
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-mediated reduction of azides under seemingly oxidising conditions: catalytic and computational studies†
Benjamin
Zelenay
ab,
Maria
Besora
 b,
Zaira
Monasterio
a,
David
Ventura-Espinosa
a,
Andrew J. P.
White
a,
Feliu
Maseras
*bc and
Silvia
Díez-González
*a
b,
Zaira
Monasterio
a,
David
Ventura-Espinosa
a,
Andrew J. P.
White
a,
Feliu
Maseras
*bc and
Silvia
Díez-González
*a
aDepartment of Chemistry, Imperial College London, Exhibition Road, South Kensington, SW7 2AZ London, UK. E-mail: s.diez-gonzalez@imperial.ac.uk
bInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Avgda. Països Catalans 16, 43007 Tarragona, Spain. E-mail: fmaseras@iciq.es
cDepartament de Química, Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain
First published on 16th October 2018
Abstract
The reduction of aryl azides in the absence of an obvious reducing agent is reported. Careful catalyst design led to the production of anilines in the presence of water and air. The reaction medium (toluene/water) is crucial for the success of the reaction, as DFT calculations support the formation of benzyl alcohol as the oxidation product. A singular catalytic cycle is presented for this transformation based on four key steps: nitrene formation through nitrogen extrusion, formal oxidative addition of water, C(sp3)–H activation of toluene and reductive elimination.
Introduction
Amines, primary ones in particular, are essential functional groups in chemistry, biology and many industries.1,2 Their importance is inevitably linked to their versatile reactivity and, in consequence, the use of protecting groups is most often required in synthetic strategies involving amines. While carbamates and amides are popular choices, azides offer a number of advantages as amino protecting groups; they are easily accessible, soluble, prevent the introduction of additional steric bulk and, crucially, azides do not react under many reaction conditions.3The Staudinger reaction is probably the most widespread methodology for the reduction of azides to amines. Originally reported in 1919,4 this transformation has notably found wide application in bioconjugation and functionalisation of polysaccharides.5 Nevertheless, this reaction requires the use of stoichiometric amounts of a phosphine or phosphite and the separation of the resulting oxidised by-products is not always straightforward.6 The development of catalytic alternatives, particularly if based on readily available and inexpensive metals, would greatly enhance the applicability of and interest in the use of azides as amino protecting groups.
In this context, several catalytic systems based on copper have been disclosed in the literature. Copper nanoparticles were found active in the hydrogenation of azides with ammonium formate as the H2 source.7 Nevertheless, 0.5 equiv. [Cu] was required and recycling tests were unsuccessful due to the air sensitive nature of the catalyst. Reductions of azides into amines in the presence of a mixture of a copper(II) salt and a (super)stoichiometric reductant are also known but barely general in scope and such a reaction outcome is most often described as ‘unexpected’ or ‘surprising’.8 It is accepted that in these examples the azide group is reduced through the oxidation of copper(I) to copper(II), which is then reduced by the reducing agent. Otherwise, a stoichiometric amount of copper(I) is needed in the absence of an external hydride source.9 In our opinion, an alternative reported pathway involving the formation of copper–nitrene,10 held better promise for the development of a robust and more sustainable catalytic system for the reduction of azides to amines. After an initial test in DMF, the reduction of nitrobenzoxadiazole azide was reported with 20 mol% of simple copper salts in degassed aqueous DMSO in the absence of any additional reducing agent (Scheme 1). The isolation of the corresponding DMSO adduct under such conditions highlighted the non-innocence of the solvent.11 Even if a single reaction was reported, the authors remarkably established the intermediacy of triplet copper–nitrene species by EPR and the importance of water and light in the reaction.
 | ||
| Scheme 1 Reported reduction of an aryl azide. | ||
Copper–nitrene derivatives are elusive species;12 nevertheless, they are accepted intermediates in catalytic alkane amination and alkene aziridination reactions.13 Their role in this aziridination process has been computationally studied,14 as well as fundamental aspects of the metal–nitrene bond.15 Some of us also have experience in the computational study of the role of copper nitrenes in olefin aziridination,16 transfer to alkynes,17 formation of oxazoles,18 and reaction with hydrocarbons.19 Taking into account the highly reactive nature of metal nitrenes, we hypothesised that the use of ligands could be instrumental in their application in reduction reactions. Herein we report our efforts in developing a chemoselective copper-based catalytic system for the reduction of aryl azides to the corresponding anilines in the absence of an additional reductant as well as our mechanistic studies to establish the identity of the hydride source.
Results and discussion
Catalytic studies
We started our optimisation studies with the screening of the [Cu(DAB)] complexes (DAB = diazabutadiene) we recently reported.20 4-Nitrophenyl azide and [Cu(DABCy)2]BF4 were chosen to test the effect of solvents in a model reaction (Table 1). All reactions were carried out in technical solvents and in the presence of air. No aniline formation was observed after heating for 20 h in THF, EtOAc, MeCN or MeOH, while moderate conversions were observed in DMSO, toluene, dioxane and water (Table 1, entries 1–4). Mixtures of these organic solvents with water were then used with varying results. While little effect was observed with dioxane, a significant drop in conversion was obtained with DMSO (Table 1, entries 5 and 7). On the other hand, a promising 30% of aniline 1a was formed when a toluene/water mixture was used as the reaction medium (Table 1, entry 6).|
|
||
|---|---|---|
| Entry | Solvent | Conva [%] |
| a 1H NMR conversions are the average of two independent reactions and are calculated with respect to 1,3,5-trimethoxybenzene as the internal standard. b 11% of the DMSO adduct also formed; see Scheme 1. c No DMSO adduct was observed. | ||
| 1 | DMSO | 28b |
| 2 | Toluene | 14 |
| 3 | Dioxane | 10 |
| 4 | Water | 19 |
| 5 | DMSO/water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
19c |
| 6 | Toluene/water (1:1) |
30 |
| 7 | Dioxane/water (1:1) |
16 |

Hence, two series of [Cu(DAB)] complexes were screened in 1:1 mixtures of toluene/water (Table 2). Cationic homoleptic complexes systematically outperformed their neutral heteroleptic counterparts (Table 2, entries 1–5 vs. 10–13) with the catalyst bearing DABAnis ligands displaying the highest catalytic activity, up to 54% conversion into 1a (Table 2, entry 4).
