
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effects of MTG catalysis on methanol mobility in ZSM-5
S. K.
Matam
 ab,
A. J.
O'Malley
bc,
C. R. A.
Catlow
bc,
Suwardiyanto
d,
P.
Collier
e,
A. P.
Hawkins
f,
A.
Zachariou
f,
D.
Lennon
f,
I.
Silverwood
g,
S. F.
Parker
bg and
R. F.
Howe
*h
ab,
A. J.
O'Malley
bc,
C. R. A.
Catlow
bc,
Suwardiyanto
d,
P.
Collier
e,
A. P.
Hawkins
f,
A.
Zachariou
f,
D.
Lennon
f,
I.
Silverwood
g,
S. F.
Parker
bg and
R. F.
Howe
*h
aDepartment of Chemistry, University College London, WC1 HOAJ, UK
bUK Catalysis Hub, Research Complex at Harwell, STFC Rutherford Appleton Laboratory, Chilton, OX11 0FA, UK
cCardiff Catalysis Institute, School of Chemistry, Cardiff University, CF10 3AT, UK
dChemistry Department, University of Jember, Jember, 68121 Indonesia
eJohnson-Matthey Technology Centre, Sonning Common, Reading RG34 9NH, UK
fSchool of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK
gISIS Facility, STFC Rutherford Appleton Laboratory, Chilton OX11 0QX, UK
hChemistry Department, University of Aberdeen, AB24 3UE, UK. E-mail: r.howe@abdn.ac
First published on 12th June 2018
Abstract
We analyse the dynamics of methanol in ZSM-5 catalysts both with and without the hydrocarbon pool, resulting from the methanol to gasoline (MTG) reaction taking place at 623 K and 673 K for three days, to determine the effects of catalyst use on molecular mobility. Using quasielastic neutron scattering (QENS), we observe that methanol is immobile on the QENS instrumental time scale in the fresh catalyst (ZSM-5-F) and in the sample used to convert methanol for 3 days at 623 K (ZSM-5-623). However, in zeolite ZSM-5-673 (MTG at 673 K for 3 days) we observe isotropic methanol rotation with an immobile fraction of 0.58 and a rotational diffusion coefficient of DR = 3 × 1010 s−1. The observed differences between the zeolites in methanol dynamics are attributed to the development of mesoporosity in ZSM-5-673 due to the high reaction temperature of 673 K, leading to dislodgement of lattice Al as is evident from NMR data.
Introduction
The catalytic conversion of methanol to hydrocarbons, either gasoline range (MTG) or light olefins (MTO), is a rapidly evolving technology for producing hydrocarbons from natural gas or coal reserves.1,2 The zeolite catalyst ZSM-5 (MFI) was first used in the Mobil MTG process commercialised in New Zealand in 1985.3 This process used fixed bed reactors which operated typically at 623 K and converted an equilibrium mixture of methanol, dimethylether and water to an aromatic rich mixture of hydrocarbons. There are three aspects of the process which have been extensively investigated over the past 30 years: first, how the first carbon–carbon bonds are formed from methanol and/or dimethylether; second, the mechanism of steady state gasoline formation; and third, the causes of catalyst deactivation.4 The chemistry of initial carbon–carbon bond formation remains a hotly debated topic,5–10 but there is widespread agreement that the steady state reaction mechanism is autocatalytic, involving a so-called hydrocarbon pool created in the zeolite pores.11–15 Catalyst deactivation is attributed to the build-up of “coke” in the zeolite pores, blocking access to the internal acid sites,16 and recent studies have suggested that formaldehyde formed from methanol is a key contributor to coke.17The diffusion of reactant methanol into the pores of the zeolite and the extent to which this is influenced by the presence of a hydrocarbon pool and/or coke deposits are important issues affecting catalyst performance which have not to date been widely addressed. Diffusion of methanol in a clean (unreacted) ZSM-5 has been previously investigated by several techniques: classical time dependent sorption,18 tracer diffusion with 14C labelled methanol,19 and quasielastic neutron scattering (QENS)20–22 There have been no previous studies, however, of methanol diffusion in used catalysts.
Neutron spectroscopy techniques are becoming an increasingly significant tool in the study of zeolite catalysed processes,23–26 and we have recently reported the characterisation by inelastic neutron scattering (INS) of a commercial ZSM-5 zeolite catalyst reacted with methanol in a 12 g scale fixed bed reactor for 3 days at 623 K and 673 K.27 The catalyst remained active for aromatic production at both reaction temperatures, although the vibrational spectra of the used catalysts from both INS and diffuse reflectance infrared spectroscopy showed some differences between the coke species present at the two temperatures.
In this paper, we present detailed QENS analysis of methanol sorption in the clean ZSM-5 catalyst and the two reacted catalysts described in ref. 27. Methanol is immobile in clean unreacted zeolite and in that reacted at 623 K. Reaction at 673 K generates significant mesoporosity in the catalyst, resulting in a higher, yet localised methanol mobility.
