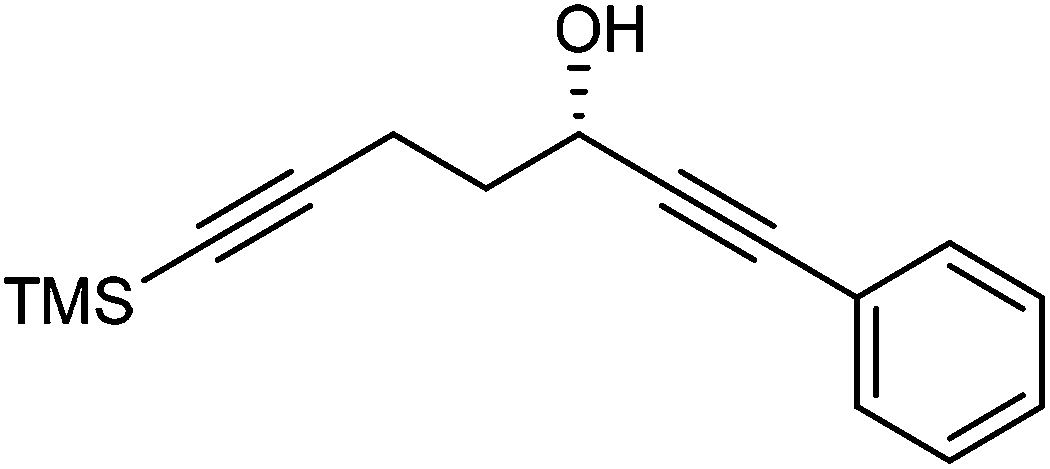 :2 mixture, 1.3 cm3) were added. After 18 hours saturated NaHCO3 aqueous solution (10 cm3) and H2O (10 cm3) were added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 20 cm3). The reunited organic layers were dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether to petroleum ether/EtOAc = 9:1) to give (S)-1-phenyl-7-(trimethylsilyl)hepta-1,6-diyn-3-ol 39 (426 mg, 1.66 mmol, yield: 89%, 94% ee) as white solid. The enantiomeric excess was determined by HPLC analysis with a ChiralPak OD column: 0.46 cm × 25 cm, mobile phase iso-propanol:hexane 9:1, flow rate 1 mL min−1, temperature 30 °C, UV detection at λ = 254 nm; tR = 6.5 min (R), tR = 13.8 min (S). 1H NMR and 13CNMR were identical to the reported data.17 M.p. 34.8–36.4 °C. [α]25D +47.6 (c 0.45, CHCl3). IR(neat) 3288 (broad), 2957, 2897, 2865, 2228, 2169, 1598, 1489, 1440, 1353, 1335, 1327, 1312, 1279, 1247. δH (500 MHz, CDCl3), 7.40–7.47 (2H, m, ArH), 7.28–7.34 (3H, m, ArH), 4.76 (1H, q, J = 5.8 Hz, CHOH), 2.41–2.59 (2H, m, CH2), 2.15 (1H, d, J = 5.3 Hz, CHOH), 2.02 (2H, q, J = 6.9 Hz, CH2), 0.16 (9H, s, CH3). δC (125 MHz, CDCl3), 131.7 (CH), 128.5 (CH), 128.3 (CH), 122.4 (C), 106.1 (C), 89.2 (C), 85.6 (C), 85.3 (C), 62.0 (CH), 36.4 (CH2), 16.0 (CH2), 0.1 (CH3).
:2 mixture, 1.3 cm3) were added. After 18 hours saturated NaHCO3 aqueous solution (10 cm3) and H2O (10 cm3) were added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 20 cm3). The reunited organic layers were dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether to petroleum ether/EtOAc = 9:1) to give (S)-1-phenyl-7-(trimethylsilyl)hepta-1,6-diyn-3-ol 39 (426 mg, 1.66 mmol, yield: 89%, 94% ee) as white solid. The enantiomeric excess was determined by HPLC analysis with a ChiralPak OD column: 0.46 cm × 25 cm, mobile phase iso-propanol:hexane 9:1, flow rate 1 mL min−1, temperature 30 °C, UV detection at λ = 254 nm; tR = 6.5 min (R), tR = 13.8 min (S). 1H NMR and 13CNMR were identical to the reported data.17 M.p. 34.8–36.4 °C. [α]25D +47.6 (c 0.45, CHCl3). IR(neat) 3288 (broad), 2957, 2897, 2865, 2228, 2169, 1598, 1489, 1440, 1353, 1335, 1327, 1312, 1279, 1247. δH (500 MHz, CDCl3), 7.40–7.47 (2H, m, ArH), 7.28–7.34 (3H, m, ArH), 4.76 (1H, q, J = 5.8 Hz, CHOH), 2.41–2.59 (2H, m, CH2), 2.15 (1H, d, J = 5.3 Hz, CHOH), 2.02 (2H, q, J = 6.9 Hz, CH2), 0.16 (9H, s, CH3). δC (125 MHz, CDCl3), 131.7 (CH), 128.5 (CH), 128.3 (CH), 122.4 (C), 106.1 (C), 89.2 (C), 85.6 (C), 85.3 (C), 62.0 (CH), 36.4 (CH2), 16.0 (CH2), 0.1 (CH3).