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Cu] | Conva [%] | Entry | [Cu] | Conva [%] |
a
1H NMR conversions are the average of two independent reactions and are calculated with respect to 1,3,5-trimethoxybenzene as the internal standard.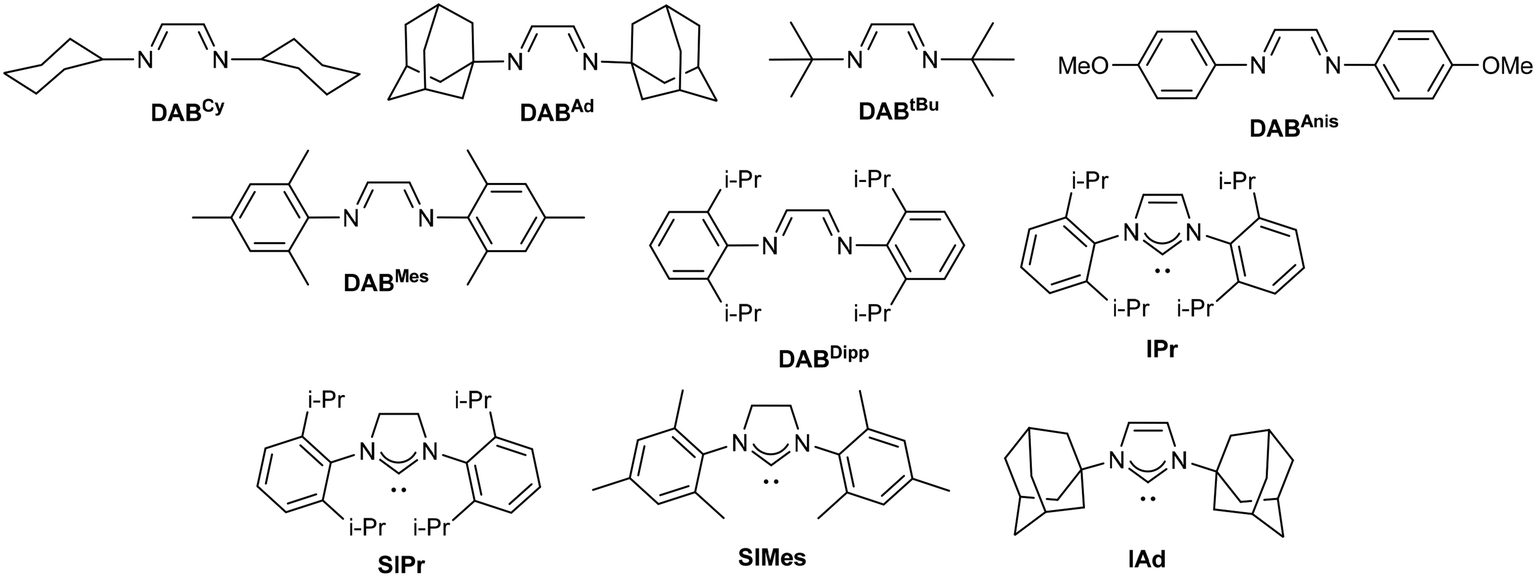
|
|||||
| 1 | [Cu(DABCy)2]BF4 | 41 | 10 | [CuCl(DABCy)] | 22 |
| 2 | [Cu(DABAd)2]BF4 | 14 | 11 | [CuCl(DABAd)] | 11 |
| 3 | [Cu(DABtBu)2]BF4 | 5 | 12 | [CuCl(DABtBu)] | 6 |
| 4 | [Cu(DABAnis)2]BF4 | 54 | 13 | [CuCl(DABAnis)] | 32 |
| 5 | [Cu(DABMes)2]BF4 | 7 | 14 | [Cu(DABDipp)(NCMe)2]BF4 | 10 |
| 6 | [Cu(IPr)2]PF6 | 16 | 15 | [CuI(IPr)] | 26 |
| 7 | [Cu(SIPr)2]PF6 | 9 | 16 | [CuI(SIPr)] | 25 |
| 8 | [Cu(SIMes)2]PF6 | 3 | 17 | [CuCl(SIPr)] | 25 |
| 9 | [Cu(IAd)2]PF6 | 19 | 18 | [CuI(IAd)] | 23 |

In an attempt to improve these results, several [Cu(NHC)] complexes (NHC = N-heterocyclic carbenes) were also screened. In this case, neutral [CuX(NHC)]21 catalysts led to better conversions to 1a than the related [Cu(NHC)2]X22 complexes. This is probably due to the absence of strong nucleophiles in the reaction mixture capable of activating the latter.22 In any case, only moderate conversions were obtained with any of these complexes.
It is important to note that under these reaction conditions no reduction occurred with simple copper salts such as [Cu(NCMe)4]BF4, CuI, or CuCl2, which confirms the importance of the ancillary ligand. It was also observed that the toluene/water ratio had an important impact on the reaction outcome with some of the catalysts tested. Notably, 84% conversion (instead of 54%) was obtained with [Cu(DABAnis)2]BF4, or 55% (instead of 25%) with [CuI(SIPr)] when the toluene/water ratio was changed from 1:1 to 1:2. Such an effect was not general, though, and it was not observed with other NHC- or DAB-bearing complexes. No further improvement, only significant decomposition, was observed with higher water ratios.
Since the best conversions so far had been obtained with a DAB ligand bearing an unencumbered electron-rich substituent on the nitrogen atoms, we next synthesised several related copper(I) complexes. Three diimine ligands bearing dimethylamino (DMA) or trimethoxy (3MeO) aryl substituents were reacted with [Cu(NCMe)4]BF4 at room temperature to obtain the corresponding homoleptic complexes 2–4. These were isolated in good to excellent yields after a simple recrystallization (Scheme 2). Alternatively, similar complexes bearing iminopyridine (ImPy) ligands were also prepared following reported procedures (5 and 6).23 The steric bulk of the iminopyridines is significantly smaller than that of the corresponding diazabutadienes, and therefore, two related complexes 7 and 8 with larger adamantyl or diisopropylphenyl groups were also synthesised for comparison.