Experimental
Zeolite ZSM-5 and catalyst evaluation
The ZSM-5 zeolite was provided by Johnson Matthey (Intercat IC16926, Si/Al = 30, surface area 371 m2 g−1). The zeolite was calcined in air at 773 K for 14 h to remove residual template. The catalytic reaction system has been described in detail in ref. 27. Briefly, it comprises a stainless steel reactor 35 mm i.d. × 65 mm length connected to a gas handling manifold and flow control system; the eluting product stream was analysed on-line with a Hiden Analytical HPR-20 mass spectrometer and liquid products collected in a down-stream trap for later off-line analysis by GC-MS. Typically, 12 g of calcined zeolite was loaded into the reactor and dehydrated in flowing helium at 623 K for several hours prior to setting the reaction temperature, either 623 K or 673 K. A methanol/helium stream was then passed through the reactor at a feed rate of 0.2 g methanol per g zeolite h−1 for three days. The reactor was then flushed with helium at the reaction temperature, cooled to room temperature in flowing helium, then transferred to an argon glove box and the zeolite samples loaded into appropriate containers for subsequent analysis.For comparative purposes, the freshly calcined zeolite was also pre-treated in He flow in the reactor at 623 K for several hours, then unloaded in the same way as described above. The zeolite samples are denoted as: ZSM-5-F (clean unreacted), ZSM-5-623 (subjected to MTG at 623 K for three days) and ZSM-5-673 (MTG at 673 K for three days). A portion of each sample was subjected to temperature programmed oxidation (TPO) up to 973 K for N2-physisorption experiments; the oxidised samples are labelled as ZSM-5-F-TPO, ZSM-5-623-TPO and ZSM-5-673-TPO.
Nitrogen Physisorption measurements
Nitrogen adsorption–desorption isotherms were measured at 77 K on a Quadrasorb EVO instrument (model QDS-30). Prior to the measurements, the samples were outgassed at 573 K for 18 h. The micro pore volume (Vmicro) and meso pore surface area (Smeso) were determined by a t-plot method, and the total surface area (Stotal) by the Brunauer–Emmett–Teller (BET) method.NMR measurements
NMR spectra were measured with a Varian Infinity Plus 400 MHz spectrometer. 7.5 mm sample rotors were loaded with catalyst in the argon glove box and sealed under argon for subsequent NMR measurements. Sample rotors were spun in dry air at typically 3 kHz. 13C spectra were recorded at 100.54 MHz using a variable amplitude cross polarisation pulse sequence and a contact time of 7 ms. Chemical shifts were externally referenced to tetramethylsilane (TMS) via a hexamethylbenzene standard, and typically 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 acquisitions averaged with a 5 s pulse delay. 27Al spectra were recorded at 104.2 MHz using a one pulse Bloch decay with a 0.5 μs pulse width (π/20) and a 5 s pulse delay. All samples were measured with the same number of 500 acquisitions to allow comparison of signal to noise, and chemical shifts externally referenced to a kaolin standard (−2.5 ppm relative to Al(H2O)63+). 29Si spectra were recorded at 79.4 MHz using a one pulse Bloch decay with proton decoupling, a π/2 pulse width of 6 μs and a 5 s pulse delay, typically 1000 acquisitions. 29Si spectra were externally referenced to TMS via a kaolin standard (−91.2 ppm).
000 acquisitions averaged with a 5 s pulse delay. 27Al spectra were recorded at 104.2 MHz using a one pulse Bloch decay with a 0.5 μs pulse width (π/20) and a 5 s pulse delay. All samples were measured with the same number of 500 acquisitions to allow comparison of signal to noise, and chemical shifts externally referenced to a kaolin standard (−2.5 ppm relative to Al(H2O)63+). 29Si spectra were recorded at 79.4 MHz using a one pulse Bloch decay with proton decoupling, a π/2 pulse width of 6 μs and a 5 s pulse delay, typically 1000 acquisitions. 29Si spectra were externally referenced to TMS via a kaolin standard (−91.2 ppm).