In an round bottom flask under a nitrogen atmosphere 1-phenyl-7-(trimethylsilyl)hepta-1,6-diyn-3-one 38 (100 mg, 0.39 mmol) was dissolved in anhydrous DCM (0.3 cm3). [(R,R)-Teth-TsDpen-RuCl] 23 (2.4 mg, 0.039 mmol) and an azeotropic mixture of formic acid/triethylamine (5
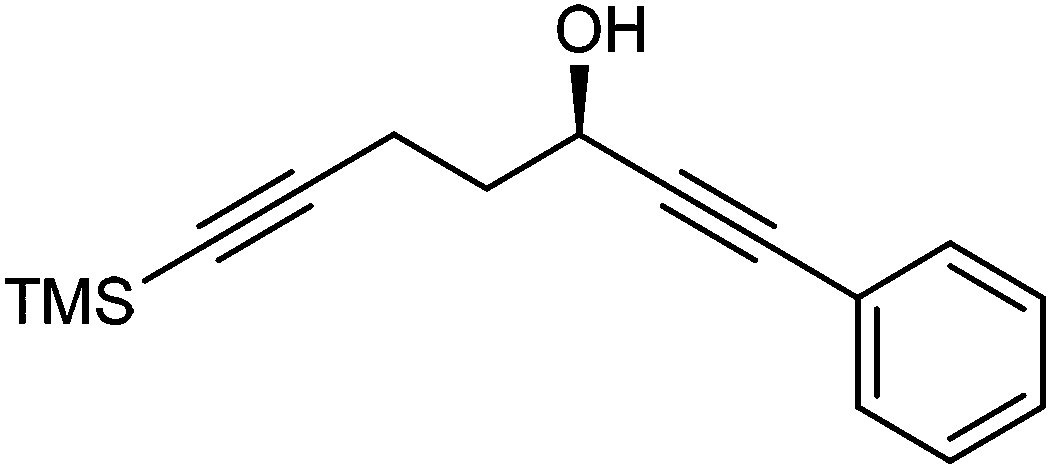 :2 mixture, 0.3 cm3) were added. After 7 hours saturated NaHCO3 aqueous solution (5 cm3) and H2O (5 cm3) were added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 20 cm3). The reunited organic layers were dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: hexane to hexane/EtOAc = 9:1) to give (R)-1-phenyl-7-(trimethylsilyl)hepta-1,6-diyn-3-ol 39 (66 mg, 0.26 mmol, yield: 66%, 95% ee) as white solid. The enantiomeric excess was determined by HPLC analysis with a ChiralPak OD column: 0.46 cm × 25 cm, mobile phase iso-propanol:hexane 9:1, flow rate 1 mL min−1, temperature 30 °C, UV detection at λ = 254 nm; tR = 6.5 min (R), tR = 13.8 min (S).
:2 mixture, 0.3 cm3) were added. After 7 hours saturated NaHCO3 aqueous solution (5 cm3) and H2O (5 cm3) were added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 20 cm3). The reunited organic layers were dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: hexane to hexane/EtOAc = 9:1) to give (R)-1-phenyl-7-(trimethylsilyl)hepta-1,6-diyn-3-ol 39 (66 mg, 0.26 mmol, yield: 66%, 95% ee) as white solid. The enantiomeric excess was determined by HPLC analysis with a ChiralPak OD column: 0.46 cm × 25 cm, mobile phase iso-propanol:hexane 9:1, flow rate 1 mL min−1, temperature 30 °C, UV detection at λ = 254 nm; tR = 6.5 min (R), tR = 13.8 min (S).
1H NMR and 13C NMR were identical to the reported data.17 M.p. 34.6–36.0 °C. [α]25D −47.3 (c 0.45, CHCl3) (lit [α]22D −36.3 (c 0.88, CHCl3), 90% ee R).
Compounds 40 and 41
These compound are novel. In an ACE pressure tube under a nitrogen atmosphere (S)-1-phenyl-7-(trimethylsilyl)hepta-1,6-diyn-3-ol 39 (300 mg, 1.16 mmol) was dissolved in anhydrous toluene (6 cm3) previously degassed by freeze–pump–thaw cycles. Fe(CO)5 (0.79 cm3, 5.86 mmol) was added, the pressure tube was sealed and the mixture was heated at 130 °C for 24 hours. After the mixture was cooled to room temperature and the pressure tube was carefully opened into the fumehood to release CO pressure. Then the mixture was diluted with EtOAc (15 cm3) and filtered through a Celite plug washing through with EtOAc (100 cm3). The volatiles were removed and the products were purified by flash chromatography on silica gel (eluent: hexane to hexane/EtOAc = 13/1) to give first the less polar 40 (198 mg, yield: 40%) as a yellow solid and then more polar 41 (136 mg, yield: 27%) as an orange solid.