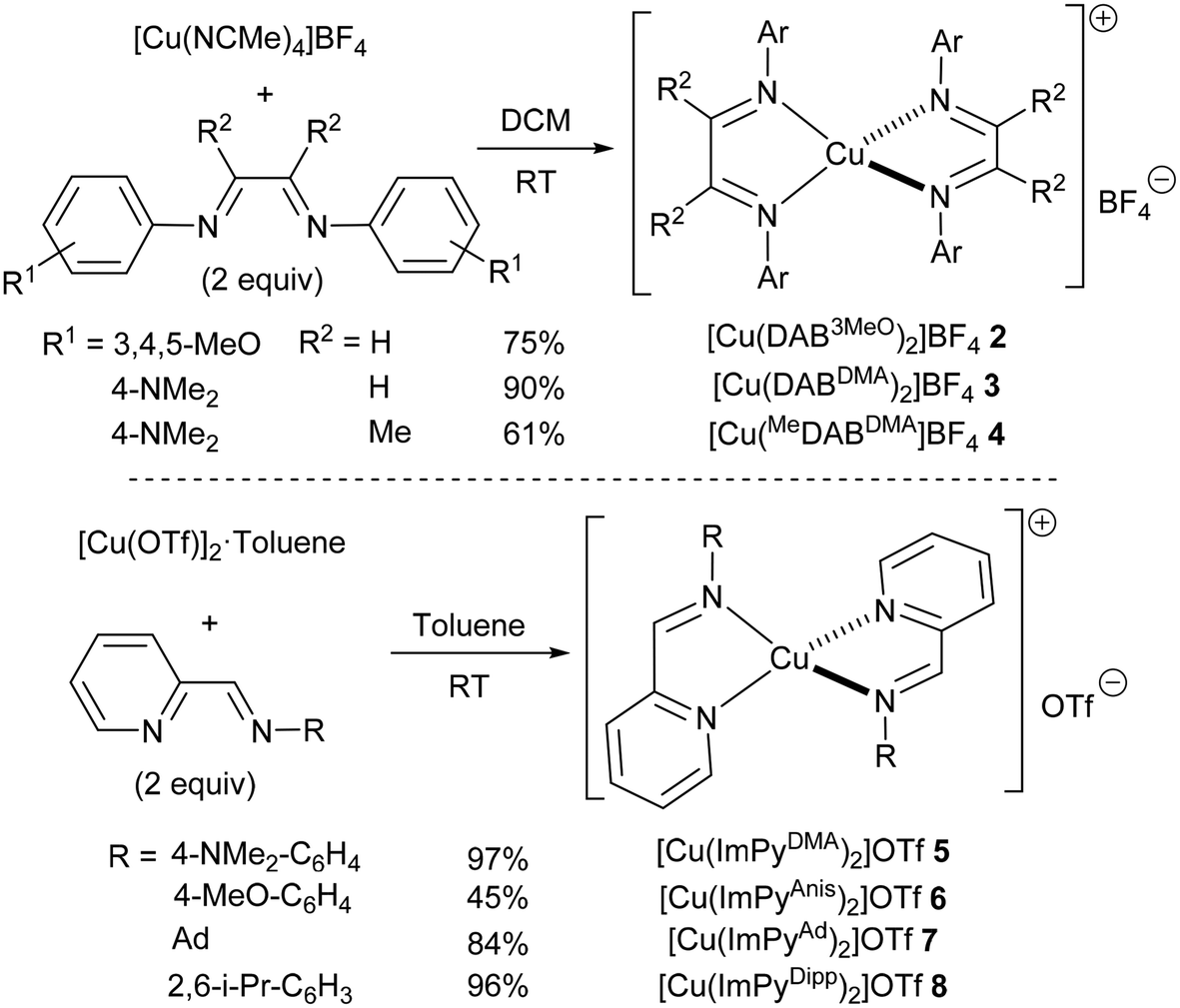 | ||
| Scheme 2 Synthesis of copper(I) complexes. | ||
Complex 2 could be handled in air but it decomposed after a few days even when kept under a nitrogen atmosphere. All other complexes were indefinitely stable towards oxygen and moisture. All complexes were fully characterised, as detailed in the ESI.† Spectroscopic data for DAB24 complexes 2–4 and ImPy25 complexes 5–8 were in good agreement with those of related known complexes.
Suitable crystals for single crystal X-ray diffraction could be obtained for complexes 3, 7 and 8 by slow diffusion of petroleum ether in DCM solutions (CCDC 1583789, 1583790 & 1583791, respectively). A summary of the crystallographic data for these compounds is provided in the ESI.† Ball and stick representations of the obtained structures are given in Fig. 1–3, and selected bond lengths are provided in the captions. Despite numerous attempts, the crystal structure of 3 is of poor quality, so only the gross features of the structure are reliable.26 Despite the evident issues, the structure derived from this data clearly confirms the reported nature of 3 as a [Cu(DABR)2]+ derivative. The structure contains two independent copper cations (3-A and 3-B), two independent BF4 anions, and two independent included dichloromethane solvent molecules (Fig. 1). The bond lengths and angles for 3-A and 3-B can be found in the corresponding CIF file.