Quasielastic neutron scattering (QENS)
All measurements were performed using the time-of-flight backscattering neutron spectrometer OSIRIS28 at the ISIS Pulsed Neutron and Muon Source. Methanol (Sigma-Aldrich, ≥99.9%) loading into the zeolite pores was conducted in the same MTG reactor at room temperature by flowing He through a room temperature methanol saturator until no further weight change was detected. The resulting methanol loadings were 0.104, 0.070 and 0.086 g g−1 of ZSM-5-F, ZSM-5-623 and ZSM-5-673, respectively. The thin walled aluminum containers of annular geometry were loaded with zeolite under an argon atmosphere in the glovebox.The cells were placed in a top-loading closed cycle refrigerator, and a resolution measurement was taken at a base temperature of 6 K. QENS measurements were then taken at 225, 275 and 325 K. Pyrolitic graphite 002 analyser crystals were used giving an energy resolution of 24.5 μeV with energy transfers measured in a window of ±0.55 meV; the detector covered measurements over a Q range of 0.2–1.7 Å−1. For each catalyst the measurement was taken of the unloaded sample and the signal then subtracted from the signal of the loaded zeolite, so that only the signal from the methanol could be extracted. In this way any scattering from the aluminium container, which is very low in comparison with the empty zeolite is also subtracted. No further corrections were necessary. The data reduction was performed using MantidPlot software and all QENS spectra were fitted using the neutron scattering analysis software DAVE.29
Results and discussion
Nitrogen adsorption–desorption
Fig. 1 shows nitrogen adsorption–desorption isotherms for each of the three zeolite catalysts before and after TPO at 973 K. The pore volumes and surface areas calculated from these data are presented in Table 1. | ||
| Fig. 1 Nitrogen adsorption–desorption isotherms of the three catalyst samples before (filled symbols) and after TPO to 973 K (open symbols). | ||
| Zeolite | V (micro) cm3 g−1 | V (total) cm3 g−1 | S (meso) m2 g−1 | S (total) m2 g−1 | Coke wt% | MeOH uptake g g−1 |
|---|---|---|---|---|---|---|
| a t-Plot. b BET method. | ||||||
| ZSM5-F | 0.148 | 0.22 | 30.6 | 387 | 0 | 0.104 |
| ZSM5-F-TPO | 0.155 | 0.24 | 35 | 406 | ||
| ZSM5-623 | 0.11 | 0.19 | 31.3 | 294 | 7.1 | 0.070 |
| ZSM5-623-TPO | 0.12 | 0.20 | 30 | 325 | ||
| ZSM5-673 | 0.146 | 0.24 | 38.6 | 384 | 4.9 | 0.086 |
| ZSM5-673-TPO | 0.18 | 0.30 | 60 | 487 | ||
The surface area and micropore volume of the fresh zeolite catalyst are comparable with values reported in the literature for ZSM-5 (e.g. micropore volumes in the range 0.13–0.23 cm3 g−1).30 The hysteresis observed above P/P0 = 0.5 indicates inter-particle mesoporosity. The catalyst used at 623 K for 3 days shows a ≈24% loss in surface area which is only partially recovered after TPO; there is however no change in the mesoporosity of this catalyst. In contrast, the catalyst used at 673 K for 3 days gives a surface area close to that of the fresh catalyst, but following TPO the total surface area and the mesoporosity are both significantly increased.
Loss of surface area in used catalysts is expected if coke deposition occurs within the micropores. Bibby et al. reported detailed measurements of nitrogen adsorption in a series of ZSM-5 catalysts reacted with methanol at different times on stream at 643 K.31 The loss of sorption capacity with increasing methanol conversion varied with the Si:Al ratio of the zeolite and the particle size. The 24% loss in surface area seen here after 3 days on stream at 623 K (≈14 g methanol reacted per g of catalyst) is, however, consistent with that reported in ref. 31 for a zeolite with comparable Si:Al ratio. The decreased capacity for methanol uptake at room temperature in the catalyst used at 623 K is also consistent with the nitrogen physisorption data. Almost complete removal of internal coke during TPO up to 973 K might be expected to restore the original pore volume and surface area, which does not happen, however, suggesting that some irreversible surface area loss has occurred either during reaction with methanol at 623 K or during the subsequent TPO up to 973 K.
In contrast, the catalyst reacted at 673 K appears to show no loss of surface area, and after TPO the surface area, micropore and mesopore volumes are all much higher than those of the fresh catalyst. This enhanced porosity cannot result from the TPO process, since it was not seen with the 623 K catalyst, but must be a result of chemistry occurring at the higher reaction temperature. Further insight into the differences between the two used zeolite catalysts was obtained from solid state NMR measurements.
NMR spectra
Fig. 2 shows 27Al and 29Si NMR spectra of the fresh and used catalysts. The fresh catalyst (a) gives an intense signal at 51 ppm characteristic of tetrahedral aluminium in the zeolite lattice plus a very weak signal at −5 ppm due to octahedral (extra-lattice) aluminium.32 Although the quadrupolar nature of 27Al precludes quantitative measurements in a one pulse NMR experiment, the spectrum of the fresh zeolite suggests that it contains very little extra-framework aluminium. After reaction at 623 K, the 51 ppm signal became noticeably less intense (Fig. 2(b)) and slightly broadened (with an increase in FWHH from 6.2 to 7.9 ppm). The amount of NMR visible tetrahedral aluminium (estimated from signal to noise ratio) fell to ∼50% of that in the fresh catalyst. After reaction at 673 K the amount of NMR visible tetrahedral aluminium remained approximately the same as that at 623 K, with no further increase in line width. | ||
| Fig. 2 27Al (left) and 29Si (right) NMR spectra of (a) ZSM-5-F; (b) ZSM-5-623; (c) ZSM-5-673. | ||
Loss of NMR visible tetrahedral aluminium was reported in the early NMR study of methanol coked ZSM-5 by Meinhold and Bibby.33 These authors attributed loss of intensity, increase in line width and decrease in chemical shift with increasing coke content to displacement of adsorbed water by coke in the zeolite pores, causing an increased electric field gradient at framework aluminium sites. Similar changes to those seen in Fig. 2 have been reported by Zhu et al.34 in the 27Al NMR spectra of ZSM-5 used for methanol conversion at 673 K. Campbell et al.35 described the almost complete loss of a tetrahedral aluminium NMR signal in ZSM-5 used in repeated MTG/regeneration cycles.