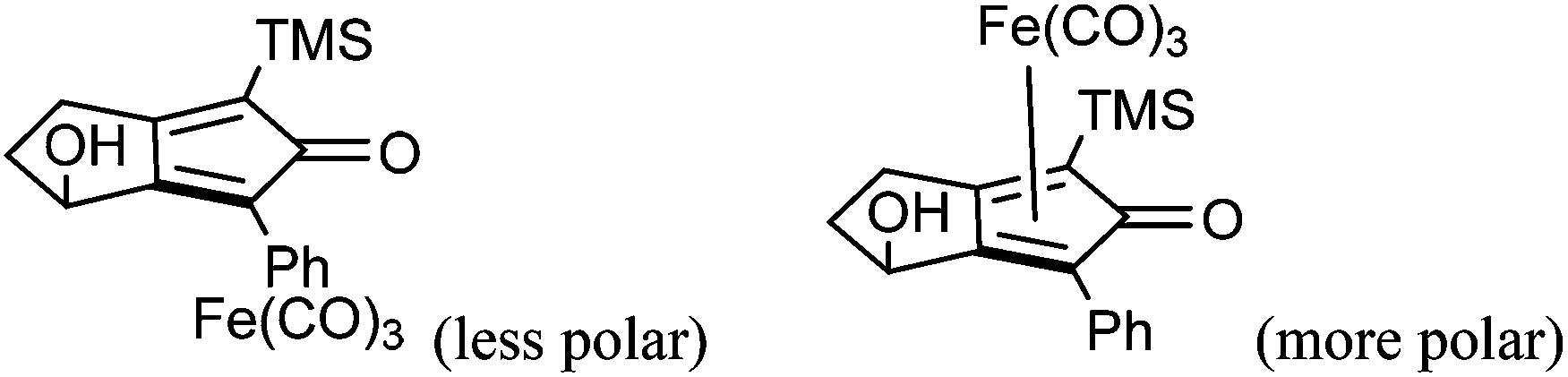
Data for 40; m.p. 193 °C dec. [α]30D −204.6 (c 0.14, CHCl3). IR(neat) 3336 (broad), 3060, 2954, 2900, 2059, 1981, 1632, 1586, 1505, 1447, 1413, 1379, 1304, 1247 cm−1. δH (500 MHz, CDCl3), 8.04 (2H, d, J = 7.9 Hz, ArH), 7.23–7.40 (3H, m, ArH), 5.33–5.44 (1H, m, CHOH), 3.02 (1H, ddd, J = 16.4, 9.3, 7.5 Hz, CHH), 2.85 (1H, br. S, OH), 2.51–2.62 (1H, m, CHH), 2.35–2.43 (1H, m, CHH), 2.23–2.35 (1H, m, CHH), 0.34 (9H, s, CH3). δC (125 MHz, CDCl3), 208.1 (C), 177.0 (C), 131.7 (C), 128.7 (CH), 128.2 (CH), 127.93 (CH), 127.90 (CH), 115.5 (C), 105.3 (C), 80.1 (C), 71.9 (CH), 69.9 (C), 36.5 (CH2), 25.4 (CH2), −0.7 (CH3). m/z (ESI) [M + H]+ 425.0; [M + Na]+ 446.9. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C20H20FeO5SiNa 447.0322; found 447.0324.
Data for 41; m.p. 196 °C dec. [α]29D +155.6 (c 0.16, CHCl3). IR(neat) 3369 (broad), 3076, 2959, 2923, 2898, 2853, 2071, 2025, 1978, 1590, 1505, 1453, 1414, 1376, 1309, 1248 cm−1. δH (500 MHz, CDCl3), 7.93 (2H, d, J = 7.5 Hz, ArH), 7.30–738 (2H, m, ArH), 7.28 (1H, d, J = 7.2 Hz, ArH), 5.51 (1H, td, J = 7.8, 4.4 Hz, CHOH), 2.52–2.78 (4H, m, CH2 and OH), 1.83–1.95 (1H, m, CHH), 0.32 (9H, s, CH3). δC (125 MHz, CDCl3), 208.1 (C), 175.8 (C), 131.2 (C), 129.0, 128.4, 128.0, 115.4 (C), 109.9 (C), 80.6 (C), 71.4 (CH), 67.5 (C), 35.7 (CH2), 24.7 (CH2), −0.7 (CH3). m/z (ESI) [M + H]+ 425.0; [M + Na]+ 446.9. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C20H20FeO5SiNa 447.0322; found 447.0325.
This compound is novel. In a Schlenk tube under a nitrogen atmosphere phenylacetylene (0.67 cm3, 4.74 mmol) was dissolved in anhydrous THF (10 cm3). The mixture was cooled at −78 °C and n-butyllithium (2.5 M in hexanes, 1.9 cm3, 4.76 mmol) was added dropwise. The cooling bath was removed and stirred for 10 minutes. Then the mixture was cooled to −78 °C and transferred via cannula dropwise to a flask containing 5-(trimethylsilyl)pent-4-ynal 43 (580 mg, 3.76 mmol) dissolved in anhydrous THF (20 cm3) at −78 °C. After 5 minutes the mixture was warmed at room temperature and stirred at this temperature for 3 hours. Then H2O (20 cm3) was added dropwise and the product was extracted with DCM (3 × 25 cm3). The reunited organic layers were washed with brine (25 cm3) and dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: hexane to hexane/Et2O = 9/1) to give 1,7-bis(trimethylsilyl)hepta-1,6-diyn-3-ol (877 mg, 3.47 mmol, 91%) as colourless oil. IR(neat) 3346 (broad), 2956, 2900, 2857, 2175, 1444, 1408, 1331, 1249 cm−1. δH (500 MHz, CDCl3), 4.51 (1H, q, J = 6.1 Hz, CHOH), 2.46 (1H, dt, J = 17.1, 7.5 Hz, CHH), 2.38 (1H, dt, J = 17.1 Hz, J = 6.9 Hz, CHH), 2.08 (1H, d, J = 5.6 Hz, OH), 1.91 (2H, q, J = 6.9 Hz, CH2), 0.18 (9H, s, CH3), 0.15 (9H, s, CH3). δC (125 MHz, CDCl3), 106.1 (C), 105.7 (C), 90.0 (C), 85.5 (C), 61.8 (CH), 36.2 (CH2), 15.9 (CH2), 0.1 (CH3), −0.2 (CH3). m/z (ESI) [M + Na]+ 274.8. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C16H18OSiNa 275.1258; found 275.1258.