 | ||
| Fig. 1 Structure of the two independent cations, 3-A (right) and 3-B (left), present in the crystal of [Cu(DABDMA)2]BF43. Most hydrogen atoms are omitted for clarity. | ||
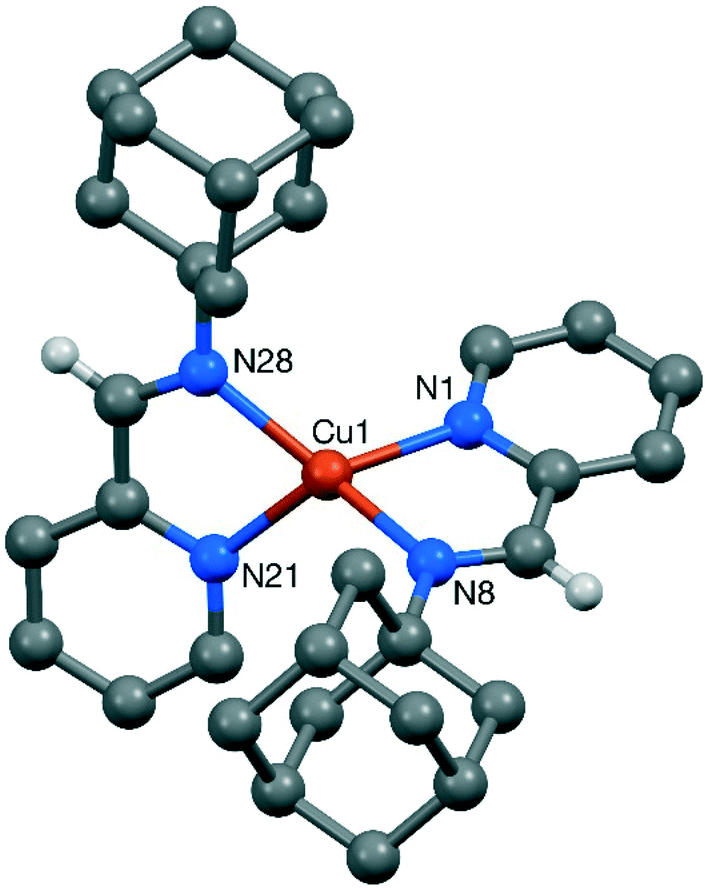 | ||
| Fig. 2 Structure of the cation present in the crystal of [Cu(ImPyAd)2]BF47. Most hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Cu(1)–N(1) 2.077(2), Cu(1)–N(8) 2.026(2), Cu(1)–N(21) 2.081(2), Cu(1)–N(28) 2.020(2); N(1)–Cu(1)–N(8) 81.25(7), N(1)–Cu(1)–N(21) 119.79(8), N(1)–Cu(1)–N(28) 119.99(7), N(8)–Cu(1)–N(21) 116.67(7), N(8)–Cu(1)–N(28) 142.27(8), N(21)–Cu(1)–N(28) 81.42(7). | ||
 | ||
| Fig. 3 Structure of the cation present in the crystal of [Cu(ImPyDipp)2]BF48. Most hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Cu(1)–N(1) 2.051(2), Cu(1)–N(8) 2.026(2), Cu(1)–N(21) 2.058(2), Cu(1)–N(28) 2.021(2); N(1)–Cu(1)–N(8) 80.98(9), N(1)–Cu(1)–N(21) 110.44(9), N(1)–Cu(1)–N(28) 127.33(9), N(8)–Cu(1)–N(21) 134.02(9), N(8)–Cu(1)–N(28) 128.58(8), N(21)–Cu(1)–N(28) 80.75(9). | ||
Complexes 7 and 8 crystallised in the monoclinic (P21/n space group) and triclinic systems (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ), respectively. As expected, they both displayed tetracoordinated copper centres in a distorted tetragonal arrangement, with the two CuN2 coordination planes inclined by 80.07(6)° in 7 and by 77.94(8)° in 8. The sp2 character of the C–N bond in the chelate rings was confirmed by the respective bond lengths of 1.275(3) and 1.284(3) Å for 7, and 1.274(3) and 1.272(3) Å for 8.27 Furthermore, no significant interaction between the counterion and the imine hydrogen or any other atom was evident in either structure.
), respectively. As expected, they both displayed tetracoordinated copper centres in a distorted tetragonal arrangement, with the two CuN2 coordination planes inclined by 80.07(6)° in 7 and by 77.94(8)° in 8. The sp2 character of the C–N bond in the chelate rings was confirmed by the respective bond lengths of 1.275(3) and 1.284(3) Å for 7, and 1.274(3) and 1.272(3) Å for 8.27 Furthermore, no significant interaction between the counterion and the imine hydrogen or any other atom was evident in either structure.
From the obtained crystallographic data, it could be confirmed that the steric pressure imposed by ImPy ligands was significantly lower than in the DAB family. The percentage buried volume (%VB)28 for ImPyAd and ImPyDipp was found to be 39.0 and 41.2% compared to 43.6% for DABAd and 44.5% for DABDipp.20
Next, the newly synthesised complexes were tested in the model reaction (Table 3). Overall, the tailored [Cu(DABR)] complexes provided improved conversions into aniline with the exception of 2, which might be attributed to the instability of this copper complex. Gratifyingly, full conversions were obtained with DAB complexes bearing a dimethylaminophenyl substituent (Table 3, entries 3 and 4). Such improvement in catalytic activity might be rationalised by the increased stability of diimine ligands and the corresponding metal complexes when bearing electron-rich substituents on the nitrogen centre.23a,29 Nevertheless, this beneficial effect was not apparent when the iminopyridine scaffold was used instead (Table 3, entries 5 and 6). Indeed, only disappointing NMR yields in 1a were obtained with these complexes, but interestingly, the best performing catalyst in this series was the one bearing a bulkier group on the imine arm, [Cu(ImPyDipp)2]OTf.
|
|
||
|---|---|---|
| Entry | [Cu] | Conva [%] |
| a 1H NMR conversions are the average of two independent reactions and are calculated with respect to 1,3,5-trimethoxybenzene as the internal standard. | ||
| 1 | [Cu(DABAnis)2]BF4 | 84 |
| 2 | [Cu(DAB3MeO)2]BF42 | 25 |
| 3 | [Cu(DABDMA)2]BF43 | >95 |
| 4 | [Cu(MeDABDMA)2]BF44 | >95 |
| 5 | [Cu(ImPyDMA)2]OTf 5 | 38 |
| 6 | [Cu(ImPyAnis)2]OTf 6 | 34 |
| 7 | [Cu(ImPyAd)2]OTf 7 | 22 |
| 8 | [Cu(ImPyDipp)2]OTf 8 | 57 |

In order to discriminate the catalytic activity of complexes 3 and 4, lower metal loadings were then tested. Nevertheless, identical conversions were obtained in all cases for both catalysts.30 Taking into account these results, catalyst 3 was chosen to investigate the reaction scope since both the ligand preparation and copper complexation steps were higher yielding than those for the preparation of 4.
The reaction scope was then investigated using 10 mol% of 3 with different aryl azides (Scheme 3). Complete conversions were actually observed with loadings as low as 1 mol% with 4-nitrophenyl azide.30 However, this is a highly activated substrate and a higher copper loading was used in all cases in an attempt to avoid undesirable thermal decomposition of the starting azides. It is important to note that in the absence of the copper catalyst no formation of the corresponding anilines was observed, only the expected degradation of the starting azides in different degrees depending on their thermal stability.31
 | ||
| Scheme 3 [Cu(DABDMA)2]BF43 mediated reduction of azides. Isolated yields are the average of two independent experiments. 1H NMR conversions are shown in brackets when complete conversions were not achieved. These are the average of two independent reactions and are calculated with respect to 1,3,5-trimethoxybenzene as the internal standard. | ||
All reactions were completely chemoselective and no other functional groups were affected by the catalytic system. Notably, tosyl azide and heteroaromatic azides were suitable substrates for this system. In addition, despite the conditions used, we never observed the formation of aromatic azo compound (or diaryl hydrazine) issue of oxidative dehydrogenative coupling of the newly formed anilines.32 Unfortunately, no anilines could be accessed from electron-rich substrates such as 4-methoxyphenyl azide, which was recovered unreacted, as well as alkyl azide such as adamantyl azide.