There is no evidence in the 27Al spectra in Fig. 2 of additional extraframework aluminium, either 5 coordinate (∼33 ppm) or 6 coordinate (∼−5 ppm),32,36 in the used catalysts. 29Si NMR spectra do show however that there is some loss of aluminium from the zeolite lattice. The 29Si spectrum of the fresh catalyst is dominated by a signal at −112 ppm due to the 24 distinct Q4(0Al) sites previously reported in high silica ZSM-5.32 A shoulder at −106 ppm is due to Q4(1Al) sites.37 The intensity of this shoulder is slightly reduced in the ZSM-5-623 sample and more noticeably so in the ZSM-5-673 sample. Similar loss of Q4(1Al) sites has been reported by Barbera et al.38 in a ZSM-5 catalyst deactivated after reaction in methanol for 500 hours at 623 K, by Zhu et al. after 55 hours at 673 K,34 and by Campbell et al. in ZSM-5 used in repeated MTG/regeneration cycles.35
We conclude from these NMR and pore volume measurements that after extended reaction for 72 hours in methanol at 673 K there is significant damage to the zeolite lattice, resulting in some loss of framework aluminium and increased mesoporosity as evident from N2-physisorption data (Fig. 1 and Table 1). Steam treatment of ZSM-5 is well known to cause framework damage. Campbell et al.39 described loss of framework aluminium from HZSM-5 subjected to steam treatment at 873 K. Ong et al.40 steamed ZSM-5 at 723 K and showed NMR evidence for hydrolysis of Al–O–Si bonds and formation of tetrahedral extraframework aluminium species. The water produced during methanol reaction over ZSM-5 may be expected to have a similar effect, although the reaction temperatures are lower than those used in the cited steaming studies.
QENS measurements
We begin by showing the QENS spectra as a function of Q at 325 K in Fig. 3–5 for the three samples ZSM-5-F, ZSM-5-623 and ZSM-5-673, respectively. The figures contain the total fit (black), and the quasielastic component of the spectra (red) given by a Lorentzian function. In Fig. 3 and 4, we note that in ZSM-5-F and ZSM-5-623, the peak intensity is described almost completely by the resolution function and the Lorentzian component is not significant enough to be characterised, suggesting that no motion is observable on the instrumental timescale (∼2–50 ps). Similar observations have been reported previously for methanol in fresh ZSM-5. Jobic et al. attributed the lack of any broadening of the elastic peak to the presence of strongly hydrogen bonded methanol.20 A similar conclusion was reached by Gupta et al.21 O'Malley et al.22 more recently also found no broadening of the elastic peak for methanol in fresh ZSM-5 measured on the same spectrometer as used in the present work. They concluded however from accompanying inelastic neutron scattering measurements of the vibrational spectrum that the lack of mobility of the methanol was due to dissociation to form methoxy groups. | ||
Fig. 3 QENS spectra as a function of Q for methanol at 325 K in ZSM-5-F. (---) is the total fit to the data points, ( ) is the quasielastic component. Alternate spectra are plotted after Q = 0.75 Å−1 for clarity. ) is the quasielastic component. Alternate spectra are plotted after Q = 0.75 Å−1 for clarity. | ||
 | ||
Fig. 4 QENS spectra as a function of Q for methanol at 325 K in ZSM-5-623. (---) is the total fit to the data points, ( ) is the quasielastic component. Alternate spectra are plotted after Q = 0.75 Å−1 for clarity. ) is the quasielastic component. Alternate spectra are plotted after Q = 0.75 Å−1 for clarity. | ||
 | ||
Fig. 5 QENS spectra as a function of Q for methanol at 325 K in ZSM-5-673. (---) is the total fit to the data points, ( ) is the quasielastic component. Alternate spectra are plotted after Q = 0.75 Å−1 for clarity. ) is the quasielastic component. Alternate spectra are plotted after Q = 0.75 Å−1 for clarity. | ||
This lack of a characterisable quasielastic component was also the case for ZSM-5-F and ZSM-5-623 and ZSM-5-673 measured at 225 and 275 K. However, the QENS spectra of ZSM-5-673 measured at 325 K revealed the presence of a more significant quasielastic component, as shown in Fig. 5.
There is also a more notable decrease in the elastic intensity with increasing Q, suggesting an increase in the total mobility of the system. It is important to note that the decay of the relative elastic intensity does not become significant until Q of >1 Å−1. Maintaining the elastic intensity in this way suggests that movement over longer length scales (such as in translational motion, as detected in previous work in zeolite HY using the same instrument41) is not occurring. However, the presence of broadening at higher Q values (as shown at Q = 1.7) in Fig. 6, when compared with ZSM-5-F and ZSM-5-623 suggests that significant localised motions (such as rotations) are present.
 | ||
Fig. 6 QENS spectra at Q = 1.7 Å−1 for methanol at 325 K in ZSM-5-F, ZSM-5-623 and ZSM-5-673. ( ) is the total fit to the data points, ( ) is the total fit to the data points, ( ) is the resolution function representing the elastic component. ) is the resolution function representing the elastic component. | ||
The localised motions can be characterised using the elastic incoherent structure factor (EISF), which is given by:
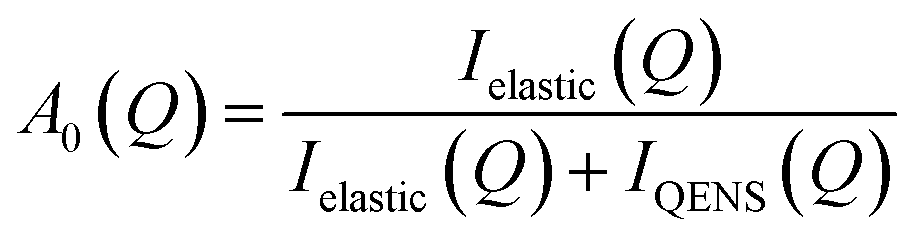 | (1) |
A number of models are available to characterise the localised motions of methanol, related to the geometrical motions of the protons in the molecule. The analysis of the EISF in this work is modelled on that carried out by Jobic20 in the analysis of methanol in a different ZSM-5 sample. We outline here the models used to fit the experimental EISF.
