
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure–function relationship during CO2 methanation over Rh/Al2O3 and Rh/SiO2 catalysts under atmospheric pressure conditions†
Natalia M.
Martin
 *a,
Felix
Hemmingsson
a,
Xueting
Wang
a,
Lindsay R.
Merte
b,
Uta
Hejral
c,
Johan
Gustafson
c,
Magnus
Skoglundh
a,
Debora Motta
Meira
d,
Ann-Christin
Dippel
e,
Olof
Gutowski
e,
Matthias
Bauer
f and
Per-Anders
Carlsson
a
*a,
Felix
Hemmingsson
a,
Xueting
Wang
a,
Lindsay R.
Merte
b,
Uta
Hejral
c,
Johan
Gustafson
c,
Magnus
Skoglundh
a,
Debora Motta
Meira
d,
Ann-Christin
Dippel
e,
Olof
Gutowski
e,
Matthias
Bauer
f and
Per-Anders
Carlsson
a
aCompetence Centre for Catalysis, Department of Chemistry and Chemical Engineering, Chalmers University of Technology, Göteborg, 412 96, Sweden. E-mail: Natalia.Martin@chalmers.se; Fax: +46 31 160062; Tel: +46 31 772 29 04
bDivision of Chemical Physics, Department of Physics, Chalmers University of Technology, Göteborg, 412 96, Sweden
cDivision of Synchrotron Radiation Research, Lund University, 22100 Lund, Sweden
dEuropean Synchrotron Radiation Facility (ESRF), 38043 Grenoble, France
eDeutsches Elektronen-Synchrotron (DESY), 22607 Hamburg, Germany
fDepartment of Chemistry, Paderborn University, 33098 Paderborn, Germany
First published on 23rd April 2018
Abstract
The effect of the support material and chemical state of Rh in Rh/A2O3 and Rh/SiO2 model catalysts during CO2 hydrogenation were studied by a combined array of in situ characterisation techniques including diffuse reflectance infrared Fourier transform spectroscopy, energy-dispersive X-ray absorption spectroscopy and high-energy X-ray diffraction at 250–350 °C and atmospheric pressure. CO2 methanation proceeds via intermediate formation of adsorbed CO species on metallic Rh, likely followed by their hydrogenation to methane. The linearly-bonded CO species is suggested to be a more active precursor in the hydrogenation compared to the bridge-bonded species, which seems to be related to particle size effects: for larger particles mainly the formation of inactive bridge-bonded CO species takes place. Further, analysis of the chemical state of Rh under the reaction conditions reveal a minor formation of RhOx from dissociation of CO2, which is a consequence of the increased activity observed over the Rh/Al2O3 catalyst.
1 Introduction
Utilisation of CO2 as a reactant for formation of valuable compounds such as hydrocarbons, oxygenates and even carbon monoxide may play an important role in the transition towards a sustainable energy system. Production of methane through CO2 hydrogenation (Sabatier reaction) is a promising route for CO2 utilization:| CO2 + 4H2 ⇌ CH4 + 2H2O, ΔH298 = −165 kJ mol−1 | (1) |
In addition, this reaction is interesting due to its application in manned space colonisation on Mars or its use in reclaiming oxygen in the International Space Station, where the oxygen resource from CO2 can be transformed into methane and water for fuel and astronaut life-support systems.1,2
Catalytic materials that facilitate CO2 hydrogenation must be able to bind and activate CO2 for reaction with dissociated hydrogen. Ni-Based catalysts have been widely investigated for industrial purposes of methane production due to their low cost and ease of availability. Ni catalysts, however, may be deactivated even at low temperatures due to sintering of Ni particles, formation of mobile Ni sub-carbonyls, or carbon deposition.3,4 Therefore several other transition metals have been investigated for the methanation of CO2 (Ru, Rh, Pd, Co, Fe, Cu, Pt, Mg, Zn, Zr, Ir, Cu, Ag, W, Mo, and Mn) (see ref. 5, 12 and 13 and references therein). Among the noble metals, Ru and Rh on various supports have been shown to be very effective catalysts for the hydrogenation of CO2 and the most selective toward methane.6–8 Rh is one of the most investigated metals for the CO2 methanation reaction. It has been reported that the support has a marked influence on the specific activity of Rh. An effective support not only provides a high surface area for the metal dispersion but it can also modify the catalytic properties of the metal nanoparticles through the so-called strong metal–support interaction (SMSI) effect.9
As pointed out by Puigdollers et al.,10 when used in catalysts, the difference between the nonreducible and reducible oxides is of fundamental importance for their chemical reactivity. Mostly reducible oxides have been reported to have a SMSI effect. In their previous work on CO2 methanation on supported Rh catalysts, Solymosi et al.7 showed that among the investigated supports (Al2O3, SiO2, MgO and TiO2), the most effective one was TiO2 and the least effective one was SiO2. The effect of the support was attributed to different extents of electronic interaction between Rh and the support, influencing the bonding and the reactivity of the chemisorbed species. Although several papers have been published on the subject during the past decades, no general consensus exists on the operating reaction mechanism and on the active phase of the catalyst.12,13
Recently, we have studied different systems consisting of metals (Pd, Rh and Ni) supported on both reducible (CeO2) and nonreducible (Al2O3 and SiO2) oxides for the activation of CO2 and its reaction with H2 under atmospheric pressure conditions and at relatively low temperatures (250–350 °C).11 Interestingly, the results showed that Rh supported on both alumina and ceria exhibits high activity in the hydrogenation of CO2 for methane production and the catalysts supported on SiO2 have negligible activity. Therefore, a clear distinction between the reactivity of catalysts supported on reducible vs. nonreducible oxides could not be made based on the kinetic data. The selectivity toward CH4 formation was higher for the alumina supported catalysts compared to the ceria supported ones at temperatures up to 350 °C. The kinetic analysis also indicated that on Rh/CeO2 small amounts of CO are formed above 250 °C, while Rh/Al2O3 shows full selectivity towards methane up to 350 °C. The reason for the high activity over Rh/Al2O3 was not fully understood. Additional in situ infrared spectroscopy (IRS) measurements suggested that the reaction mechanism for the ceria and alumina supported Rh catalysts is different. The likely reaction pathway was found to proceed via formation of formates at the Rh–ceria interface or via CO2 dissociation and formation of Rh–CO on the Rh/Al2O3 sample. However, the correlation between the activity/selectivity and surface chemistry of rhodium and the structural properties of both rhodium and ceria or alumina was not accessible with this technique.