Computational studies
Due to the unusual nature of this transformation and the lack of an obvious hydride source, we carried out a computational study of the mechanism of the reaction of [Cu(DABR)] complexes with aromatic azides. Calculations were conducted with phenyl azide and [Cu(DABMe)2]+ as the model system with the BP86-D3 functional and a double-zeta plus polarisation basis set.26 A collection data set of all computational data is accessible in the ioChem-BD repository.33 All energies presented correspond to free energies in solution at 373.15 K unless stated otherwise.Prior to the actual mechanistic study, we needed to identify the oxidised product and reducing agent. No oxidation of the complex or ligand was observed experimentally, and therefore, only toluene and/or H2O could act as reductants. Both toluene and water may undergo different oxidation processes, as depicted in Fig. 4. Even if all of them were found thermodynamically feasible, some could be ruled out for different reasons. The formation of H2O2 under catalytic conditions would result in the oxidation of anilines to their corresponding hydroxylanilines,34 which was never observed in our reactions. Water splitting leading to the formation of O2 would have been particularly interesting, but preliminary calculations of possible dinuclear intermediates35 led to unviable energies.26
 | ||
| Fig. 4 Overview of the energies of different oxidation products. Free energies at 373.15 K in kcal mol−1. | ||
Overall, oxidation products from toluene appear more realistic thermodynamically. However, neither benzylic alcohol, benzaldehyde, nor bibenzyl could be detected at the end of our catalytic reactions. While bibenzyl was found stable under catalytic conditions, complete decomposition of benzyl alcohol or benzaldehyde was observed after heating them overnight in the presence of [Cu(DABDMA)2]BF4 and aniline. No decomposition products could be identified from these reactions but an insoluble sludge was recovered, indicative of significant oligomerisation/polymerisation. In an attempt to reduce the decomposition of the oxidation product, the model reaction was carried out at 70 °C. While aniline formation dropped to 42% at this temperature, the GC-MS analysis of the organic phase now showed that benzyl alcohol was present even if it was only a minor product. No alcohol was detected when the organic phase was concentrated under reduced pressure before GC-MS analysis, which confirms how sensitive benzyl alcohol is even in the presence of traces of our copper catalyst or its derivatives. Accordingly, the model reaction for the DFT studies contained benzyl alcohol as the oxidation product (Scheme 4).
 | ||
| Scheme 4 Model reaction for computational studies. | ||
The complete reaction cycle was computed and an overview with selected intermediates is shown in Scheme 5 and their corresponding energies are in Fig. 5. The full reaction cycle is depicted in the ESI† (Scheme S1).
 | ||
| Scheme 5 Overview of the proposed catalytic cycle with selected intermediates. | ||
 | ||
| Fig. 5 Free energy diagram for the proposed catalytic cycle with selected intermediates at 373.15 K and in kcal mol−1. | ||
Starting from [Cu(DABMe)2]+I.1, one of the DABMe ligands is displaced by water and aniline to form a tetracoordinated copper intermediate I.3. Considering the low configurational stability in solution of these complexes,20 such displacement is expected to be straightforward when diluted. DABDMA partially hydrolysed under catalytic conditions (hot toluene/water mixture) forming traces of 4-(dimethylamino)aniline, which would be enough for the first catalyst turnover to take place. Methyl amine would be formed from model [Cu(DADMe)2]+, however, aniline was expected to mimic better the behaviour of 4-dimethylaminoaniline that is formed experimentally. Phenyl azide would then coordinate the copper(I) centre by displacing the water molecule to give intermediate II, which is very close in energy to I.3 (Scheme 5). Dinitrogen is then extruded in TS(II) with an activation energy of 30.8 kcal mol−1 (Fig. 5). This is a relatively high energy barrier for a reaction that takes place at 373.15 K, but the irreversibility of this step contributes to the progress of the reaction. It is noteworthy that the uncatalysed extrusion of N2 from phenyl azide has been calculated to proceed through a transition state above 45 kcal mol−1.36 From TS(II), a singlet nitrene intermediate III is formed to then undergo a spin-cross over to give the more stable triplet species of III (Scheme 5). The minimum energy conversion point (MECP) for the spin-crossover was located at 2.7 kcal mol−1, close to the singlet species with III at 3.2 kcal mol−1. The MECP has a free energy 0.5 kcal mol−1 below the singlet; this is due to thermal and entropic contributions as its potential energy is actually 1.2 kcal mol−1 above.
Next, coordination of a water molecule forms IV, where water acts as a proton shuttle to form a diamido intermediate V, 2.5 kcal mol−1 below the triplet III. This water molecule then undergoes a formal oxidative addition through TS(V) to form a copper(III) hydroxyl amido aniline complex VI.1. This penta-coordinated copper(III) intermediate can interact with a toluene molecule through the aromatic ring. However, for the amido ligand to be able to receive a hydrogen from the bound toluene, it needs to be in an equatorial position. The conversion from VI.1 to VI.5 shows the required rearrangement to bring the amido group to an equatorial position and the aniline group to an axial one (Scheme 6). This process is assisted by an additional water molecule that is present in the second coordination sphere of the copper centre.
 | ||
| Scheme 6 Rearrangement sequence to VI.5. Free energies at 373.15 K and in kcal mol−1. | ||
Following this rearrangement, toluene binds through π-interactions to form a copper(III) complex VII. Then, an sp3 hydrogen from toluene is transferred to the amido ligand with a very reasonable activation barrier of 11.3 kcal mol−1. This transfer seems to be a radical process rather than a deprotonation as a Mulliken spin-density of 0.57 was observed on the nitrogen of the former nitrene before the transfer, decreasing to 0.34 on TS(VII) and to 0.11 on VIII in the triplet spin state. Toluene has no spin-density on the sp3 carbon in complex VII and the spin-density increases to 0.32 on TS(VII) and to 0.53 on VIII in the triplet spin state. These values indicate the movement of the unpaired electron from the nitrogen to carbon or in other words, the transfer of a hydrogen radical.
Following this hydrogen abstraction, the triplet copper(III)–benzyl intermediate VIII readily undergoes a spin-crossover (barrier of 1.1 kcal mol−1) to form a much more stable singlet species (−17.3 kcal mol−1 compared to −5.5 kcal mol−1, Scheme 5). Aniline, the model reaction product, is then displaced by a water molecule without a significant increase in free energy, after which benzyl alcohol is reductively eliminated and the active catalytic species I.3 is regenerated. As previously mentioned, benzyl alcohol is unstable under our catalytic conditions, which explains its absence in the reaction mixtures.