Isotropic rotation is characterised by a molecule whose reorientation takes place through a series of small angle, random rotations so that no most probable orientation exists on a time average, as depicted in Scheme 1. The scattering law as derived by Sears42 for this form of rotation has an EISF A0(Q) given as
| A0(Q) = j20(Qr) | (2) |
 | (3) |
 | ||
| Scheme 1 Isotropic rotation of a methanol molecule with a radius of rotation (r). | ||
To deduce the radius of total molecule rotation, r, the bond lengths and bond angles from microwave spectroscopy were implemented (O–H = 0.95 Å, C–O =1.425 Å, C–H = 1.094 Å while θH–C–H = 108.3° and θC–O–H = 108.4°) leading to a mean radius of gyration of 1.48 Å. The model is plotted against the experimental EISF as the bold black line in Fig. 7, and falls below the experimental points at all Q values.
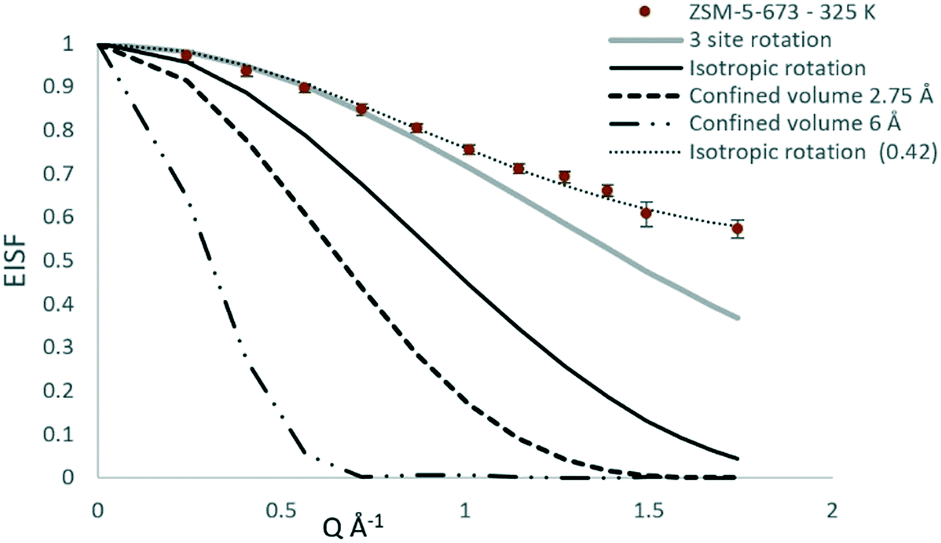 | ||
| Fig. 7 The experimental EISF of methanol in ZSM-5-673 at 325 K, plotted against the models of localised motions described in the text. | ||
The next consideration is that of a methanol molecule which is fixed through adsorption to the zeolite surface, either through physisorption due to H-bonding via the O–H group to the catalyst surface, or due to methoxylation such that the only motion observable is that of rotating methyl groups. We consider a model which can be used to describe methyl rotation is a jump rotation model between three equidistant sites on a circle with a radius (r) as depicted in Scheme 2.
 | ||
| Scheme 2 Methyl rotation described by a three site jump model around a circle (red arrows). | ||
The elastic incoherent structure factor is given as
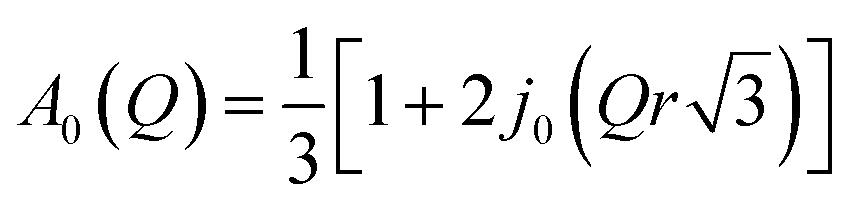 | (4) |
Thus, we now consider translational motion of methanol but localised to a confining volume. Volino and Dianoux43 developed a model to describe a scattering molecule undergoing translational motions in a confined spherical volume of radius rconf. as shown in Scheme 3.
