
Synergistic effect of VOx and MnOx surface species for improved performance of V2O5/Ce0.5Ti0.5−xMnxO2−δ catalysts in low-temperature NH3-SCR of NO†

Abstract
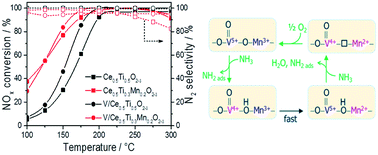
Supported V2O5/Ce0.5Ti0.5−xMnxO2−δ (x = 0, 0.01, 0.05, 0.1, and 0.2) catalysts and bare supports were investigated in the selective catalytic reduction (SCR) of NO by NH3 between 100 and 300 °C. Incorporation of Mn into Ce0.5Ti0.5O2 increased the NO removal efficiency, reaching both NO and NH3 conversions above 85% between 175 to 300 °C at remarkably high N2 selectivities of ≥95%. Moreover, Mn-modified supports were also much more stable against deactivation in the presence of water. Deposition of vanadia on the Ce0.5Ti0.5−xMnxO2−δ supports enhanced the N2 selectivity, reaching ≥98% in the whole temperature range for Mn percentages of ≤0.1. Characterization results revealed that incorporation of Mn leads to supports with higher lattice disorder and smaller crystallite size and enhances the oxygen mobility and the reducibility of the supports. Operando EPR studies show that the synergistic effect of VOx and MnOx surface species in moieties containing Mn3+/Mn2+ and VO3+/VO2+ in close vicinity contributes to the prevention of undesired NH3 oxidation, thus improving the N2 selectivity of these catalysts.
 Please wait while we load your content...
Please wait while we load your content...