In the present report, we give a detailed account of the catalytic behaviour of Rh supported on Al2O3 and SiO2 under in situ conditions. Special attention is paid to the identification of surface species and the effects of alumina and silica supports during the CO2 methanation reaction. The scope of the work is to observe chemical/structural changes of both Rh and the support's phases including chemisorbed species and to relate them to their catalytic behavior. For simplicity, we focus on Rh supported on nonreducible oxides in the present work. In situ energy-dispersive X-ray absorption spectroscopy (ED-XAS) and high-energy X-ray diffraction (HE-XRD) have been employed to characterise the dynamic response of supported Rh catalysts when exposed to varying CO2 hydrogenation conditions. The results reveal reversible structural changes of the catalysts under transient operation conditions. The observable changes occur mainly in the Rh phase during the measurements and no significant changes are observed in the alumina or silica phases. The metal–support interaction cannot be excluded for the alumina supported catalyst since a test on the alumina sample showed no measurable methane production under the same reaction conditions. The interaction, however, may be considerably lower as in the case of reducible oxides.7 The complementary Fourier transform infrared (FTIR) spectroscopy results during the methanation reaction reveal the intermediate surface species formed on the Rh supported catalysts. Differences are observed for Rh/Al2O3 and Rh/SiO2 and the enhanced activity over Rh/alumina is related to the formation of adsorbed CO species linearly bonded to Rh as well as some RhOx species resulting from the dissociation of CO2.
The theoretical metal loading is expected to be very close to the actual loading as previously reported for incipient wetness impregnation.16
2 Experimental section
2.1 Catalyst preparation and ex situ characterization
The catalysts were prepared by incipient wetness impregnation using powder samples of alumina (Puralox SBa200, Sasol) and silica (Kromasil Silica KR-300-10, Akzo Nobel Eka Chemicals). The powder samples were calcined in air at 600 °C for 2 h starting from room temperature with a heating rate of 5° min−1 to remove carbonaceous impurities and stabilise the structure of the support materials. Precursor solutions of rhodium were prepared by dissolving Rh(NO3)3 × 2H2O (Alfa Aesar) salt in Milli-Q water (18 MΩ cm). To increase the solubility of the rhodium salt, 25 droplets of 70% HNO3 were added. A specific amount of precursor solution to obtain 3 wt% of the metal was added to 3 g of each support. The impregnated alumina and silica samples were instantly frozen in liquid nitrogen, freeze-dried for 24 h and finally calcined in air at 550 °C for 1 h.The specific surface area of the catalysts was determined by nitrogen sorption at 77 K (Micrometrics Tristar 3000) using the Brunauer–Emmett–Teller (BET) method.14 The samples were dried under vacuum at 230 °C for 3 h prior to the measurements. The results are summarized in Table 1.
| Sample | Rh loading (wt%) | Calcination temperature (°C) | Specific surface area (m2 g−1) | CO2 conversion11 (350 °C) |
|---|---|---|---|---|
| Rh/SiO2 | 3.0 | 550 | 310 | ∼10% |
| Rh/Al2O3 | 3.0 | 550 | 180 | ∼40% |
2.2 In situ measurements
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 220 eV. The experimental set-up included a specially designed reaction cell developed at ID24 to meet established practice on simultaneous ED-XAS and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) during transient feeding of reactants. The cell has a small reactor volume in which a sample cup with a diameter of 5 mm and a depth of 2.5 mm loaded with about 40 mg of the powder sample is positioned. The gas composition was controlled by Bronkhorst mass flow controllers and introduced to the cell via air actuated high-speed gas valves (Valco, VICI), all in all facilitating rapid gas composition changes over the catalyst sample.
220 eV. The experimental set-up included a specially designed reaction cell developed at ID24 to meet established practice on simultaneous ED-XAS and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) during transient feeding of reactants. The cell has a small reactor volume in which a sample cup with a diameter of 5 mm and a depth of 2.5 mm loaded with about 40 mg of the powder sample is positioned. The gas composition was controlled by Bronkhorst mass flow controllers and introduced to the cell via air actuated high-speed gas valves (Valco, VICI), all in all facilitating rapid gas composition changes over the catalyst sample.
The XAS measurements included both the X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) regions. Energy calibration was performed using a Rh metal foil. After energy calibration the XANES spectra were normalized using the Athena software.17 The XAS data were further processed and analysed using Athena and Larch software.18 Fourier transformation of the k2-weighted EXAFS data to the R space was done between k = 3 and k = 10 Å−1.
Oxidation and reduction measurements were performed in situ at 350, 300 and 250 °C, respectively, with alternating pulses of 2 vol% O2 and 2 vol% H2 (4 min long pulses) by measuring the Rh K-edge. The pulses were repeated 6 times to give a total duration of the experiment of 48 min. The introduction of the first O2 pulse triggered the recording of the XAS spectra. He was used as a carrier gas, and the total gas flow was kept constant at 75 mL min−1.