 | ||
| Scheme 3 Translational motion of methanol confined to a spherical volume of radius rconf. | ||
This scattering model is based on the general problem of a particle diffusing in a potential field of spherical symmetry, where the potential is low inside the sphere's volume but infinite outside of it. The EISF in this model is given as:
 | (5) |
 | (6) |
However, we may also consider that only a fraction of molecules are mobile on the timescale of the instrument, with the remaining molecules considered as being static. We can calculate an effective EISF which takes this into consideration, given by:
| A0eff(Q) = px[A0(Q)] + (1 − px) | (7) |
 | ||
| Fig. 8 The experimental EISF of methanol in ZSM-5-673 at 325 K, plotted against the models of localised motions after fitting with an immobile fraction. The optimum px value is listed in brackets. | ||
The fitting of this model with an immobile fraction suggests that in the most dealuminated zeolite ZSM-5-673, a major fraction (0.58) of methanol molecules remain strongly adsorbed to the catalyst (either through methoxylation, or H-bonding to Brønsted acid sites), but that a significant fraction are also not bound to the catalyst surface and are freely rotating. The presence of this freely rotating fraction may be due to loss of a fraction of Brønsted acid sites arising from the dealumination as evident from NMR (Fig. 2) which resulted in mesopores (Fig. 1 and Table 1) in zeolite that allow for space for the fraction of molecules to rotate freely. Further evidence for rotational motion being observed at 325 K in the ZSM-5-673 sample is given by the broadenings of the Lorentzian component of S(Q, ω), plotted as a function of Q in Fig. 9.
 | ||
| Fig. 9 FWHM at 325 K as a function of Q2 of methanol in ZSM-5-673 sample. | ||
The broadenings are shown to be independent of Q, as would be expected upon observing rotational motions, as opposed to the Q2 dependence shown with Fickian diffusion, or fitting to Q dependence models associated with jump diffusion, or diffusion in confined volumes.44,45 The widths of the Lorentzian component can then be used to calculate the isotropic rotational diffusion coefficient DR. The total incoherent scattering law for our isotropic rotational diffusion is given as:
 | (8) |
| Al(Q) = (2l + 1)j2l(Qr) | (9) |
The elastic (l = 0) and quasielastic structure factors calculated from eqn 2 and 9 respectively are plotted in Fig. 10.
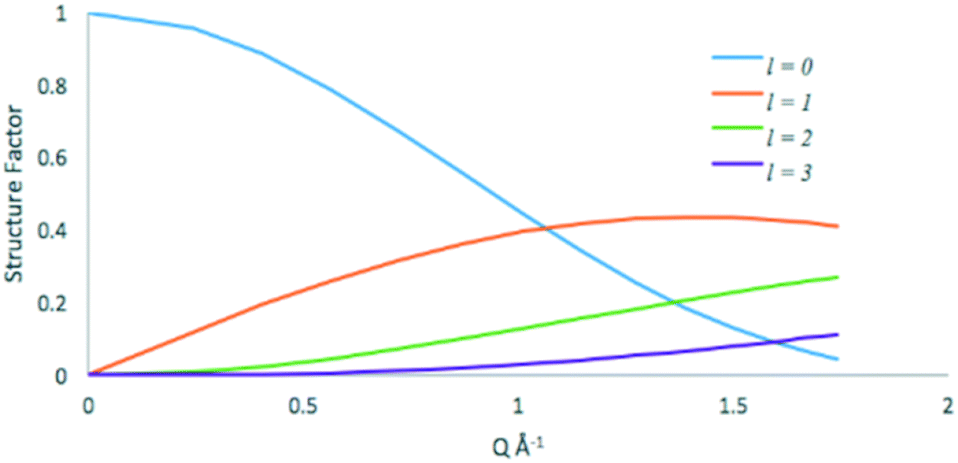 | ||
| Fig. 10 Elastic (l = 0) and quasielastic structure factors for the isotropic rotational diffusion of methanol in ZSM-5. | ||
At low Q values the vast majority of the quasielastic contribution comes from the l = 1 term, thus using eqn 9, the FWHM of the broadening is 4 DR. We then calculate DR = 3 × 1010 s−1 for methanol in ZSM-5-673 at 325 K. This value is lower by a factor of ∼35 than that obtained for propane in NaY at 300 K (ref. 46) illustrating the effects of both pore diameter, and strength of the adsorbate:adsorbent interaction.
Implications for MTH catalysis
The work presented here together with our earlier spectroscopic study,27 allows us to draw the following inferences on the catalytic materials studied. First it is clear that the MTG reaction leads to dislodgement of lattice Al, and the higher the reaction temperature the more severe the dislodgement as evident from 27Al- and 29Si- NMR. These observations are consistent with previous studies on the susceptibility of the zeolite lattice to damage by steam, which increases with increasing temperature.39,40 In agreement with this finding, N2-physisorption data show significantly increased mesoporosity in ZSM-5-673 while pore plugging is noted in ZSM-5-623 due to differences in the nature of hydrocarbon deposits in the pores.27 This work does not provide any new information on the fate of aluminium dislodged from the lattice, although we note an earlier NMR study suggesting that the extraframework species are aluminosilicate rather than amorphous alumina.50Secondly, we find that methanol in a clean ZSM-5-F catalyst is strongly adsorbed to the point of being immobile on the QENS time scale (∼2–50 ps for the spectrometer used here). This result agrees with earlier reports. However there remains an apparent contradiction between the interpretation of INS measurements, which show complete dissociation of methanol at room temperature26 and infrared47,48 and NMR measurements48,49 which indicate the presence of hydrogen bonded methanol at room temperature. Work is in progress to resolve this problem.