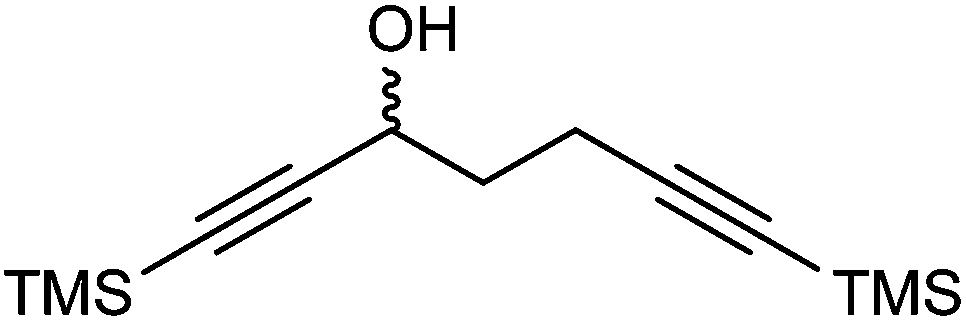
This compound is novel. In an round bottom flask 1,7-bis(trimethylsilyl)hepta-1,6-diyn-3-ol (877 mg, 3.47 mmol) was dissolved DCM (20 cm3). Then PCC (1.12 g, 6.40 mmol) and molecular sieves were added. After 14 hours the mixture was filtered through a Celite plug washing through with DCM (50 cm3). The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: hexane/EtOAc = 97/3) to give 1,7-bis(trimethylsilyl)hepta-1,6-diyn-3-one 44 (771 mg, 3.08 mmol, 86%) as a colourless oil. IR(neat) 2960, 2901, 2178, 2149, 1680, 1409, 1355, 1333, 1250, 1224 cm−1. δH (500 MHz, CDCl3), 2.77–2.83 (2H, m, CH2), 2.50–2.56 (2H, m, CH2), 0.23 (9H, s, CH3), 0.12 (9H, s, CH3). δC (125 MHz, CDCl3), 185.4 (C), 133.1 (CH), 130.9 (CH), 128.6 (CH), 119.8 (C), 104.7 (C), 91.6 (C), 87.3 (C), 85.6 (C), 44.2 (CH2), 14.7 (CH2), 0.0 (CH3). 13C NMR (126 MHz, chloroform-d) δ = 185.1 (C), 104.6 (C), 101.4 (C), 98.7 (C), 85.5 (C), 44.0 (CH2), 14.5 (CH2), 0.0 (CH3), −0.8 (CH3). m/z (ESI) [M + Na]+ 272.8. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C13H22OSi2Na 273.1101; found 273.1111.
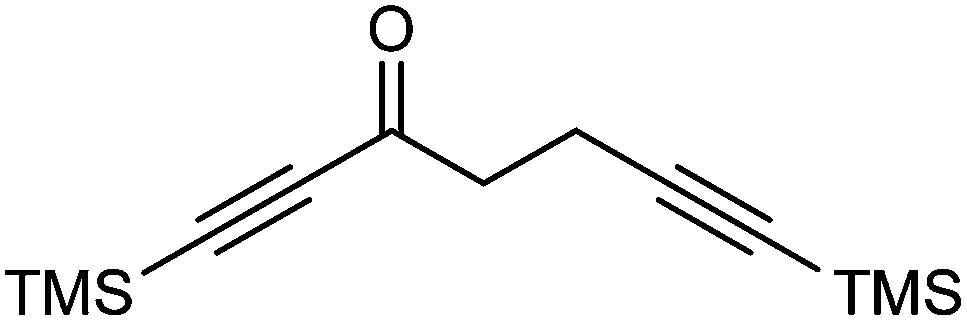
In an ACE pressure tube under a nitrogen 1,7-bis(trimethylsilyl)hepta-1,6-diyn-3-one 44 (745 mg, 2.97 mmol) was dissolved/suspended in anhydrous toluene (5 cm3) previously degassed by freeze–pump–thaw cycles. Fe(CO)5 (2.0 cm3, 14.83 mmol) was added, the pressure tube was sealed and the mixture was heated at 130 °C for 20 hours. After the mixture was cooled to room temperature and the pressure tube was carefully opened into the fumehood to release CO pressure. Then the mixture was diluted with EtOAc (15 cm3) and filtered through a Celite plug washing through with EtOAc (100 cm3). The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: petroleum ether to petroleum ether /EtOAc = 9
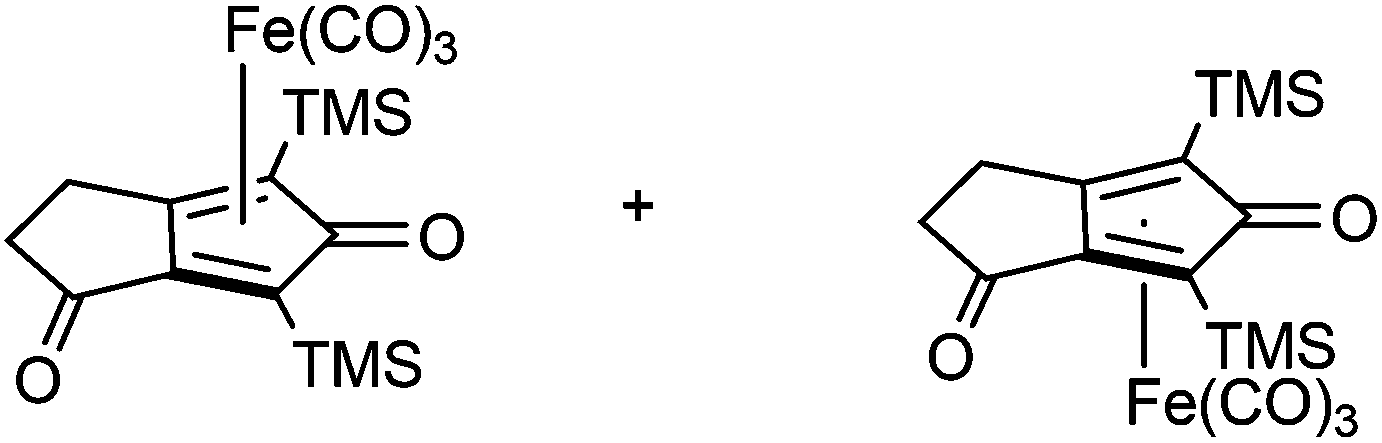 :1) to give tricarbonyl-4,6-bis(trimethylsilyl)-2,3-dihydropentalene-1,5-dione iron 42 (789 mg, 1.89 mmol, yield: 63%) as yellow solid. 1H NMR and 13CNMR were identical to the reported data.2c M.p. 105.5 °C dec. IR(neat) 2958, 2900, 2071, 2016, 1994, 1717, 1626, 1446, 1426, 1404, 1347, 1263, 1244, 1226, 1203 cm−1. δH (500 MHz, CDCl3), 3.15 (1H, dt, J = 17.2, 6.9 Hz, CHH), 3.02 (1H, dt, J = 17.4, 5.0 Hz, CHH), 2.78 (2H, dd, J = 6.9, 5.0 Hz, CH2), 0.34 (9H, s, CH3), 0.32 (9H, s, CH3). δC (125 MHz, CDCl3), 206.8 (C), 204.5 (C), 183.4 (C), 129.8 (C), 98.0 (C), 78.9 (C), 77.3 (C), 76.8 (C), 67.4 (C), 36.9 (CH2), 23.5 (CH2), −0.4 (CH3), −0.8 (CH3).