The methanation reaction was performed under transient operation conditions by introducing H2 pulses (2 vol%) to an otherwise constant flow of CO2 (0.5 vol%) at 350, 300 and 250 °C, respectively. The pulses were 10 min long and were repeated 5 times to give a total duration of the experiment of 100 min. Prior to the reaction the catalysts had been reduced in H2 for 10 min at the corresponding temperature at which the reaction was performed. For both the oxidation/reduction and CO2 methanation measurements, the spectra were recorded with a time resolution of 0.87 seconds.
Similar to the ED-XAS experiments, the gas feed composition was controlled by mass flow controllers and introduced to the cell via air actuated high-speed gas valves. The methanation reaction was performed under transient operation conditions by introducing H2 pulses (2 vol%) to an otherwise constant flow of CO2 (0.5 vol%) at 350, 300 and 250 °C, respectively. The pulses were 10 min long and were repeated 5 times to give a total duration of the experiment of 100 min. The time resolution of the measurements was 0.5 seconds. For these measurements, a specially designed reaction cell was employed which is described in detail elsewhere.20
3 Results and discussion
3.1 In situ characterization of the redox behavior
When the methanation reaction is performed, the catalyst is exposed to a reductant (H2) and an oxidant (CO2). Since the catalyst may change its oxidation state during this reaction, especially under transient operation conditions, it is important to elucidate how the catalyst behaves under oxidation or reduction conditions. Transient oxidation and reduction of the Rh-based catalysts was studied by in situ ED-XAS. Fig. 1 shows the evolution of the XAFS spectra for the Rh/Al2O3 sample at 350 °C during the oxidation–reduction cycling experiment in either 2 vol% O2 or 2 vol% H2. The experiment starts with a 4 min oxidation pulse. The left panel (a) shows the XAS spectra recorded at the end of the oxidation and reduction periods, while the right panel shows the recorded XAS spectra as a function of time (b) together with the white line intensity during the experiment (c). The strong peak above the edge, i.e. the white line, corresponds to the 1s–4d transition at the Rh K-edge and is directly related to the density of vacant d-orbital states. Therefore we have used the white line intensity to monitor the oxidation of Rh. However, other effects, such as that of particle size, can also contribute to the intensity of the white line.21,22 As observed in the figure, there is a sharp increase in the white line intensity during the O2 pulse which increases further during the first minute of the oxidation cycle, later reaching a maximum intensity, and there is a fast decrease in the white line intensity when the O2 supply is switched off and H2 is introduced to the feed. This indicates a fast oxidation/reduction kinetics of the Rh nanoparticles and since the white line intensity seems to be stable under both the oxidation and reduction conditions a change in the particle size is not likely to occur. | ||
| Fig. 1 XAS spectra of Rh K-edge recorded during consecutive 4 min oxidising (2 vol% O2) and reducing (2 vol% H2) periods over Rh/Al2O3 at 350 °C. (a) XAS spectra recorded at the end of the oxidising and reducing periods. (b) Color-coded intensities of XAS spectra (blue: low intensity; red: high intensity) and (c) the XAS white line intensity at 23240 eV. The time dependent spectra in (b) are presented as difference spectra implying that a couple of initial spectra have been subtracted from all spectra in order to facilitate the observation of the major changes occurring during the measurements. | ||