The reaction mechanism in Scheme 5 and Fig. 5 discussed above is one of a variety of different possibilities that were computationally explored. Other potentially active species for the nitrogen extrusion step are reported in the ESI,† as well as an alternative reaction mechanism without aniline coordination.
Overall, the rate limiting step of this mechanism is the nitrogen extrusion to form the reactive nitrene. In the model reaction, this is associated with a rather high barrier of 30.8 kcal mol−1. We have verified experimentally that phenyl azide does not react with our system and that the presence of an EWG on the substrate is key for the reduction to take place. Calculations using 4-nitrophenyl azide for this key step show that the presence of the nitro group significantly lowers the barrier for nitrogen extrusion to 24.1 kcal mol−1. This barrier is fully consistent with a reaction taking place at 373.15 K and explains the need for an EWG on the substrate. This proposed mononuclear mechanism is further supported by preliminary kinetic studies to determine the reaction order with respect to the catalyst. Indeed, 4-cyanophenyl azide was reacted under optimal conditions with either 5 or 10 mol% of [Cu(DABDMA)2]BF4, confirming a reaction order of 1 for the catalyst.26 On the other hand, the intermediacy of species such as VII and IX.2 can be directly linked with the original complex screening (see Table 2 and Scheme 5). These intermediates are necessary for the C–H activation and reductive elimination steps and because of their high coordination numbers, they are susceptible to be destabilised/unattainable when bulkier ligands (particularly NHCs) are coordinated to the copper centre.
Finally, important similarities can be drawn between our catalytic system and the one shown in Scheme 1. Both mechanisms appear to have triplet nitrenes as key intermediates and be accelerated by standard room light. Indeed, when the reduction of 4-nitrophenyl azide was carried out in the dark only 39% of the expected aniline 1a was observed by 1H NMR spectroscopy. Nevertheless, significant differences can also be found: our reactions are insensitive to the presence or absence of dioxygen and the inclusion of water in the reaction medium greatly accelerates the reaction.
Conclusions
A careful choice of copper(I) catalyst has led to the development of a new protocol for the reduction of azides under very simple conditions. This reduction was performed in the presence of air and water; still no undesired oxidation by-products were formed. Complex 3 is readily available and indefinitely stable towards air and moisture. This stability, together with its remarkable activity, was instrumental for the development of a robust and user-friendly catalytic system. DFT calculations suggest that the presence of an EWG group on the substrate accelerates the rate limiting step and support the intermediacy of a triplet carbene and the role of toluene as a hydride source through the abstraction of one of its sp3 hydrogen atoms. This is in agreement with the experimental lack of reactivity of electron-rich aryl azides, which were recovered unreacted with no sign of C(sp3)–H activation.The importance of these results is twofold. First, it represents a necessary step towards the development of a viable and general catalytic alternative to the Staudinger reaction. Secondly, the promise of copper-mediated oxidation of aliphatic C–H bonds under such simple conditions is of high interest.37 Of note, benzyl alcohol is stable in the presence of anilines in hot toluene/water as well as with catalyst 3 in the absence of a base. Hence, the observed instability of benzyl alcohol in these reactions is directly linked to the presence of equimolar amounts of an aniline in the reaction mixture and not the copper complex itself. With an improved mechanistic understanding in hand, we are currently working on broadening the azide scope of our system through catalyst design. Efforts to isolate a stable oxidation product are also ongoing since it would not only confirm the proposed mechanism but also allow its application to challenging C–H activation processes. Results will be reported in due course.
Experimental section
Experimental details
All chemicals were obtained from commercial sources and used without further purification. Air and moisture sensitive manipulations were performed using standard Schlenk line techniques. Anhydrous solvents were dried by passing them through columns of molecular sieves in a solvent purification system. NMR spectra were measured on Bruker AVANCE 400 spectrometers (1H: 400 MHz, 13C: 101 MHz, 19F: 377 MHz) at 20 °C unless stated otherwise. The chemical shifts (δ) are given in ppm relative to a tetramethylsilane standard or the residual solvent signal. The multiplicity is given in br, s, d, t, q, sept, and m for broad, singlet, doublet, triplet, quartet, septet, and multiplet. Mass spectra (MS) were recorded on a Micromass AutoSpec Premier, Micromass LCT Premier or a VG Platform II spectrometer using EI or ESI techniques at the Mass Spectroscopy Service of Imperial College London. Infrared spectra were recorded using a PerkinElmer 100 series FT-IR spectrometer, equipped with a beam-condensing accessory (samples were sandwiched between diamond compressor cells). Melting points (uncorrected) were determined on a Gallenkamp electrothermal apparatus. Single crystal X-ray diffraction patterns were collected using Agilent Xcalibur PX Ultra A and Excalibur 3 E diffractometers, and the structures were refined using the SHELXTL, and SHELX-2013 program systems.Computational details