:1) to give tricarbonyl-4,6-bis(trimethylsilyl)-2,3-dihydropentalene-1,5-dione iron 42 (789 mg, 1.89 mmol, yield: 63%) as yellow solid. 1H NMR and 13CNMR were identical to the reported data.2c M.p. 105.5 °C dec. IR(neat) 2958, 2900, 2071, 2016, 1994, 1717, 1626, 1446, 1426, 1404, 1347, 1263, 1244, 1226, 1203 cm−1. δH (500 MHz, CDCl3), 3.15 (1H, dt, J = 17.2, 6.9 Hz, CHH), 3.02 (1H, dt, J = 17.4, 5.0 Hz, CHH), 2.78 (2H, dd, J = 6.9, 5.0 Hz, CH2), 0.34 (9H, s, CH3), 0.32 (9H, s, CH3). δC (125 MHz, CDCl3), 206.8 (C), 204.5 (C), 183.4 (C), 129.8 (C), 98.0 (C), 78.9 (C), 77.3 (C), 76.8 (C), 67.4 (C), 36.9 (CH2), 23.5 (CH2), −0.4 (CH3), −0.8 (CH3).
In an round bottom flask tricarbonyl-4,6-bis(trimethylsilyl)-2,3-dihydropentalene-1,5-dione iron 42 (100 mg, 0.24 mmol) was dissolved in EtOH (12 cm3) NaBH4 (11.5 mg, 0.30 mmol) was added and the reaction was stirred for 1 hours. Then H2O (1 cm3) was added and the mixture was stirred 30 minutes. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: pentane/EtOAc = 19
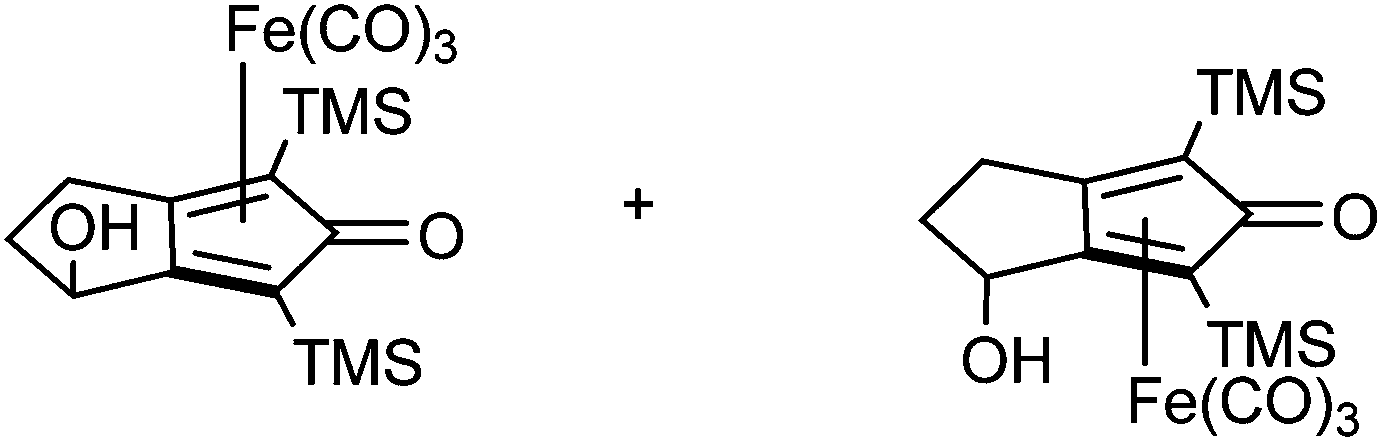 :1 to 9:1) to give tricarbonyl-4-hydroxy-1,3-bis(trimethylsilyl)-5,6-dihydropentalen-2(4H)-one iron 45 (75 mg, 0.18 mmol, 75%) as a yellow solid. 1H NMR and 13C NMR were identical to the reported data.2c M.p. 172.9–173.7 °C. [α]25D −88.8 (c 0.49, CHCl3). IR(neat) 3289 (broad), 2961, 2934, 2900, 2064, 2013, 1975, 1583, 1453, 1406, 1380, 1356, 1312, 1247 cm−1. δH (500 MHz, CDCl3), 5.21 (1H, td, J = 7.6, 5.0 Hz, CHOH), 2.60–2.69 (2H, m, CH2), 2.51–2.60 (1H, m, CHH), 2.24–2.34 (1H, m, CHOH), 1.72–1.83 (1H, m, CHH), 0.31 (9H, s, CH3), 0.26 (9H, s, CH3). δC (125 MHz, CDCl3), 208.4 (C), 183.1 (C), 118.9 (C), 117.7 (C), 71.3 (CH), 70.5 (C), 68.2 (C), 35.4 (CH2), 24.9 (CH2), −0.1 (CH3), −0.7 (CH3). m/z (ESI) [M + H]+, 421.1; [M + Na]+, 443.0. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C17H24FeO5Si2Na 443.0404; found 443.0406.