The XAS spectra in Fig. 1(a) are clearly different when they are recorded at the end of an oxidising period compared to the end of a reducing period, whereas the spectra recorded at different oxidising or reducing periods are very similar to one another. Similar results are obtained for the Rh/SiO2 sample. Fig. 2 displays the Rh K-edge XANES and EXAFS spectra of the Rh/Al2O3 and Rh/SiO2 catalysts during the end of the oxidising and reducing periods at 250, 300 and 350 °C, respectively. The XANES spectra (left panels) show a decreased white line intensity for the H2 treated samples, indicating that Rh is in a reduced state, while the white line intensity increases during the O2 treatment, indicating the oxidation of Rh. The left panels show the XANES spectra recorded from the reduced or oxidised samples at different temperatures. A similar behavior to that of Rh/Al2O3 is observed for the Rh/SiO2 sample, i.e. a decreased white line intensity under the reducing conditions and an increased intensity under oxidising conditions. For the Rh/SiO2 sample, oxidising treatment at 350 °C results in a higher increase of the white line intensity compared to the oxidising treatment at lower temperatures, suggesting a thicker oxide formation or a change in the particle size. By comparing the XAS spectra of the alumina and silica supported Rh catalysts it can be observed that the spectra for Rh/SiO2 in general show a higher intensity of the white line and of the EXAFS oscillations. This indicates an increased number of neighboring atoms, in line with our recent XRD results for the as-prepared catalyst which suggested larger particles of Rh oxide for the silica supported sample.11
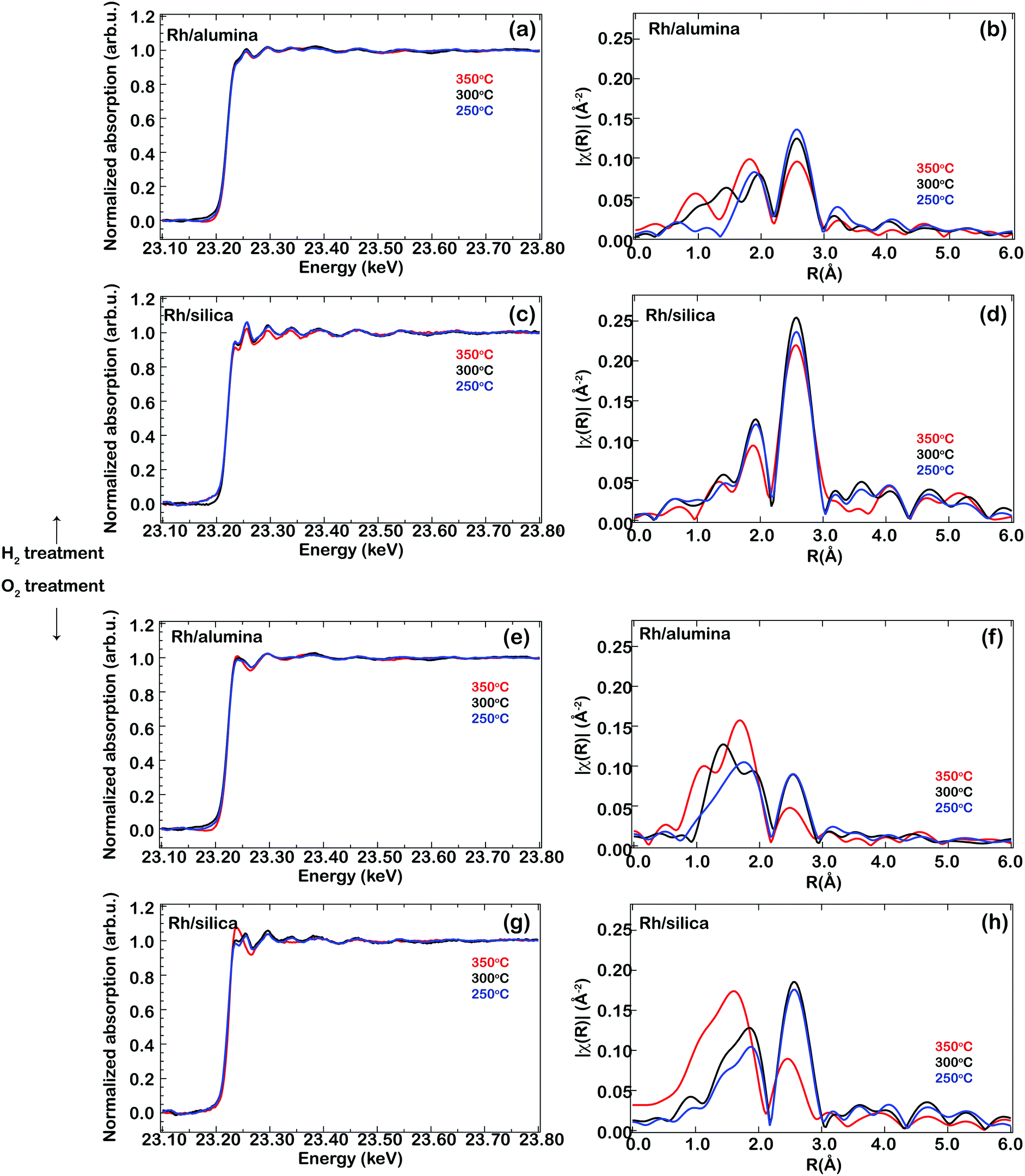 | ||
| Fig. 2 Rh K-edge in situ XAS spectra (XANES and EXAFS) for the reduced (a–d) and oxidised (e–h) Rh-based catalysts at 350 (red), 300 (black) and 250 °C (blue), respectively. | ||
To gain more information about the local structure surrounding the Rh atoms, a qualitative EXAFS analysis was performed. The associated Fourier transforms are shown on the right panels in Fig. 2, where R represents the radial distance from the absorbing atom. As no phase shift correction was applied, the peaks are observed at distances shorter than those of the actual positions.
The EXAFS spectra recorded from the samples exposed to reducing conditions (Fig. 2(b and d)) show the presence of two coordination shells: a peak at a radial distance of 2.57 Å, which is attributed to metal–metal scattering (Rh–Rh bond, first coordination shell) and a low-intensity peak at lower coordination distances, below 2 Å (1.94 Å), attributed to Rh–O scattering from the first shell of Rh2O3.23,24,26 The presence of a Rh–O coordination shell during exposure to reducing conditions indicates that the samples contain a fraction of irreducible Rh.
When the samples are exposed to oxidising conditions, the EXAFS spectra show a decrease in intensity of the Rh–Rh scattering peak (∼2.5 Å) and an increase of the peak at lower R, below 2 Å (due to Rh–O scattering). The weakness of the Rh–O peak intensity at temperatures below 350 °C and the absence of the Rh–O–Rh peak at 2.7 and 3.3 Å as expected for other shells of Rh2O3 (ref. 23, 24 and 26) suggest that small Rh oxide particles exist on the support materials, and/or an interaction between Rh and O atoms in the support. Contributions from both Rh–O and Rh–Rh scattering under reducing or oxidising conditions are observed at all temperatures for both samples indicating that, for both samples, Rh is not fully reduced or oxidised under the present experimental conditions. Since XAS is a bulk sensitive technique and the presence of the Rh–Rh coordination shell for both samples indicates that the samples can be envisaged as a metal core with an oxidic outer layer (its thickness depends on the temperature for the oxygen exposure, i.e. thicker oxide at 350 °C for both Rh/Al2O3 and Rh/SiO2).
According to the results presented in Fig. 2, the proportion of reduced Rh after exposure to reducing conditions is higher for Rh/SiO2 than for Rh/Al2O3. It is known that Rh particles supported on alumina can interact with the surface avoiding reduction of the particles as reported previously by Hwang et al.27 Therefore, the results suggest that there is a higher degree of interaction with the support for the alumina-supported sample as compared to the silica-supported one. However, there are no contributions observed from a Rh–O–Al shell which shall appear at a distance above 2.5 Å (R ∼ 2.5–3 Å), suggesting that the metal–support interaction is somehow weaker than, for example, in the case of ceria-supported Rh (ref. 23 and 24) or aged alumina-supported Rh (ref. 25 and 26) which have shown such contributions.