All calculations were carried out with the Gaussian09 program package38 using the methods of density functional theory (DFT). The selected functional was BP8639 with the empirical dispersion correction of Grimme (BP86-D3).40 The selected basis set was 6-31G(d) for C, N, O and H,41 and SDD for Cu with the corresponding electron core potentials.42 The validity of the description was confirmed by benchmarking with a variety of functionals, as reported in the ESI.† All geometry optimisations were carried out in water solvent (SMD) without symmetry restrictions. We confirmed the nature of all computed stationary points as minima or transition states through vibrational frequency calculations. Systematic conformation analyses were carried out for all steps and therefore only results with the most stable conformers are discussed. Free energy corrections were calculated at 373.15 K and 105 Pa pressure, including zero point energy corrections (ZPE). Minimum energy crossing points (MECP) were computed with the program supplied by Harvey.43 The reported MECP free energies correspond to an average of those for the two intervening states. A collection data set of all computational data is accessible in the ioChem-BD repository,33 and can be accessed viahttps://doi.org/10.19061/iochem-bd-1-97.Bis(N,N′-bis(4-N,N-dimethylaminophenyl)-1,4-diazabuta-1,3-dien)copper(I) tetrafluoroborate [Cu(DABDMA)2]BF4 (2)
[Cu(NCMe)4]BF4 (78.9 mg, 0.25 mmol) and DABDMA (159 mg, 0.50 mmol) were suspended in dry, degassed DCM (15 mL) under a N2 atmosphere and stirred at room temperature for 16 h. The reaction mixture was then filtered through Celite and concentrated to a volume of ∼5 mL under reduced pressure, followed by the addition of petroleum ether. The formed precipitate was collected, washed with petroleum ether, and dried under reduced pressure to give 2 as a light green solid (145 mg, 79%). Mp: 293 °C. 1H NMR (400 MHz, CDCl3): δ 8.95 (s, 4H), 7.51 (d, J = 9.0 Hz, 8H), 6.66 (d, J = 9.0 Hz, 8H), 2.92 (s, 24H). 13C{1H} NMR (101 MHz, CDCl3): δ 151.7, 147.3, 135.5, 124.9, 112.8, 40.2. 19F NMR (377 MHz, CDCl3): δ −148.3 (s). IR: 2857 (C–H st), 2803 (C–H st), 1600, 1554 (C–N asym st), 1515, 1439, 1358 (C–N sym st), 1297, 1165, 1047 (BF4−), 945, 812, 635 cm−1. HRMS calcd for C36H44N8Cu: 651.2985, found 651.2984 ([Cu(DABDMA)2]+).General procedure for the reduction of azides
In a microwave vial, azide (0.5 mmol) and [Cu(DABDMA)2]BF43 (37 mg, 10 mol%) were suspended in toluene (0.33 mL) and water (0.66 mL) and sealed using a crimped cap. This mixture was heated to 100 °C for 20 h before being cooled and filtered through Celite and washed with EtOAc. The organic filtrate was washed with saturated, aqueous Na4EDTA (3 × 10 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The reported yields are isolated yields and are the average of at least two independent runs.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
This research was financially supported by Imperial College London, the Engineering and Physical Sciences Research Council (EPSRC) (EP/K030760/1), the CERCA Programme (Generalitat de Catalunya) and MINECO (grant CTQ2017-87792-R and Severo Ochoa Excellence Accreditation 2014-2018 SEV-2013-0319). BZ acknowledges SNF for a travel grant (# PS1SKP2-168439) and DVE thanks MECD for a doctoral fellowship. Lalita Radtanajiravong is sincerely acknowledged for carrying out some final experiments.Notes and references
- S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge, 2004 Search PubMed.
- (a) P. Roose, K. Eller, E. Henkes, R. Rossbacher and H. Höke, Amines, Aliphatic. Ullmann's Encyclopedia of Industrial Chemistry, 2015, pp. 1–55 Search PubMed; (b) P. F. Vogt and J. J. Gerulis, Amines, Aromatic. Ullmann's Encyclopedia of Industrial Chemistry, 2000 Search PubMed; (c) K. Drauz, I. Grayson, A. Kleemann, H.-P. Krimmer, W. Leuchtenberger and C. Weckbecker, Amino Acids. Ullmann's Encyclopedia of Industrial Chemistry, 2007 Search PubMed.
- (a) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed; (b) E. F. V. Scriven and K. Turnbull, Chem. Rev., 1988, 88, 297–368 CrossRef CAS.
- H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 635–646 CrossRef CAS.
- (a) C. I. Schilling, N. Jung, M. Biskup, U. Schepers and S. Bräse, Chem. Soc. Rev., 2011, 40, 4840–4871 RSC; (b) S. Liu and K. J. Edgar, Biomacromolecules, 2015, 16, 2556–2571 CrossRef CAS PubMed.
- A catalytic version on the Staudinger reaction still requires an excess of hydride source and harsher reaction conditions, see: H. A. van Kalkeren, J. J. Bruins, F. P. J. T. Rutjes and F. L. van Delft, Adv. Synth. Catal., 2012, 354, 1417–1421 CrossRef CAS.
- S. Ahammed, A. Saha and B. C. Ranu, J. Org. Chem., 2011, 76, 7235–7239 CrossRef CAS PubMed For a more recent and promising heterogeneous system, see: Á. Georgiádes, S. B. Ötvös and F. Fülöp, Adv. Synth. Catal., 2018, 360, 1841–1849 CrossRef.
- (a) Y. Xia, W. Li, F. Qu, Z. Fan, X. Liu, C. Berro, E. Rauzy and L. Peng, Org. Biomol. Chem., 2007, 5, 1695–1701 RSC; (b) Y. A. Cho, D.-S. Kim, H. R. Ahn, B. Canturk, G. A. Molander and J. Ham, Org. Lett., 2009, 11, 4330–4333 CrossRef CAS PubMed; (c) Y. Goriya and C. V. Ramana, Tetrahedron, 2010, 66, 7642–7650 CrossRef CAS.
- J. T. Markiewicz, O. Wiest and P. Helquist, J. Org. Chem., 2010, 75, 4887–4890 CrossRef CAS PubMed.
- H. Peng, K. H. Dornevil, A. B. Draganov, W. Chen, C. Dai, W. H. Nelson, A. Liu and B. Wang, Tetrahedron, 2013, 69, 5079–5085 CrossRef CAS PubMed.
- For related examples of non-innocent solvents (under basic conditions), see: (a) H. Zhao, H. Fu and R. Qiao, J. Org. Chem., 2010, 75, 3311–3316 CrossRef CAS PubMed; (b) S. R. Lanke and B. M. Bhanage, Synth. Commun., 2014, 44, 399–407 CrossRef CAS.
- For characterised examples, see: (a) Y. M. Badiei, A. Krishnaswamy, M. M. Melzer and T. H. Warren, J. Am. Chem. Soc., 2006, 128, 15056–15057 CrossRef CAS PubMed; (b) Y. M. Badei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2008, 47, 9961–9964 CrossRef PubMed; (c) S. Kundu, E. Miceli, E. Farquhar, F. F. Pfaff, U. Kuhlmann, P. Hildebrandt, B. Braun, C. Greco and K. Ray, J. Am. Chem. Soc., 2012, 134, 14710–14713 CrossRef CAS PubMed; (d) F. Dielmann, D. M. Andrada, G. Frenking and G. Bertrand, J. Am. Chem. Soc., 2014, 136, 3800–3802 CrossRef CAS PubMed; (e) T. Corona, L. Ribas, M. Rovira, E. R. Farquhar, X. Ribas, K. Ray and A. Company, Angew. Chem., Int. Ed., 2016, 55, 14005–14008 CrossRef CAS PubMed; (f) A. Bakhoda, Q. Jiang, J. A. Bertke, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2017, 56, 6426–6430 CrossRef CAS PubMed.
- (a) R. T. Gephart III and T. H. Warren, Organometallics, 2012, 31, 7728–7752 CrossRef; (b) M. M. Díaz-Requejo and P. J. Pérez, Chem. Rev., 2008, 108, 3379–3394 CrossRef PubMed; (c) P. Müller and C. Fruit, Chem. Rev., 2003, 103, 2905–2919 CrossRef PubMed.