:1 to 9:1) to give tricarbonyl-4-hydroxy-1,3-bis(trimethylsilyl)-5,6-dihydropentalen-2(4H)-one iron 45 (75 mg, 0.18 mmol, 75%) as a yellow solid. 1H NMR and 13C NMR were identical to the reported data.2c M.p. 172.9–173.7 °C. [α]25D −88.8 (c 0.49, CHCl3). IR(neat) 3289 (broad), 2961, 2934, 2900, 2064, 2013, 1975, 1583, 1453, 1406, 1380, 1356, 1312, 1247 cm−1. δH (500 MHz, CDCl3), 5.21 (1H, td, J = 7.6, 5.0 Hz, CHOH), 2.60–2.69 (2H, m, CH2), 2.51–2.60 (1H, m, CHH), 2.24–2.34 (1H, m, CHOH), 1.72–1.83 (1H, m, CHH), 0.31 (9H, s, CH3), 0.26 (9H, s, CH3). δC (125 MHz, CDCl3), 208.4 (C), 183.1 (C), 118.9 (C), 117.7 (C), 71.3 (CH), 70.5 (C), 68.2 (C), 35.4 (CH2), 24.9 (CH2), −0.1 (CH3), −0.7 (CH3). m/z (ESI) [M + H]+, 421.1; [M + Na]+, 443.0. HRMS (ESI-Q-TOF) m/z: [M + Na]+ calcd for C17H24FeO5Si2Na 443.0404; found 443.0406.
In an round bottom flask under a nitrogen atmosphere, racemic tricarbonyl-4-hydroxy-1,3-bis(trimethylsilyl)-5,6-dihydropentalen-2(4H)-one iron 42 (500 mg, 1.19 mmol) was dissolved in anhydrous DCM (2 cm3). [(S,S)-Teth-TsDpen-RuCl] 23 (26 mg, 0.12 mmol) and an azeotropic mixture of formic acid/triethylamine (5
 :2 mixture, 0.8 cm3) were added. After 7 hours saturated NaHCO3 aqueous solution (10 cm3) and H2O (15 cm3) were added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 30 cm3). The reunited organic layers were washed with brine (100 cm3) and dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: pentane to pentane/EtOAc = 9:1) to give alcohol (−)-45 (204 mg, 0.49 mmol, yield: 40.6%, 97.6% ee) as yellow solid and ketone (+)-42 (175 mg, 0.42 mmol, yield: 35.0%, 88.2% ee) as yellow solid. The enantiomeric excesses ere determined by HPLC analysis with a ChiralPak IA column: 0.46 cm × 25 cm, mobile phase iso-propanol:hexane 3:97, flow rate 1 mL min−1, temperature 30 °C, UV detection at λ = 254 nm; tR = 6.1 min (ketone), tR = 7.0 min (ketone), tR = 11.4 min (alcohol), tR = 14.3 min (alcohol). Alcohol (−)-45; [α]24D −37.1 (c 0.48, CHCl3). Ketone (+)-42; [α]24D +98.2 (c 0.11, CHCl3).
:2 mixture, 0.8 cm3) were added. After 7 hours saturated NaHCO3 aqueous solution (10 cm3) and H2O (15 cm3) were added and the mixture was stirred 30 minutes. The product was extracted with DCM (3 × 30 cm3). The reunited organic layers were washed with brine (100 cm3) and dried over Na2SO4. The volatiles were removed and the product was purified by flash chromatography on silica gel (eluent: pentane to pentane/EtOAc = 9:1) to give alcohol (−)-45 (204 mg, 0.49 mmol, yield: 40.6%, 97.6% ee) as yellow solid and ketone (+)-42 (175 mg, 0.42 mmol, yield: 35.0%, 88.2% ee) as yellow solid. The enantiomeric excesses ere determined by HPLC analysis with a ChiralPak IA column: 0.46 cm × 25 cm, mobile phase iso-propanol:hexane 3:97, flow rate 1 mL min−1, temperature 30 °C, UV detection at λ = 254 nm; tR = 6.1 min (ketone), tR = 7.0 min (ketone), tR = 11.4 min (alcohol), tR = 14.3 min (alcohol). Alcohol (−)-45; [α]24D −37.1 (c 0.48, CHCl3). Ketone (+)-42; [α]24D +98.2 (c 0.11, CHCl3).