3.2 In situ studies of the CO2 methanation reaction under atmospheric pressure conditions
Structural changes of the Rh and support phases during CO2 hydrogenation over Rh/Al2O3 and Rh/SiO2 under atmospheric pressure conditions in the temperature range 250–350 °C were monitored in time-resolved in situ mode with ED-XAS and HE-XRD and the results are presented below. The activity of the catalysts measured by mass spectrometry showed results comparable to the ones reported on our previous publication11 and summarized in Table 1 and, for simplicity, we focus here on the structural changes of the Rh and support phases occurring during the CO2 methanation reaction.:4 ratio of CO2:H2) at 300 °C are presented in Fig. S1 in the ESI.† The data are presented as difference spectra implying that a couple of initial spectra have been subtracted from all spectra in order to facilitate the observation of the major changes occurring during the measurements. No clear changes are observed in the XAS spectra of the Rh K-edge (nor in the white line intensity) when the catalysts are exposed to CO2 or a CO2 + H2 mixture at 300 °C. However, some differences are observed in the EXAFS analysis. Spectra recorded at the end of the CO2 hydrogenation and CO2 pulses at 350 and 300 °C, including both XANES and EXAFS, are presented in Fig. 3 below.
 | ||
| Fig. 3 Rh K-edge in situ XAS spectra (XANES – upper panels and EXAFS – lower panels) for the Rh-based catalysts supported on either alumina (a and b) or silica (c and d) recorded during the transient hydrogenation of CO2. The selected spectra were recorded after the samples had been exposed to pulses of either 2 vol% H2 + 0.5 vol% CO2 or 0.5 vol% CO2 for 10 min at different temperatures. | ||
For the Rh/Al2O3 sample, the decreased white line intensity of the Rh K-edge XANES spectra in Fig. 3(a) indicates that the state of Rh is predominantly metallic. The Fourier transforms of the Rh K-edge EXAFS spectra of the Rh/Al2O3 catalyst during methanation reaction at different temperatures are presented in Fig. 3(b). The spectra feature a peak at about 2.5 Å indicative of a Rh–Rh bond. However, an additional peak below 2 Å is observed at all temperatures, indicative of Rh–O bonding, similar to the results obtained under reducing conditions and presented above. The EXAFS analysis of the spectra recorded at 300 °C shows no clear differences between Rh/Al2O3 exposed to CO2 and CO2 + H2. However, when the measurements were performed at 350 °C, the EXAFS analysis shows a decrease of the Rh–O bond peak intensity during the methanation of CO2 suggesting a decrease in the oxidation state of Rh as compared to when exposed to CO2. This coincides with the high CO2 conversion and the increased methane selectivity observed for this catalyst under the present experimental conditions. We cannot exclude that some Rh atoms are still bonded to O during the methanation reaction since some Rh–O bond scattering is still visible in the spectra. This is most likely due to some chemisorbed O or from the interaction with the alumina support and its origin will be discussed further below (section 3.2.4). This difference has been observed to be reversible when switching back and forward between the CO2 and CO2 + H2 pulses.
For the Rh/SiO2 sample the XANES data presented in Fig. 3(c) show an overall higher white line intensity of the Rh K-edge, which may indicate an increased oxidation state of the Rh atoms, or larger particles, similar to the results from the oxidising/reducing treatments presented above. The EXAFS analysis shown in Fig. 3(d) shows a strong peak at around 2.5 Å indicative of a Rh–Rh bond at both temperatures and a lower intensity peak just below 2 Å indicative of a Rh–O bond. Thus the larger particle size for the silica supported catalyst is a more plausible explanation for the increased white line intensity. The intensity of the Rh–O peak decreases at higher temperatures, similar to the results obtained during the treatment in a reducing atmosphere. By comparing the EXAFS spectra recorded during the exposure to CO2 + H2 with the spectra recorded during CO2 exposure, even though a slight increase in the Rh–O bond peak intensity is observed during the CO2 methanation at 350 °C, the relative intensity of the Rh–Rh and Rh–O peaks is still higher indicating that Rh is present in a more metallic state in Rh/SiO2 as compared to that in the Rh/Al2O3 sample.
 | ||
| Fig. 4 Transient hydrogenation of 0.5 vol% CO2 over Rh/Al2O3 during periodic variation of the feed gas composition between 2 vol% H2 + 0.5 vol% CO2 and 0.5 vol% CO2 at 350 °C for 10 min recorded using HE-XRD. The top panel shows the color coded intensities (blue corresponds to low intensity, red to high intensity) of the XRD patterns in the region 0–8 Å−1versus time. The bottom panel shows selected XRD patterns at the end of selected CO2 (red) or CO2 + H2 (black) pulses. The inset indicates a zoom in between 2.7 and 3.7 Å−1. | ||
| Structural formula | Bragg plane | d (Bragg spacing) (Å) | ICSD ref no. |
|---|---|---|---|
| Rh | (111) | 2.07 | #171677 |
| Rh2O3 | (002) | 2.7 | #41534 |
| RhO2 | (111) | 2.8 | #251565 |
Comparing the analysis of the diffraction pattern recorded during the CO2 pulse to the pattern recorded during the CO2 + H2 pulse it is observed that the RhOx reflection broadens during the CO2 pulse and is sharper during the methanation reaction. This difference is only observed at 350 °C (see Fig. S2, ESI†) and can be explained by a change in the apparent crystallite size: larger crystallites give sharper reflections. This is in line with the XAS results presented above which indicated a reduced amount of oxidised Rh during the CO2 hydrogenation at 350 °C which is likely due to the increased particle size. However, the discrepancy in the amount of Rh oxide formed during the methanation at 350 °C over the Rh/Al2O3 sample between the XAS and XRD results may be related to disordered vs. well-ordered phases. The RhOx species resulting from CO2 dissociation in the presence of H2 seem more ordered as compared to the oxide species determined by XAS during the CO2 pulse.