Next we observe that zeolite ZSM-5-623 is indistinguishable from the fresh ZSM-5-F as far as the QENS of adsorbed methanol is concerned, other than a reduced intensity due to the lower methanol uptake/loading. Infrared and INS measurements indicate that the hydrocarbon “coke” species in this zeolite are a mixture of sp2 and sp3 carbons.27 The infrared spectrum also shows retention of ∼50% of the acidic hydroxyl groups in the used catalyst. The reduced pore volume and methanol uptake at room temperature are fully consistent with partial blockage of the internal pores by “coke”, and the behaviour of adsorbed methanol in the remaining (∼70%) porosity is similar to that in the fresh zeolite.
Zeolite ZSM-5-673 contains less “coke” than its 623 K counterpart, and the infrared and INS measurements indicate a higher aromatic content of the hydrocarbon “coke”.27 This catalyst still retains a significant population of acidic hydroxyl groups, but the infrared spectrum also shows an enhanced contribution from AlOH species associated with extra-lattice aluminium.27 Reaction at 673 K is clearly causing damage to the zeolite lattice and generating mesoporosity which not only reduces the pore blockage by coke but also modifies the mobility of adsorbed methanol at low temperatures. Thus, the significant quasielastic broadening for ZSM5-673 is measured at 325 K. The experimental EISF is explained by an isotropic methanol rotation model with a mobile fraction of 0.42 and a rotational diffusion coefficient of DR = 3 × 1010 s−1.
Conclusions
Our study has enhanced understanding of the framework degradation, coke formation and methanol dynamics in ZSM-5 catalysts. The immobilisation of methanol in freshly prepared catalysts found in our earlier study is confirmed. Future work will reconcile and combine these observations with other spectroscopic and catalytic studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The UK Catalysis Hub is thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC (grants EP/I038748/1, EP/I019693/1, EP/K014706/1, EP/K014668/1, EP/K014854/1, EP/K014714/1 and EP/M013219/1). A. J. O. M. would like to acknowledge the Ramsay Trust for provision of the Ramsay Trust Memorial Fellowship. The STFC Rutherford Appleton Laboratory is thanked for access to neutron beam facilities. Johnson Matthey plc is thanked for studentship support via the EPSRC Industrial CASE scheme (A. Z. and A. H.) and for the provision of the ZSM5 catalyst and its characterisation.Notes and references
- F. J. Keil, Microporous Mesoporous Mater., 1999, 29, 49 CrossRef.
- M. Stocker, Microporous Mesoporous Mater., 1999, 29, 3 CrossRef.
- C. D. Chang, Catal. Today, 1992, 13, 103 CrossRef.
- U. Olsbye, S. Svelle, K. P. Lillerud, Z. H. Wei, Y. Y. Chen, J. F. Li, J. G. Wang and W. B. Fan, Chem. Soc. Rev., 2015, 44, 7155 RSC.
- G. H. Hutchings and R. Hunter, Catal. Today, 1990, 6, 279 CrossRef.
- S. D. Hellring, K. D. Schmitt and C. D. Chang, J. Chem. Soc., Chem. Commun., 1987, 1320 RSC.
- P. Salvador and W. Kladnig, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 1153 RSC.
- L. Kubelkova, J. Novakova and K. Nedomova, J. Catal., 1990, 124, 441 CrossRef.
- J. Li, Y. Chen, B. Jing, Y. He, M. Dong, H. Jiao, X. Li, Z. Qin, J. Wang and W. Fan, J. Catal., 2014, 317, 277 CrossRef.
- Y. Liu, S. Mueller, D. Berger, J. Jelic, K. Reuter, M. Tonigold, M. Sanchez-Sanchez and J. Lercher, Angew. Chem., Int. Ed., 2016, 55, 5723 CrossRef PubMed.
- I. M. Dahl and S. Kolboe, Catal. Lett., 1993, 20, 329 CrossRef.
- S. Svelle, J. Joensen, J. Nerlov, U. Olsbye, K. P. Lillerud, S. Kolboe and M. Bjorgen, J. Am. Chem. Soc., 2006, 128, 14770 CrossRef PubMed.
- M. Bjorgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195 CrossRef.
- U. Olsbye, S. Svelle, M. Bjorgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810 CrossRef PubMed.
- S. Ilias and A. Bhan, ACS Catal., 2013, 3, 18 CrossRef.
- D. M. Bibby, R. F. Howe and G. D. McLellan, Appl. Catal., A, 1992, 93, 1 CrossRef.
- S. Mueller, Y. Liu, M. Vishnuvarthan, X. Sun, A. C. Van Veen, G. L. Haller, M. Sanchez-Sanchez and J. Lercher, J. Catal., 2015, 325, 48 CrossRef.
- A. Zhokh and P. Strizhak, J. Chem. Phys., 2017, 146, 124704 CrossRef PubMed.
- K. K. Pitale, R. A. Rajadhyaksha, A. S. Pendharkar and A. C. Eapen, Curr. Sci., 1988, 57, 355 Search PubMed.
- H. Jobic, A. Renouprez, M. Bec and C. Poinsignon, J. Phys. Chem., 1986, 90, 1059 CrossRef.
- N. M. Gupta, D. Kumar, V. S. Kamble, S. Mitra, R. Mukhopadhyay and V. B. Kartha, J. Phys. Chem. B, 2006, 110, 4815 CrossRef PubMed.