- (a) P. Brandt, M. J. Sodergren, P. G. Andersson and P. O. Norrby, J. Am. Chem. Soc., 2000, 122, 8013–8020 CrossRef CAS; (b) P. Comba, C. L. Lang, C. L. de Laorden, A. Muruganantham, G. Rajaraman, H. Wadepold and M. Zajaczkowski, Chem. – Eur. J., 2008, 14, 5313–5328 CrossRef CAS PubMed; (c) K. P. Hou, D. A. Hrovat and X. G. Bao, Chem. Commun., 2015, 51, 15414–15417 RSC.
- T. R. Cundari, A. Dinescu and A. B. Kazi, Inorg. Chem., 2008, 47, 10067–10072 CrossRef CAS PubMed.
- L. Maestre, W. M. C. Sameera, M. M. Diaz-Requejo, F. Maseras and P. J. Perez, J. Am. Chem. Soc., 2013, 135, 1338–1348 CrossRef CAS PubMed.
- M. R. Rodriguez, A. Beltran, A. L. Mudarra, E. Alvarez, F. Maseras, M. M. Diaz-Requejo and P. J. Perez, Angew. Chem., Int. Ed., 2017, 56, 12842–12847 CrossRef CAS PubMed.
- E. Haldon, M. Besora, I. Cano, X. C. Cambeiro, M. A. Pericas, F. Maseras, M. C. Nicasio and P. J. Perez, Chem. – Eur. J., 2014, 20, 3463–3474 CrossRef CAS PubMed.
- M. Besora, A. A. C. Braga, W. M. C. Sameera, J. Urbano, M. R. Fructos, P. J. Perez and F. Maseras, J. Organomet. Chem., 2015, 784, 2–12 CrossRef CAS.
- B. Zelenay, R. Frutos-Pedreño, J. Markalain-Barta, E. Vega-Isa, A. J. P. White and S. Díez-González, Eur. J. Inorg. Chem., 2016, 4649–4658 CrossRef CAS.
- S. Díez-González, E. C. Escudero-Adán, J. Benet-Buchholz, E. D. Stevens, A. M. Slawin and S. P. Nolan, Dalton Trans., 2010, 39, 7595–7606 RSC.
- S. Díez-González, E. D. Stevens, N. M. Scott, J. L. Petersen and S. P. Nolan, Chem. – Eur. J., 2008, 14, 158–168 CrossRef PubMed.
- (a) D. Schultz and J. R. Nitschke, J. Am. Chem. Soc., 2006, 128, 9887–9892 CrossRef CAS PubMed; (b) M. Lal Saha and M. Schmittel, Inorg. Chem., 2016, 55, 12366–12375 CrossRef PubMed.
- See ref. 20 and references therein..
- (a) G. C. van Stein, G. van Koten, K. Vrieze, C. Brevard and A. L. Spek, J. Am. Chem. Soc., 1984, 106, 4486–4492 CrossRef CAS; (b) D. M. Haddleton, D. J. Duncalf, D. Kukulj, M. C. Crossman, S. G. Jackson, S. A. F. Bon, A. J. Clark and A. J. Shooter, Eur. J. Inorg. Chem., 1998, 1799–1806 CrossRef CAS; (c) S. Dehghanpour, N. Bouslimani, R. Welter and F. Mojahed, Polyhedron, 2007, 26, 154–162 CrossRef CAS.
- See ESI† for further details..
- M. Burke-Laing and M. Laing, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 3216–3224 CrossRef.
- A simple on-line tool calculates the percentage of the volume of a metal-centred sphere of defined radius that is occupied by a given ligand: www.molnac.unisa.it/OMtools/sambvca.php..
- W. D. Kerber, D. L. Nelsen, P. S. White and M. R. Gagne, Dalton Trans., 2005, 1948–1951 RSC.
- 5 mol% [Cu]: >95% for 3 and 4; 1 mol% [Cu]: >95% for 3 and 4; 0.5 mol% [Cu]: 13% for 3 and 15% for 4..
- A noteworthy reported exception to this behaviour is the formation of azepines in the presence of primary and secondary amines. For leading reports, see: (a) R. Huisgen, D. Vossius and M. Appl, Chem. Ber., 1958, 91, 1–12 CrossRef CAS; (b) R. Huisgen and M. Appl, Chem. Ber., 1958, 91, 12–31 CrossRef CAS.
- C. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2010, 49, 6174–6177 CrossRef CAS PubMed.
- M. Álvarez-Moreno, C. de Graaf, N. López, F. Maseras, J. M. Poblet and C. Bo, J. Chem. Inf. Model., 2015, 55, 95–103 CrossRef PubMed.
- S. Fountoulaki, P. L. Gkizis, T. S. Symeonidis, E. Kaminioti, A. Karina, I. Tamiolakis, G. S. Armatas and I. N. Lykakis, Adv. Synth. Catal., 2016, 358, 1500–1508 CrossRef CAS.
- (a) S. Y. Tee, K. Y. Win, W. S. Teo, L. D. Koh, S. Liu, C. P. Tengand and M. Y. Han, Adv. Sci., 2017, 4, 1600337 CrossRef PubMed; (b) S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498–502 CrossRef CAS PubMed.
- L. K. Dyall, Pyrolysis of Aryl Azides, in Halides, Pseudo-Halides and Azides, ed. S. Patai and Z. Rappoport, John Wiley & Sons, Ltd., Chichester, UK, 1983, vol. 1 Search PubMed.
- For the use of (highly sensitive) dicopper(II) peroxo complexes in this context, see: (a) H. R. Lucas, L. Li, A. A. Narducci Sarjeant, M. A. Vance, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2009, 131, 3230–3245 CrossRef CAS PubMed; (b) C. Würtele, O. Sander, V. Lutz, T. Waitz, F. Tuczek and S. Schindler, J. Am. Chem. Soc., 2009, 131, 7544–7545 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision A.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1998, 38, 3098–3100 CrossRef; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- H. Stoll, P. Fuentealba, P. Schwerdtfeger, J. Flad, L. V. Szentpály and H. Preuss, J. Chem. Phys., 1984, 81, 2732–2736 CrossRef CAS.
- J. N. Harvey, M. Aschi, H. Schwarz and W. Koch, Theor. Chem. Acc., 1998, 99, 95–99 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1583789–1583791. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cy00515j |
| This journal is © The Royal Society of Chemistry 2018 |