Based on the XRD results it can be concluded that crystalline RhOx forms during the methanation of CO2 and is likely linked to the increased activity of the Rh/Al2O3 catalyst. No metallic Rh reflections are observed at any time in the XRD patterns during the CO2 or CO2 + H2 pulses, in contrast to the EXAFS analysis where some Rh–Rh components are visible at all times. XRD classically provides information on structures with long-range order for phase identification and the estimation of apparent crystallite/particle size, and therefore, non-crystalline Rh or very small Rh particles will not be identified by XRD. The formation of some Rh-carbide has been questioned under the present experimental conditions, possibly resulting from the dissociation of adsorbed CO species. However, the in situ XAS and XRD measurements do not show any evidence of carbide formation and, therefore, it is likely that its formation does not occur.
The DRIFTS results from a steady-state measurement obtained after the Rh/Al2O3 and Rh/SiO2 catalysts have been exposed to a flow of 0.2 vol% CO2 and 0.8 vol% H2 at 350 °C for 20 min are presented in Fig. 5. The interaction of Rh-based catalysts with a 0.2 vol% CO2 and 0.8 vol% H2 mixture at 350 °C results in the development of bands around 2020 and 1800 cm−1, indicating dissociation of CO2 and formation of Rh-bonded carbonyl species. This fact suggests that CO2 hydrogenation proceeds via the dissociation of CO2 forming adsorbed CO and O as an intermediate reaction step. Some differences are observed between the two investigated catalysts.
 | ||
| Fig. 5 IR absorption bands in the wavenumber region 1100–3800 cm−1 for the Rh/Al2O3 (red) and Rh/SiO2 (black) catalysts exposed to 0.2 vol% CO2 and 0.8 vol% H2 at 350 °C for 20 min. | ||
For the Rh/Al2O3 sample, a strong absorption band centred around 2020 cm−1 corresponding to linearly adsorbed CO species on Rh (Rh–CO) appears during CO2 hydrogenation, suggesting CO2 dissociation, in agreement with previous reports on CO2 methanation over Rh/Al2O3.28 A broad band around 1780 cm−1 is also detected, which is close to the reported values of bridge-bonded CO on Rh (Rh02-CO, ∼1800 cm−1).33,34 Some additional weaker peaks in the region 1700–1500 cm−1 are also visible corresponding to carbonate or formate-like species on the alumina support.
The positions of adsorbed carbonyl bands are shifted to lower vibrational frequencies during the CO2 hydrogenation as compared to reported values for adsorbed carbonyl species. This trend has previously been reported in the literature and can be attributed to the formation of Rh carbonyl hydrides (Rh–CO–H).6,7,28–30 A reduction of the surface coverage of these species can produce a decrease in CO(ads) dipole–dipole coupling and may also contribute to the observed shift.
For the Rh/SiO2 sample the band associated with bridge-bonded CO on Rh increases in intensity during the CO2 hydrogenation, while the band assigned to linearly adsorbed CO on Rh has a much lower intensity. The preferred adsorption of CO in bridge coordination is likely due to the increased particle size for this sample compared to the alumina supported sample, as previously observed for Rh/TiO2.35 The adsorption and dissociation of CO2 is enhanced by the presence of H2 since experiments with exposure to CO2 only result in less intense absorption bands (not shown). The decreased content of linearly adsorbed CO species on Rh may be responsible for the low activity observed over this sample, which may indicate that the bridge-bonded CO on Rh only is a spectator species in the CO2 hydrogenation. Generally a decrease of CO adsorbed species is observed for the silica supported catalyst likely due to the larger particles and/or an increased difficulty of the silica supported catalyst to dissociate CO2.
No vibrations from adsorbed CHx species (2800–3000 cm−1) could be seen for Rh/Al2O3 or Rh/SiO2. Further we do not see the presence of CO species adsorbed on Rh+ sites, which previously has been reported in the case of CO2 hydrogenation over Rh/Al2O3.36 Therefore, our combined XAS, XRD and DRIFTS results indicate that the first step in the reaction mechanism for CO2 hydrogenation over supported Rh is dissociative adsorption of CO2 forming adsorbed CO and O. The CO adsorbs on the metallic Rh while some other Rh atoms can interact with the O formed by the dissociation of CO2 forming RhOx as supported by in situ XAS and XRD results.
There are several mechanistic and kinetic studies in the literature on CO2 hydrogenation. However, little effort has been made to characterize the surface structure of the catalysts under the reaction conditions and their relation to the catalytic activity for CO2 methanation. In fact, to our knowledge, there are only a few studies available in the open literature on the characterization of the surface structure and its relation to the catalytic activity for Rh-based catalysts on the CO2 methanation reaction, mostly based on DRIFT spectroscopy. There are some speculations regarding the chemical state of Rh during the methanation reaction, based on ex situ studies. Some ex situ XPS studies on Rh/Al2O3 have suggested formation of Rh3+ species during the methanation reaction. However, the in situ DRIFTS results suggested that the active state of Rh during the reaction is very likely to be Rh0.28 Further, Karelovic et al.28 reported bridge-bonded CO to be more reactive than linearly-bonded CO at low temperatures (150 and 200 °C).