The alcohols formed by reductions have been reported and our procedures and characterisation followed the protocols reported.11,18
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for generous financial support of this work through a project grant to fund ADG (grant no. EP/M006670/1) and Johnson Matthey Catalysis and Chiral Technologies for a gift of TethTsDpenRuCl catalyst 23. The X-ray diffraction instrument was obtained through the Science City Project with support from the AWM and part funded by the ERDF. Warwick University are thanked for support of MChem student AEC.References
- Reviews: (a) K. Junge, K. Schroder and M. Beller, Chem. Commun., 2011, 47, 4849–4859 RSC; (b) M. Darwish and M. Wills, Catal. Sci. Technol., 2012, 2, 243–255 RSC; (c) K. Gopalaiah, Chem. Rev., 2013, 113, 3248–3296 CrossRef CAS PubMed; (d) A. Quintard and J. Rodriquez, Angew. Chem., Int. Ed., 2014, 53, 4044–4055 CrossRef CAS PubMed; (e) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- Early examples of cyclopentatdienone iron complexes: (a) G. N. Schrauzer, J. Am. Chem. Soc., 1959, 81, 5307–5310 CrossRef CAS; (b) A. J. Pearson, R. J. Shively Jr. and R. A. Dubbert, Organometallics, 1992, 11, 4096–4104 CrossRef CAS; (c) A. J. Pearson and R. J. Shively Jr., Organometallics, 1994, 13, 578–584 CrossRef CAS; (d) A. J. Pearson and J. B. Kim, Org. Lett., 2002, 4, 2837–2840 CrossRef CAS PubMed.
- (a) H.-J. Knölker, J. Heber and C. H. Mahler, Synlett, 1992, 1002–1004 CrossRef; (b) H.-J. Knölker, E. Baum, H. Goesman and R. Klauss, Angew. Chem., Int. Ed., 1999, 38, 2064–2066 CrossRef; (c) H.-J. Knölker, E. Baum, H. Goesmann and R. Klauss, Angew. Chem., Int. Ed., 1999, 38, 702–705 CrossRef.
- Hydrogenation: (a) C. P. Casey and H. Guan, J. Am. Chem. Soc., 2007, 129, 5816–5817 CrossRef CAS PubMed; (b) C. P. Casey and H. Guan, J. Am. Chem. Soc., 2009, 131, 2499–2507 CrossRef CAS PubMed; (c) C. P. Casey and H. Guan, Organometallics, 2011, 31, 2631–2638 CrossRef PubMed; (d) S. Fleischer, S. Zhou, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 5120–5124 CrossRef CAS PubMed; (e) X. Lu, R. Cheng, N. Turner, Q. Liu, M. Zhang and X. Sun, J. Org. Chem., 2014, 79, 9355–9364 CrossRef CAS PubMed; (f) H. Ge, X. Chen and X. Yang, Chem. – Eur. J., 2017, 23, 8850–8856 CrossRef CAS PubMed; (g) A. Rosas-Hernández, H. Junge, M. Beller, M. Roemelt and R. Francke, Catal. Sci. Technol., 2017, 7, 459–465 RSC; (h) S. V. Facchini, J.-M. NeuDorfl, L. Pignataro, M. Cettolin, C. Gennari, A. Berkessel and U. Piarulli, ChemCatChem, 2017, 9, 1461–1468 CrossRef.
- Transfer hydrogenation: (a) T. C. Johnson, G. J. Clarkson and M. Wills, Organometallics, 2011, 30, 1859–1868 CrossRef CAS; (b) S. A. Moyer and T. W. Funk, Tetrahedron Lett., 2010, 51, 5430–5433 CrossRef CAS; (c) M. G. Coleman, A. N. Brown, B. A. Bolton and H. Guan, Adv. Synth. Catal., 2010, 352, 967–970 CrossRef CAS; (d) M. K. Thorson, K. L. Klinkel, J. Wang and T. J. Williams, Eur. J. Inorg. Chem., 2009, 295–302 CrossRef CAS; (e) T. N. Plank, J. L. Drake, D. K. Kim and T. W. Funk, Adv. Synth. Catal., 2012, 354, 597–601 CrossRef CAS; (f) H. H. Zhang, D. Z. Chen, Y. H. Zhang, G. Q. Zhang and J. B. Liu, Dalton Trans., 2010, 39, 1972–1978 RSC; (g) K. Natte, W. Li, S. Zhou, H. Neumann and X.-F. Wu, Tetrahedron Lett., 2015, 56, 1118–1121 CrossRef CAS.
- Reductive amination: (a) A. Quintard, T. Constantieux and J. Rodriquez, Angew Chem., Int. Ed., 2013, 52, 12883–12887 CrossRef CAS PubMed; (b) S. Moulin, H. Dentel, A. Pagnoux-Ozherelyeva, S. Galliard, A. Poater, L. Cavallo, J.-F. Lohier and J.-L. Renaud, Chem. – Eur. J., 2013, 19, 17881–17890 CrossRef CAS PubMed; (c) A. Pagnoux-Ozherelyeva, N. Pannetier, M. D. Mbaye, S. Gaillard and J.-L. Renaud, Angew. Chem., Int. Ed., 2012, 51, 4976–4980 CrossRef CAS PubMed; (d) D. S. Merel, M. Elie, J.-L. Lohier, S. Gaillard and J.-L. Renaud, ChemCatChem, 2013, 5, 2939–2945 CrossRef CAS; (e) T.-T. Thai, D. S. Mérel, A. Poater, S. Gaillard and J.-L. Renaud, Chem. – Eur. J., 2015, 21, 7066–7070 CrossRef CAS PubMed; (f) L.-Q. Lu, Y. Li, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 2763–2768 CrossRef CAS PubMed.