- A. J. O'Malley, S. F. Parker, A. Chutia, M. R. Farrow, I. P. Silverwood, V. Garcia-Sakai and C. R. A. Catlow, Chem. Commun., 2016, 52, 2897 RSC.
- A. J. O'Malley, S. F. Parker and C. R. A. Catlow, Chem. Commun., 2017, 53, 12164 RSC.
- A. J. O'Malley, I. Hitchcock, M. Sarwar, I. P. Silverwood, S. Hindocha, C. R. A. Catlow, A. P. E. York and P. J. Collier, Phys. Chem. Chem. Phys., 2016, 18, 17159 RSC.
- M. E. Potter, A. J. O'Malley, S. Chapman, J. Kezina, S. H. Newland, I. P. Silverwood, S. Mukhopadhyay, M. Carravetta, T. M. Mezza, S. F. Parker, C. R. A. Catlow and R. Raja, ACS Catal., 2017, 7, 2926 CrossRef.
- M. E. Potter, S. Chapman, A. J. O'Malley, A. Levy, M. Carravetta, T. M. Mezza, S. F. Parker and R. Raja, ChemCatChem, 2017, 9, 1897 CrossRef.
- Suwardiyanto, R. F. Howe, E. K. Gibson, C. R. A. Catlow, A. Hameed, J. McGregor, P. Collier, S. F. Parker and D. Lennon, Faraday Discuss., 2017, 197, 447 RSC.
- M. T. Telling and K. H. Andersen, Phys. Chem. Chem. Phys., 2005, 7, 1255 RSC.
- R. T. Azuah, L. R. Kneller, Y. Qiu, P. L. Tregenna-Piggot, C. M. Brown, J. R. Copley and R. M. Dimeo, J. Res. Natl. Inst. Stand. Technol., 2000, 114, 341 CrossRef.
- A. Sayari, E. Crusson, S. Kaliaguine and J. R. Brown, Langmuir, 1991, 7, 314 CrossRef.
- D. M. Bibby, N. B. Milestone, J. E. Patterson and L. P. Aldridge, J. Catal., 1986, 97, 493 CrossRef.
- J. Jiao, W. Wang, B. Sulikowski, J. Weitkamp and M. Hunger, Microporous Mesoporous Mater., 2006, 90, 246 CrossRef.
- R. H. Meinhold and D. M. Bibby, Zeolites, 1990, 10, 146 CrossRef.
- L. Zhu, S. Yin, X. Wang, Y. Liu and S. Wong, RSC Adv., 2016, 6, 82515 RSC.
- S. M. Campbell, D. M. Bibby, J. M. Coddington and R. F. Howe, J. Catal., 1996, 161, 350 CrossRef.
- E. Brunner, H. Ernst, D. Freude, T. Frohlich, M. Hunger and H. Pfeifer, J. Catal., 1991, 127, 34 CrossRef.
- C. A. Fyfe, Y. Feng, H. Grondey, G. T. Kokatailo and H. Giess, Chem. Rev., 1991, 91, 1525 CrossRef.
- K. Barbera, S. Sorensen, S. Bordiga, J. Skibsted, H. Fordsmand, P. Beato and T. V. W. Janssen, Catal. Sci. Technol., 2012, 2, 1196 Search PubMed.
- S. M. Campbell, D. M. Bibby, J. M. Coddington, R. F. Howe and R. H. Meinhold, J. Catal., 1996, 161, 338 CrossRef.
- L. H. Ong, M. Doe, R. Okado, A. C. van Veen and J. A. Lercher, Microporous Mesoporous Mater., 2012, 164, 9 CrossRef.
- A. J. O'Malley, V. G. Sakai, I. P. Silverwood, N. Dimitratos, S. F. Parker and C. R. A. Catlow, Phys. Chem. Chem. Phys., 2016, 18, 17294 RSC.
- V. F. Sears, Can. J. Phys., 1966, 44, 1279 CrossRef.
- F. Volino and A. J. Dianoux, Mol. Phys., 1980, 41, 271 CrossRef.
- A. J. O'Malley and C. R. A. Catlow, in Experimental Methods in the Physical Sciences, Academic Press, 2017, vol. 49, pp. 349–401 Search PubMed.
- M. Bee, Quasielastic neutron scattering: principles and applications in solid state chemistry, Biology and Materials Science, Adam Hilger, Bristol, 1988 Search PubMed.
- R. Mukhopadhyay, A. Sayeed, S. Mitra, A. A. Kumar, M. N. Rao, S. Yashonath and S. L. Chaplot, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2002, 66(6), 061201 CrossRef PubMed.
- A. Zecchina, S. Bordiga, G. Spoto, D. Scarano, G. Spano and F. Geobaldo, J. Chem. Soc., Faraday Trans., 1996, 92, 4863 RSC.
- S. M. Campbell, X. Z. Jian and R. F. Howe, Microporous Mesoporous Mater., 1999, 29, 91 CrossRef.
- M. W. Anderson and J. Klinowski, J. Am. Chem. Soc., 1990, 112, 10 CrossRef.
- Y. Long, M. Jin, Y. Suh, T. Wu, L. Wang and L. Fei, J. Chem. Soc., Faraday Trans., 1996, 92, 1647 RSC.
| This journal is © The Royal Society of Chemistry 2018 |