The present in situ XAS and XRD results obtained for the highly active Rh/Al2O3 provide evidence that Rh is slightly oxidised during the methanation reaction and not fully reduced. Comparing the XAS spectra obtained during the CO2 hydrogenation (Fig. 3) to the spectra obtained during the reduction period (Fig. 2), one can observe that at 350 °C the Rh/Al2O3 catalyst is more reduced during CO2 hydrogenation as compared to when it is exposed to an H2 treatment. A possible explanation for the increased oxidation during the H2 treatment may be the smaller particle size and therefore there is a stronger interaction with the support which in turn hinders the reduction of the small particles.
Nevertheless, the active state of Rh during the reaction is very likely Rh0. As observed by in situ DRIFTS experiments, only Rh0–(CO)x (x = 0.5, 1) species are detected and the linearly adsorbed CO on Rh is found to be the intermediate species in the reaction. Rh–CO species are proposed to be associated with H forming Rh carbonyl hydride species (Rh–CO–H). CO adsorbed on oxidised Rh, which would appear in the 2090–2135 cm−1 region,37 was not detected in our measurements. It is shown that the linearly-bonded CO species are responsible for the increased activity for CO2 hydrogenation compared to the bridge-bonded species under the investigated reaction conditions. These are related to the particle size effects: for larger particles mainly the formation of inactive bridge-bonded CO species takes place. However, some Rh–O bonds are detected by XAS and are likely due to oxidised Rh from the O atoms from the dissociation of CO2 or from internal layers of the particles (i.e., close to the alumina surface). These results are in agreement with previous reports suggesting the reduced Rh species to play an important role in the reaction mechanism.36,38
By comparing the results obtained on a highly active catalyst (Rh/Al2O3) to the ones obtained on a less active one (Rh/SiO2), it can be concluded that the effect of the support on the methanation reaction for nonreducible oxides provides a high dispersion of the active phase. Some electronic interaction between the Rh and the support, influencing the bonding and the reactivity of the chemisorbed species, cannot be excluded and it is shown that the alumina support interacts with Rh facilitating the adsorption and dissociation of CO2. The results are in agreement with a previous study by Rönsch et al. who reported that CO2 conversion can be enhanced by a supporting material that fosters high CO2 coverage (e.g., Al2O3).13
Regarding the reaction mechanism for CO2 methanation, two reaction mechanisms have previously been reported in the literature. The first mechanism involves the adsorption of CO2 on the support and its reaction with H(ads) species formed on the metal which leads to the formate intermediate (COOH) at the metal–support interface. The formates can give rise to CO(ads) species which are subsequently hydrogenated to CH4.39 The second mechanism involves the direct dissociation of CO2 to CO(ads) and O(ads) on the metal surface, with CO(ads) being subsequently hydrogenated to CH4.33,36,38,40 In our case the direct dissociation of CO2 seems to be evidenced. We have confirmed experimentally that the first step in the mechanism by which the reaction occurs is dissociative adsorption of CO2 on the surface of the catalyst. On Rh/Al2O3 CO2 adsorption takes place preferably on the metal–support interface, while CO2 dissociation takes place on the active Rh surface and the O can interact with some other Rh atoms forming RhOx. However, the mechanism by which the hydrogen reacts with the dissociated species is not determined in this work and further experiments are needed to clarify the next steps of the reaction.
4 Conclusions
In situ ED-XAS and HE-XRD studies on Rh/Al2O3 during the process of CO2 methanation under atmospheric pressure conditions and at relatively low temperatures (below 350 °C) reveal reversible structural changes of the catalysts under transient operation conditions. Such changes were not observed for the Rh/SiO2 catalyst, which also exhibits lower CO2 conversion. Some of the Rh atoms are found to be in a low oxidation state (RhOx) for the highly active Rh/Al2O3 during the methanation reaction. The in situ DRIFTS results indicate that mainly the linearly- and bridge-bonded CO species are formed on the Rh surface during CO2 hydrogenation over both alumina and silica supported catalysts suggesting that a metallic Rh phase is required for the dissociation of CO2. However, the oxygen resulting from the CO2 dissociation reacts with some other Rh atoms and form RhOx as revealed by XAS and XRD measurements. The linearly adsorbed CO species on metallic Rh are responsible for the higher activity of Rh/Al2O3. Thus, it is concluded that CO2 methanation over Rh/Al2O3 proceeds by the dissociative adsorption of CO2 giving rise to linearly adsorbed CO species on reduced Rh (Rh–COlin) while the adsorbed O can interact with other Rh atoms forming RhOx.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank ESRF for providing the beamtime. Parts of this research were carried out at PETRA III at DESY, a member of the Helmholtz Association (HGF). P. Velin and E. C. Adams are acknowledged for their contribution to sample preparation. This work was financially supported by the Swedish Research Council through the Röntgen–Ångström collaboration “Time-resolved in situ methods for design of catalytic sites within sustainable chemistry” (No. 349-2013-567), the Swedish Foundation for Strategic Research through the project “Novel two-dimensional systems obtained on SiC as a template, for electronics, sensing and catalysis” (RMA15-0024), and the Competence Centre for Catalysis, which is financially supported by Chalmers University of Technology, the Swedish Energy Agency and the member companies: AB Volvo, ECAPS AB, Haldor Topsøe A/S, Volvo Car Corporation, Scania CV AB, and Wärtsilä Finland Oy.References
- A. Shima, M. Sakurai, Y. Sone, M. Ohnishi and T. Abe, Development of a CO2 reduction catalyst for the Sabatier reaction, in 42nd International Conference on Environmental Systems 15-19, San Diego, California, AIAA, July 2012, pp. 2012–3552 Search PubMed.
- G. A. Du, S. Lim, Y. H. Yang, C. Wang, L. Pfefferle and G. L. Haller, J. Catal., 2007, 249, 370–379 CrossRef CAS.
