
Revisiting the iridacycle-catalyzed hydrosilylation of enolizable imines†

Abstract
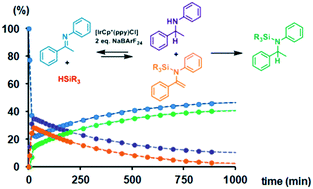
The hydrosilylation of enolizable imines using iridacycle catalysts has been investigated using a family of half-sandwich iridium(III) complexes. Several pitfalls had been detected when using standard procedures, and the use of in situ NMR spectroscopy appeared decisive to assess the actual reaction pathway. The results obtained clearly demonstrate that iridacycle-catalyzed hydrosilylation of enolizable imines with monohydrosilanes should be described as a domino reaction sequence, analogous to the one described by Oestreich et al. for boron-based Lewis acid catalysts. Equimolecular quantities of amine (b) and N-silylated enamine (c) were formed in the first stage, consuming 0.5 eq. of hydrosilane, which were subsequently converted into N-silylated amine (d) through a consecutive catalytic cycle using an additional 0.5 eq. of hydrosilane. However, when polyhydrosilanes were used as hydrosilylating agents, N-silylated amine (d) was directly obtained as the reaction product, but after a fast initial conversion, the reaction suffered from product inhibition. Additionally, iridacycles were also found to be active in the dehydrocoupling of Et3SiH and amines and in the hydrolysis of hydrosilanes, which can be competitive processes. These findings lead to an intricate mechanistic picture containing interrelated processes, which need to be taken into account for the development of more efficient, selective or enantioselective systems.
 Please wait while we load your content...
Please wait while we load your content...

