
A new approach to construct a hydrodesulfurization catalyst from a crystalline precursor: ligand-induced self-assembly, sulfidation and hydrodesulfurization†
 ‡ab
Daofeng
Sun,
a
‡ab
Daofeng
Sun,
a
Abstract
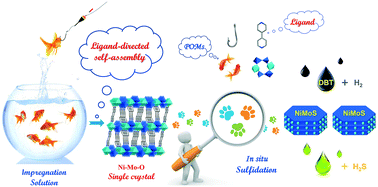
This paper proposes a new approach for investigating the mechanism of the formation of the active phase of a hydrodesulfurization (HDS) catalyst via crystalline polyoxometalate (POM) precursors. The proposed strategy induces the crystallization of small Ni–Mo–O clusters in an impregnating solution by the coordinate bonding and supramolecular interaction of organic ligands to form POMs. By exploiting the “ligand-induced self-assembly” strategy, two Ni–Mo binary POMs with different frameworks, namely, Mo2Ni and PMo11Ni, were isolated from the impregnating solution by means of 4,4′-bpy. The sulfidation process of the precursors and the formation mechanism of the NiMoS active phase were fully characterized by a multi-technique approach that comprised, in particular, in situ FT-IR spectroscopy, XRD and Raman spectroscopy for different degrees of sulfidation. The results of the characterization revealed the structure-directing effects (framework effect, promoting effect and ligand effect) of the POM precursors on the structure of the active phase and even its HDS performance. MoS2 was formed at 200 °C from Mo2Ni, and the Ni species interacted with the edges of MoS2 to form the NiMoS active phase, whereas PMo11Ni formed MoS2 at 300 °C. The structure-directing effects enabled a higher content and better dispersion of the NiMoS active phase, which explains the higher HDS reactivity of sulfided Mo2Ni. The bottom-up self-assembly approach not only provides a better understanding of the composition of the impregnating solution and the formation mechanism of the NiMoS active phase but also sheds light on the rational design and controllable preparation of NiMoS catalysts with high performance.
 Please wait while we load your content...
Please wait while we load your content...