- M. Agneli, M. Kolb and C. Mirodatos, J. Catal., 1994, 148, 9–21 CrossRef.
- M. Agneli, H. Swaan, C. Marquez-Alvarez, G. Martin and C. Mirodatos, J. Catal., 1998, 175, 117–128 CrossRef.
- I. Fechete and J. C. Vedrine, Molecules, 2015, 20, 5638–5666 CrossRef CAS PubMed.
- F. Solymosi and A. Erdöhelyi, J. Mol. Catal., 1980, 8, 471–474 CrossRef CAS.
- F. Solymosi, A. Erdöhelyi and T. Bansagi, J. Catal., 1981, 68, 371–382 CrossRef CAS.
- T. Iizuka, Y. Tanaka and K. Tanabe, J. Mol. Catal., 1982, 17, 381–389 CrossRef CAS.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- A. R. Puigdollers, P. Schlexer, S. Tosoni and G. Pacchioni, ACS Catal., 2017, 7, 6493–6513 CrossRef.
- M. N. Martin, P. Velin, M. Skoglundh, M. Bauer and P.-A. Carlsson, Catal. Sci. Technol., 2017, 7, 1086–1094 Search PubMed.
- P. Frontera, A. Macario, M. Ferraro and P. L. Antonucci, Catalysis, 2017, 7, 59–87 Search PubMed.
- S. Rönsch, J. Schneider, S. Matthische, M. Schluter, M. Götz, J. Lefebvre, P. Prabhakaran and S. Bajohr, Fuel, 2016, 166, 276–296 CrossRef.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- S. Pascarelli, et al. , J. Synchrotron Radiat., 1999, 6, 146–148 CrossRef PubMed.
- E. C. Adams, M. Skoglundh, M. Folic, E. C. Bendixen, P. Gabrielsson and P.-A. Carlsson, Appl. Catal., B, 2015, 165, 10–19 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- M. Newville, J. Phys.: Conf. Ser., 2013, 430, 012007–012014 CrossRef.
- N. Schell, A. King, F. Beckmann, H.-U. Ruhnau, R. Kirchhof, R. Kiehn, M. Müller, A. Schreyer, R. Garrett and I. Gentle, et al., The high energy materials science beamline (hems) at petra iii, AIP Conf. Proc., 2010, 1234(1), 391 CrossRef CAS.
- C. Zhang, J. Gustafson and L. R. Merte, et al. , Rev. Sci. Instrum., 2015, 86, 033112 CrossRef PubMed.
- D. Bazin, D. Sayers, J. J. Rehr and C. Mottet, J. Phys. Chem. B, 1997, 101, 5332–5336 CrossRef CAS.
- D. C. Bazin, D. A. Sayers and J. J. Rehr, J. Phys. Chem. B, 1997, 101, 11040–11050 CrossRef CAS.
- A. Gayen, K. R. Priolkar, P. R. Sarode, V. Jayaram, M. S. Hegde, G. N. Subbanna and S. Emura, Chem. Mater., 2004, 16, 2317–2328 CrossRef CAS.
- S. Hosokawa, M. Taniguchi, Z. Utani, H. Kanai and S. Imamura, Appl. Catal., A, 2005, 289, 115–120 CrossRef CAS.
- V. Marchionni, M. A. Newton, A. Kambolis, S. K. Matan, A. Weidenkaff and D. Ferri, Catal. Today, 2014, 229, 80–87 CrossRef CAS.
- H. Hirata, K. Kishita, Y. Nagai, K. Dohmae, H. Shinjoh and S. Matsumoto, Catal. Today, 2011, 164, 467–473 CrossRef CAS.
- C. P. Hwang, C. T. Yeh and Q. M. Zhu, Catal. Today, 1999, 51, 93–101 CrossRef CAS.
- A. Karelovic and P. Ruiz, Appl. Catal., B, 2012, 113–114, 237–249 CrossRef CAS.
- F. Solymosi and H. Knosinger, J. Catal., 1990, 122, 166–177 CrossRef CAS.
- M. A. Henderson and S. D. Worley, J. Phys. Chem., 1985, 89, 1417–1423 CrossRef CAS.
- W. H. Bragg and W. L. Bragg, Proc. R. Soc. London, Ser. A, 1913, 88(605), 428–438 CrossRef CAS.
- https://icsd.fiz-karlsruhe.de (accessed January 2018).
- I. A. Fisher and A. T. Bell, J. Catal., 1996, 162, 54–65 CrossRef CAS.
- H. Y. Luo, H. W. Zhou, L. W. Lin, D. B. Liang, C. Li, D. Fu and Q. Xin, J. Catal., 1994, 145, 232–234 CrossRef CAS.
- A. Karelovic and P. Ruiz, J. Catal., 2013, 301, 141–153 CrossRef CAS.
- A. Beuls, C. Swalus, M. Jacquemin, G. Heyen, A. Karelovic and P. Ruiz, Appl. Catal., B, 2012, 113–114, 2–10 CrossRef CAS.
- S. S. C. Chuang, R. W. Stevens and R. Khatri, Top. Catal., 2005, 32, 225–232 CrossRef CAS.
- M. Jacquemin, A. Beuls and P. Ruiz, Catal. Today, 2010, 157, 462–466 CrossRef CAS.
- D. I. Kondarides, P. Panagiotopoulou and X. E. Verykios, J. Phys. Chem. C, 2011, 115, 1220–1230 Search PubMed.
- L. F. Liotta, G. A. Martin and G. Deganello, J. Catal., 1996, 164, 322–333 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2. See DOI: 10.1039/c8cy00516h |
| This journal is © The Royal Society of Chemistry 2018 |