- Hydrogen borrowing: (a) T. Yan, B. L. Feringa and K. Barta, Nat. Commun., 2014, 5, 5602 CrossRef CAS PubMed; (b) T. Yan, B. L. Feringa and K. Barta, ACS Catal., 2016, 6, 381–388 CrossRef CAS; (c) H.-J. Pan, T. Ng and Y. Zhao, Chem. Commun., 2015, 51, 11907–11910 RSC; (d) K. P. J. Gustafson, A. Guõmundsson, K. Lewis and J.-E. Bäckvall, Chem. – Eur. J., 2017, 23, 1048–1051 CrossRef CAS PubMed; (e) O. El-Sepelgy, N. Alandini and M. Reuping, Angew. Chem., Int. Ed., 2016, 55, 13602–13605 CrossRef CAS PubMed; (f) A. J. Rawlings, L. J. Diorazio and M. Wills, Org. Lett., 2015, 17, 1086–1089 CrossRef CAS PubMed.
- (a) X. Lu, Y. Zhang, P. Yun, M. Zhang and T. Li, Org. Biomol. Chem., 2013, 11, 5264–5277 RSC; (b) X. Lu, Y. Zhang, N. Turner, M. Ahang and T. Li, Org. Biomol. Chem., 2014, 12, 4361–4371 RSC; (c) X. Lu, R. Cheng, N. Turner, Q. Liu, M. Zhang and X. Sun, J. Org. Chem., 2014, 79, 9355–9364 CrossRef CAS PubMed.
- Asymmmetric reductions: (a) A. Berkessel, S. Reichau, A. Von der Höh, N. Leconte and J.-M. Nordörfl, Organometallics, 2011, 30, 3880–3887 CrossRef CAS; (b) A. Berkessel and A. Van der Höh, ChemCatChem, 2011, 3, 861–867 CrossRef; (c) J. P. Hopewell, J. E. D. Martins, T. C. Johnson, J. Godfrey and M. Wills, Org. Biomol. Chem., 2012, 10, 134–145 RSC; (d) P. Gajewski, M. Renom-Carrasco, S. V. Facchini, L. Pignataro, L. Lefort, J. G. de Vries, R. Ferraccioli, A. Forni, U. Piarulli and C. Gennari, Eur. J. Org. Chem., 2015, 1887–1893 CrossRef CAS.
- (a) A. Tlili, J. Schranck, H. Neumann and M. Beller, Chem. – Eur. J., 2012, 18, 15935–15939 CrossRef CAS PubMed; (b) S. Fleischer, S. Zhou, S. Werkmeister, K. Junge and M. Beller, Chem. – Eur. J., 2013, 4997–5003 CrossRef CAS PubMed; (c) S. Fleischer, S. Werkmeister, S. Zhou, C. Junge and M. Beller, Chem. – Eur. J., 2012, 18, 9005–9010 CrossRef CAS PubMed.
- R. C. Hodgkinson, A. Del Grosso, G. J. Clarkson and M. Wills, Dalton Trans., 2016, 45, 3992–4005 RSC.
- Z. Fang and M. Wills, J. Org. Chem., 2013, 78, 8594–8605 CrossRef CAS PubMed.
- (a) T.-Y. Luh, Coord. Chem. Rev., 1984, 60, 255–276 CrossRef CAS; (b) N. A. Bailey, V. S. Jassal, R. Vefghi and C. White, J. Chem. Soc., Dalton Trans., 1987, 2815–2822 RSC; (c) H.-J. Knölker, E. Baum and J. Heber, Tetrahedron Lett., 1992, 36, 7647–7650 CrossRef; (d) B. Dasgupta and W. A. Donaldson, Tetrahedron Lett., 1998, 39, 343–346 CrossRef CAS; (e) A. J. Pearson and Y. Kwak, Tetrahedron Lett., 2005, 46, 5417–5419 CrossRef CAS.
- Y. Yamamoto, K. Yamashita and M. Nakamura, Organometallics, 2010, 29, 1472–1478 CrossRef CAS.
- R. Liu, G. Zhou, T. H. Hall, G. J. Clarkson, M. Wills and W. Chen, Adv. Synth. Catal., 2015, 357, 3453–3457 CrossRef CAS.
- H. Miyamoto, T. Hirano, Y. Okawa, A. Nakazaki and S. Kobayashi, Tetrahedron, 2013, 69, 9481–9493 CrossRef CAS.
- J. Ying, K. B. Brown, M. J. Sandridge, B. A. Hering, M. Sabat and L. Pu, J. Org. Chem., 2015, 80, 3195–3202 CrossRef CAS PubMed.
- R. Hodgkinson, V. Jurčík, A. Zanotti-Gerosa, H. G. Nedden, A. Blackaby, G. J. Clarkson and M. Wills, Organometallics, 2014, 33, 5517–5524 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra, GC examples. X-ray data. Chiral HPLC of the ligands prior to complexation. CCDC 1567218–1567222. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt03250a |
| This journal is © The Royal Society of Chemistry 2018 |